 Anne-Sophie Laramée1,2
Anne-Sophie Laramée1,2 Hannah Raczkowski1,2Peng Shao1,2Carolina Batista1,2Devanshi Shukla1Li Xu1,2
Hannah Raczkowski1,2Peng Shao1,2Carolina Batista1,2Devanshi Shukla1Li Xu1,2 S. M. Mansour Haeryfar1,3Yodit Tesfagiorgis1
S. M. Mansour Haeryfar1,3Yodit Tesfagiorgis1 Steven Kerfoot1
Steven Kerfoot1 Rodney DeKoter1,2*
Rodney DeKoter1,2*- 1Department of Microbiology and Immunology, Center for Human Immunology, Schulich School of Medicine and Dentistry, Western University, London, ON, Canada
- 2Division of Genetics and Development, Children’s Health Research Institute, Lawson Research Institute, London, ON, Canada
- 3Division of Clinical Immunology and Allergy, Department of Medicine, Western University, London, ON, Canada
Generation of specific antibodies during an immune response to infection or vaccination depends on the ability to rapidly and accurately select clones of antibody-secreting B lymphocytes for expansion. Antigen-specific B cell clones undergo the cell fate decision to differentiate into antibody-secreting plasma cells, memory B cells, or germinal center B cells. The E26-transformation-specific (ETS) transcription factors Spi-B and Spi-C are important regulators of B cell development and function. Spi-B is expressed throughout B cell development and is downregulated upon plasma cell differentiation. Spi-C is highly related to Spi-B and has similar DNA-binding specificity. Heterozygosity for Spic rescues B cell development and B cell proliferation defects observed in Spi-B knockout mice. In this study, we show that heterozygosity for Spic rescued defective IgG1 secondary antibody responses in Spib–/– mice. Plasma cell differentiation was accelerated in Spib–/– B cells. Gene expression, ChIP-seq, and reporter gene analysis showed that Spi-B and Spi-C differentially regulated Bach2, encoding a key regulator of plasma cell and memory B cell differentiation. These results suggest that Spi-B and Spi-C oppose the function of one another to regulate B cell differentiation and function.
Introduction
Naïve B cells possess the capacity to differentiate into heterogeneous cellular subsets that promote the resolution of both current and future infections. These subsets include Plasma Cells (PC), memory B cells, and germinal center (GC) B cells (1, 2). The formation of GCs by GC B cells is crucial for the generation of robust humoral responses to T cell-dependent antigens, as this specialized microenvironment allows B cells to undergo class-switch recombination and affinity maturation (3, 4). Within the dark zone of the GC, rapidly cycling B cells accumulate point mutations in the variable regions of BCR-encoding immunoglobulin genes (5). This stochastic process, known as somatic hypermutation, diversifies the range of antigenic affinities exhibited by GC B cells (6).
The molecular determinants responsible for regulating B cell fate decisions following GC entry have become the focus of intense scrutiny. The transcription factors Bcl-6 and Blimp-1 (encoded by Bcl6 and Prdm1, respectively) exert opposing activities on B cell differentiation. Blimp-1 coordinates the silencing of B cell gene expression patterns by repressing Pax5 (7, 8). In contrast, Bcl6 promotes the generation and maintenance of GCs by repressing Prdm1 (9, 10). Interferon regulatory factors 4 (IRF4) and 8 (IRF8) govern the fate of activated B cells in a concentration-dependent manner (11). High intracellular abundance of IRF4 (paired with low levels of IRF8) promote the generation of plasmablasts and PCs, while high IRF8 and correspondingly low IRF4 expression promote the GC fate (11). Therefore, transcription factors regulate PC differentiation versus GC differentiation through networks involving mutually cross-antagonistic activity.
Spi-C (encoded by Spic), Spi-B (encoded by Spib) and PU.1 (encoded by Spi1) are related E26-transformation-specific (ETS) transcription factors that are critically important at multiple stages of B cell development (12, 13). PU.1 and Spi-B function as complementary transcriptional activators of genes involved in B cell development and function (14–16). PU.1 and Spi-B expression are required for secondary Ig responses in mice (17, 18). In contrast, down-regulation of PU.1 and Spi-B expression is required for PC differentiation (19, 20). Forced expression of Spi-B has been shown to inhibit PC differentiation (20). Spi-C is expressed in pre-B and mature B cells as well as in red pulp macrophages (21–23). Spi-C functions primarily as a transcriptional repressor, and may antagonize the activities of PU.1 and Spi-B (24–27). Deletion of one allele of Spic was found to partially rescue B cell development, and proliferation of cultured Spib–/– B cells in response to LPS or anti-IgM (28). However, the role of Spi-C in regulating B cell function including antibody responses has not been investigated.
