 Sebastian Baasch
Sebastian Baasch Zsolt Ruzsics
Zsolt Ruzsics Philipp Henneke
Philipp Henneke- 1Institute for Immunodeficiency, Center for Chronic Immunodeficiency (CCI), Medical Center - University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 2Center for Pediatrics and Adolescent Medicine, University of Freiburg, Freiburg, Germany
- 3Institute of Virology, Medical Center - University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
Starting at birth, newborn infants are exposed to numerous microorganisms. Adaptation of the innate immune system to them is a delicate process, with potentially advantageous and harmful implications for health development. Cytomegaloviruses (CMVs) are highly adapted to their specific mammalian hosts, with which they share millions of years of co-evolution. Throughout the history of mankind, human CMV has infected most infants in the first months of life without overt implications for health. Thus, CMV infections are intertwined with normal immune development. Nonetheless, CMV has retained substantial pathogenicity following infection in utero or in situations of immunosuppression, leading to pathology in virtually any organ and particularly the central nervous system (CNS). CMVs enter the host through mucosal interfaces of the gastrointestinal and respiratory tract, where macrophages (MACs) are the most abundant immune cell type. Tissue MACs and their potential progenitors, monocytes, are established target cells of CMVs. Recently, several discoveries have revolutionized our understanding on the pre- and postnatal development and site-specific adaptation of tissue MACs. In this review, we explore experimental evidences and concepts on how CMV infections may impact on MAC development and activation as part of host-virus co-adaptation.
Introduction
The human body harbors diverse communities of microorganisms, in particular bacteria and fungi colonizing the outer and inner surfaces of the body (microbiome), as well as latently infecting viruses (virome) (1). This ecosystem is subject to influences, e.g. nutrient supply, interspecies competition and diffusible immunological effector molecules. At the same time, microbiome and virome shape host immunity via direct interaction with immune and non-immune cells, and indirectly, e.g. via secreted metabolites (2). In contrast to extracellular bacteria and fungi, which are largely controlled on the population level, viruses can be expected to rely on reciprocal adaptations with the individually infected host cell. The genus of cytomegaloviruses (CMVs), which belong to the subfamily of betaherpesvirinae, have co-evolved with their mammalian hosts for millions of years (3). In humans, infection with human CMV (HCMV) usually occurs in the first months of life, although infection has been pushed toward later life in highly industrialized societies (4–6). Therefore, HCMV is part of a “physiological” virome in immunocompetent individuals. Infants are infected via smear infections or via HCMV-containing milk as seropositive mothers reactivate HCMV locally (7) and transmit the virus to their children in more than 30% of cases (8).
CMVs have co-evolved with their specific hosts. Therefore, cross species infection models to study virus-host interactions are not available in vivo. Murine CMV (MCMV) and HCMV share only 45% of their genes (9), but have many similarities in cell tropism and immune modulatory properties. Hence, MCMV infection of mice is regarded as a useful experimental model to understand HCMV pathology (10). In the subsequent text we will use the abbreviation “CMV” in the case of general statements and if features are shared by the CMVs, which were studied.
Intraperitoneal and subcutaneous (foot pad) infections have provided valuable information on MCMV biology in the complex in vivo situation. However, since breast milk and saliva are regarded as important HCMV and MCMV sources, intragastric and intranasal infections have more recently been exploited (11). HCMV may infect cells of the mouth/upper gastro-intestinal tract, or it may reach the intestine. Moreover, HCMV may infect the respiratory tract via aspiration of virus containing milk. MACs and their potential progenitors, circulating monocytes, are well-known target cells for CMV (12–16). In the barrier tissues of intestinal and respiratory tracts, MACs represent the most abundant immune cells. However, tissue resident MACs are highly heterogeneous and undergo age specific changes during the individual host development, with respect to their origin and the tissue they inhere (17). For example, lamina propria MACs (LpMAC) in the intestine and microglia in the CNS represent two extremes with and without replenishment by monocytes, respectively. Models on how the phenotypic and functional MAC diversity impacts on CMV infections and vice versa are still in infancy.
In this review we focus on the ability of MACs to recognize CMV early after infection, and the known cellular consequences of infected MACs with regard of cytokine production and polarization. We summarize mechanisms of how CMV exploits monocyte influx and discuss potential consequences in putative target tissues. We propose that early CMV infections train the monocyte-macrophage-axis and are therefore beneficial in the immunocompetent host. Finally, we highlight the central role of monocytes and MACs in CMV infection serving as latent reservoirs and reactivation sites.
CMV recognition by macrophages and monocytes
The high frequency of tissue MACs in CMV entry sites, e.g. the lamina propria (intestinal tract) or alveolar spaces, allows for a potent response to epithelial barrier disruption and invasion of microorganisms, such as bacteria, or viruses. In order to cover a huge variety of pathogens with distinct extracellular or intracellular lifestyles, MACs and monocytes are equipped with pattern recognition receptors on plasma and endosomal membranes and in the cytosol. Together, these receptors recognize conserved microbial molecules or alterations in host structures, such as nucleic acids occurring at atypical sites. The engagement of pattern recognition receptors leads to the formation of cytokines, which are suited to initiate an appropriate immune response. During viral infections, type I interferons (IFN I) play an important role in creating a hostile cellular environment for viral replication and spread (18). Accordingly, mice deficient in the IFN I receptor (IFNAR−/−) succumb to CMV infection (19). Furthermore, inflammasome-dependent secretion of interleukin 18 (IL-18) augments NK-cell function in MCMV infections (20).
CMV and Toll-Like Receptors
Upon ligand binding Toll-like receptors (TLRs) transduce signals via the cytosolic adapter molecule myeloid differentiation primary response 88 (MyD88). In this respect, TLR3 is an exception, since it uses TIR-domain-containing adapter inducing interferon-β (TRIF) and TRIF-related adaptor molecule (TRAM) as sole adapters (21). To induce IFN I transcription, dimerization of transcription factors interferon regulatory factor (IRF) 3 (through TLR3-TRIF) and/or IRF7 (through TLR9-MyD88) is essential. Accordingly, peritoneal MAC from IRF3 and IRF7 double knockout mice do not produce IFN-β when infected with MCMV (22). The role of upstream MyD88 in IFN I production in MCMV infection was confirmed in several studies (23–25). A loss-of-function frameshift mutation in TRIF increases susceptibility and diminishes circulating IFN I in MCMV infection (26). Moreover, bone marrow cells from mice with a combined deficiency in MyD88 and TRIF, showed an impaired IFN I formation in MCMV infection in vitro. However, residual IFN I formation in these cells suggests the existence of a TLR-independent pathway (27).
The strictly intracellular lifestyle of CMV requires expression of host cell receptors that provide docking sites for viral ligands and facilitate cellular entry. Complexes of the CMV glycopoteins B and H (gB, gH) mediate host cell entry (28). Although, the entry-mediating host receptors for these protein complexes are still controversial (29), TLR2, which is expressed on the cell membrane, is known to interact with gB/ gH (30). TLR2 binding drives HCMV-induced nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB)-dependent production of inflammatory cytokines in MACs (31) (Figure 1) and mediates the control of CMV in immunocompromised humans and mice (32, 33). Interestingly, TLR2-dependent IFN I production has been found to be specific for inflammatory monocytes (27) (Figure 1), while dendritic cells did not mount an IFN I response through TLR2 (27).
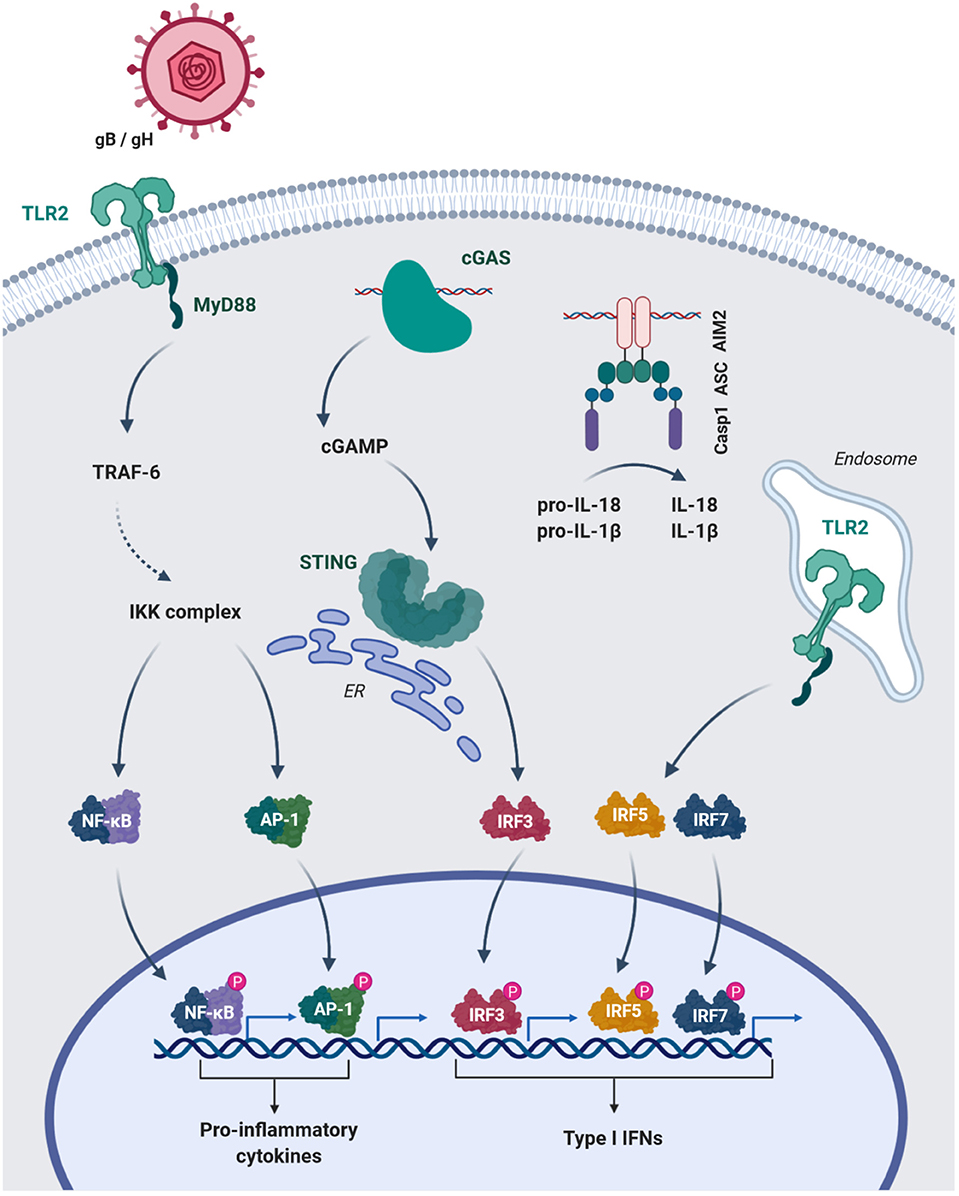
Figure 1. HCMV glycoproteins B and H (gB/gH) engange TLR2 of MAC on the surface and activate NFκB and AP-1 mediated transcription of pro-inflammatory cytokines. The cytosolic sensors cGAS and AIM2 recognize CMV-DNA. cGAS produces the signaling mediator cGAMP, which leads to STING activation and IRF3-dependent type I IFN transcription. Engagement of DNA by the HIN domain of AIM2 leads to interaction with the adaptor ASC (PYD-PYD) and subsequent recruitment of pro-caspase 1 via their CARD domains. Activated caspase 1 (Casp1) can cleave the pro-forms of IL-18 and IL-1β converting them into their mature bioactive forms. Additionally, inflammatory monocytes can uptake DNA viruses into endosomal compartments and induce type I IFN via IRF5 and 7. TRAF-6, TNF receptor associated factor-6; IKK-complex, IκB kinase-complex; AP-1, Activator protein-1; ER, endoplasmatic reticulum; ASC, Apoptosis-asspciated speck-like protein containing a CARD; PYD, pyrin domain.
