 Zhenkun Wang1
Zhenkun Wang1 Fenglin Cao
Fenglin Cao Joseph A. Bellanti
Joseph A. Bellanti Song Guo Zheng
Song Guo Zheng- 1Central Laboratory of Hematology and Oncology, First Affiliated Hospital, Harbin Medical University, Harbin, China
- 2College of Life Science, Northeast Agricultural University, Harbin, China
- 3Departments of Pediatrics and Microbiology-Immunology, The International Center for Interdisciplinary Studies of Immunology (ICISI), Georgetown University Medical Center, Washington, DC, United States
- 4Department of Hematology, First Affiliated Hospital, Harbin Medical University, Harbin, China
- 5Department of Internal Medicine, Ohio State University College of Medicine, Columbus, OH, United States
Conditions in which abnormal or excessive immune responses exist, such as autoimmune diseases (ADs), graft-versus-host disease, transplant rejection, and hypersensitivity reactions, are serious hazards to human health and well-being. The traditional immunosuppressive drugs used to treat these conditions can lead to decreased immune function, a higher risk of infection, and increased tumor susceptibility. As an alternative therapeutic approach, cell therapy, in which generally intact and living cells are injected, grafted, or implanted into a patient, has the potential to overcome the limitations of traditional drug treatment and to alleviate the symptoms of many refractory diseases. Cell therapy could be a powerful approach to induce immune tolerance and restore immune homeostasis with a deeper understanding of immune tolerance mechanisms and the development of new techniques. The purpose of this review is to describe the current panoramic scope of cell therapy for immune-mediated disorders, discuss the advantages and disadvantages of different types of cell therapy, and explore novel directions and future prospects for these tolerogenic therapies.
Immune Homeostasis
It is now clear that many factors such as cytokines, immune cells, immune checkpoints, and microbiota are important in maintaining the immune balance between the external and internal environment and that they accomplish their function by either promoting or inhibiting inflammatory responses (1–3). For example, balanced production of pro- and anti-inflammatory cytokines in response to a foreign configuration results in immunological equilibrium and defines the immune system in health in contrast to the immunologic imbalance seen when an overproduction of pro-inflammatory cytokines and/or inadequate production of anti-inflammatory cytokines creates disequilibrium, which represents the immune system in disease (Figure 1).
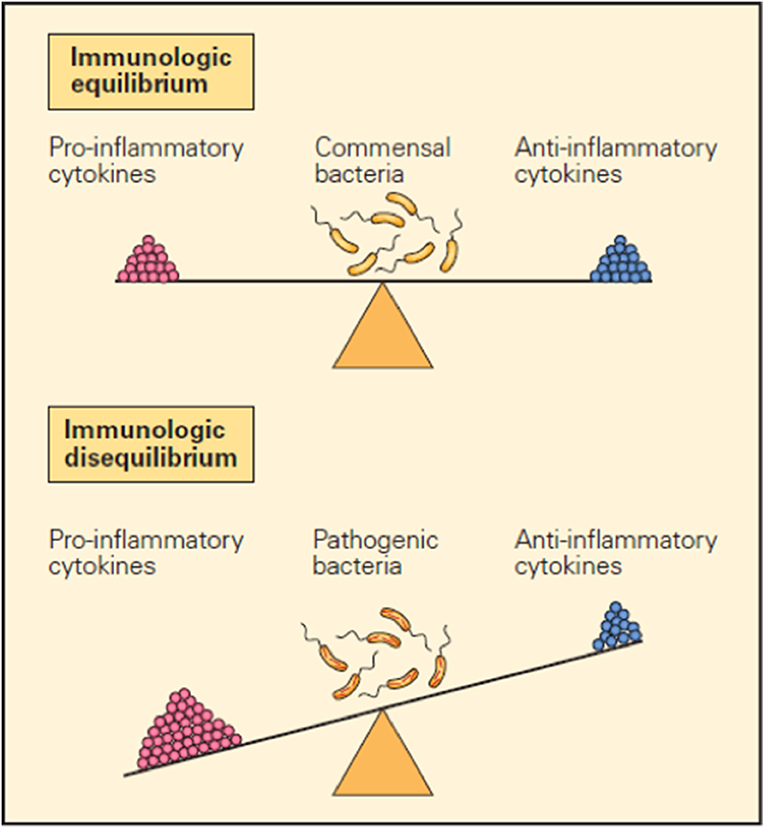
Figure 1. Schematic representation of the opposing pro- and anti-inflammatory functions of cytokines in maintaining immunological equilibrium. A balanced production of pro- and anti-inflammatory cytokines in response to a foreign configuration is one of the mechanisms that contribute to immunological equilibrium (upper panel). Conversely, immunological imbalance or disease is seen when there is overproduction of pro-inflammatory cytokines and/or inadequate production of anti-inflammatory cytokines (lower panel) (1).
A healthy immune system should, therefore, exist in a state of dynamic equilibrium, with a balance between pro-inflammatory and anti-inflammatory responses, to achieve immune homeostasis. Ideally, the immune system normally maintains its non-responsiveness to autoantigens and various harmless environmental entities (allergens, commensal microbiota, etc.) through complex mechanisms of central and peripheral tolerance. During the course of pathogen invasion or the appearance of a malignant proliferation of autologous cells, the body needs to recognize and generate a targeted and effective immune response to the abnormality. After successfully eliminating these deleterious challenges, the immune response normally self-adjusts and returns to its beneficial state of immune homeostasis. However, if immune homeostasis is not effectively achieved, continuous immune hyperactivity or inhibition might contribute to two types of disease occurrence: autoimmune disease or cancer (4–7). On the other hand, the hypo-functioning immune system with deficiencies in immune recognition or effects will increase the possibility of infection, chronic inflammation, and tumors. Additionally, abnormal autoimmune responses to autoantigens might occur if the central tolerance or peripheral tolerance of the immune system is damaged (Figure 2) (8, 9). As two types of contrasting immune dysfunction disorders, their different mechanisms of pathogenesis can serve as a useful reference for designing counter-treatment strategies (10). For example, tumor cells are “altered cells” and are generally perceived as “self” and non-dangerous, with the privileges of normal healthy tissues. Treating autoimmune diseases might help to understand how tumor cells lose these properties, making them target the immune system, and gain such properties, making them appear non-dangerous (11).
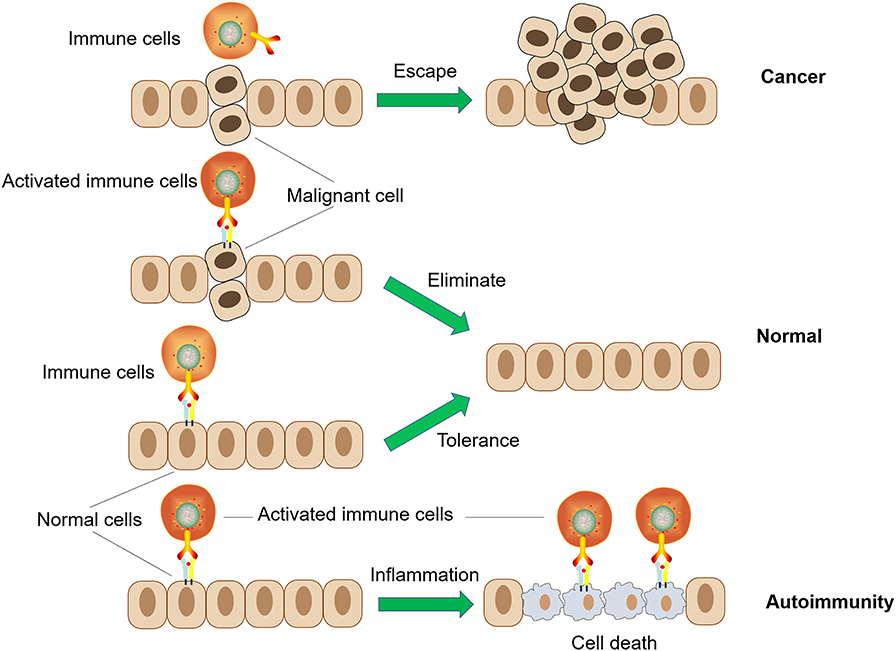
Figure 2. Two types of immune dysfunction: cancer and autoimmunity.
In addition to autoimmune diseases (ADs), excessive inflammatory reactions caused by organ transplantation and hypersensitivity reactions can be life-threatening for patients. Therefore, restoring immune tolerance to autoantigens or inducing immune tolerance to specific antigens is of considerable importance for the clinical treatment of many diseases (12, 13).
Autoimmune responses are associated with imbalances between effector and inhibitory cells. When a pathological immune response begins, the effector cells in the tissue accumulate, eliciting an autoimmune response, which may be accompanied by a relative decrease in the number of inhibitory cells or an increased number of dysfunctional inhibitory cells (14). Imbalances in effector and regulatory cells can lead to autoimmunity or even tissue damage, and the patients often experience remission and recurrence due to persistent interactions between pathogenic responses and regulation (15). Thus, the development of strategies to eliminate autoimmune responses and recover the balance of effector and regulatory cells is becoming an important task for achieving immune tolerance and disease prevention.
Traditional drugs are often limited in their ability to induce immune tolerance due to complex immunoregulatory processes (16). Since cell–cell interactions are fundamental for immune responses, cell-based therapeutic techniques have developed rapidly (17, 18) as alternative approaches to traditional drugs. Cell therapy has become the “hot topic” in cancer research; in particular, chimeric antigen receptor T (CAR-T) cells have made significant breakthroughs in the treatment of hematologic malignancies (19, 20). In contrast, cell therapy for ADs has progressed slowly in recent years. Although cytokine antagonists have altered the course of many ADs, most current therapeutic agents target the terminal phase of inflammation and do not address the fundamental problems that are responsible for the initiation and progression of the autoimmune process (15). ADs are often characterized by chronic conditions, which in most cases require sustained, even life-long treatment, which imposes a heavy strain and financial burden on patients and significantly increases their risk of malignant tumors and infectious complications (21–23). As cell immune therapy technology can induce immune tolerance through various immunosuppressive pathways and is expected to achieve long-term relief of the disease (24–26), it is becoming a promising technology in the field of immune tolerability.
