 Dongshan Wan
Dongshan Wan Wei Jiang
Wei Jiang Junwei Hao
Junwei Hao- 1Department of Neurology, Xuanwu Hospital, Capital Medical University, Beijing, China
- 2Department of Neurology, Tianjin Neurological Institute, Tianjin Medical University General Hospital, Tianjin, China
Double-stranded DNA (dsDNA) sensor cyclic-GMP-AMP synthase (cGAS) along with the downstream stimulator of interferon genes (STING) acting as essential immune-surveillance mediators have become hot topics of research. The intrinsic function of the cGAS-STING pathway facilitates type-I interferon (IFN) inflammatory signaling responses and other cellular processes such as autophagy, cell survival, senescence. cGAS-STING pathway interplays with other innate immune pathways, by which it participates in regulating infection, inflammatory disease, and cancer. The therapeutic approaches targeting this pathway show promise for future translation into clinical applications. Here, we present a review of the important previous works and recent advances regarding the cGAS-STING pathway, and provide a comprehensive understanding of the modulatory pattern of the cGAS-STING pathway under multifarious pathologic states.
Introduction
Pattern-recognition receptors (PRRs) serve as innate cellular sensors of danger signals, such as pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs), and yield cellular-stress response. DNA molecules are vital genetic components within cells, which are compartmentalized restrictively into specific regions. The occasionally misplaced DNA is degraded rapidly by scavenger cells and extracellular or intracellular ribonucleases. Aberrant accumulation of DNA is relevant to tissue damage (1).
In 2008, several research teams discovered a new protein on the endoplasmic reticulum (ER) which can be activated by immune-stimulatory DNA (ISD) and initiate type-I interferon (IFN) responses, which was named “stimulator of interferon genes” (STING, also known as MITA, ERIS) (2–4). STING does not bind to DNA directly, and bacteria-derived cyclic di-guanylate monophosphate (c-dGMP) or cyclic di-adenosine monophosphate (c-dAMP) were confirmed to be ligands for STING (5, 6). Subsequently, it was found that some DNA sensors can facilitate STING activation, such as interferon gamma inducible protein 16 (IFI16) (7). However, STING activation could not be fully explained by the upstream factors/ligands that had been found. It was postulated that an unknown upstream regulator might be responsible for STING activation.
In 2013, Wu and Sun found that cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) was a novel secondary messenger serving as a ligand of STING (8). Beside it, they purified a new protein named “cyclic-GMP-AMP synthase” (cGAS) that had cytosolic DNA-sensing ability and can synthesize cGAMP (8). Also, they found that the cGAS-cGAMP-STING pathway was indispensable for host anti-viral immunity (9). Their work filled in the gaps missing from upstream of STING.
Stimulator of interferon genes or cyclic-GMP-AMP synthase is expressed widely in a broad spectrum of cells including immune, non-immune, cancer cells (10). Mounting evidence has demonstrated that the cGAS-STING pathway is important for mediating cellular immune sensing, and shows particular responses pattern to the ISD distinguished from other nucleotide-sensing pathways. It is also regulated delicately by several molecules or feedback loops to maintain cellular homeostasis. Nevertheless, cGAS-cGAMP-STING pathway itself has distinctive or even opposing effects under different conditions.
In this review, we cover the roles of cGAS-STING pathway in cellular type-I IFN immune response, and several cellular processes including autophagy, survival and senescence. We also summarize the literature on intrinsic cellular mechanisms modulating cGAS-STING pathway as well as its cross-regulations with other DNA-sensing pathways. Moreover, the inflammation-modulation capacities of this pathway in infectious disease, inflammation and cancers have been elucidated too, and a pervasive pattern of this pathway has been described, which could provide a plausible explanation of the contradictory findings of studies. Finally, current or prospective therapeutic strategies targeting the pathway, and issues that need to be addressed in the future, are discussed.
cGas Recognizes Cytosolic DNA and Produces cGamp
Cyclic-GMP-AMP synthase belongs to the structurally conserved cGAS/DncV-like nucleotidyltransferase (CD-Ntases) superfamily. The latter is expressed universally in prokaryotes and eukaryotes, and can use purines or pyrimidines selectively as substrates for the production of linear or cyclic di- or even tri-nucleotide compounds, which act as secondary intracellular messengers (11).
Cyclic-GMP-AMP synthase is distributed mainly in the cytosol (also nucleus in some specific conditions) (8). Generally speaking, cGAS is activated upon the recognition of B-type double-stranded DNA (dsDNA) without sequence-specificity but not A-type dsDNA or RNA (12, 13). Hybrid DNA:RNA or stem-like single-stranded DNA (ssDNA) are also low-affinity ligands for cGAS (14, 15). After binding with ligands, cGAS undergoes an allosteric structural change, and subsequently catalyzes its substrates guanosine triphosphate (GTP) and adenosine triphosphate (ATP) to produce a mixed phosphodiester-linked cyclic dinucleotide: G(2′–5′)pA(3′–5′)p cGAMP (abbreviated as 2′,3′-cGAMP or cGAMP) (16). cGAS also catalyze the synthesis of linear dinucleotides such as AMP-2′-ATP, GMP-2′-GTP, and AMP-2′-GTP as intermediate products (17).
There are two major dsDNA-binding sites on opposite sides of the catalytic pocket: A and B site. Site A is the primary contact surface for dsDNA, whereas site B is complementary, binding another dsDNA. It allows for cGAS to the formation of a 2:2 cGAS:dsDNA complex structure directed into two orientations with dsDNA at least 20 bp (18–20) (Figure 1A). Increased numbers of back-to-back dimers of cGAS hold the two dsDNA molecules together and permit successive recruitment of cGAS which, consequently, forms a 2n:2 cGAS:dsDNA higher-ordered “ladder-like” oligomerization, with cGAS arrayed “head to head/tail to tail” (19, 21). The DNA-binding protein HU, mitochondrial transcription factor A (TFAM), or bacterial high mobility group box 1 protein (HMGB1) can bend the dsDNA into a U-shaped structure and, thus, facilitate binding of cGAS dimers to the same strand as it travels in opposite directions (21) (Figure 1B). Human cGAS, unlike mouse cGAS, is prone to formation of this ladder-like network with long dsDNA, because of the human-specific residues K187 and L195. These two dsDNA-interfacing residues of site A loosen the interaction of dsDNA with cGAS, leading to dsDNA curving and allowing more convenient binding for the next adjacent cGAS (20, 21) (Figure 1C). Finally, accumulated cGAS-dsDNA complexes can go through a liquid-phase separation and condense into gel-like droplets as a reaction unit (Figure 1D). This conformation requires a sufficiently long dsDNA strand to form multivalent interaction positions, also requires the function of the N-terminal tail of cGAS and a recently discovered dsDNA-binding site in the catalytic domain of cGAS (site C) (22, 23). Meanwhile, the N-terminal tail of cGAS mediates cGAS localization onto the membrane by binding to phosphatidylinositol 4,5-bisphosphate (PI (4, 5) P2) and prevents liberation of cGAS and oligomerization, but can release cGAS during cell stress (24). The structure of cGAS determines long strand dsDNA (>500–1,000 bp) could potentially stimulate the enzyme activity and cGAMP production of cGAS (25). The ability of human cGAS to discriminate long dsDNA strands from shorter dsDNA may contribute to the specific sensing and recognition of the “danger DNA” of pathogens, necrotic cells or cancer cells rather than irrelevant shorter dsDNA, thereby enhancing the immunity against them specifically.
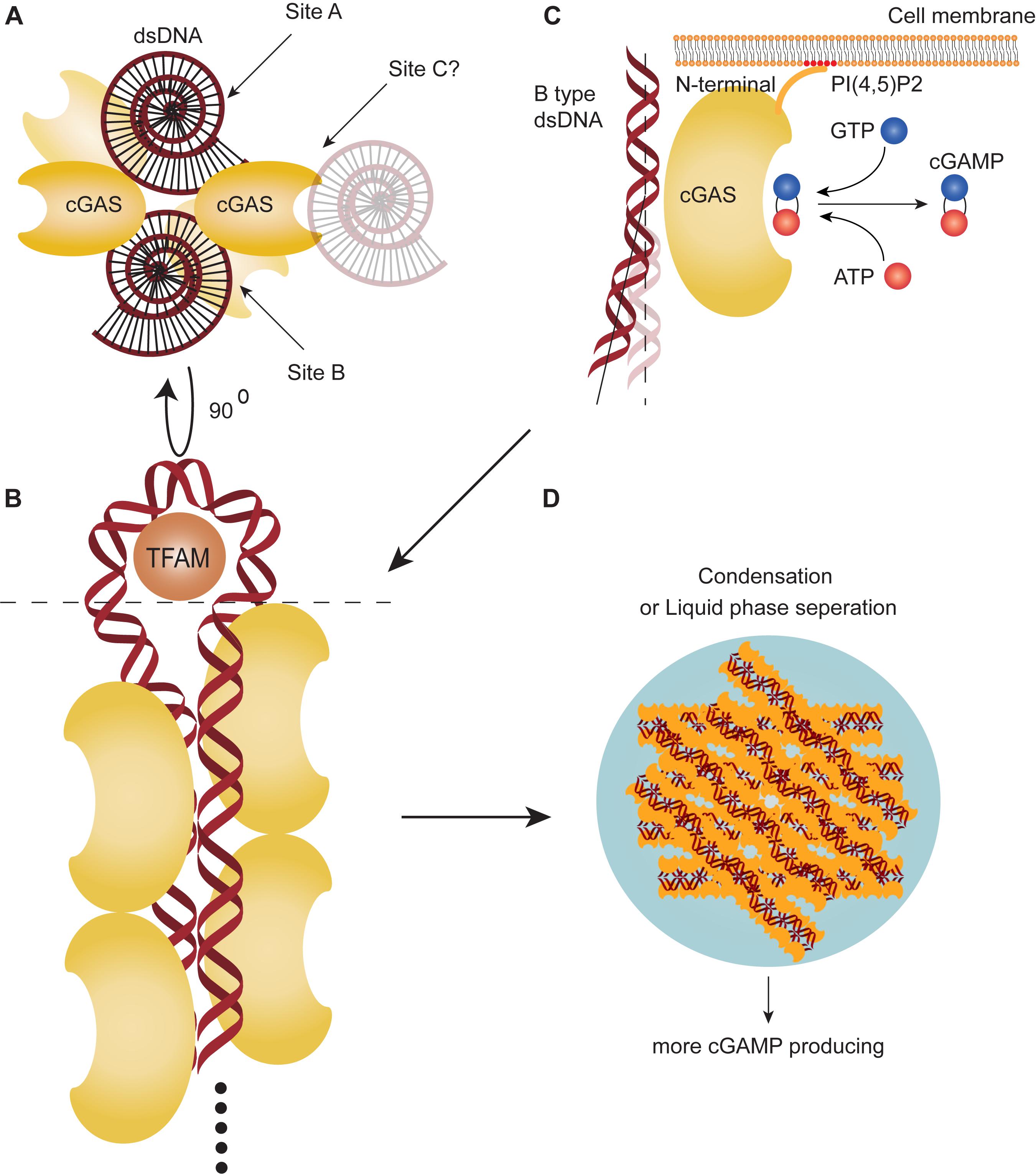
Figure 1. cGAS can recognize cytosolic DNA and produce cGAMP. (A) There are two DNA-binding sites on opposite sides of the catalytic pocket (site A,B) and a proposed DNA-binding site at the catalytic domain of cyclic-GMP-AMP synthesis (cGAS) (site C). (B) Multiple cGAS molecules can bind two double-stranded DNAs (dsDNA) to form a 2n:2 cGAS:dsDNA higher-ordered “ladder-like” oligomerization. Mitochondrial transcription factor A (TFAM) can bend the dsDNA into a U-shaped structure and promote polymerization. (C) cGAS can recognize B-type dsDNA. In humans, the cGAS DNA-interfacing residue of site A loosens the interaction of dsDNA to curve dsDNA away for more convenient binding with next adjacent cGAS. cGAS can catalyze GTP and ATP to synthesize cyclic guanosine monophosphate-adenosine monophosphate (cGAMP). The N-terminal tail binds to the cell membrane, associating with phosphatidylinositol 4,5-bisphosphate [PI (4,5)P2]. (D) Accumulation of cGAS-DNA complex goes through a liquid-phase separation and condenses into gel-like droplets.
Activation of The cGas-Sting Pathway
Double-stranded DNA is restricted into the nucleus or mitochondria and is rarely present in the cytoplasm. Extrinsic dsDNA from pathogens such as viruses, bacteria, transcellular vesicles or rupture of dying cells can be internalized into the cytosol in several diverse ways (26–28). These extrinsic dsDNA sources are engulfed by endosome through phagocytosis and digested immediately by DNaseII when fusing with lysosomes (29, 30). However, some escaping mechanisms under certain conditions could help protect them from being degraded. For example, antimicrobial peptide LL37 could efficiently transports self-DNA from endosome into cytosol of monocytes (28). Cell oxidative stress can lead to phagosomal acidification delay and probably release endosome context including dsDNA owing to increased membrane permeability (27, 31).
The intrinsic self-dsDNA can also be segregated inaccurately and released into the cytosol (32, 33). For example, genomic DNA (gDNA) injury as a result of genotoxic stress and DNA self-instability or replication errors leads to double-strand breaks (DSBs) and can be repaired by several ways (34). Impaired mediators of DNA-damage repair response mediators, such as ataxia telangiectasia mutated (ATM)-RAD3, poly ADP-ribose polymerase (PARP) and breast cancer1/2 (BRCA1/2) are associated with persisting DSBs and accumulation of cytosolic DNA (35–37). Extra-nuclear micronuclei formation during mitosis is a source of cytosolic dsDNA caused by DSBs (32, 38). Followed by homologous recombination repair of collapsed replication forks, DNA cleavage by methyl methanesulphonate (MMS) and ultraviolet-sensitive 81 (MUS81) also lead to cytosolic dsDNA presenting (39). Furthermore, manually Cre/loxP recombination technology can induce dsDNA damage during DNA cleavage, which results in the accumulation of cytoplasmic dsDNA (40). In normal cellular mitotic processes, chromosomal DNA can be exposed to the cytoplasm, while it is hard to bind and trigger cGAS (41).
In addition, mitochondrial DNA (mtDNA) is also a considerable ligand of cGAS and can be released into the cytosol under mitochondrial stress or dysfunction of proteins which participates in maintaining mitochondrial operations (33, 42, 43) (Figure 2A).
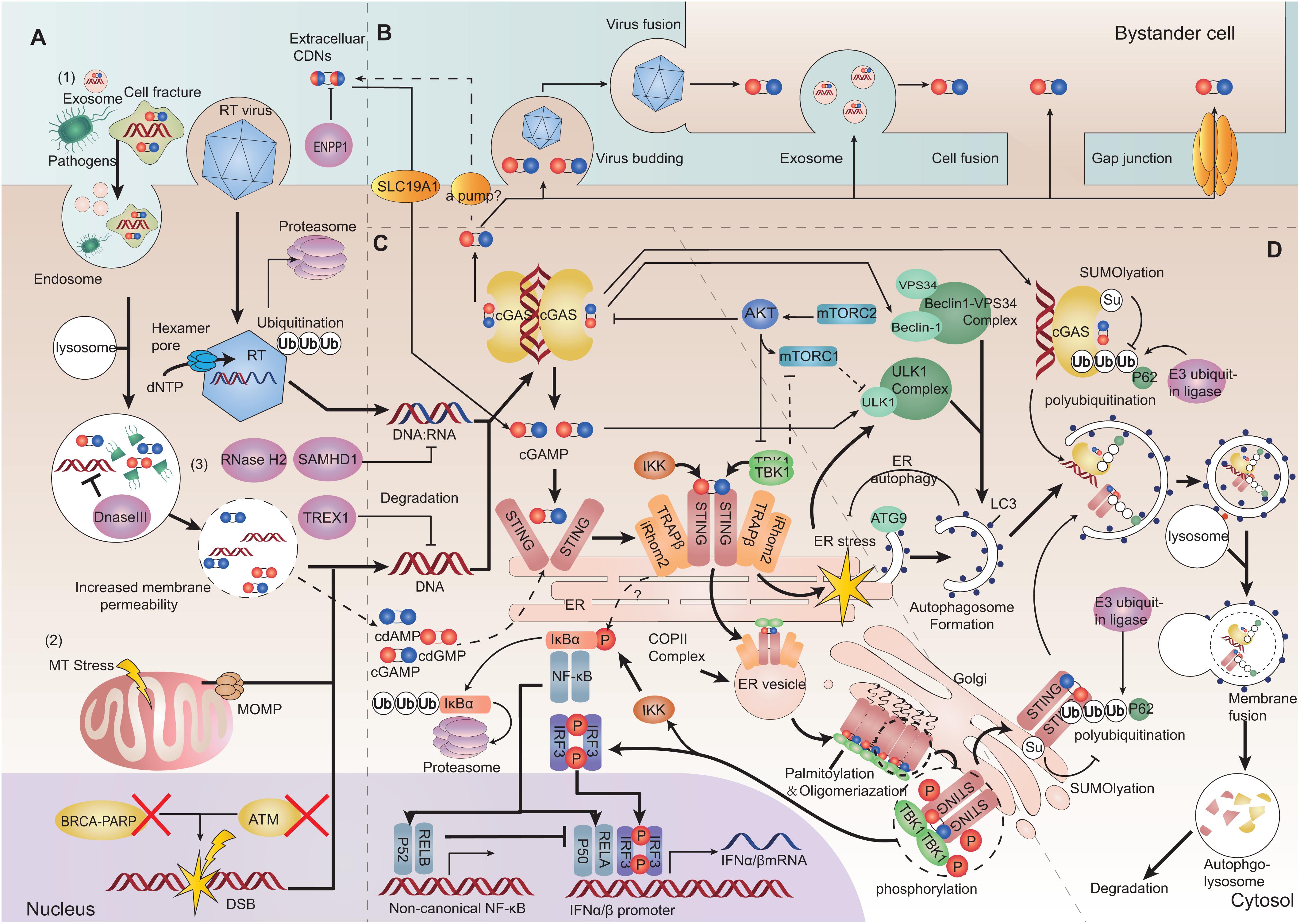
Figure 2. Depiction of the cGAS-STING pathway. (A) Cytoplasmic DNA challenge. (1) Extrinsic DNA source: extracellular DNA can be taken up into endosomes for digestion. Increased membrane permeability allows endolysosomal pathogenic DNA release into the cytosol. Viruses can release virons into the cytosol. The hexamer pore facilitates movement of nucleotides into the capsid. The latter can also be ubiquitinated and degraded. (2) Intrinsic DNA source: self-DNA from the nucleus with dysfunctional chromatin proteins can lead to chromatin DNA injury and gDNA presenting in the cytoplasm. Mitochondrial outer membrane permeabilization (MOMP) induced by mitochondrial stress can release oxidized mitochondrial DNA (mtDNA). (3) Three-prime repair exonuclease 1 (TREX1), DNase II, SAM domain, HD domain-containing protein 1 (SAMHD1) and RNaseH2 can restrict cytosolic DNA and DNA/RNA hybrids. cGAMP can be hydrolyzed by ectonucleotide pyrophosphatase/phosphodiesterase family member 1 (ENPP1). (B) cGAMP transferring between cells. cGAMP can be transferred between cells through viruses, membrane fusion, extracellular vesicles and cell gap junctions. Extracellular cyclic dinucleotides (CDNs) can transfer directly into cells by SLC19A1. CDNs (including cGAMP) might be exported by some ways. (C) Inflammatory signaling mediated by the cGAS-STING pathway. After sensing DNA, cGAS produces cGAMP and extracellular CDNs, promoting stimulator of interferon genes (STING) to undergo dimerization. STING can exit from the endoplasmic reticulum (ER), and be translocated from the ER to the ER-Golgi vesicle, and arrives at the Golgi. STING and TANK binding kinase 1 (TBK1) can be oligomerized and cluster at the Golgi. The STING-TBK1/IκB kinase (IKK) signalosome forms a scaffold to phosphorylate interferon regulatory factor 3 (IRF3) and inhibitor of NF-κBα (IκBα). Then, dimerized IRF3 and the activated canonical NF-κB p50/p65 complex can be translocated into the nucleus as transcription factors to promote transcription of type-I IFN. (D) Autophagy initiation and degradation. STING activation on ER triggers ER stress and mechanistic target of rapamycin complex1 (mTORC1) dysfunction. ER stress and mTORC1 dysfunction can stimulate the Unc-51 like autophagy activating kinase (ULK1) complex and beclin1-phosphatidylinositol 3-kinase catalytic subunit type 3 (PI3KC3) complex. Autophagy-related protein 9 (ATG9) and light chain 3 (LC3) are associated with genesis and elongation of the autophagosome. After autophagy initiation, cGAS-STING is ubiquitinated and binds with p62. Then, they are packaged into autophagosomes and terminally sorted to lysosomes (Bold arrows represent main signaling pathways, thinner arrows represent regulatory signaling pathways, and dashed arrows represent bypass or suspicious pathways).