In addition, endosomal TLRs, e.g. TLR 3, 7 and 9, are involved in MCMV recognition, since a missense mutation in their adapter UNC-93B leads to substantially decreased formation of interferon gamma (IFN-γ), IL-12, tumor necrosis factor (TNF) and IFN I in the plasma of mice after intraperitoneal MCMV infection (34).
Although the cell specific contribution of individual TLRs remains unclear, interaction of MCMV with TLR2 on monocytes and MACs may contribute to the rapid mounting of inflammatory and antiviral mediators in case of viremia. In contrast, TLR9 in dendritic cells seems to be crucial for IFN I in later stages of infection (24, 34–37).
CMV and Cytosolic Sensors
The observation that MACs, which accumulate DNA in phagosomes due to a DNAse II-deficiency, induce interferon-mediated anemia, which was reversed by deletion of IFNAR (38), suggested a TLR9-independent DNA sensor in the cytosol. Moreover, MACs deficient in MyD88, TRIF and mitochondrial antiviral signaling (MAVS) protein maintained a IFN I response in MCMV infection (39). The identification of the stimulator of interferon genes (STING) (40, 41) and cyclic guanosin monophosphate-adenosine monophosphate synthase (cGAS) (42), the upstream sensor for cytosolic DNA, provided mechanisms for the recognition of self and microbial DNA, e.g. from herpesviruses (43, 44). Tegtmeyer et al. recently demonstrated the importance of cGAS-STING-signaling in MCMV infection (45). STING-mediated IFN I was induced as early as 4 h post infection (hpi) (45). Kupffer cells, the liver resident MACs, were a main source of STING-dependent IFN I and restrained viral dissemination to the lymphnodes (45). These results support data generated in vitro in immortalized bone marrow-derived MACs, which responded to MCMV infection with IFN-β in a cGAS-STING-dependent fashion (46). Accordingly, HCMV induces IFN I in human monocyte-derived MACs via cGAS and STING (47) (Figure 1).
The DNA-dependent activator of IFN-regulatory factors (DAI) has been identified as another cytosolic sensor of nucleic acids (48). DAI interacts with TANK binding kinase 1 (TBK1) and IRF3, suggesting IRF3-mediated transcription of IFN I (48). Moreover, DAI can activate NFκB via RHIM/RIP1 and 3 recruitment (49, 50). However, the importance of DAI-mediated activation of NFκB and IRF3 in the IFN I response to cytosolic DNA seems marginal (51). Moreover, DAI has been recently challenged as a specific DNA sensor, since it was shown to interact with genomic RNA of influenza A virus (52) and with newly synthesized MCMV RNA (53), ultimately leading to necroptosis of fibroblasts (52, 53). This is in line with previous studies, where infection with MCMV, lacking the viral inhibitor of RIP activation (vIRA/m45) (54–56), induced DAI-RIPK3-dependent necroptosis in MACs (57). These findings support a role of DAI-dependent cell death induction in infected cells, rather than in direct innate immune activation.
The interaction of CMV with intracellular sensors appears to induce cellular activation beyond IFN I. A prominent example is the engagement of the absent in melanoma 2 (AIM2) by MCMV DNA in MACs, which leads to the maturation of IL-1β and IL-18 via caspase 1 cleavage (22) (Figure 1). Inflammasome activation and IL-18 secretion is essential for NK cell activation, as the formation of IFN-γ by these cells is reduced in ASC−/− and AIM2−/− mice (22).
Overall, MACs recognize CMV particles and form IFN I to restrict viral spread within hours after infections (45, 58). In vitro data generated in MACs further suggest that they induce NK-cell recruitment and IFN-γ production via inflammasome activation during the early response (Figure 1). On the other hand, MCMV and HCMV have evolved numerous strategies to evade the IFN I response (59–62). As an example, the MCMV protein M35 antagonizes IFN I induction downstream of STING and TLRs and thus ensures MCMV replication in MACs (46) (Table 1).
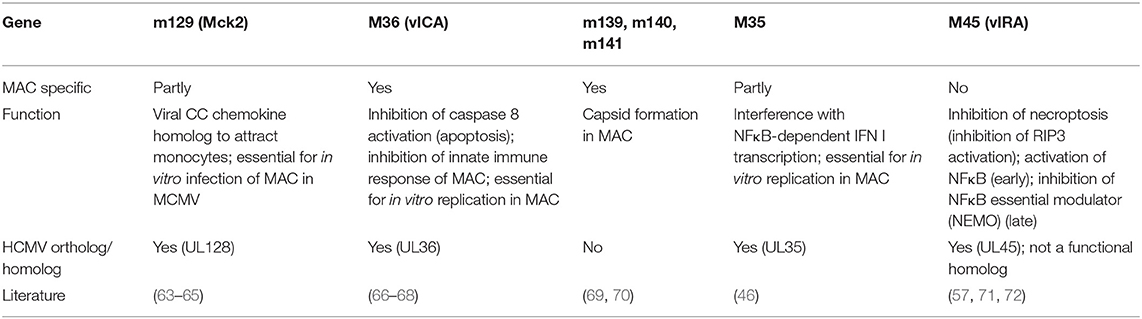
Table 1. Genes of MCMV and their HCMV homologs in this review.
Cytomegalovirus Affects Dynamics of Monocyte Recruitment to Infected Tissues
As outlined above, MACs can be a major source of IFN I in CMV infections. IFN I induce CCL2 and, to a lesser extent, CCL7 and 12 in the liver and the bone-marrow in MCMV infections (73, 74). A gradient of CCL2, which binds to its receptor CCR2, facilitates the egress of inflammatory monocytes from the bone marrow into the blood stream and the entry into infected tissues (75–79). Notably, all CMV species encode for viral chemokines located directly downstream to their major immediate early locus, corresponding to the UL128-UL131A region in HCMV. In MCMV, the viral chemokine is encoded by the m131 and m129 ORFs, which are fused by splicing. The resulting transcript encodes for a single protein called MCK-2 (80), which cooperates with CCL2 to attract monocytes (63) (Table 1). In HCMV, the chemokine homolog is encoded by the UL128 gene. The chemokine function of this gene product has not been well-studied yet. This may change in the near future by exploiting primate CMV models (81).
At the site of infection, Ly6Chi inflammatory monocytes can differentiate into monocyte-derived dendritic cells or MACs, which express the inducible nitric oxide synthase iNOS (82, 83). Nitric oxide formed by inflammatory monocytes inhibits CD8+ T-cells and thus modulates adaptive immunity in MCMV infection (84). Additionally, recruitment of inflammatory monocytes increases the number of NK-cells via CCL3 in the liver (85). NK-cells contain the T-cell response by killing of infected antigen presenting cells (86). This process contributes to MCMV persistence (86).
In a foot pad infection model with MCMV, a second subset of monocytes, Ly6Clo CX3CR1hi patrolling monocytes, are rapidly recruited (64). Intravital microscopy revealed that patrolling monocytes crawl along the inner lining of blood vessels (87) to remove particles inside the vessel lumen (88). Accordingly, patrolling monocytes may acquire MCMV from Tie2+ endothelial cells (89) and outmatch inflammatory monocytes as primary targets of MCMV. They harbor viral DNA and serve as vehicles to disseminate MCMV to the spleen and salivary gland (64). However, the roles of the chemokine receptor CX3CR1, which is highly expressed on patrolling monocytes, and MCK-2, which supports the recruitment of patrolling monocytes, are controversial in the context of viral spread (64, 90). In contrast to an earlier study (64), Farrell et al. did not find significant differences in MCMV salivary gland titers between CX3CR1-deficient and control mice after foot pad infection (90). Furthermore, MCK-2-deficient MCMV spreads in similar magnitude as wt-MCMV to the salivary gland 5 days after lung infection. A significant role of MCK-2 in MCMV dissemination was found only late, i.e. 10 days post infection (90). These findings highlight the value of kinetic experiments especially in elaborate in vivo experiments.
Collectively, MCMV infection leads to the recruitment of inflammatory and patrolling monocytes. While this appears to assist viral spread during initial infection, the monocyte influx changes the tissue-specific cell composition and might thus ultimately affect adaptive immunity and tissue integrity.
Monocyte Contribution to Tissue Macrophages in Steady State and Infections
It has been appreciated for a long time that circulating monocytes can be the direct progenitors of tissue MACs (91). However, in the last decade, substantial heterogeneity of MACs in different organs with respect to origin, renewal and immunophenotype has been uncovered (92–94). Resident MACs are seeded already in the embryo, either directly from the yolk-sac or via fetal liver-derived monocytes (17, 95). Postnatally, with increasing age and adaptation to the outer world, monocytes replenish MACs of the heart (96), the skin (97) and the intestine (98) even in steady state. This situation changes during inflammation or infection, when monocytes are recruited in great numbers also to other tissues (17, 99).
The depletion of MACs, e.g. via lytic infection of CMV, opens niches in the resident tissue MAC pool, which may be filled by invading monocytes (100). In mice, Ly6Chi monocytes give rise to MACs in the skin (97). Patrolling monocytes, on the other hand, fail to populate the intestine after depletion of CD11c+ MACs (101). Accordingly, inflammatory and not patrolling monocytes are considered to be the source of tissue MACs under described conditions. However, it seems highly context and tissue dependent, whether monocyte-derived MACs fully adapt and turn into long lived resident MACs or act as “transitory” cells (102). Monocytes cease to engraft into some tissues once inflammation has resolved (103–105). While they poorly perform tissue specific functions to prevent pulmonary alveolar proteinosis in the lung (106), they successfully replace and functionally restore resident cells in other organs, e.g. Kupffer cells in the liver (107). Therefore, origin and time of tissue invasion can impact on MAC function. Interestingly, monocyte-derived MACs may also show context and tissue-specific functional properties during inflammation (108, 109). As examples, monocytes ensure tissue regeneration after skeletal muscle injury (109), yet they show high inflammatory activity during DSS-induced colitis (108). This suggests that fine tuning of MAC function is largely influenced by local cues of the target tissue (110).
In summary, CMV infections and subsequent monocyte recruitment most likely have tissue specific consequences for the resident MAC population and therefore function, which can be either beneficial or deleterious. Thus, all organs, which are targeted in CMV infection deserve individual investigation.
Macrophages—a special target for CMV?
MACs are defined by morphology, phenotype and function, i.e. phagocytosis and cytokine secretion (111). Tissue MACs are terminally differentiated, however they retain plasticity to react on changing environmental cues, like those induced in infections (112). Next to their prominent role as a first line of defense, MACs in barrier tissues also bear central functions to maintain an anti-inflammatory state in homeostasis. Attempts to grasp the response capacity of MACs to different stimuli have led to a conceptionally useful, but oversimplifying view of pro-inflammatory (M1) and anti-inflammatory, or regulatory (M2) MAC polarization states (113).
Interestingly, polarization of MACs toward either a pro-inflammatory or regulatory state before exposure to HCMV and MCMV alters their susceptibility to infection in vitro (114, 115). With respect to putative MCMV virulence factors, MCK-2 deficiency limits CMV infectivity of MACs in vitro (65) and in vivo (116). MCK-2 appears to be incorporated in infectious particles via binding to virion glycoproteins gH and gL. Since gH/gL is curtail for cell entry, MCK-2 binding to gH/gL has a potential to modulate viral tropism (65) (Table 1). In contrast to the relatively clear in vitro phenotype, MCK-2 deficiency does not show a strong tropism phenotype in vivo (117). In HCMV, the MCK-2 homolog UL128 participates in the formation of the alternative gH/gL envelop glycoprotein complex, which also appears to influence viral host cell tropism (118).
HCMV can induce inflammatory transcriptional programs in MACs as soon as 4 hpi, with upregulation of genes of the ontology “Anti-viral response” (119). Within 24 hpi inflammatory cytokines are secreted (114) involving NFκB, phosphoinositide 3-kinase (PI3K) (120) and IRF signal transduction. The interplay of NFκB and PI3K seems necessary for early transcription of inflammatory cytokines, e.g. TNFα, but also anti-inflammatory cytokines, e.g. IL-10 (120). Accordingly, MCMV induces IL-10 production in peritoneal MACs leading to downregulation of MHCII (121). HCMV and rhesus CMV encode for a viral IL-10 homolog, which reduces migration of dendritic cells to the lymph node, as well as T-cell activation (122, 123). On the other hand, IL-10 producing CD4+ T-cells are induced via IFN I signaling in MACs during MCMV infection (124). IL-10 dampens inflammatory cytokines, such as IFN-γ and IL-6, attenuates tissue damage after MCMV infection (125, 126) and promotes persistence of infection in the salivary gland (127). Finally, TGFβ is secreted by infected human fibroblasts and in rat splenocytes after infection with the respective CMV species (128, 129). Thus, there is strong evidence that CMV induces immunoregulatory cytokines in MACs in addition to viral IL-10, e.g. in HCMV.
