 Kevin R. Parducho1,2
Kevin R. Parducho1,2 Brent Beadell1
Brent Beadell1 Tiffany K. Ybarra1
Tiffany K. Ybarra1 Mabel Bush1
Mabel Bush1 Erick Escalera1Aldo T. Trejos1
Erick Escalera1Aldo T. Trejos1 Andy Chieng2Marlon Mendez1Chance Anderson1
Andy Chieng2Marlon Mendez1Chance Anderson1 Hyunsook Park1
Hyunsook Park1 Yixian Wang2
Yixian Wang2 Wuyuan Lu3
Wuyuan Lu3 Edith Porter1*
Edith Porter1*- 1Department of Biological Sciences, California State University, Los Angeles, Los Angeles, CA, United States
- 2Department of Chemistry and Biochemistry, California State University, Los Angeles, Los Angeles, CA, United States
- 3Institute of Human Virology and Department of Biochemistry and Molecular Biology, University of Maryland School of Medicine, Baltimore, MD, United States
Biofilm production is a key virulence factor that facilitates bacterial colonization on host surfaces and is regulated by complex pathways, including quorum sensing, that also control pigment production, among others. To limit colonization, epithelial cells, as part of the first line of defense, utilize a variety of antimicrobial peptides (AMPs) including defensins. Pore formation is the best investigated mechanism for the bactericidal activity of AMPs. Considering the induction of human beta-defensin 2 (HBD2) secretion to the epithelial surface in response to bacteria and the importance of biofilm in microbial infection, we hypothesized that HBD2 has biofilm inhibitory activity. We assessed the viability and biofilm formation of a pyorubin-producing Pseudomonas aeruginosa strain in the presence and absence of HBD2 in comparison to the highly bactericidal HBD3. At nanomolar concentrations, HBD2 – independent of its chiral state – significantly reduced biofilm formation but not metabolic activity, unlike HBD3, which reduced biofilm and metabolic activity to the same degree. A similar discrepancy between biofilm inhibition and maintenance of metabolic activity was also observed in HBD2 treated Acinetobacter baumannii, another Gram-negative bacterium. There was no evidence for HBD2 interference with the regulation of biofilm production. The expression of biofilm-related genes and the extracellular accumulation of pyorubin pigment, another quorum sensing controlled product, did not differ significantly between HBD2 treated and control bacteria, and in silico modeling did not support direct binding of HBD2 to quorum sensing molecules. However, alterations in the outer membrane protein profile accompanied by surface topology changes, documented by atomic force microscopy, was observed after HBD2 treatment. This suggests that HBD2 induces structural changes that interfere with the transport of biofilm precursors into the extracellular space. Taken together, these data support a novel mechanism of biofilm inhibition by nanomolar concentrations of HBD2 that is independent of biofilm regulatory pathways.
Introduction
Biofilms are composed of microbial communities encased in a protective layer of self-produced, extracellular polymers. Biofilms are formed on both abiotic and biotic surfaces and play a significant role in a variety of settings such as aquaculture (1), the food industry, and the clinical field as a factor for antimicrobial drug resistance. Biofilms can colonize body surfaces and mechanisms regarding how our bodies prevent biofilm formation are under extensive investigation (2). In part, biofilms provide tolerance to host immune factors and antibiotics through impeding their diffusion. Furthermore, biofilms enhance bacterial resistance to these factors by altering bacterial metabolism resulting from the decreased oxygen levels in the center of the biofilm mass as well as the acidification of the local microenvironment (2–5). The biofilm matrix is primarily composed of exopolysaccharide, proteins, and extracellular DNA and has been particularly well studied in Pseudomonas aeruginosa, a ubiquitous, opportunistic, Gram-negative bacterium. The major structural polysaccharides of P. aeruginosa biofilms are Pel, which is composed of positively charged amino sugars, and Psl, which is a polymer of glucose, rhamnose, and mannose; and in certain strains, alginate – an anionic polysaccharide (6–8). Proteinaceous components of biofilm include type 4 pili and cup fimbriae serving attachment and various proteins that connect matrix components adding strength to the biofilm (9). Extracellular DNA (eDNA), which is released via cell lysis (10), plays an important role in priming surfaces for the initial adhesion of the bacteria as well as in maintaining the structural integrity of the polysaccharide fibers (3, 6, 11–14).
Multiple regulatory networks govern the complex process of biofilm formation (15), which progresses from initial attachment mediated by the flagella and the production of pili, to downregulation of flagellar genes, upregulation of the production and secretion of matrix components, maturation, and eventual reappearance of flagella and dispersion. For P. aeruginosa, biofilm regulation has been well studied and several regulatory systems have been identified including the Las, Rhl, and quinolone quorum sensing systems, the GacA/GacS two-component system, and c-di-GMP controlled pathways. Key quorum sensing molecules for Las, Rhl, and quinolone systems are N-(3-oxododecanoyl)-homoserine lactone (3-oxo-C12-HSL), N-butanoyl-homoserine lactone (C4-HSL), and 2-heptyl-3-hydroxy-4-quinolone (known as Pseudomonas Quorum Sensing molecule or PQS), respectively (16, 17). These overlapping regulatory systems not only control the production of biofilm but also the production of pigment and various other virulence factors (17, 18). Genes whose expression is modulated during biofilm formation include flgF, which encodes for the basal rod in bacterial flagellin, and pslA, which is the first gene in the polysaccharide synthesis locus (19, 20).
In addition to being able to produce biofilm, P. aeruginosa possesses potent virulence factors such as: a type III secretion system, which allows it to directly deliver exotoxins to host cells (21); rhamnolipids, which enable P. aeruginosa to disrupt the tight junctions of respiratory epithelia (22); and pigments with diverse functions in metal-chelation, competitive inhibition of other bacteria, and resistance to oxidative stress (23–25). All of these virulence factors and resistance mechanisms contribute to P. aeruginosa being one of the leading isolates in healthcare-associated pneumonia in intensive care units and chronic lung infection in patients with cystic fibrosis, a genetic disorder characterized by impaired anion transport and increased mucous viscosity (26). Yet, despite its ubiquity in nature and its prevalence in healthcare-associated infections, P. aeruginosa is not known to cause lung infection in healthy adults, suggesting that humans possess effective innate defense mechanisms in the airways against this organism.
Antimicrobial peptides (AMPs) are small, highly conserved effector molecules that play a key role in innate immunity (27, 28). Present in plants, insects, and mammals, most AMPs are between 2 and 5 kDa in size and are cationic with varying degrees of hydrophobicity. Upon the detection of microbial components via pattern recognition receptors, AMPs can be synthesized by epithelial cells and myeloid cells as part of the first line of defense against microbes (29–33). A wealth of research has been performed on the ability of AMPs to displace cations bound to bacterial membranes, which are rich in either negatively charged lipopolysaccharides or lipoteichoic acids in addition to anionic phospholipids (34). After binding to bacterial membranes, AMPs can perturb the membrane structure and form pores mediated by hydrophobic and electrostatic forces. In addition to the charge of the membrane, phospholipid species and the presence or absence of cholesterol, which is absent in bacterial membranes, also affect the binding and orientation of AMPs and hence, their pore-forming capabilities (35–40). While pore-formation has been a widely studied mechanism of action, an increasing body of research suggests that the antimicrobial activity of AMPs may also depend on other mechanisms – disruption of cell wall synthesis, metabolic activity, ATP and nucleic acid synthesis, and amino acid uptake (33, 41). Furthermore, certain AMPs interact with the eukaryotic host cells and have immunoregulatory functions in addition to their antimicrobial activity. A notable example is that LL-37 can also: act as a chemotactic agent to recruit other immune cells and modulate cytokine and chemokine expression in host cells, bind bacterial lipopolysaccharide, and dysregulate the expression of genes involved in biofilm formation (42–46). Other AMPs have also shown multi-functional capabilities, in particular human beta-defensin 2 (HBD2) and 3 (HBD3), which have been proven to possess mechanisms of action that are more complex than simple pore formation and membrane perturbation (47–49). In fact, HBD2 was the first human beta defensin to demonstrate chemotactic activity (50). Beta-defensins are characterized by three, antiparallel β-strands stabilized by three conserved disulfide linkages preceded by an α-helical domain near the N-terminus (51–53). Although HBD2 and HBD3 share amino acid sequence and some structural similarities, their overall net charge, hydrophobicity, and charge distribution differ significantly (Table 1) and may play a role in their unique and distinct mechanisms of action. Expression of HBD2 and HBD3 is low or absent during steady state but both peptides are induced in airway epithelial tissues during infection or inflammation (31, 32, 48, 54).

Table 1. Human beta defensins-2 and -3 physicochemical properties.
Due to their lasting potency for millions of years and the feasibility of modifying AMP structures, AMPs continue to be in the spotlight as potential antimicrobial agents (33). The importance of biofilm in the infection process and in their resistance to antimicrobial agents has been recognized, yet there is a lack of drugs that interfere with biofilm. Therefore, knowledge on the structure-function relationships of AMPs, and the effects of AMPs on bacterial biofilm formation may benefit rational engineering and design of novel AMP variants and therapeutic regimens that are effective against microbial biofilms (55). Considering the induction of HBD2 and HBD3 and their secretion to the epithelial surface in response to bacteria and their products, we hypothesized that HBD2 and HBD3 have biofilm inhibitory activity. We discovered that biofilm and metabolic inhibition are proportionally reduced by HBD3 but not by HBD2. At low concentrations, HBD2 inhibits biofilm production, but not metabolic activity. We undertook multiple approaches to delineate the underlying mechanism for the selective biofilm inhibitory effects of HBD2. This research may lead to the identification of novel targets for the engineering of antimicrobials, which, in the era of increasing multi-drug resistance, is of great importance.
Materials and Methods
Antimicrobial Peptides
Chemical synthesis and purification of human beta-defensin 2 (HBD2/L-HBD2), its D- form (D-HBD2) comprised entirely of D-amino acids, its linearized mutant (Linear HBD2 with alanine replacing all cysteine residues), and human beta-defensin 3 (HBD3, in L-form) have been described previously (56, 57). Table 1 summarizes their physicochemical properties. Stock solutions (500 μM) were prepared in 0.01% acetic acid and stored at −20°C. For experiments, peptides were used as 10-fold concentration in 0.01% acetic acid.
Bacterial Culture
For this study, a pyorubin-producing P. aeruginosa strain (a cystic fibrosis isolate previously obtained from Dr. Michael J. Welsh, University of Iowa, Iowa City) and Acinetobacter baumannii ATCC 19606 were used. For each experiment, snap-frozen 18 h cultures in Tryptic Soy Broth (TSB) (Oxoid) were quickly thawed, subcultured into prewarmed TSB (750 μL into 50 mL), and brought to mid-log growth phase (3 h at 37°C, 200 rpm). Bacterial cells were then sedimented and washed with 140 mM NaCl by centrifugation for 10 min at 805 × g in a precooled centrifuge (4°C), and resuspended in 500 μL 140 mM NaCl. For gene expression analysis, the suspended bacteria were used directly. For all other assays, the concentration of bacteria was first adjusted to 5 × 107CFU/mL in 140 mM NaCl, and then further diluted as needed.
Biofilm Quantification
In a round bottom 96-well polystyrene microtiter plate (Costar #3795), 90 μL mid-logarithmic growth phase bacteria were added to 10 μL of 10-fold concentrated defensin or 0.01% acetic acid as solvent control to yield the following final assay conditions: 1 × 106 CFU/mL, 10% Mueller-Hinton broth (Oxoid, without cations), and 140 mM NaCl. Samples were incubated for 18 h at 37°C and biofilms were quantified according to Merritt et al. (58). Briefly, the content of sample wells containing non-adherent bacteria (planktonic and/or dead) was carefully discarded without disturbing the biofilm, and the well walls were rinsed three times with dH2O (200 μL/well) followed by addition of 125 μL of 0.1% crystal violet (Sigma-Aldrich, St. Louis, MO, United States). After 10 min incubation at RT, the crystal violet solution was removed, wells were rinsed three times with dH2O (200 μL/well) and air dried for at least 30 min. To solubilize crystal violet bound to biofilm, 200 μL of 30% acetic acid was added to each well and after 15 min incubation at RT 125 μL was transferred to optically clear flat-bottom 96-well polystyrene microtiter plates (Perkin Elmer from Waltham, MA, United States). Absorbance was read at 570 nm using a Victor X3 Plate Reader (Perkin Elmer). Wells containing only 125 μL of 30% acetic acid were used to subtract baseline absorbance values from samples for analysis.
Metabolic Activity Measurement
Resazurin reduction was employed as a measure of bacterial metabolic activity (59, 60). Metabolites accumulating during bacterial growth reduce the weakly fluorescent resazurin to the highly fluorescent resorufin. Samples were prepared as described above but with resazurin (Sigma) added to the assay buffer to obtain a final concentration of 0.01% resazurin (w/v). Relative fluorescent units (RFU) were measured every 3 h with a preheated Victor X3 Plate Reader (Perkin Elmer) at 530 nm excitation and 616 nm emission wavelength and a top read.
ATP Quantification
ATP concentrations of non-adherent bacteria were determined using the BacTiterGlo kit (Promega), with ATP standard curves prepared according to the manufacturer’s instructions. Bacteria were prepared and incubated with defensins for 18 h as described for the biofilm assay. Then, the entire well contents were transferred to a new 96 well plate, thoroughly resuspended, and of this 75 μL from each well was transferred to a black 96-well half area plate (Perkin-Elmer). After addition of 75 μL ATP substrate solution to each well and 5 min mixing on an orbital shaker, luminescence was quantified with a Victor X3 plate reader. Seventy-five μL aliquots of serially diluted ATP standard were treated in the same way.