Bach1 and Bach2 are related basic region leucine zipper proteins that function as transcriptional repressors (29). Bach1 and Bach2 de-repress target genes upon interaction with heme (30). Bach1 is highly expressed in the myeloid lineage, while Bach2 is expressed in the B cell and T cell lineages (31). Bach2 is required for the GC and memory B cell fates (32, 33), while down-regulation of Bach2 is required for PC differentiation (34, 35). Bach1 and Bach2 transcriptionally repress the myeloid gene program in macrophages and lymphocytes (36, 37). Bach1 represses Spic transcription in myeloid cells (38). De-repression of Spic transcription by heme-induced Bach1 degradation is required for differentiation into red pulp macrophages (38). The Heme-Bach1-Spi-C pathway has emerged as an paradigm for how an external signal can instruct lineage cell fate decisions through a cell type specific transcription factor (21, 38).
In this study, we show that deletion of one allele of Spic rescued IgG1 secondary antibody responses in Spib–/– mice. Differentiation of Spib–/– B cells into plasmablasts was accelerated in culture. Gene expression and ChIP-seq analysis showed that Spi-B and Spi-C differentially regulated the target gene Bach2 that is a key regulator of secondary antibody responses and PC differentiation. These results suggest that Spi-C is a negative regulator of Spi-B activity, and that both proteins are important regulators of B cell fate decisions.
Materials and Methods
Mice
Spib–/– and Spib–/–Spic + /− mice were maintained as previously described (28). C57BL/6 (WT) mice were purchased from Charles River Laboratories (Pointe-Claire, QC, Canada). All animals were housed under specific pathogen-free conditions at the West Valley facility (London, ON, Canada), and were monitored in accordance with an animal use protocol approved by the Western University Council on Animal Care and the Animal Care Committee. Genotyping was performed by PCR, as previously described (12, 21). All experiments performed in this study used 6–10 week old mice. Mice were immunized i.p. with 100 μg of 4-Hydroxy-3-nitrophenylacetyl hapten (NP)-conjugated ovalbumin or keyhole limpet hemocyanin (KLH) (conjugation ratios of 15:1 NP:OVA, 8:1 NP:KLH) (Biosearch Technologies, Novato, CA, United States) adjuvanted with 50% (vol/vol) of ImjectTM alum (ThermoFisher Scientific, Rochester, NY, United States). For experiments involving secondary responses, mice were re-immunized by i.p. injection on day 30 following the primary challenge with a boosting dose identical to that of the prime.
B Cell Enrichment and Culture
Splenocytes were enriched for B cells by negative selection using the VarioMACS magnet, LD depletion columns, streptavidin microbeads (Miltenyi Biotec, Germany) and biotin-conjugated mouse anti-CD43 (S7). Enriched B cells were stained using the CellTrace Violet Cell Proliferation Kit (ThermoFisher) at a concentration of 1.5 μM. Enriched B cells or murine 38B9 cells were cultured in were cultured in complete RPMI-1640 containing 10% fetal bovine serum (Wisent, St. Bruno, QC, Canada), 5 × 10–5 M β-mercaptoenthanol (Sigma-Aldrich, St. Louis, MO, United States), 0.01 M HEPES (Sigma-Aldrich), and 1X penicillin/streptomycin/L-glutamine (Wisent). WEHI-279 cells were cultured in complete DMEM medium containing 4.5 g/L glucose (Wisent). Cells were maintained in 5% CO2 at 37°C. B cells were stimulated with LPS 0111:B4 (10 μg/ml, List Biological Laboratories, Campbell, CA, United States), 100 ng/ml CD40L (R&D Systems, Minneapolis, MI, United States), 10 ng/ml Interleukin-4 (R&D Systems), and/or 10 ng/ml Interleukin-5 (R&D Systems). Cultured plasmablasts were analyzed by flow cytometry on day 3, 4, or 5 of culture.
Luciferase Assays
Bach2 region of interest 1 (ROI 1) was PCR amplified from murine genomic DNA using Q5 high-fidelity DNA polymerase (New England Biolabs, Ipswich, MA, United States). PCR products were cloned using the StrataClone Blunt PCR cloning kit (Agilent Technologies, La Jolla, CA, United States). ROI 1 was ligated in the forward orientation into the KpnI/SacI sites of the luciferase reporter pGL3-promoter (Promega, Madison, WI, United States) and confirmed by sequencing. Mutation of the predicted ROI 1 ETS site (GGAA → GGCC) was performed using the Q5 site directed mutagenesis kit (New England Biolabs). pRL-TK (Renilla luciferase), pGL3-basic, pGL3-promoter, pGL3-promoter-ROI 1, and pGL3-promoter ROI 1 mutant vectors were transfected into 4 × 106 WEHI-279 cells by electroporation using a Gene Pulser II with capacitance extender at 220V and 950 mF (Bio-Rad, Mississauga, ON, United States). Luciferase activity was measured 24 h after transfection using the Dual Luciferase Assay Kit (Promega). Luminescence was determined using a Cytation 5 plate reader (BioTek, Winooski, VT, United States).