CMV inhibits apoptosis and necroptosis in MACs via its proteins pM36 (66) and pM45 (57), respectively (Table 1). The M36 gene is conserved among all CMVs and encodes for a cytosolic protein, binds to and blocks the activation of caspase-8 (66). Mutants lacking the M36 gene fail to inhibit apoptosis, show poor viral growth in MAC cell cultures, and loose in vivo fitness (67, 130, 131). vIRA/m45 and the cell death regulator vICA/M36 (66, 132) are essential for CMV replication in MACs. After intraperitoneal infection F4/80+ MACs seeded MCMV into the blood and brown adipose tissue, while CD11c+ myeloid cells, which can be expected to comprise dendritic cells, MACs and monocytes, were necessary for dissemination to the salivary gland after lung infection (133, 134). These findings indicate a migratory character of otherwise resident MACs. Interestingly, MCMV mutants lacking M36 (ΔM36) cannot disseminate after peripheral infection (67), and they grow normally in most of the cell types in vitro except for MACs. The growth impairment of MCMV mutants lacking M36 in mice with a defect adaptive immunity was rescued by the depletion of MACs. Accordingly, activated MACs were sufficient to impair ΔM36 growth in normally permissive MEFs in vitro. This could be reverted by caspase inhibition. TNFα from activated MAC synergized with IFN-γ in MEFs to inhibit ΔM36 growth. Hence, the altered ΔM36 growth in MAC and probably the altered virulence of this mutant does not reflect a defect in tropism, but rather a defect in the suppression of innate immune mediators secreted by infected and/or bystander MACs (67). The vICA in HCMV is encoded by the UL36 gene. The protein pUL36 also binds to pro-caspase-8, inhibits apoptosis and allows for viral replication in THP-1 cells (135). In vivo studies on the function of UL36 are limited due to the strict host specificity of CMVs. However, the cloning of UL36 into ΔM36 MCMV completely rescues the viral function both in vitro and in vivo (68). This indicates functional conservation of vICA in MCMV and HCMV.
Similar to M36, the complex of the products of MCMV genes m139, m140, and m141 is dispensable for the viral growth in fibroblasts, but it is essential for lytic MAC infection (66, 69) (Table 1). The products of these genes aid efficient capsid formation, which is apparent only in MACs (70). The underlying mechanism is not clear. It was proposed that the complex of pm139/pm140/pm141 influences cell type specific regulation of transport processes, which are important for assembly of infectious particles (69).
Together, cytokines formed by CMV-infected MACs, such as IFN I, TNFα and IFN-γ, help to contain viral infection (45, 58, 136), while viral and host IL-10 ensure replication and persistence of CMV. Simultaneously, IL-10 maintains a tolerogenic environment, which prevents tissue damage and may benefit the host during CMV infection. Regulatory or “unprimed” MACs can be considered to be more susceptible to CMV infections. This and the notion that neonatal innate immune cells produce lower amounts of IFN I (137), may explain, why newborn infants shed HCMV in higher concentrations than adults in primary HCMV infection (138). Furthermore, CMVs encode viral gene products (Table 1) to specifically target MACs and modulate their functions. This is decisive for viral dissemination and confers a central role to MACs in CMV infections.
CMV infection of the respiratory tract, the intestine and the CNS
In the lung, a potential CMV entry site, two major MAC types can be discriminated: Alveolar MACs (aMACs), which reside in the alveolar space to safeguard the lung from inhaled particles or pathogens (139), and interstitial MACs (iMACs) (Table 2). Fetal liver-monocytes colonize the lung to differentiate and mature into aMACs in the first week of life (140). Under steady state conditions, the aMAC population does not receive a monocyte influx (140, 157). IMACs are a heterogeneous population of at least two sub-populations (145–147). One subset bears significant self-renewal properties, whereas the other is constantly replenished by patrolling monocytes (146), which is unique for tissue resident MACs. Under steady state conditions, iMACs constantly produce IL-10, which alters DC function and maintains regulatory T-cells (104, 108, 146, 148–150, 158–160). At the same time, aMAC secrete TGFβ to induce differentiation of naïve T-cells into FoxP3+ regulatory T-cells (141, 161, 162). Thus, both aMACs and iMACs contribute to an anti-inflammatory tolerogenic environment in homeostasis (141, 142, 148, 149, 161, 162). AMACs are primarily targeted after intranasal infection with MCMV and their depletion leads to higher viral burden (163). After infection Ly6Chi monocytes infiltrate the lung in high numbers and are infected too (163). However, data to further define the consequences of monocyte recruitment, i.e. the fate of infected and uninfected monocytes, in intranasal MCMV infections are missing. Interestingly, inflammatory monocytes are able to enter the tissue and traffic to the draining lymph nodes (164), a sequence also described after intranasal MCMV infection (133).
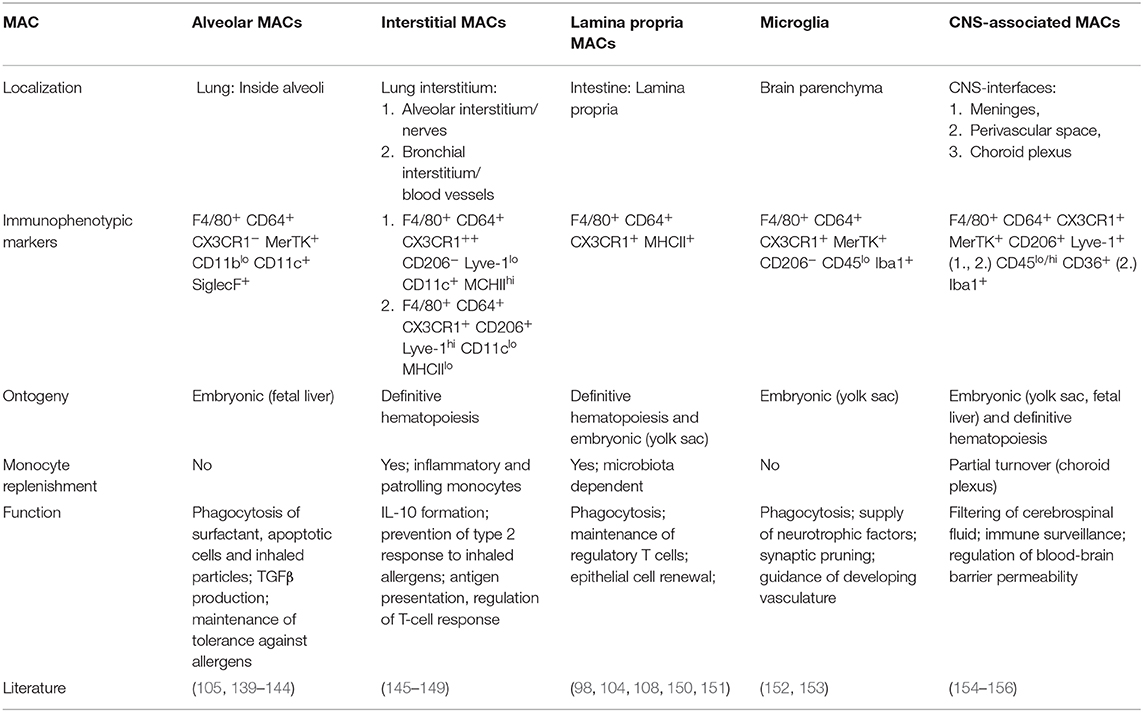
Table 2. MAC heterogeneity in CMV target tissues (steady state).
In MCMV latency, the infection may be reactivated in immunosuppressive conditions also in the lung (165). Thus, monocytes (64, 166) could carry CMV to the lung, where they can reactivate the virus upon differentiation into iMACs. Accordingly, CMV may exploit the physiological recruitment of patrolling monocytes (167–169), which serve as vehicles in MCMV infections (64).
Intestinal LpMACs represent the largest MAC subset in the mouse (170) (Table 2). They form a dense network close to the basal site of epithelial cells. Moreover, LpMACs directly reach into the intestinal lumen with their protrusions (171). The majority of LpMACs are constantly replenished by circulating inflammatory monocytes (Ly6Chi) after week 3 of life, i.e. starting with weaning (98). Accordingly, CCR2-deficient mice, which are impaired in monocyte egress from the bone-marrow, are deficient in LpMACs (98, 108). This process is dependent on the microbiota (98).
Once monocytes enter the lamina propria they differentiate and mature into LpMACs, which are characterized by a site specific response program to TLR-stimuli (172). Moreover, maturation of intestinal tolerogenic LpMACs and subsequent tolerance of the gut, similar to the lung, depends on IL-10 and TGFβ signaling (104, 108, 150, 158, 173). Inflammation interferes with this maturation process and leads to the formation of inflammatory effector cells (174), which control neutrophil activation and limit commensal-mediated tissue damage (175). In neonatal mice, an enteral challenge with MCMV-containing milk leads to viral dissemination (176). Yet, adult mice seem largely resistant to this infection mode (11). In HCMV associated intestinal inflammation, CD14+ monocytes, the putative human analog of mouse Ly6Chi monocytes (177), upregulate the TGFβ antagonist Smad7, which leads to the acquisition of inflammatory properties of intestinal MACs (178). It is tempting to speculate that early postnatal infections with CMV promote the monocyte influx into the intestine and thus protect against the invasion of commensals and opportunistic pathogens (179).
CMV-infections in fetuses cause severe symptoms and can lead to permanent damage of the CNS with immediate consequences and late sequelae, such as hearing loss. Microglia, the resident MACs of the brain parenchyma, are yolk sac-derived and maintain their population size exclusively via self-renewal (180) (Table 2). Monocyte-derived MACs only populate the brain after blood-brain-barrier disruption (181). In contrast to microglia, CNS-associated MACs include resident tissue MACs of barriers and interfaces of the CNS parenchyma and the periphery, such as perivascular space, meninges and the choroid plexus (154) (Table 2). They are also predominantly embryonically seeded and mainly self-renew (155, 182). Congenital transmission of CMV is best modeled by early postnatal intraperitoneal infection of mice (PND0-2), because a cell-free or cell-associated viremia is preceding focal encephalitis (183, 184) similar to the situation in humans (185). Thus, regions around blood vessels are infected first, including the choroid plexus of the periventricular region (186) and the meninges (187). In these regions MACs identified via F4/80+ (186) and Iba1+ (ionized calcium-binding adaptor molecule 1) (187) are infected or activated, respectively. In human brain aggregate culture systems, microglia or monocyte-derived MACs also appear to be initially infected (188). From the periventricular region, the meningoencephalitis caused by MCMV spreads to the hippocampus and cortex (186). In mice, MCMV-encephalitis leads to monocyte recruitment, diapedesis and subsequent differentiation into monocyte-derived MACs (189), which were also found to be infected. Thereby, monocyte-derived MACs might represent a potential way of viral dissemination from CNS interfaces into the brain parenchyma (186, 190). Accordingly, infection of MACs in the CNS may lead to heavy reorganization of otherwise tightly regulated immune cell populations, which may contribute to different pathologies (191–193).
Since resident MACs ensure the structural and functional integrity of their respective tissue (194), an exchange with monocyte-derived MACs after postnatal CMV infection bears opportunities, but also risks. On the one hand, tissue resident MACs are terminally differentiated and less plastic compared to monocytes (106). Postnatal infection could induce an early turnover of MACs in tissues like the intestine (98) and more distal, the skin (97). Thereby monocyte-derived MAC may foster maturation of the immune system and change the Th2-biased immune state (138) to a more inflammatory state, which may promote resistance to future infections. On the other hand, in organs, where MACs are largely maintained through self-renewal, invasion of monocytes could lead to exaggerated inflammation and subsequently tissue damage.