Cell Therapy Approaches to ADs
ADs comprise more than 80 different immune disorders including rheumatoid arthritis (RA), type 1 diabetes (T1DM), systemic lupus erythematosus (SLE), multiple sclerosis (MS), Amyotrophic lateral sclerosis (ALS), etc., which collectively affect ~4.5% of the population (27, 28). The consensus is that a mixture of genetic susceptibility and environmental factors are involved in the development of autoimmunity. Large-scale genome-wide association studies (GWASs) and their meta-analyses with a large sample size have elucidated the disease-susceptible genes and disease-causing pathways of many ADs (29). However, potential target autoantigens remain unknown in some of the most important human ADs, and the precise mechanism of disease pathogenesis remains elusive (30).
ADs are generally characterized by the presence of autoreactive immune lymphocytes, leading to intermittent or sustained inflammation (15). In terms of genetic factors, typically, polymorphic HLA genes are associated with antigen presentation pathways, and gene polymorphisms of FOXP3, immune checkpoints, cytokines, and cytokine receptors are all related to immune regulation (31–34). In terms of environmental factors, smoking and intestinal microbes, among others, have been implicated in triggering autoimmunity reactions (28, 35–39). A simple hypothesis that has been put forward to explain the pathogenesis of autoimmune disease is that polymorphisms in various genes result in defective regulation or reduced thresholds for lymphocyte activation, and environmental factors initiate or augment the activation of self-reactive lymphocytes that have escaped control and are poised to react against self-constituents (15). A combination of triggering factors leads to the diversity of immune disorders, including cellular immunity, humoral immunity, neuroendocrine networks, and cytokine networks (21). There are individual differences in immune disorder indicators among different patients, and the immune disorder status of the same patient can change as the AD progresses, presenting a difficult clinical challenge for the treatment of complex, individualized conditions. These different modes of pathogenesis combined with different courses of disease progression are often associated with treatment failure, particularly when the patients are treated with a single drug regimen (40).
The occurrence and development of different ADs share the same common features of lost immune tolerance to autoantigens. Therefore, the fundamental way to treat the underlying problem of AD is to restore the patient's immune tolerance to autoantigens. The mechanisms through which cells maintain immune tolerance are diverse, and with the study of cellular functions and mechanisms, cell-based therapeutic techniques might provide broader prospects than conventional drugs. Several cell therapies have been developed and tested to treat ADs, allograft rejection, and graft-versus-host disease (GVHD).
Strategy 1: Hematopoietic Stem Cell Transplantation (HSCT) for Immune Reconstitution
In 1977, Baldwin et al. (41) observed that RA symptoms were relieved in patients with gold salt-induced aplastic anemia (AA) following allogeneic bone marrow transplantation. In 1995, HSCT was clinically applied to severe ADs where conventional therapy had proved ineffective (42). There were two purposes for using HSCT: one was to eliminate autoreactive immune cells, and the other was to rebuild the self-tolerant immune system. Several clinical trials showed that autologous HSCT was superior to conventional treatments for the treatment of severe ADs, including multiple sclerosis and systemic sclerosis (43–46), and induces long-term disease remission without immunosuppressive drugs. However, some patients still experienced AD recurrence after transplantation (42, 47).
Most hematopoietic stem cells currently used to treat AD have been autologous using either an infusion of unpurified lymphocytes or immunopurified CD34+ HSCs. For several refractory ADs, autologous HSCT can be an effective treatment that induces long-term, drug-free, and asymptomatic remission (42). However, HSCT is still a high-risk treatment, with the occurrence of transplant failure, recurrence of disease, infection, and other complications that might decrease the survival rate of patients. Therefore, HSCT is only considered suitable for patients with serious disease and risk of death. It should be noted that incomplete eradication of autoreactive memory cells or the infusion of unpurified lymphocytes could cause disease recurrence. Encouragingly, a method for long-term ex vivo HSC expansion has been developed recently (48) that might make homogeneous HSCT easier. Moreover, some patients might develop disease flares resulting from the re-induction of autoimmunity driven by genetic predisposition, and allogeneic HSCT could be used as an alternative therapy (49). The further development of more effective and safer HSCT methods remains the next challenge in cell therapy so that this approach can be used more widely in the future for patients with ADs.
Strategy 2: Adoptive Immunotherapy to Eliminate Autoreactive Immune Cells
Autoimmunity is characterized by the presence of autoantibodies and autoreactive T cells directed against normal components of an individual. T-cell vaccination (TCV) therapy is a type of autologous, personalized cell-based therapy in which attenuated autoreactive T cells are administered as immunogenic agents and targeted T-cells are deleted or inactivated (Figure 3A). The concept of TCV was first raised by Ben-nun et al. (50, 51) in 1981, based on the finding that irradiated T lymphocyte cells reactive against myelin basic protein (MBP) can induce a vaccination against experimental autoimmune encephalomyelitis (EAE). Vaccination with the attenuated anti-MBP T cells led to resistance to later attempts to induce EAE by active immunization to MBP in adjuvant (52). Subsequent research on the mechanisms of TCV has revealed a complicated anti-idiotypic and anti-ergotypic network to be responsible for the pathogenic procedure (53, 54). The subject responds to own vaccine T cells by activating regulatory networks of T cells, which, in turn, arrests the damaging inflammation that causes the autoimmune disease (55, 56). Over the past decades, the effect of TCV has been justified in several animal models of autoimmune diseases and graft rejection, including experimental autoimmune encephalomyelitis, lupus, autoimmune uveoretinitis, autoimmune diabetes, autoimmune thyroiditis, collagen-induced arthritis (CIA), and so on (57–62).
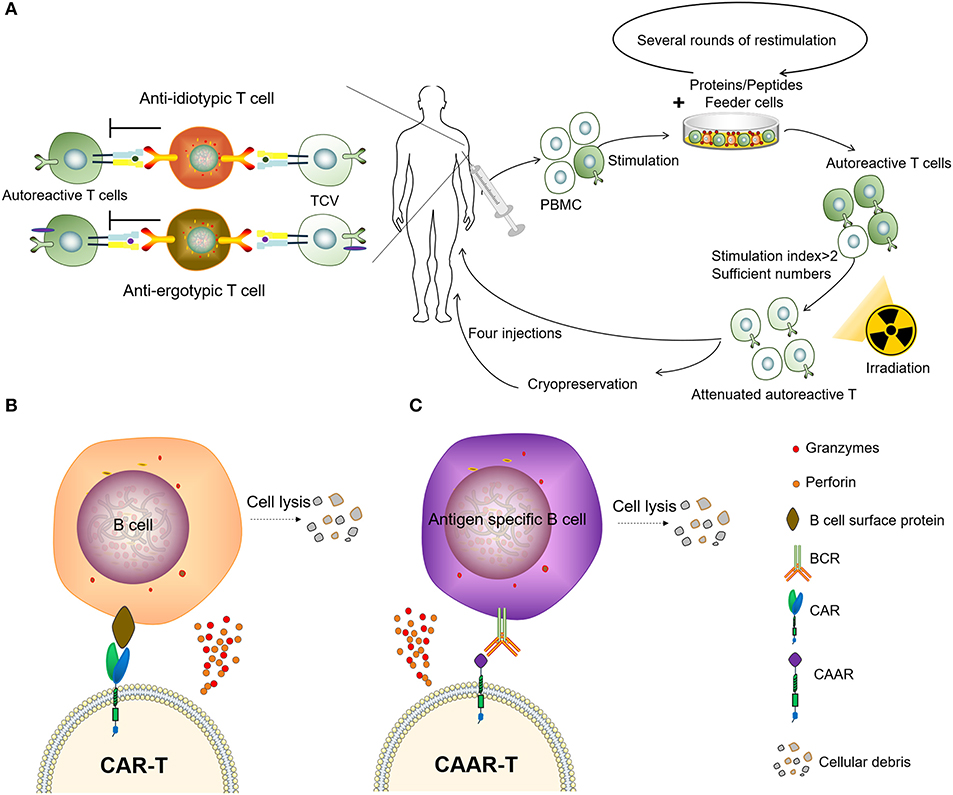
Figure 3. Two types of adoptive immunotherapy to eliminate autoreactive immune cells. (A) Patients receive TCV. (B) Chimeric antigen receptor T (CAR-T) cells targeting B-lineage antigens to kill all B cells. (C) Autoantigen-based chimeric immunoreceptors direct T cells to kill autoreactive B lymphocytes through the specificity of the B cell receptor (BCR).
Moreover, TCV has shown safety and effectiveness in various clinical trials, mostly for patients with MS but also for RA, SLE, and ALS (63–66). Achiron et al. (67) evaluated the efficacy of TCV therapy for 20 patients with aggressive relapsing-remitting MS. TCV treatment had a favorable impact on both annual relapse rate and progression to disability. Seledtsova et al. (68) conducted a study where 39 patients with progressive (chronic) MS were multiply immunized with autological polyclonal TCVs. In the TCV-treated patients, sustained reduction in plasma IFN-γ levels and concomitant increases in IL-4 levels were documented. Indeed, polyclonal T-cell vaccination led to a considerable reduction of proliferative responses of T cells to myelin-associated antigens. Huang et al. (66) enrolled 16 patients with systemic lupus erythematosus (SLE). They found that TCV was associated with remissions in clinical symptoms, reductions in Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) and anti-ds-DNA antibodies, and increases in complement component 3 (C3) and C4. In addition, it is helpful in lowering the glucocorticoid doses of patients' regular usage. Unfortunately, TCV has been somewhat ignored in the past due to standard pharmaceutical avoidance of cell-based and individualized treatments. Nonetheless, cell therapy appears to be coming of age, and TCV has been granted fast-track status by the FDA for the treatment of some types of multiple sclerosis (10).
The presence of autoantibodies is a feature of many ADs and has been widely used to aid the diagnosis of such diseases. B cell/plasma cells have been recognized as an important target for the treatment of some ADs (69). Several drugs that target B cells are in clinical use or are currently being developed, including monoclonal antibodies to target CD19, CD20, and CD22, which are expected to effectively treat various ADs (69). Rituximab depletes B cells by complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC) effects. This drug is now approved for the treatment of RA, granulomatosis with polyangiitis (GPA), and microscopic polyangiitis (MPA), as well as several systemic inflammatory autoimmune diseases (SIADs) (70, 71). However, such antibody therapy requires repeated administration, and drug resistance can occur during long-term administration (72).