Cells have several types of nucleases to restrict cytosolic DNA to avoid cGAS activation. For example, three-prime repair exonuclease 1 (TREX1 also known as DNaseIII) is a cytosolic DNA exonuclease which removes unprotected dsDNA from the cytosol (44). RNaseH2 locates to the nucleus and specifically degrades the RNA in RNA:DNA hybrids participating in DNA replication (45). DNaseII is a lysosomal DNase which degrades undigested DNA in endosomes or autophagosomes to prevent their entry into the cytoplasm (30). SAM domain and HD domain-containing protein 1 (SAMHD1) is characterized as a dNTPase and restricts reverse transcription of the RNA virus (46, 47). SAMHD1 can also stimulate the exonuclease (but not the endonuclease) activity of MRE11 to degrade nascent ssDNA, and start DNA-repair responses at stalled replication forks (48). Depletion of SAMHD1 leads to the cleaving of nascent ssDNA by the activity of MRE11 endonuclease and cytosolic translocation of gDNA (48). Deficiency of any of these nucleases can lead to accumulation of self-DNA in the cytoplasm, thereby activating the cGAS-STING pathway against DNA molecules (30) (Figure 2A).
cGamp Can Transfer Between Cells and Activate Sting
The production of asymmetrically linked 2′,3′-cGAMP catalyzed by cGAS has the highest affinity for STING to promotes STING dimerization (49, 50). cGAMP as a second messenger can be also transferred among cells in several ways to pass danger signaling of cytosolic DNA. Intercellular gap junction consists of two docking hexamer channels formed by different connexins, which allows many small molecules, including cGAMP, to pass bi-directionally through cells. And intercellular transfer of cGAMP through gap junction is largely dependent on connexin 43 (51–53). Additionally, cGAMP can be packaged into virons and pre-notify newly infected cells (54, 55). Cell fusion is a distinct manner for intracellular transmission of the human immunodeficiency virus (HIV); cGAMP also enter membrane-fused bystander cells in this way (56). Extracellular vesicles such as exosomes can contain cGAMP along with viral DNA, host gDNA or mtDNA, and mediate cells communication (57, 58). There were no evidences that cGAMP could be pumped out to extracellular space by a channel/transporter. However, it was found that SLC19A1 can transmit cyclic dinucleotides (CDNs) into cell plasma (59, 60). Notably, ectonucleotide pyrophosphatase/phosphodiesterase family member 1 (ENPP-1) can degrade extracellular cGAMP (61) (Figure 2B). Besides triggering STING, these exogenous cGAMP can directly bind to cGAS and prompt its activation as well (62).
After binding to cGAMP, the “lid” region of the STING dimer undergoes a conformational change that converts STING from an inactive “open” formation to an active “closed” formation. Following that, the STING dimer translocates from the ER to perinuclear ER-Golgi intermediate compartment (ERGIC) vesicles, finally arriving at the Golgi to form punctuate structures with downstream molecules (2, 63). ER-retention of STING caused by mutations results in reduced IFN signaling (64, 65). The translocon-associated protein β (TRAPβ) recruited by inactive rhomboid protein 2 (iRhom2) initially forms the TRAP translocon complex that mediates STING exit from the ER (2, 66). They both assist cytoplasmic coat protein complex-II (COPII) to drive ER-vesicle formation and carry the STING complex to the Golgi (67, 68).
Trafficking STING can bind directly to and be phosphorylated by TANK binding kinase 1 (TBK1) dimer or IκB kinase (IKK) complex (3, 69, 70). The C-terminal tail (CTT) of STING is a linear unfolded segment, which determines the optimization of combination specificity. STING CTT in mammals tends to bind TBK1, whereas in fish it tends to activate nuclear factor-kappaB (NF-κB) signal (71). The STING phosphorylation site Ser366 in the CTT cannot reach the kinase-domain active site of its directly bound TBK1, instead can reach the kinase-domain active site of the next adjacent TBK1 binding with another STING and be phosphorylated, while TBK1 phosphorylate each other (72, 73). Hence, STING and TBK1 can aggregate on the Golgi to form the STING signalosome. Clustering STING undergoes palmitoylation and full activation (74). It is also possible for STING-IKK to cluster and form the STING signalosome in this manner. The STING-TBK1/IKK signalosome produces a scaffold to phosphorylate interferon regulatory factor 3 (IRF3) or inhibitor of NF-κBα (IκBα). Activated IRF3 undergo dimerization (70). The activation of IκBα leads to its polyubiquitination and degradation by the proteosome, thereby eliminating its inhibition of NF-κB. There is also evidence suggesting that NF-κB activation might not require STING trafficking from the ER (75). Then, the dimerized IRF3 or activated NF-κB p50/p65 (p65 is also known as RelA) complex are translocated into the nucleus as transcription factors and bind to the promoter of type-I IFN to aid the transcription of type-I IFN (2, 3, 70). Meanwhile, activation of NF-κB p52/RelB can prevent recruitment of p65 and inhibit the p50/p65 signal (76) (Figure 2C).
Sting Downstream Signaling Promotes Ifnα/β Expression and Autophagy
Expressed type-I IFN can propagate among cells in paracrine or autocrine manners. The binding of IFNα/β with its receptor triggers janus kinase (JAK) and signal transducer and activator of transcription (STAT) pathways, then induce transcription of type-I IFN-stimulated genes (ISGs), which have IFN-sensitive response elements (ISREs) in their 5′-untranslated regions (UTRs) (77). IRF3 can also bind partially to several ISREs alone (78). Herein, the expression of some ISGs including interferon-induced protein with tetratricopeptide repeats (IFIT) and pro-inflammatory cytokines such as tumor necrosis factor α (TNFα), interleukin (IL)-6, C-X-C motif chemokine ligand 10 (CXCL10) and C-C motif chemokine ligand 5 (CCL5) is increased remarkably by the cGAS-STING pathway (79). Furthermore, cGAS and STING are both ISGs, suggesting a positive feedback loop in spreading of the IFN signal (80, 81).
Stimulator of interferon genes activation on the ER also triggers an ER stress response with an “unfolded protein response (UPR) motif” on the C-terminus of STING, which leads to and ER stress-mediated autophagy (82, 83). STING-TBK1 activation and ER stress also induce mechanistic target of rapamycin complex 1 (mTORC1) dysfunction (84). ER stress or reduced mTORC1 signaling activates Unc-51-likeautophagy activating kinase (ULK1) complex and the Beclin-1-class III phosphatylinositol 3-kinase (PI3KC3 also known as VPS34) complex, which promotes initiation of the classical autophagy path (85). cGAS can also interact directly with the autophagy protein beclin-1-PI3KC3 complex and trigger autophagy (86). Furthermore, cGAS-dsDNA polymer can form a liquid-phase condensate (as mentioned above), which could theoretically be an initiator of autophagy (87). After autophagy initiation, autophagy-related protein 9 (ATG9) undertakes the genesis of the autophagosome along with light chain3 (LC3) undergoing lipidation, thereby resulting in elongation of the autophagosome (88). LC3 can also be recruited directly by ERGIC-loading STING and bypass the classical autophagy pathway (68, 89).
cGAS-STING-mediated autophagy can spread to the whole cell and help the elimination of intracellular microorganisms, subcellular organelles or misfolded proteins, as well as the ER itself that loads the STING signalosome (90–92) (Figure 2D). cGAS-STING-mediated autophagy is also indispensable for removing cytosolic DNA and inflammatory signaling factors to restrict the inflammatory response raised by the pathway itself (93). Excessive signaling of the autophagy cascade can lead to irreversible apoptosis termed “autophagic cell death” (94). Consequently, oligomerized cGAS or STING undergoes ubiquitination and is packaged into autophagosomes with the help of p62, to be terminally sorted into lysosomes (79, 83, 95, 96). cGAS or STING is digested immediately in the autophagolysosome after transient activation of downstream signaling (68, 79, 83, 89). Autophagy functions as a negative feedback loop which ensures transient cGAS-STING signaling and avoids consistent over-reaction of the pathway. Thus, impairment of autophagy may give rise to destructive inflammatory diseases (31).
Regulation of the cGas-Sting Pathway
We cataloged factors in the literature that could potentially up- or down-regulate expression of cGAS/cGAMP/STING in pre-translational or post-translational stages (Tables 1, 2). The regulatory mechanisms of TBK1, IRF, and NF-κB in signaling pathways associated with expression of type-I IFN are outside the scope of this review.
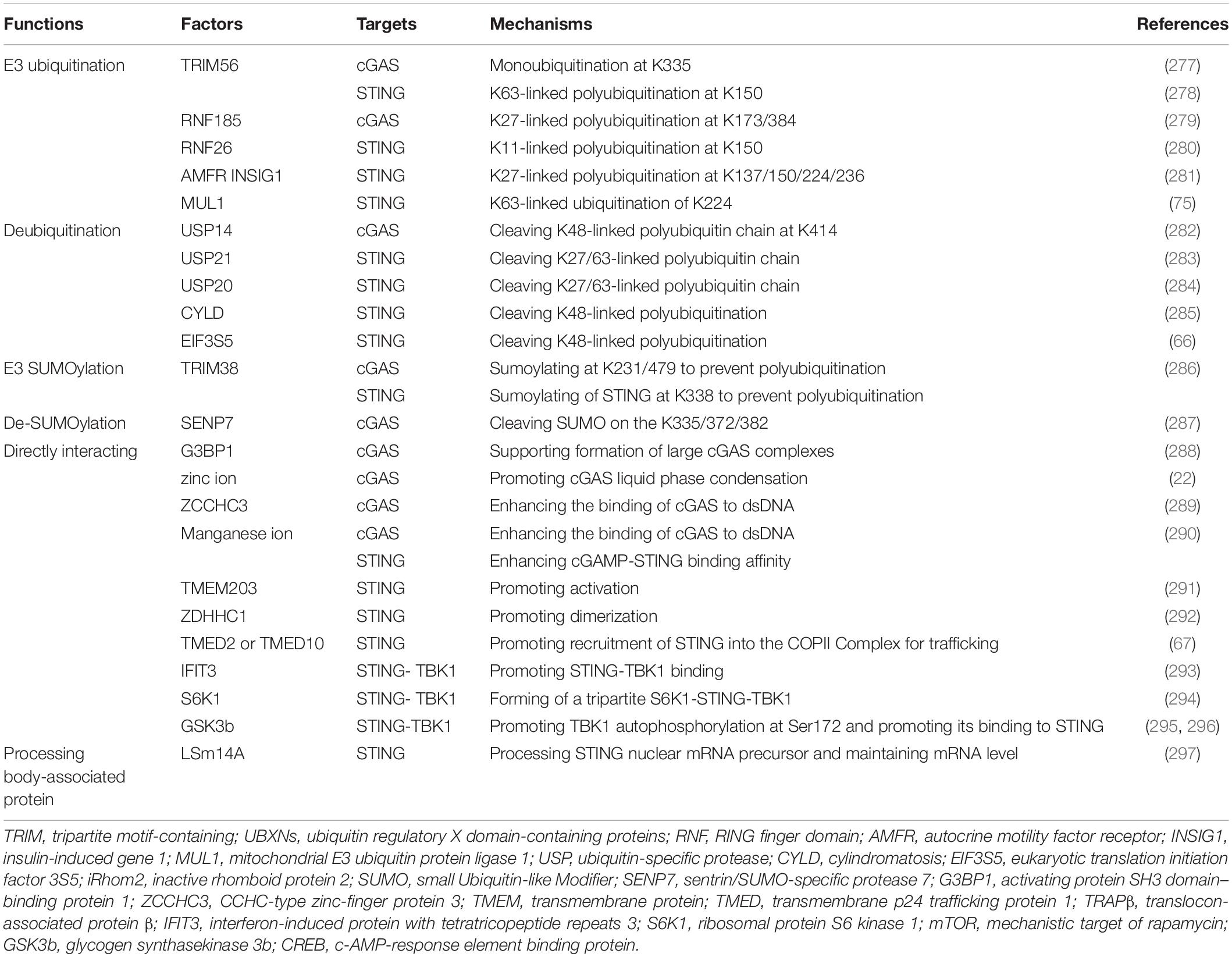
Table 1. Factors promoting cGAS-STING pathway.
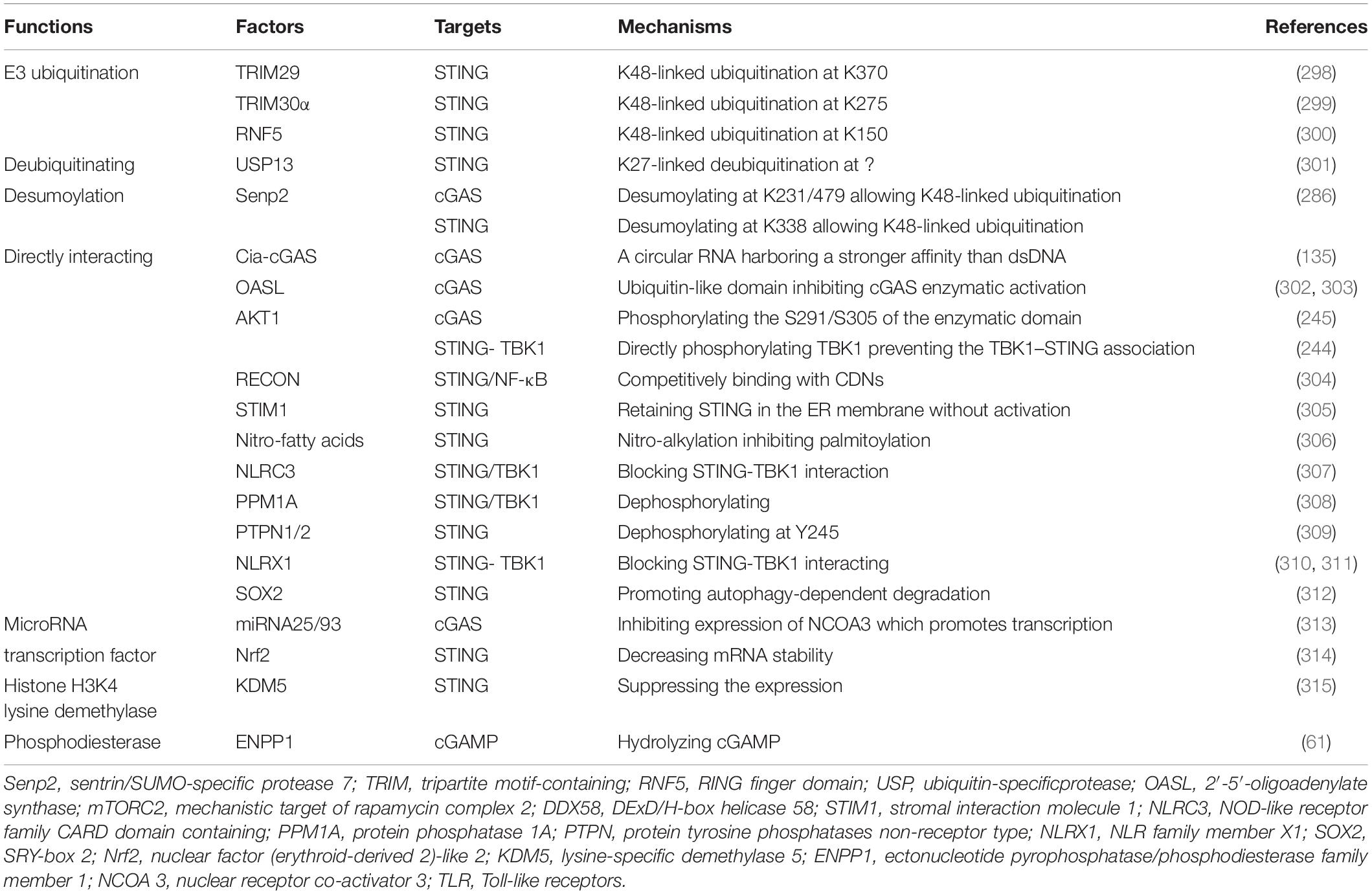
Table 2. Factors inhibiting cGAS-STING pathway.
Cross-Regulation of the cGas-Sting Pathway With Other DNA-Sensing Pathways
PYHIN family member absent in melanoma 2 (AIM2) is a cytoplasmic dsDNA sensor. It can recruit apoptosis-associated speck-like protein containing a CARD (ASC) by its PYHIN domain and form the AIM2 inflammasome. The inflammasome activates caspase-1, which activates IL-1 and trigger pyroptosis (97). The AIM2 pathway could counteract the cGAS-STING pathway (98). First, cGAS is a target for caspase-1 cleavage (99). Second, gasdermin D activated by caspase-1 can lead to potassium ion (K+) efflux which inhibits cGAS (100). Conversely, the cGAS-STING pathway can trigger the AIM2 inflammasome or NLR family pyrin domain containing 3 (NLRP3) by several means, and the process lags behind canonical IFN signaling (96, 101). In this way, the inhibitory nucleic-acid sensor NLR family CARD domain containing 3 (NLRC3) can counteract STING by binding and occupying it, but viral DNA as a possible NLRC3 ligand can reverse its occupation of STING (102) (Figure 3A).
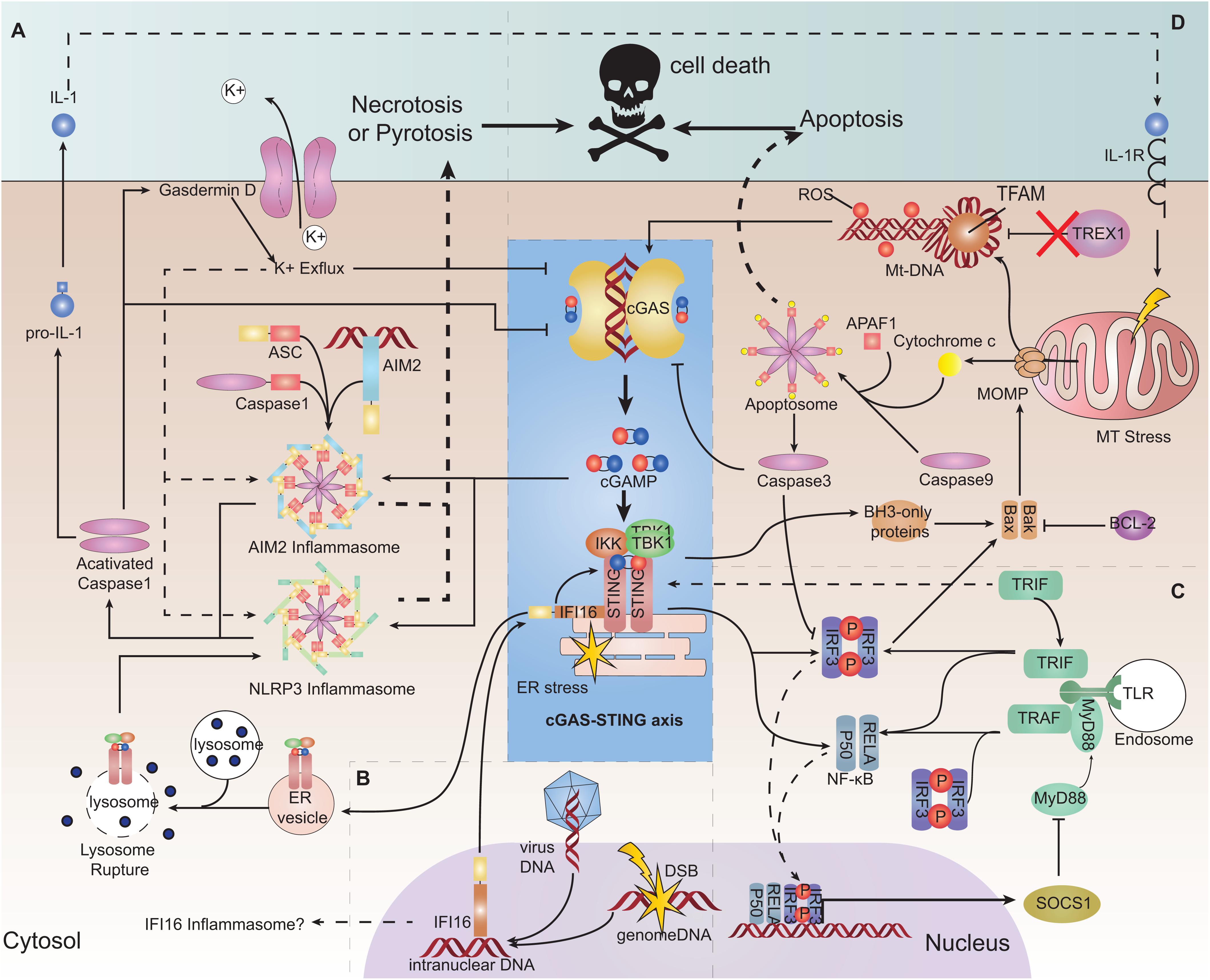
Figure 3. Interaction of the cGAS-STING pathway with other DNA-sensing pathways and its role in cell survival. (A) Absent in melanoma2 (AIM2) pathway and pyroptosis and necroptosis. AIM2 can be triggered by cGAMP and form an inflammasome, consequently triggering interleukin (IL)-1 production and pyroptosis. Stimulator of interferon genes (STING) trafficking to the lysosome ruptures the lysosome membrane, resulting in K+ efflux and activation of the NLRP3 inflammasome, leading to pyroptosis. Cyclic-GMP-AMP synthase (cGAS) and interferon regulatory factor 3 (IRF3) can be a target for caspase-1 cleaving. Gasdermin D can lead to K+ efflux and inhibition of cGAS. (B) Interferon gamma inducible protein 16 (IFI16). IFI16 can be transported to the cytoplasm to help to recruit STING and TANK binding kinase 1 (TBK1). IFI16 as a PYHIN family protein may form the inflammasome only in theory. (C) Toll-like receptor (TLR) pathway. TIR domain-containing adaptor-inducing IFNβ (TRIF) may be responsible for helping the dimerization of STING. STING signaling can induce suppressor of cytokine signaling 1 (SOCS1) expression, which negatively regulates MyD88 activity. (D) Apoptosis. Mitochondrial outer membrane permeabilization (MOMP) formed by BAX/BAK induced by mitochondrial stress can release oxidized mitochondrial DNA (mtDNA) and cytochrome c into the cytosol. Oxidized mtDNA is a suitable ligand for cGAS recognition and is resistant to DNaseIII (TREX1) degradation. Cytochrome c binds to apoptotic protease-activating factor 1 (APAF1) and initiates the formation of an apoptosome cooperatively with caspase-9 to activate caspase-3, which can induce apoptosis. Caspase-3 can cleave cGAS.