Do CMV infections protect against consecutive challenges or allergy?—Immunomodulation of the innate immune system
Individual immune memory is conventionally attributed to the adaptive immune system. However, it has been known for decades that innate immune cells can be primed by infection for long-lasting alterations in the response to subsequent challenges. These changes in activation programs were variably coined immune priming, immune tolerance, and most recently trained immunity (195–197). As an example for the latter, Rag1−/− mice without T- and B-cells, but not CCR2−/− mice, which are deficient in circulating monocytes, survived a lethal dose of Candida albicans, when they had been subjected to a low dose fungal infection one week before (198). Human monocyte and MAC training have been found to involve altered cytokine formation and epigenetic changes (198, 199).
In case of herpesvirus infection models, latent murid herpesvirus 4 (MuHV4) infection leads to the replacement of aMACs with regulatory monocyte-derived MACs, which generate tolerance to house dust mite extracts (200). Furthermore, peritoneal MACs of latently (>28 days) MCMV or mouse gamma-herpesvirus 68 infected mice showed an activated phenotype with increased MHC II expression and a higher killing capacity when re-infected with Listeria monocytogenes ex vivo. Latently infected mice were protected against infections with a lethal dose of L. monocytogenes. This mode of host resistance was dependent on IFN-γ, but differed from classical IFN-γ-induced protection with respect to both, duration and quality (201).
The fetal and neonatal immune systems have been suggested to be polarized toward protection against extracellular pathogens, which may render them especially vulnerable to viral infections, e.g. by HCMV (138). In contrast, postnatal HCMV infections often pass without overt symptoms and lead to a latent infection with sporadic viral reactivation. It constitutes an attractive model that reactivation occurs, when cues (e.g. interferons) from initial postnatal CMV infection wear off allowing for viral replication (202, 203). In other words, immune priming and recruitment of regulatory myeloid cells may quite rapidly fade. Subsequently, CMV reactivation and containment may induce a further wave of protection/tolerance by innate immune cells without provoking overt disease in immunocompetent individuals. Therefore, infection with CMV may keep the immune system in an alert state, which allows for a rapid response against potentially harmful agents. At the same time, recruitment of regulatory monocytes may maintain tissue integrity and tolerance at mucocutaneous surfaces.
Myelopoiesis, latency, and reactivation in CMV infections
The development along the monocyte-macrophage-axis may be involved in lifelong persistence of CMV (Figure 2). Human CD34+ hematopoietic stem and progenitor cells (HSPC) and CD14+ monocytes can be latently infected without ongoing replication and virus release (166, 205–209). The proportion of mononuclear cells carrying HCMV genome in latently infected individuals is rather low (1:104-105) (210). Yet, CD14+ CD74lo MHCIIlo monocytes contain more virus genomes as compared to CD14+ CD74+ and MHCII+ cells (211) (Figure 2). Furthermore, new techniques have allowed for the enrichment and characterization of latency-associated transcripts (212). Expression of US28 and UL138 in HCMV is important to establish latency in HSPC (213–215). Notably, in vitro infected HSPC and peripheral blood mononuclear cells (PBMC) from clinical samples showed similar HCMV transcriptome profiles (212), pointing to a potent antiviral program already in immature cells.
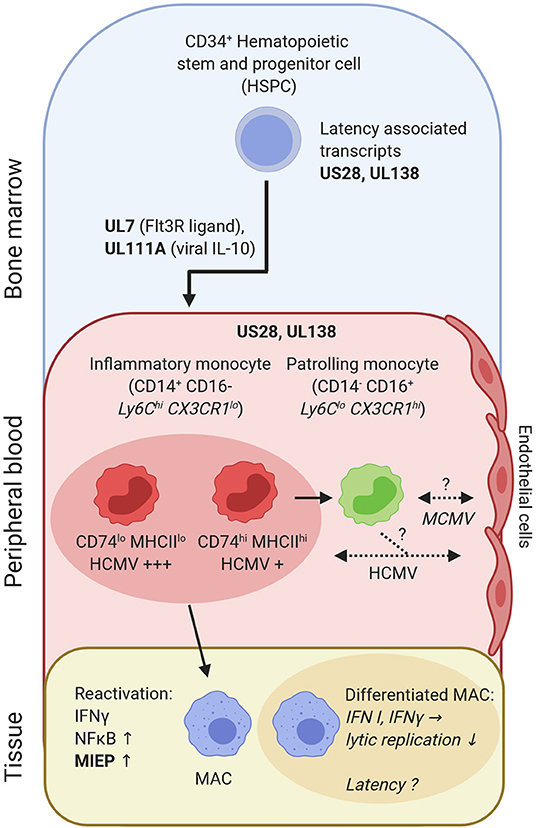
Figure 2. HCMV-derived transcripts (US28 and UL138) are needed to establish latency in CD34+ HSPC and CD14+ monocytes. HCMV gene products (pUL7, pUL111A) promote preferential differentiation of HSPC into monocytes. CD14+ inflammatory monocytes can be subdivided into CD74hi and CD74lo cells, which differ in latent HCMV content and reactivation. Inflammatory monocytes can differentiate into patrolling monocytes. Patrolling monocytes may transfer CMV to uninfected endothelial cells or acquire infection from previously infected endothelial cells [as shown for human CD14+ monocytes in vitro (204)]. Upon tissue entry and differentiation into monocyte-derived MACs reactivation and lytic replication occurs, once MIEP is activated. IFN-γ and NFκB are central in this process. In contrast, IFN I and IFN-γ protect against active/lytic replication of MCMV in differentiated MACs, which may explain viral latent states observed in MACs. Bold letters, CMV transcripts/genes; italic, mouse data; non-italic, human data; consistent arrow, differentiation; dashed arrow, CMV transmission.
The HCMV protein pUL7 binds to the Fms-like tyrosine kinase 3 receptor (Flt3R) and further steers HSPC toward myeloid monocyte commitment (216). Moreover, pUL111A, a viral IL-10 analog, impairs HSPC differentiation into dendritic cells (217) (Figure 2). This is in line with recent single cell sequencing data, where HCMV-infected HSPC predominantly differentiated into monocytes (211). Thus, during the course of infection, viral IL-10 ensures a supply of monocytes, which may aid in HCMV dissemination.
In the absence of acute inflammation, inflammatory monocytes recirculate to the bone marrow (101), where they differentiate into patrolling monocytes (218, 219). Interestingly, inflammatory monocytes are short lived (half-life: 20h in mice; ~1d in humans), yet they control the lifespan of patrolling monocytes (half-life: ~2.2d in mice; ~7.5d in humans) via M-CSF consumption in mice (219, 220). Thus, it seems conceivable that inflammatory monocytes can be infected, harbor CMV and continue to differentiate into patrolling monocytes. This may lead to complex changes in the composition of circulating monocytes. Moreover, infected monocytes could pass CMV on to endothelial cells (204), another cell type discussed for life-long latency in mice (221) and persistent infection in humans (222) (Figure 2). In a mouse model of latency, IFN-β prevents immediate early (IE) gene expression, which confers protection of lytic MCMV replication in endothelial cells. Reactivation of lytic infection occurred, once the effect of IFN-β wore off (203).
Upon activation and differentiation of monocytes into MACs, viral replication can restart (223, 224). Ex vivo infection and culturing of primary human CD34+ cells until differentiation into MACs, resembling the sequence of myelopoiesis and MAC determination, was associated with HCMV reactivation (225). Transcriptional activation of the major immediate early promoter (MIEP) and subsequent expression of IE1, IE2 (HCMV) or ie1, ie3 (MCMV) genes is a key switch to lytic infection. The enhancer of the MIE locus contains binding sites for NFκB (226). Hence, inflammation and cytokine production, e.g. TNF, may lead to reactivation, which is controlled in immunocompetent individuals. However, in case of an impaired inflammation control, reactivation causes complications as seen in the gut, lung or skin (227–230). Interestingly, in HCMV seropositive individuals IFN-γ producing T-cells appear to be more frequent as in seronegative individuals (231). IFN-γ represents a crucial factor for viral reactivation during the differentiation of human monocyte-derived MACs (224) (Figure 2). However, the presence of IFN-γ also leads to MAC activation and confers protection against lytic MCMV infection in already differentiated MACs (202) (Figure 2).
Early studies suggested that MACs are also a cellular reservoir for viral latency. After administering MCMV into the abdominal cavity, peritoneal MACs were found to bear MCMV DNA 3–9 months after infection. Furthermore, co-culturing with mouse embryonic fibroblasts resulted in reactivation of lytic viral replication, arguing for latently infected MACs (232). Another study used PCR in situ hybridization (PISH) to label viral DNA in tissue sections 6 months after peritoneal infection with MCMV. LAMP-2+ (CD107b+/Mac-3+) bona fide lung MACs were found to carry MCMV genome. However, the association of MCMV PISH- and LAMP-2-positivity were based on colocalisation in interalveolar tissue and not determined on the single cell level, which hampers the interpretation of these data (233).
In summary, CMV latency in myeloid cells may provide solutions for several puzzles in CMV disease progression and control, yet further data are required to robustly establish this scenario. In particular the discrimination between human inflammatory/classical and patrolling/non-classical monocytes could serve well to translate murine in vivo models into the human system.
Conclusions
In CMV infection, barrier tissue MACs are both targets and effector cells. The early formation of antiviral IFNs, which control several thousand of genes (234), is essential for regulating the immune response. The expression of numerous IFN inhibitory proteins by both HCMV and MCMV (235) is in full support of a model, where the armament of host and virus ultimately serves both sides. Subsequent signaling events, including the formation of IL-10, impact on restricting CMV-induced immunopathology and antiviral immunity, thus allowing for reestablishment of tissue immune homeostasis, as well as viral latency for years. When CMV infection occurs very early in life, as it has in most of human history, antiviral immunity and individual development of myeloid cells are intertwined. This is particular true for organs with high turnover of MACs, since monocytes as MAC progenitors integrate cues from CMV into the site specific differentiation program. Accordingly, in the case of an immunocompetent host, CMV and tissue MACs are primarily not foes. On the contrary, given the ancient CMV adaptation to mammalian hosts, it is a relationship with reciprocal benefits, e.g. the tuning of basal activation for a better response against more harmful microbial invaders, the renewal of tissue resident cells and modulation of autoimmunity (as it has been shown for gamma herpesviruses). At the same time, CMV has developed strategies to manipulate host immunity for lifelong persistence and inter-individual spread. Therefore, adverse consequences of CMV in the elderly, e.g. T-cell inflation (236) may be due to a CMV-human co-evolution tailored for a shorter host lifespan. Currently, direct evidence for such “mutual friendship” is just emerging. Yet, a scenario, where the benefits and harms of CMV infection are tissue and context specific, is highly attractive.
Author Contributions
SB, ZR, and PH wrote and edited the manuscript.