Pyorubin Quantification
Pyorubin is a collection of pigments produced by certain P. aeruginosa strains including our test strain. Although its full chemical composition is unknown, it consists of at least two, water-soluble, red-colored pigments (61). Pyorubin quantification was based on Hosseinidoust et al. (23). Briefly, bacteria were grown for 18 h in 10% Mueller-Hinton and 140 mM NaCl in the presence of 0.125–1 μM of HBD2 or solvent control in final assay volumes of 1 mL in 12-well microtiter plate (non-tissue culture treated, Costar). After 18 h incubation, well contents were collected and centrifuged at 5,000 × g for 10 min at 4°C to remove non-adherent bacteria. Equivolume mixtures of cell free supernatant (900 μL) and chloroform (900 μL) were mixed and centrifuged at 12,000 × g for 15 min at 4°C to separate the aqueous and organic phases and remove cell debris and other molecules. The aqueous phase containing pyorubin was lyophilized, dissolved in 125 μL volume of dH2O. From this, 100 μL were transferred to a 96-well flat bottom plate (Perkin Elmer) followed by an absorbance reading at 535 nm using a Victor X3 Plate Reader (Perkin Elmer).
In silico Molecular Docking Studies
The in silico modeling of binding between QS molecules and HBD2 was performed using Autodock Vina (The Scripps Research Institute) through the UCSF Chimera program1. LasR receptor (RSCB 3IX3) and HBD2 (RSCB 1FQQ) were considered as rigid receptors and were docked with N-(3-oxododecanoyl) homoserine lactone (3-oxo-C12-HSL), N-butanoyl homoserine lactone (C4-HSL), and 2-heptyl-3-hydroxy-4-quinolone (PQS) as ligands. Phosphorylcolamine (NEtP) was used as a negative control. Free energy of binding was used to calculate dissociation constants using Eq. (1) with R = 0.00198 kcal/(mol K) and T = 37°C = 310.15 K (62).
Gene Expression Analysis
Mid-logarithmic growth phase bacteria were prepared and washed as described above. The assay was up-scaled using 12-well polystyrene flat bottom plates with non-reversible lids with condensation rings (Genesee Scientific, San Diego, CA, United States). Twenty μL of the washed bacteria was added to HBD2 or solvent (100 μL of 10-fold concentrated defensin in 0.01% acetic acid or 0.01% acetic acid, respectively, diluted in 900 μL 10% Mueller Hinton/140 mM NaCl) yielding about 1 × 108 CFU/mL. After incubation at 37°C for the specified time points, biofilm and planktonic phase bacteria were homogenized by 10 min vortexing with 1 mm glass beads and tightly secured lids (Sigma-Aldrich, St. Louis, MO, United States). RNA extraction was performed on the homogenized samples using an RNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s enzymatic lysis and mechanical disruption protocol with acid-washed 425–600 μm glass beads (Sigma-Aldrich). Residual genomic DNA was removed with in-solution TurboDNase treatment (2 U/μL, Invitrogen, Carlsbad, CA, United States) according to the manufacturer’s recommendations followed by purification and concentration of RNA samples with RNA Clean and Concentrator-5 kit (Zymo Research, Irvin, CA, United States). Purity of RNA was confirmed by lack of amplification in SsoAdvancedTM Universal SYBR® Green (Bio-Rad, Hercules, CA, United States) real-time PCR using the RNA samples as template and primers for the housekeeping gene gapA (see Table 2). Confirmed pure RNA samples were reverse transcribed with iScript Reverse Transcription Supermix (Bio-Rad) and resulting cDNA was diluted to 25 ng/μL in nuclease free water. SsoAdvancedTM Universal SYBR® Green real-time PCR was performed with target primers for pslA and flgF and housekeeping gene gapA as reference gene (see Table 2, used at 0.75 μM final concentrations) in 10 μL reaction volumes and 12.5 ng cDNA input. Primers (Integrated DNA Technology’s, IDT, Coralville, IA, United States) were designed using IDT’s primerQuest Tool. Quantitative PCR (qPCR) and subsequent melt curve was performed using BIO-RAD’s CFX96 Real Time Thermocycler following standard conditions with annealing/extension at 60°C. CT values and relative gene expression were determined with BIO-RAD’s CFX Maestro Version 1.1. Amplified products were verified through size determination via standard agarose gel electrophoresis and melt curve analysis. Each time point was assessed in three independent experiments conducted in duplicates for a total n of 6. Initially, 16S rRNA was considered as a second housekeeping gene. However, its CT values (around 5) were substantially earlier than the CT values for the target genes and gapA (at or above 20) and thus, 16S rRNA gene expression was not further evaluated in this study.
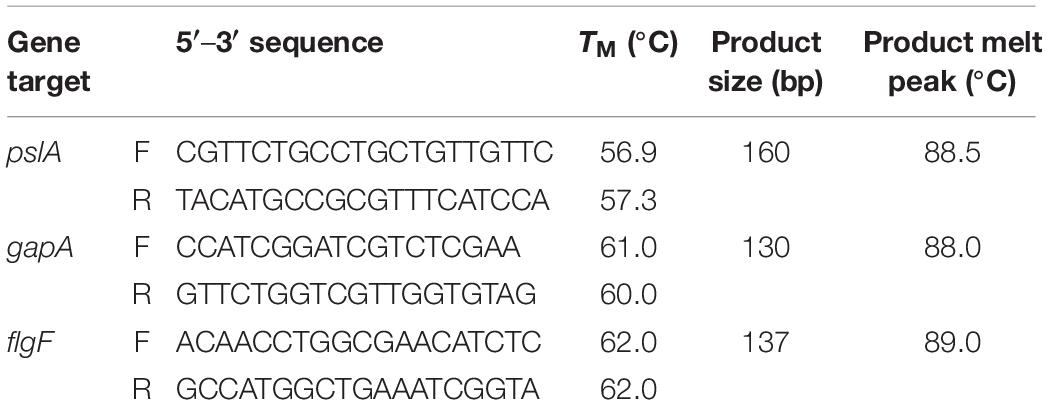
Table 2. Primers used in this study.
Outer Membrane Protein Profile Analysis
Pseudomonas aeruginosa outer membranes were harvested after incubation with HBD2 or solvent control according to Park et al. (63) with minor modifications. Briefly, bacteria were prepared as above and then grown for 18 h in 10% Mueller-Hinton and 140 mM NaCl in the presence of 0.125 to 1 μM of HBD2 or solvent control in final assay volumes of 1 mL in 12-well microtiter plate (Costar® not treated, Corning). After 18 h incubation, the well contents were resuspended, transferred into microfuge tubes, and centrifuged at 5,000 × g for 10 min at 4°C to pellet the bacterial cells. Cells were then resuspended in 80 μL of 0.2 M Tris–HCl, pH 8.0. Then, 120 μL lysis buffer was added to the resuspended cells (final conditions were 200 μg/mL hen egg white lysozyme (Sigma-Aldrich), 20 mM sucrose and 0.2 mM EDTA in 0.2 M Tris-HCl, pH 8.0). After a 10 min incubation at RT, 2 μL of Protease Inhibitor Cocktail (Sigma Aldrich P8340) was added followed by 202 μL of extraction buffer (10 μg/mL DNAse I [Sigma-Aldrich DN25] in 50 mM Tris–HCl/10 mM MgCl2/2% Triton X-100). After 1.5 h incubation on a rocker at 4°C, samples were centrifuged at 1500 × g at 4°C for 5 min. The resulting supernatants from triplicate samples, which contain the outer membranes, were pooled and placed into 4 mL ultrafiltration tubes with 5 kDa cut off molecular weight (Amicon Ultracel, 5k, Millipore). PBS was added to yield a volume of 4 mL, and then the tubes were centrifuged at 2400 × g until about 500 μL residual volume was obtained. The outer membranes in this residual were then washed by suspending in 3.5 mL PBS and then centrifuging at 2400 × g for 25 min at RT, yielding a residual volume of approximately 200 μL. Of this, 4 μL were subjected to standard SDS-PAGE using Bio-Rad 16.5% Mini-Protean Tris-Tricine gels followed by silver stain. Images were acquired with Versadoc (Bio-Rad) and analyzed with Image Lab version 6.01 software from Bio-Rad Laboratories.
Atomic Force Microscopy
P. aeruginosa (1 × 106 CFU/mL inoculum) was incubated in 10% Mueller Hinton broth/140 mM NaCl/12.5 mM sodium phosphate pH 7.0 with and without HBD2 (0.25 μM), on glass coverslips (Borosilicate glass square coverslips, Thermo Fisher Scientific) in 6-well plates (Corning) for 18 h at 37°C. As negative controls for HBD2 the peptide solvent 0.01% acetic acid was included, respectively. Coverslips were then transferred into wells of a fresh six-well plate and adherent bacteria were fixed with 2.5% glutaraldehyde (Ted Pella, CA; 25%, electron microscopy grade) diluted in PBS for 20 min at 4°C followed by washing with deionized water according to Chao and Zhang, 2011 (64), and stored at 4°C until imaging by atomic force microscopy (AFM).
All AFM tests (65) were carried out with a NX12 AFM system (Park System) using an aluminum coated PPP NCHR (Park systems) cantilever with a spring constant of 42 N/m, a resonance frequency of 330 kHz, and a nominal tip radius of <10 nm. At least five images were acquired per sample in air with non-contact mode (NCM) with settings of 256 pixels/line and 0.75 Hz scan rate and continuous monitoring of the tip integrity. The images were first order flattened and the roughness and height of all bacteria were measured using XEI software (Park Systems). Specifically, roughness of each bacterium was calculated from the root mean square value (RMS, i.e., standard deviation of the distribution of height over the whole bacterium surface).
Data and Statistical Analysis
Data graphs were generated using Microsoft Excel® 2016 or GraphPad Prism 7.04 Software. Statistical analyses were performed using IBM SPSS version 24 or GraphPad Prism 7.04 Software. A p-value < 0.05 was considered statistically significant.
Results
At Low Concentrations, HBD2 Does Not Reduce Metabolic Activity but Inhibits Biofilm Production by P. aeruginosa, Unlike HBD3
To compare the antimicrobial activities of HBD2 and HBD3, P. aeruginosa was exposed to either peptide at various concentrations over a period of 18 h. Viability was assessed by measuring metabolic activity every 3 h via quantification of resazurin reduction to the highly fluorescent resorufin by bacterial metabolites. Biofilm was assessed at 18 h post-incubation via quantification of crystal violet staining through absorbance readings. The resazurin reduction assay showed that both HBD2 and HBD3 reduced metabolic activity in a dose-dependent manner, with HBD3 being more effective on a per molar basis, producing around a 30% reduction at 0.5 μM compared to the 4 μM needed by HBD2 at 18 h for the same effect (Figure 1). However, when comparing the effect on biofilm production between the two peptides, a notable difference was observed. At concentrations of 0.25 and 0.5 μM, HBD2 reduced P. aeruginosa biofilm to ∼75% of the control without significantly reducing the metabolic activity (Figure 2A). In contrast, at these concentrations, HBD3 reduced the formation of P. aeruginosa biofilm in a dose dependent manner that was directly proportional to the cumulative effect on metabolic activity and further reduced both biofilm and resorufin production to nearly undetectable levels at a concentration of 1 μM (Figure 2B) consistent with direct microbicidal activity. ATP concentrations measured at the end of the 18 h incubation period corroborated the resazurin data (Figure 3), showing maintained ATP levels in HBD2 treated bacteria but a significant reduction of ATP levels in HBD3 treated P. aeruginosa (at 2 μM defensin, 17.65 ± 5.31 nM ATP compared to 3.6 ± 2.88 nM ATP, respectively, p = 0.011). These data suggest a differential mechanism for the antimicrobial activity between HBD2 and HBD3, and that HBD2 selectively inhibits biofilm formation at low concentrations.
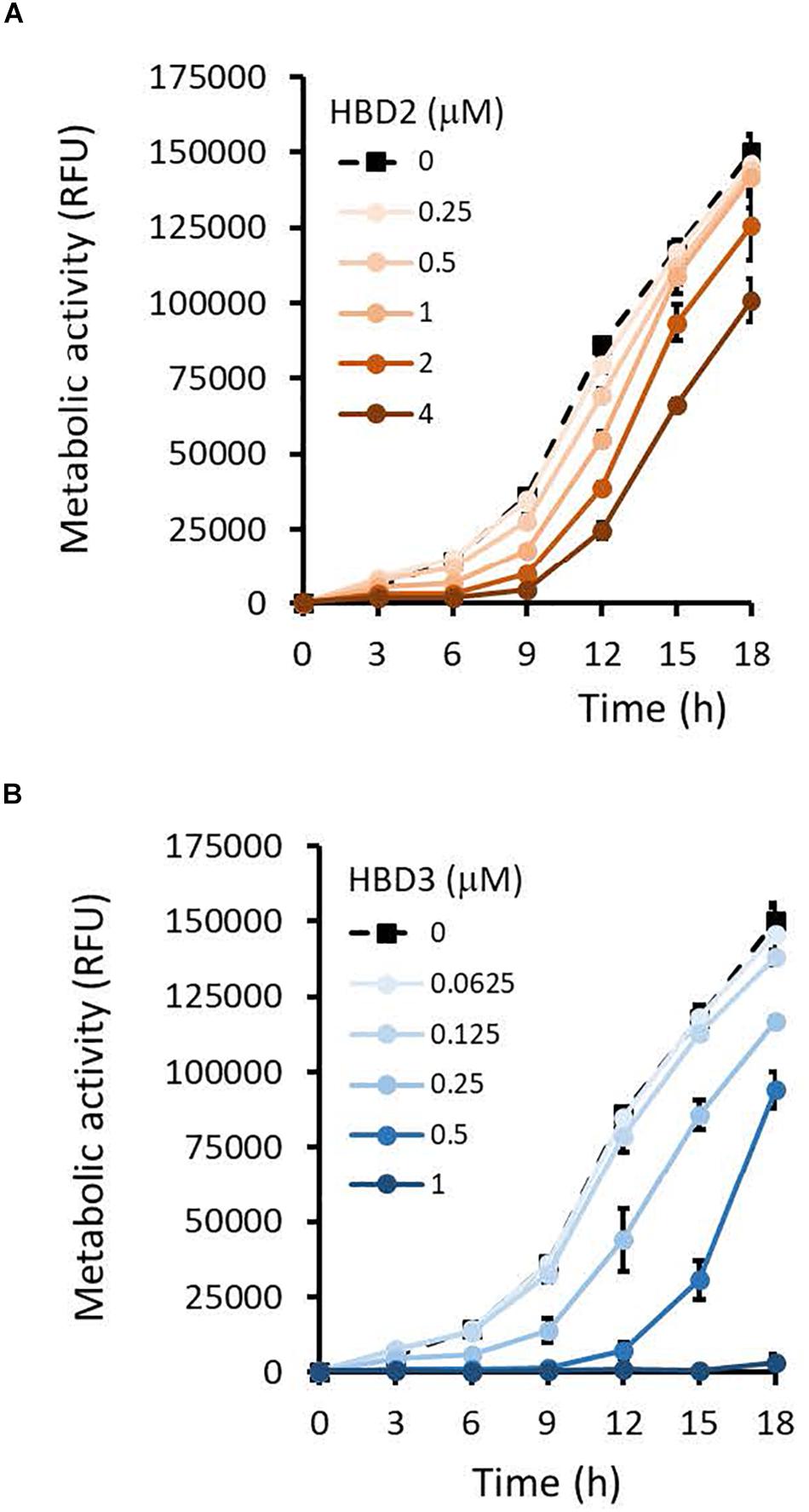
Figure 1. Metabolic activity of P. aeruginosa in the presence and absence of HBD2 and HBD3 over 18 h. Bacteria were incubated in 10% Mueller-Hinton/140 mM NaCl supplemented with 0.01% resazurin and fluorescence emitted by resorufin reflecting the production of reducing metabolites was measured every 3 h (530 nmex, 616 nmem). Shown are the means ± SD of three independent experiments conducted in duplicates. RFU: relative fluorescence units. p < 0.001 for HBD2 (A) at 1, 2, and 4 μM and for HBD3 (B) at 0.25, 0.5, and 1 μM compared to the solvent control in univariate ANOVA with Bonferrroni post hoc analysis. All other concentrations were not significantly different from the solvent controls.
