 Dorothee Viemann1,2,3*
Dorothee Viemann1,2,3*- 1Department of Pediatric Pneumology, Allergy and Neonatology, Hannover Medical School, Hanover, Germany
- 2Cluster of Excellence RESIST (EXC 2155), Hannover Medical School, Hanover, Germany
- 3PRIMAL Consortium, Hanover, Germany
The restricted capacity of newborn infants to mount inflammatory responses toward microbial challenges has traditionally been linked to the high risk of septic diseases during the neonatal period. In recent years, substantial evidence has been provided that this characteristic of the neonatal immune system is actually a meaningful physiologic state that is based on specific transiently active cellular and molecular mechanisms and required for a favorable course of postnatal immune adaptation. The identification of physiologically high amounts of S100-alarmins in neonates has been one of the crucial pieces in the puzzle that contributed to the change of concept. In this context, innate immune immaturity could be redefined and assigned to the epigenetic silence of adult-like cell-autonomous regulation at the beginning of life. S100-alarmins represent an alternative age-specific mechanism of immune regulation that protects neonates from hyperinflammatory immune responses. Here, we summarize how infants are provided with S100-alarmins and why these allow an uneventful clash between the innate immune system and the extrauterine world. The mode of action of S100-alarmins is highlighted including their tuning functions at multiple levels for establishing a state of homeostasis with the environment in the newborn individual.
Introduction
After transition from the largely sterile intrauterine environment, the immune system encounters a flood of antigenic stimuli in the new extrauterine world. This represents an enormous challenge for the newborn infant and initiates a process of immune adaptation. Ideally, early-life immune adaptation is a training by new antigens that goes along with the acquisition of protecting immune memory. Especially during the neonatal window of life, responses to new antigens have lifelong imprinting effects by transcriptional and epigenetic reprogramming of systemic and mucosal immunity (1–8). Therefore, early-life immune adaptation critically determines the long-term development of health and disease. Successful immune adaptation achieves an energy-efficient state of homeostasis which balances tolerizing and defending activities of the immune system toward the environment. If immune adaptation fails, the activity of the immune system remains unbalanced which sooner or later leads to the development of chronic inflammatory diseases and an increased susceptibility to infections (2, 3, 5, 9–14). With respect to systemic immunity, important direct reprogramming immune challenges are commensal microbes colonizing the host's barrier sites including thereof translocating metabolites, infections, vaccines, and metabolized food components (2, 3, 5, 7). Therefore, in terms of promotion of successful immune adaptation, a myriad of excellent work has been done and still goes into identifying the ideal pattern of developing microbiota compositions, dissecting “bad” and “good” early-life infections, testing optimized kinds, and combinations of vaccines and defining the most favorable diet including probiotics for infants and young children. On the other side, the mechanisms and molecular options, the newborn infant has to impact on the process of immune adaptation, are still barely elucidated. The interindividual differences in coping with the flood of new antigens after birth and the postnatal development of immunity indicate that host factors must play an important role for the outcome of immune adaptation. Host factors might exert direct imprinting effects and/or determine how the neonate meets its new environment by regulating the neonatal immune response. In this respect, the alarmins S100A8 and S100A9 have been identified as important host factors warranting uneventful and favorable postnatal immune adaptation. In this review, we highlight the current knowledge about the sources of S100A8/A9 supply to the newborn infant and the mode of action of S100A8/A9 in controlling postnatal innate immune adaptation.
Protein Biochemistry and Signaling of S100A8/A9
S100A8 (also named calgranulin A; myeloid-related protein 8, MRP8) and S100A9 (calgranulin B; MRP14) are two members of the S100 protein family specifically linked to innate immune functions. They are calcium-binding proteins characterized by two calcium binding EF-hand motifs, which are connected by a central hinge region. Physiologically, S100A8 and S100A9 exist only as heterodimer S100A8/A9 (termed calprotectin) that can form heterotetramers in the presence of calcium or zinc, but not or at most very little as S100A8/A8 or S100A9/A9 homodimers (15, 16). The reason for it is the instability of the homodimers. Two independent laboratories have shown that myeloid cells of the S100a9-knockout mouse express S100a8 mRNA but no S100a8 protein, suggesting that the posttranscriptional in vivo stability of S100a8 protein is dependent on S100A9 expression (17, 18). Interestingly, the deletion of the mouse S100a8 gene results in an embryonically lethal phenotype without detectability of S100A9 homodimers, pointing to an important role of S100A8 during embryogenesis (19). Once released to the extracellular space, S100A8/A9 impacts on immune cells through binding to different cell surface receptors.
In inflammatory disease states, S100A8/A9 has been shown to bind to endothelial cells (ECs) and chondrocytes by a mechanism involving heparan sulfate proteoglycans and carboxylated N-glycans (20–22). The multiligand receptor for advanced glycation end products (RAGE) was also proposed to act as an S100A8/A9 receptor (23, 24). However, in healthy conditions, the physiological relevance is debatable as RAGE is widespread (on vascular ECs, neutrophils, monocytes/macrophages, lymphocytes, dendritic cells, cardiomyocytes, and neurons) but relatively low expressed (25). Moreover, the binding of S100A8/A9 to RAGE is rather weak and was shown to rely on S100A9 and the presence of Ca2+ or Zn2+ ions (26). Therefore, it is still controversial and questionable that RAGE mediates S100A8/A9 signal transduction.
The main signaling pathway of S100A8/A9 has been delineated by Vogl et al. who demonstrated that S100A8/A9 is a ligand of the Toll-like receptor 4 (TLR4) (27–30). A direct binding of S100A8 and S100A9 to the TLR4–myeloid differentiation factor 2 (MD2) complex was confirmed by surface plasmon resonance studies (29) and comprehensive binding assays (30). The discovery of S100A8/A9 as endogenous TLR4 ligand has been groundbreaking as before TLR4 was only known as a pattern recognition receptor (PRR) for the exogenous TLR4 ligand lipopolysaccharide (LPS), the integral part of the outer membrane of gram-negative bacteria. Fueled by this work, the concept of endogenous damage-associated molecular pattern molecules (DAMPs, also termed alarmins) evolved in analogy to the exogenous microbe-associated respective pathogen-associated pattern molecules (MAMPs respective PAMPs) as activators of PRRs. The canonical downstream signaling induced by TLR4 ligation through S100A8/A9 and LPS has a huge overlap (27–29). Similar to LPS (31, 32), the binding of S100A8/A9 induces the translocation of the adaptor molecule MyD88 from the cytosol to the TLR4 receptor complex which subsequently activates interleukin-1 receptor–associated kinase-1 (IRAK-1) and the transcription factor nuclear factor (NF)-κB p65/p50, thereby increasing the expression of a classical pro-inflammatory gene program including, e.g., tumor necrosis factor (TNF), interleukin (IL)6, IL1B, C-X-C motif chemokine ligand 2 (CXCL2), and C-C motif chemokine ligand 2 (CCL2) (8, 27–30). However, in contrast to LPS, at least in neonatal monocytes, it was shown that S100A8/A9 does not activate the TLR4 adaptor molecule TIR domain-containing adaptor protein-inducing interferon-β (TRIF)-TRIF-related adaptor molecule (TRAM) (8). The reason for it has not been completely elucidated yet, but S100A8/A9 seems to bind different domains of the TLR4/MD2 receptor complex than LPS (29, 30). TRIF-dependent TLR4 signaling shifts the transcription factors IFN regulatory factor (IRF)3 and IRF7 into the nucleus and activates a regulatory gene program including, e.g., CCL5, CXCL9, CXCL10, CD80, and IFN-response genes that in turn regulates the LPS response in a cell-autonomous manner and also links the innate to the adaptive immune system (33–36).