Funding
Funding was provided by the Else-Kröner-Fresenius Foundation, the German Ministry of Education and Research (grant Nos: 01EO0803, 01GL1746A and 01EK1602A to PH) and the German Research Council (grant Nos: HE3127/9-1, HE3127/12-1, and SFB/TRR167 to PH and 406922110 to ZR). The article processing charge was funded by the University of Freiburg in the funding programme Open Access Publishing.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wylie KM, Weinstock GM, Storch GA. Emerging view of the human virome. Transl Res. (2012) 160:283–90. doi: 10.1016/j.trsl.2012.03.006
2. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. (2012) 336:1268–73. doi: 10.1126/science.1223490
3. McGeoch DJ, Rixon FJ, Davison AJ. Topics in herpesvirus genomics and evolution. Virus Res. (2006) 117:90–104. doi: 10.1016/j.virusres.2006.01.002
4. Bello C, Whittle H. Cytomegalovirus infection in Gambian mothers and their babies. J Clin Pathol. (1991) 44:366–9. doi: 10.1136/jcp.44.5.366
5. Lanzieri TM, Dollard SC, Bialek SR, Grosse SD. Systematic review of the birth prevalence of congenital cytomegalovirus infection in developing countries. Int J Infect Dis. (2014) 22:44–8. doi: 10.1016/j.ijid.2013.12.010
6. Voigt S, Schaffrath Rosario A, Mankertz A. Cytomegalovirus Seroprevalence among children and adolescents in Germany: data from the German health interview and examination survey for children and adolescents (KiGGS), 2003-2006. Open forum Infect Dis. (2016) 3:ofv193. doi: 10.1093/ofid/ofv193
7. Stagno S, Cloud GA. Working parents: the impact of day care and breast-feeding on cytomegalovirus infections in offspring. Proc Natl Acad Sci USA. (1994) 91:2384–9. doi: 10.1073/pnas.91.7.2384
8. Hamprecht K, Maschmann J, Vochem M, Dietz K, Speer CP, Jahn G. Epidemiology of transmission of cytomegalovirus from mother to preterm infant by breastfeeding. Lancet. (2001) 357:513–8. doi: 10.1016/S0140-6736(00)04043-5
9. Rawlinson WD, Farrell HE, Barrell BG. Analysis of the complete DNA sequence of murine cytomegalovirus. J Virol. (1996) 70:8833–49. doi: 10.1128/JVI.70.12.8833-8849.1996
10. Reddehase MJ. Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat Rev Immunol. (2002) 2:831–44. doi: 10.1038/nri932
11. Oduro JD, Redeker A, Lemmermann NA, Ebermann L, Marandu TF, Dekhtiarenko I, et al. Murine cytomegalovirus (CMV) infection via the intranasal route offers a robust model of immunity upon mucosal CMV infection. J Gen Virol. (2016) 97:185–95. doi: 10.1099/jgv.0.000339
12. Smith PD, Saini SS, Raffeld M, Manischewitz JF, Wahl SM. Cytomegalovirus induction of tumor necrosis factor-alpha by human monocytes and mucosal macrophages. J Clin Invest. (1992) 90:1642–8. doi: 10.1172/JCI116035
13. Sinzger C, Plachter B, Grefte A, The TH, Jahn G. Tissue macrophages are infected by human cytomegalovirus in vivo. J Infect Dis. (1996) 173:240–5. doi: 10.1093/infdis/173.1.240
14. Mims CA, Gould J. The role of macrophages in mice infected with murine cytomegalovirus. J Gen Virol. (1978) 41:143–53. doi: 10.1099/0022-1317-41-1-143
15. Brautigam AR, Dutko FJ, Olding LB, Oldstone MB. Pathogenesis of murine cytomegalovirus infection: the macrophage as a permissive cell for cytomegalovirus infection, replication and latency. J Gen Virol. (1979) 44:349–59. doi: 10.1099/0022-1317-44-2-349
16. Ibanez CE, Schrier R, Ghazal P, Wiley C, Nelson JA. Human cytomegalovirus productively infects primary differentiated macrophages. J Virol. (1991) 65:6581–8. doi: 10.1128/JVI.65.12.6581-6588.1991
17. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. (2016) 44:439–49. doi: 10.1016/j.immuni.2016.02.024
18. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
19. Presti RM, Pollock JL, Dal Canto AJ, O'Guin AK, Virgin IV HW. Interferon γ regulates acute and latent murine cytomegalovirus infection and chronic disease of the great vessels. J Exp Med. (1998) 188:577–88. doi: 10.1084/jem.188.3.577
20. Madera S, Sun JC. Cutting edge: stage-specific requirement of IL-18 for antiviral NK cell expansion. J Immunol. (2015) 194:1408–12. doi: 10.4049/jimmunol.1402001
21. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
22. Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. (2010) 11:395–402. doi: 10.1038/ni.1864
23. Delale T, Paquin A, Asselin-Paturel C, Dalod M, Brizard G, Bates EE, et al. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J Immunol. (2005) 175:6723–32. doi: 10.4049/jimmunol.175.10.6723
24. Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, et al. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. (2004) 21:107–19. doi: 10.1016/j.immuni.2004.06.007
25. Hokeness-Antonelli KL, Crane MJ, Dragoi AM, Chu WM, Salazar-Mather TP. IFN-alphabeta-mediated inflammatory responses and antiviral defense in liver is TLR9-independent but MyD88-dependent during murine cytomegalovirus infection. J Immunol. (2007) 179:6176–83. doi: 10.4049/jimmunol.179.9.6176
26. Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. (2003) 424:743–8. doi: 10.1038/nature01889
27. Barbalat R, Lau L, Locksley RM, Barton GM. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol. (2009) 10:1200–7. doi: 10.1038/ni.1792
28. Nguyen CC, Kamil JP. Pathogen at the gates: human cytomegalovirus entry and cell tropism. Viruses. (2018) 10:704. doi: 10.3390/v10120704
29. Ryckman BJ, Chase MC, Johnson DC. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc Natl Acad Sci USA. (2008) 105:14118–23. doi: 10.1073/pnas.0804365105
30. Boehme KW, Guerrero M, Compton T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J Immunol. (2006) 177:7094–102. doi: 10.4049/jimmunol.177.10.7094
31. Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, et al. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol. (2003) 77:4588–96. doi: 10.1128/JVI.77.8.4588-4596.2003
32. Kijpittayarit S, Eid AJ, Brown RA, Paya CV, Razonable RR. Relationship between Toll-like receptor 2 polymorphism and cytomegalovirus disease after liver transplantation. Clin Infect Dis. (2007) 44:1315–20. doi: 10.1086/514339
33. Szomolanyi-Tsuda E, Liang X, Welsh RM, Kurt-Jones EA, Finberg RW. Role for TLR2 in NK cell-mediated control of murine cytomegalovirus in vivo. J Virol. (2006) 80:4286–91. doi: 10.1128/JVI.80.9.4286-4291.2006
34. Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. (2006) 7:156–64. doi: 10.1038/ni1297
35. Andoniou CE, van Dommelen SL, Voigt V, Andrews DM, Brizard G, Asselin-Paturel C, et al. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat Immunol. (2005) 6:1011–9. doi: 10.1038/ni1244
36. Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA. (2004) 101:3516–21. doi: 10.1073/pnas.0400525101
37. Puttur F, Francozo M, Solmaz G, Bueno C, Lindenberg M, Gohmert M, et al. Conventional dendritic cells confer protection against mouse cytomegalovirus infection via TLR9 and MyD88 signaling. Cell Rep. (2016) 17:1113–27. doi: 10.1016/j.celrep.2016.09.055
38. Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol. (2005) 6:49–56. doi: 10.1038/ni1146
39. Doring M, Lessin I, Frenz T, Spanier J, Kessler A, Tegtmeyer P, et al. M27 expressed by cytomegalovirus counteracts effective type I interferon induction of myeloid cells but not of plasmacytoid dendritic cells. J Virol. (2014) 88:13638–50. doi: 10.1128/JVI.00216-14
40. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. (2008) 455:674–8. doi: 10.1038/nature07317
41. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. (2013) 339:786–91. doi: 10.1126/science.1232458
42. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, et al. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature. (2013) 498:380–4. doi: 10.1038/nature12306
43. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. (2009) 461:788–92. doi: 10.1038/nature08476
44. Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. (2014) 505:691–5. doi: 10.1038/nature12862
45. Tegtmeyer PK, Spanier J. STING induces early IFN-beta in the liver and constrains myeloid cell-mediated dissemination of murine cytomegalovirus. Nat Commun. (2019) 10:2830. doi: 10.1038/s41467-019-10863-0
46. Chan B, Goncalves Magalhaes V. The murine cytomegalovirus M35 protein antagonizes type I IFN induction downstream of pattern recognition receptors by targeting NF-kappaB mediated transcription. PLoS pathogens. (2017) 13:e1006382. doi: 10.1371/journal.ppat.1006382
47. Paijo J, Doring M, Spanier J, Grabski E, Nooruzzaman M, Schmidt T, et al. cGAS senses human cytomegalovirus and induces type I interferon responses in human monocyte-derived cells. PLoS Pathog. (2016) 12:e1005546. doi: 10.1371/journal.ppat.1005546
48. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. (2007) 448:501–5. doi: 10.1038/nature06013
49. Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. (2009) 10:916–22. doi: 10.1038/embor.2009.109
50. Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol. (2008) 181:6427–34. doi: 10.4049/jimmunol.181.9.6427
51. Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. (2008) 451:725–9. doi: 10.1038/nature06537
52. Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, et al. DAI senses influenza a virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe. (2016) 20:674–81. doi: 10.1016/j.chom.2016.09.014
53. Maelfait J, Liverpool L. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. (2017) 36:2529–43. doi: 10.15252/embj.201796476
54. Brune W, Menard C, Heesemann J, Koszinowski UH. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science. (2001) 291:303–5. doi: 10.1126/science.291.5502.303
55. Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. (2010) 7:302–13. doi: 10.1016/j.chom.2010.03.006
56. Koehler HS, Feng Y, Mandal P, Mocarski ES. Recognizing limits of Z-nucleic acid binding protein (ZBP1/DAI/DLM1) function. FEBS J. (2020). doi: 10.1111/febs.15242. [Epub ahead of print].
57. Daley-Bauer LP, Roback L, Crosby LN, McCormick AL, Feng Y, Kaiser WJ, et al. Mouse cytomegalovirus M36 and M45 death suppressors cooperate to prevent inflammation resulting from antiviral programmed cell death pathways. Proc Natl Acad Sci USA. (2017) 114:E2786–95. doi: 10.1073/pnas.1616829114
58. Farrell HE, Bruce K, Lawler C, Cardin RD, Davis-Poynter NJ. Type 1 interferons and NK cells limit murine cytomegalovirus escape from the lymph node subcapsular sinus. PLoS Pathog. (2016) 12:e1006069. doi: 10.1371/journal.ppat.1006069
59. Zimmermann A, Trilling M, Wagner M, Wilborn M, Bubic I, Jonjic S, et al. A cytomegaloviral protein reveals a dual role for STAT2 in IFN-γ signaling and antiviral responses. J Exp Med. (2005) 201:1543–53. doi: 10.1084/jem.20041401
60. Browne EP, Shenk T. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc Natl Acad Sci USA. (2003) 100:11439–44. doi: 10.1073/pnas.1534570100
61. Taylor RT, Bresnahan WA. Human cytomegalovirus immediate-early 2 gene expression blocks virus-induced beta interferon production. J Virol. (2005) 79:3873–7. doi: 10.1128/JVI.79.6.3873-3877.2005
62. Stempel M, Chan B, Juranic Lisnic V, Krmpotic A, Hartung J, Paludan SR, et al. The herpesviral antagonist m152 reveals differential activation of STING-dependent IRF and NF-kappaB signaling and STING's dual role during MCMV infection. EMBO J. (2019) 38:e100983. doi: 10.15252/embj.2018100983
63. Saederup N, Lin YC, Dairaghi DJ, Schall TJ, Mocarski ES. Cytomegalovirus-encoded beta chemokine promotes monocyte-associated viremia in the host. Proc Natl Acad Sci USA. (1999) 96:10881–6. doi: 10.1073/pnas.96.19.10881
64. Daley-Bauer LP, Roback LJ, Wynn GM, Mocarski ES. Cytomegalovirus hijacks CX3CR1(hi) patrolling monocytes as immune-privileged vehicles for dissemination in mice. Cell Host Microbe. (2014) 15:351–62. doi: 10.1016/j.chom.2014.02.002
65. Wagner FM, Brizic I, Prager A, Trsan T, Arapovic M, Lemmermann NA, et al. The viral chemokine MCK-2 of murine cytomegalovirus promotes infection as part of a gH/gL/MCK-2 complex. PLoS Pathog. (2013) 9:e1003493. doi: 10.1371/journal.ppat.1003493
66. Menard C, Wagner M, Ruzsics Z, Holak K, Brune W, Campbell AE, et al. Role of murine cytomegalovirus US22 gene family members in replication in macrophages. J Virol. (2003) 77:5557–70. doi: 10.1128/JVI.77.10.5557-5570.2003
67. Ebermann L, Ruzsics Z, Guzman CA, van Rooijen N, Casalegno-Garduno R, Koszinowski U, et al. Block of death-receptor apoptosis protects mouse cytomegalovirus from macrophages and is a determinant of virulence in immunodeficient hosts. PLoS Pathog. (2012) 8:e1003062. doi: 10.1371/journal.ppat.1003062
68. Chaudhry MZ, Kasmapour B, Plaza-Sirvent C, Bajagic M, Casalegno Garduño R, Borkner L, et al. UL36 rescues apoptosis inhibition and in vivo replication of a Chimeric MCMV Lacking the M36 Gene. Front Cell Infect Microbiol. (2017) 7:312. doi: 10.3389/fcimb.2017.00312
69. Hanson LK, Slater JS, Karabekian Z, Ciocco-Schmitt G, Campbell AE. Products of US22 genes M140 and M141 confer efficient replication of murine cytomegalovirus in macrophages and spleen. J Virol. (2001) 75:6292–302. doi: 10.1128/JVI.75.14.6292-6302.2001
70. Hanson LK, Slater JS, Cavanaugh VJ, Newcomb WW, Bolin LL, Nelson CN, et al. Murine cytomegalovirus capsid assembly is dependent on US22 family gene M140 in infected macrophages. J Virol. (2009) 83:7449–56. doi: 10.1128/JVI.00325-09
71. Krause E, de Graaf M, Fliss PM, Dölken L, Brune W. Murine cytomegalovirus virion-associated protein M45 mediates rapid NF-κB activation after infection. J Virol. (2014) 88:9963–75. doi: 10.1128/JVI.00684-14
72. Fliss PM, Jowers TP, Brinkmann MM, Holstermann B, Mack C, Dickinson P, et al. Viral mediated redirection of NEMO/IKKγ to autophagosomes curtails the inflammatory cascade. PLoS Pathog. (2012) 8:e1002517. doi: 10.1371/journal.ppat.1002517
73. Hokeness KL, Kuziel WA, Biron CA, Salazar-Mather TP. Monocyte chemoattractant protein-1 and CCR2 interactions are required for IFN-alpha/beta-induced inflammatory responses and antiviral defense in liver. J Immunol. (2005) 174:1549–56. doi: 10.4049/jimmunol.174.3.1549
74. Crane MJ, Hokeness-Antonelli KL, Salazar-Mather TP. Regulation of inflammatory monocyte/macrophage recruitment from the bone marrow during murine cytomegalovirus infection: role for type I interferons in localized induction of CCR2 ligands. J Immunol. (2009) 183:2810–7. doi: 10.4049/jimmunol.0900205
75. Huffnagle GB, Strieter RM, Standiford TJ, McDonald RA, Burdick MD, Kunkel SL, et al. The role of monocyte chemotactic protein-1 (MCP-1) in the recruitment of monocytes and CD4+ T cells during a pulmonary Cryptococcus neoformans infection. J Immunol. (1995) 155:4790–7.
76. Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, et al. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. (1998) 187:601–8. doi: 10.1084/jem.187.4.601
77. Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV Jr, Broxmeyer HE, et al. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest. (1997) 100:2552–61. doi: 10.1172/JCI119798
78. Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, et al. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci USA. (1997) 94:12053–8. doi: 10.1073/pnas.94.22.12053
79. Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. (2006) 7:311–7. doi: 10.1038/ni1309
80. MacDonald MR, Burney MW, Resnick SB, Virgin HW. Spliced mRNA encoding the murine cytomegalovirus chemokine homolog predicts a β chemokine of novel structure. J Virol. (1999) 73:3682–91. doi: 10.1128/JVI.73.5.3682-3691.1999
81. Assaf BT, Mansfield KG, Strelow L, Westmoreland SV, Barry PA, Kaur A. Limited dissemination and shedding of the UL128 complex-intact, UL/b'-defective rhesus cytomegalovirus strain 180.92. J Virol. (2014) 88:9310–20. doi: 10.1128/JVI.00162-14
82. Menezes S, Melandri D, Anselmi G, Perchet T, Loschko J, Dubrot J, et al. The heterogeneity of Ly6Chi monocytes controls their differentiation into iNOS+ macrophages or monocyte-derived dendritic cells. Immunity. (2016) 45:1205–18. doi: 10.1016/j.immuni.2016.12.001
83. Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Ann Rev Immunol. (2009) 27:669–92. doi: 10.1146/annurev.immunol.021908.132557
84. Daley-Bauer LP, Wynn GM, Mocarski ES. Cytomegalovirus impairs antiviral CD8+ T cell immunity by recruiting inflammatory monocytes. Immunity. (2012) 37:122–33. doi: 10.1016/j.immuni.2012.04.014
85. Salazar-Mather TP, Orange JS, Biron CA. Early murine cytomegalovirus (MCMV) infection induces liver natural killer (NK) cell inflammation and protection through macrophage inflammatory protein 1alpha (MIP-1alpha)-dependent pathways. J Exp Med. (1998) 187:1–14. doi: 10.1084/jem.187.1.1
86. Andrews DM, Estcourt MJ, Andoniou CE, Wikstrom ME, Khong A, Voigt V, et al. Innate immunity defines the capacity of antiviral T cells to limit persistent infection. J Exp Med. (2010) 207:1333–43. doi: 10.1084/jem.20091193
87. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. (2007) 317:666–70. doi: 10.1126/science.1142883
88. Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell. (2013) 153:362–75. doi: 10.1016/j.cell.2013.03.010
89. Sacher T, Podlech J, Mohr CA, Jordan S, Ruzsics Z, Reddehase MJ, et al. The major virus-producing cell type during murine cytomegalovirus infection, the hepatocyte, is not the source of virus dissemination in the host. Cell Host Microbe. (2008) 3:263–72. doi: 10.1016/j.chom.2008.02.014
90. Farrell HE, Bruce K, Redwood AJ, Stevenson PG. Murine cytomegalovirus disseminates independently of CX3CR1, CCL2 or its m131/m129 chemokine homologue. J Gen Virol. (2019) 100:1695–700. doi: 10.1099/jgv.0.001333
91. van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med. (1968) 128:415–35. doi: 10.1084/jem.128.3.415
92. Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science. (2013) 342:1242974. doi: 10.1126/science.1242974
93. Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. (2016) 17:34–40. doi: 10.1038/ni.3324
94. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. (2014) 14:392–404. doi: 10.1038/nri3671
95. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518:547–51. doi: 10.1038/nature13989
96. Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, et al. Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med. (2014) 211:2151–8. doi: 10.1084/jem.20140639
97. Kolter J, Feuerstein R, Zeis P, Hagemeyer N, Paterson N, d'Errico P, et al. A subset of skin macrophages contributes to the surveillance and regeneration of local nerves. Immunity. (2019) 50:1482–97.e7. doi: 10.1016/j.immuni.2019.05.009
98. Bain CC, Bravo-Blas A, Scott CL, Gomez Perdiguero E. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. (2014) 15:929–37. doi: 10.1038/ni.2967
99. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. (2011) 11:762–4. doi: 10.1038/nri3070
100. Guilliams M, Scott CL. Does niche competition determine the origin of tissue-resident macrophages? Nat Rev Immunol. (2017) 17:451–60. doi: 10.1038/nri.2017.42
101. Varol C, Landsman L, Fogg DK, Greenshtein L, Gildor B, Margalit R, et al. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J Exp Med. (2007) 204:171–80. doi: 10.1084/jem.20061011
102. Perdiguero EG, Geissmann F. The development and maintenance of resident macrophages. Nat Immunol. (2016) 17:2–8. doi: 10.1038/ni.3341
103. Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. (2011) 14:1142–9. doi: 10.1038/nn.2887
104. Zigmond E, Bernshtein B, Friedlander G, Walker CR, Yona S, Kim KW, et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity. (2014) 40:720–33. doi: 10.1016/j.immuni.2014.03.012
105. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
106. van de Laar L, Saelens W, De Prijck S, Martens L, Scott CL, Van Isterdael G, et al. Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity. (2016) 44:755–68. doi: 10.1016/j.immuni.2016.02.017
107. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun. (2016) 7:10321. doi: 10.1038/ncomms10321
108. Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. (2013) 6:498–510. doi: 10.1038/mi.2012.89
109. Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med. (2007) 204:1057–69. doi: 10.1084/jem.20070075
110. Okabe Y, Medzhitov R. Tissue biology perspective on macrophages. Nat Immunol. (2016) 17:9–17. doi: 10.1038/ni.3320
111. Gunther P, Schultze JL. Mind the map: technology shapes the myeloid cell space. Front Immunol. (2019) 10:2287. doi: 10.3389/fimmu.2019.02287
112. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. (2011) 11:750–61. doi: 10.1038/nri3088
113. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
114. Bayer C, Varani S, Wang L, Walther P, Zhou S, Straschewski S, et al. Human cytomegalovirus infection of M1 and M2 macrophages triggers inflammation and autologous T-cell proliferation. J Virol. (2013) 87:67–79. doi: 10.1128/JVI.01585-12
115. Presti RM, Popkin DL, Connick M, Paetzold S, Virgin IV HW. Novel cell type–specific antiviral mechanism of interferon γ action in macrophages. J Exp Med. (2001) 193:483–96. doi: 10.1084/jem.193.4.483
116. Noda S, Aguirre SA, Bitmansour A, Brown JM, Sparer TE, Huang J, et al. Cytomegalovirus MCK-2 controls mobilization and recruitment of myeloid progenitor cells to facilitate dissemination. Blood. (2006) 107:30–8. doi: 10.1182/blood-2005-05-1833
117. Lemmermann NA, Krmpotic A, Podlech J, Brizic I, Prager A, Adler H, et al. Non-redundant and redundant roles of cytomegalovirus gH/gL complexes in host organ entry and intra-tissue spread. PLoS Pathog. (2015) 11:e1004640. doi: 10.1371/journal.ppat.1004640
118. Ryckman BJ, Rainish BL, Chase MC, Borton JA, Nelson JA, Jarvis MA, et al. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J Virol. (2008) 82:60–70. doi: 10.1128/JVI.01910-07
119. Chan G, Bivins-Smith ER, Smith MS, Smith PM, Yurochko AD. Transcriptome analysis reveals human cytomegalovirus reprograms monocyte differentiation towards a M1 macrophage. J Immunol. (2008) 181:698–711. doi: 10.4049/jimmunol.181.1.698
120. Chan G, Bivins-Smith ER, Smith MS, Yurochko AD. NF-κB and phosphatidylinositol 3-kinase activity mediates the HCMV-induced Atypical M1/M2 polarization of monocytes. Virus Res. (2009) 144:329–33. doi: 10.1016/j.virusres.2009.04.026
121. Redpath S, Angulo A, Gascoigne NR, Ghazal P. Murine cytomegalovirus infection down-regulates MHC class II expression on macrophages by induction of IL-10. J Immunol. (1999) 162:6701–7.