The CAR-T has been a major breakthrough for the treatment of B cell malignancies, and these CAR-T cells can eliminate B cells in vivo for an extended period of time (73). It has been reported that the CD19-targeted CAR-T cells persistently depleted CD19+ B cells, eliminated autoantibody production and extended life spans in a mouse lupus model (74). However, as normal B cells can also be being killed by CAR-T cells targeting B-lineage antigens, this significantly impairs the body's ability to fight disease, resulting in the requirement of immunoglobulin injections to continuously maintain immunity. Therefore, it might not be practical to treat autoimmune diseases using CAR-T to clear B-lineage lymphocytes. To circumvent this problem, the chimeric autoantibody receptor (CAAR) has been constructed based on autoantigens, this is capable of binding to the BCR of autoreactive B lymphocytes and therefore has the ability to specifically eliminate autoantigen-reactive B cells. It was reported that CAAR-T cells with the extracellular domain desmoglein3 (Dsg3) were able to eliminate Dsg3-specific B cells in preclinical models of pemphigus vulgaris (PV) (75). Compared to other B-cell depletion therapies, CAAR technology only removes autoreactive B cells, avoiding a decline in immunoglobulins and the opportunistic infections caused by B-cell clearance, and might provide an effective and universal strategy for the specific targeting of autoreactive B cells in antibody-mediated autoimmune disease (Figures 3B,C). However, there are multiple autoantigens that are attacked by the immune system and individualized differences in ADs. Therefore, not only the coverage of disease-associated antigens but also the molecular weight and spatial epitope of each antigen should be considered in the design and construction of effective CAARs to treat such diseases. Recently we observed that the depletion of PD1+ cells mainly removes autoreactive cells and results in a therapeutic effect in several animal models of inflammatory diseases (76), providing an encouraging approach to combat autoimmune diseases.
Strategy 3: Rebuilding Autoimmune Tolerance Using Various Immunoregulatory Cells
Antigen-presenting cells (APCs), including dendritic cells (DCs) and monocytes/macrophages, play an important role in the regulation of innate and acquired immunity, as well as bidirectional regulation in both antigen-specific immunity and immune tolerance. Tolerogenic antigen-presenting cells (tolAPCs) comprise a specific type of APC with immunoregulatory functions (77, 78).
Tolerogenic DCs (tolDCs) are a heterogeneous pool of dendritic cells with immunosuppressive properties. In autoimmunity, DCs tend to produce pro-inflammatory cytokines and lead to the activation of autologous antigen-reactive T cells (79). The DC-induced immune response or immunotolerance is determined by the maturation state of DCs, and both immature DCs (iDCs) and semi-mature DCs have been shown to be tolerogenic (80–84). TolDCs modulate adaptive immune responses and restore tolerance through different mechanisms that involve anergy, the generation of regulatory lymphocyte populations, or the deletion of potentially harmful inflammatory T cell subsets (Figure 4A) (85–88). Dhodapkar et al. (80) first reported the ability of tolDCs to induce antigen-specific immune tolerance in healthy volunteers, using keyhole limpet hemocyanin (KLH) and influenza matrix peptide (MP) in combination with iDC injection, which led to the specific inhibition of MP-specific CD8+T cell effector function and the appearance of MP-specific interleukin 10-producing cells. Giannoukakis et al. (81) reported the safety of tolDCs in adult type 1 diabetic patients. C-peptide levels in some of the subjects became detectable, whereas prior to and at the time of enrollment, they were undetectable. Amezaga et al. (82) reported the treatment of refractory Crohn's disease using tolDCs characterized by high levels of IL-10 production in response to lipopolysaccharide and gram-negative bacteria, wherein three of the nine patients exhibited improved outcomes, with an increase in circulating regulatory T cells (Tregs) and a decrease in IFN-γ. Benham et al. (83) used four different citrulline peptide-loaded NF-κB inhibitor (BAY11-7082)-treated tolDCs, “Rheumavax,” for subcutaneous injection to RA patients. One month post-treatment, patients exhibited lower DAS28 scores, decreased effector T cells, increased FOXP3+ Tregs, and reduced serum C reactive protein, IL-15, IL-29, CX3CL1, and CXCL11, confirming the efficacy and safety of Rheumavax for the treatment of RA. In a further demonstration, Bell et al. (84) injected autologous synovial fluid antigen-loaded tolDCs into the joint cavity to treat rheumatic and inflammatory arthritis, leading to reduced inflammation and pain. Researchers have also established various optimized protocols to maintain the stability of tolDCs and enhance their lymph node-homing capacity (Table 1). Several modulating actors, such as dexamethasone and vitamin A or vitamin D3 were proven to induce DCs with stable regulatory capacity, whereas partial maturation endows tolDCs with robust antigen presentation and lymph node-homing capacity (82, 84, 89).
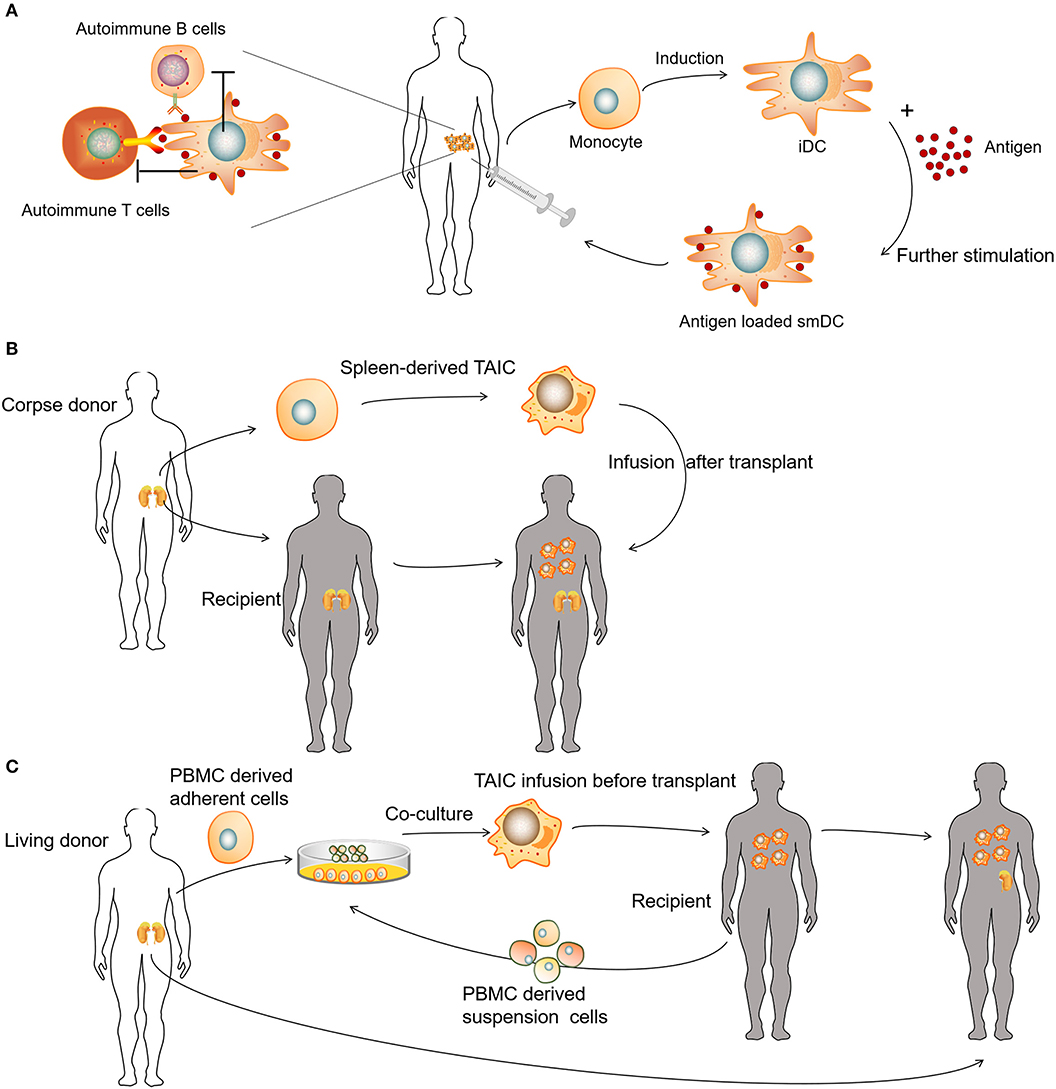
Figure 4. Clinical uses of various tolerogenic antigen-presenting cell (tolAPC) methods. (A) Patients receive autoantigen-loaded tolDCs. (B) Patients receive kidney transplants and corpse donor spleen-derived transplant acceptance-inducing cell (TAIC) treatment. (C) Patients receive living donor PBMC-derived TAICs before kidney transplants.
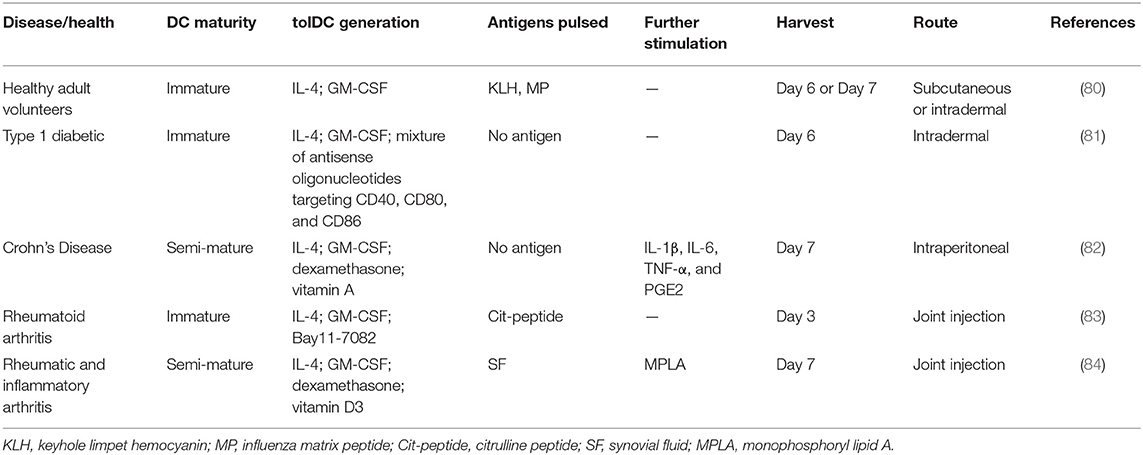
Table 1. Tolerogenic dendritic cell (tolDC)-inducing protocols.