Another PYHIN family member, IFI16, is a DNA sensor located in the nucleus. IFI16 can bind to viral DNA sequences or damaged chromatin DNA and be translocated to the cytoplasm to recruit STING cooperatively with TNF receptor associated factor 6 (TRAF6) and p53 (103, 104). Several studies have shown that IFI16 (which can stimulate the phosphorylation and recruitment of STING and TBK1) is required for the full response of STING (105, 106) (Figure 3B). Conversely, cGAS can partially enter the nucleus and interact with IFI16 to promote its stability (107). Therefore, it is inferred that during viral infection, IFI16 can facilitate recognition of decapsidated viral DNA in the nucleus, while cGAS in the cytoplasm engages with viral gene transcription products (104, 108). However, STING signaling can trigger IFI16 degradation by tripartite motif-containing 21 (TRIM21) ubiquitination (109).
TLR is also an important PRR for multiple PAMPs (110). TIR domain-containing adaptor-inducing IFNβ (TRIF) is downstream of several subtypes of TLRs (including TLR3). TRIF may be responsible for interacting with STING and helping the dimerization of STING (111). During viral infection, the TLR9-myeloid differentiation primary response 88 (MYd88)-IRF3/7 pathway is necessary for mouse monocytes recruitment to lymph nodes, whereas the STING pathway is necessary for local production of type-I IFN (112). However, STING signaling can induce suppressor of cytokine signaling1 (SOCS1) expression, which can negatively regulate MyD88 activity (113) (Figure 3C).
cGas-Sting Pathway in Cell Survival
Oxidized mtDNA can be released into the cytoplasm during cell stress elicited by hypoxia, viral infection and mitochondrial damage, etc.; oxidized mtDNA is resistant to degradation by the cytosolic nuclease TREX1 (114). In addition, mtDNA accompanied with TFAM (a mtDNA-binding protein that can bend mtDNA) is also a reasonable target for recognition by cGAS (21, 33). However, during regulated cell death (as represented by apoptosis), it undergoes mtDNA release but has certain mechanisms to ensure a minimal cGAS-STING-mediated immune response. Mitochondrial outer membrane permeabilization (MOMP) activation, which is executed by BCL-2-associated X protein (BAX) and BCL-2 antagonist or killer (BAK), is a highly controlled conserved process in regulated cell death. BAK and BAX activated by apoptosis signals cooperatively form a pore-like conformation on the mitochondrial outer membrane, leading to a permeability change of outer and also inner membranes (115, 116). Consequently, the mitochondrial matrix, including cytochrome C and oxidized mtDNA-TFAM, is released into the cytoplasm (115, 117). Cytochrome C binds to apoptotic protease-activating factor 1 (APAF1) and initiates the formation of the apoptosome cooperatively with caspase-9, which further triggers the intrinsic apoptosis program (117). In vivo and in vitro studies have shown that an absence of caspase-9 is associated with greater release of type-I IFN (43, 117). This occurs because caspase-9 and its downstream caspase-3 can cleave cGAS and IRF3 to restrain deleterious inflammation (118) (Figure 3D).
The cGAS-STING pathway can also initiate programmed cell death. Activation of STING enhances phosphorylation and activation of receptor interacting serine/threonine kinase 3 (RIP3) and mixed lineage kinase domain-like pseudokinase (MLKL). Proapoptotic BCL2 binding component 3 (PUMA), a member of BH3-only family, is subsequently activated in a RIP3/MLKL-dependent manner, which promotes leakage of mtDNA by MOMP (119, 120). Activated IRF3 can bind directly to BAX to form IRF3/BAX complex and induce apoptosis (47). Excessive cGAS-STING-mediated autophagy signaling can cause “autophagic cell death” and prevent malignant transformation of cells through DNA damage (94, 121). STING trafficking to the lysosome can broaden permeabilization of the lysosome membrane, thereby rupturing the lysosome and releasing its contents, resulting in “lysosomal cell death (LCD)”. LCD further triggers K+ efflux and NLRP3 activation, ultimately resulting in pyroptosis (96, 101) (Figure 3D). Moreover, stimulating STING-dependent type-I IFN and TNFα signals simultaneously can lead to necroptosis of tumor cells (122, 123).
cGas-Sting Pathway in Cell Senescence
Cell senescence is recognized as a permanent arrest of the cell cycle, and is common in aging, immunity, ontogenesis and infectious defense (124). It lacks a specific biomarker but can be identified by the expression of several anti-proliferative molecules (representatively Rb-p16 andp53-p21 pathway) (125). During senescence, changes in the nuclear structure and loss of the nuclear lamina protein disrupt the integrity of the nuclear envelope, leading ultimately to DNA damage and cytoplasmic chromatin fragments (126). Cellular senescence can be accelerated by accumulation of cytoplasmic chromatin in turn (127). These senescent cells produce the senescence-associated secretory phenotype (SASP), which shapes an inflammatory microenvironment (128). The cGAS-STING pathway has been reported to be involved in the recognition of cytoplasmic chromatin fragments from senescence-related DNA damage, and mediate the expression of SASP genes (129–132). Along with these actions, the expression of TREX1 and DNaseII is inhibited by DNA damage through the inhibition of E2F/DP (a potential transcription factor of TREX1 and DNaseII) (130).
For hematopoietic stem cells (HSCs), DNA damage can promote excessive secretion of type-I IFN in the HSC niche and activate p53 pathway, both of which can lead to long-term senescence and exhaustion of HSCs (133, 134). HSCs expressing a circular RNA named “cia-cGAS” in the nucleus, however, are protected from this exhaustion as a result of cia-cGAS having stronger affinity than that of self-DNA, which prevents it from being sensed (135). It implied a novel target to manipulate the immune environment in bone marrow and help for finding treatment approaches for hematopoiesis-based diseases, such as aplastic anemia. Utilizing cellular senescence to restrain tumor growth is discussed below.
cGas-Sting Pathway in Infection
cGAS-STING signaling has an essential role in defense against a broad spectrum of intracellular DNA and RNA viruses (9, 26, 50). HIV is a typical RNA retrovirus: there is neither dsDNA in its genome, nor production of nucleic acids (50). Nevertheless, cGAS can detect the presence of HIV. RNA:DNA hybrids synthesized during reverse transcription that can be sensed by cGAS explain (at least in part) this phenomenon (14). cGAS may be triggered by endogenous DNA broken and released during HIV infection as well theoretically. However, some studies found the new mechanisms. The early reverse-transcription production of HIV-1 can flank short stem loops with paired base, which lead to the production of Y-type DNA containing unpaired guanosines that can activate cGAS well (15). Moreover, nucleolus protein non-POU domain-containing octamer-binding protein (NONO), as a sensor of capsid components of HIV, can help cGAS to be translocated to the nucleus and assist cGAS to sense HIV DNA accompanied by polyglutamine-binding protein 1 (PQBP1) (136, 137). The assistance proffered by NONO in assisting cGAS to sense DNA is also associated with its role in constructing a ribonuclear complex with DNA-dependent protein kinase (DNA-PK) subunits around hexamethylene bis-acetamide-inducible protein1 (HEXIM1), termed as “HEXIM1- DNA-PK - paraspeckle components-ribonuclear protein complex (HDP-RNP),” which also has a role in repair of DNA damage and transduction of genotoxic signals (138). This complex is also required to accompany cGAS-PQBP1 in sensing DNA virus, such as Kaposi’s sarcoma-associated herpes virus (139). In addition, during virus infection, STING activation can lead to global suppression of translation in cells, which restricts viral replication (140).
Compared with HIV-1, HIV-2 is less infective because it can infect dendritic cells (DCs) and elicit an anti-virus immune response. As a result, HIV-2 can cross-protect against HIV-1 (141). This phenomenon has been attributed to the fact that HIV-2 (instead of HIV-1) can encode protein Vpx, which overcomes the SAMHD1 restriction of dNTP in DCs (46, 142). HIV-1 can infect DCs via Vpx presentation, nevertheless, HIV-1 still cannot be fully sensed and induce an efficient immune response owing to certain escape mechanisms. Whether it is HIV-1 or HIV-2, a completely robust IFN response is required at pre- and post-integration sensing stages (143). cGAS in DCs can detect reverse-transcribed cDNA of HIV-2 before and after integration, whereas HIV-1 sensing is after genome integration owing to its capsid protection (144, 145). It was suggested that during initial infection by HIV-1, nucleotides are recruited into the intact capsid through the hexamer pores on the HIV-1 capsid. Therefore, the capsid-coated HIV-1 virus prevents the encapsidated reverse-transcription production from being sensed by the cytosolic nucleic-acid sensors (146). HIV-1 capsids can be ubiquitinated and then degraded by the host E3 ubiquitin ligase function of TRIM5, which leads to detection of viral DNA, meanwhile HIV-1 could use some host protein like cyclophilins to evade the sensing (147, 148) (Figure 2A).
Similarly, other viruses also have evasion mechanisms to escape cGAS-STING pathway surveillance (Table 3). Therefore, identifying and preventing such viral-evasion factors could be a viable means to design novel anti-viral drugs.
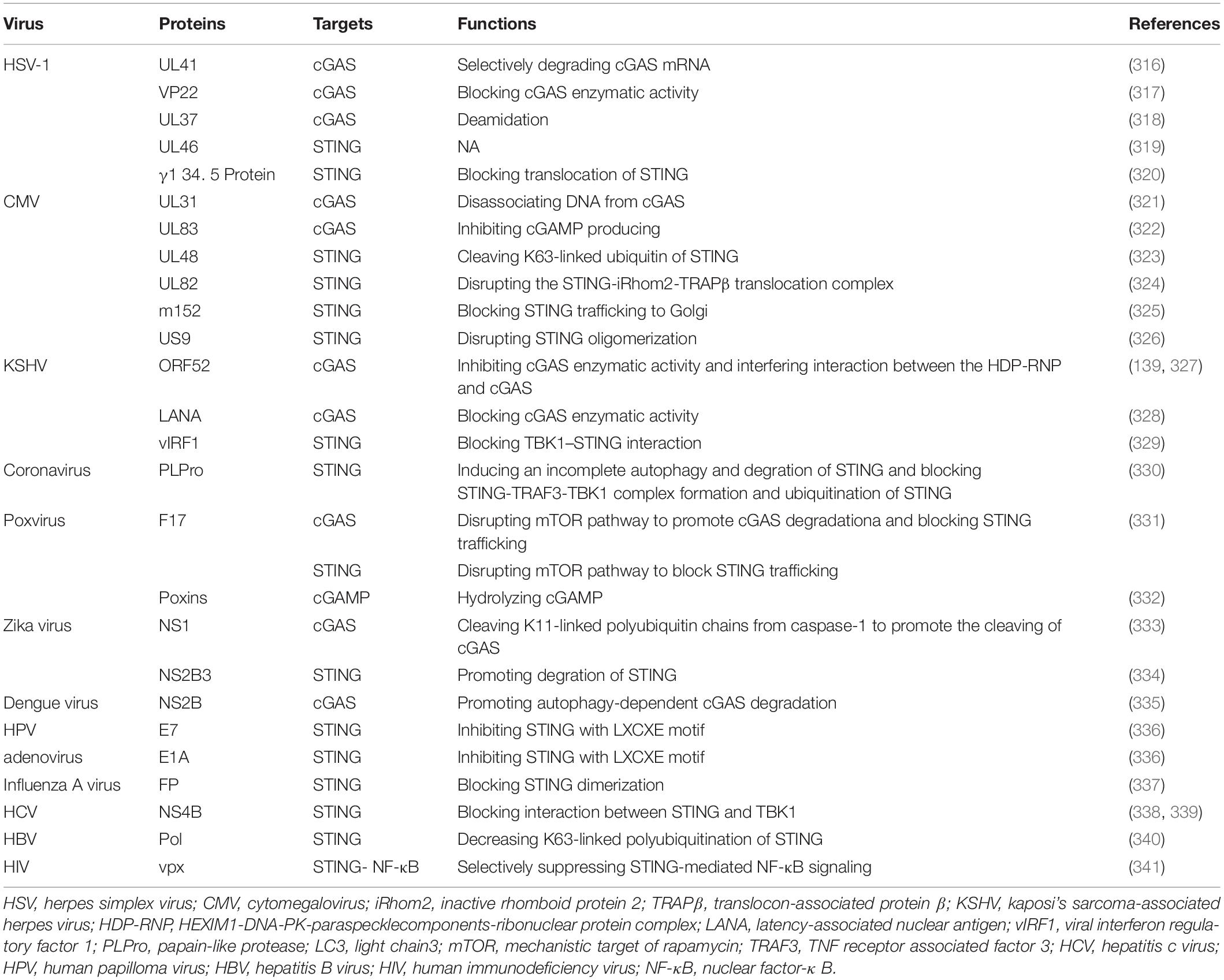
Table 3. Virus productions associated with evasion from cGAS-STING pathway.
cGAS-STING pathway is responsible to protect against intracellular or extracellular bacterial infection (especially intracellular infections). CDNs (e.g., c-dGMP, c-dAMP, and cGAMP) produced by bacteria are essential for the regulation of bacterial function, such as biofilm formation, colonization, and reproduction (149, 150). As ligands for STING, CDNs can bind directly to and activate STING independently of cGAS, which contributes to several immune responses from bacteria (151). Usually, bacteria can enter or be engulfed by the cell through the endophagosome and be sequestered from the cytosolic sense receptor. Some bacteria, such as Mycobacterium tuberculosis (Mtb), can survive in vacuoles, resulting in an insufficient cellular immune response to defend against it (152). In contrast, the ESX-1 secretion system of the mycobacterium can translocate the phagosomal vacuolar matrix including bacterial genome molecules into the cytoplasm and trigger the cGAS-sensing pathway (153). For other bacteria, such as Legionella pneumophila or and Brucella abortus, the host guanylate binding proteins (GBPs) facilitate rupture of phagosome vacuoles and are indispensable for controlling their infection (154, 155). Autophagy signaling mediated by cGAS/STING is also involved in microorganism clearance mentioned above (90, 91).
Bacteria have evolved strategies to confront this pathway too. Bacterial phosphodiesterase CdnP produced by Mtb or group-B streptococci can degrade CDNs (156, 157). CpsA (a type of Mtb LytR-CpsA-Psr domain-containing protein) can prevent autophagy responses for eliminating pathogens (90). Chlamydia trachomatis inclusion membrane proteins can maintain the stability of the inclusion membrane and avoid inclusion lysis (leading to pathogen antigens leaking out and being detected by the host cell) (158, 159). Yersinia outer protein J (YopJ) deubiquitinates STING and impedes the formation of the STING signalosome (160). The cGAS-STING pathway activation even impedes the elimination of Listeria monocytogenes because bacterial DNA can be packaged into EVs and transferred into T cells, where it induces apoptosis of T cells (161, 162).
Several protozoans, such as Toxoplasma gondii and malaria parasites, have an intracellular period in their lifecycle. T. gondii could engage cGAS-STING exclusively (163). However, IRF3 activation inducing ISG expression promotes T. gondii development independently of IFN expression (163). P. falciparum can target erythrocytes, lacking a nucleus and unable produce IFN, but infected erythrocytes can secrete EVs containing parasitic gDNA to monocytes and trigger cGAS (164).
cGas-Sting Pathway in Autoimmune or Inflammatory Diseases
The immune system is regulated by a complicated network. Disorder of immune signaling can elicit non-infectious inflammatory or autoimmune diseases. Excessive, uncontrolled production of type-I IFN can lead to a spectrum of inflammation diseases termed “type-I interferonopathies,” which have some common manifestations (165). cGAS-STING is the one of main sources of type-I IFN, acts as a cellular immune-sensing signaling axis, and is involved in type-I interferonopathies.
Stimulator of interferon genes -associated vasculopathy with onset in infancy (SAVI) is a typical STING-related hereditary inflammatory type-I interferonopathy, and is manifested by interstitial lung disease, dermatomyositis and arthritis. Its pathology is featured by leukocytoclastic vasculitis and microthrombotic angiopathy of small dermal vessels (166, 167) and patients can also suffer from lymphopenia (166–169). The etiology of SAVI is a gain of function (GOF) mutant in STING which leads to constitutive STING activation without CDNs stimulation (166). Currently, several mutant amino acids residues have been found in or close to the dimerization domain (V155M, N154S, G166E, V147L, and V147M) (64, 166, 168, 170), as well as R284G, R284S, R281Q, and C206Y in the cGAMP-binding domain (171).
Other types of type-I interferonopathies, such as systemic lupus erythematosus (SLE) and Aicardi–Goutières syndrome (AGS), have relationships with defective clearance of cytosolic nucleic acids caused by congenital dysfunction of TREX1, RNASEH2, and SAMHD1. SLE is a heterogeneous autoimmune disease which has prominent type-I and also -II IFN signatures (172). AGS comprises some systemic autoimmune syndromes overlapping with SLE, and can be classified as a “lupus-like disease” (173). Additionally, AGS also causes severe developmental neurological disorders, including cerebral calcifications, encephalopathy and cerebral atrophy.
Systemic lupus erythematosus is a representative model for elucidating the mechanism of type-I interferonopathies. In SLE, the level of self-DNA which is packaged into apoptosis-derived membrane vesicles along with the level of anti-dsDNA antibody is increased in the serum of patients (174). A study revealed increasing levels of ISGs (including cGAS/STING) as well as the cGAMP-detected ratio in peripheral-blood mononuclear cells of SLE patients (175). As innate immune cells, DCs have essential roles in antigen presentation, cytokine secretion, and priming the adaptive response of immune cells (176). Plasmacytoid DCs (pDCs) can internalize and recognize self-DNA and they are the main source of type-I IFN in serum during SLE (177). IFNα/β is essential for complete function of immature pDCs (178). IFNα/β and IL-1 can induce mitochondrial oxidative stress in DCs and decrease ATP production, which blocks proton-pump function and increases pH of lysosomes. This process inhibits mitochondrial degradation and blocks mtDNA clearance, which engages the cGAS-STING pathway (31, 179). Moreover, monocytes may sense mtDNA through cGAS-STING and differentiate into DCs (31, 180). Neutrophil extracellular traps (NETs) are complexes released by neutrophils exposed to stimuli or autoantibody immune complexes. NETs comprise extracellularly released chromatin, myeloperoxidase enzymes, and also oxidized mtDNA. In lupus-like diseases, NETs can be induced by IFNα/β and may play a major part in priming pDCs (181, 182). All mechanisms stated above contribute to a more aggravated type-IIFN response and exacerbate disease. A similar phenomenon can be observed in SAVI, ataxia telangiectasia (AT) and Artemis deficiency (183). However, compared with SAVI, DCs in SLE can prime T-cell maturation significantly and increasing secretion of pro-inflammatory cytokines, such as IL-6 and TNFα can also lead to activation of adaptive immunity (Figure 4A).