ELISpot Assays
Single-cell suspensions of splenocytes were red blood cell-depleted using ammonium-chloride-potassium (ACK) lysis, washed in MACS buffer and counted using the Moxi Z Mini Automated Cell Counter (Orflo, Ketchum, ID, United States). Splenocytes were serially diluted and incubated for 5 h at 37°C (5% CO2) in 10% FBS-containing RPMI 1640X. Alkaline phosphatase-conjugated goat anti-mouse IgM (Mabtech), IgG1, IgG2b, and IgG2c (Jackson Immunoresearch, Westgrove, PA, United States) were incubated overnight in corresponding wells. 5-bromo-4-chloro-3-indolyl-phosphate (BCIP, Sigma-Aldrich) in 3% low-melt agarose gel (NuSieveTM GTGTM Agarose, Lonza, Basel, Switzerland) was used to develop spots. Plates were imaged using the ImmunoSpot® S6 Analyzer and counted using the ImmunoSpot® software (Cellular Technology Limited, Cleveland, OH). Spot counts from triplicate wells in individual mice were averaged, then plotted against the corresponding dilution for each antibody. Data points were calculated from logarithmic functions, obtained by plotting mean values of spots from triplicate wells against corresponding dilutions for each mouse. Logarithmic regressions were performed in Excel, and curves of best fit were used to calculate adjusted frequencies of ASCs per 1 × 106 cells.
Flow Cytometry
Anti-Fc-γ receptor blocking was performed using purified anti-CD16/CD32 (Mouse BD Fc Block). Cell-surface staining was performed using the following Abs, purchased from eBioscience (San Diego, CA, United States), BD Bioscience (Franklin Lakes, NJ, United States) or Biolegend (San Diego, CA, United States): Brilliant Violet 421 and allophycocyanin (APC)-conjugated anti-CD45R/B220 (clone Ra3-6B2); biotin, phycoerythrin (PE), or brilliant violet 421 (BV421)-conjugated anti-CD138 (clone 281-2); PE, fluorescein (FITC), or Alexa Fluor 488-conjugated CD19 (6D5); PE-conjugated anti-CD38 (clone 90); PE-Cy5-conjugated anti-CD4 (clone RM4-5); PE-Cy7 anti-CD95 (clone Jo2); and PE-conjugated streptavidin. Live/dead discrimination was performed using fixable viability dye eFluor 506 (eBioScience). Analysis was performed using BD FACSCanto or LSR II instruments (BD Immunocytometry Systems, San Jose, CA, United States). Cell sorting was performed using a FACSAria III instrument (BD Immunocytometry Systems) at the London, Ontario Regional Flow Cytometry Facility. Analysis was performed using FlowJo 10.4 (TreeStar, Ashland, OR, United States).
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Splenic B cells were enriched from WT, Spib–/– and Spib–/–Spic + /− mice and cultured in complete RPMI supplemented with IL-4, IL-5, and CD40L for 4 days. Cultured B cells were enriched into CD138+ or CD138– fractions using biotinylated anti-CD138 (281-2) and the Miltenyi system (Miltenyi Biotec, Germany). Total RNA was extracted using TRIzol (ThermoFisher) or the RNeasy Mini Prep Kit (Qiagen, Hilden, Germany). Following cDNA synthesis (iScript cDNA Synthesis Kit, Bio-Rad), RT-qPCR analysis was conducted using the SensiFAST SYBR No-ROX Kit (Bioline, Singapore) on the QuantStudio 5 Real-Time PCR System (ThermoFisher). Analyses were conducted in duplicates, with relative expression of target genes normalized to Tata-binding protein (Tbp), and calculated as fold change using the comparative threshold cycle [2(–ΔΔCT)] method (39). The selection of Tbp as a reference gene was carried out on the basis of its relative stability and high expression, by re-analysis of previously published RNA-seq data (GEO accession code: GSE60927) (40), in which the variance in log2FPKM values from sorted FO B cells, GC B cells, plasmablast and PC subsets was compared. Amplification efficiencies were calculated for each primer pair (Supplementary Table S1) using calibration curves generated by triplicate doubling dilutions of total splenocyte cDNA. Primer pairs with efficiencies ranging from 90 to 110% were used in the study.