Unlike autologous tolDCs, donor-derived regulatory macrophages (Mregs) have also been used to induce allogeneic tolerance during organ transplantation. Transplant acceptance-inducing cells (TAICs) are primarily considered a class of immunoregulatory macrophages (90–92). In the TAIC-I clinical trial, 12 patients received renal transplants and corpse donor spleen-derived TAIC treatment (Figure 4B). An immunosuppressive regimen (including tacrolimus and sirolimus triple) was used, following TAIC treatment 5 days post-transplantation. In the majority of patients (10 of 12), the amount of supporting immunosuppressant was gradually reduced, whereas eight patients discontinued steroids at 8 weeks, and six of eight patients discontinued sirolimus at 12 weeks; only low-dose tacrolimus maintenance therapy was required subsequently (91). In the TAIC-II clinical trial, the investigators co-cultured adherent cells derived from living donor peripheral blood mononuclear cells (PBMCs) with suspension cells from the recipient PBMCs to obtain TAICs for renal transplantation (Figure 4C). Clinically, it was possible to treat four of five with tacrolimus monotherapy rather than conventional immunosuppressive regimens, whereas three of five patients were able to tolerate low-dose tacrolimus monotherapy. Although the trial failed to demonstrate the effectiveness of TAICs, treatment was able to eliminate several allogeneic reactions (92). For example, post-TAIC transplantation, the anti-donor HLA-specific antibody disappeared and, in some cases, serum immunoglobulin returned to normal levels, indicating that TAIC treatment could eliminate donor-HLA-specific antibodies (90).
In another clinical trial (93), the investigators adjusted the TAIC-II clinical trial to trace the presence of Mregs in the body. Mregs were initially trapped in the lung, and thereafter, cells were detected in circulation after 2.5 h and began to accumulate in the liver and spleen. After 22 h of Mreg administration, the signal was markedly reduced in the lung and increased in the liver, spleen, and hematopoietic active bone marrow. No signal was detected from the patient's urinary tract throughout the examination period, indicating that most infused Mregs remained alive. There is a risk of pulmonary embolism in cell therapies, and the risk is theoretically related to the number and size of the cells. However, human Mregs are larger in volume than other cells used in cell therapy, having a diameter of ~15–30 μm. As the average diameter of pulmonary capillaries is 7.5 ± 2.3 μm, it is important to consider the risk of pulmonary embolism associated with Mreg treatments (94).
Tregs are important regulatory cells that maintain peripheral immune tolerance (95–98). Claudio et al. (99) first used cultured cord blood polyclonal Tregs to treat acute GVHD (aGVHD), and 23 patients treated with Tregs exhibited a lower incidence of grade II-IV aGVHD compared to that in 108 historical controls (43 vs. 61%, P = 0.05); this was associated with no toxicity after infusion and no detrimental effects of infection or instances of relapse or early death. Mauro et al. (100) evaluated the effects of GVHD prevention and immune reconstitution through the early infusion of freshly isolated Tregs for 28 high-risk hematologic malignancy patients treated with HLA-haploidentical HSCT. Their findings indicated that the adoptive transfer of Tregs prevented GVHD in the absence of any post-transplantation immunosuppression, promoted lymphoid reconstitution, improved immunity to opportunistic pathogens, and did not diminish the graft-versus-leukemia effect. Natalia et al. (101) treated 12 children with type 1 diabetes (T1D) with autologous expanded Tregs. After transfusion, the number of Tregs in peripheral blood increased, and no serious side effects were observed. Most patients exhibited increased C-peptide levels and lower demand for insulin, whereas two patients were completely insulin-independent at 1 year. In another study, similar results were observed in adult subjects (102). However, a phase II clinical trial (NCT02691247) showed that autologous ex vivo polyclonally expanded Treg infusion failed to preserve C-peptide production 1 year after the start of treatment in 113 newly diagnosed adolescents with T1D (103). One major problem is that transferring a large number of Tregs of broad undefined specificity can potentially suppress protective immunity against tumors and infectious diseases (104). Evidence from animal models of Treg therapy has clearly shown that antigen-specific Tregs are vastly superior to polyclonal Tregs, meaning that fewer cells are needed for the desired therapeutic effect (105).
In recent years, studies of Tregs engineered with chimeric antigen receptors (CAR-Tregs) have made great progress in optimizing anti-inflammatory and immune-tolerogenic responses (104, 106, 107). There are several examples of this as follows. (i) Elinav et al. (108) constructed a CAR-Treg specific for 2,4,6-trinitrophenol and demonstrated the CAR-Treg-specific effect pattern in different colitis mouse models. (ii) Fransson et al. (109) constructed CAR-Treg targeting myelin oligodendrocyte glycoproteins, which exerted a continual inhibitory effect for the treatment of EAE in mice. (iii) Dan et al. (110) constructed CAR-Tregs to target carcinoembryonic antigen to treat colitis in mice and found that CAR-Tregs can accumulate at the inflammation site in mice and inhibit colitis development. (iv) MacDonald et al. (111) constructed an HLA-A2-specific CAR (A2-CAR) and showed the inhibition of GVHD caused by HLA-A2+ T cells. (v) Boardman et al. (112) and Noyan et al. (113) also demonstrated that A2-CAR-Tregs could completely prevent allogeneic transplant rejection without using other immunosuppressant intervention in humanized mice. (iv) Yoon et al. (114) constructed CAR-Tregs targeting factor FVIII, which effectively inhibited T cell and B cell responses to FVIII. These results showed that specifically designed CAR-Tregs can induce immune tolerance not only by targeting cell surface proteins but also by targeting secretory proteins (Figure 5). In conclusion, CAR-Tregs display the hallmarks of cell therapy, including being target-specific, long-lasting, chemotaxic, and highly efficient for broad application in the treatment of ADs, graft-versus-host diseases, and transplant rejection. We believe that CAR-Tregs will be the next frontier in the development of anti-inflammatory and tolerogenic therapies.
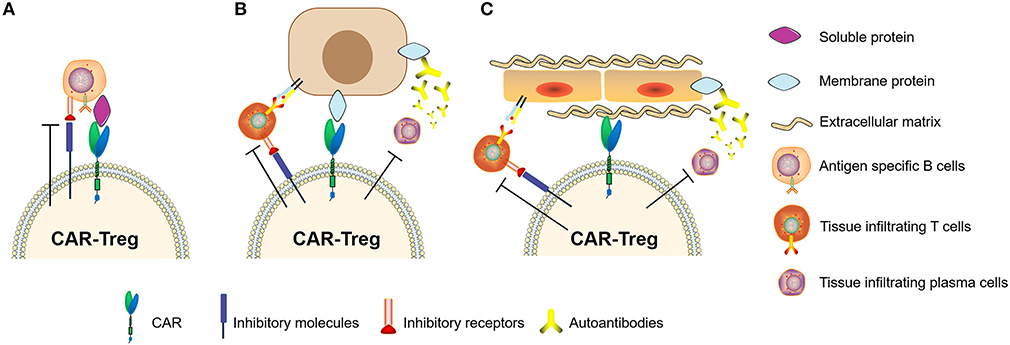
Figure 5. Chimeric antigen receptor regulatory T cells (CAR-Tregs) can target different types of antigens. (A) CAR-Tregs targeting soluble proteins, (B) cell membrane proteins, and inhibiting autoimmune B cells or/and T cells. (C) We hypothesized that CAR-Tregs could also target structural proteins to achieve tissue-specific inhibition.
Mesenchymal stromal cells (MSCs), widely distributed in various connective tissues, have multi-directional differentiation potential and strong immune regulation and tissue repair functions. Therefore, MSCs naturally play a role in modulating ADs, organ transplantation, and GVHD. MSC-related clinical trials include the treatment of diabetes, RA, Behcet's disease, organ transplantation, systemic lupus erythematosus, and others (115–119). Among them, it has been shown that MSC infusion promotes implantation in HSCT, reduces the incidence of chronic GVHD, and also plays an important role in the treatment of aGVHD (120–123). In a meta-analysis of over 200 patients to evaluate the efficacy of locally injected MSCs as a treatment for inflammatory bowel disease (IBD), more than half of patients displayed complete remission, and more than two-thirds of patients showed positive treatment responses, indicating that MSCs have considerable potential for IBD treatment (124). However, the majority of MSC-based trials for ADs are still in early phases I or II. Some have promising results and no reported toxicity to date, but phase III studies will be needed to confirm their efficacy (125, 126).
MSC sources currently used for clinical trials include bone marrow, perinatal tissue, dental pulp, and adipose tissue (127–132). Differences associated with tissue source, such as donor-related variability, cell culture system, passage number, and reagent formulation, might be important factors for the biological characteristics of MSC. These characteristics mainly include the homing ability of MSCs, viability in vivo, and immunosuppressive ability, which exert important influences on the therapeutic effect of tolerability. Intravenous injection is one of the most commonly used infusion methods for MSCs, which can play a systemic regulatory role. Whether MSCs can home to the site of inflammation is an important factor influencing their therapeutic effect, as these cells require interactions between tissue-specific chemokines and the corresponding receptors. In a mouse model of contact hypersensitivity (CHS), CXCR5-overexpressing MSCs show significantly increased migration ability toward CXCL13, which is highly upregulated in inflamed ears of CHS mice (133). Under certain pathological conditions, ischemic or inflammatory environments might be detrimental to the survival of MSCs. Researchers have used some proinflammatory cytokine pretreatments, such as IFN-γ, TNF-α, and IL-17, among others, to promote MSC expression of IDO and NO, which increases their immunosuppressive abilities (18, 134–138).