When released secondarily during a primary acute inflammatory process like sepsis and infection or autoimmune and autoinflammatory reactions, S100A8/A9 amplifies the primary pro-inflammatory response by activating the MyD88-dependent TLR4 signaling pathway (29, 37–42) (Figure 1). In adults, the sepsis amplifying effect was proposed to be pathogenetically relevant. In a small study in 17 septic shock patients, S100A8/A9 levels decreased in surviving patients during recovery while non-survivors were characterized by high S100A8/A9 serum levels (43). This could be validated in another cohort of 49 septic shock patients with high plasma levels of S100A8/A9 being associated with a higher risk of death (42). However, under sterile stress conditions with no underlying primary inflammatory stimulus, S100A8/A9 does not induce an inflammatory disease state but contrary a state of inflammatory hyporesponsiveness to subsequent inflammatory stimuli, particularly microbial challenges (Figure 1) (8, 27, 44). However, S100A8/A9 was shown to tolerize not only for a subsequent TLR4 signaling but also TLR2 signaling (27). This work led to the introduction of the concept of “stress tolerance” by endogenous DAMPs and completed the proof of principle that DAMPs similar as PAMPs are able to act in a dual manner, i.e., pro-inflammatory when released secondary and regulatory under sterile conditions. No matter whether pro-inflammatory or regulatory, the S100a8 homodimer proved in several studies as much more potent than the S100a9 homodimer or the heterodimer S100A8/A9 (8, 27, 45). The question of the reason for it could be solved recently. S100A8 and S100A9 bind to the TLR4 complex via specific peptide sequences (29, 30). It was shown that calcium-induced (S100A8/A9)2 tetramer formation hides the TLR4-binding site and blocks the ability of the heterodimer to further bind to TLR4 which prevents undesirable systemic effects (30). This work provided the explanation for how S100A8/A9 effects are locally restricted in sterile settings.
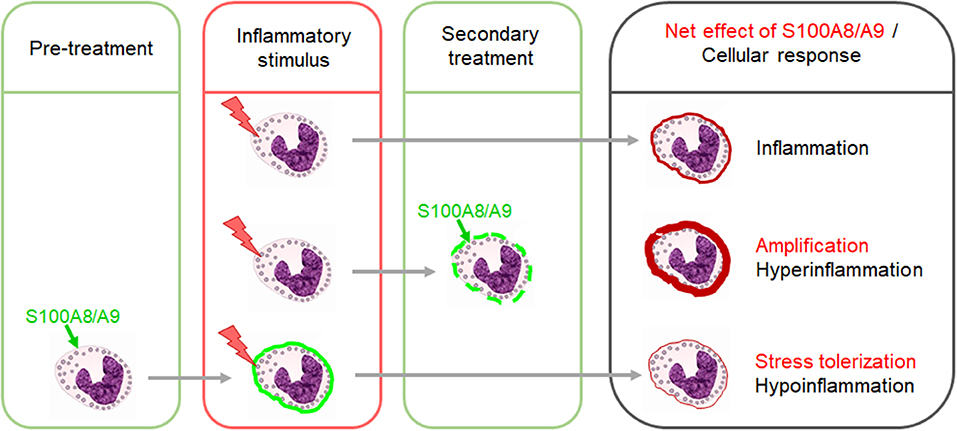
Figure 1. Dual impact of S100A8/A9 on the outcome of inflammatory responses. The secondary release of S100A8/A9 during an inflammatory response upon a preceding stimulus has amplifying effects. Excessive S100A8/A9 release in such settings increases the risk of hyperinflammation. In contrast, pretreatment of immune cells with S100A8/A9 induces a state of hyporesponsiveness of innate signaling pathways which dampens the response to subsequent inflammatory stimuli.
An important molecular mechanism of stress tolerance induction is the S100A8/A9-TLR4-MyD88-mediated preactivation of NF-κB. After activation, cytosolic NF-κB shifts into the nucleus and is then subjected to rapid proteasomal degradation (8, 46, 47). Thus, after S100A8/A9-conditioning, cytosolic NF-κB is no longer available for the canonical signaling cascades of subsequently activated innate signaling pathways which except for TLR3 all depend essentially on NF-κB (31). Yet, recently, it could be revealed that S100A8/A9 priming induces still more in-depth reprogramming of immune cells than only “NF-κB consumption.” In human as well as murine monocytes/macrophages, two major pathways responsible for the S100A8/A9-primed hyporesponsiveness could be identified (44). Firstly, S100A8/A9 induced a lasting inactivation of the phosphatidylinositol 3-kinase (PI3K)/AKT/GSK-3β pathway which resulted in an accumulation of NF-κB inhibitors. Secondly, IL-10-dependent STAT3 activation and nuclear BCL-3 accumulation were identified as master regulators of S100A8/A9-induced tolerance, with the latter resulting in an inhibition of NF-κB transactivation.
Systemic Supply of the Newborn Infant With S100A8/A9
In health, significant expression of S100A8 and S100A9 has been found only in cells of myeloid origin, i.e., granulocytes, monocytes, and macrophages (48, 49). S100A8 and S100A9 constitute up to 40% of cytosolic proteins in granulocytes and 5% in monocytes (50). Inducible expression in microvascular ECs has been described (51, 52) in experimental lung cancer but not under physiologic conditions (51). In psoriasis and other inflammatory skin diseases, S100A8 and S100A9 are highly overexpressed in keratinocytes, while levels are low in normal epidermis (53, 54). Moreover, S100A8/A9 has been identified as a differentiation marker of mammary epithelial cells suppressing malignant transition (55).