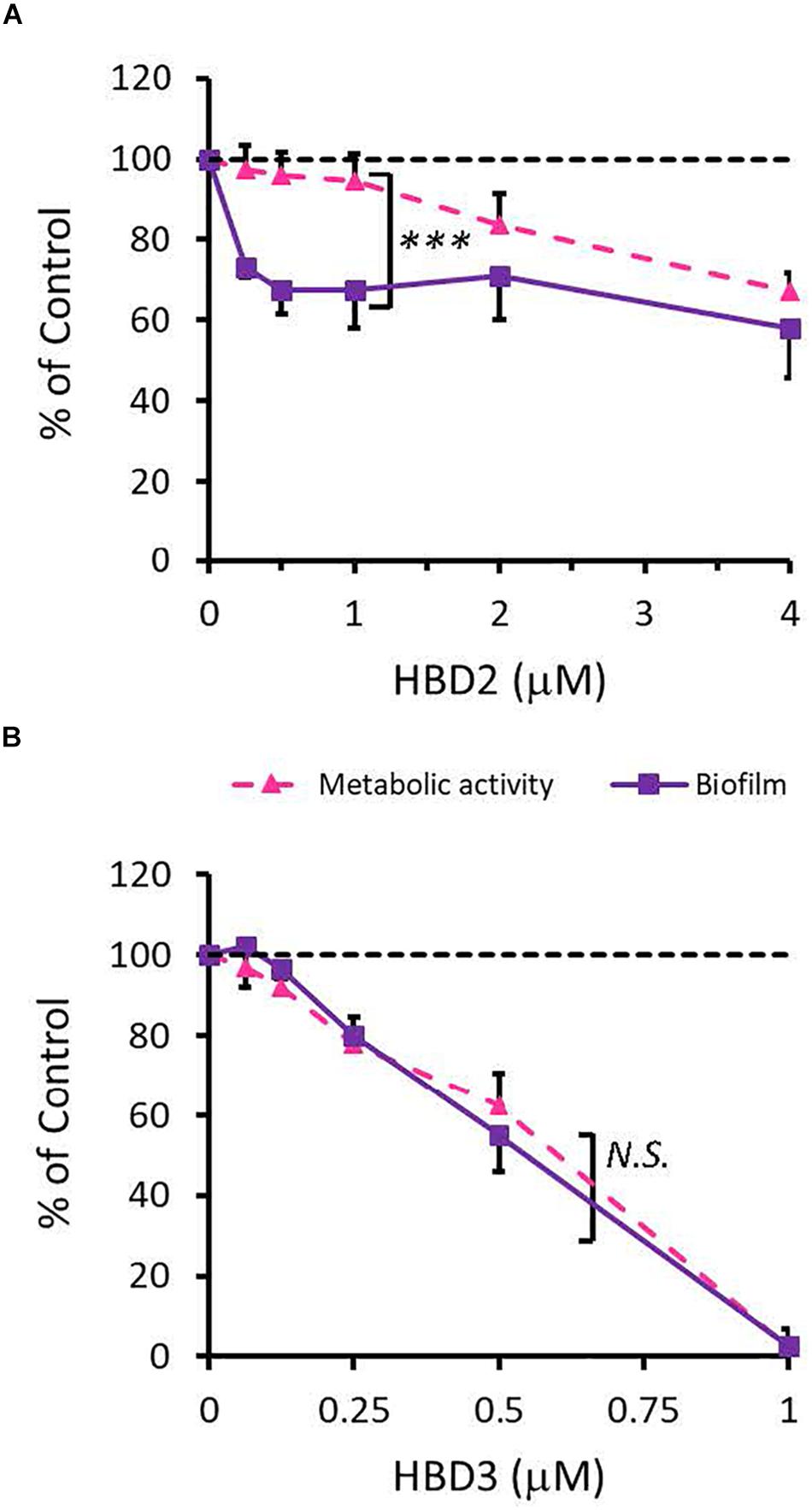
Figure 2. Comparative effects of HBD2 and HBD3 on P. aeruginosa biofilm and metabolic activity. Shown are biofilm formation and accumulated resorufin fluorescence after 18 h of incubation with HBD2 (A) and HBD3 (B) at the concentrations given. Data are expressed relative to the control and represent means ± SD of three independent experiments conducted in triplicates. ***p = 0.0004 in Two-way ANOVA. N.S: not significant (p = 0.7721).
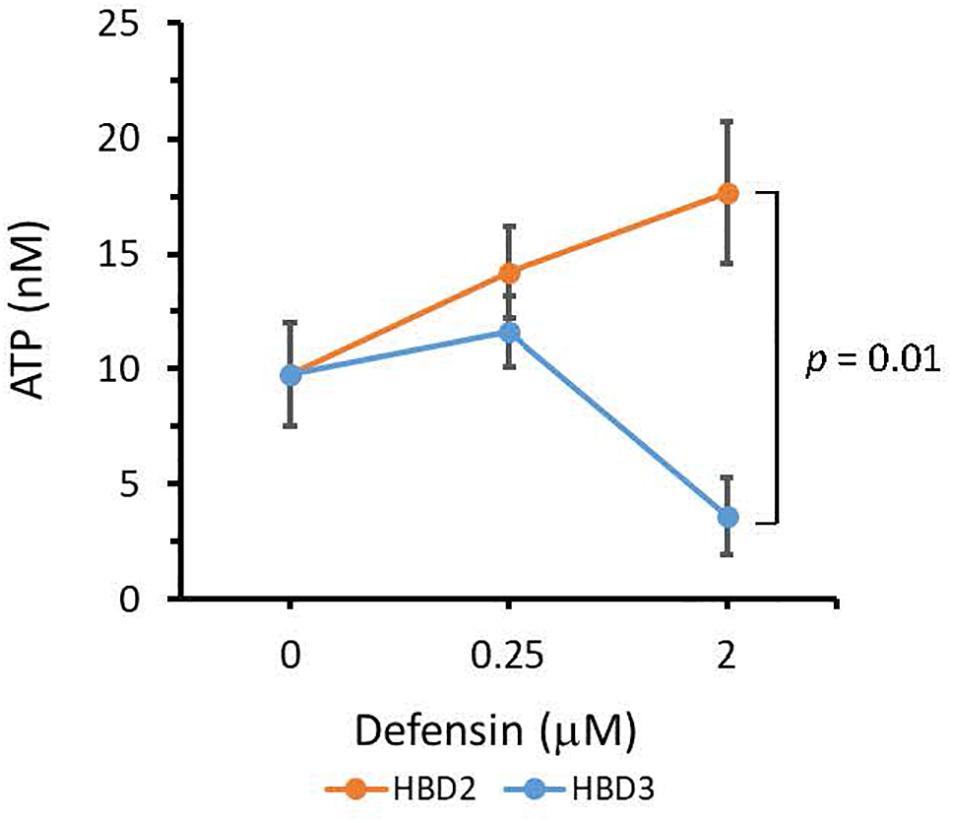
Figure 3. ATP quantification in P. aeruginosa after 18 h incubation in the presence or absence of HBD2 and HBD3 at the concentrations given. ATP concentrations are in nM and were calculated based on a standard curve. Shown are means ± SD of three independent experiments conducted in duplicates. p = 0.01 in One way ANOVA with Bonferroni post hoc analysis for 2 μM HBD2 compared to 2 μM HBD3.
HBD2 Similarly Inhibits Biofilm Production by A. baumannii Without Reducing Metabolic Activity at Lower Concentrations
To rule out that the observed differential biofilm reducing activity of HBD2 activity was strain-specific and restricted to P. aeruginosa, we also subjected A. baumannii-another opportunistic Gram-negative rod of clinical relevance – to varying doses of HBD2 and determined resazurin reduction and biofilm production after 18 h incubation. As shown in Figure 4, at low concentrations, HBD2 similarly inhibited biofilm formation while not reducing metabolic activity of A. baumannii. For example, at 1 μM, HBD2 effected a significant reduction of biofilm to 51.77 ± 2.93% of the control (p < 0.001) while resazurin reduction was still at 115 ± 0.67% (p = 1.0) of the control (means ± SD, n = 3). At higher concentrations though, HBD2 appeared to have greater effects on A. baumannii compared to P. aeruginosa as both biofilm and metabolic activity were reduced to less than 2 and 20% of the control at 4 μM HBD2, respectively (1.23 ± 0.48 and 18.71 ± 10.43%, means ± SD, n = 3).
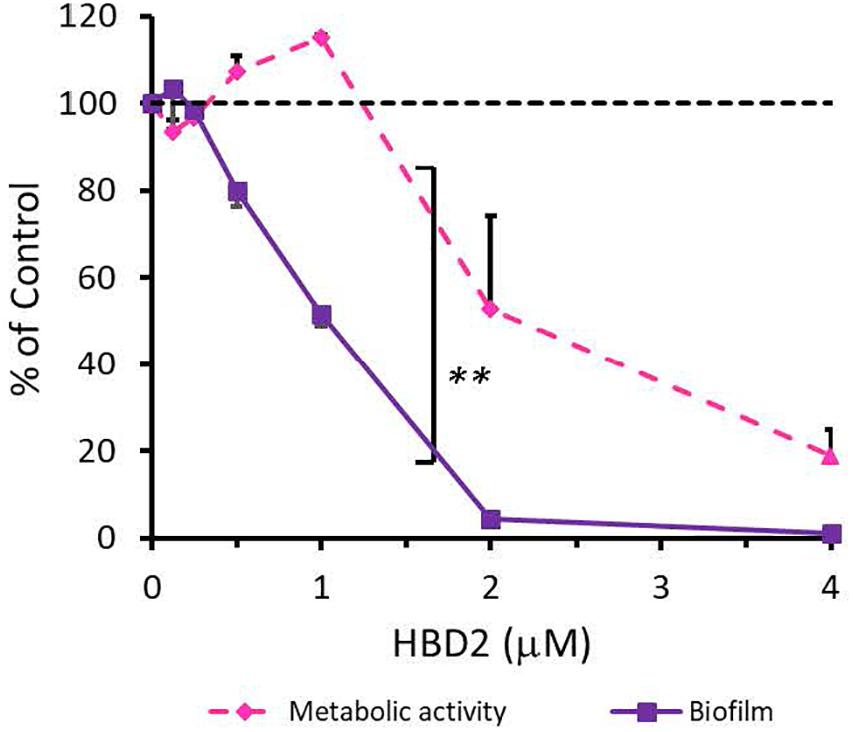
Figure 4. Effects of HBD2 on A. baumannii biofilm formation and metabolic activity. Shown are crystal violet absorbance and accumulated resorufin fluorescence expressed as % of the control after 18 h of incubation with HBD2 at the concentrations given. Data represent means ± SD of three independent experiments conducted in triplicates. **p = 0.004 for biofilm reduction versus reduction of metabolic activity in two tailed Paired Samples Test. In Oneway ANOVA with Bonferroni post hoc analysis, p = 0.001 for resazurin reduction at 4 μM HBD2, and p < 0.001 for biofilm reduction at 1, 2, and 4 μM HBD2, compared to the solvent control. All other data points were not significantly different from the control.
HBD2 Biofilm Inhibitory Activity Does Not Depend on Chirality but on Folding State
Since HBD2 appeared to selectively reduce biofilm formation and it has been known to bind to chemokine receptors on eukaryotic cells (66, 67), it was possible that the effects of HBD2 were due to binding to receptors involved in the biofilm regulatory pathway such as the GacA/GacS system. To test this, we assessed the activity of the D-form of HBD2, which, due to mismatched chirality, does not bind to proteinaceous receptors of L-HBD2. Like L-HBD2, D-HBD2 effected a significant reduction of biofilm production by P. aeruginosa without reducing metabolic activity (Figure 5A). Thus, this suggests that the observed HBD2 effect on P. aeruginosa biofilm production was not due to binding to receptors important for biofilm regulatory pathways.