Conclusion and Perspectives
Most immunoregulatory cells exhibit issues regarding survivability, stability, plasticity, and homing capacity. Cells for tolerogenic treatment can be derived from an autologous or allogeneic source. Further, the fact that autologous cells might contain gene-related defects, resulting in less effective tolerogenic therapy, cannot be ignored. Whereas, the short survival period of allogeneic cells, because of rejection, might lead to poor treatment efficacy, the prolonged persistence of allogeneic cells will raise safety concerns. After infusion, the avoidance of the transformation of immunoregulatory cells into effector cells is essential for the long-term efficacy of tolerogenic therapy. In particular, if the regulatory cells used for targeted cell therapy (e.g., CAR-Treg) are unstable and transform into inflammatory cells, they might exert an adverse effect on the disease. For treatment with Tregs, the memory Tregs induce a stronger inflammatory response than naïve Tregs (139). Therefore, not only the choice between autologous or allogeneic cells, as well as the cell type, needs to be considered, but also the stable subpopulation of cells for treatment should be considered.
Genetically modified immune cell therapy technology, such as CAR-Treg, could present a breakthrough for the treatment of ADs. Genetic modification can compensate for genetic defects to a certain extent and has advantages for therapeutic cell targeting, chemotaxis, and enhancement. Similarly, the safety of gene therapy should be carefully considered, including genetically modifying vectors, the recombination of internal and exogenous genes, the controllability of gene expression, and even ethical issues.
In addition, we propose two directions: immortalized cell lines and cell-like materials. Immortalized NK92 cells have been used to treat malignant tumors in several clinical trials (140). Immortalized cell lines can be mass-produced, and the product quality is easier to control. To date, there have been no studies using immortal cells for immune tolerance therapy. With the development of genetic modification technology, the immortalized cell line can be genetically engineered to express a certain level of death ligands/inhibitory checkpoint molecules/membrane-bound inhibitory cytokines or ectoenzymes to exert potent immunosuppressive effects under targeted conditions. Gamma-ray treatment of immortalized cells can induce a loss of proliferative ability while maintaining biological activity for a short period of time. Functions of regulatory cells such as Tregs do not depend on self-proliferation, and apoptotic Tregs display stronger immunosuppressive effects (141). The function of inflammatory cells might be inhibited and immunosuppressive cell functions can be enhanced by contact with artificial immortalized cells, thereby achieving the induction of immune tolerance. Therefore, immortalized cells might serve as an alternative choice for tolerogenic cell therapy (Figure 6).
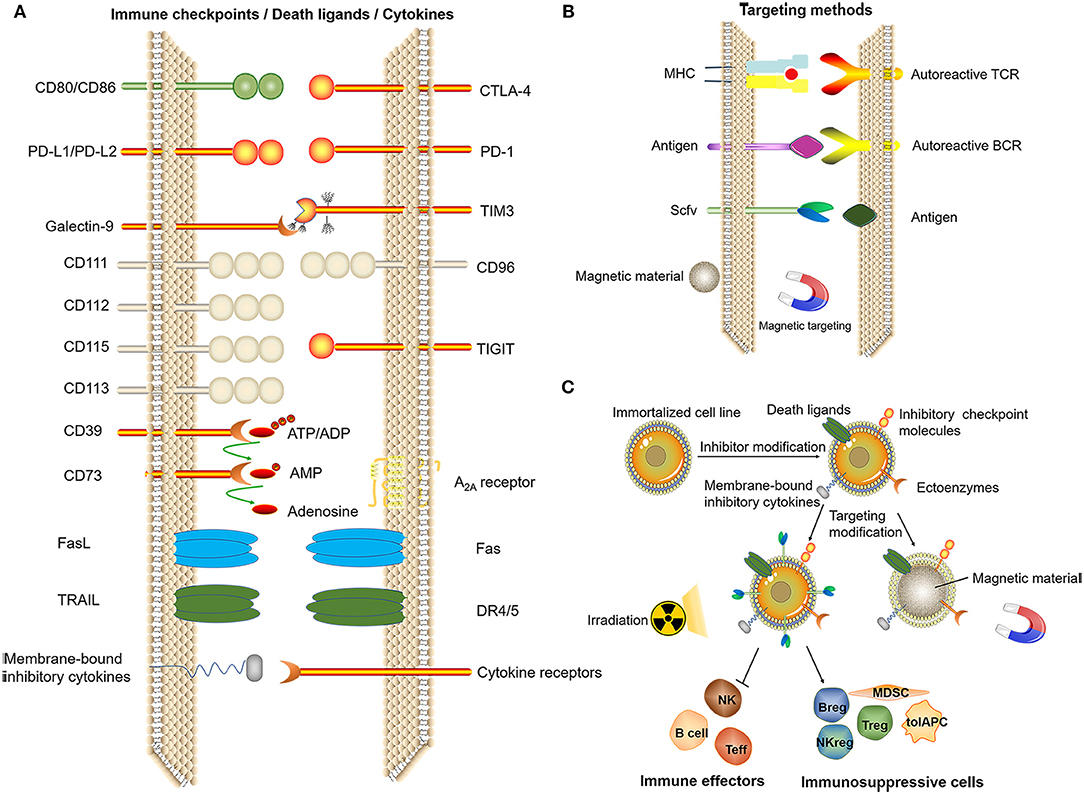
Figure 6. Related technologies for tolerogenic cell therapy that can be mass-produced. (A) Induction of immune tolerance by immune checkpoints, death ligands, (artificial) membrane-type cytokines. (B) Targeting methods. (C) Genetic modification of immortalized cell lines and screening to obtain cells with the ability to induce immune tolerance after irradiation for treatment or by using the cell membrane to bind magnetic materials, thus exerting an immunomodulatory effect at a specific site under the action of a magnetic field.
As the mechanism through which various inhibitory cytokines or ligands exert immunosuppressive effects are being gradually discovered, one effective strategy might be the construction of cell-like particles based on the immune checkpoints of these known mechanisms. Some researchers have studied tolerogenic therapies using nanomaterials (142–144). Cell-like materials with individualized and regulated functions could be synthesized through biochemical methods, which can induce the apoptosis of autoreactive immune cells, thereby achieving the recovery of immune tolerance.
In conclusion, there are still many problems to be solved regarding current tolerogenic cell therapy technology. However, accumulating evidence indicates that cell-based tolerogenic treatment will undoubtedly play an important role in future medical technology.
Author Contributions
ZW and XL wrote the manuscript. SZ, JZ, FC, and JB reviewed and edited the manuscript. All authors read and approved the manuscript.
Funding
This work was, in part, supported by Grant QC2017099 from the Natural Science Foundation of Heilongjiang Province (Q.Z).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bellanti JA (Ed) (2012). Immunology IV: Clinical Applications in Health and Disease. Bethesda, MD: I Care Press.
2. Riva A, Chokshi S. Immune checkpoint receptors: homeostatic regulators of immunity. Hepatol Int. (2018) 12:223–36. doi: 10.1007/s12072-018-9867-9
3. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. (2014) 157:121–41. doi: 10.1016/j.cell.2014.03.011
4. Huntington ND, Gray DH. Immune homeostasis in health and disease. Immunol Cell Biol. (2018) 96:451–2. doi: 10.1111/imcb.12043
5. Luo Y, Zheng SG. Hall of fame among pro-inflammatory cytokines: interleukin-6 gene and its transcriptional regulation mechanisms. Front Immunol. (2016) 7:604. doi: 10.3389/fimmu.2016.00604
6. Horwitz DA, Zheng SG, Gray JD. Natural and TGF-beta-induced Foxp3(+)CD4(+) CD25(+) regulatory T cells are not mirror images of each other. Trends Immunol. (2008) 29:429–35. doi: 10.1016/j.it.2008.06.005
7. Pan HF, Leng RX, Li XP, Zheng SG, Ye DQ. Targeting T-helper 9 cells and interleukin-9 in autoimmune diseases. Cytokine Growth Factor Rev. (2013) 24:515–22. doi: 10.1016/j.cytogfr.2013.09.001
8. Zou Y, Xu S, Xiao Y, Qiu Q, Shi M, Wang J, et al. Long noncoding RNA LERFS negatively regulates rheumatoid synovial aggression and proliferation. J Clin Invest. (2018) 128:4510–24. doi: 10.1172/JCI97965
9. Liu Y, Pan YF, Xue YQ, Fang LK, Guo XH, Guo X, et al. uPAR promotes tumor-like biologic behaviors of fibroblast-like synoviocytes through PI3K/Akt signaling pathway in patients with rheumatoid arthritis. Cell Mol Immunol. (2018) 15:171–81. doi: 10.1038/cmi.2016.60
10. Cohen IR. Activation of benign autoimmunity as both tumor and autoimmune disease immunotherapy: a comprehensive review. J Autoimmun. (2014) 54:112–7. doi: 10.1016/j.jaut.2014.05.002
11. Maueröder C, Munoz LE, Chaurio RA, Herrmann M, Schett G, Berens C. Tumor immunotherapy: lessons from autoimmunity. Front Immunol. (2014) 5:212. doi: 10.3389/fimmu.2014.00212
12. Chen Y, Colello J, Jarjour W, Zheng SG. Cellular metabolic regulation in the differentiation and function of regulatory T cells. Cells. (2019) 8:188. doi: 10.3390/cells8020188
13. Xu A, Liu Y, Chen W, Wang J, Xue Y, Huang F, et al. TGF-beta-induced regulatory T cells directly suppress B cell responses through a noncytotoxic mechanism. J Immunol. (2016) 196:3631–41. doi: 10.4049/jimmunol.1501740
14. Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. (2010) 10:849–59. doi: 10.1038/nri2889
15. Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest. (2015) 125:2228–33. doi: 10.1172/JCI78088
16. Polachek A, Li S, Polachek IS, Chandran V, Gladman D. Therapeutic tolerance in autoimmune disease. Semin Arthritis Rheum. (2018) 48:558–62. doi: 10.1016/j.semarthrit.2018.09.008
17. Gu J, Ni X, Pan X, Lu H, Lu Y, Zhao J, et al. Human CD39(hi) regulatory T cells present stronger stability and function under inflammatory conditions. Cell Mol Immunol. (2017) 14:521–8. doi: 10.1038/cmi.2016.30
18. Su W, Wan Q, Huang J, Han L, Chen X, Chen G, et al. Culture medium from TNF-α-stimulated mesenchymal stem cells attenuates allergic conjunctivitis through multiple antiallergic mechanisms. J Allergy Clin Immunol. (2015) 136:423–32.e8. doi: 10.1016/j.jaci.2014.12.1926
19. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. (2018) 359:1361–5. doi: 10.1126/science.aar6711