In healthy neonates, the concentration of S100A8/A9 in the serum is massively increased in the first days of life up to mean levels of >3,000 ng/ml that in adults are only detectable in inflammatory diseases (27). The high serum levels of S100A8/A9 after birth rapidly decrease but reach normal adult levels (≤ 330 ng/ml) not before the second week of life. Hitherto, two sources have been identified that contribute to the high serum levels of S100A8/A9 in newborn infants. Firstly, neonatal blood monocytes and macrophages overexpress and strongly release S100A8/A9, while expression in the corresponding cell types in healthy adult individuals is low (8, 27, 45). Neonatal granulocytes probably also express S100A8 and S100A9 at high levels, but this still needs to be demonstrated. Secondly, human breast milk contains the highest amounts of S100A8/A9 hitherto found under physiologic conditions (56). In healthy term newborns, the levels in breast milk are in average at least six times higher than the already elevated S100A8/A9 serum levels. The kinetics of S100A8/A9 breast milk levels resemble much that in the neonate's serum, i.e., levels are highest in the colostrum and then gradually decrease over time, reaching normal adult serum levels 1 month after birth. In exclusively breast milk-fed human infants, fecal levels of S100A8/A9 were shown to be significantly higher than in formula-fed ones (57). In mice, enterally supplied S100A8/A9 was demonstrated to be systemically bioavailable in the blood circulation (56). In humans, the dependence of the height of S100A8/A9 levels in the serum on the supply by breast milk awaits quantification. Maternal myeloid cells certainly represent a major production site of the high amounts of S100A8/A9 in human breast milk (58) as levels are three times higher if breast milk samples are analyzed without prior depletion of cells (unpublished data of the authors). To what extent mammary epithelial cells (55) increase the production of S100A8/A9 in the lactating human breast and release it into the milk is currently unknown.
A remarkable aspect related to the physiologically high levels of S100A8/A9 in newborn infants is its unsuitability as an inflammatory biomarker, underlining once more the importance and different role of S100A8/A9 in the neonatal period as compared to later childhood and adulthood. In children and adults, the serum level of S100A8/A9 is clinically used as an excellent biomarker in inflammatory processes like sepsis (42, 43), rheumatoid arthritis, juvenile idiopathic arthritis, and autoinflammatory diseases (40, 59–62). In contrast, in neonates, attempts to establish S100A8/A9 as a clinical biomarker of neonatal sepsis remained without tangible success. Two groups proposed serum S100A8/A9 as a promising sepsis marker in very low birth weight infants (63) respective of infants of all gestational ages (64). Both groups surprisingly claimed that S100A8/A9 values were not influenced by postnatal age and the area under receiver operating characteristic (ROC) curve of 0.6 at a cutoff level of 2,200 ng/ml (64) unveiled S100A8/A9 as a rather poor discriminator.
The release of S100A8/A9 from myeloid cells is a specific and energy-dependent process (65). Any sort of stress and cell damage triggers the release of S100A8/A9 like infections, malignancies, burns, and trauma (22, 27, 29, 37–39, 41, 51, 54, 59–62, 66, 67). The interaction of activated endothelium with phagocytes was also described as an important stimulus for S100A8/A9 secretion (66). A study in marathon runners was the first one revealing that heavy exercise represents a physiologic stress trigger leading to increased S100A8/A9 serum levels during the early post-exercise period that returned to normal levels 1 day after the run (68). Labor and birth are certainly one of the most exhausting conditions in life, imposing heavy stress on the mother as well as the newborn infant. Thus, birth-related stress is probably the main trigger of perinatal S100A8/A9 release, and the gradual resolution of stress after birth well explains the postnatal decrease of S100A8/A9 in breast milk (56) and the infant's blood circulation (27). Therewith in line is a previous report that glucocorticoids can induce the expression of S100-alarmins (69). S100A8-producing macrophages were significantly elevated in rheumatoid arthritis patients treated with high-dose steroids. Further trigger for the release of S100A8/A9 in the perinatal context are thinkable but would need validation, e.g., the hormonal changes upon birth giving like the strong increase of estrogens and oxytocin or the withdrawal of progesterone (70).
The major determinant of birth-induced stress is the extent of labor and uterine contractions which is absent in elective cesarean section (CS) (71). This explains why S100A8/A9 levels in breast milk were significantly higher after vaginal delivery (VD) compared to delivery by CS (56). Moreover, the gestational age has an influence on the height of S100A8/A9 levels. S100A8/A9 levels in breast milk of mothers who gave birth to term babies were significantly higher than of mothers with premature born infants (56). The same holds true for S100A8/A9 levels in cord blood of preterm infants compared to term ones (8). Various other parameters like birth weight, gender, Apgar score, hormonal status of the neonate and the mother, maternal body mass index, and perinatal medications might also influence the early-life S100A8/A9 levels and are currently analyzed in the frame of a prospective multicenter clinical study (BMBF 01GL1746B, DRKS00013197) (72).
Summarized, clearly identified states of early-life S100A8/A9 deficiency are premature birth, elective CS, and formula feeding. The manifold reported associations of all these conditions with an unfavorable short-term as well as long-term immune adaptation including an increased susceptibility to infections (5, 9, 10, 14) and a higher risk for developing chronic inflammatory diseases (2, 3, 11, 12, 14) are a strong indicator for S100-alarmins being a possible common molecular denominator setting the stage for successful postnatal immune adaptation.
Role of S100A8/A9 for the Postnatal Adaptation of Systemic Immunity
The neonatal immune system has traditionally been regarded as “deficient” and the high susceptibility to infections has been understood as a “general weakness.” This concept referred to numerous experimental studies that found impaired inflammatory responses of neonatal immune cells to different microbial challenges (5, 13, 14, 73). The expansion of certain suppressive cell types, e.g., regulatory T cells (Tregs) and granulocytic myeloid suppressor cells (MDSCs) observed in neonates was interpreted as part of this concept (74, 75). However, the hallmark of neonatal sepsis is an extremely rapid course with a hyperinflammatory immune response (76), and the inconsistency of experimental and clinical findings remained unsolved for a long time. The disclosure of diverse molecular and cellular effects of physiologically high amounts of S100-alarmins on the neonatal immune system (Figure 2) significantly contributed to the change of concept that the previously misunderstood characteristics of neonatal immunity actually represent an essential programming that warrants postnatal immune adaptation.