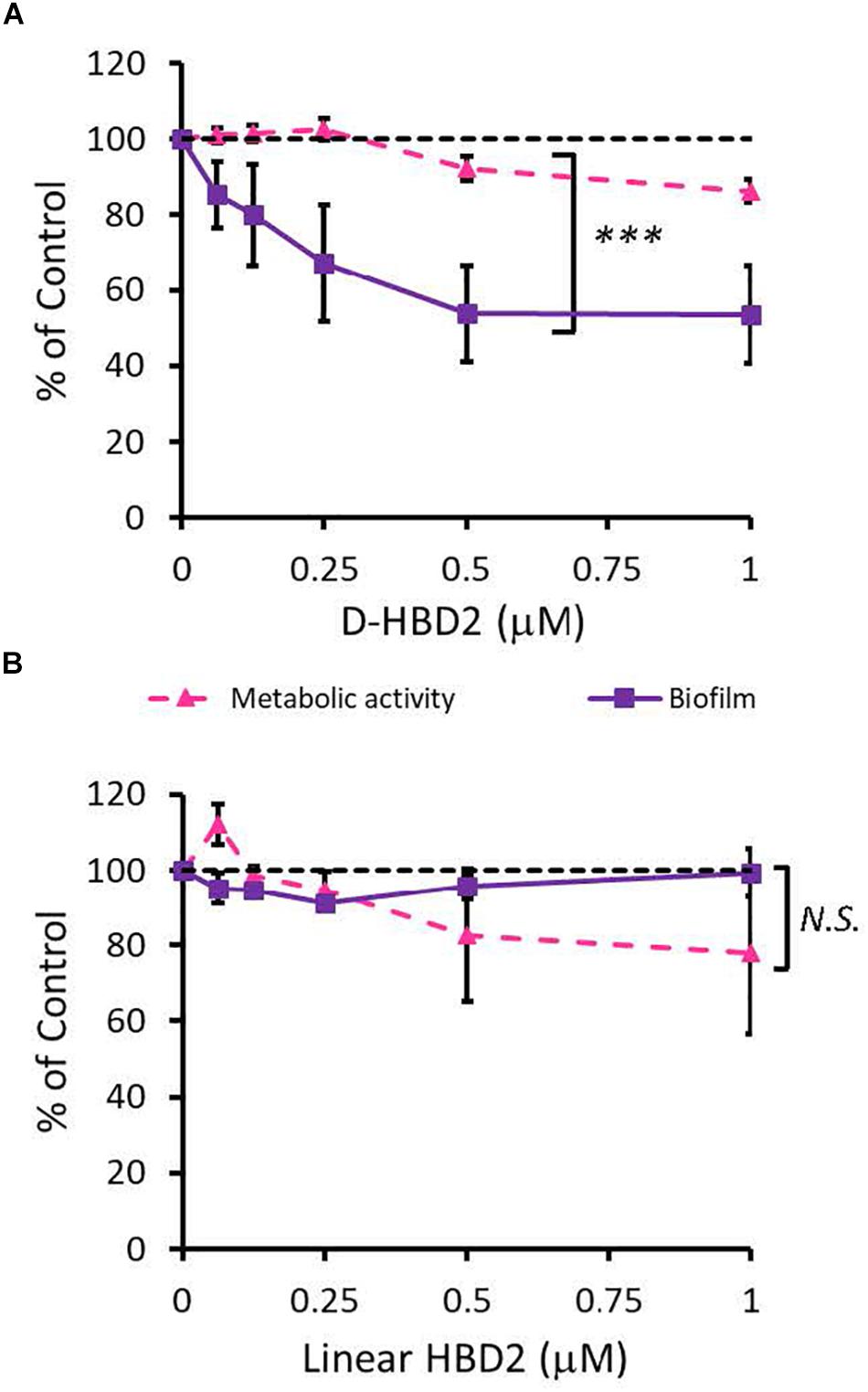
Figure 5. Comparative effects of D-HBD2 and linear HBD2 on P. aeruginosa biofilm and metabolic activity. Shown are biofilm formation and accumulated resorufin fluorescence expressed as % of the control after 18 h of incubation with all D-HBD2 (A) and linear HBD2 (B) at the concentrations given. Data represent means ± SD of three independent experiments conducted in triplicates. In paired T test comparing biofilm reduction and reduction of metabolic activity, ***p < 0.001 for D-HBD2 (A) and not significant (N.S.) for linear HBD2 (B). In Oneway ANOVA with Bonferroni post hoc analysis, biofilm formation (p = 0.033) but not metabolic activity (p = 0.473) is significantly reduced by D-HBD2. For linear HBD2, none of the data is significantly different from the solvent control.
Upon proper folding, defensins form three intramolecular disulfide bridges, which stabilize an amphipathic structure where cationic and hydrophobic amino acid residues are spatially segregated. To assess the importance of the structure and thus, charge distribution of HBD2 for its observed activity, a comparison was made between wildtype HBD2 and a linearized HBD2 mutant (Linear HBD2) with cysteine residues replaced by alanine residues. Loss of the cysteine residues prevents the formation of stabilizing disulfide bonds, drastically limits proper folding, and disrupts the organization of charged domains thought to be critical for AMP activity (68–70). As shown in Figure 5B, linearization of HBD2 resulted in a pronounced loss of activity.
Taken together, these data provided evidence for a receptor-independent activity that requires proper sequestration of charged and hydrophobic residues. We next asked whether HBD2 disrupts regulatory pathways of biofilm production through QS molecule binding. To answer this question, we took a three-pronged approach and performed in silico docking studies with known QS molecules involved in biofilm regulation, employed qPCR probing for genes differentially expressed during biofilm formation, and quantified pyorubin, a pigment regulated by the pathways that also affect biofilm production.
HBD2 Binding to QS Molecules Is Unlikely Based on Autodock Vina Prediction
QS molecules are small and flexible molecules with a potential for hydrogen bonding and hydrophobic interactions. Thus, they may bind to and be sequestered by HBD2. To explore this further, Autodock Vina was used (Figure 6) to predict HBD2 binding to known P. aeruginosa QS molecules representing three different QS systems, namely 3-oxo-C12-HSL – as the major QS molecule for P. aeruginosa utilized by the Las system, C4-HSL primarily utilized by the Rhl system, and PQS a key sensing molecule in the 4-quinolone system (71). As a positive control, Autodock Vina was also used to match the known binding pocket of the QS molecule 3-oxo-C12-HSL to its receptor LasR that has been previously assessed by X-ray diffraction (RSCB 3IX3) (72). Phosphorylcolamine (NEtP), which is not expected to bind to either LasR receptor or HBD2, was used as a negative control. Using the same methodology that confirmed binding of 3-oxo-C12-HSL to LasR here (Figure 6A) no binding of 3-oxo-C12-HSL to HBD2 was found (Figure 6B). Furthermore, we calculated the free energy of binding and found for LasR values corresponding to those reported in the literature (62, 73). Employing a −6 kcal/mol threshold for likely binding between ligand and receptor, binding between LasR and 3-oxo-C12-HSL, C4-HSL, and PQS was much more favorable (Figure 6C) than binding between HBD2 and these sensing molecules (Figure 6D).
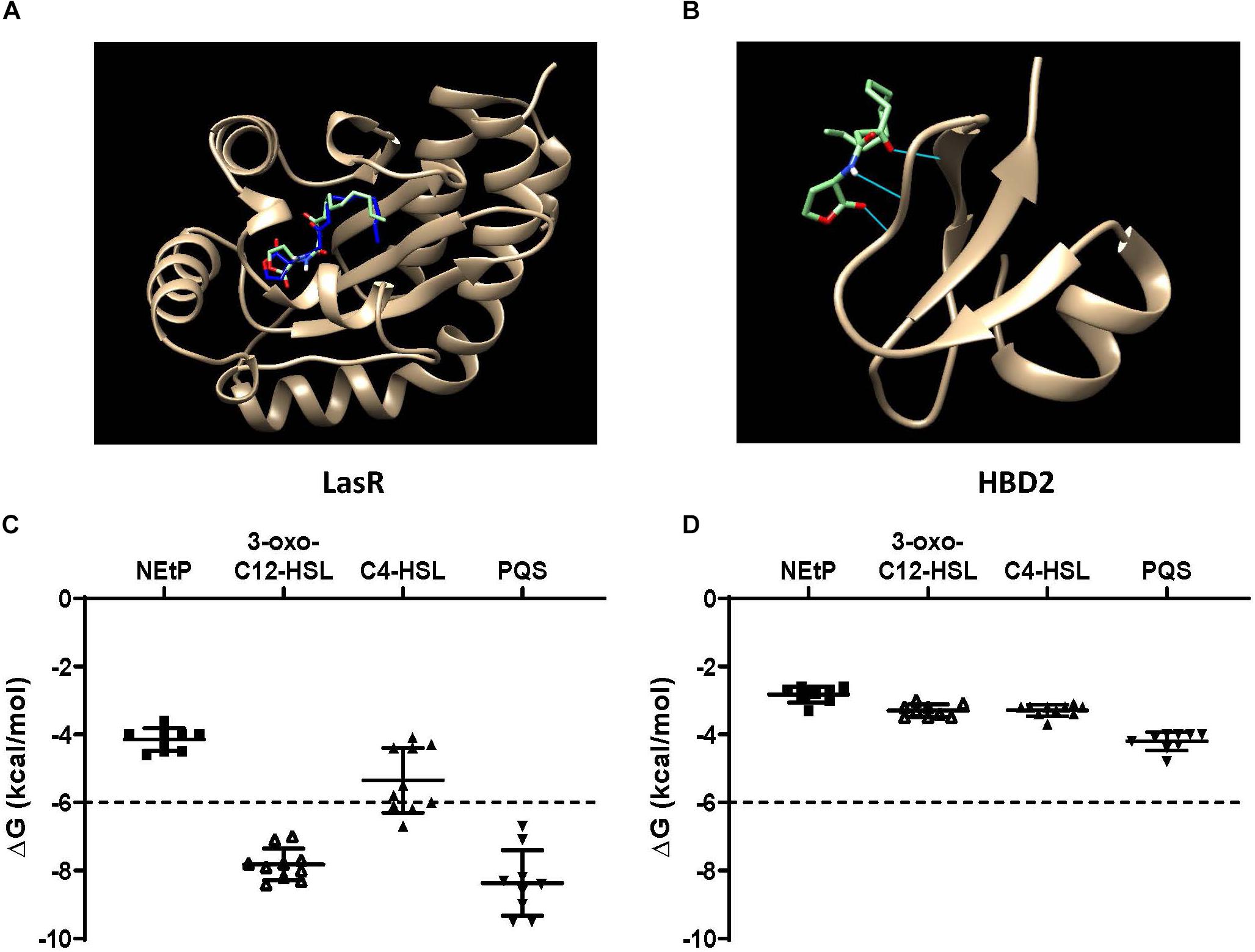
Figure 6. In silico docking and binding energies (ΔG) of various QS molecules calculated for LasR and HBD2. AutoDock Vina was used to predict binding sites and potential hits for HBD2 and quorum sensing molecules in comparison to LasR. (A) Test N-(3-oxohexanoyl) homoserine lactone (3-oxo-C12-HSL, green) lies inside the LasR binding pocket in the same region as co-crystallized 3-oxo-C12-HSL (blue) with LasR (RSCB 3IX3). (B) HBD2 does not contain a binding pocket for test 3-oxo-C12-HSL (green). Free energy of binding (ΔG) for various hits were determined for phosphorylcolamine (NEtP), 3-oxo-C12-HSL, N-butyryl homoserine lactone (C4-HSL), and 2-heptyl-3-hydroxy-4-quinolone (PQS) as ligands with either LasR (C) or HBD2 (D) as rigid receptors. Dashed lines indicate the –6 kcal/mol threshold for actively bound molecules.
Using Eq. (1), the dissociation constants (KD) for the most favorable binding pair between either LasR or HBD2 with each QS molecule was calculated (Table 3). This method predicted the KD of 3-oxo-C12-HSL and LasR (1.15 μM) near that of previously reported values (∼5.5 μM) (74). Furthermore, KD values for LasR binding with all three P. aeruginosa QS molecules were consistently two to three orders of magnitude lower than those of HBD2 binding with any of these QS molecules. This suggests that it is unlikely for HBD2 at physiological concentrations (75–77) to significantly bind these QS molecules.

Table 3. Dissociation constants for quorum sensing molecules calculated using AutoDock Vina measurements.
Gene Expression of flgF and pslA Is Not Affected by HBD2
During biofilm formation, motility and production of exopolysaccharide are reciprocally regulated with reduction of the expression of flagella-related genes and increase in the expression of genes contributing to polysaccharide synthesis including Psl polysaccharide. Thus, we compared the expression of flgF (Figure 7A) and pslA (Figure 7B) in P. aeruginosa treated with 0.25 μM HBD2 or solvent at various timepoints for up to 12 h. For solvent treated control bacteria, as expected, flgF gene expression decreased within 2 h reaching statistical significance after 6 h and the expression of pslA was signficantly increased after 2 h compared to the later time points (p < 0.01 and p < 0.05 in multivariate ANOVA with Bonferroni post hoc analysis). As observed for control bacteria, flgF gene expression decreased over time and was significantly reduced in HBD2 treated bacteria (p < 0.05) though changes in pslA gene expression did not reach statistical significance. However, there was overall no statistical significant difference between solvent and HBD2 treated bacteria. Thus, expression analysis of genes altered early in the biofilm production process does not support that HBD2 interference with biofilm production occurs at the transcriptional level.