21. Coronel-Restrepo N, Posso-Osorio I, Naranjo-Escobar J, Tobón GJ. Autoimmune diseases and their relation with immunological, neurological and endocrinological axes. Autoimmun Rev. (2017) 16:684–92. doi: 10.1016/j.autrev.2017.05.002
22. Talotta R, Atzeni F, Batticciotto A, Benucci M, Bongiovanni S, Sarzi-Puttini P. Biological agents in rheumatoid arthritis: a cross-link between immune tolerance and immune surveillance. Curr Rheumatol Rev. (2018) 14:131–9. doi: 10.2174/1573397112666161230125317
23. Her M, Kavanaugh A. Alterations in immune function with biologic therapies for autoimmune disease. J Allergy Clin Immunol. (2016) 137:19–27. doi: 10.1016/j.jaci.2015.10.023
24. Zheng SG, Meng L, Wang JH, Watanabe M, Barr ML, Cramer DV, et al. Transfer of regulatory T cells generated ex vivo modifies graft rejection through induction of tolerogenic CD4+CD25+ cells in the recipient. Int Immunol. (2006) 18:279–89. doi: 10.1093/intimm/dxh368
25. Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA. Natural and induced CD4+CD25+ cells educate CD4+CD25- cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J Immunol. (2004) 172:5213–21. doi: 10.4049/jimmunol.172.9.5213
26. Liu Y, Lan Q, Lu L, Chen M, Xia Z, Ma J, et al. Phenotypic and functional characteristic of a newly identified CD8+ Foxp3- CD103+ regulatory T cells. J Mol Cell Biol. (2014) 6:81–92. doi: 10.1093/jmcb/mjt026
27. Hayter SM, Cook MC. Updated assessment of the prevalence, spectrum and case definition of autoimmune disease. Autoimmun Rev. (2012) 11:754–65. doi: 10.1016/j.autrev.2012.02.001
28. Parks CG, Miller FW, Pollard KM, Selmi C, Germolec D, Joyce K, et al. Expert panel workshop consensus statement on the role of the environment in the development of autoimmune disease. Int J Mol Sci. (2014) 15:14269–97. doi: 10.3390/ijms150814269
29. Kochi Y. Genetics of autoimmune diseases: perspectives from genome-wide association studies. Int Immunol. (2016) 28:155–61. doi: 10.1093/intimm/dxw002
30. Floreani A, Leung PS, Gershwin ME. Environmental basis of autoimmunity. Clin Rev Allergy Immunol. (2016) 50:287–300. doi: 10.1007/s12016-015-8493-8
31. Gutierrez-Arcelus M, Rich SS, Raychaudhuri S. Autoimmune diseases - connecting risk alleles with molecular traits of the immune system. Nat Rev Genet. (2016) 17:160–74. doi: 10.1038/nrg.2015.33
32. Katoh H, Zheng P, Liu Y. FOXP3: genetic and epigenetic implications for autoimmunity. J Autoimmun. (2013) 41:72–8. doi: 10.1016/j.jaut.2012.12.004
33. Zamani MR, Aslani S, Salmaninejad A, Javan MR, Rezaei N. PD-1/PD-L and autoimmunity: a growing relationship. Cell Immunol. (2016) 310:27–41. doi: 10.1016/j.cellimm.2016.09.009
34. Vandenbroeck K. Cytokine gene polymorphisms and human autoimmune disease in the era of genome-wide association studies. J Interferon Cytokine Res. (2012) 32:139–51. doi: 10.1089/jir.2011.0103
35. Anaya JM, Ramirez-Santana C, Alzate MA, Molano-Gonzalez N, Rojas-Villarraga A. The autoimmune ecology. Front Immunol. (2016) 7:139. doi: 10.3389/fimmu.2016.00139
36. Jörg S, Grohme DA, Erzler M, Binsfeld M, Haghikia A, Müller DN, et al. Environmental factors in autoimmune diseases and their role in multiple sclerosis. Cell Mol Life Sci. (2016) 73:4611–22. doi: 10.1007/s00018-016-2311-1
37. Nielsen PR, Kragstrup TW, Deleuran BW, Benros ME. Infections as risk factor for autoimmune diseases - A nationwide study. J Autoimmun. (2016) 74:176–81. doi: 10.1016/j.jaut.2016.05.013
38. Wang H, Chen W, Li D, Yin X, Zhang X, Olsen N, et al. Vitamin D and chronic diseases. Aging Dis. (2017) 8:346–53. doi: 10.14336/AD.2016.1021
39. Zhou L, Wang J, Li J, Li T, Chen Y, June RR, et al. 1,25-Dihydroxyvitamin D3 ameliorates collagen-induced arthritis via suppression of Th17 cells through miR-124 mediated inhibition of IL-6 signaling. Front Immunol. (2019) 10:178. doi: 10.3389/fimmu.2019.00178
40. Chandrashekara S. The treatment strategies of autoimmune disease may need a different approach from conventional protocol: a review. Indian J Pharmacol. (2012) 44:665–71. doi: 10.4103/0253-7613.103235
41. Baldwin JL, Storb R, Thomas ED, Mannik M. Bone marrow transplantation in patients with gold-induced marrow aplasia. Arthritis Rheum. (1977) 20:1043–8. doi: 10.1002/art.1780200502
42. Swart JF, Delemarre EM, Van Wijk F, Boelens JJ, Kuball J, Van Laar JM, et al. Haematopoietic stem cell transplantation for autoimmune diseases. Nat Rev Rheumatol. (2017) 13:244–56. doi: 10.1038/nrrheum.2017.7
43. Snowden JA, Badoglio M, Labopin M, Giebel S, McGrath E, Marjanovic Z, et al. Evolution, trends, outcomes, and economics of hematopoietic stem cell transplantation in severe autoimmune diseases. Blood Adv. (2017) 1:2742–55. doi: 10.1182/bloodadvances.2017010041
44. Muraro PA, Pasquini M, Atkins HL, Bowen JD, Farge D, Fassas A, et al. Long-term outcomes after autologous hematopoietic stem cell transplantation for multiple sclerosis. JAMA Neurol. (2017) 74:459–69. doi: 10.1001/jamaneurol.2016.5867
45. Alexander T, Bondanza A, Muraro PA, Greco R, Saccardi R, Daikeler T, et al. SCT for severe autoimmune diseases: consensus guidelines of the European Society for Blood and Marrow Transplantation for immune monitoring and biobanking. Bone Marrow Transplant. (2015) 50:173–80. doi: 10.1038/bmt.2014.251
46. van Laar JM, Farge D, Sont JK, Naraghi K, Marjanovic Z, Larghero J, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA. (2014) 311:2490–8. doi: 10.1001/jama.2014.6368
47. Malmegrim KCR, Lima-Júnior JR, Arruda LCM, de Azevedo JTC, de Oliveira GLV, Oliveira MC. Autologous hematopoietic stem cell transplantation for autoimmune diseases: from mechanistic insights to biomarkers. Front Immunol. (2018) 9:2602. doi: 10.3389/fimmu.2018.02602
48. Wilkinson AC, Ishida R, Kikuchi M, Sudo K, Morita M, Crisostomo RV, et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature. (2019) 571:117–21. doi: 10.1038/s41586-019-1244-x
49. Greco R, Labopin M, Badoglio M, Veys P, Furtado Silva JM, Abinun M, et al. Allogeneic HSCT for autoimmune diseases: a retrospective study from the EBMT ADWP, IEWP, and PDWP working parties. Front Immunol. (2019) 10:1570. doi: 10.3389/fimmu.2019.01570
50. Ben-Nun A, Wekerle H, Cohen IR. The rapid isolation of clonable antigen-specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur J Immunol. (1981) 11:195–9. doi: 10.1002/eji.1830110307
51. Ben-Nun A, Wekerle H, Cohen IR. Vaccination against autoimmune encephalomyelitis with T-lymphocyte line cells reactive against myelin basic protein. Nature. (1981) 292:60–1. doi: 10.1038/292060a0
52. Holoshitz J, Naparstek Y, Ben-Nun A, Cohen IR. Lines of T lymphocytes induce or vaccinate against autoimmune arthritis. Science. (1983) 219:56–8. doi: 10.1126/science.6336851
53. Lider O, Reshef T, Beraud E, Ben-Nun A, Cohen IR. Anti-idiotypic network induced by T cell vaccination against experimental autoimmune encephalomyelitis. Science. (1988) 239:181–3. doi: 10.1126/science.2447648
54. Lohse AW, Mor F, Karin N, Cohen IR. Control of experimental autoimmune encephalomyelitis by T cells responding to activated T cells. Science. (1989) 244:820–2. doi: 10.1126/science.2471264
55. Cohen IR, Quintana FJ, Mimran A. Tregs in T cell vaccination: exploring the regulation of regulation. J Clin Invest. (2004) 114:1227–32. doi: 10.1172/JCI200423396
56. Wang M, Yang L, Sheng X, Chen W, Tang H, Sheng H, et al. T-cell vaccination leads to suppression of intrapancreatic Th17 cells through Stat3-mediated RORgammat inhibition in autoimmune diabetes. Cell Res. (2011) 21:1358–69. doi: 10.1038/cr.2011.74
57. De Alborán IM, Gutierrez JC, Gonzalo JA, Andreu JL, Marcos MA, Kroemer G, et al. lpr T cells vaccinate against lupus in MRL/lpr mice. Eur J Immunol. (1992) 22:1089–93. doi: 10.1002/eji.1830220432
58. Beraud E, Kotake S, Caspi RR, Oddo SM, Chan CC, Gery I, et al. Control of experimental autoimmune uveoretinitis by low dose T cell vaccination. Cell Immunol. (1992) 140:112–22. doi: 10.1016/0008-8749(92)90180-W
59. Kakimoto K, Katsuki M, Hirofuji T, Iwata H, Koga T. Isolation of T cell line capable of protecting mice against collagen-induced arthritis. J Immunol. (1988) 140:78–83.