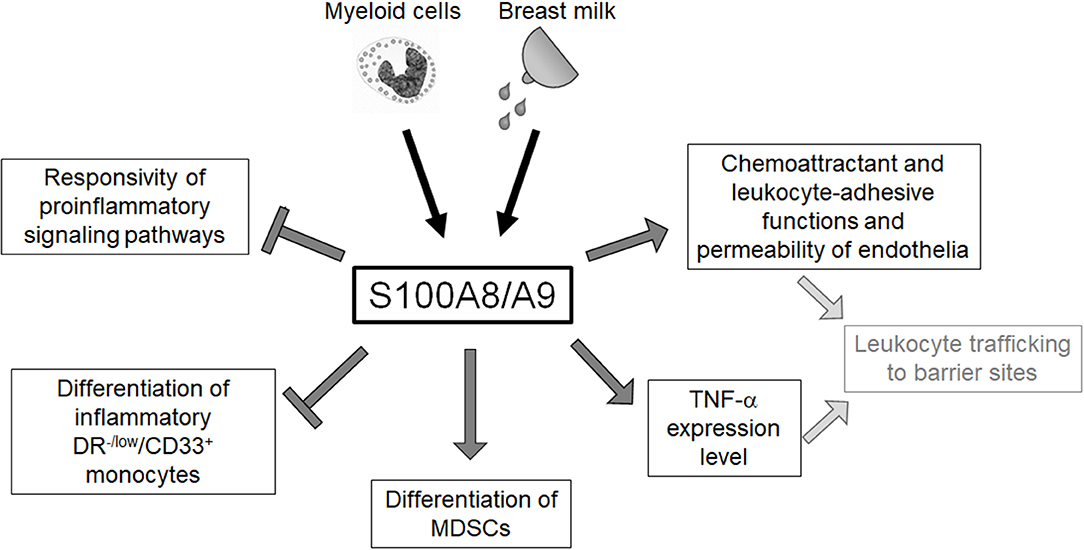
Figure 2. Overview of the regulatory impact of S100A8/A9 on the neonatal innate immune system. Neonatal blood monocytes and breast milk are sources of S100A8/A9 that contribute to the high serum levels of these alarmins in newborn infants. S100A8/A9 constrains systemic inflammatory responses of neonates by tolerizing innate signaling pathways and regulating the expansion of inflammatory and anti-inflammatory myeloid subpopulations. S100A8/A9 impacts directly and indirectly on the leukocyte-recruiting functions of the vascular endothelium S100A8/A9 which suggests an important role in warranting the essential postnatal trafficking of leukocytes to barrier sites.
First of all, it was recognized that the high release and supply of S100A8/A9 directly at the beginning of life corresponds to a conditioning of the neonatal immune system with S100-alarmins and that such pretreatment with S100-alarmins results in stress tolerization of immune cells toward subsequent microbial stimuli (27). The importance and biological relevance of this specific mechanism in newborn infants was further elucidated by Ulas et al. (8) who could link the impaired response of neonatal monocytes to LPS to an activated initial state of MyD88-dependent gene expression at baseline. They further identified S100A8/A9 as an inducer of this specific activation state of neonatal monocytes that was transcriptionally as well as epigenetically fixed as long as serum levels of S100A8/A9 were high. Withdrawal or blocking of S100A8/A9 terminated the activation of the MyD88-dependent gene program and came along with an increased inflammatory response toward microbial stimuli in vitro and in vivo in experimental sepsis models (8, 27, 45). Furthermore, S100A8/A9 also caused a metabolic programming in cord blood macrophages that was characterized by an impaired glycolysis pathway and suppressed mammalian target of rapamycin (mTOR) activation linked to the inflammatory hyporesponsiveness (77). Importantly, S100A8/A9 regulated the inflammatory responsivity without impairing the antimicrobial functions of neonatal phagocytes (8), a decisive requirement to achieve the critical balance between unimpaired defense and uneventful tolerance development. Consequently, the pretreatment of S100a9-knockout mice with the S100A8/A9 heterodimer or the more potent S100A8 homodimer directly after birth rescued murine neonates from fatal courses of later sepsis (8, 45). In human neonates, S100A8/A9 serum levels were negatively associated with the risk of sepsis and significantly higher in term compared to preterm infants. Additionally, in preterm infants, levels higher than 2,000 ng/ml in cord blood were associated with a 25-fold lower risk of late-onset sepsis compared with levels <330 ng/ml (8). This finding closed the gap regarding the seeming discrepancy between the in vitro inflammatory hyporesponsiveness of S100-conditioned neonatal immune cells and the clinical characteristic of hyperdynamic neonatal sepsis with overshooting inflammatory responses in S100-deficient states (Figure 3).
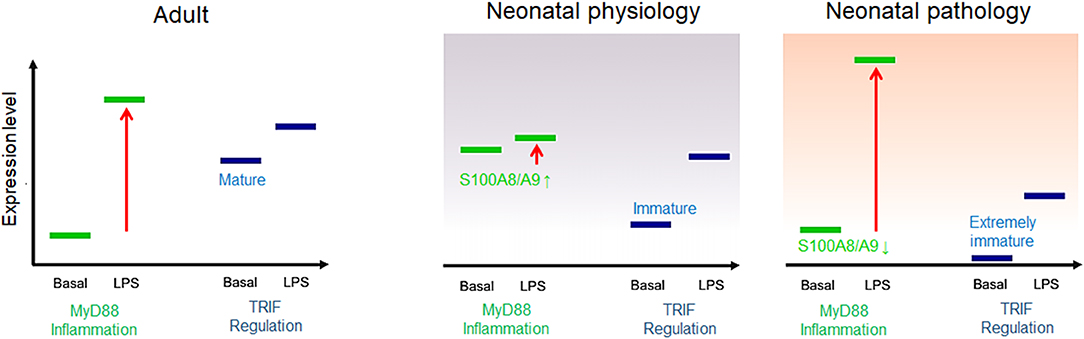
Figure 3. Model of adult and neonatal regulation of lipopolysaccharide (LPS)-responsive gene expression. In healthy adults, MyD88-dependent pro-inflammatory genes are not expressed at baseline but strongly induced by LPS; the basal expression tonus of regulatory TIR domain-containing adaptor protein-inducing interferon (IFN)-β (TRIF)-dependent genes in adults is high and moderately increased by LPS stimulation. In healthy neonates, MyD88-dependent gene expression is physiologically pre-activated by S100A8/A9 and thereby tolerized toward subsequent LPS activation. TRIF-dependent genes are barely expressed at baseline but initiated by a first LPS challenge after birth. In S100A8/A9-deficient neonates, insufficient tolerization of the MyD88-dependent pro-inflammatory response and/or an immature expression tonus or altered postnatal initiation of TRIF-dependent regulatory gene programs results in unregulated hyperinflammatory LPS responses, increasing the risk of severe sepsis.
This relationship became even more important when evidence was provided that TRIF-dependent regulatory gene programs including IFN-response genes are low expressed and epigenetically silent in neonatal monocytes (8). Their expression tonus was not affected by S100A8/A9 and only gradually increased over a prolonged period of time during the first year of life, which suggests environment-dependent initiation. In parallel, another group demonstrated that preterm infants specifically differ from term infants by an even lower baseline expression of regulatory IFN-response genes (78). The profound immaturity of cell-autonomous regulation together with the impaired age-specific alternative kind of regulation by S100A8/A9 provided an explanation for the massively increased sepsis risk of individual preterm infants (Figure 3). Moreover, identifying this period as a critical phase in which the regulation of inflammatory responsiveness shifts from S100A8/A9-programming to an adult-like endogenous regulation allowed understanding why the incidence of late-onset sepsis is highest in the first 2 weeks of life (79).