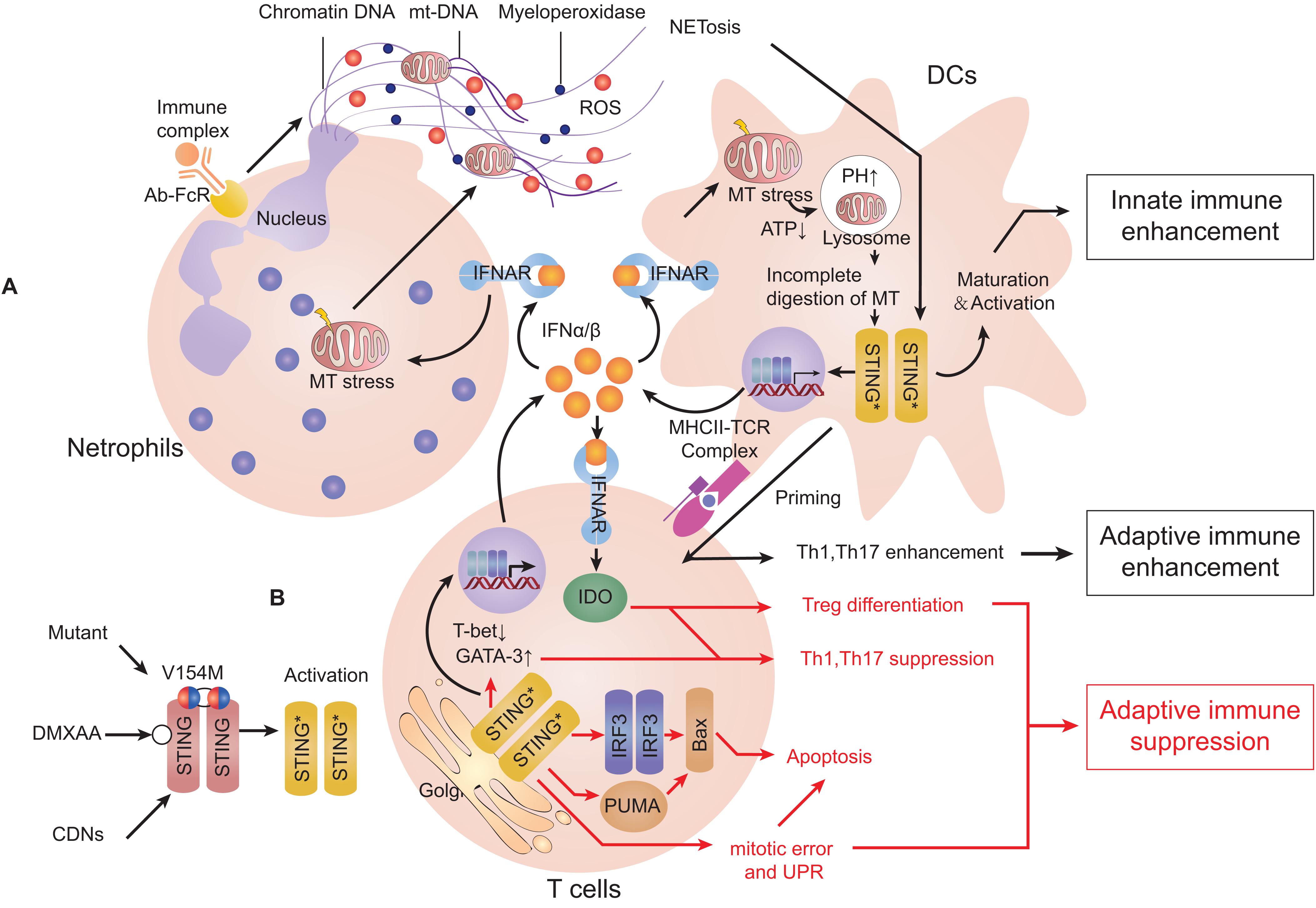
Figure 4. Activation of the cGAS-STING pathway can induce type-I interferonopathies or adaptive immune deficiency. Stimulator of interferon genes (STING) can be stimulated by cyclic dinucleotides (CDNs), agonist compounds and mutants. (A) STING activation in dendritic cells (DCs). When exposed to type-I interferon (IFN), neutrophils can release neutrophil extracellular traps (NETs). DCs can internalize self-DNA and trigger the cyclic-GMP-AMP synthase (cGAS)-STING pathway to secrete type-I IFN, which activates DC maturation. Type-I IFN in turn induces mitochondrial stress and decreases ATP production that increases the pH of lysosomes. Incomplete degradation of mitochondria leads to accumulation of mtDNA in the cytosol and activates STING. Mature DCs can present antigen and co-activate signaling to prime T cell-mediated adaptive immune responses. (B) STING activation in T cells. IFNα/β enhances differentiation of T helper 2 (Th2) cells but suppresses differentiation of Th1 cells. Activation of STING can induce mitotic delay and unfolded protein response (UPR), leading to reduced proliferation and promotion of apoptosis. All of the actions stated above suppress the functions of Th1 cells and lead to suppression of adaptive immune responses (Red arrows represent mechanisms limiting cellular immune responses, and black arrows represent mechanisms promoting cellular immune responses).
The cGAS-STING pathway can mediate systemic inflammation as well as autoimmune activation. However, it is also involved in the local inflammation of multiple tissues. With regard to ischemic myocardial infarction (MI), cardiac macrophages can sense dying ruptured cells and lead to fatal post-MI cardiac inflammation, which is reversed by ablation of cGAS/STING/IRF3 (184, 185). In a non-alcoholic steatohepatitis (NASH) model induced by a methionine- and choline-deficient or high-fat diet, lipotoxicity can cause mitochondrial damage and up-regulate STING/IRF3 expression in hepatocytes, which in turn promotes lipid accumulation and inhibits glycogen synthesis. All above bring out hepatic inflammation and hepatocytes apoptosis (186). In this model, mice with deficiency of STING presents alleviated insulin resistance and lower levels of low-density lipoprotein in serum, and also decreased hepatic inflammation and fibrosis/steatosis, in which hepatic macrophages/kupffer cells may take a big part (187, 188). Lipotoxicity can induce p62 to be phosphorylated through the cGAS-STING-TBK1 pathway, which causes aggravated protein inclusions in hepatocytes and it indicates that p62 could be a biomarker for NASH prognosis (189). mtDNA-dependent inflammation induced by lipotoxicity also occurs in adipose tissue and endothelial cells of blood vessels, which contributes to tissue inflammation, insulin resistance, and cardiovascular diseases (42, 190). In traumatic brain injury (TBI), local injury initiates breakdown of the blood–brain barrier and global neuroinflammation (191). STING expression is up-regulated in TBI and can lead to increased expression of pro-inflammatory cytokines and enlargement of secondary injury (192). Reduced autophagy-associated protein expression induced by STING may contribute to the dysfunction of autophagy and dampen the elimination of necrotic tissue, thereby intensifying inflammation (192). During silicosis, silica can yield cytosolic dsDNA release and engage cGAS-STING, which activates DCs and macrophages to cause severe lung inflammation. It also leads to death of epithelial cells through the NLRP3 pathway and pulmonary fibrosis (193). Similarly, mtDNA release in renal tubule cells has been found to be associated with acute kidney injury by cytotoxic drugs and chronic renal fibrosis (194, 195).
Neurodegenerative diseases are correlated with local inflammation (196). In the central nervous system (CNS), microglia is considered to be the main source of cGAS-STING-dependent IFN expression (197). In neurodegenerative diseases, levels of the marker of microglia activation-cluster of differentiation 68 (CD68), and pro-inflammatory cytokines are increased (198). A significant feature of Parkinson’s disease (PD) is the neuronal loss of cerebral nuclei (especially dopaminergic neurons in the substantianigra). Serine/threonine-protein kinase PINK1 and E3 ubiquitin-protein ligase parkin are ubiquitin-related factors that take part in removing damaged mitochondria by autophagy, and their dysfunction lead to the early onset of PD (199). Parkin–/– and PINK1–/– mice, following exhaustive exercise, show inflammation and loss of dopaminergic neurons, which can be rescued by loss of STING (200). Similarly, AT is a genetic disease caused by missense mutation of a DNA-repair protein: ATM. AT patients usually show neurodegenerative defects (especially ataxia) complicated with telangiectasia on their eyes or body, deficiency of adaptive immune cells, and predisposition to cancer (201). Nevertheless, up-regulation of expression of type-I IFN can also be found in AT patients and mice, causing them to be prone to autoimmune diseases (36, 183, 202). This syndrome is related to p53-mediated senescence but also the chronic inflammation mediated by the cGAS-STING pathway which engages cytosolic uncombined broken gDNA caused by ATM dysfunction (127). In addition, accumulation of cellular mtDNA occurs in age-related macular degeneration characterized by retinal pigmented epithelium (RPE) degeneration. This can trigger chronic inflammation by cGAS-STING pathway, in which NLRP3 inflammasomes, inflammatory/apoptotic caspases are also involved (203, 204).
With regard to other diseases in which adaptive immune cells prime, cGAS-STING has a different role. Multiple sclerosis (MS) is a local inflammatory disease of CNS. MS is characterized by over-reactive microglia, infiltration of self-reactive T cells, demyelization of nerve fibers and hyperplasia of gliocytes. Autoantibodies against proteins expressed in immune-privileged regions of CNS also contribute to its pathogenesis (205). In MS, IFNα/β can attenuate disease severity effectively. This implies a protective role for type-I IFN in CNS, which is considered to counteract the pro-inflammatory IFNγ (206). Using experimental autoimmune encephalitis (EAE) as a MS model, STING was found to be indispensable for amelioration of type-I IFN-mediated neuroinflammation, and it could be induced by a conventional anti-viral drug ganciclovir (207). Ultraviolet (UV) radiation is a factor inversely related to the morbidity of MS (208). It was found that UV-B irradiation can recruit inflammatory monocytes and produce type-I IFN in a STING-dependent manner (209). All above indicate that cGAS-STING-IFNα/β pathway may have a beneficial effect on some CNS inflammatory diseases such as MS.
A possible reason for the observed effect above is due to indoleamine 2,3-dioxygenase (IDO), which can catabolize tryptophan (Trp) oxidatively. Trp withdrawal or Trpoxidative catabolites can interact with general control non-derepressible 2 (GCN2) and mTOR, of which both can control cellular amino-acid metabolism and suppress T helper 1 (Th1) cells immunity (210). IFNα/β is a potent IDO inducer to suppress proliferation of CD4+ Th1 cells and promote differentiation of Foxp3+ regulatory T (Treg) cells, which are believed to suppress CNS-specific autoimmunity (210, 211). In addition, DNA released from dying cells can be internalized directly by T cells and sensed by cGAS-STING pathway, which leads to enhancement of the Th2 transcription factor GATA3 but suppression of the Th1 transcription factor T-bet. Consequently, this process polarizes naive T cells toward Th2 differentiation (212). Studies mentioned above may (at least in part) explain why the cGAS-STING signal is a negative regulator of MS.
The inhibitory role of cGAS-STING in inflammation is also attributed to its apoptosis-triggering role. In some subtypes of SAVI and mouse models, apoptosis of blood-vessel endothelial cells or bronchial epithelial cells and leucopenia can be observed (especially T-cell lymphopenia) (166, 169, 170). When the STING signal is stimulated, apoptosis occurs more frequently in normal or cancerous T cells (119). Also, bone-marrow chimeras and gene-knockout studies have shown that T cells defect in SAVI are not associated with type-I IFN signaling or cGAS (213, 214). Localization of STING at the Golgi can cause delay of T-cell mitosis and reduced proliferation independently of IRF3 and TBK1 (215). Furthermore, a “UPR motif” on the C-terminus of STING can cause ER stress and UPR, resulting in Ca2+ overloading and T-cell death (82). A controversial view is that B cells express STING variously and may undergo apoptosis through this way (166, 216, 217). However, simultaneous signaling by STING and the B-cell antigen receptor can promote B-cell activation and antibody production independently of type-I IFN (217) (Figure 4B).
As for some diseases with inflammatory responses involved, the acute phase of pancreatitis causes dying acinar cells to produce free dsDNA, which activates cGAS-STING signaling in macrophages, and exacerbates inflammation severity (218). However, in the chronic phase of pancreatitis, cGAS-STING activation decreases pancreatic inflammation, which may be mediated by limiting Th17 response (219). For gut mucosal immunity, transient stimulation of STING could strengthen the function of antigen-presenting cells (APCs) and promote Th1 and Th17 immune responses against microbes (220). Chronic STING signaling, however, elicits an IL-10 response to control the inflammation and avoids inflammatory enterocolitis such as bowel disease (221). STING knockout mice present reduced numbers of goblet cells, a decreased ratio of commensal versus harmful bacteria and compromised Treg cells in the gut, making it prone to enterocolitis (222).
cGas-Sting Pathway in Cancer
Chromosomal instability (CIN) is an intrinsic feature of cancer, and results spontaneously from errors in chromosome segregation during the mitosis of cancer cells. CIN can also be induced manually by radiotherapy or chemotherapy, which causes DSBs. It results in micronuclei formation outside the nucleus, of which rupture brings out irrepressible accumulation of cytosolic self-DNA and engages cGAS (32, 38, 223). However, normal mitotic processes involve exposure of chromosomal DNA to the cytoplasm, but this cannot initiate a substantial inflammatory reaction or apoptosis because nucleosomes can suppress dsDNA-cGAS binding in a competitive manner (41).
An appropriate immune response against tumors via a type-I IFN plays an indispensable part in limiting tumors and prolonging host survival (224). It was found STING-deficient mice are prone to developing several types of cancer and have poor survival under a tumor burden, whereas stimulation of STING can elicit robust immunity to tumors (225–227). A mechanism is many cancer cells expressing cGAS can recognize cytosolic DNA and produce cGAMP to stimulate secretion of type-I IFN through STING (228, 229). Excessive expression of TREX1 in cancer cells, which can be induced by radiotherapy, attenuates this progression (228). cGAS-STING can also promote senescence of cancer cells through the p53-p21 pathway (129). cGAS-STING-mediated autophagy contributes to autophagic cell death if mitotic crisis occurs to avoid transformation of cancer cells (121). Melanoma cells can also transfer cGAMP produced by them to proximal non-cancerous host cells through gap-junction channels and activate STING in these cells, which contributes to the recruitment of tumor-infiltrating immune cells such as natural killer (NK) cells (51, 230). Expression of the NK cell-specific ligand NKG2D retinoic acid early transcript 1 (RAE1) on cancer cells is highly up-regulated by STING once NK cells permeate into tumor tissue (231). The activation of STING in the endothelium within the tumor microenvironment (TME) could contribute to the remodeling of tumor vasculatures, and may have positive effects on tumor regression (232).
Dendritic Cells are the main source of type-I IFN in several types of TMEs and are dependent on STING signaling (229). More preferentially than macrophages, infiltrating DCs take up tumor-derived DNA or cGAMP from dying cell fragments by phagocytosis (27, 29, 129, 226, 233). Moreover, cancer cells can package DNA into exosomes and transfer DNA to DCs (234). Produced cGAMP by cancer cells can also be transferred to DCs through forming gap junction (53). By activating cGAS-STING signal in DCs, CD8α+ subtype DCs secret chemokines such as CCL5 and CXCL10 and cross-prime infiltrating anti-tumor CD8+ T cells (29, 226, 235–237). In contrast, numbers of immune-suppressing cells such as Treg cells, myeloid-derived suppressor monocytes and M2 macrophages have been reported to be decreased (225, 238, 239). Expression of IL-15/IL-15Rα complex is up-regulated in myeloid cells with the help of STING/type-I IFN and promotes tumor regression (240).
Tumor cells can evade intrinsic cellular surveillance in different ways. In various cancer cell lines, cGAS, STING, TBK1, and IRF3 are mutated frequently and their decreased expressions are also related to the high level of methylation (241). STING expression has been shown to be suppressed by the alternative lengthening of telomeres (ALT) pathway, which is responsible for prolonging the telomere length and maintaining the proliferation of tumor cells (242). A hypoxic environment in tumor cells can lead to accumulation of lactic acid and is associated with the inhibition of tumor-conditional DCs and reduced expression of IFN signaling molecules (243). In breast cancer, functional up-regulation of expression of human epidermal growth factor receptor 2 (HER2), a ligand-independent receptor tyrosine kinase (RTK), can arrest the expression of RAC-alpha serine/threonine-protein kinase (AKT1) (a key factor in the mTOR pathway), which is reported to inhibit the activation of cGAS and TBK1 (244, 245).
Patients with lung adenocarcinoma have a low probability of survival if they have reduced expression of cGAS (132). Thus, expression of the cGAS-STING and DNA-damage marker histone γH2AX in tumor cells could be considered as independent prognostic factors to predict therapy response and clinical outcome, and could be superior to that of traditional markers like immunogenic cell death and T cells number (246).
However, some scholars have arrived at opposite conclusions. When DSBs occur in cancer cells, cGAS can be relocated to the nucleus and obstruct the formation of the PARP1-Timeless complex, thereby inhibiting homologous recombination repair and maintaining CIN, which potentiates tumor evolution (35, 223). It has also been reported that cGAS recognizing CIN activates non-canonical NF-κB signaling and potentiates cellular metastasis programs (247). Furthermore, STING–/– mice are resistant to skin carcinogenesis in a 7,12-dimethylbenz(a)anthracene (DMBA)-treatment model. It has been demonstrated that when DMBA-induced nuclear DNA leaks into the cytoplasm, STING can induce chronic inflammatory stimulation that contributes to cancer development (248). During brain metastasis, cGAMP transferred to bystander cells (e.g., astrocytes) can also produce IFNα and TNFα in the TME but, in this context, it will support tumor development and chemoresistance (249). Coordinating with myeloid cells penetrating into the tumor, myeloid-derived suppressor cells can also be recruited through the C-C chemokine receptor type 2 (CCR2) (250). Another study found that microparticles yielded by tumor cells can turn macrophages into the M2 type through cGAS-STING-TBK1, contrary to previous findings (251).
Immune-system interactions with tumor cells are complicated. The effect of cGAS-STING on cancer is dependent on the type of tumor, host immune state, activated cell types, therapeutic intervention, and the magnitude of cGAS-STING activation. Like inflammation generated by cGAS-STING, a time-dependent inflammatory anti-tumor response mediated by cGAS-STING may be present. Temporary activation of cGAS-STING in innate immune cells could enhance the anti-tumor effect, whereas sustained activation of cGAS-STING might induce immune tolerance of the tumor. More investigations are necessary to ascertain the exact role of cGAS-STING in oncology, and elucidate the specific advantages and adverse effects of targeting the cGAS-STING pathway in cancer therapy (Figure 5).