60. Maron R, Zerubavel R, Friedman A, Cohen IR. T lymphocyte line specific for thyroglobulin produces or vaccinates against autoimmune thyroiditis in mice. J Immunol. (1983) 131:2316–22.
61. Volovitz I, Marmor Y, Mor F, Flügel A, Odoardi F, Eisenbach L, et al. T cell vaccination induces the elimination of EAE effector T cells: analysis using GFP-transduced, encephalitogenic T cells. J Autoimmun. (2010) 35:135–44. doi: 10.1016/j.jaut.2010.05.003
62. Elias D, Tikochinski Y, Frankel G, Cohen IR. Regulation of NOD mouse autoimmune diabetes by T cells that recognize a TCR CDR3 peptide. Int Immunol. (1999) 11:957–66. doi: 10.1093/intimm/11.6.957
63. Breedveld FC, Struyk L, van Laar JM, Miltenburg AM, de Vries RR, van den Elsen PJ. Therapeutic regulation of T cells in rheumatoid arthritis. Immunol Rev. (1995) 144:5–16. doi: 10.1111/j.1600-065X.1995.tb00063.x
64. Moviglia GA, Moviglia-Brandolino MT, Varela GS, Albanese G, Piccone S, Echegaray G, et al. Feasibility, safety, and preliminary proof of principles of autologous neural stem cell treatment combined with T-cell vaccination for ALS patients. Cell Transplant. (2012) 21(Suppl. 1):S57–63. doi: 10.3727/096368912X633770
65. Karussis D, Shor H, Yachnin J, Lanxner N, Amiel M, Baruch K, et al. T cell vaccination benefits relapsing progressive multiple sclerosis patients: a randomized, double-blind clinical trial. PLoS ONE. (2012) 7:e50478. doi: 10.1371/journal.pone.0050478
66. Huang L, Yang Y, Kuang Y, Wei D, Li W, Yin Q, et al. The Impact of T Cell vaccination in alleviating and regulating systemic lupus erythematosus manifestation. J Immunol Res. (2016) 2016:5183686. doi: 10.1155/2016/5183686
67. Achiron A, Lavie G, Kishner I, Stern Y, Sarova-Pinhas I, Ben-Aharon T, et al. T cell vaccination in multiple sclerosis relapsing-remitting nonresponders patients. Clin Immunol. (2004) 113:155–60. doi: 10.1016/j.clim.2004.06.004
68. Seledtsova GV, Ivanova IP, Shishkov AA, Seledtsov VI. Immune responses to polyclonal T-cell vaccination in patients with progressive multiple sclerosis. J Immunotoxicol. (2016) 13:879–84. doi: 10.1080/1547691X.2016.1223767
69. Hofmann K, Clauder AK, Manz RA. Targeting B cells and plasma cells in autoimmune diseases. Front Immunol. (2018) 9:835. doi: 10.3389/fimmu.2018.00835
70. MacIsaac J, Siddiqui R, Jamula E, Li N, Baker S, Webert KE, et al. Systematic review of rituximab for autoimmune diseases: a potential alternative to intravenous immune globulin. Transfusion. (2018) 58:2729–35. doi: 10.1111/trf.14841
71. Vikse J, Jonsdottir K, Kvaløy JT, Wildhagen K, Omdal R. Tolerability and safety of long-term rituximab treatment in systemic inflammatory and autoimmune diseases. Rheumatol Int. (2019) 39:1083–90. doi: 10.1007/s00296-019-04272-1
72. Atreya R, Neurath MF. Mechanisms of molecular resistance and predictors of response to biological therapy in inflammatory bowel disease. Lancet Gastroenterol Hepatol. (2018) 3:790–802. doi: 10.1016/S2468-1253(18)30265-6
73. Seidel K, Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med. (2019) 25:1341–55. doi: 10.1038/s41591-019-0564-6
74. Kansal R, Richardson N, Neeli I, Khawaja S, Chamberlain D, Ghani M, et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci Transl Med. (2019) 11:eaav1648. doi: 10.1126/scitranslmed.aav1648
75. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. (2016) 353:179–84. doi: 10.1126/science.aaf6756
76. Zhao P, Wang P, Dong S, Zhou Z, Cao Y, Yagita H, et al. Depletion of PD-1-positive cells ameliorates autoimmune disease. Nat Biomed Eng. (2019) 3:292–305. doi: 10.1038/s41551-019-0360-0
77. Lan Q, Zhou X, Fan H, Chen M, Wang J, Ryffel B, et al. Polyclonal CD4+Foxp3+ Treg cells induce TGF β-dependent tolerogenic dendritic cells that suppress the murine lupus-like syndrome. J Mol Cell Biol. (2012) 4:409–19. doi: 10.1093/jmcb/mjs040
78. Ramalingam R, Larmonier CB, Thurston RD, Midura-Kiela MT, Zheng SG, Ghishan FK, et al. Dendritic cell-specific disruption of TGF-β receptor II leads to altered regulatory T cell phenotype and spontaneous multiorgan autoimmunity. J Immunol. (2012) 189:3878–93. doi: 10.4049/jimmunol.1201029
79. Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat Rev Immunol. (2013) 13:566–77. doi: 10.1038/nri3477
80. Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. (2001) 193:233–8. doi: 10.1084/jem.193.2.233
81. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care. (2011) 34:2026–32. doi: 10.2337/dc11-0472
82. Jauregui-Amezaga A, Cabezón R, Ramírez-Morros A, España C, Rimola J, Bru C, et al. Intraperitoneal administration of autologous tolerogenic dendritic cells for refractory crohn's disease: a phase i study. J Crohns Colitis. (2015) 9:1071–8. doi: 10.1093/ecco-jcc/jjv144
83. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Transl Med. (2015) 7:290ra87. doi: 10.1126/scitranslmed.aaa9301
84. Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA, et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheum Dis. (2017) 76:227–34. doi: 10.1136/annrheumdis-2015-208456
85. Qian L, Qian C, Chen Y, Bai Y, Bao Y, Lu L, et al. Regulatory dendritic cells program B cells to differentiate into CD19hiFcyIIbhi regulatory B cells through IFN-β and CD40. Blood. (2012) 120:581–91. doi: 10.1182/blood-2011-08-377242
86. Huang H, Dawicki W, Zhang X, Town J, Gordon JR. Tolerogenic dendritic cells induce CD4+CD25hiFoxp3+ regulatory T cell differentiation from CD4+CD25-/loFoxp3- effector T cells. J Immunol. (2010) 185:5003–10. doi: 10.4049/jimmunol.0903446
87. Steinbrink K, Graulich E, Kubsch S, Knop J, Enk AH. CD4(+) and CD8(+) anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood. (2002) 99:2468–76. doi: 10.1182/blood.V99.7.2468
88. García-González P, Ubilla-Olguín G, Catalán D, Schinnerling K, Aguillón JC. Tolerogenic dendritic cells for reprogramming of lymphocyte responses in autoimmune diseases. Autoimmun Rev. (2016) 15:1071–80. doi: 10.1016/j.autrev.2016.07.032
89. Huang Z, Liu Y, Qi G, Brand D, Zheng SG. Role of Vitamin A in the immune system. J Clin Med. (2018) 7:258. doi: 10.3390/jcm7090258
90. Hutchinson JA, Roelen D, Riquelme P, Brem-Exner BG, Witzke O, Philipp T, et al. Preoperative treatment of a presensitized kidney transplant recipient with donor-derived transplant acceptance-inducing cells. Transpl Int. (2008) 21:808–13. doi: 10.1111/j.1432-2277.2008.00712.x
91. Hutchinson JA, Riquelme P, Brem-Exner BG, Schulze M, Matthäi M, Renders L, et al. Transplant acceptance-inducing cells as an immune-conditioning therapy in renal transplantation. Transpl Int. (2008) 21:728–41. doi: 10.1111/j.1432-2277.2008.00680.x
92. Hutchinson JA, Brem-Exner BG, Riquelme P, Roelen D, Schulze M, Ivens K, et al. A cell-based approach to the minimization of immunosuppression in renal transplantation. Transpl Int. (2008) 21:742–54. doi: 10.1111/j.1432-2277.2008.00692.x
93. Hutchinson JA, Riquelme P, Sawitzki B, Tomiuk S, Miqueu P, Zuhayra M, et al. Cutting edge: immunological consequences and trafficking of human regulatory macrophages administered to renal transplant recipients. J Immunol. (2011) 187:2072–8. doi: 10.4049/jimmunol.1100762
94. Hutchinson JA, Ahrens N, Riquelme P, Walter L, Gruber M, Böger CA, et al. Clinical management of patients receiving cell-based immunoregulatory therapy. Transfusion. (2014) 54:2336–43. doi: 10.1111/trf.12641
95. Sakaguchi S. Regulatory T cells: history and perspective. Methods Mol Biol. (2011) 707:3–17. doi: 10.1007/978-1-61737-979-6_1
96. Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. (2007) 178:2018–27. doi: 10.4049/jimmunol.178.4.2018
97. Gao Y, Tang J, Chen W, Li Q, Nie J, Lin F, et al. Inflammation negatively regulates FOXP3 and regulatory T-cell function via DBC1. Proc Natl Acad Sci USA. (2015) 112:E3246–54. doi: 10.1073/pnas.1421463112
98. Su W, Fan H, Chen M, Wang J, Brand D, He X, et al. Induced CD4+ forkhead box protein-positive T cells inhibit mast cell function and established contact hypersensitivity through TGF-β1. J Allergy Clin Immunol. (2012) 130:444–52.e7. doi: 10.1016/j.jaci.2012.05.011
99. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. (2011) 117:1061–70. doi: 10.1182/blood-2010-07-293795
100. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. (2011) 117:3921–8. doi: 10.1182/blood-2010-10-311894
101. Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścinska J, et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin Immunol. (2014) 153:23–30. doi: 10.1016/j.clim.2014.03.016
102. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
103. Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next-generation regulatory T cell therapy. Nat Rev Drug Discov. (2019) 18:749–69. doi: 10.1038/s41573-019-0041-4