Next to the outlined molecular effects of S100 programming, S100A8/A9 has influence on the differentiation of several cell types of the innate immune system. In humans and mice, high levels of S100A8/A9 prevented the expansion a specific subpopulation of inflammatory blood monocytes in neonates (DR−/low/CD33+ monocytes in humans and CD11b+/Gr-1int/Ly6G−/Ly6Chi cells in mice) which promoted systemic hyperinflammatory responses (45). Treating S100a9-knockout neonates directly after birth with S100A8/A9 prevented excessive expansion of this inflammatory monocyte population and death from septic shock. Among the rapidly growing list of mechanisms controlling overshooting inflammation in newborn infants are granulocytic MDSCs. Similar as S100A8/A9 levels, MDSCs are expanded in breast milk and the blood circulation of newborn infants and drop down to low adult-like levels 1 month after birth (75, 80, 81). Initially considered as a sign of immune immaturity, their suppressive activity on the inflammatory phenotype of neonatal T cells and monocytes has meanwhile been included into the concept of alternative immune regulation during the neonatal period (81–83). S100A8 and S100A9 are two of the most important positive regulators of the number and function of MDSCs by acting on hematopoietic progenitor cells (84–88). It was recently shown that S100A8/A9 triggers the suppressive activity and also the antibacterial activity of neonatal MDSCs, thereby controlling inflammation (82). Another important process in the neonatal period is the exchange of fetal tissue-resident leukocytes by hematopoietic blood-derived leukocytes, which is an integral part of immune adaptation and results in the reorganization of the leukocyte profiles at barrier sites (89–92). In this context, the impact of S100A8/A9 on the vascular endothelium as well as on monocytes/macrophages might be of biological relevance. S100A8/A9 induces a specific endothelial response characterized by the induction of chemokines and adhesion molecules and a loss of cell–cell contacts that increases the vascular permeability and promotes leukocyte recruitment (67). Furthermore, TNF-α has been demonstrated to be a pivotal mediator maintaining the postnatal trafficking of leukocyte to barrier sites in the neonatal period (93). By increasing the expression of TNF-α in neonatal monocytes/macrophages (8) together with its impact on the chemoattractant and leukocyte-adhesive function and the permeability of endothelia S100A8/A9 might be crucially involved in orchestrating the extravasation and redistribution of leukocytes in the neonatal period.
Finally, it should be noted that glucocorticoids induce the expression and release of S100-alarmins (69). Antenatal corticosteroids for women at risk of imminent preterm birth are the major perinatal intervention to reduce the incidence of respiratory distress syndrome and neonatal mortality associated with preterm birth (94). The effects of steroid-induced S100-alarmins on neonatal immunity are probably part of the beneficial impact of antenatal corticosteroids in promoting fetal maturation and improving postnatal adaptation.
Concluding Remarks
A large body of evidence suggests that S100A8/A9 programs the inflammatory responsivity of systemic innate immunity of newborn infants at multiple molecular and cellular levels. Current data suggest that the tuning of the initial programming of immunity by S100-alarmins is an important determinant of how newborn infants react toward the new antigenic challenges in the extrauterine world. Deficient priming by S100-alarmins increases the risk of life-threatening systemic inflammatory response syndromes, which hampers uneventful immune adaptation and regulated reprogramming of immunity by the environment. Better understanding of how the host contributes to the postnatal development of immunity is an opportunity to exploit endogenous mechanisms like S100-alarmins for intervention strategies, which ensure favorable immune adaptation and could benefit health not only in infancy but also in adulthood.
Author Contributions
DV conceptualized and composed the manuscript.
Funding
The Volkswagen Foundation (Az 90 005), the Appenrodt Foundation and the Deutsche Forschungsgemeinschaft (DFG) (VI 538/6-1), and the DFG under Germany's Excellence Strategy–EXC 2155 RESIST–Project ID 39087428 supported research in this area.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Garand M, Cai B, Kollmann TR. Environment impacts innate immune ontogeny. Innate Immun. (2017) 23:3–10. doi: 10.1177/1753425916671018
2. Gensollen T, Blumberg RS. Correlation between early-life regulation of the immune system by microbiota and allergy development. J Allergy Clin Immunol. (2017) 139:1084–91. doi: 10.1016/j.jaci.2017.02.011
3. Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. (2008) 359:61–73. doi: 10.1056/NEJMra0708473
4. Hornef MW, Torow N. 'Layered immunity' and the 'neonatal window of opportunity' - timed succession of non-redundant phases to establish mucosal host-microbial homeostasis after birth. Immunology. (2020) 159:15–25. doi: 10.1111/imm.13149
5. Kollmann TR, Kampmann B, Mazmanian SK, Marchant A, Levy O. Protecting the newborn and young infant from infectious diseases: lessons from immune ontogeny. Immunity. (2017) 46:350–63. doi: 10.1016/j.immuni.2017.03.009
6. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
7. Renz H, Adkins BD, Bartfeld S, Blumberg RS, Farber DL, Garssen J, et al. The neonatal window of opportunity-early priming for life. J Allergy Clin Immunol. (2018) 141:1212–4. doi: 10.1016/j.jaci.2017.11.019
8. Ulas T, Pirr S, Fehlhaber B, Bickes MS, Loof TG, Vogl T, et al. S100-alarmin-induced innate immune programming protects newborn infants from sepsis. Nat Immunol. (2017) 18:622–32. doi: 10.1038/ni.3745
9. Burgner DP, Doherty D, Humphreys J, Currie A, Simmer K, Charles A, et al. Maternal chorioamnionitis and postneonatal respiratory tract infection in ex-preterm infants. J Pediatr. (2017) 184:62–7.e2. doi: 10.1016/j.jpeds.2017.01.037
10. Doyle LW, Ford G, Davis N. Health and hospitalistions after discharge in extremely low birth weight infants. Semin Neonatol. (2003) 8:137–45. doi: 10.1016/S1084-2756(02)00221-X
11. Harju M, Keski-Nisula L, Georgiadis L, Raisanen S, Gissler M, Heinonen S. The burden of childhood asthma and late preterm and early term births. J Pediatr. (2014) 164:295–9.e1. doi: 10.1016/j.jpeds.2013.09.057
12. Holsti A, Adamsson M, Hagglof B, Farooqi A, Serenius F. Chronic conditions and health care needs of adolescents born at 23 to 25 weeks' gestation. Pediatrics. (2017) 139:e20162215–25. doi: 10.1542/peds.2016-2215
13. Kan B, Razzaghian HR, Lavoie PM. An immunological perspective on neonatal sepsis. Trends Mol Med. (2016) 22:290–302. doi: 10.1016/j.molmed.2016.02.001
14. Zhang X, Zhivaki D, Lo-Man R. Unique aspects of the perinatal immune system. Nat Rev Immunol. (2017) 17:495–507. doi: 10.1038/nri.2017.54
15. Vogl T, Gharibyan AL, Morozova-Roche LA. Pro-inflammatory S100A8 and S100A9 proteins: self-assembly into multifunctional native and amyloid complexes. Int J Mol Sci. (2012) 13:2893–917. doi: 10.3390/ijms13032893
16. Vogl T, Leukert N, Barczyk K, Strupat K, Roth J. Biophysical characterization of S100A8 and S100A9 in the absence and presence of bivalent cations. Biochim Biophys Acta. (2006) 1763:1298–306. doi: 10.1016/j.bbamcr.2006.08.028
17. Manitz M-P, Horst B, Seeliger S, Strey A, Skryabin BV, Gunzer M, et al. Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol Cell Biol. (2003) 23:1034–43. doi: 10.1128/MCB.23.3.1034-1043.2003
18. Hobbs JA, May R, Tanousis K, McNeill E, Mathies M, Gebhardt C, et al. Myeloid cell function in MRP-14 (S100A9) null mice. Mol Cell Biol. (2003) 23:2564–76. doi: 10.1128/MCB.23.7.2564-2576.2003
19. Passey RJ, Williams E, Lichanska AM, Wells C, Hu S, Geczy CL, et al. A null mutation in the inflammation-associated S100 protein S100A8 causes early resorption of the mouse embryo. J Immunol. (1999) 163:2209–16.