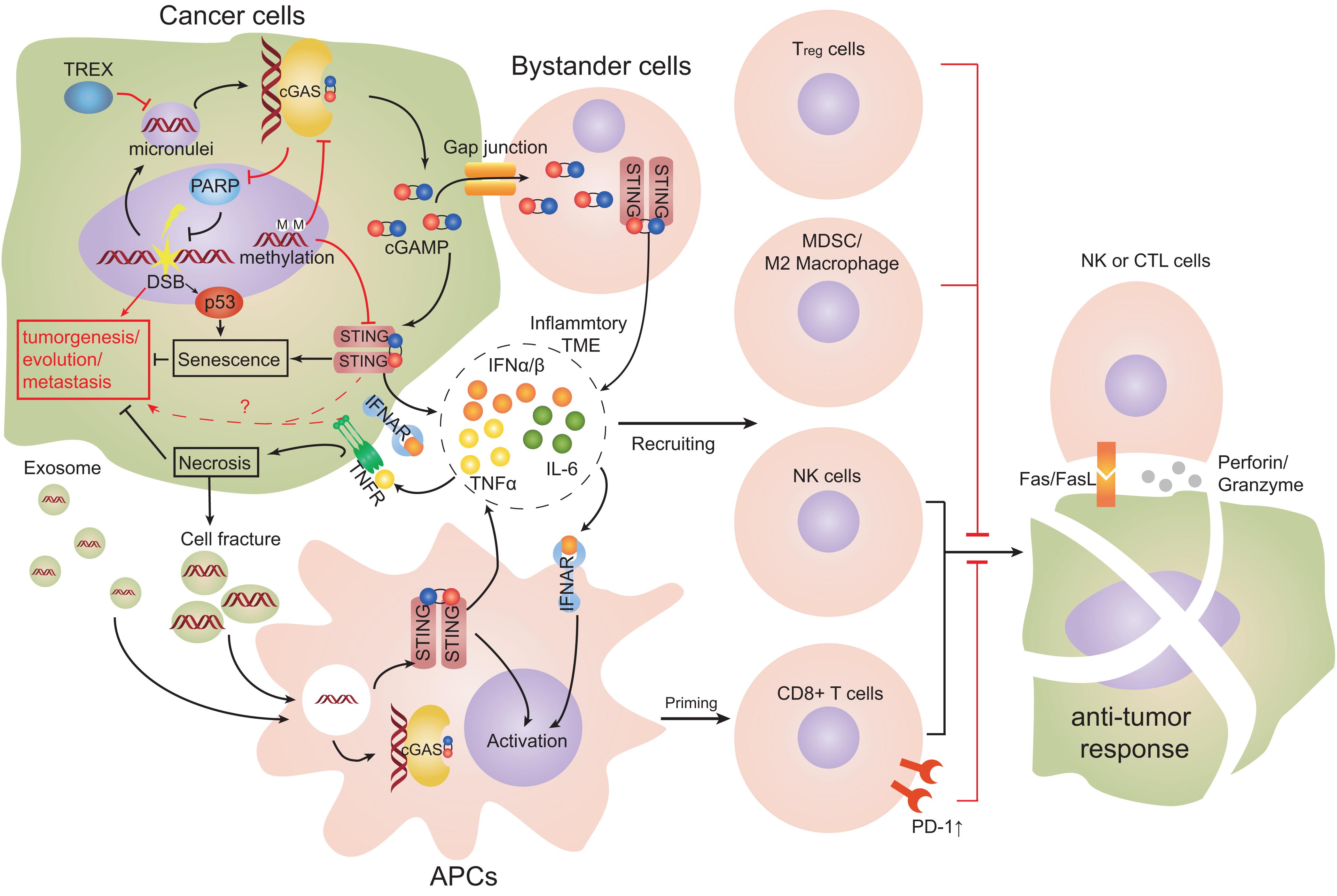
Figure 5. Cell interactions in TME engaging the cGAS-STING pathway. Double-stranded breaks (DSBs) can lead to cytosolic self-DNA that engages the cyclic-GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway and produces type-I interferon (IFN). STING promotes the senescence of cancer cells. Cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) can transfer to proximal host non-cancer cells through gap-junction channels and promote the formation of an inflammatory tumor microenvironment (TME). Dendritic cells (DCs) are the main source of type-I IFN and tumor necrosis factor α (TNFα), and promote an inflammatory TME. Infiltrating DCs take up tumor-derived DNA or cGAMP from dying cell fragments or exosomes. DCs cross-prime and activate anti-tumor cluster of differentiation 8 (CD8)+ T cells and constrict cancer growth effectively. Co-stimulation of type-I IFN and TNFα signaling simultaneously leads to remarkable necroptosis of tumor cells. cGAS can relocate to the nucleus and obstruct polymerase 1 (PARP1), which suppresses DNA repair. Chronic levels of IFNα and TNFα in TME could support tumor development and cause type-2 macrophages and regulatory T cells to be recruited (Red arrows represent mechanisms promoting tumors; black arrows represent mechanisms limiting tumors).
Targeting the cGas-Sting Pathway for Treatment
Considering the pivotal role of the cGAS-STING pathway in infection, inflammation and cancer, positive modulation of the pathway signaling is a promising way to enhance the immune state and restrict microorganisms or heterogeneous cells, whereas negative modulation can control aberrant inflammation.
Radiotherapy or chemotherapy drugs such as cisplatin or cyclophosphamide can induce DSBs and micronuclei, then trigger the cGAS-STING pathway to enhance tumor immunogenicity (252–254). In addition, PARP inhibitors such as olaparib have promising effects on cancer cells lacking BRCA2 because of their cooperative DNA-repair functions (253). Although cGAS activation is inhibited by nucleosomes, taxol can induce mitotic cell-cycle arrest and sustain divided chromatin in the cytosol to activate the cGAS-STING pathway slowly, and accumulation of signaling could stimulate apoptosis of cancer cells (41). Inhibitors of topoisomerase 1 or 2 used conventionally as chemotherapy drugs trigger minor damage to DNA and accumulation of cytosolic DNA, which can engage cGAS and enhance the anti-tumor or anti-infection responses of cells (255–257).
cGAS-STING is essential on anti-tumor immune checkpoints therapies. For example, blockade of CD47-signal regulatory protein α (SIRPα) signaling on DCs can activate NADPH oxidase 2 (NOX2) and increase the pH in phagosomes along with incomplete degradation of mtDNA, which can trigger cGAS-STING (129). STING deficiency in mice abrogates the anti-tumor effect of CD47 blockade (258). A similar phenomenon also can be seen in anti-programmed cell death-1 (PD-1) therapy (259). There is greater infiltration of IFNγ+ cells and CD8+ T cells and PD-1/PD-1 ligand 1 (PD-L1) expression in TME treated by STING-ligand derivatives (260). Therefore, in several types of tumors, combined administration of a STING agonist and immune-checkpoint antibody could elicit a more curative outcome compared with one therapy alone (238, 261).
Viruses can infect cells lacking cGAS-STING more effectively, and have higher oncolytic activity compared with virus therapy alone. Hence, the use of oncolytic viruses such as talimogene laherparepvec is beneficial for treating tumors with low expression of cGAS/STING. STING expression can be regarded as a prognostic measurement for such therapy (262).
Some artificial analog molecules of CDNs, such as 5,6-dimethylxanthenone-4-acetic acid (DMXAA) and 10-carboxymethyl-9 (10H) acridone (CMA) can bind the CDN pocket of mouse-specific STING dimers and promote conformational transition of STING from inactive “open” to an active “closed” state (263, 264). DMXAA showed convincing efficacy in restraining tumors in mice (265). However, DMXAA is restricted in activating mouse-specific STING but not human-specific STING, which could be an explanation for the failure of DMXAA in treating 1299 non-small-cell lung cancer patients in a phase-III clinical trial (NCT00662597) (266). Nonetheless, with three substitutions (G230I, Q266I, and S162A), human STING can also be induced by DMXAA to undergo conformational transition (264). Another new compound, amidobenzimidazole, has been found to be an agonist of STING without lid closure and has potential therapeutic value (267).
Cyclic dinucleotides and their derivates that can stimulate STING directly are candidate adjuvants to restrain tumors. Intratumoral administration of c-dAMP, c-dGMP, or cGAMP analogs alone or combined with other adjuvants or tumor antigens have shown anti-tumor effects (259, 268); phase-I or II clinical trials (NCT02675439, NCT03172936, and NCT03937141) of dithio-(RP,RP)-c-dAMP (known as ADU-S100) are ongoing (261). To avoid the degradation and ensure maximal phagocyte internalization of CDNs, endosomolytic nanoparticles have been designed to package and deliver CDNs. For example, pH-sensitive nanoparticles (e.g., STING-nanoparticles) can release their contents if located in acidic endosomal environments (269).
For treatment of type-I interferonopathies, lessons can be taken from the treatment of canonical autoimmune disease such as SLE, but there are several differences. For example, it was found that corticosteroid pulse therapy, γ-immunoglobulins, disease-modifying anti-rheumatic drugs, anti-CD20, and some immunosuppressants (e.g., methotrexate) have limited efficacy against SAVI (166, 171). JAK inhibitors such as ruxolitinib, tofacitinib and baricitinib that reduce type-I IFN downstream signaling have shown therapeutic value against SAVI, but further verification of their efficacy is needed (270). Moreover, novel immune therapies, such as anti-IFNα and anti-IFNAR immunoglobin, are in clinical trials for SLE. These could also be tested against SAVI in the future (165).
Pharmaceutically screening has revealed that some anti-malaria drugs, such as suramin, have an inhibitory effect on cGAS by blockade of interaction between DNA and cGAS (271). In addition, novel small molecules such as RU320521 or G150 can occupy the enzymatic pocket of species-specific cGAS to abrogate cGAMP synthesis (272, 273). Recently, a study found that aspirin can acetylate cGAS at three lysine residues and block cGAS activity (274). With regard to STING, the cyclopeptide Astin C can block IRF3 recruitment onto the STING signalosome (275). The molecule H-151 can block the palmitoylation of human-STING (276). All of these agents are potential candidates for alleviating type-I interferonopathies.
Concluding Remarks and Future Perspectives
The cGAS-STING pathway is primarily responsible for the modulation of immune response in cells when facing cytosolic dsDNA challenge. Moreover, it is complicatedly cross-regulated by other cellular processes or cellular signaling networks. The exact fundamental mechanism of the pathway in cells and the effect on the whole organism in specific states is not completely clear and requires further investigation.
In conclusion, the cGAS-STING pathway has dichotomous roles in the immune system. In general, cGAS-STING-type-I IFN signaling can promote the innate immune response in myeloid cells but alleviate the adaptive immune response exerted by T cells and B cells. cGAS shows high expression in APCs such as macrophages and DCs, but STING is expressed in most cells (10). cGAS-STING signaling corresponding to cytoplasmic dsDNA in APCs can boost innate and adaptive immunity transiently. In this situation, the DNA challenge signal is limited to only macrophages and DCs. Their pro-inflammatory and antigen-presenting functions to adaptive immune cells are promoted in the short-term. If the signal spreads to other bystander cells, such as T cells, B cells, local resident cells by means of cGAMP transfer, or just aberrant STING activation by GOF, it causes apoptosis in bystander cells or adaptive immune cells and immune tolerance in the long-term. Therefore, it is reasonable to conclude that the intrinsic function of the cGAS-STING pathway is essential for the innate immune system responses of the host immediately after pathogen invasion or abnormal cell appearing. Once the challenge persists, the cGAS-STING pathway controls the adaptive immune system to avoid chronic, detrimental inflammatory reactions or autoimmune diseases.
The inflammatory response exists universally in almost all physiologic and pathologic progressions. cGAS-STING is pivotal in modulating cellular inflammation, so it is promising to extend our conception of the cGAS-STING pathway onto more diseases with inflammatory responses involved, especially CNS-based diseases such as stroke, in which the inflammatory reaction exists but was recognized less.
Moreover, for targeting the cGAS-STING pathway for therapeutic purposes, drugs should be optimized to augment the desirable effect and prevent its unwanted effects. For example, to eliminate tumor cells or infectious agents, agonists of cGAS-STING would be a rational option if designed to target APCs exclusively but not T cells or B cells. In this scheme, the anti-tumor immune response is enhanced while avoiding apoptosis of adaptive immune cells and infiltration of immune suppressor cells. Also, most research on the cGAS-STING pathway has focused on its IFN-expressing role but overlooked autophagy and cell-death roles, which are also main downstream signaling of the pathway. Therefore, some drugs, such as emricasan, are potential apoptosis inhibitors that may have a complementary effect on ameliorating apoptosis of blood-vessel endothelial cells or bronchial epithelial cells, and lymphopenia, in SAVI.
Until now, studies of the cGAS-STING pathway have been done mainly in the laboratory but it has large space to be explored in clinical or translational fields. Additionally More PRRs and cellular immune-surveillance pathways may remain to be discovered to piece together the molecular puzzles of the cell.
Author Contributions
DW drafted the manuscript and drew the figures. WJ and JH conceived and revised the review.
Funding
This work was supported by the National Natural Science Foundation of China (81571600, 81322018, and 81273287 to JH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank K. wood from Barrow Neurological Institute for discussions and editing.
References
1. Roers A, Hiller B, Hornung, V. Recognition of endogenous nucleic acids by the innate immune system. Immunity. (2016) 44:739–54. doi: 10.1016/j.immuni.2016.04.002
2. Ishikawa H, Barber, GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. (2008) 455:674–8. doi: 10.1038/nature07317
3. Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. (2008) 29:538–50. doi: 10.1016/j.immuni.2008.09.003
4. Sun W, Li Y, Chen L, Chen H, You F, Zhou X, et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci USA. (2009) 106:8653–8. doi: 10.1073/pnas.0900850106
5. Woodward JJ, Iavarone AT, Portnoy, DA. C-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science. (2010) 328:1703–5. doi: 10.1126/science.1189801
6. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. (2011) 478:515–8. doi: 10.1038/nature10429
7. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. (2010) 11:997–1004. doi: 10.1038/ni.1932
8. Sun L, Wu J, Du F, Chen X, Chen, ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. (2013) 339:786–91. doi: 10.1126/science.1232458
9. Li XD, Wu J, Gao D, Wang H, Sun L, Chen, ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. (2013) 341:1390–4. doi: 10.1126/science.1244040
10. Mabbott NA, Baillie JK, Brown H, Freeman TC, Hume, DA. An expression atlas of human primary cells: inference of gene function from coexpression networks. BMC Genomics. (2013) 14:632. doi: 10.1186/1471-2164-14-632
11. Whiteley AT, Eaglesham JB, de Oliveira Mann CC, Morehouse BR, Lowey B, Nieminen EA, et al. Bacterial cGAS-like enzymes synthesize diverse nucleotide signals. Nature. (2019) 567:194–9. doi: 10.1038/s41586-019-0953-5
12. Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. (2013) 498:332–7. doi: 10.1038/nature12305
13. Kranzusch PJ, Lee AS, Berger JM, Doudna, JA. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. (2013) 3:1362–8. doi: 10.1016/j.celrep.2013.05.008
14. Mankan AK, Schmidt T, Chauhan D, Goldeck M, Honing K, Gaidt M, et al. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J. (2014) 33:2937–46. doi: 10.15252/embj.201488726
15. Herzner AM, Hagmann CA, Goldeck M, Wolter S, Kubler K, Wittmann S, et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat Immunol. (2015) 16:1025–33. doi: 10.1038/ni.3267
16. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. (2013) 498:380–4. doi: 10.1038/nature12306
17. Hall J, Ralph EC, Shanker S, Wang H, Byrnes LJ, Horst R, et al. The catalytic mechanism of cyclic GMP-AMP synthase (cGAS) and implications for innate immunity and inhibition. Protein Sci. (2017) 26:2367–80. doi: 10.1002/pro.3304
18. Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. (2014) 6:421–30. doi: 10.1016/j.celrep.2014.01.003
19. Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity. (2013) 39:1019–31. doi: 10.1016/j.immuni.2013.10.019
20. Zhou W, Whiteley AT, de Oliveira Mann CC, Morehouse BR, Nowak RP, Fischer ES, et al. Structure of the human cGAS-DNA complex reveals enhanced control of immune surveillance. Cell. (2018) 174:300–11.e11. doi: 10.1016/j.cell.2018.06.026
21. Andreeva L, Hiller B, Kostrewa D, Lassig C, de Oliveira Mann CC, Jan Drexler D, et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature. (2017) 549:394–8. doi: 10.1038/nature23890
22. Du M, Chen, ZJ. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science. (2018) 361:704–9. doi: 10.1126/science.aat1022
23. Xie W, Lama L, Adura C, Tomita D, Glickman JF, Tuschl T, et al. Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proc Natl Acad Sci USA. (2019) 116:11946–55. doi: 10.1073/pnas.1905013116
24. Barnett KC, Coronas-Serna JM, Zhou W, Ernandes MJ, Cao A, Kranzusch PJ, et al. Phosphoinositide interactions position cGAS at the plasma membrane to ensure efficient distinction between self- and viral DNA. Cell. (2019) 176:1432–46.e11. doi: 10.1016/j.cell.2019.01.049
25. Luecke S, Holleufer A, Christensen MH, Jonsson KL, Boni GA, Sorensen LK, et al. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. (2017) 18:1707–15. doi: 10.15252/embr.201744017
26. Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. (2014) 505:691–5. doi: 10.1038/nature12862
27. Klarquist J, Hennies CM, Lehn MA, Reboulet RA, Feau S, Janssen, EM. STING-mediated DNA sensing promotes antitumor and autoimmune responses to dying cells. J Immunol. (2014) 193:6124–34. doi: 10.4049/jimmunol.1401869
28. Chamilos G, Gregorio J, Meller S, Lande R, Kontoyiannis DP, Modlin RL, et al. Cytosolic sensing of extracellular self-DNA transported into monocytes by the antimicrobial peptide LL37. Blood. (2012) 120:3699–707. doi: 10.1182/blood-2012-01-401364
29. Ahn J, Xia T, Rabasa Capote A, Betancourt D, Barber, GN. Extrinsic phagocyte-dependent STING signaling dictates the immunogenicity of dying cells. Cancer Cell. (2018) 33:862–73.e5. doi: 10.1016/j.ccell.2018.03.027
30. Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci USA. (2015) 112:E5699–705. doi: 10.1073/pnas.1516465112
31. Gkirtzimanaki K, Kabrani E, Nikoleri D, Polyzos A, Blanas A, Sidiropoulos P, et al. IFNalpha impairs autophagic degradation of mtDNA promoting autoreactivity of SLE monocytes in a STING-dependent fashion. Cell Rep. (2018) 25:921–33.e5. doi: 10.1016/j.celrep.2018.09.001
32. Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. (2017) 548:461–5. doi: 10.1038/nature23449
33. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. (2015) 520:553–7. doi: 10.1038/nature14156
34. Price BD, D’Andrea, AD. Chromatin remodeling at DNA double-strand breaks. Cell. (2013) 152:1344–54. doi: 10.1016/j.cell.2013.02.011
35. Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature. (2018) 563:131–6. doi: 10.1038/s41586-018-0629-6
36. Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. (2015) 42:332–43. doi: 10.1016/j.immuni.2015.01.012
37. Heijink AM, Talens F, Jae LT, van Gijn SE, Fehrmann RSN, Brummelkamp TR, et al. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat Commun. (2019) 10:100. doi: 10.1038/s41467-018-07927-y
38. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg, RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. (2017) 548:466–70. doi: 10.1038/nature23470
39. Ho SS, Zhang WY, Tan NY, Khatoo M, Suter MA, Tripathi S, et al. The DNA structure-specific endonuclease MUS81 mediates DNA sensor STING-dependent host rejection of prostate cancer cells. Immunity. (2016) 44:1177–89. doi: 10.1016/j.immuni.2016.04.010
40. Pepin G, Ferrand J, Honing K, Jayasekara WS, Cain JE, Behlke MA, et al. Cre-dependent DNA recombination activates a STING-dependent innate immune response. Nucleic Acids Res. (2016) 44:5356–64. doi: 10.1093/nar/gkw405
41. Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, Funabiki, H. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell. (2019) 178:302–15.e23. doi: 10.1016/j.cell.2019.05.035
42. Bai J, Cervantes C, Liu J, He S, Zhou H, Zhang B, et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc Natl Acad Sci USA. (2017) 114:12196–201. doi: 10.1073/pnas.1708744114
43. White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. (2014) 159:1549–62. doi: 10.1016/j.cell.2014.11.036
44. Snyder HR, Kaiser RH, Warren SL, Heller, W. Obsessive-compulsive disorder is associated with broad impairments in executive function: a meta-analysis. Clin Psychol Sci. (2015) 3:301–30. doi: 10.1177/2167702614534210
45. Mackenzie KJ, Carroll P, Lettice L, Tarnauskaite Z, Reddy K, Dix F, et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. (2016) 35:831–44. doi: 10.15252/embj.201593339
46. Maelfait J, Bridgeman A, Benlahrech A, Cursi C, Rehwinkel, J. Restriction by SAMHD1 limits cGAS/STING-dependent innate and adaptive immune responses to HIV-1. Cell Rep. (2016) 16:1492–501. doi: 10.1016/j.celrep.2016.07.002
47. Sze A, Belgnaoui SM, Olagnier D, Lin R, Hiscott J, van Grevenynghe, J. Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe. (2013) 14:422–34. doi: 10.1016/j.chom.2013.09.009
48. Coquel F, Silva MJ, Techer H, Zadorozhny K, Sharma S, Nieminuszczy J, et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature. (2018) 557:57–61. doi: 10.1038/s41586-018-0050-1
49. Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell. (2013) 51:226–35. doi: 10.1016/j.molcel.2013.05.022
50. Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. (2013) 341:903–6. doi: 10.1126/science.1240933
51. Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, et al. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. (2013) 503:530–4. doi: 10.1038/nature12640
52. Pepin G, De Nardo D, Rootes CL, Ullah TR, Al-Asmari SS, Balka KR, et al. Connexin-dependent transfer of cGAMP to phagocytes modulates antiviral responses. mBio. (2020) 11:e03187-19. doi: 10.1128/mBio.03187-19
53. Schadt L, Sparano C, Schweiger NA, Silina K, Cecconi V, Lucchiari G, et al. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep. (2019) 29:1236–48.e7. doi: 10.1016/j.celrep.2019.09.065
54. Bridgeman A, Maelfait J, Davenne T, Partridge T, Peng Y, Mayer A, et al. Viruses transfer the antiviral second messenger cGAMP between cells. Science. (2015) 349:1228–32. doi: 10.1126/science.aab3632
55. Gentili M, Kowal J, Tkach M, Satoh T, Lahaye X, Conrad C, et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. (2015) 349:1232–6. doi: 10.1126/science.aab3628
56. Xu S, Ducroux A, Ponnurangam A, Vieyres G, Franz S, Musken M, et al. cGAS-mediated innate immunity spreads intercellularly through HIV-1 env-induced membrane fusion sites. Cell Host Microbe. (2016) 20:443–57. doi: 10.1016/j.chom.2016.09.003
57. Torralba D, Baixauli F, Villarroya-Beltri C, Fernandez-Delgado I, Latorre-Pellicer A, Acin-Perez R, et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat Commun. (2018) 9:2658. doi: 10.1038/s41467-018-05077-9
58. Kalamvoki M, Du T, Roizman, B. Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, microRNAs. Proc Natl Acad Sci USA. (2014) 111:E4991–6. doi: 10.1073/pnas.1419338111
59. Ritchie C, Cordova AF, Hess GT, Bassik MC, Li, L. SLC19A1 is an importer of the immunotransmitter cGAMP. Mol Cell. (2019) 75:372–81.e5. doi: 10.1016/j.molcel.2019.05.006
60. Luteijn RD, Zaver SA, Gowen BG, Wyman SK, Garelis NE, Onia L, et al. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature. (2019) 573:434–8. doi: 10.1038/s41586-019-1553-0
61. Li L, Yin Q, Kuss P, Maliga Z, Millan JL, Wu H, et al. Hydrolysis of 2′3′-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat Chem Biol. (2014) 10:1043–8. doi: 10.1038/nchembio.1661
62. Liu H, Moura-Alves P, Pei G, Mollenkopf HJ, Hurwitz R, Wu X, et al. cGAS facilitates sensing of extracellular cyclic dinucleotides to activate innate immunity. EMBO Rep. (2019) 20:e46293. doi: 10.15252/embr.201846293
63. Ishikawa H, Ma Z, Barber, GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. (2009) 461:788–92. doi: 10.1038/nature08476
64. Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan, NSTING. Activation by translocation from the ER is associated with infection and Autoinflammatory disease. Cell Host Microbe. (2015) 18:157–68. doi: 10.1016/j.chom.2015.07.001
65. Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA. (2009) 106:20842–6. doi: 10.1073/pnas.0911267106
66. Luo WW, Li S, Li C, Lian H, Yang Q, Zhong B, et al. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat Immunol. (2016) 17:1057–66. doi: 10.1038/ni.3510
67. Sun MS, Zhang J, Jiang LQ, Pan YX, Tan JY, Yu F, et al. TMED2 potentiates cellular IFN responses to DNA viruses by reinforcing MITA dimerization and facilitating its trafficking. Cell Rep. (2018) 25:3086–98.e3. doi: 10.1016/j.celrep.2018.11.048
68. Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. (2019) 567:262–6. doi: 10.1038/s41586-019-1006-9
69. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS STING, TRIF induces IRF3 activation. Science. (2015) 347:aaa2630. doi: 10.1126/science.aaa2630
70. Tanaka Y, Chen, ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. (2012) 5:ra20. doi: 10.1126/scisignal.2002521
71. de Oliveira Mann CC, Orzalli MH, King DS, Kagan JC, Lee ASY, Kranzusch, PJ. Modular architecture of the STING C-terminal tail allows interferon and NF-kappaB signaling adaptation. Cell Rep. (2019) 27:1165–75.e5. doi: 10.1016/j.celrep.2019.03.098
72. Zhang C, Shang G, Gui X, Zhang X, Bai XC, Chen, ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature. (2019) 567:394–8. doi: 10.1038/s41586-019-1000-2
73. Zhao B, Du F, Xu P, Shu C, Sankaran B, Bell SL, et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature. (2019) 569:718–22. doi: 10.1038/s41586-019-1228-x
74. Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, et al. Activation of STING requires palmitoylation at the Golgi. Nat Commun. (2016) 7:11932. doi: 10.1038/ncomms11932
75. Ni G, Konno H, Barber, GN. Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci Immunol. (2017) 2:eaah7119. doi: 10.1126/sciimmunol.aah7119
76. Hou Y, Liang H, Rao E, Zheng W, Huang X, Deng L, et al. Non-canonical NF-kappaB antagonizes STING sensor-mediated DNA sensing in radiotherapy. Immunity. (2018) 49:490–503.e4. doi: 10.1016/j.immuni.2018.07.008
77. Gonzalez-Navajas JM, Lee J, David M, Raz, E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. (2012) 12:125–35. doi: 10.1038/nri3133
78. Andrilenas KK, Ramlall V, Kurland J, Leung B, Harbaugh AG, Siggers, T. DNA-binding landscape of IRF3, IRF5 and IRF7 dimers: implications for dimer-specific gene regulation. Nucleic Acids Res. (2018) 46:2509–20. doi: 10.1093/nar/gky002
79. Prabakaran T, Bodda C, Krapp C, Zhang BC, Christensen MH, Sun C, et al. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J. (2018) 37:e97858. doi: 10.15252/embj.201797858
80. Ma F, Li B, Liu SY, Iyer SS, Yu Y, Wu A, et al. Positive feedback regulation of type I IFN production by the IFN-inducible DNA sensor cGAS. J Immunol. (2015) 194:1545–54. doi: 10.4049/jimmunol.1402066
81. Ma F, Li B, Yu Y, Iyer SS, Sun M, Cheng, G. Positive feedback regulation of type I interferon by the interferon-stimulated gene STING. EMBO Rep. (2015) 16:202–12. doi: 10.15252/embr.201439366
82. Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ, et al. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med. (2019) 216:867–83. doi: 10.1084/jem.20182192
83. Moretti J, Roy S, Bozec D, Martinez J, Chapman JR, Ueberheide B, et al. STING senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell. (2017) 171:809–23.e13. doi: 10.1016/j.cell.2017.09.034
84. Hasan M, Gonugunta VK, Dobbs N, Ali A, Palchik G, Calvaruso MA, et al. Chronic innate immune activation of TBK1 suppresses mTORC1 activity and dysregulates cellular metabolism. Proc Natl Acad Sci USA. (2017) 114:746–51. doi: 10.1073/pnas.1611113114
85. Saxton RA, Sabatini, DM. mTOR signaling in growth metabolism, disease. Cell. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
86. Liang Q, Seo GJ, Choi YJ, Kwak MJ, Ge J, Rodgers MA, et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe. (2014) 15:228–38. doi: 10.1016/j.chom.2014.01.009
87. Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol. (2018) 20:521–7. doi: 10.1038/s41556-018-0092-5
88. Clarke AJ, Simon, AK. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol. (2019) 19:170–83. doi: 10.1038/s41577-018-0095-2
89. Liu D, Wu H, Wang C, Li Y, Tian H, Siraj S, et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. (2019) 26:1735–49. doi: 10.1038/s41418-018-0251-z
90. Koster S, Upadhyay S, Chandra P, Papavinasasundaram K, Yang G, Hassan A, et al. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc Natl Acad Sci USA. (2017) 114:E8711–20. doi: 10.1073/pnas.1707792114
91. Watson RO, Manzanillo PS, Cox, JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. (2012) 150:803–15. doi: 10.1016/j.cell.2012.06.040
92. Senft D, Ronai, ZA. UPR autophagy, mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. (2015) 40:141–8. doi: 10.1016/j.tibs.2015.01.002
93. Lan YY, Londono D, Bouley R, Rooney MS, Hacohen, N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep. (2014) 9:180–92. doi: 10.1016/j.celrep.2014.08.074
94. Marino G, Niso-Santano M, Baehrecke EH, Kroemer, G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. (2014) 15:81–94. doi: 10.1038/nrm3735
95. Gonugunta VK, Sakai T, Pokatayev V, Yang K, Wu J, Dobbs N, et al. Trafficking-mediated STING degradation requires sorting to acidified endolysosomes and can be targeted to enhance anti-tumor response. Cell Rep. (2017) 21:3234–42. doi: 10.1016/j.celrep.2017.11.061
96. Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, et al. The DNA inflammasome in human myeloid cells is initiated by a STING-Cell death program upstream of NLRP3. Cell. (2017) 171:1110–24.e18. doi: 10.1016/j.cell.2017.09.039
97. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. (2009) 458:514–8. doi: 10.1038/nature07725
98. Corrales L, Woo SR, Williams JB, McWhirter SM, Dubensky, TW Jr., and Gajewski, TF. Antagonism of the STING pathway via activation of the AIM2 inflammasome by intracellular DNA. J Immunol. (2016) 196:3191–8. doi: 10.4049/jimmunol.1502538
99. Wang Y, Ning X, Gao P, Wu S, Sha M, Lv M, et al. Inflammasome activation triggers caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity. (2017) 46:393–404. doi: 10.1016/j.immuni.2017.02.011
100. Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, et al. Gasdermin D restrains Type I interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity. (2018) 49:413–26.e5. doi: 10.1016/j.immuni.2018.07.006
101. Swanson KV, Junkins RD, Kurkjian CJ, Holley-Guthrie E, Pendse AA, El Morabiti R, et al. A noncanonical function of cGAMP in inflammasome priming and activation. J Exp Med. (2017) 214:3611–26. doi: 10.1084/jem.20171749
102. Li X, Deng M, Petrucelli AS, Zhu C, Mo J, Zhang L, et al. Viral DNA binding to NLRC3, an inhibitory nucleic acid sensor, unleashes STING, a cyclic dinucleotide receptor that activates Type I interferon. Immunity. (2019) 50:591–9.e6. doi: 10.1016/j.immuni.2019.02.009
103. Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jonsson KL, et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-kappaB signaling after nuclear DNA damage. Mol Cell. (2018) 71:745–60.e5. doi: 10.1016/j.molcel.2018.07.034
104. Orzalli MH, DeLuca NA, Knipe, DM. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci USA. (2012) 109:E3008–17. doi: 10.1073/pnas.1211302109
105. Jonsson KL, Laustsen A, Krapp C, Skipper KA, Thavachelvam K, Hotter D, et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat Commun. (2017) 8:14391. doi: 10.1038/ncomms14391
106. Almine JF, O’Hare CA, Dunphy G, Haga IR, Naik RJ, Atrih A, et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun. (2017) 8:14392. doi: 10.1038/ncomms14392
107. Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, et al. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci. (2015) 112:E1773–81. doi: 10.1073/pnas.1424637112
108. Diner BA, Lum KK, Toettcher JE, Cristea, IM. Viral DNA sensors IFI16 and cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, apoptotic responses during herpesvirus infection. mBio. (2016) 7:e01553-16. doi: 10.1128/mBio.01553-16
109. Li D, Wu R, Guo W, Xie L, Qiao Z, Chen S, et al. STING-mediated IFI16 degradation negatively controls Type I interferon production. Cell Rep. (2019) 29:1249–60.e4. doi: 10.1016/j.celrep.2019.09.069
110. Gay NJ, Symmons MF, Gangloff M, Bryant, CE. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol. (2014) 14:546–58. doi: 10.1038/nri3713
111. Wang X, Majumdar T, Kessler P, Ozhegov E, Zhang Y, Chattopadhyay S, et al. STING requires the adaptor TRIF to trigger innate immune responses to microbial infection. Cell Host Microbe. (2016) 20:329–41. doi: 10.1016/j.chom.2016.08.002
112. Xu RH, Wong EB, Rubio D, Roscoe F, Ma X, Nair S, et al. Sequential activation of two pathogen-sensing pathways required for Type I Interferon expression and resistance to an acute DNA virus infection. Immunity. (2015) 43:1148–59. doi: 10.1016/j.immuni.2015.11.015
113. Yu X, Cai B, Wang M, Tan P, Ding X, Wu J, et al. Cross-regulation of two Type I interferon signaling pathways in plasmacytoid dendritic cells controls anti-malaria immunity and host mortality. Immunity. (2016) 45:1093–107. doi: 10.1016/j.immuni.2016.10.001
114. Gehrke N, Mertens C, Zillinger T, Wenzel J, Bald T, Zahn S, et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity. (2013) 39:482–95. doi: 10.1016/j.immuni.2013.08.004
115. Tait SW, Green, DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. (2010) 11:621–32. doi: 10.1038/nrm2952
116. Riley JS, Quarato G, Cloix C, Lopez J, O’Prey J, Pearson M, et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. (2018) 37:e99238. doi: 10.15252/embj.201899238
117. Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, et al. Apoptotic caspases prevent the induction of Type I interferons by mitochondrial DNA. Cell. (2014) 159:1563–77. doi: 10.1016/j.cell.2014.11.037
118. Ning X, Wang Y, Jing M, Sha M, Lv M, Gao P, et al. Apoptotic caspases suppress Type I interferon production via the cleavage of cGAS MAVS, IRF3. Mol Cell. (2019) 74:19–31.e7. doi: 10.1016/j.molcel.2019.02.013
119. Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, et al. Signalling strength determines proapoptotic functions of STING. Nat Commun. (2017) 8:427. doi: 10.1038/s41467-017-00573-w
120. Chen D, Tong J, Yang L, Wei L, Stolz DB, Yu J, et al. PUMA amplifies necroptosis signaling by activating cytosolic DNA sensors. Proc Natl Acad Sci USA. (2018) 115:3930–5. doi: 10.1073/pnas.1717190115
121. Nassour J, Radford R, Correia A, Fuste JM, Schoell B, Jauch A, et al. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature. (2019) 565:659–63. doi: 10.1038/s41586-019-0885-0
122. Schock SN, Chandra NV, Sun Y, Irie T, Kitagawa Y, Gotoh B, et al. Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ. (2017) 24:615–25. doi: 10.1038/cdd.2016.153
123. Francica BJ, Ghasemzadeh A, Desbien AL, Theodros D, Sivick KE, Reiner GL, et al. TNFalpha and radioresistant stromal cells are essential for therapeutic efficacy of cyclic dinucleotide STING agonists in nonimmunogenic tumors. Cancer Immunol Res. (2018) 6:422–33. doi: 10.1158/2326-6066.CIR-17-0263
124. Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti, A. Cellular senescence: aging. Cancer Injury Physiol Rev. (2019) 99:1047–78. doi: 10.1152/physrev.00020.2018
125. Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. (2006) 443:453–7. doi: 10.1038/nature05092
126. Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, et al. Autophagy mediates degradation of nuclear lamina. Nature. (2015) 527:105–9. doi: 10.1038/nature15548
127. Lan YY, Heather JM, Eisenhaure T, Garris CS, Lieb D, Raychowdhury R, et al. Extranuclear DNA accumulates in aged cells and contributes to senescence and inflammation. Aging Cell. (2019) 18:e12901. doi: 10.1111/acel.12901
128. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
129. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. (2017) 550:402–6. doi: 10.1038/nature24050
130. Takahashi A, Loo TM, Okada R, Kamachi F, Watanabe Y, Wakita M, et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun. (2018) 9:1249. doi: 10.1038/s41467-018-03555-8
131. Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. (2017) 19:1061–70. doi: 10.1038/ncb3586
132. Yang H, Wang H, Ren J, Chen Q, Chen, ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci USA. (2017) 114:E4612–20. doi: 10.1073/pnas.1705499114
133. Yu Q, Katlinskaya YV, Carbone CJ, Zhao B, Katlinski KV, Zheng H, et al. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep. (2015) 11:785–97. doi: 10.1016/j.celrep.2015.03.069
134. Kobayashi H, Kobayashi CI, Nakamura-Ishizu A, Karigane D, Haeno H, Yamamoto KN, et al. Bacterial c-di-GMP affects hematopoietic stem/progenitors and their niches through STING. Cell Rep. (2015) 11:71–84. doi: 10.1016/j.celrep.2015.02.066
135. Xia P, Wang S, Ye B, Du Y, Li C, Xiong Z, et al. A circular RNA protects dormant hematopoietic stem cells from DNA sensor cGAS-mediated exhaustion. Immunity. (2018) 48:688–701.e7. doi: 10.1016/j.immuni.2018.03.016
136. Yoh SM, Schneider M, Seifried J, Soonthornvacharin S, Akleh RE, Olivieri KC, et al. PQBP1 Is a proximal sensor of the cGAS-dependent innate response to HIV-1. Cell. (2015) 161:1293–305. doi: 10.1016/j.cell.2015.04.050
137. Lahaye X, Gentili M, Silvin A, Conrad C, Picard L, Jouve M, et al. NONO detects the nuclear HIV capsid to promote cGAS-mediated innate immune activation. Cell. (2018) 175:488–501.e22. doi: 10.1016/j.cell.2018.08.062
138. Adriaens C, Standaert L, Barra J, Latil M, Verfaillie A, Kalev P, et al. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat Med. (2016) 22:861–8. doi: 10.1038/nm.4135
139. Morchikh M, Cribier A, Raffel R, Amraoui S, Cau J, Severac D, et al. HEXIM1 and NEAT1 long non-coding RNA form a multi-subunit complex that regulates DNA-mediated innate immune response. Mol Cell. (2017) 67:387–99.e5. doi: 10.1016/j.molcel.2017.06.020
140. Franz KM, Neidermyer WJ, Tan YJ, Whelan SPJ, Kagan, JC. STING-dependent translation inhibition restricts RNA virus replication. Proc Natl Acad Sci USA. (2018) 115:E2058–67. doi: 10.1073/pnas.1716937115
141. Esbjornsson J, Mansson F, Kvist A, Isberg PE, Nowroozalizadeh S, Biague AJ, et al. Inhibition of HIV-1 disease progression by contemporaneous HIV-2 infection. N Engl J Med. (2012) 367:224–32. doi: 10.1056/NEJMoa1113244
142. Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. (2011) 474:654–7. doi: 10.1038/nature10117
143. Johnson JS, Lucas SY, Amon LM, Skelton S, Nazitto R, Carbonetti S, et al. Reshaping of the dendritic cell chromatin landscape and interferon pathways during HIV infection. Cell Host Microbe. (2018) 23:366–81.e9. doi: 10.1016/j.chom.2018.01.012
144. Lahaye X, Satoh T, Gentili M, Cerboni S, Conrad C, Hurbain I, et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. (2013) 39:1132–42. doi: 10.1016/j.immuni.2013.11.002
145. Kumar S, Morrison JH, Dingli D, Poeschla, E. HIV-1 activation of innate immunity depends strongly on the intracellular level of TREX1 and sensing of incomplete reverse transcription products. J Virol. (2018) 92:e00001-18. doi: 10.1128/JVI.00001-18
146. Jacques DA, McEwan WA, Hilditch L, Price AJ, Towers GJ, James, LC. HIV-1 uses dynamic capsid pores to import nucleotides and fuel encapsidated DNA synthesis. Nature. (2016) 536:349–53. doi: 10.1038/nature19098
147. Ganser-Pornillos BK, Pornillos, O. Restriction of HIV-1 and other retroviruses by TRIM5. Nat Rev Microbiol. (2019) 17:546–56. doi: 10.1038/s41579-019-0225-2
148. Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. (2013) 503:402–5. doi: 10.1038/nature12769
149. Jenal U, Reinders A, Lori, C. Cyclic di-GMP: second messenger extraordinaire. Nat Rev Microbiol. (2017) 15:271–84. doi: 10.1038/nrmicro.2016.190
150. Cohen D, Melamed S, Millman A, Shulman G, Oppenheimer-Shaanan Y, Kacen A, et al. Cyclic GMP-AMP signalling protects bacteria against viral infection. Nature. (2019) 574:691–5. doi: 10.1038/s41586-019-1605-5
151. Dey B, Dey RJ, Cheung LS, Pokkali S, Guo H, Lee JH, et al. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat Med. (2015) 21:401–6. doi: 10.1038/nm.3813
152. Philips JA, Ernst, JD. Tuberculosis pathogenesis and immunity. Annu Rev Pathol. (2012) 7:353–84. doi: 10.1146/annurev-pathol-011811-132458
153. Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, et al. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe. (2015) 17:799–810. doi: 10.1016/j.chom.2015.05.003