104. Bluestone JA, Tang Q. Treg cells-the next frontier of cell therapy. Science. (2018) 362:154–5. doi: 10.1126/science.aau2688
105. Dawson NAJ, Levings MK. Antigen-specific regulatory T cells: are police CARs the answer? Transl Res. (2017) 187:53–8. doi: 10.1016/j.trsl.2017.06.009
106. Boardman D, Maher J, Lechler R, Smyth L, Lombardi G. Antigen-specificity using chimeric antigen receptors: the future of regulatory T-cell therapy? Biochem Soc Trans. (2016) 44:342–8. doi: 10.1042/BST20150247
107. Dawson NAJ, Vent-Schmidt J, Levings MK. Engineered tolerance: tailoring development, function, and antigen-specificity of regulatory T Cells. Front Immunol. (2017) 8:1460. doi: 10.3389/fimmu.2017.01460
108. Elinav E, Waks T, Eshhar Z. Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology. (2008) 134:2014–24. doi: 10.1053/j.gastro.2008.02.060
109. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation. (2012) 9:112. doi: 10.1186/1742-2094-9-112
110. Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther. (2014) 22:1018–28. doi: 10.1038/mt.2014.41
111. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. (2016) 126:1413–24. doi: 10.1172/JCI82771
112. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. (2017) 17:931–43. doi: 10.1111/ajt.14185
113. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am J Transplant. (2017) 17:917–30. doi: 10.1111/ajt.14175
114. Yoon J, Schmidt A, Zhang AH, Königs C, Kim YC, Scott DW. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVII. Blood. (2017) 129:238–45. doi: 10.1182/blood-2016-07-727834
115. Qi Y, Ma J, Li S, Liu W. Applicability of adipose-derived mesenchymal stem cells in treatment of patients with type 2 diabetes. Stem cell research & therapy. (2019) 10:274. doi: 10.1186/s13287-019-1362-2
116. Ansboro S, Roelofs AJ, De Bari C. Mesenchymal stem cells for the management of rheumatoid arthritis: immune modulation, repair or both? Curr Opin Rheumatol. (2017) 29:201–7. doi: 10.1097/BOR.0000000000000370
117. Li Y, Wang Z, Zhao Y, Luo Y, Xu W, Marion TN, et al. Successful mesenchymal stem cell treatment of leg ulcers complicated by Behcet disease: A case report and literature review. Medicine. (2018) 97:e0515. doi: 10.1097/MD.0000000000010515
118. Casiraghi F, Perico N, Remuzzi G. Mesenchymal stromal cells for tolerance induction in organ transplantation. Hum Immunol. (2018) 79:304–13. doi: 10.1016/j.humimm.2017.12.008
119. Wang D, Zhang H, Liang J, Wang H, Hua B, Feng X, et al. A Long-Term follow-up study of allogeneic mesenchymal stem/stromal cell transplantation in patients with drug-resistant systemic lupus erythematosus. Stem Cell Rep. (2018) 10:933–41. doi: 10.1016/j.stemcr.2018.01.029
120. Zhao L, Chen S, Yang P, Cao H, Li L. The role of mesenchymal stem cells in hematopoietic stem cell transplantation: prevention and treatment of graft-versus-host disease. Stem Cell Res Ther. (2019) 10:182. doi: 10.1186/s13287-019-1287-9
121. Elgaz S, Kuçi Z, Kuçi S, Bönig H, Bader P. Clinical use of mesenchymal stromal cells in the treatment of acute graft-versus-host disease. Transfus Med Hemother. (2019) 46:27–34. doi: 10.1159/000496809
122. Moritani K, Miyawaki R, Tokuda K, Ochi F, Eguchi-Ishimae M, Tauchi H, et al. Mesenchymal stem cell therapy overcomes steroid resistance in severe gastrointestinal acute graft-versus-host disease. Case Rep Transplant. (2019) 2019:7890673. doi: 10.1155/2019/7890673
123. Wang Z, Yu H, Cao F, Liu Z, Liu Z, Feng W, et al. Donor-derived marrow mesenchymal stromal cell co-transplantation following a haploidentical hematopoietic stem cell transplantation trail to treat severe aplastic anemia in children. Ann Hematol. (2019) 98:473–9. doi: 10.1007/s00277-018-3523-2
124. Grégoire C, Lechanteur C, Briquet A, Baudoux É, Baron F, Louis E, et al. Review article: mesenchymal stromal cell therapy for inflammatory bowel diseases. Aliment Pharmacol Ther. (2017) 45:205–21. doi: 10.1111/apt.13864
125. Mastrolia I, Foppiani EM, Murgia A, Candini O, Samarelli AV, Grisendi G, et al. Challenges in clinical development of mesenchymal stromal/stem cells: concise review. Stem Cells Transl Med. (2019) 8:1135–48. doi: 10.1002/sctm.19-0044
126. Galipeau J, Sensebe L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell. (2018) 22:824–33. doi: 10.1016/j.stem.2018.05.004
127. Noriega DC, Ardura F, Hernández-Ramajo R, Martín-Ferrero MÁ, Sánchez-Lite I, Toribio B, et al. Intervertebral disc repair by allogeneic mesenchymal bone marrow cells: a randomized controlled trial. Transplantation. (2017) 101:1945–51. doi: 10.1097/TP.0000000000001484
128. Can A, Celikkan FT, Cinar O. Umbilical cord mesenchymal stromal cell transplantations: a systemic analysis of clinical trials. Cytotherapy. (2017) 19:1351–82. doi: 10.1016/j.jcyt.2017.08.004
129. Ferrarotti F, Romano F, Gamba MN, Quirico A, Giraudi M, Audagna M, et al. Human intrabony defect regeneration with micrografts containing dental pulp stem cells: a randomized controlled clinical trial. J Clin Periodontol. (2018) 45:841–50. doi: 10.1111/jcpe.12931
130. Lightner AL, Dozois EJ, Dietz AB, Fletcher JG, Friton J, Butler G, et al. Matrix-delivered autologous mesenchymal stem cell therapy for refractory rectovaginal crohn's fistulas. Inflamm Bowel Dis. (2019) 26:670–7. doi: 10.1093/ibd/izz215
131. Luo Y, Wu W, Gu J, Zhang X, Dang J, Wang J, et al. Human gingival tissue-derived MSC suppress osteoclastogenesis and bone erosion via CD39-adenosine signal pathway in autoimmune arthritis. EBioMedicine. (2019) 43:620–31. doi: 10.1016/j.ebiom.2019.04.058
132. Chen M, Su W, Lin X, Guo Z, Wang J, Zhang Q, et al. Adoptive transfer of human gingiva-derived mesenchymal stem cells ameliorates collagen-induced arthritis via suppression of Th1 and Th17 cells and enhancement of regulatory T cell differentiation. Arthritis Rheum. (2013) 65:1181–93. doi: 10.1002/art.37894
133. Zhang X, Huang W, Chen X, Lian Y, Wang J, Cai C, et al. CXCR5-Overexpressing mesenchymal stromal cells exhibit enhanced homing and can decrease contact hypersensitivity. Mol Ther. (2017) 25:1434–47. doi: 10.1016/j.ymthe.2017.04.004
134. Kim DS, Jang IK, Lee MW, Ko YJ, Lee DH, Lee JW, et al. Enhanced immunosuppressive properties of human mesenchymal stem cells primed by interferon-γ. EBioMedicine. (2018) 28:261–73. doi: 10.1016/j.ebiom.2018.01.002
135. Bai M, Zhang L, Fu B, Ba J, Zhang Y, Cai G, et al. IL-17A improves the efficacy of mesenchymal stem cells in ischemic-reperfusion renal injury by increasing Treg percentages by the COX-2/PGE2 pathway. Kidney internat. (2018) 93:814–25. doi: 10.1016/j.kint.2017.08.030
136. Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. (2008) 2:141–50. doi: 10.1016/j.stem.2007.11.014
137. Bai X, Xi J, Bi Y, Zhao X, Bing W, Meng X, et al. TNF-α promotes survival and migration of MSCs under oxidative stress via NF-κB pathway to attenuate intimal hyperplasia in vein grafts. J Cell Mol Med. (2017) 21:2077–91. doi: 10.1111/jcmm.13131
138. Mizukami A, Caliari-Oliveira C, Cominal JG, Rocha JL, Covas DT, Swiech K, et al. Priming approaches to improve the efficacy of mesenchymal stromal cell-based therapies. Stem Cell Res Ther. (2019) 10:131. doi: 10.1186/s13287-019-1259-0
139. Hoffmann P, Eder R, Boeld TJ, Doser K, Piseshka B, Andreesen R, et al. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood. (2006) 108:4260–7. doi: 10.1182/blood-2006-06-027409
140. Klingemann H, Boissel L, Toneguzzo F. Natural killer cells for immunotherapy - advantages of the NK-92 cell line over blood NK cells. Front Immunol. (2016) 7:91. doi: 10.3389/fimmu.2016.00091
141. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. (2017) 18:1332–41. doi: 10.1038/ni.3868
142. Kishimoto TK, Maldonado RA. Nanoparticles for the induction of antigen-specific immunological tolerance. Front Immunol. (2018) 9:230. doi: 10.3389/fimmu.2018.00230
143. Li PY, Fan Z, Cheng H. Cell membrane bioconjugation and membrane-derived nanomaterials for immunotherapy. Bioconjug Chem. (2018) 29:624–34. doi: 10.1021/acs.bioconjchem.7b00669
Keywords: autoimmune diseases, immune homeostasis, cell therapy, immune tolerance, immune reconstitution
Citation: Wang Z, Liu X, Cao F, Bellanti JA, Zhou J and Zheng SG (2020) Prospects of the Use of Cell Therapy to Induce Immune Tolerance. Front. Immunol. 11:792. doi: 10.3389/fimmu.2020.00792
Received: 25 December 2019; Accepted: 07 April 2020;
Published: 12 May 2020.
Edited by:
Irun R. Cohen, Weizmann Institute of Science, IsraelReviewed by:
Willem Van Eden, Utrecht University, NetherlandsHartmut Wekerle, Max-Planck-Gesellschaft (MPG), Germany
Copyright © 2020 Wang, Liu, Cao, Bellanti, Zhou and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin Zhou, emhvdWppbjExMTFAMTI2LmNvbQ==; Song Guo Zheng, c29uZ2d1b3poZW5nMjAxM0B5YWhvby5jb20=