20. Robinson MJ, Tessier P, Poulsom R, Hogg N. The S100 family heterodimer, MRP-8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J Biol Chem. (2002) 277:3658–65. doi: 10.1074/jbc.M102950200
21. Srikrishna G, Panneerselvam K, Westphal V, Abraham V, Varki A, Freeze HH. Two proteins modulating transendothelial migration of leukocytes recognize novel carboxylated glycans on endothelial cells. J Immunol. (2001) 166:4678–88. doi: 10.4049/jimmunol.166.7.4678
22. van Lent PL, Grevers LC, Blom AB, Arntz OJ, van de Loo FA, van der Kraan P, et al. Stimulation of chondrocyte-mediated cartilage destruction by S100A8 in experimental murine arthritis. Arthritis Rheum. (2008) 58:3776–87. doi: 10.1002/art.24074
23. Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis. (2008) 29:2035–43. doi: 10.1093/carcin/bgn188
24. Gebhardt C, Riehl A, Durchdewald M, Nemeth J, Furstenberger G, Muller-Decker K, et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. (2008) 205:275–85. doi: 10.1084/jem.20070679
25. Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. (2010) 28:367–88. doi: 10.1146/annurev.immunol.021908.132603
26. Bjork P, Bjork A, Vogl T, Stenstrom M, Liberg D, Olsson A, et al. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. (2009) 7:e97. doi: 10.1371/journal.pbio.1000097
27. Austermann J, Friesenhagen J, Fassl SK, Petersen B, Ortkras T, Burgmann J, et al. Alarmins MRP8 and MRP14 induce stress tolerance in phagocytes under sterile inflammatory conditions. Cell Rep. (2014) 9:2112–23. doi: 10.1016/j.celrep.2014.11.020
28. Fassl SK, Austermann J, Papantonopoulou O, Riemenschneider M, Xue J, Bertheloot D, et al. Transcriptome assessment reveals a dominant role for TLR4 in the activation of human monocytes by the alarmin MRP8. J Immunol. (2015) 194:575–83. doi: 10.4049/jimmunol.1401085
29. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen, et al. Mrp8 and Mrp14 are endogenous activators of toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. (2007) 13:1042–9. doi: 10.1038/nm1638
30. Vogl T, Stratis A, Wixler V, Voller T, Thurainayagam S, Jorch SK, et al. Autoinhibitory regulation of S100A8/S100A9 alarmin activity locally restricts sterile inflammation. J Clin Invest. (2018) 128:1852–66. doi: 10.1172/JCI89867
31. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
32. Escoubet-Lozach L, Benner C, Kaikkonen MU, Lozach J, Heinz S, Spann NJ, et al. Mechanisms establishing TLR4-responsive activation states of inflammatory response genes. PLoS Genet. (2011) 7:e1002401. doi: 10.1371/journal.pgen.1002401
33. Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, et al. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med. (2003) 198:1043–55. doi: 10.1084/jem.20031023
34. Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. (2009) 30:475–87. doi: 10.1016/j.it.2009.07.009
35. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. (2003) 301:640–3. doi: 10.1126/science.1087262
36. Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, et al. TRAM is specifically involved in the toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. (2003) 4:1144–50. doi: 10.1038/ni986
37. van Zoelen MA, Vogl T, Foell D, van Veen SQ, van Till JW, Florguin S, et al. Expression and role of myeloid-related protein-14 in clinical and experimental sepsis. Am J Respir Crit Care Med. (2009) 180:1098–106. doi: 10.1164/rccm.200810-1552OC
38. Frosch M, Ahlmann M, Vogl T, Wittkowski H, Wulffraat N, Foell D, et al. The myeloid-related proteins 8 and 14 complex, a novel ligand of toll-like receptor 4, and interleukin-1beta form a positive feedback mechanism in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. (2009) 60:883–91. doi: 10.1002/art.24349
39. Frosch M, Vogl T, Seeliger S, Wulffraat N, Kuis W, Viemann D, et al. Expression of myeloid-related proteins 8 and 14 in systemic-onset juvenile rheumatoid arthritis. Arthritis Rheum. (2003) 48:2622–6. doi: 10.1002/art.11177
40. Chan JK, Roth J, Oppenheim JJ, Tracey KJ, Vogl T, Feldmann M, et al. Alarmins: awaiting a clinical response. J Clin Invest. (2012) 122:2711–9. doi: 10.1172/JCI62423
41. Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in inflammation. Front Immunol. (2018) 9:1298. doi: 10.3389/fimmu.2018.01298
42. Dubois C, Marce D, Faivre V, Lukaszewicz A-C, Junot C, Fenaille F, et al. High plasma level of S100A8/S100A9 and S100A12 at admission indicates a higher risk of death in septic shock patients. Sci Rep. (2019) 9:15660. doi: 10.1038/s41598-019-52184-8
43. Payen D, Lukaszewicz A-C, Belikova I, Faivre V, Gelin C, Russwurm S, et al. Gene profiling in human blood leucocytes during recovery from septic shock. Intensive Care Med. (2008) 34:1371–6. doi: 10.1007/s00134-008-1048-1
44. Freise N, Burghard A, Ortkras T, Daber N, Imam Chasan A, Jauch S-L, et al. Signaling mechanisms inducing hyporesponsiveness of phagocytes during systemic inflammation. Blood. (2019) 134:134–46. doi: 10.1182/blood.2019000320
45. Heinemann AS, Pirr S, Fehlhaber B, Mellinger L, Burgmann J, Busse M, et al. In neonates S100A8/S100A9 alarmins prevent the expansion of a specific inflammatory monocyte population promoting septic shock. FASEB J. (2017) 31:1153–64. doi: 10.1096/fj.201601083R
46. Tanaka T, Grusby MJ, Kaisho T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol. (2007) 8:584–91. doi: 10.1038/ni1464
47. Geng H, Wittwer T, Dittrich-Breiholz O, Kracht M, Schmitz ML. Phosphorylation of NF-kappaB p65 at Ser468 controls its COMMD1-dependent ubiquitination and target gene-specific proteasomal elimination. EMBO Rep. (2009) 10:381–6. doi: 10.1038/embor.2009.10
48. Lagasse E, Clerc RG. Cloning and expression of two human genes encoding calcium-binding proteins that are regulated during myeloid differentiation. Mol Cell Biol. (1988) 8:2402–10. doi: 10.1128/MCB.8.6.2402
49. Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). Biochem Biophys Res Commun. (2004) 322:1111–22. doi: 10.1016/j.bbrc.2004.07.096
50. Edgeworth J, Gorman M, Bennett R, Freemont P, Hogg N. Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J Biol Chem. (1991) 266:7706–13.
51. Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. (2006) 8:1369–75. doi: 10.1038/ncb1507
52. Yen T, Harrison CA, Devery JM, Leong S, Iismaa SE, Yoshimura T, et al. Induction of the S100 chemotactic protein, CP-10, in murine microvascular endothelial cells by proinflammatory stimuli. Blood. (1997) 90:4812–21. doi: 10.1182/blood.V90.12.4812.4812_4812_4821
53. Gabrielsen TO, Dale I, Brandtzaeg P, Hoel PS, Fagerhol MK, Larsen TE, et al. Epidermal and dermal distribution of a myelomonocytic antigen (L1) shared by epithelial cells in various inflammatory skin diseases. J Am Acad Dermatol. (1986) 15(2 Pt 1):173–9. doi: 10.1016/S0190-9622(86)70152-7
54. Kunz M, Roth J, Sorg C, Kolde G. Epidermal expression of the calcium binding surface antigen 27E10 in inflammatory skin diseases. Arch Dermatol Res. (1992) 284:386–90. doi: 10.1007/BF00372067
55. Cormier K, Harquail J, Ouellette RJ, Tessier PA, Guerrette R, Robichaud GA. Intracellular expression of inflammatory proteins S100A8 and S100A9 leads to epithelial-mesenchymal transition and attenuated aggressivity of breast cancer cells. Anticancer Agents Med Chem. (2014) 14:35–45. doi: 10.2174/18715206113136660333
56. Pirr S, Richter M, Fehlhaber B, Pagel J, Hartel C, Roth J, et al. High amounts of S100-alarmins confer antimicrobial activity on human breast milk targeting pathogens relevant in neonatal sepsis. Front Immunol. (2017) 8:1822. doi: 10.3389/fimmu.2017.01822
57. Savino F, Castagno E, Calabrese R, Viola S, Oggero R, Miniero R. High faecal calprotectin levels in healthy, exclusively breast-fed infants. Neonatology. (2010) 97:299–304. doi: 10.1159/000255161
58. Witkowska-Zimny M, Kaminska-El-Hassan E. Cells of human breast milk. Cell Mol Biol Lett. (2017) 22:11. doi: 10.1186/s11658-017-0042-4
59. Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. (2007) 81:28–37. doi: 10.1189/jlb.0306170
60. Foell D, Roth J. Proinflammatory S100 proteins in arthritis and autoimmune disease. Arthritis Rheum. (2004) 50:3762–71. doi: 10.1002/art.20631
61. Frosch M, Roth J. New insights in systemic juvenile idiopathic arthritis–from pathophysiology to treatment. Rheumatology. (2008) 47:121–5. doi: 10.1093/rheumatology/kem271
62. Holzinger D, Frosch M, Kastrup A, Prince FH, Otten MH, Van Suijlekom-Smit, et al. The toll-like receptor 4 agonist MRP8/14 protein complex is a sensitive indicator for disease activity and predicts relapses in systemic-onset juvenile idiopathic arthritis. Ann Rheum Dis. (2012) 71:974–80. doi: 10.1136/annrheumdis-2011-200598
63. Terrin G, Passariello A, Manguso F, Salvia G, Rapacciuolo L, Messina F, et al. Serum calprotectin: an antimicrobial peptide as a new marker for the diagnosis of sepsis in very low birth weight newborns. Clin Dev Immunol. (2011) 2011:291085. doi: 10.1155/2011/291085
64. Decembrino L, De Amici M, Pozzi M, De Silvestri A, Stronati M. Serum calprotectin: a potential biomarker for neonatal sepsis. J Immunol Res. (2015) 2015:147973. doi: 10.1155/2015/147973
65. Rammes A, Roth J, Goebeler M, Klempt M, Hartmann M, Sorg C. Myeloid-related protein (MRP) 8 and MRP14, calcium-binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin-dependent pathway. J Biol Chem. (1997) 272:9496–502. doi: 10.1074/jbc.272.14.9496
66. Frosch M, Strey A, Vogl T, Wulffraat NM, Kuis W, Sunderkotter C, et al. Myeloid-related proteins 8 and 14 are specifically secreted during interaction of phagocytes and activated endothelium and are useful markers for monitoring disease activity in pauciarticular-onset juvenile rheumatoid arthritis. Arthritis Rheum. (2000) 43:628–37. doi: 10.1002/1529-0131(200003)43:3<628::AID-ANR20>3.0.CO;2-X
67. Viemann D, Strey A, Janning A, Jurk K, Klimmek K, Vogl T, et al. Myeloid-related proteins 8 and 14 induce a specific inflammatory response in human microvascular endothelial cells. Blood. (2005) 105:2955–62. doi: 10.1182/blood-2004-07-2520
68. Mooren FC, Lechtermann A, Fobker M, Brandt B, Sorg C, Volker K, et al. The response of the novel pro-inflammatory molecules S100A8/A9 to exercise. Int J Sports Med. (2006) 27:751–8. doi: 10.1055/s-2005-872909
69. Harrison CA, Raftery MJ, Walsh J, Alewood P, Iismaa SE, Thliveris S, et al. Oxidation regulates the inflammatory properties of the murine S100 protein S100A8. J Biol Chem. (1999) 274:8561–9. doi: 10.1074/jbc.274.13.8561
70. Kota SK, Gayatri K, Jammula S, Kota SK, Krishna SV, Meher LK, et al. Endocrinology of parturition. Indian J Endocrinol Metab. (2013) 17:50–9. doi: 10.4103/2230-8210.107841
71. Kiriakopoulos N, Grigoriadis S, Maziotis E, Philippou A, Rapani A, Giannelou P, et al. Investigating stress response during vaginal delivery and elective cesarean section through assessment of levels of cortisol, interleukin 6 (IL-6), growth hormone (GH) and insulin-like growth factor 1 (IGF-1). J Clin Med. (2019) 8:1112–27. doi: 10.3390/jcm8081112