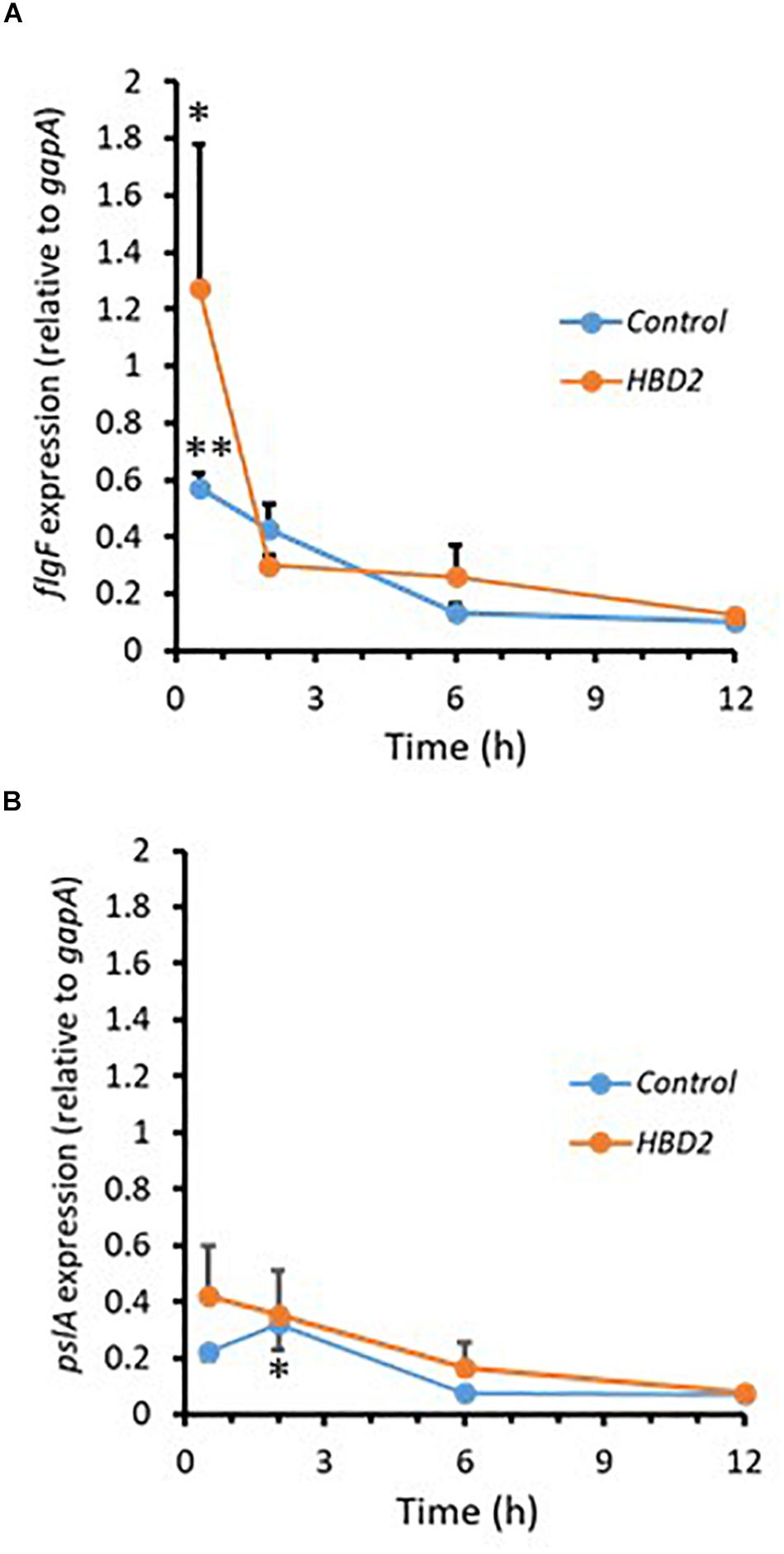
Figure 7. Relative gene expression of flgF and pslA in the presence and absence of 0.25 μM HBD2 as determined by qPCR. Gene expression of flgF (A) and pslA (B) in P. aeruginosa was calculated relative to the reference gene gapA after incubation in the presence or absence of HBD2 for up to 12 h. Shown are means ± SEM, n = 6. In multivariate ANOVA with Bonferroni post hoc analysis (*p < 0.05 and **p < 0.01), gene expression of flgF and pslA changed over time (Control: p < 0.01 for flgF 0.5 h versus 6 h and 12 h, and p < 0.05 for pslA 2 h versus 6 h and 12 h; HBD2: p < 0.05 for flgF 0.5 h versus 12 h) but there was no significant difference between the control and HBD2 treated bacteria.
Pyorubin Accumulation Is Not Reduced in Media Collected From HBD2 Treated P. aeruginosa
Pigment production in P. aeruginosa has been shown to be also regulated by QS (24, 61). To further corroborate that HBD2 does not interfere with QS, we quantified pyorubin released into culture supernatants in the presence and absence of HBD2. At 0.125 and 0.25 μM HBD2 there was no difference in pyorubin accumulation compared to the control (data not shown). In the presence of 0.5 and 1 μM HBD2, there was a slight increase of pyorubin (109.5 ± 4.9 and 109.9 ± 5.8% of the control, respectively, p < 0.01 in univariate ANOVA with Bonferroni post hoc adjustment). This finding further supports that HBD2 does not inhibit quorum sensing and next, we explored whether HBD2 may induce structural changes in the outer membrane that could interfere with the transport of biofilm precursors to the extracellular space.
HBD2 Alters the Outer Membrane Protein Profile of P. aeruginosa
Outer membrane proteins participate in the process of biofilm formation (78). Hence, we probed whether incubation with HBD2 leads to changes in the outer membrane protein profile of P. aeruginosa (Figure 8). A representative image of outer membrane preparations resolved by silver stained SDS-PAGE is depicted in Figure 8A. Numerous bands are detected ranging from about 10 kDa to over 200 kDa with the most dominant bands appearing above 25 kDa, in particular a band around 35 kDa similar to the molecular weights of previously reported P. aeruginosa outer membrane proteins (79). Two weaker bands around 10 kDa are consistently visible only in the outer membrane preparations from control bacteria. Overall, the outer membranes from HBD2 treated bacteria appear to contain less proteins between 35 and 75 kDa. A prominent band between 10 and 15 kDa is detected in all samples, including the medium control, consistent with the molecular weight of the lysozyme (14 kDa) added during the extraction procedure. Figure 8B summarizes the protein profiles of the outer membrane preparations from control bacteria and HBD2 treated bacteria. To control for variations during the ultrafiltration process and gel loading, the band intensities of the various proteins were normalized with the presumptive lysozyme band intensity. HBD2 appears to affect a decrease in outer membrane proteins in particular at about 22, 34, 40, 45, and 50 kDa, with the changes noticeable at all concentrations tested.
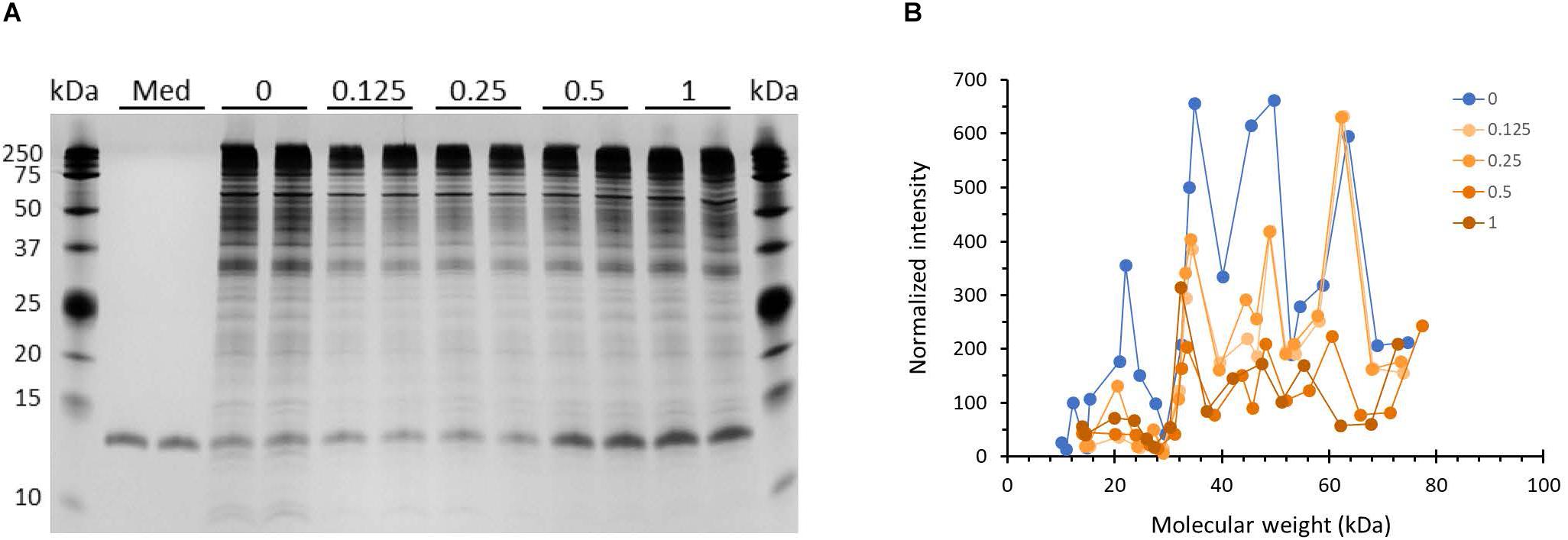
Figure 8. Outer membrane protein profile of P. aeruginosa after 18 h incubation in the presence and absence of HBD2. (A) Four μL of concentrated outer membrane preparations from HBD2 treated (0.125–1 μM) or solvent control exposed bacteria (0) were resolved by SDS Tris-Tricine PAGE and visualized by silver stain. (Med) indicates medium only processed like bacteria-containing samples. The band migrating between 10 and 15 kDa in all samples is consistent with the expected molecular weight of lysozyme (14 kDa) that was added to the extraction buffer. (B) Approximate molecular weight and intensities of bands were quantified with Image Lab software and band intensities detected in both replicates were normalized to the intensity of the presumptive lysozyme band. Each data point represents the average of replicates. Each line represents the protein profile for the indicated HBD2 concentration (in μM).
Atomic Force Microscopy Reveals Ultrastructural Changes in HBD2 Treated Bacteria Reflected in Increased Surface Roughness
We also assessed whether the changes at the outer membrane induced by HBD2 resulted in topographical changes and employed atomic force microscopy to measure bacterial height and roughness after incubation for 18 h in the presence or absence of 0.25 μM HBD2 (Figure 9). Representative images of control and HBD2 treated bacteria are shown in Figure 9A. The surface of control bacteria appears smoother compared to the surface of HBD2 treated bacteria, the latter showing irregular dents. While the overall bacterial height is not significantly different in HBD2 treated bacteria compared to solvent only exposed bacteria (215.22 ± 3.96 nm versus 220.24 ± 3.23 nm, means ± SEM, n = 85 and n = 69, respectively, p = 0.343), there is a significant increase in roughness in HBD2 treated samples (Figure 9B) consistent with a structurally altered surface (43.39 ± 1.52 versus 51.86 ± 1.5 nm, means ± SEM, n = 85 and n = 69, p < 0.001 in independent samples T test).
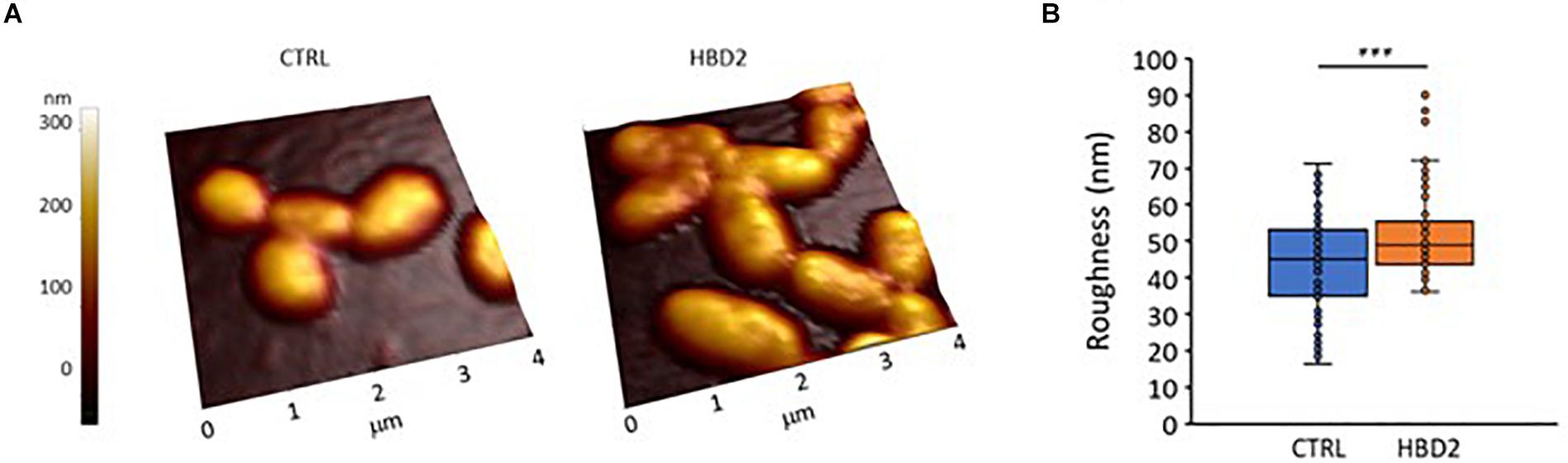
Figure 9. Atomic force microscopy of P. aeruginosa after 18 h incubation in the presence and absence of 0.25 μM HBD2. Bacteria were incubated on glass slides and fixed with 2.5% glutaraldehyde prior to imaging. Images taken with the atomic force microscope were first order flattened before extracting measurement for bacterial roughness. (A) Representative images. CTRL: solvent control exposed bacteria. (B) Box and whisker chart (with inner points and outliers) of roughness measurements from multiple images of solvent exposed control bacteria (CTRL, n = 85) and 0.25 μM HBD2 treated bacteria (n = 69). ***p < 0.001 in independent samples T test.