154. Costa Franco MM, Marim F, Guimaraes ES, Assis NRG, Cerqueira DM, Alves-Silva J, et al. Brucella abortus triggers a cGAS-independent STING pathway to induce host protection that involves guanylate-binding proteins and inflammasome activation. J Immunol. (2018) 200:607–22. doi: 10.4049/jimmunol.1700725
155. Rajani S, Archana R, Indla YR, Rajesh, P. Beneficial effects of Yogasanas and Pranayama in limiting the cognitive decline in Type 2 Diabetes. Natl J Physiol Pharm Pharmacol. (2017) 7:232–5. doi: 10.5455/njppp.2017.7.0825508092016
156. Andrade WA, Firon A, Schmidt T, Hornung V, Fitzgerald KA, Kurt-Jones EA, et al. Group B Streptococcus degrades cyclic-di-AMP to modulate STING-dependent Type I interferon production. Cell Host Microbe. (2016) 20:49–59. doi: 10.1016/j.chom.2016.06.003
157. Dey RJ, Dey B, Zheng Y, Cheung LS, Zhou J, Sayre D, et al. Inhibition of innate immune cytosolic surveillance by an M. tuberculosis phosphodiesterase. Nat Chem Biol. (2017) 13:210–7. doi: 10.1038/nchembio.2254
158. Weber MM, Lam JL, Dooley CA, Noriea NF, Hansen BT, Hoyt FH, et al. Absence of specific Chlamydia trachomatis inclusion membrane proteins triggers premature inclusion membrane lysis and host cell death. Cell Rep. (2017) 19:1406–17. doi: 10.1016/j.celrep.2017.04.058
159. Sixt BS, Bastidas RJ, Finethy R, Baxter RM, Carpenter VK, Kroemer G, et al. The Chlamydia trachomatis inclusion membrane protein CpoS counteracts STING-mediated cellular surveillance and suicide programs. Cell Host Microbe. (2017) 21:113–21. doi: 10.1016/j.chom.2016.12.002
160. Cao Y, Guan K, He X, Wei C, Zheng Z, Zhang Y, et al. Yersinia YopJ negatively regulates IRF3-mediated antibacterial response through disruption of STING-mediated cytosolic DNA signaling. Biochim Biophys Acta. (2016) 1863:3148–59. doi: 10.1016/j.bbamcr.2016.10.004
161. Nandakumar R, Tschismarov R, Meissner F, Prabakaran T, Krissanaprasit A, Farahani E, et al. Intracellular bacteria engage a STING-TBK1-MVB12b pathway to enable paracrine cGAS-STING signalling. Nat Microbiol. (2019) 4:701–13. doi: 10.1038/s41564-019-0367-z
162. Archer KA, Durack J, Portnoy, DA. STING-dependent type I IFN production inhibits cell-mediated immunity to Listeria monocytogenes. PLoS Pathog. (2014) 10:e1003861. doi: 10.1371/journal.ppat.1003861
163. Majumdar T, Chattopadhyay S, Ozhegov E, Dhar J, Goswami R, Sen GC, et al. Induction of interferon-stimulated genes by IRF3 promotes replication of Toxoplasma gondii. PLoS Pathog. (2015) 11:e1004779. doi: 10.1371/journal.ppat.1004779
164. Sisquella X, Ofir-Birin Y, Pimentel MA, Cheng L, Abou Karam P, Sampaio NG, et al. Malaria parasite DNA-harbouring vesicles activate cytosolic immune sensors. Nat Commun. (2017) 8:1985. doi: 10.1038/s41467-017-02083-1
165. Muskardin TLW, Niewold, TB. Type I interferon in rheumatic diseases. Nat Rev Rheumatol. (2018) 14:214–28. doi: 10.1038/nrrheum.2018.31
166. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
167. Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest. (2014) 124:5516–20. doi: 10.1172/JCI79100
168. Munoz J, Rodiere M, Jeremiah N, Rieux-Laucat F, Oojageer A, Rice GI, et al. Stimulator of interferon genes-associated vasculopathy with onset in infancy: a mimic of childhood Granulomatosis with Polyangiitis. JAMA Dermatol. (2015) 151:872–7. doi: 10.1001/jamadermatol.2015.0251
169. Bouis D, Kirstetter P, Arbogast F, Lamon D, Delgado V, Jung S, et al. Severe combined immunodeficiency in stimulator of interferon genes (STING) V154M/wild-type mice. J Allergy Clin Immunol. (2019) 143:712–25.e5. doi: 10.1016/j.jaci.2018.04.034
170. Warner JD, Irizarry-Caro RA, Bennion BG, Ai TL, Smith AM, Miner CA, et al. STING-associated vasculopathy develops independently of IRF3 in mice. J Exp Med. (2017) 214:3279–92. doi: 10.1084/jem.20171351
171. Melki I, Rose Y, Uggenti C, Van Eyck L, Fremond ML, Kitabayashi N, et al. Disease-associated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J Allergy Clin Immunol. (2017) 140:543–52.e5. doi: 10.1016/j.jaci.2016.10.031
172. Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell. (2016) 165:1548–50. doi: 10.1016/j.cell.2016.05.057
173. Crow YJ, Manel, N. Aicardi-goutieres syndrome and the type I interferonopathies. Nat Rev Immunol. (2015) 15:429–40. doi: 10.1038/nri3850
174. Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum Dis. (2018) 77:1507–15. doi: 10.1136/annrheumdis-2018-212988
175. An J, Durcan L, Karr RM, Briggs TA, Rice GI, Teal TH, et al. Expression of cyclic GMP-AMP synthase in patients with systemic lupus erythematosus. Arthritis Rheumatol. (2017) 69:800–7. doi: 10.1002/art.40002
176. Ganguly D, Haak S, Sisirak V, Reizis, B. The role of dendritic cells in autoimmunity. Nat Rev Immunol. (2013) 13:566–77. doi: 10.1038/nri3477
177. Bode C, Fox M, Tewary P, Steinhagen A, Ellerkmann RK, Klinman D, et al. Human plasmacytoid dentritic cells elicit a Type I interferon response by sensing DNA via the cGAS-STING signaling pathway. Eur J Immunol. (2016) 46:1615–21. doi: 10.1002/eji.201546113
178. Laustsen A, Bak RO, Krapp C, Kjaer L, Egedahl JH, Petersen CC, et al. Interferon priming is essential for human CD34+ cell-derived plasmacytoid dendritic cell maturation and function. Nat Commun. (2018) 9:3525. doi: 10.1038/s41467-018-05816-y
179. Aarreberg LD, Esser-Nobis K, Driscoll C, Shuvarikov A, Roby JA, Gale, MJr Interleukin-1beta induces mtDNA release to activate innate immune signaling via cGAS-STING. Mol Cell. (2019) 74:801–15.e6. doi: 10.1016/j.molcel.2019.02.038
180. Blanco P, Palucka AK, Gill M, Pascual V, Banchereau, J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. (2001) 294:1540–3. doi: 10.1126/science.1064890
181. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
182. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
183. Gul E, Sayar EH, Gungor B, Eroglu FK, Surucu N, Keles S, et al. Type I IFN-related NETosis in ataxia telangiectasia and Artemis deficiency. J Allergy Clin Immunol. (2018) 142:246–57. doi: 10.1016/j.jaci.2017.10.030
184. Cao DJ, Schiattarella GG, Villalobos E, Jiang N, May HI, Li T, et al. Cytosolic DNA sensing promotes macrophage transformation and governs myocardial ischemic injury. Circulation. (2018) 137:2613–34. doi: 10.1161/CIRCULATIONAHA.117.031046
185. King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng, RP Jr. et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med. (2017) 23:1481–7. doi: 10.1038/nm.4428
186. Qiao JT, Cui C, Qing L, Wang LS, He TY, Yan F, et al. Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism. (2018) 81:13–24. doi: 10.1016/j.metabol.2017.09.010
187. Luo X, Li H, Ma L, Zhou J, Guo X, Woo SL, et al. Expression of STING Is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology. (2018) 155:1971–84.e4. doi: 10.1053/j.gastro.2018.09.010
188. Yu Y, Liu Y, An W, Song J, Zhang Y, Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J Clin Invest. (2019) 129:546–55. doi: 10.1172/JCI121842
189. Cho CS, Park HW, Ho A, Semple IA, Kim B, Jang I, et al. Lipotoxicity induces hepatic protein inclusions through TANK binding kinase 1-mediated p62/sequestosome 1 phosphorylation. Hepatology. (2018) 68:1331–46. doi: 10.1002/hep.29742
190. Mao Y, Luo W, Zhang L, Wu W, Yuan L, Xu H, et al. STING-IRF3 Triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in diet-induced obesity. Arterioscler Thromb Vasc Biol. (2017) 37:920–9. doi: 10.1161/ATVBAHA.117.309017
191. Witcher KG, Eiferman DS, Godbout, JP. Priming the inflammatory pump of the CNS after traumatic brain injury. Trends Neurosci. (2015) 38:609–20. doi: 10.1016/j.tins.2015.08.002
192. Abdullah A, Zhang M, Frugier T, Bedoui S, Taylor JM, Crack, PJ. STING-mediated type-I interferons contribute to the neuroinflammatory process and detrimental effects following traumatic brain injury. J Neuroinflammation. (2018) 15:323. doi: 10.1186/s12974-018-1354-7
193. Benmerzoug S, Rose S, Bounab B, Gosset D, Duneau L, Chenuet P, et al. STING-dependent sensing of self-DNA drives silica-induced lung inflammation. Nat Commun. (2018) 9:5226. doi: 10.1038/s41467-018-07425-1
194. Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, et al. Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab. (2019) 30:784–99.e5. doi: 10.1016/j.cmet.2019.08.003
195. Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H, et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep. (2019) 29:1261–73.e6. doi: 10.1016/j.celrep.2019.09.050
196. Ransohoff, RM. How neuroinflammation contributes to neurodegeneration. Science. (2016) 353:777–83. doi: 10.1126/science.aag2590
197. Reinert LS, Lopusna K, Winther H, Sun C, Thomsen MK, Nandakumar R, et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun. (2016) 7:13348. doi: 10.1038/ncomms13348
198. Nazmi A, Field RH, Griffin EW, Haugh O, Hennessy E, Cox D, et al. Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia. (2019) 67:1254–76. doi: 10.1002/glia.23592
199. Pickrell AM, Youle, RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. (2015) 85:257–73. doi: 10.1016/j.neuron.2014.12.007
200. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. (2018) 561:258–62. doi: 10.1038/s41586-018-0448-9
201. McKinnon, PJ. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol. (2012) 7:303–21. doi: 10.1146/annurev-pathol-011811-132509
202. Ammann AJ, Hong, R. Autoimmune phenomena in ataxia telangiectasia. J Pediatr. (1971) 78:821–6. doi: 10.1016/s0022-3476(71)80353-0
203. Dib B, Lin H, Maidana DE, Tian B, Miller JB, Bouzika P, et al. Mitochondrial DNA has a pro-inflammatory role in AMD. Biochim Biophys Acta. (2015) 1853(11 Pt A):2897–906. doi: 10.1016/j.bbamcr.2015.08.012
204. Kerur N, Fukuda S, Banerjee D, Kim Y, Fu D, Apicella I, et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat Med. (2018) 24:50–61. doi: 10.1038/nm.4450
205. Reich DS, Lucchinetti CF, Calabresi, PA. Multiple sclerosis. N Engl J Med. (2018) 378:169–80. doi: 10.1056/NEJMra1401483
206. Deczkowska A, Baruch K, Schwartz, M. Type I/II interferon balance in the regulation of brain physiology and pathology. Trends Immunol. (2016) 37:181–92. doi: 10.1016/j.it.2016.01.006
207. Mathur V, Burai R, Vest RT, Bonanno LN, Lehallier B, Zardeneta ME, et al. Activation of the STING-dependent Type I interferon response reduces microglial reactivity and neuroinflammation. Neuron. (2017) 96:1290–302.e6. doi: 10.1016/j.neuron.2017.11.032
208. Hart PH, Gorman S, Finlay-Jones, JJ. Modulation of the immune system by UV radiation: more than just the effects of vitamin D? Nat Rev Immunol. (2011) 11:584–96. doi: 10.1038/nri3045
209. Sontheimer C, Liggitt D, Elkon, KB. Ultraviolet b irradiation causes stimulator of interferon genes-dependent production of protective Type I interferon in mouse skin by recruited inflammatory monocytes. Arthritis Rheumatol. (2017) 69:826–36. doi: 10.1002/art.39987
210. Huang L, Lemos HP, Li L, Li M, Chandler PR, Baban B, et al. Engineering DNA nanoparticles as immunomodulatory reagents that activate regulatory T cells. J Immunol. (2012) 188:4913–20. doi: 10.4049/jimmunol.1103668
211. Lemos H, Huang L, Chandler PR, Mohamed E, Souza GR, Li L, et al. Activation of the STING adaptor attenuates experimental autoimmune encephalitis. J Immunol. (2014) 192:5571–8. doi: 10.4049/jimmunol.1303258
212. Imanishi T, Ishihara C, Badr Mel S, Hashimoto-Tane A, Kimura Y, Kawai T, et al. Nucleic acid sensing by T cells initiates Th2 cell differentiation. Nat Commun. (2014) 5:3566. doi: 10.1038/ncomms4566
213. Luksch H, Stinson WA, Platt DJ, Qian W, Kalugotla G, Miner CA, et al. STING-associated lung disease in mice relies on T cells but not type I interferon. J Allergy Clin Immunol. (2019) 144:254–66.e8. doi: 10.1016/j.jaci.2019.01.044
214. Motwani M, Pawaria S, Bernier J, Moses S, Henry K, Fang T, et al. Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models. Proc Natl Acad Sci USA. (2019) 116:7941–50. doi: 10.1073/pnas.1818281116
215. Cerboni S, Jeremiah N, Gentili M, Gehrmann U, Conrad C, Stolzenberg MC, et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J Exp Med. (2017) 214:1769–85. doi: 10.1084/jem.20161674
216. Tang CH, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, et al. Agonist-Mediated activation of STING induces apoptosis in malignant B Cells. Cancer Res. (2016) 76:2137–52. doi: 10.1158/0008-5472.CAN-15-1885
217. Walker MM, Crute BW, Cambier JC, Getahun, A. B cell-Intrinsic STING signaling triggers cell activation, synergizes with B cell receptor signals, promotes antibody responses. J Immunol. (2018) 201:2641–53. doi: 10.4049/jimmunol.1701405
218. Zhao Q, Wei Y, Pandol SJ, Li L, Habtezion, A. STING signaling promotes inflammation in experimental acute pancreatitis. Gastroenterology. (2018) 154:1822–35.e2. doi: 10.1053/j.gastro.2018.01.065
219. Zhao Q, Manohar M, Wei Y, Pandol SJ, Habtezion, A. STING signalling protects against chronic pancreatitis by modulating Th17 response. Gut. (2019) 68:1827–37. doi: 10.1136/gutjnl-2018-317098
220. Van Dis E, Sogi KM, Rae CS, Sivick KE, Surh NH, Leong ML, et al. STING-activating adjuvants elicit a Th17 immune response and protect against Mycobacterium tuberculosis infection. Cell Rep. (2018) 23:1435–47. doi: 10.1016/j.celrep.2018.04.003
221. Ahn J, Son S, Oliveira SC, Barber, GN. STING-dependent signaling underlies IL-10 controlled inflammatory colitis. Cell Rep. (2017) 21:3873–84. doi: 10.1016/j.celrep.2017.11.101
222. Canesso MCC, Lemos L, Neves TC, Marim FM, Castro TBR, Veloso ES, et al. The cytosolic sensor STING is required for intestinal homeostasis and control of inflammation. Mucosal Immunol. (2018) 11:820–34. doi: 10.1038/mi.2017.88
223. Bakhoum SF, Cantley, LC. The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell. (2018) 174:1347–60. doi: 10.1016/j.cell.2018.08.027
224. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. (2017) 17:97–111. doi: 10.1038/nri.2016.107
225. Ohkuri T, Ghosh A, Kosaka A, Zhu J, Ikeura M, David M, et al. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol Res. (2014) 2:1199–208. doi: 10.1158/2326-6066.CIR-14-0099
226. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. (2014) 41:830–42. doi: 10.1016/j.immuni.2014.10.017
227. Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING associated with LKB1 Loss in KRAS-driven lung cancer. Cancer Discov. (2019) 9:34–45. doi: 10.1158/2159-8290.CD-18-0689
228. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. (2017) 8:15618. doi: 10.1038/ncomms15618
229. Andzinski L, Spanier J, Kasnitz N, Kroger A, Jin L, Brinkmann MM, et al. Growing tumors induce a local STING dependent Type I IFN response in dendritic cells. Int J Cancer. (2016) 139:1350–7. doi: 10.1002/ijc.30159
230. Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet, DH. Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity. (2018) 49:754–63.e4. doi: 10.1016/j.immuni.2018.09.016
231. Lam AR, Bert NL, Ho SS, Shen YJ, Tang LF, Xiong GM, et al. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res. (2014) 74:2193–203. doi: 10.1158/0008-5472.CAN-13-1703
232. Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest. (2019) 130:4350–64. doi: 10.1172/JCI125413
233. Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, et al. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross-priming through signal regulatory protein alpha signaling. Immunity. (2017) 47:363–73.e5. doi: 10.1016/j.immuni.2017.07.016
234. Diamond JM, Vanpouille-Box C, Spada S, Rudqvist NP, Chapman JR, Ueberheide BM, et al. Exosomes shuttle TREX1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol Res. (2018) 6:910–20. doi: 10.1158/2326-6066.CIR-17-0581
235. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. (2011) 208:2005–16. doi: 10.1084/jem.20101159
236. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced Type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. (2014) 41:843–52. doi: 10.1016/j.immuni.2014.10.019
237. Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. (2019) 9:646–61. doi: 10.1158/2159-8290.CD-18-1020
238. Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran, MA. Intratumoral STING activation with T-cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol Res. (2017) 5:676–84. doi: 10.1158/2326-6066.CIR-17-0049
239. Zhang CX, Ye SB, Ni JJ, Cai TT, Liu YN, Huang DJ, et al. STING signaling remodels the tumor microenvironment by antagonizing myeloid-derived suppressor cell expansion. Cell Death Differ. (2019) 26:2314–28. doi: 10.1038/s41418-019-0302-0
240. Santana Carrero RM, Beceren-Braun F, Rivas SC, Hegde SM, Gangadharan A, Plote D, et al. IL-15 is a component of the inflammatory milieu in the tumor microenvironment promoting antitumor responses. Proc Natl Acad Sci USA. (2019) 116:599–608. doi: 10.1073/pnas.1814642116
241. Konno H, Yamauchi S, Berglund A, Putney RM, Mule JJ, Barber, GN. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene. (2018) 37:2037–51. doi: 10.1038/s41388-017-0120-0
242. Chen YA, Shen YL, Hsia HY, Tiang YP, Sung TL, Chen, LY. Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS-STING DNA sensing pathway. Nat Struct Mol Biol (2017) 24(12):1124–31. doi: 10.1038/nsmb.3498
243. Caronni N, Simoncello F, Stafetta F, Guarnaccia C, Ruiz-Moreno JS, Opitz B, et al. Downregulation of membrane trafficking proteins and lactate conditioning determine loss of dendritic cell function in lung cancer. Cancer Res. (2018) 78:1685–99. doi: 10.1158/0008-5472.CAN-17-1307
244. Wu S, Zhang Q, Zhang F, Meng F, Liu S, Zhou R, et al. HER2 recruits AKT1 to disrupt STING signalling and suppress antiviral defence and antitumour immunity. Nat Cell Biol. (2019) 21:1027–40. doi: 10.1038/s41556-019-0352-z
245. Seo GJ, Yang A, Tan B, Kim S, Liang Q, Choi Y, et al. Akt kinase-mediated checkpoint of cGAS DNA sensing pathway. Cell Rep. (2015) 13:440–9. doi: 10.1016/j.celrep.2015.09.007
246. Laengle J, Stift J, Bilecz A, Wolf B, Beer A, Hegedus B, et al. DNA damage predicts prognosis and treatment response in colorectal liver metastases superior to immunogenic cell death and T cells. Theranostics. (2018) 8:3198–213. doi: 10.7150/thno.24699
247. Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. (2018) 553:467–72. doi: 10.1038/nature25432
248. Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber, GN. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun. (2014) 5:5166. doi: 10.1038/ncomms6166
249. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. (2016) 533:493–8. doi: 10.1038/nature18268
250. Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun. (2017) 8:1736. doi: 10.1038/s41467-017-01566-5
251. Ma R, Ji T, Chen D, Dong W, Zhang H, Yin X, et al. Tumor cell-derived microparticles polarize M2 tumor-associated macrophages for tumor progression. Oncoimmunology. (2016) 5:e1118599. doi: 10.1080/2162402X.2015.1118599
252. Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J Clin Invest. (2019) 129:1211–28. doi: 10.1172/JCI123319
253. Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, et al. PARP inhibitor efficacy depends on CD8(+) T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. (2019) 9:722–37. doi: 10.1158/2159-8290.CD-18-1218