72. Marissen J, Haiss A, Meyer C, van Rossum T, Bunte LM, Frommhold D, et al. Efficacy of bifidobacterium longum, B. infantis and Lactobacillus acidophilus probiotics to prevent gut dysbiosis in preterm infants of 28+0-32+6 weeks of gestation: a randomised, placebo-controlled, double-blind, multicentre trial: the PRIMAL Clinical Study protocol. BMJ Open. (2019) 9:e032617. doi: 10.1136/bmjopen-2019-032617
73. Levy O. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat Rev Immunol. (2007) 7:379–90. doi: 10.1038/nri2075
74. Pagel J, Hartz A, Figge J, Gille C, Eschweiler S, Petersen K, et al. Regulatory T cell frequencies are increased in preterm infants with clinical early-onset sepsis. Clin Exp Immunol. (2016) 185:219–27. doi: 10.1111/cei.12810
75. Rieber N, Gille C, Kostlin N, Schafer I, Spring B, Ost M, et al. Neutrophilic myeloid-derived suppressor cells in cord blood modulate innate and adaptive immune responses. Clin Exp Immunol. (2013) 174:45–52. doi: 10.1111/cei.12143
76. Zhao J, Kim KD, Yang X, Auh S, Fu Y-X, Tang H. Hyper innate responses in neonates lead to increased morbidity and mortality after infection. Proc Natl Acad Sci USA. (2008) 105:7528–33. doi: 10.1073/pnas.0800152105
77. Dreschers S, Ohl K, Lehrke M, Mollmann J, Denecke B, Costa I, et al. Impaired cellular energy metabolism in cord blood macrophages contributes to abortive response toward inflammatory threats. Nat Commun. (2019) 10:1685. doi: 10.1038/s41467-019-09359-8
78. Singh VV, Chauhan SK, Rai R, Kumar A, Singh SM, Rai G. Decreased pattern recognition receptor signaling, interferon-signature, and bactericidal/permeability-increasing protein gene expression in cord blood of term low birth weight human newborns. PLoS ONE. (2013) 8:e62845. doi: 10.1371/journal.pone.0062845
79. Brook B, Harbeson D, Ben-Othman R, Viemann D, Kollmann TR. Newborn susceptibility to infection vs. disease depends on complex in vivo interactions of host and pathogen. Semin Immunopathol. (2017) 39:615–25. doi: 10.1007/s00281-017-0651-z
80. Schwarz J, Scheckenbach V, Kugel H, Spring B, Pagel J, Hartel C, et al. Granulocytic myeloid-derived suppressor cells (GR-MDSC) accumulate in cord blood of preterm infants and remain elevated during the neonatal period. Clin Exp Immunol. (2018) 191:328–37. doi: 10.1111/cei.13059
81. Kostlin N, Schoetensack C, Schwarz J, Spring B, Marme A, Goelz R, et al. Granulocytic myeloid-derived suppressor cells (GR-MDSC) in breast milk (BM) GR-MDSC accumulate in human BM and modulate T-Cell and monocyte function. Front Immunol. (2018) 9:1098. doi: 10.3389/fimmu.2018.01098
82. He Y-M, Li X, Perego M, Nefedova Y, Kossenkov AV, Jensen EA, et al. Transitory presence of myeloid-derived suppressor cells in neonates is critical for control of inflammation. Nat Med. (2018) 24:224–31. doi: 10.1038/nm.4467
83. Kostlin N, Vogelmann M, Spring B, Schwarz J, Feucht J, Hartel C, et al. Granulocytic myeloid-derived suppressor cells from human cord blood modulate T-helper cell response towards an anti-inflammatory phenotype. Immunology. (2017) 152:89–101. doi: 10.1111/imm.12751
84. Chen X, Eksioglu EA, Zhou J, Zhang L, Djeu J, Fortenbery N, et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J Clin Invest. (2013) 123:4595–611. doi: 10.1172/JCI67580
85. Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. (2008) 205:2235–49. doi: 10.1084/jem.20080132
86. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
87. Petersen B, Wolf M, Austermann J, van Lent P, Foell D, Ahlmann M, et al. The alarmin Mrp8/14 as regulator of the adaptive immune response during allergic contact dermatitis. EMBO J. (2013) 32:100–11. doi: 10.1038/emboj.2012.309
88. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. (2008) 181:4666–75. doi: 10.4049/jimmunol.181.7.4666
89. Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. (2016) 17:34–40. doi: 10.1038/ni.3324
90. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
91. Perdiguero EG, Geissmann F. The development and maintenance of resident macrophages. Nat Immunol. (2016) 17:2–8. doi: 10.1038/ni.3341
92. van de Laar L, Saelens W, De Prijck S, Martens L, Scott CL, van Isterdael G, et al. Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity. (2016) 44:755–68. doi: 10.1016/j.immuni.2016.02.017
93. Bickes MS, Pirr S, Heinemann AS, Fehlhaber B, Halle S, Vollger L, et al. Constitutive TNF-α signaling in neonates is essential for the development of tissue-resident leukocyte profiles at barrier sites. FASEB J. (2019) 33:10633–47. doi: 10.1096/fj.201900796R
Keywords: neonate, S100A8/A9, systemic immunity, innate immunity, trained immunity, immune adaptation, inflammatory diseases
Citation: Viemann D (2020) S100-Alarmins Are Essential Pilots of Postnatal Innate Immune Adaptation. Front. Immunol. 11:688. doi: 10.3389/fimmu.2020.00688
Received: 17 December 2019; Accepted: 26 March 2020;
Published: 30 April 2020.
Edited by:
Andrew Currie, Murdoch University, AustraliaReviewed by:
Markus Sperandio, Ludwig Maximilian University of Munich, GermanyDaniel Scott-Algara, Institut Pasteur, France
Copyright © 2020 Viemann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dorothee Viemann, VmllbWFubi5Eb3JvdGhlZUBtaC1oYW5ub3Zlci5kZQ==