Taken together, our data demonstrate that at low concentrations L- and D-forms of HBD2 inhibit biofilm formation while not reducing metabolic activity in Gram-negative bacteria of two different genera, Pseudomonas and Acinetobacter. Furthermore, this activity appears to be receptor-independent and not mediated by interference with quorum sensing or other regulatory pathways of biofilm production at the transcriptional level. Instead, our data are consistent with structural changes induced by HBD2 that interfere with the transport of biofilm precursors into the extracellular space suggesting a novel mechanism of action for the antimicrobial peptide HBD2.
Discussion
In this study, we demonstrate that, HBD2, at nanomolar concentrations, and independent of its chiral state, significantly reduced biofilm formation of P. aeruginosa without affecting metabolic activity. This was unlike HBD3, which equally reduced biofilm and metabolic activity at nanomolar concentrations. HBD2 similarly affected A. baumannii, another Gram-negative bacterium, at low concentrations. In silico modeling did not support direct binding of HBD2 to QS molecules, the release of a QS regulated pigment was not inhibited, and the expression of biofilm-related genes was not influenced by HBD2. However, the outer membrane protein profile was altered in HBD2 treated bacteria with reduced representation of several proteins, which was accompanied by increased roughness of the bacterial surface. Taken together, these data support a novel mechanism of biofilm inhibition by HBD2 at low concentrations that is independent of biofilm regulatory pathways but involves structural changes induced by HBD2 that may interfere with the transport of biofilm precursors into the extracellular space.
HBD2 has been previously reported to reduce bacterial survival in existing biofilm cultures of Lactobacillus ssp., Gram-positive bacteria, at higher, micromolar concentrations (80). However, inhibition of biofilm formation by HBD2 has not been reported previously to the best of our knowledge. Considering the rapid induction of HBD2 in epithelial cells’ response to proinflammatory cytokines and bacterial challenge (81), the ability to interfere with biofilm formation at low concentrations adds importance to the role of HBD2 in innate host defense during the early interaction between host and pathogen. Bacteria are more susceptible to host-derived and exogenous antimicrobial agents while they are metabolically active in the planktonic state prior to biofilm production. Thus, HBD2 may amplify host defenses early in the attempted infection process and could improve the action of antibiotics in a clinical setting (82). Synergism studies will be able to address this experimentally in the future.
Anti-biofilm activity of HBD2 in the absence of inhibition of metabolic activity of P. aeruginosa occurred only at low concentrations. A concentration dependent mechanism of action has been well documented for the lantibiotic nisin, which, at nanomolar concentrations, preferentially binds to lipid II disrupting cell wall synthesis and, at micromolar concentrations, embeds into the bacterial membrane causing pore formation (83–87). More recently, the alpha-defensin human neutrophil peptide 1 (HNP1) has been added to the list of AMPs that initially interact with lipid II, and when concentrations increase, with the bacterial cell membrane (88). Binding of HBD3 to lipid II has also been described, albeit at higher concentrations, in the micromolar range (47). It is conceivable that HBD2 could similarly interfere with membrane-embedded proteins responsible for the transport of biofilm components (17) at low concentrations followed by membrane perturbation at higher concentrations.
The differential effect of HBD2 on biofilm production and metabolic activity of P. aeruginosa was not observed in the related beta-defensin HBD3, which was active at lower concentrations than HBD2 and equally reduced biofilm and metabolic activity reflecting a strong bactericidal activity. These differences in their activity could be at least in part attributed to the differences in their physicochemical properties with respect to net charge, surface charge distribution, hydrophobicity index, and behavior in solution (51, 89). Biofilm is a complex matrix with numerous components that can be affected in different ways by HBD2 and HBD3. For example, alginate has been shown to affect antimicrobial peptide conformation inducing alpha-helices contigent on the hydrophobicity (90), and HBD2 and HBD3 substantially differ in their hydrophobicity with HBD2 being more hydrophobic than HBD3.
HBD2, at low concentrations, similarly inhibited biofilm production in A. baumannii without reducing metabolic activity suggesting the observed effects are not strain specific. However, at higher HBD2 concentrations differences between the effects on P. aeruginosa and A. baumannii emerged as reflected in a near complete inhibition of biofilm production of A. baumannii contrasting the stalled biofilm inhibition of P. aeruginosa. The lesser susceptibility of P. aeruginosa to HBD2 may be due to a greater outer membrane vesicle production in P. aeruginosa that may sequester HBD2 before it reaches the bacterial cell (91).
Like other defensins, HBD2 forms three intramolecular disulfide bridges and linearization of the peptide can reveal the importance of its structure for its antimicrobial activity (92). Here, linearization of full length HBD2 led to a pronounced loss of both its antimicrobial and biofilm inhibitory activity. This contrasts reports for other defensins including HBD3 and could be attributed to a lack of accumulation of positively charged amino acid residues at the C-terminus of HBD2 compared to HBD3. Chandrababu et al. (93) have shown that positively charged residues cluster in the C-terminal segment of a linearized form of HBD3 allowing them to interact with the negatively charged phospholipids of micelles. The inherent antimicrobial activity of this patch of cationic residues is also reflected in studies with HBD3 analogs truncated to the C-terminal region (94). The here observed loss of activity after linearization could indicate that HBD2 functions through a receptor (56). However, D- and L forms of HBD2 did not differ in their activity and thus, we interrogated the possibility that HBD2 interferes with regulatory networks of biofilm production.
QS molecules are key to the regulation of virulence factor production including biofilm and pigment in P. aeruginosa. They are small hydrophobic molecules (95) and thus, we interrogated possible binding of HBD2 to QS molecules in silico. We found favorable binding of LasR to not only its cognate ligand 3-oxo-C12-HSL but also to C4-HSL and PQS. This is in line with a recent study describing LasR as promiscuis for binding a variety of QS molecules (96). The unfavorable binding energies derived for HBD2 suggest that interference of QS-dependent processes through direct HBD2 binding to individual QS molecules is unlikely. Another type of QS molecule, (2S,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuran-borate (S-THMF-borate), has been shown to increase biofilm formation in P. aeruginosa (97, 98). However, although S-THMF-borate – a molecule with a distinct structure from major Gram-negative QS molecules – has been identified in some Gram-positive and Gram-negative bacteria (99), P. aeruginosa does not encode the luxS gene required for its synthesis (100) and binding to this S-THMF-borate should not be further considered as an underlying mechanism for the observed biofilm inhibition.
In agreement with the in silico data, HBD2 did not affect the expression of flgF and pslA. Thus, interference of HBD2 with regulatory networks at the transcriptional level is not likely to account for its biofilm inhibitory activity. However, we cannot rule out that HBD2 has posttranscriptional effects through interference with the two component signal transduction system GacS/GacA (71, 101). GacS is a transmembrane sensor kinase phosphorylating GacA, which in turn induces the expression of small RNA molecules that antagonize the protein RsmA, a translational repressor interfering with psl translation and known to normally block exopolysaccharide production (102). It is conceivable that HBD2 could interfere with GacS upon inserting into the bacterial membrane. Finally, HBD2 might bind to the secondary messenger molecule c-di-GMP, which regulates biofilm formation in P. aeruginosa at multiple levels (103). Previously, de la Fuente-Nunez et al. (104) demonstrated that peptide 1018, derived from the antimicrobial peptide bovine Bac2a (105), inhibited biofilm formation in P. aeruginosa while not affecting planktonic growth by binding to the second messenger p(pp)Gpp and promoting its degradation. A similar mode of action could apply to HBD2.
Further supporting that HBD2 does not act through interference with regulatory networks is our finding that pyorubin accumulation in the extracellular fluid was not diminished after incubation with HBD2. Pyorubin is composed of several pigments including aeruginosin A, which is a phenazine, like the much better studied P. aeruginosa pigment pyocyanin (106). Phenazines typically traverse the bacterial membrane freely and their production is under the same controls that govern biofilm production (107, 108).
Considering the lack of evidence for interference with regulatory networks and the stereoisometry independent activity of HBD2, we conceived that the observed HBD2 mediated inhibition of biofilm production is most likely due to embedding in the bacterial membrane and disruption of transport of biofilm precursor molecules across the membrane. An increasing amount of research suggests that AMPs can target discrete loci in bacterial membranes and thereby disrupt biological processes (109). For example, AMPs are known to impair the assembly of multicomponent enzyme complexes in the bacterial cell membrane (110) or disrupt periplasmic protein-protein interaction interfering with molecular transport (111). In 2013, Kandaswamy et al. showed that HBD2 localizes to the mid-cell region of the Gram-positive bacterium E. faecalis (112). The authors determined that this mid-cell region is rich in anionic phospholipids and that HBD2 delocalized the spatial organization of protein translocase SecA and sortases, both of which are important for pilus biogenesis (112, 113). It is possible that HBD2 targets similar machinery in P. aeruginosa to impair biofilm formation. SecA also plays a role in the transport of outer membrane proteins in Gram-negative bacteria (114) and outer membrane proteins have been shown to participate in biofilm formation, including the 11 kDa LecB protein and the 38 kDa OprF (115, 116). Consistent with this we found an altered outer membrane protein profile in HBD2 treated bacteria with a paucity of proteins around 10 kDa and proteins around the molecular weights of previously reported outer membrane proteins. This may indicate structural changes of the outer membrane, which was further supported by our atomic force microscopy data demonstrating an increased roughness of the bacterial surface after HBD2 treatment. It is important to note, however, that increased roughness could also represent changes in the LPS profile. Atomic force microscopy has been previously employed elsewhere to demonstrate outer membrane damages in P. aeruginosa (117). Resolving the outer membrane proteins by 2D gel electrophoresis could further delineate the observed changes in future experiments, which should also revisit the action of the D-form of HBD2 and effects on the outer membrane of A. baumannii. Finally, outer membrane vesicles have been recognized to take part in the formation of biofilm by interacting with extracellular DNA and HBD2 interference with proper outer membrane formation may disrupt this process (118).
Conclusion
This study reveals distinct activity of two epithelial beta-defensins, HBD2 and HBD3, and provides evidence for a novel antibacterial action of HBD2. At low concentrations in the nanomolar range, HBD2 reduced biofilm formation without reducing the metabolic activity of P. aeruginosa. Biofilm production of A. baumannii was similarly affected, indicating that the observed HBD2 activity is not strain specific. This activity is unlikely mediated through a receptor-dependent interference with regulatory networks but contingent on preservation of HBD2 structure. Our findings are consistent with a membrane-targeted action of HBD2 that affects proper function of membrane-associated proteins involved in biofilm precursor transport into the extracellular environment. Future studies dissecting the molecular basis for the described HBD2 activity may inform the development of new methods for the manipulation of biofilms in aquaculture, in the food industry, and in the healthcare setting, which is in particular of interest for the latter considering the rise of multidrug resistance.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
KP, BB, TY, MB, EE, AT, AC, and YW: acquisition of the data. KP, BB, TY, MB, EE, AT, AC, HP, YW, WL, and EP: analysis and interpretation of the data. KP, MM, and CA: method development. KP and EP: statistical analysis. KP: molecular docking. KP, HP, YW, WL, and EP: conceptual and experimental design. KP, HP, and EP: drafted the manuscript. KP, BB, TY, MB, EE, AT, AC, MM, CA, HP, YW, WL, and EP: critical revision of the manuscript for important intellectual content. All authors approved the final manuscript submission.
Funding
This work was supported by the National Institutes of Health (Grants NIH SC1 GM096916, NIH RISE GM061331, and NIH LA Basin Bridges to Ph.D. GM054939), the National Science Foundation (Grant NSF-MRI 1828334), the California State University Library Open Access Author Fund, and the College of Natural and Social Sciences at California State University Los Angeles (NSS Research and Scholarship Award 2018). The funders provided no input to the study design nor the collection, analyses and interpretation of data.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past co-authorship with one of the authors WL.
Acknowledgments
We thank Susan Cohen for helpful discussions. Parts of the result presented here have been included in the Master’s Thesis of KP (119) and presented at the Microbe 2019 General Meeting of the American Society for Microbiology in San Francisco, CA, June 20–24, 2019.
Footnotes
References
1. Qian PY, Lau SC, Dahms HU, Dobretsov S, Harder T. Marine biofilms as mediators of colonization by marine macroorganisms: implications for antifouling and aquaculture. Mar Biotechnol (N. Y.). (2007) 9:399–410.
2. Taylor PK, Yeung AT, Hancock RE. Antibiotic resistance in Pseudomonas aeruginosa biofilms: towards the development of novel anti-biofilm therapies. J Biotechnol. (2014) 191:121–30. doi: 10.1016/j.jbiotec.2014.09.003
3. Wilton M, Charron-Mazenod L, Moore R, Lewenza S. Extracellular DNA acidifies biofilms and induces aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother. (2016) 60:544–53. doi: 10.1128/AAC.01650-15
4. Donlan RM, Costerton JW. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev. (2002) 15:167–93.
5. Hoiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O. Antibiotic resistance of bacterial biofilms. Int J Antimicrob Agents. (2010) 35:322–32. doi: 10.1016/j.ijantimicag.2009.12.011
6. Jennings LK, Storek KM, Ledvina HE, Coulon C, Marmont LS, Sadovskaya I, et al. Pel is a cationic exopolysaccharide that cross-links extracellular DNA in the Pseudomonas aeruginosa biofilm matrix. Proc Natl Acad Sci USA. (2015) 112:11353–8. doi: 10.1073/pnas.1503058112
7. Colvin KM, Irie Y, Tart CS, Urbano R, Whitney JC, Ryder C, et al. The Pel and Psl polysaccharides provide Pseudomonas aeruginosa structural redundancy within the biofilm matrix. Environ Microbiol. (2012) 14:1913–28. doi: 10.1111/j.1462-2920.2011.02657.x
8. May TB, Shinabarger D, Maharaj R, Kato J, Chu L, DeVault JD, et al. Alginate synthesis by Pseudomonas aeruginosa: a key pathogenic factor in chronic pulmonary infections of cystic fibrosis patients. Clin Microbiol Rev. (1991) 4:191–206.