254. Baird JR, Friedman D, Cottam B, Dubensky, TW Jr., Kanne DB, Bambina S, et al. Radiotherapy combined with novel STING-targeting Oligonucleotides results in regression of established tumors. Cancer Res. (2016) 76:50–61. doi: 10.1158/0008-5472.CAN-14-3619
255. Pepin G, Nejad C, Ferrand J, Thomas BJ, Stunden HJ, Sanij E, et al. Topoisomerase 1 inhibition promotes cyclic GMP-AMP synthase-dependent antiviral responses. mBio. (2017) 8:1–9. doi: 10.1128/mBio.01611-17
256. Luthra P, Aguirre S, Yen BC, Pietzsch CA, Sanchez-Aparicio MT, Tigabu B, et al. Topoisomerase II inhibitors induce DNA damage-dependent interferon responses circumventing ebola virus immune evasion. mBio. (2017) 8:e00368-17. doi: 10.1128/mBio.00368-17
257. Wang Z, Chen J, Hu J, Zhang H, Xu F, He W, et al. cGAS/STING axis mediates a topoisomerase II inhibitor-induced tumor immunogenicity. J Clin Invest. (2019) 130:4850–62. doi: 10.1172/JCI127471
258. Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. (2015) 21:1209–15. doi: 10.1038/nm.3931
259. Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci USA. (2015) 112:15408–13. doi: 10.1073/pnas.1512832112
260. Foote JB, Kok M, Leatherman JM, Armstrong TD, Marcinkowski BC, Ojalvo LS, et al. A STING agonist given with OX40 receptor and PD-L1 modulators primes immunity and reduces tumor growth in tolerized mice. Cancer Immunol Res. (2017) 5:468–79. doi: 10.1158/2326-6066.CIR-16-0284
261. Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, et al. Magnitude of therapeutic STING activation determines CD8(+) T Cell-mediated anti-tumor immunity. Cell Rep. (2018) 25:3074–85.e5. doi: 10.1016/j.celrep.2018.11.047
262. Xia T, Konno H, Barber, GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. (2016) 76:6747–59. doi: 10.1158/0008-5472.CAN-16-1404
263. Cavlar T, Deimling T, Ablasser A, Hopfner KP, Hornung, V. Species-specific detection of the antiviral small-molecule compound CMA by STING. EMBO J. (2013) 32:1440–50. doi: 10.1038/emboj.2013.86
264. Gao P, Zillinger T, Wang W, Ascano M, Dai P, Hartmann G, et al. Binding-pocket and lid-region substitutions render human STING sensitive to the species-specific drug DMXAA. Cell Rep. (2014) 8:1668–76. doi: 10.1016/j.celrep.2014.08.010
265. Weiss JM, Guerin MV, Regnier F, Renault G, Galy-Fauroux I, Vimeux L, et al. The STING agonist DMXAA triggers a cooperation between T lymphocytes and myeloid cells that leads to tumor regression. Oncoimmunology. (2017) 6:e1346765. doi: 10.1080/2162402X.2017.1346765
266. Lara, PN Jr., Douillard JY, Nakagawa K, von Pawel J, McKeage MJ, Albert I, et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J Clin Oncol. (2011) 29:2965–71. doi: 10.1200/JCO.2011.35.0660
267. Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. (2018) 564:439–43. doi: 10.1038/s41586-018-0705-y
268. Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, et al. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol Res. (2014) 2:901–10. doi: 10.1158/2326-6066.CIR-13-0123
269. Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S, et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat Nanotechnol. (2019) 14:269–78. doi: 10.1038/s41565-018-0342-5
270. Fremond ML, Rodero MP, Jeremiah N, Belot A, Jeziorski E, Duffy D, et al. Efficacy of the Janus kinase ½ inhibitor ruxolitinib in the treatment of vasculopathy associated with TMEM173-activating mutations in 3 children. J Allergy Clin Immunol. (2016) 138:1752–5. doi: 10.1016/j.jaci.2016.07.015
271. Wang M, Sooreshjani MA, Mikek C, Opoku-Temeng C, Sintim, HO. Suramin potently inhibits cGAMP synthase cGAS, in THP1 cells to modulate IFN-beta levels. Future Med Chem. (2018) 10:1301–17. doi: 10.4155/fmc-2017-0322
272. Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y, et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun. (2017) 8:750. doi: 10.1038/s41467-017-00833-9
273. Lama L, Adura C, Xie W, Tomita D, Kamei T, Kuryavyi V, et al. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat Commun. (2019) 10:2261. doi: 10.1038/s41467-019-08620-4
274. Dai J, Huang YJ, He X, Zhao M, Wang X, Liu ZS, et al. Acetylation Blocks cGAS Activity and Inhibits Self-DNA-Induced Autoimmunity. Cell. (2019) 176:1447–60.e14. doi: 10.1016/j.cell.2019.01.016
275. Li S, Hong Z, Wang Z, Li F, Mei J, Huang L, et al. The cyclopeptide astin C specifically inhibits the innate immune CDN sensor STING. Cell Rep. (2018) 25:3405–21.e7. doi: 10.1016/j.celrep.2018.11.097
276. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. Targeting STING with covalent small-molecule inhibitors. Nature. (2018) 559:269–73. doi: 10.1038/s41586-018-0287-8
277. Seo GJ, Kim C, Shin WJ, Sklan EH, Eoh H, Jung, JU. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat Commun. (2018) 9:613. doi: 10.1038/s41467-018-02936-3
278. Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, et al. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity. (2010) 33:765–76. doi: 10.1016/j.immuni.2010.10.013
279. Wang Q, Huang L, Hong Z, Lv Z, Mao Z, Tang Y, et al. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog. (2017) 13:e1006264. doi: 10.1371/journal.ppat.1006264
280. Qin Y, Zhou MT, Hu MM, Hu YH, Zhang J, Guo L, et al. RNF26 temporally regulates virus-triggered type I interferon induction by two distinct mechanisms. PLoS Pathog. (2014) 10:e1004358. doi: 10.1371/journal.ppat.1004358
281. Wang Q, Liu X, Cui Y, Tang Y, Chen W, Li S, et al. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity. (2014) 41:919–33. doi: 10.1016/j.immuni.2014.11.011
282. Chen M, Meng Q, Qin Y, Liang P, Tan P, He L, et al. TRIM14 inhibits cGAS degradation mediated by Selective autophagy receptor p62 to promote innate immune responses. Mol Cell. (2016) 64:105–19. doi: 10.1016/j.molcel.2016.08.025
283. Chen Y, Wang L, Jin J, Luan Y, Chen C, Li Y, et al. p38 inhibition provides anti-DNA virus immunity by regulation of USP21 phosphorylation and STING activation. J Exp Med. (2017) 214:991–1010. doi: 10.1084/jem.20161387
284. Zhang M, Zhang MX, Zhang Q, Zhu GF, Yuan L, Zhang DE, et al. USP18 recruits USP20 to promote innate antiviral response through deubiquitinating STING/MITA. Cell Res. (2016) 26:1302–19. doi: 10.1038/cr.2016.125
285. Zhang L, Wei N, Cui Y, Hong Z, Liu X, Wang Q, et al. The deubiquitinase CYLD is a specific checkpoint of the STING antiviral signaling pathway. PLoS Pathog. (2018) 14:e1007435. doi: 10.1371/journal.ppat.1007435
286. Hu MM, Yang Q, Xie XQ, Liao CY, Lin H, Liu TT, et al. Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA Virus. Immunity. (2016) 45:555–69. doi: 10.1016/j.immuni.2016.08.014
287. Cui Y, Yu H, Zheng X, Peng R, Wang Q, Zhou, Y et al. SENP7 Potentiates cGAS Activation by Relieving SUMO-Mediated Inhibition of Cytosolic DNA Sensing. PLoS Pathog (2017) 13(1):e1006156. doi: 10.1371/journal.ppat.1006156
288. Liu ZS, Cai H, Xue W, Wang M, Xia T, Li WJ, et al. G3BP1 promotes DNA binding and activation of cGAS. Nat Immunol. (2019) 20:18–28. doi: 10.1038/s41590-018-0262-4
289. Lian H, Wei J, Zang R, Ye W, Yang Q, Zhang XN, et al. ZCCHC3 is a co-sensor of cGAS for dsDNA recognition in innate immune response. Nat Commun. (2018) 9:3349. doi: 10.1038/s41467-018-05559-w
290. Wang C, Guan Y, Lv M, Zhang R, Guo Z, Wei X, et al. Manganese increases the sensitivity of the cGAS-STING pathway for double-stranded DNA and is required for the host defense against DNA viruses. Immunity. (2018) 48:675–87.e7. doi: 10.1016/j.immuni.2018.03.017
291. Li Y, James SJ, Wyllie DH, Wynne C, Czibula A, Bukhari A, et al. TMEM203 is a binding partner and regulator of STING-mediated inflammatory signaling in macrophages. Proc Natl Acad Sci USA. (2019) 116:16479–88. doi: 10.1073/pnas.1901090116
292. Zhou Q, Lin H, Wang S, Wang S, Ran Y, Liu Y, et al. The ER-associated protein ZDHHC1 is a positive regulator of DNA virus-triggered, MITA/STING-dependent innate immune signaling. Cell Host Microbe. (2014) 16:450–61. doi: 10.1016/j.chom.2014.09.006
293. Wang J, Dai M, Cui Y, Hou G, Deng J, Gao X, et al. Association of Abnormal Elevations in IFIT3 With Overactive Cyclic GMP-AMP Synthase/Stimulator of Interferon Genes Signaling in Human Systemic Lupus Erythematosus Monocytes. Arthritis Rheumatol (2018) 70(12):2036–45. doi: 10.1002/art.40576
294. Wang F, Alain T, Szretter KJ, Stephenson K, Pol JG, Atherton MJ, et al. S6K-STING interaction regulates cytosolic DNA-mediated activation of the transcription factor IRF3. Nat Immunol (2016) 17(5):514–22. doi: 10.1038/ni.3433
295. Lei CQ, Zhong B, Zhang Y, Zhang J, Wang S, Shu, HB. Glycogen synthase kinase 3beta regulates IRF3 transcription factor-mediated antiviral response via activation of the kinase TBK1. Immunity. (2010) 33:878–89. doi: 10.1016/j.immuni.2010.11.021
296. Qin Y, Liu Q, Tian S, Xie W, Cui J, Wang, RF. TRIM9 short isoform preferentially promotes DNA and RNA virus-induced production of type I interferon by recruiting GSK3beta to TBK1. Cell Res. (2016) 26:613–28. doi: 10.1038/cr.2016.27
297. Liu TT, Yang Q, Li M, Zhong B, Ran Y, Liu LL, et al. LSm14A plays a critical role in antiviral immune responses by regulating MITA level in a cell-specific manner. J Immunol. (2016) 196:5101–11. doi: 10.4049/jimmunol.1600212
298. Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng MS, et al. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun. (2017) 8:945. doi: 10.1038/s41467-017-00101-w
299. Wang Y, Lian Q, Yang B, Yan S, Zhou H, He L, et al. TRIM30alpha is a negative-feedback regulator of the intracellular DNA and DNA virus-triggered response by targeting STING. PLoS Pathog. (2015) 11:e1005012. doi: 10.1371/journal.ppat.1005012
300. Zhong B, Zhang L, Lei C, Li Y, Mao AP, Yang Y, et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity. (2009) 30:397–407. doi: 10.1016/j.immuni.2009.01.008
301. Sun H, Zhang Q, Jing YY, Zhang M, Wang HY, Cai Z, et al. USP13 negatively regulates antiviral responses by deubiquitinating STING. Nat Commun. (2017) 8:15534. doi: 10.1038/ncomms15534
302. Ghosh A, Shao L, Sampath P, Zhao B, Patel NV, Zhu J, et al. Oligoadenylate-synthetase-family protein OASL inhibits activity of the DNA sensor cGAS during DNA virus infection to limit interferon production. Immunity. (2019) 50:51–63.e5. doi: 10.1016/j.immuni.2018.12.013
303. Lum KK, Song B, Federspiel JD, Diner BA, Howard T, Cristea, IM. Interactome and proteome dynamics uncover immune modulatory associations of the pathogen sensing factor cGAS. Cell Syst. (2018) 7:627–42.e6. doi: 10.1016/j.cels.2018.10.010
304. McFarland AP, Luo S, Ahmed-Qadri F, Zuck M, Thayer EF, Goo YA, et al. Sensing of bacterial cyclic dinucleotides by the oxidoreductase RECON promotes NF-kappaB activation and shapes a proinflammatory antibacterial state. Immunity. (2017) 46:433–45. doi: 10.1016/j.immuni.2017.02.014
305. Srikanth S, Woo JS, Wu B, El-Sherbiny YM, Leung J, Chupradit K, et al. The Ca(2+) sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol. (2019) 20:152–62. doi: 10.1038/s41590-018-0287-8
306. Hansen AL, Buchan GJ, Ruhl M, Mukai K, Salvatore SR, Ogawa E, et al. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc Natl Acad Sci USA. (2018) 115:E7768–75. doi: 10.1073/pnas.1806239115
307. Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, et al. NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity. (2014) 40:329–41. doi: 10.1016/j.immuni.2014.01.010
308. Li Z, Liu G, Sun L, Teng Y, Guo X, Jia J, et al. PPM1A regulates antiviral signaling by antagonizing TBK1-mediated STING phosphorylation and aggregation. PLoS Pathog. (2015) 11:e1004783. doi: 10.1371/journal.ppat.1004783
309. Xia T, Yi XM, Wu X, Shang J, Shu, HB. PTPN1/2-mediated dephosphorylation of MITA/STING promotes its 20S proteasomal degradation and attenuates innate antiviral response. Proc Natl Acad Sci USA. (2019) 116:20063–9. doi: 10.1073/pnas.1906431116
310. Guo H, Konig R, Deng M, Riess M, Mo J, Zhang L, et al. NLRX1 sequesters STING to negatively regulate the interferon response, thereby facilitating the replication of HIV-1 and DNA viruses. Cell Host Microbe. (2016) 19:515–28. doi: 10.1016/j.chom.2016.03.001
311. Feng H, Lenarcic EM, Yamane D, Wauthier E, Mo J, Guo H, et al. NLRX1 promotes immediate IRF1-directed antiviral responses by limiting dsRNA-activated translational inhibition mediated by PKR. Nat Immunol. (2017) 18:1299–309. doi: 10.1038/ni.3853
312. Tan YS, Sansanaphongpricha K, Xie Y, Donnelly CR, Luo X, Heath BR, et al. Mitigating SOX2-potentiated immune escape of head and neck squamous cell carcinoma with a STING-inducing nanosatellite vaccine. Clin Cancer Res. (2018) 24:4242–55. doi: 10.1158/1078-0432.CCR-17-2807
313. Wu MZ, Cheng WC, Chen SF, Nieh S, O’Connor C, Liu CL, et al. miR-25/93 mediates hypoxia-induced immunosuppression by repressing cGAS. Nat Cell Biol. (2017) 19:1286–96. doi: 10.1038/ncb3615
314. Olagnier D, Brandtoft AM, Gunderstofte C, Villadsen NL, Krapp C, Thielke AL, et al. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat Commun. (2018) 9:3506. doi: 10.1038/s41467-018-05861-7
315. Wu L, Cao J, Cai WL, Lang SM, Horton JR, Jansen DJ, et al. KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol. (2018) 16:e2006134. doi: 10.1371/journal.pbio.2006134
316. Su C, Zheng, C. Herpes simplex virus 1 abrogates the cGAS/STING-mediated cytosolic DNA-sensing pathway via its virion host shutoff protein, UL41. J Virol. (2017) 91:e02414-16. doi: 10.1128/JVI.02414-16
317. Huang J, You H, Su C, Li Y, Chen S, Zheng, C. Herpes simplex virus 1 Tegument Protein VP22 abrogates cGAS/STING-mediated antiviral innate immunity. J Virol. (2018) 92:e00841-18. doi: 10.1128/JVI.00841-18
318. Zhang J, Zhao J, Xu S, Li J, He S, Zeng Y, et al. Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe. (2018) 24:234–48.e5. doi: 10.1016/j.chom.2018.07.004
319. Deschamps T, Kalamvoki, M. Evasion of the STING DNA-sensing pathway by VP11/12 of Herpes simplex virus 1. J Virol. (2017) 91:e00535-17. doi: 10.1128/JVI.00535-17
320. Pan S, Liu X, Ma Y, Cao Y, He, B. Herpes simplex virus 1 gamma134.5 protein inhibits STING activation that restricts viral replication. J Virol. (2018) 92:e01015-18. doi: 10.1128/JVI.01015-18
321. Huang ZF, Zou HM, Liao BW, Zhang HY, Yang Y, Fu YZ, et al. Human Cytomegalovirus Protein UL31 Inhibits DNA Sensing of cGAS to Mediate Immune Evasion. Cell Host Microbe (2018) 24(1):69–80 e4. doi: 10.1016/j.chom.2018.05.007
322. Biolatti M, Dell’Oste V, Pautasso S, Gugliesi F, von Einem J, Krapp C, et al. Human Cytomegalovirus Tegument Protein pp65 (pUL83) dampens Type I interferon production by inactivating the DNA sensor cGAS without affecting STING. J Virol. (2018) 92:e01774-17. doi: 10.1128/JVI.01774-17
323. Kumari P, Saha I, Narayanan A, Narayanan S, Takaoka A, Kumar NS, et al. Essential role of HCMV deubiquitinase in promoting oncogenesis by targeting anti-viral innate immune signaling pathways. Cell Death Dis. (2017) 8:e3078. doi: 10.1038/cddis.2017.461
324. Fu YZ, Su S, Gao YQ, Wang PP, Huang ZF, Hu MM, et al. Human Cytomegalovirus Tegument Protein UL82 inhibits STING-mediated signaling to evade antiviral immunity. Cell Host Microbe. (2017) 21:231–43. doi: 10.1016/j.chom.2017.01.001
325. Stempel M, Chan B, Juranic Lisnic V, Krmpotic A, Hartung J, Paludan SR, et al. The herpesviral antagonist m152 reveals differential activation of STING-dependent IRF and NF-kappaB signaling and STING’s dual role during MCMV infection. EMBO J. (2019) 38:e100983. doi: 10.15252/embj.2018100983
326. Choi HJ, Park A, Kang S, Lee E, Lee TA, Ra EA, et al. Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type I interferon immune responses. Nat Commun. (2018) 9:125. doi: 10.1038/s41467-017-02624-8
327. Wu JJ, Li W, Shao Y, Avey D, Fu B, Gillen J, et al. Inhibition of cGAS DNA Sensing by a Herpesvirus Virion protein. Cell Host Microbe. (2015) 18:333–44. doi: 10.1016/j.chom.2015.07.015
328. Zhang G, Chan B, Samarina N, Abere B, Weidner-Glunde M, Buch A, et al. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc Natl Acad Sci USA. (2016) 113:E1034–43. doi: 10.1073/pnas.1516812113
329. Ma Z, Jacobs SR, West JA, Stopford C, Zhang Z, Davis Z, et al. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci USA. (2015) 112:E4306–15. doi: 10.1073/pnas.1503831112
330. Chen X, Yang X, Zheng Y, Yang Y, Xing Y, Chen, Z. SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell. (2014) 5:369–81. doi: 10.1007/s13238-014-0026-3
331. Meade N, Furey C, Li H, Verma R, Chai Q, Rollins MG, et al. Poxviruses evade cytosolic sensing through disruption of an mTORC1-mTORC2 regulatory circuit. Cell. (2018) 174:1143–57.e17. doi: 10.1016/j.cell.2018.06.053
332. Eaglesham JB, Pan Y, Kupper TS, Kranzusch, PJ. Viral and metazoan poxins are cGAMP-specific nucleases that restrict cGAS-STING signalling. Nature. (2019) 566:259–63. doi: 10.1038/s41586-019-0928-6
333. Zheng Y, Liu Q, Wu Y, Ma L, Zhang Z, Liu T, et al. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J. (2018) 37:e99347. doi: 10.15252/embj.201899347
334. Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. (2012) 8:e1002934. doi: 10.1371/journal.ppat.1002934
335. Aguirre S, Luthra P, Sanchez-Aparicio MT, Maestre AM, Patel J, Lamothe F, et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat Microbiol. (2017) 2:17037. doi: 10.1038/nmicrobiol.2017.37
336. Lau L, Gray EE, Brunette RL, Stetson, DB. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science. (2015) 350:568–71. doi: 10.1126/science.aab3291
337. Holm CK, Rahbek SH, Gad HH, Bak RO, Jakobsen MR, Jiang Z, et al. Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat Commun. (2016) 7:10680. doi: 10.1038/ncomms10680
338. Ding Q, Cao X, Lu J, Huang B, Liu YJ, Kato N, et al. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J Hepatol. (2013) 59:52–8. doi: 10.1016/j.jhep.2013.03.019
339. Nitta S, Sakamoto N, Nakagawa M, Kakinuma S, Mishima K, Kusano-Kitazume A, et al. Hepatitis C virus NS4B protein targets STING and abrogates RIG-I-mediated type I interferon-dependent innate immunity. Hepatology. (2013) 57:46–58. doi: 10.1002/hep.26017
340. Liu Y, Li J, Chen J, Li Y, Wang W, Du X, et al. Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J Virol. (2015) 89:2287–300. doi: 10.1128/JVI.02760-14
Keywords: inflammation cGAS, STING cGAMP, type I interferonopathies
Citation: Wan D, Jiang W and Hao J (2020) Research Advances in How the cGAS-STING Pathway Controls the Cellular Inflammatory Response. Front. Immunol. 11:615. doi: 10.3389/fimmu.2020.00615
Received: 23 January 2020; Accepted: 17 March 2020;
Published: 28 April 2020.
Edited by:
Haichao Wang, The Feinstein Institute for Medical Research, United StatesReviewed by:
Nicolas Riteau, UMR7355 Immunologie et Neurogénétique Expérimentales et Moléculaires (INEM), FranceMichael Paul Gantier, Hudson Institute of Medical Research, Australia
Copyright © 2020 Wan, Jiang and Hao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junwei Hao, aGFvanVud2VpQHZpcC4xNjMuY29t