122. Avdic S, Cao JZ, McSharry BP, Clancy LE, Brown R, Steain M, et al. Human cytomegalovirus interleukin-10 polarizes monocytes toward a deactivated M2c phenotype to repress host immune responses. J Virol. (2013) 87:10273–82. doi: 10.1128/JVI.00912-13
123. Chang WW, Barry PA. Attenuation of innate immunity by cytomegalovirus IL-10 establishes a long-term deficit of adaptive antiviral immunity. Proc Natl Acad Sci USA. (2010) 107:22647–52. doi: 10.1073/pnas.1013794108
124. Clement M, Marsden M, Stacey MA, Abdul-Karim J, Brias SG, Bento DC, et al. Cytomegalovirus-specific IL-10-producing CD4+ T cells are governed by type-I IFN-induced IL-27 and promote virus persistence. PLoS Pathog. (2016) 12:e1006050. doi: 10.1371/journal.ppat.1006050
125. Oakley OR, Garvy BA, Humphreys S, Qureshi MH, Pomeroy C. Increased weight loss with reduced viral replication in interleukin-10 knock-out mice infected with murine cytomegalovirus. Clin Exp Immunol. (2008) 151:155–64. doi: 10.1111/j.1365-2249.2007.03533.x
126. Tang-Feldman YJ, Lochhead GR, Lochhead SR, Yu C, Pomeroy C. Interleukin-10 repletion suppresses pro-inflammatory cytokines and decreases liver pathology without altering viral replication in murine cytomegalovirus (MCMV)-infected IL-10 knockout mice. Inflammation Res. (2011) 60:233–43. doi: 10.1007/s00011-010-0259-4
127. Humphreys IR, De Trez C, Kinkade A, Benedict CA, Croft M, Ware CF. Cytomegalovirus exploits IL-10–mediated immune regulation in the salivary glands. J Exp Med. (2007) 204:1217–25. doi: 10.1084/jem.20062424
128. Michelson S, Alcami J, Kim S, Danielpour D, Bachelerie F, Picard L, et al. Human cytomegalovirus infection induces transcription and secretion of transforming growth factor beta 1. J Virol. (1994) 68:5730–7. doi: 10.1128/JVI.68.9.5730-5737.1994
129. Haagmans B, Teerds K, van den Eijnden-van Raaij A, Horzinek M, Schijns V. Transforming growth factor beta production during rat cytomegalovirus infection. J Gen Virol. (1997) 78:205–13. doi: 10.1099/0022-1317-78-1-205
130. Cičin-Šain L, Podlech J, Messerle M, Reddehase MJ, Koszinowski UH. Frequent coinfection of cells explains functional in vivo complementation between cytomegalovirus variants in the multiply infected host. J Virol. (2005) 79:9492–502. doi: 10.1128/JVI.79.15.9492-9502.2005
131. Cicin-Sain L, Ruzsics Z, Podlech J, Bubic I, Menard C, Jonjic S, et al. Dominant-negative FADD rescues the in vivo fitness of a cytomegalovirus lacking an antiapoptotic viral gene. J Virol. (2008) 82:2056–64. doi: 10.1128/JVI.01803-07
132. Skaletskaya A, Bartle LM, Chittenden T, McCormick AL, Mocarski ES, Goldmacher VS. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc Natl Acad Sci USA. (2001) 98:7829–34. doi: 10.1073/pnas.141108798
133. Farrell HE, Bruce K, Lawler C, Stevenson PG. Murine cytomegalovirus spread depends on the infected myeloid cell type. J Virol. (2019) 93:e00540–19. doi: 10.1128/JVI.00540-19
134. Farrell HE, Bruce K, Lawler C, Oliveira M, Cardin R, Davis-Poynter N, et al. Murine cytomegalovirus spreads by dendritic cell recirculation. mBio. (2017) 8:e01264–17. doi: 10.1128/mBio.01264-17
135. McCormick AL, Roback L, Livingston-Rosanoff D, Clair CS. The human cytomegalovirus UL36 gene controls caspase-dependent and-independent cell death programs activated by infection of monocytes differentiating to macrophages. J Virol. (2010) 84:5108–23. doi: 10.1128/JVI.01345-09
136. Farrell HE, Davis-Poynter N, Bruce K, Lawler C, Dolken L, Mach M, et al. Lymph node macrophages restrict murine cytomegalovirus dissemination. J Virol. (2015) 89:7147–58. doi: 10.1128/JVI.00480-15
137. Kollmann TR, Crabtree J, Rein-Weston A, Blimkie D, Thommai F, Wang XY, et al. Neonatal innate TLR-mediated responses are distinct from those of adults. J Immunol. (2009) 183:7150–60. doi: 10.4049/jimmunol.0901481
138. Prendergast AJ, Klenerman P, Goulder PJ. The impact of differential antiviral immunity in children and adults. Nat Rev Immunol. (2012) 12:636–48. doi: 10.1038/nri3277
139. Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature. (2014) 506:503–6. doi: 10.1038/nature12902
140. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
141. Soroosh P, Doherty TA, Duan W, Mehta AK, Choi H, Adams YF, et al. Lung-resident tissue macrophages generate Foxp3+ regulatory T cells and promote airway tolerance. J Exp Med. (2013) 210:775–88. doi: 10.1084/jem.20121849
142. Holt PG, Oliver J, Bilyk N, McMenamin C, McMenamin PG, Kraal G, et al. Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. J Exp Med. (1993) 177:397–407. doi: 10.1084/jem.177.2.397
143. Watanabe Y, Hashimoto Y, Shiratsuchi A, Takizawa T, Nakanishi Y. Augmentation of fatality of influenza in mice by inhibition of phagocytosis. Biochem Biophys Res Commun. (2005) 337:881–6. doi: 10.1016/j.bbrc.2005.09.133
144. Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. (2013) 49:503–10. doi: 10.1165/rcmb.2013-0086MA
145. Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, et al. Three unique interstitial macrophages in the murine lung at steady state. Am J Respir Cell Mol Biol. (2017) 57:66–76. doi: 10.1165/rcmb.2016-0361OC
146. Schyns J, Bai Q. Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nat Commun. (2019) 10:3964. doi: 10.1038/s41467-019-11843-0
147. Chakarov S, Lim HY. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science. (2019) 363:eaau0964. doi: 10.1126/science.aau0964
148. Bedoret D, Wallemacq H, Marichal T, Desmet C, Quesada Calvo F, Henry E, et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J Clin Investig. (2009) 119:3723–38. doi: 10.1172/JCI39717
149. Kawano H, Kayama H, Nakama T, Hashimoto T, Umemoto E, Takeda K. IL-10-producing lung interstitial macrophages prevent neutrophilic asthma. Int Immunol. (2016) 28:489–501. doi: 10.1093/intimm/dxw012
150. Shouval DS, Biswas A, Goettel JA, McCann K, Conaway E, Redhu NS, et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. (2014) 40:706–19. doi: 10.1016/j.immuni.2014.03.011
151. Kayama H, Ueda Y, Sawa Y, Jeon SG, Ma JS, Okumura R, et al. Intestinal CX3C chemokine receptor 1high (CX3CR1high) myeloid cells prevent T-cell-dependent colitis. Proc Natl Acad Sci USA. (2012) 109:5010–5. doi: 10.1073/pnas.1114931109
152. Kolter J, Kierdorf K, Henneke P. Origin and differentiation of nerve-associated macrophages. J Immunol. (2020) 204:271–9. doi: 10.4049/jimmunol.1901077
153. Prinz M, Erny D, Hagemeyer N. Ontogeny and homeostasis of CNS myeloid cells. Nat Immunol. (2017) 18:385–92. doi: 10.1038/ni.3703
154. Kierdorf K, Masuda T. Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat Rev Neurosci. (2019) 20:547–62. doi: 10.1038/s41583-019-0201-x
155. Goldmann T, Wieghofer P, Jordão MJC, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. (2016) 17:797–805. doi: 10.1038/ni.3423
156. Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-dimensional single-cell mapping of Central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity. (2018) 48:380–95.e6. doi: 10.1016/j.immuni.2018.01.011
157. Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, et al. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog. (2014) 10:e1004053. doi: 10.1371/journal.ppat.1004053
158. Schridde A, Bain CC, Mayer JU, Montgomery J, Pollet E, Denecke B, et al. Tissue-specific differentiation of colonic macrophages requires TGFbeta receptor-mediated signaling. Mucosal Immunol. (2017) 10:1387–99. doi: 10.1038/mi.2016.142
159. Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity. (2011) 34:237–46. doi: 10.1016/j.immuni.2011.01.016
160. Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. (2009) 10:1178–84. doi: 10.1038/ni.1791
161. Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. (1999) 96:319–28. doi: 10.1016/S0092-8674(00)80545-0
162. Lloyd CM, Hawrylowicz CM. Regulatory T cells in asthma. Immunity. (2009) 31:438–49. doi: 10.1016/j.immuni.2009.08.007
163. Farrell HE, Lawler C, Oliveira MT, Davis-Poynter N, Stevenson PG. Alveolar macrophages are a prominent but nonessential target for murine cytomegalovirus infecting the lungs. J Virol. (2015) 90:2756–66. doi: 10.1128/JVI.02856-15
164. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. (2013) 39:599–610. doi: 10.1016/j.immuni.2013.08.007
165. Polić B, Hengel H, Krmpotić A, Trgovcich J, Pavić I, Lučin P, et al. Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J Exp Med. (1998) 188:1047–54. doi: 10.1084/jem.188.6.1047
166. Taylor-Wiedeman J, Sissons JG, Borysiewicz LK, Sinclair JH. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J Gen Virol. (1991) 72:2059–64. doi: 10.1099/0022-1317-72-9-2059
167. Smith MS, Bentz GL, Alexander JS, Yurochko AD. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J Virology. (2004) 78:4444–53. doi: 10.1128/JVI.78.9.4444-4453.2004
168. Chan G, Nogalski MT, Yurochko AD. Human cytomegalovirus stimulates monocyte-to-macrophage differentiation via the temporal regulation of caspase 3. J Virol. (2012) 86:10714–23. doi: 10.1128/JVI.07129-11
169. Noriega VM, Haye KK, Kraus TA, Kowalsky SR, Ge Y, Moran TM, et al. Human cytomegalovirus modulates monocyte-mediated innate immune responses during short-term experimental latency in vitro. J Virol. (2014) 88:9391–405. doi: 10.1128/JVI.00934-14
170. Lee SH, Starkey PM, Gordon S. Quantitative analysis of total macrophage content in adult mouse tissues. Immunochemical studies with monoclonal antibody F4/80. J Exp Med. (1985) 161:475–89. doi: 10.1084/jem.161.3.475
171. Cerovic V, Bain CC, Mowat AM, Milling SW. Intestinal macrophages and dendritic cells: what's the difference? Trends Immunol. (2014) 35:270–7. doi: 10.1016/j.it.2014.04.003
172. Hirotani T, Lee PY, Kuwata H, Yamamoto M, Matsumoto M, Kawase I, et al. The nuclear IkappaB protein IkappaBNS selectively inhibits lipopolysaccharide-induced IL-6 production in macrophages of the colonic lamina propria. J Immunol. (2005) 174:3650–7. doi: 10.4049/jimmunol.174.6.3650
173. Maheshwari A, Kelly DR, Nicola T, Ambalavanan N, Jain SK, Murphy-Ullrich J, et al. TGF-beta2 suppresses macrophage cytokine production and mucosal inflammatory responses in the developing intestine. Gastroenterology. (2011) 140:242–53. doi: 10.1053/j.gastro.2010.09.043
174. Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity. (2012) 37:1076–90. doi: 10.1016/j.immuni.2012.08.026
175. Grainger JR, Wohlfert EA, Fuss IJ, Bouladoux N, Askenase MH, Legrand F, et al. Inflammatory monocytes regulate pathologic responses to commensals during acute gastrointestinal infection. Nat Med. (2013) 19:713–21. doi: 10.1038/nm.3189
176. Wu CA, Paveglio SA, Lingenheld EG, Zhu L, Lefrancois L, Puddington L. Transmission of murine cytomegalovirus in breast milk: a model of natural infection in neonates. J Virol. (2011) 85:5115–24. doi: 10.1128/JVI.01934-10
177. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. (2003) 19:71–82. doi: 10.1016/S1074-7613(03)00174-2
178. Dennis EA, Smythies LE, Grabski R, Li M, Ballestas ME, Shimamura M, et al. Cytomegalovirus promotes intestinal macrophage-mediated mucosal inflammation through induction of Smad7. Mucosal Immunol. (2018) 11:1694–704. doi: 10.1038/s41385-018-0041-4
179. Kolter J, Henneke P. Codevelopment of microbiota and innate immunity and the risk for group B streptococcal disease. Front Immunol. (2017) 8:1497. doi: 10.3389/fimmu.2017.01497
180. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
181. Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch U-K, Mack M, et al. Microglia in the adult brain arise from Ly-6C hi CCR2+ monocytes only under defined host conditions. Nat Neurosci. (2007) 10:1544. doi: 10.1038/nn2015
182. Van Hove H, Martens L, Scheyltjens I, de Vlaminck K, Antunes ARP, de Prijck S, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci. (2019) 22:1021–35. doi: 10.1038/s41593-019-0393-4
183. Cekinovic D, Golemac M, Pugel EP, Tomac J, Cicin-Sain L, Slavuljica I, et al. Passive immunization reduces murine cytomegalovirus-induced brain pathology in newborn mice. J Virol. (2008) 82:12172–80. doi: 10.1128/JVI.01214-08
184. Koontz T, Bralic M, Tomac J, Pernjak-Pugel E, Bantug G, Jonjic S, et al. Altered development of the brain after focal herpesvirus infection of the central nervous system. J Exp Med. (2008) 205:423–35. doi: 10.1084/jem.20071489
185. Renard T, Daumas-Duport B, Auffray-Calvier E, Bourcier R, Desal H. Cytomegalovirus encephalitis: undescribed diffusion-weighted imaging characteristics. Original aspects of cases extracted from a retrospective study, and from literature review. J Neuroradiol. (2016) 43:371–7. doi: 10.1016/j.neurad.2016.03.004
186. Kosugi I, Kawasaki H, Arai Y, Tsutsui Y. Innate immune responses to cytomegalovirus infection in the developing mouse brain and their evasion by virus-infected neurons. Am J Pathol. (2002) 161:919–28. doi: 10.1016/S0002-9440(10)64252-6
187. Kosmac K, Bantug GR, Pugel EP, Cekinovic D, Jonjic S, Britt WJ. Glucocorticoid treatment of MCMV infected newborn mice attenuates CNS inflammation and limits deficits in cerebellar development. PLoS Pathog. (2013) 9:e1003200. doi: 10.1371/journal.ppat.1003200
188. Pulliam L. Cytomegalovirus preferentially infects a monocyte derived macrophage/microglial cell in human brain cultures: neuropathology differs between strains. J Neuropathol Exp Neurol. (1991) 50:432–40. doi: 10.1097/00005072-199107000-00004
189. Seleme MC, Kosmac K, Jonjic S, Britt WJ. TNFalpha induced recruitment of inflammatory mononuclear cells leads to inflammation and altered brain development in MCMV infected newborn mice. J Virol. (2017) 91:JVI.01983-16. doi: 10.1128/JVI.01983-16
190. Bentz GL, Jarquin-Pardo M, Chan G, Smith MS, Sinzger C, Yurochko AD. Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J Virol. (2006) 80:11539–55. doi: 10.1128/JVI.01016-06
191. Djukic M, Mildner A, Schmidt H, Czesnik D, Brück W, Priller J, et al. Circulating monocytes engraft in the brain, differentiate into microglia and contribute to the pathology following meningitis in mice. Brain. (2006) 129:2394–403. doi: 10.1093/brain/awl206
192. Gres V, Kolter J, Erny D, Henneke P. The role of CNS macrophages in streptococcal meningoencephalitis. J Leukoc Biol. (2019) 106:209–18. doi: 10.1002/JLB.4MR1118-419R
193. Jordao MJC, Sankowski R. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science. (2019) 363:eaat7554. doi: 10.1126/science.aat7554
194. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. (2013) 496:445–55. doi: 10.1038/nature12034
195. Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. (2007) 447:972–8. doi: 10.1038/nature05836
196. Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe. (2011) 9:355–61. doi: 10.1016/j.chom.2011.04.006
197. Netea MG, Domínguez-Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol. (2020). doi: 10.1038/s41577-020-0285-6. [Epub ahead of print].
198. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. (2012) 12:223–32. doi: 10.1016/j.chom.2012.06.006
199. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. (2014) 345:1251086. doi: 10.1126/science.1251086
200. Machiels B, Dourcy M, Xiao X, Javaux J, Mesnil C, Sabatel C, et al. A gammaherpesvirus provides protection against allergic asthma by inducing the replacement of resident alveolar macrophages with regulatory monocytes. Nat Immunol. (2017) 18:1310–20. doi: 10.1038/ni.3857
201. Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, et al. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. (2007) 447:326–9. doi: 10.1038/nature05762
202. Kropp KA, Robertson KA, Sing G, Rodriguez-Martin S, Blanc M, Lacaze P, et al. Reversible inhibition of murine cytomegalovirus replication by gamma interferon (IFN-γ) in primary macrophages involves a primed type I IFN-signaling subnetwork for full establishment of an immediate-early antiviral state. J Virol. (2011) 85:10286–99. doi: 10.1128/JVI.00373-11
203. Dag F, Dolken L, Holzki J, Drabig A, Weingartner A, Schwerk J, et al. Reversible silencing of cytomegalovirus genomes by type I interferon governs virus latency. PLoS Pathog. (2014) 10:e1003962. doi: 10.1371/journal.ppat.1003962
204. Waldman WJ, Knight DA, Huang EH, Sedmak DD. Bidirectional transmission of infectious cytomegalovirus between monocytes and vascular endothelial cells: an in vitro model. J Infect Dis. (1995) 171:263–72. doi: 10.1093/infdis/171.2.263
205. Mendelson M, Monard S, Sissons P, Sinclair J. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J Gen Virol. (1996) 77:3099–102. doi: 10.1099/0022-1317-77-12-3099
206. von Laer D, Meyer-Koenig U, Serr A, Finke J, Kanz L, Fauser AA, et al. Detection of cytomegalovirus DNA in CD34+ cells from blood and bone marrow. Blood. (1995). 86:4086–90. doi: 10.1182/blood.V86.11.4086
207. Taylor-Wiedeman J, Sissons P, Sinclair J. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J Virol. (1994) 68:1597–604. doi: 10.1128/JVI.68.3.1597-1604.1994
208. Sindre H, Tjoonnfjord G, Rollag H, Ranneberg-Nilsen T, Veiby OP, Beck S, et al. Human cytomegalovirus suppression of and latency in early hematopoietic progenitor cells. Blood. (1996) 88:4526–33. doi: 10.1182/blood.V88.12.4526.bloodjournal88124526
209. Kondo K, Kaneshima H, Mocarski ES. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc Natl Acad Sci USA. (1994) 91:11879–83. doi: 10.1073/pnas.91.25.11879
210. Slobedman B, Mocarski ES. Quantitative analysis of latent human cytomegalovirus. J Virol. (1999) 73:4806–12. doi: 10.1128/JVI.73.6.4806-4812.1999
211. Shnayder M, Nachshon A, Rozman B, Bernshtein B, Lavi M, Fein N, et al. Single cell analysis reveals human cytomegalovirus drives latently infected cells towards an anergic-like monocyte state. eLife. (2020) 9:e52168. doi: 10.7554/eLife.52168.sa2
212. Cheng S, Caviness K, Buehler J, Smithey M, Nikolich-Žugich J, Goodrum F. Transcriptome-wide characterization of human cytomegalovirus in natural infection and experimental latency. Proc Natl Acad Sci USA. (2017) 114:E10586–95. doi: 10.1073/pnas.1710522114
213. Humby MS, O'Connor CM. Human cytomegalovirus US28 is important for latent infection of hematopoietic progenitor cells. J Virol. (2016) 90:2959–70. doi: 10.1128/JVI.02507-15
214. Petrucelli A, Rak M, Grainger L, Goodrum F. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J Virol. (2009) 83:5615–29. doi: 10.1128/JVI.01989-08
215. Weekes MP, Tan SY, Poole E, Talbot S, Antrobus R, Smith DL, et al. Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science. (2013) 340:199–202. doi: 10.1126/science.1235047
216. Crawford LB, Kim JH, Collins-McMillen D, Lee B-J, Landais I, Held C, et al. Human cytomegalovirus encodes a novel FLT3 receptor ligand necessary for hematopoietic cell differentiation and viral reactivation. mBio. (2018) 9:e00682–18. doi: 10.1128/mBio.00682-18
217. Avdic S, Cao JZ, Cheung AK, Abendroth A, Slobedman B. Viral interleukin-10 expressed by human cytomegalovirus during the latent phase of infection modulates latently infected myeloid cell differentiation. J Virol. (2011) 85:7465–71. doi: 10.1128/JVI.00088-11
218. Mildner A, Schonheit J, Giladi A, David E, Lara-Astiaso D, Lorenzo-Vivas E, et al. Genomic characterization of murine monocytes reveals C/EBPbeta transcription factor dependence of Ly6C- cells. Immunity. (2017) 46:849–62.e7. doi: 10.1016/j.immuni.2017.04.018
219. Patel AA, Zhang Y. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med. (2017) 214:1913–23. doi: 10.1084/jem.20170355
220. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2013.05.008
221. Seckert CK, Renzaho A, Tervo HM, Krause C, Deegen P, Kuhnapfel B, et al. Liver sinusoidal endothelial cells are a site of murine cytomegalovirus latency and reactivation. J Virol. (2009) 83:8869–84. doi: 10.1128/JVI.00870-09
222. Fish KN, Soderberg-Naucler C, Mills LK, Stenglein S, Nelson JA. Human cytomegalovirus persistently infects aortic endothelial cells. J Virol. (1998) 72:5661–8. doi: 10.1128/JVI.72.7.5661-5668.1998
223. Soderberg-Naucler C, Streblow DN, Fish KN, Allan-Yorke J, Smith PP, Nelson JA. Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J Virol. (2001) 75:7543–54. doi: 10.1128/JVI.75.16.7543-7554.2001
224. Soderberg-Naucler C, Fish KN, Nelson JA. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell. (1997) 91:119–26. doi: 10.1016/S0092-8674(01)80014-3
225. Zhuravskaya T, Maciejewski JP, Netski DM, Bruening E, Mackintosh FR, St Jeor S. Spread of human cytomegalovirus (HCMV) after infection of human hematopoietic progenitor cells: model of HCMV latency. Blood. (1997) 90:2482–91. doi: 10.1182/blood.V90.6.2482
226. Dorsch-Hasler K, Keil GM, Weber F, Jasin M, Schaffner W, Koszinowski UH. A long and complex enhancer activates transcription of the gene coding for the highly abundant immediate early mRNA in murine cytomegalovirus. Proc Natl Acad Sci USA. (1985) 82:8325–9. doi: 10.1073/pnas.82.24.8325
227. Riddell SR. Pathogenesis of cytomegalovirus pneumonia in immunocompromised hosts. Semin Respir Infect. (1995) 10:199–208.
228. Goodgame RW. Gastrointestinal cytomegalovirus disease. Ann Int Med. (1993) 119:924–35. doi: 10.7326/0003-4819-119-9-199311010-00010
229. Lee JY. Cytomegalovirus infection involving the skin in immunocompromised hosts: a clinicopathologic study. Am J Clin Pathol. (1989) 92:96–100. doi: 10.1093/ajcp/92.1.96
230. Smyth RL, Scott JP, Borysiewicz LK, Sharples LD, Stewart S, Wreghitt TG, et al. Cytomegalovirus infection in heart-lung transplant recipients: risk factors, clinical associations, and response to treatment. J Infect Dis. (1991) 164:1045–50. doi: 10.1093/infdis/164.6.1045
231. Dunn HS, Haney DJ, Ghanekar SA, Stepick-Biek P, Lewis DB, Maecker HT. Dynamics of CD4 and CD8 T cell responses to cytomegalovirus in healthy human donors. J Infect Dis. (2002) 186:15–22. doi: 10.1086/341079
232. Pollock JL, Presti RM, Paetzold S, Virgin HWt. Latent murine cytomegalovirus infection in macrophages. Virology. (1997) 227:168–79. doi: 10.1006/viro.1996.8303
233. Koffron AJ, Hummel M, Patterson BK, Yan S, Kaufman DB, Fryer JP, et al. Cellular localization of latent murine cytomegalovirus. J Virol. (1998) 72:95–103. doi: 10.1128/JVI.72.1.95-103.1998
234. Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, et al. Interferome v2. 0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. (2012) 41:D1040–6. doi: 10.1093/nar/gks1215
235. Hengel H, Koszinowski UH, Conzelmann K-K. Viruses know it all: new insights into IFN networks. Trends Immunol. (2005) 26:396–401. doi: 10.1016/j.it.2005.05.004
Keywords: macrophage, monocyte, CMV (cytomegalovirus), innate immunity, pathogen-host coevolution, mucosal immune barrier, virus-host adaptation, macrophage heterogeneity
Citation: Baasch S, Ruzsics Z and Henneke P (2020) Cytomegaloviruses and Macrophages—Friends and Foes From Early on? Front. Immunol. 11:793. doi: 10.3389/fimmu.2020.00793
Received: 18 December 2019; Accepted: 07 April 2020;
Published: 12 May 2020.
Edited by:
Tobias Strunk, King Edward Memorial Hospital, AustraliaReviewed by:
Cecilia Söderberg-Naucler, Karolinska Institutet (KI), SwedenLuka Cicin-Sain, Helmholtz Association of German Research Centers (HZ), Germany
Copyright © 2020 Baasch, Ruzsics and Henneke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philipp Henneke, cGhpbGlwcC5oZW5uZWtlQHVuaWtsaW5pay1mcmVpYnVyZy5kZQ==