9. Fong JNC, Yildiz FH. Biofilm matrix proteins. Microbiol spectr. (2015) 3:MB-0004-2014. doi: 10.1128/microbiolspec.MB-0004-2014
10. Turnbull L, Toyofuku M, Hynen AL, Kurosawa M, Pessi G, Petty NK, et al. Whitchurch, explosive cell lysis as a mechanism for the biogenesis of bacterial membrane vesicles and biofilms. Nat Commun. (2016) 7:11220. doi: 10.1038/ncomms11220
11. Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. Extracellular DNA required for bacterial biofilm formation. Science (N. Y.). (2002) 295:1487.
12. Das T, Sehar S, Manefield M. The roles of extracellular DNA in the structural integrity of extracellular polymeric substance and bacterial biofilm development. Environ Microbiol Rep. (2013) 5:778–86. doi: 10.1111/1758-2229.12085
13. Petrova OE, Sauer K. Sticky situations: key components that control bacterial surface attachment. J Bacteriol. (2012) 194:2413–25. doi: 10.1128/JB.00003-12
14. Stojkovic B, Sretenovic S, Dogsa I, Poberaj I, Stopar D. Viscoelastic properties of levan-DNA mixtures important in microbial biofilm formation as determined by micro- and macrorheology. Biophys J. (2015) 108:758–65. doi: 10.1016/j.bpj.2014.10.072
15. Cowles KN, Gitai Z. Surface association and the MreB cytoskeleton regulate pilus production, localization and function in Pseudomonas aeruginosa. Mol Microbiol. (2010) 76:1411–26. doi: 10.1111/j.1365-2958.2010.07132.x
16. Pearson JP, Passador L, Iglewski BH, Greenberg EP. A second N-acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proc Natl Acad Sci USA. (1995) 92:1490–4.
17. Laverty G, Gorman SP, Gilmore BF. Biomolecular mechanisms of Pseudomonas aeruginosa and Escherichia coli biofilm formation. Pathogens. (2014) 3:596–632. doi: 10.3390/pathogens3030596
18. Rutherford ST, Bassler BL. Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb Perspect Med. (2012) 2:a012427. doi: 10.1101/cshperspect.a012427
19. Overhage J, Schemionek M, Webb JS, Rehm BH. Expression of the psl operon in Pseudomonas aeruginosa PAO1 biofilms: PslA performs an essential function in biofilm formation. Appl Environ Microbiol. (2005) 71:4407–13.
20. Irie Y, Starkey M, Edwards AN, Wozniak DJ, Romeo T, Parsek MR. Pseudomonas aeruginosa biofilm matrix polysaccharide Psl is regulated transcriptionally by RpoS and post-transcriptionally by RsmA. Mol Microbiol. (2010) 78:158–72. doi: 10.1111/j.1365-2958.2010.07320.x
21. Hauser AR. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol. (2009) 7:654–65. doi: 10.1038/nrmicro2199
22. Zulianello L, Canard C, Kohler T, Caille D, Lacroix JS, Meda P. Rhamnolipids are virulence factors that promote early infiltration of primary human airway epithelia by Pseudomonas aeruginosa. Infect Immun. (2006) 74:3134–47.
23. Hosseinidoust Z, Tufenkji N, van de Ven TG. Predation in homogeneous and heterogeneous phage environments affects virulence determinants of Pseudomonas aeruginosa. Appl Environ Microbiol. (2013) 79:2862–71. doi: 10.1128/AEM.03817-12
24. Naik V, Mahajan G. Quorum sensing: a non-conventional target for antibiotic discovery. Nat Prod Commun. (2013) 8:1455–8.
25. Orlandi VT, Bolognese F, Chiodaroli L, Tolker-Nielsen T, Barbieri P. Pigments influence the tolerance of Pseudomonas aeruginosa PAO1 to photodynamically induced oxidative stress. Microbiology. (2015) 161:2298–309. doi: 10.1099/mic.0.000193
26. Driscoll JA, Brody SL, Kollef MH. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs. (2007) 67:351–68.
27. Zasloff M. Antibiotic peptides as mediators of innate immunity. Curr Opini Immunol. (1992) 4:3–7.
28. Bevins CL. Antimicrobial peptides as effector molecules of mammalian host defense. Contrib Microbiol. (2003) 10:106–48.
29. Martin E, Ganz T, Lehrer RI. Defensins and other endogenous peptide antibiotics of vertebrates. J Leukoc Biol. (1995) 58:128–36.
30. Oren A, Ganz T, Liu L, Meerloo T. In human epidermis, beta-defensin 2 is packaged in lamellar bodies. Exp Mol Pathol. (2003) 74:180–2.
31. Liu L, Wang L, Jia HP, Zhao C, Heng HH, Schutte BC, et al. Structure and mapping of the human beta-defensin HBD-2 gene and its expression at sites of inflammation. Gene. (1998) 222:237–44.
32. Hertz CJ, Wu Q, Porter EM, Zhang YJ, Weismuller KH, Godowski PJ, et al. Activation of Toll-like receptor 2 on human tracheobronchial epithelial cells induces the antimicrobial peptide human beta defensin-2. J Immunol (Baltimore Md 1950). (2003) 171:6820–6.
33. Wang G. Human antimicrobial peptides and proteins. Pharmaceuticals (Basel Switzerland). (2014) 7:545–94.
34. Matsuzaki K. Membrane permeabilization mechanisms. Adv Exp Med Biol. (2019) 1117:9–16. doi: 10.1007/978-981-13-3588-4_2
35. Bozelli JC Jr., Sasahara ET, Pinto MR, Nakaie CR, Schreier S. Effect of head group and curvature on binding of the antimicrobial peptide tritrpticin to lipid membranes. Chem Phys Lipid. (2012) 165:365–73. doi: 10.1016/j.chemphyslip.2011.12.005
36. Strandberg E, Tiltak D, Ehni S, Wadhwani P, Ulrich AS. Lipid shape is a key factor for membrane interactions of amphipathic helical peptides. Biochim Biophys Acta. (2012) 1818:1764–76.
37. Strandberg E, Zerweck J, Wadhwani P, Ulrich AS. Synergistic insertion of antimicrobial magainin-family peptides in membranes depends on the lipid spontaneous curvature. Biophys J. (2013) 104:L9–11. doi: 10.1016/j.bpj.2013.01.047
38. Afonin S, Glaser RW, Sachse C, Salgado J, Wadhwani P, Ulrich AS. (19)F NMR screening of unrelated antimicrobial peptides shows that membrane interactions are largely governed by lipids. Biochim Biophys Acta. (2014) 1838:2260–8. doi: 10.1016/j.bbamem.2014.03.017
39. Perrin BS Jr., Sodt AJ, Cotten ML, Pastor RW. The curvature induction of surface-bound antimicrobial peptides Piscidin 1 and Piscidin 3 varies with lipid chain length. J Membr Biol. (2015) 248:455–67. doi: 10.1007/s00232-014-9733-1
40. Paterson DJ, Tassieri M, Reboud J, Wilson R, Cooper JM. Lipid topology and electrostatic interactions underpin lytic activity of linear cationic antimicrobial peptides in membranes. Proc Natl Acad Sci USA. (2017) 114:E8324–32. doi: 10.1073/pnas.1704489114
41. Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. (2005) 3:238–50.
42. Fabisiak A, Murawska N, Fichna J. LL-37: cathelicidin-related antimicrobial peptide with pleiotropic activity. Pharmacol Rep. (2016) 68:802–8.
43. Overhage J, Campisano A, Bains M, Torfs EC, Rehm BH, Hancock RE. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect Immun. (2008) 76:4176–82. doi: 10.1128/IAI.00318-08
44. Nagant C, Pitts B, Nazmi K, Vandenbranden M, Bolscher JG, Stewart PS, et al. Identification of peptides derived from the human antimicrobial peptide LL-37 active against biofilms formed by Pseudomonas aeruginosa using a library of truncated fragments. Antimicrob Agents Chemother. (2012) 56:5698–708. doi: 10.1128/AAC.00918-12
45. Koro C, Hellvard A, Delaleu N, Binder V, Scavenius C, Bergum B, et al. Carbamylated LL-37 as a modulator of the immune response. Innate Immun. (2016) 22:218–29. doi: 10.1177/1753425916631404
46. Kai-Larsen Y, Agerberth B. The role of the multifunctional peptide LL-37 in host defense. Front Biosci. (2008) 13:3760–7. doi: 10.2741/2964
47. Sass V, Schneider T, Wilmes M, Korner C, Tossi A, Novikova N, et al. Human beta-defensin 3 inhibits cell wall biosynthesis in Staphylococci. Infect Immun. (2010) 78:2793–800. doi: 10.1128/IAI.00688-09
48. Kanda N, Kamata M, Tada Y, Ishikawa T, Sato S, Watanabe S. Human beta-defensin-2 enhances IFN-gamma and IL-10 production and suppresses IL-17 production in T cells. J Leukoc Biol. (2011) 89:935–44. doi: 10.1189/jlb.0111004
49. Estrela AB, Rohde M, Gutierrez MG, Molinari G, Abraham WR. Human beta-defensin 2 induces extracellular accumulation of adenosine in Escherichia coli. Antimicrob Agents Chemother. (2013) 57:4387–93. doi: 10.1128/AAC.00820-13
50. Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, et al. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science (N. Y.). (1999) 286:525–8.
51. Spudy B, Sonnichsen FD, Waetzig GH, Grotzinger J, Jung S. Identification of structural traits that increase the antimicrobial activity of a chimeric peptide of human beta-defensins 2 and 3. Biochem Biophys Res Commun. (2012) 427:207–11. doi: 10.1016/j.bbrc.2012.09.052
52. Hoover DM, Wu Z, Tucker K, Lu W, Lubkowski J. Antimicrobial characterization of human beta-defensin 3 derivatives. Antimicrob Agents Chemother. (2003) 47:2804–9.
53. Dhople V, Krukemeyer A, Ramamoorthy A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim Biophys Acta. (2006) 1758:1499–512.
54. Silva ON, Porto WF, Ribeiro SM, Batista I, Franco OL. Host-defense peptides and their potential use as biomarkers in human diseases. Drug Discov Today. (2018) 23:1666–71. doi: 10.1016/j.drudis.2018.05.024
55. Galdiero E, Lombardi L, Falanga A, Libralato G, Guida M, Carotenuto R. Biofilms: novel strategies based on antimicrobial peptides. Pharmaceutics. (2019) 11:322.
56. Wu Z, Hoover DM, Yang D, Boulegue C, Santamaria F, Oppenheim JJ, et al. Engineering disulfide bridges to dissect antimicrobial and chemotactic activities of human beta-defensin 3. Proc Natl Acad Sci USA. (2003) 100:8880–5.
57. Wei G, de Leeuw E, Pazgier M, Yuan W, Zou G, Wang J, et al. Through the looking glass, mechanistic insights from enantiomeric human defensins. J Biol Chem. (2009) 284:29180–92. doi: 10.1074/jbc.M109.018085
58. Merritt JH, Kadouri DE, O’Toole GA. Growing and analyzing static biofilms. Curr Protoc Microbiol. (2005) Chapter 1:Unit 1B.1. doi: 10.1002/9780471729259.mc01b01s00
59. Shiloh MU, Ruan J, Nathan C. Evaluation of bacterial survival and phagocyte function with a fluorescence-based microplate assay. Infect Immun. (1997) 65:3193–8.
60. Martinez JG, Waldon M, Huang Q, Alvarez S, Oren A, Sandoval N, et al. Membrane-targeted synergistic activity of docosahexaenoic acid and lysozyme against Pseudomonas aeruginosa. Biochem J. (2009) 419:193–200. doi: 10.1042/BJ20081505
61. Abu EA, Su S, Sallans L, Boissy RE, Greatens A, Heineman WR, et al. Cyclic voltammetric, fluorescence and biological analysis of purified aeruginosin A, a secreted red pigment of Pseudomonas aeruginosa PAO1. Microbiology. (2013) 159:1736–47. doi: 10.1099/mic.0.065235-0
62. Shityakov S, Forster C. In silico predictive model to determine vector-mediated transport properties for the blood-brain barrier choline transporter. Adv Appl Bioinform Chem. (2014) 7:23–36. doi: 10.2147/AABC.S63749
63. Park M, Yoo G, Bong JH, Jose J, Kang MJ, Pyun JC. Isolation and characterization of the outer membrane of Escherichia coli with autodisplayed Z-domains. Biochim Biophys Acta. (2015) 1848:842–7. doi: 10.1016/j.bbamem.2014.12.011
64. Chao Y, Zhang T. Optimization of fixation methods for observation of bacterial cell morphology and surface ultrastructures by atomic force microscopy. Appl Microbiol Biotechnol. (2011) 92:381–92. doi: 10.1007/s00253-011-3551-5
65. Dufrene YF, Ando T, Garcia R, Alsteens D, Martinez-Martin D, Engel A, et al. Imaging modes of atomic force microscopy for application in molecular and cell biology. Nat Nanotechnol. (2017) 12:295–307. doi: 10.1038/nnano.2017.45
66. Rohrl J, Yang D, Oppenheim JJ, Hehlgans T. Human beta-defensin 2 and 3 and their mouse orthologs induce chemotaxis through interaction with CCR2. J Immunol (Baltimore Md 1950). (2010) 184:6688–94. doi: 10.4049/jimmunol.0903984
67. Rohrl J, Yang D, Oppenheim JJ, Hehlgans T. Specific binding and chemotactic activity of mBD4 and its functional orthologue hBD2 to CCR6-expressing cells. J Biol Chem. (2010) 285:7028–34. doi: 10.1074/jbc.M109.091090
68. Hoover DM, Rajashankar KR, Blumenthal R, Puri A, Oppenheim JJ, Chertov O, et al. The structure of human beta-defensin-2 shows evidence of higher order oligomerization. J Biol Chem. (2000) 275:32911–8.
69. Trivedi MV, Laurence JS, Siahaan TJ. The role of thiols and disulfides on protein stability. Curr Protein Pept Sci. (2009) 10:614–25.
70. Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem. (1997) 48:545–600.
71. Jimenez PN, Koch G, Thompson JA, Xavier KB, Cool RH, Quax WJ. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol Mol Biol Rev. (2012) 76:46–65.
72. Zou Y, Nair SK. Molecular basis for the recognition of structurally distinct autoinducer mimics by the Pseudomonas aeruginosa LasR quorum-sensing signaling receptor. Chem Biol. (2009) 16:961–70. doi: 10.1016/j.chembiol.2009.09.001
73. Le CF, Yusof MY, Hassan MA, Lee VS, Isa DM, Sekaran SD. In vivo efficacy and molecular docking of designed peptide that exhibits potent antipneumococcal activity and synergises in combination with penicillin. Sci Rep. (2015) 5:11886. doi: 10.1038/srep11886
74. Moore JD, Rossi FM, Welsh MA, Nyffeler KE, Blackwell HE. A comparative analysis of synthetic quorum sensing modulators in Pseudomonas aeruginosa: new insights into mechanism, active efflux susceptibility, phenotypic response, and next-generation ligand design. J Am Chem Soc. (2015) 137:14626–39. doi: 10.1021/jacs.5b06728
75. Choi IJ, Rhee CS, Lee CH, Kim DY. Effect of allergic rhinitis on the expression of human beta-defensin 2 in tonsils. Ann Allergy Asthma Immunol. (2013) 110:178–83. doi: 10.1016/j.anai.2012.12.020
76. Hiratsuka T, Nakazato M, Date Y, Ashitani J, Minematsu T, Chino N, et al. Identification of human beta-defensin-2 in respiratory tract and plasma and its increase in bacterial pneumonia. Biochem Biophys Res Commun. (1998) 249:943–7.
77. Hiratsuka T, Mukae H, Iiboshi H, Ashitani J, Nabeshima K, Minematsu T, et al. Increased concentrations of human beta-defensins in plasma and bronchoalveolar lavage fluid of patients with diffuse panbronchiolitis. Thorax. (2003) 58:425–30.
78. Chevalier S, Bouffartigues E, Bodilis J, Maillot O, Lesouhaitier O, Feuilloley MGJ, et al. Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol Rev. (2017) 41:698–722. doi: 10.1093/femsre/fux020
79. Hancock RE, Siehnel R, Martin N. Outer membrane proteins of Pseudomonas. Mol Microbiol. (1990) 4:1069–75.
80. Goeke JE, Kist S, Schubert S, Hickel R, Huth KC, Kollmuss M. Sensitivity of caries pathogens to antimicrobial peptides related to caries risk. Clin Oral Investig. (2018) 22:2519–25. doi: 10.1007/s00784-018-2348-7
81. O’Neil DA, Cole SP, Martin-Porter E, Housley MP, Liu L, Ganz T, et al. Regulation of human beta-defensins by gastric epithelial cells in response to infection with Helicobacter pylori or stimulation with interleukin-1. Infect Immun. (2000) 68:5412–5.
82. Crabbe A, Ostyn L, Staelens S, Rigauts C, Risseeuw M, Dhaenens M, et al. Host metabolites stimulate the bacterial proton motive force to enhance the activity of aminoglycoside antibiotics. PLoS Pathog. (2019) 15:e1007697. doi: 10.1371/journal.ppat.1007697
83. Scherer KM, Spille JH, Sahl HG, Grein F, Kubitscheck U. The lantibiotic nisin induces lipid II aggregation, causing membrane instability and vesicle budding. Biophys J. (2015) 108:1114–24. doi: 10.1016/j.bpj.2015.01.020
84. van Kraaij C, Breukink E, Noordermeer MA, Demel RA, Siezen RJ, Kuipers OP, et al. Pore formation by nisin involves translocation of its C-terminal part across the membrane. Biochemistry. (1998) 37:16033–40.
85. Winkowski K, Ludescher RD, Montville TJ. Physiochemical characterization of the nisin-membrane interaction with liposomes derived from Listeria monocytogenes. Appl Environ Microbiol. (1996) 62:323–7.
86. Garcera MJ, Elferink MG, Driessen AJ, Konings WN. In vitro pore-forming activity of the lantibiotic nisin. Role of protonmotive force and lipid composition. Eur J Biochemi. (1993) 212:417–22.
87. Bierbaum G, Sahl HG. Lantibiotics: mode of action, biosynthesis and bioengineering. Curr Pharm Biotechnol. (2009) 10:2–18.
88. de Leeuw E, Li C, Zeng P, Li C, Diepeveen-de Buin M, Lu WY, et al. Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett. (2010) 584:1543–8. doi: 10.1016/j.febslet.2010.03.004
89. Schibli DJ, Hunter HN, Aseyev V, Starner TD, Wiencek JM, McCray PB Jr. et al. The solution structures of the human beta-defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus. J Biol Chem. (2002) 277:8279–89.
90. Chan C, Burrows LL, Deber CM. Helix induction in antimicrobial peptides by alginate in biofilms. J Biol Chem. (2004) 279:38749–54.
91. Perez-Cruz C, Delgado L, Lopez-Iglesias C, Mercade E. Outer-inner membrane vesicles naturally secreted by gram-negative pathogenic bacteria. PLoS One. (2015) 10:e0116896. doi: 10.1371/journal.pone.0116896
92. Mathew B, Nagaraj R. Antimicrobial activity of human alpha-defensin 6 analogs: insights into the physico-chemical reasons behind weak bactericidal activity of HD6 in vitro. J Pept Sci. (2015) 21:811–8. doi: 10.1002/psc.2821
93. Chandrababu KB, Ho B, Yang D. Structure, dynamics, and activity of an all-cysteine mutated human beta defensin-3 peptide analogue. Biochemistry. (2009) 48:6052–61. doi: 10.1021/bi900154f
94. Krishnakumari V, Nagaraj R. Interaction of antibacterial peptides spanning the carboxy-terminal region of human beta-defensins 1-3 with phospholipids at the air-water interface and inner membrane of E. coli. Peptides. (2008) 29:7–14.
95. Mashburn-Warren L, Howe J, Garidel P, Richter W, Steiniger F, Roessle M, et al. Interaction of quorum signals with outer membrane lipids: insights into prokaryotic membrane vesicle formation. Mol Microbiol. (2008) 69:491–502.
96. McCready AR, Paczkowski JE, Henke BR, Bassler BL. Structural determinants driving homoserine lactone ligand selection in the Pseudomonas aeruginosa LasR quorum-sensing receptor. Proc Natl Acad Sci USA. (2019) 116:245–54. doi: 10.1073/pnas.1817239116
97. Tavender TJ, Halliday NM, Hardie KR, Winzer K. LuxS-independent formation of AI-2 from ribulose-5-phosphate. BMC Microbiol. (2008) 8:98. doi: 10.1186/1471-2180-8-98
98. Li H, Li X, Wang Z, Fu Y, Ai Q, Dong Y, et al. Autoinducer-2 regulates Pseudomonas aeruginosa PAO1 biofilm formation and virulence production in a dose-dependent manner. BMC Microbiol. (2015) 15:192. doi: 10.1186/s12866-015-0529-y
100. Duan K, Dammel C, Stein J, Rabin H, Surette MG. Modulation of Pseudomonas aeruginosa gene expression by host microflora through interspecies communication. Mol Microbiol. (2003) 50:1477–91.
101. Wei Q, Ma LZ. Biofilm matrix and its regulation in Pseudomonas aeruginosa. Int J Mol Sci. (2013) 14:20983–1005. doi: 10.3390/ijms141020983
102. Mikkelsen H, Sivaneson M, Filloux A. Key two-component regulatory systems that control biofilm formation in Pseudomonas aeruginosa. Environ Microbiol. (2011) 13:1666–81. doi: 10.1111/j.1462-2920.2011.02495.x
103. Ha DG, O’Toole GA. c-di-GMP and its effects on biofilm formation and dispersion: a Pseudomonas Aeruginosa review. Microbiol Spectr. (2015) 3:MB-0003-2014. doi: 10.1128/microbiolspec.MB-0003-2014
104. de la Fuente-Nunez C, Reffuveille F, Haney EF, Straus SK, Hancock RE. Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog. (2014) 10:e1004152. doi: 10.1038/srep43321
105. Steinstraesser L, Hirsch T, Schulte M, Kueckelhaus M, Jacobsen F, Mersch EA, et al. Innate defense regulator peptide 1018 in wound healing and wound infection. PLoS One. (2012) 7:e39373. doi: 10.1371/journal.pone.0039373
106. Byng GS, Eustice DC, Jensen RA. Biosynthesis of phenazine pigments in mutant and wild-type cultures of Pseudomonas aeruginosa. J Bacteriol. (1979) 138:846–52.
107. Bala A, Kumar L, Chhibber S, Harjai K. Augmentation of virulence related traits of pqs mutants by Pseudomonas quinolone signal through membrane vesicles. J Basic Microbiol. (2015) 55:566–78. doi: 10.1002/jobm.201400377
108. Lo YL, Shen L, Chang CH, Bhuwan M, Chiu CH, Chang HY. Regulation of motility and phenazine pigment production by FliA is cyclic-di-GMP dependent in Pseudomonas aeruginosa PAO1. PLoS One. (2016) 11:e0155397. doi: 10.1371/journal.pone.0155397
109. Rashid R, Veleba M, Kline KA. Focal targeting of the bacterial envelope by antimicrobial peptides. Front Cell Dev Biol. (2016) 4:55. doi: 10.3389/fcell.2016.00055
110. Wenzel M, Chiriac AI, Otto A, Zweytick D, May C, Schumacher C, et al. Small cationic antimicrobial peptides delocalize peripheral membrane proteins. Proc Natl Acad Sci USA. (2014) 111:E1409–18. doi: 10.1073/pnas.1319900111
111. Vetterli SU, Zerbe K, Muller M, Urfer M, Mondal M, Wang SY, et al. Thanatin targets the intermembrane protein complex required for lipopolysaccharide transport in Escherichia coli. Sci Adv. (2018) 4:eaau2634. doi: 10.1126/sciadv.aau2634
112. Kandaswamy K, Liew TH, Wang CY, Huston-Warren E, Meyer-Hoffert U, Hultenby K, et al. Focal targeting by human beta-defensin 2 disrupts localized virulence factor assembly sites in Enterococcus faecalis. Proc Natl Acad Sci USA. (2013) 110:20230–5. doi: 10.1073/pnas.1319066110
113. Nielsen HV, Flores-Mireles AL, Kau AL, Kline KA, Pinkner JS, Neiers F, et al. Pilin and sortase residues critical for endocarditis- and biofilm-associated pilus biogenesis in Enterococcus faecalis. J Bacteriol. (2013) 195:4484–95. doi: 10.1128/JB.00451-13
114. Ma Q, Zhai Y, Schneider JC, Ramseier TM, Saier MHJr. Protein secretion systems of Pseudomonas aeruginosa and P fluorescens. Biochim Biophys Acta. (2003) 1611:223–33.
115. Tielker D, Hacker S, Loris R, Strathmann M, Wingender J, Wilhelm S, et al. Pseudomonas aeruginosa lectin LecB is located in the outer membrane and is involved in biofilm formation. Microbiology. (2005) 151:1313–23. doi: 10.1099/mic.0.27701-0
116. Cassin EK, Tseng BS. Pushing beyond the envelope: the potential roles of OprF in Pseudomonas aeruginosa biofilm formation and pathogenicity. J Bacteriol. (2019) 201:e00050-19. doi: 10.1128/JB.00050-19
117. Li A, Lee PY, Ho B, Ding JL, Lim CT. Atomic force microscopy study of the antimicrobial action of Sushi peptides on Gram negative bacteria. Biochim Biophys Acta. (2007) 1768:411–8.
118. Schooling SR, Hubley A, Beveridge TJ. Interactions of DNA with biofilm-derived membrane vesicles. J Bacteriol. (2009) 191:4097–102. doi: 10.1128/JB.00717-08
119. Parducho KMR. Airway Peptides and Lipids- Effects on Pseudomonas aeruginosa Morphology, Biofilm Formation, and Quorum Sensing, Chemistry & Biochemistry. Los Angeles, CA: California State University Chancellor’s Office (2018).
120. Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. (1982) 157:105–32.
Keywords: airways, antimicrobial peptides, biofilm, cystic fibrosis, epithelial cells, innate immunity, mucosa, Pseudomonas aeruginosa
Citation: Parducho KR, Beadell B, Ybarra TK, Bush M, Escalera E, Trejos AT, Chieng A, Mendez M, Anderson C, Park H, Wang Y, Lu W and Porter E (2020) The Antimicrobial Peptide Human Beta-Defensin 2 Inhibits Biofilm Production of Pseudomonas aeruginosa Without Compromising Metabolic Activity. Front. Immunol. 11:805. doi: 10.3389/fimmu.2020.00805
Received: 05 November 2019; Accepted: 08 April 2020;
Published: 08 May 2020.
Edited by:
Charles Lee Bevins, University of California, Davis, United StatesReviewed by:
Mark Ambrose, University of Tasmania, AustraliaRicardo Oropeza, National Autonomous University of Mexico, Mexico
Copyright © 2020 Parducho, Beadell, Ybarra, Bush, Escalera, Trejos, Chieng, Mendez, Anderson, Park, Wang, Lu and Porter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edith Porter, ZXBvcnRlckBjYWxzdGF0ZWxhLmVkdQ==