Production of Retrovirus and Primary B Cell Transduction
MIG-3XFLAG-SpiB and MIG-3XFLAG-SpiC retroviral vectors (15) were packaged by transient transfection of Platinum-E (Plat-E) retroviral packaging cells using polyethyleminine (PEIpro, PolyPlus, Illkirch, France) (41). Plat-E supernatant containing viral particles was harvested after 48 h, and transfection efficiency was analyzed by flow cytometry. Primary B cells were stimulated in CD40L+IL-4+IL-5 (R&D Systems) overnight. Transduction of stimulated, enriched B cells was performed by centrifugal infection at 3000 × g for 2 h at 32°C. Following transduction, primary B cells were cultured for 3 days in complete RPMI (Wisent) containing CD40L+IL-4+IL-5 (R&D Systems), as described above.
Chromatin Immunoprecipitation
Chromatin was prepared from pellets of 1 × 106 transduced, cultured B cells as described in (12). Cross-linking was performed using 1% formaldehyde (Millipore-Sigma, Darmstadt, Germany) and halted using glycine. Pellets were flash-frozen in liquid nitrogen prior to sonication. Thawed pellets were lysed in lysis buffer supplemented with Halt Protease Inhibitor (ThermoFisher Scientific, Rochester, NY, United States), and sonicated for 25 cycles using the Bioruptor UCD-300 (Diagenode, Sparta, NJ, United States). Immunoprecipitation of FLAG-bound chromatin was performed using anti-FLAG M2 magnetic beads (MilliporeSigma, Darmstadt, Germany). Eluted DNA was purified with QIAquick PCR Purification Kit (Qiagen, Hilden, Germany). qPCR on purified DNA was performed as described above, using primers shown in Supplementary Table S1. Threshold cycle values were used to calculate enrichment, represented as percent input. ROIs were identified by analysis of published ChIP-seq data (GEO accession code: GSE58128) (14). ChIP-seq was performed as described in Solomon et al. (14). Quality control for chromatin enriched by anti-FLAG antibody was performed by qPCR analysis for association with the IgH intronic enhancer. Sequencing was performed by Genome Quebec on two independent replicates of anti-FLAG ChIP chromatin as well as on input chromatin DNA.
Bioinformatic and Statistical Analysis
ChIP-seq analysis was performed using the Galaxy Suite of bioinformatic tools (42). Bowtie2 was used to merge the two experimental samples and align reads to mouse genome Mm9 (43). Peaks were called using MACS (44) with the input as control, using a tag size of 70, a band width of 300, and a p-value cutoff of 1e–5. Peak-to-gene association was called using Cistrome, with a 15,000 bp cutoff (45). Gene Ontology analysis was performed using DAVID (46). Motif analysis was performed using MEME Suite 4.11.0 (47). The sequence of the Bach2 locus was analyzed for multi-species conservation analysis (PhastCons46wayPlacental) using ORCAtk (Version 1.0.0), with the following settings: minimum conservation 70%, minimum conserved region 20. ChIP-seq data is available from the Gene Expression Omnibus accession GSE115593. Statistical analyses were performed using Prism 8.2 (GraphPad, San Diego, CA, United States) using specific tests described in figure legends.
Results
Heterozygosity for Spi-C Rescues Spib–/– Defect in Secondary T Cell-Dependent B Cell Responses
Mice homozygous for a null allele of Spib on a BALB/c background exhibit reduced titers of anti-nitrophenyl (NP) antibodies following secondary challenge with nitrophenyl (NP) conjugated to keyhole limpet hemocyanin (KLH) (18). We previously reported that heterozygosity for Spic rescued B cell development and proliferation of Spib–/– B cells in response to TLR stimulation. We sought to determine whether heterozygosity for Spic could rescue the impairment in antibody-secreting cell (ASC) frequencies in Spib–/– mice on a C57BL/6 background immunized with NP conjugated to ovalbumin (OVA). Spib–/– Spic + /− mice or Spic + /− mice could not be included due to the high frequency of embryonic lethality in these mice (28). WT, Spib–/–, and Spib–/–Spic + /− mice were immunized intraperitoneally with a priming dose of alum-precipitated NP-OVA. An identical challenge was administered at day 30 post-immunization, and splenic frequencies of ASCs were assessed by Enzyme-Linked ImmunoSpot (ELISpot) at day 37 post-immunization. For Spib–/– and Spib–/–Spic + /− immunized littermates, we observed IgG1 responses of greater magnitude than IgM, and very low frequencies of B cells of other isotypes, which was expected based on the known dominance of IgG1 utilizing VH186.2-DFL16.1-JH2 and l1 in anti-NP responses (48) (Figure 1A and Supplementary Figures S1A–C). There was a significant reduction in frequencies of NP-reactive, IgG1-forming or IgM-forming ASCs in immunized Spib–/– mice relative to WT controls (Figure 1B and Supplementary Figure S1A). Heterozygosity for Spic in Spib–/–Spic + /− mice increased IgG1 ASC frequencies to WT levels (Figure 1B), but did not increase IgM ASC frequencies.
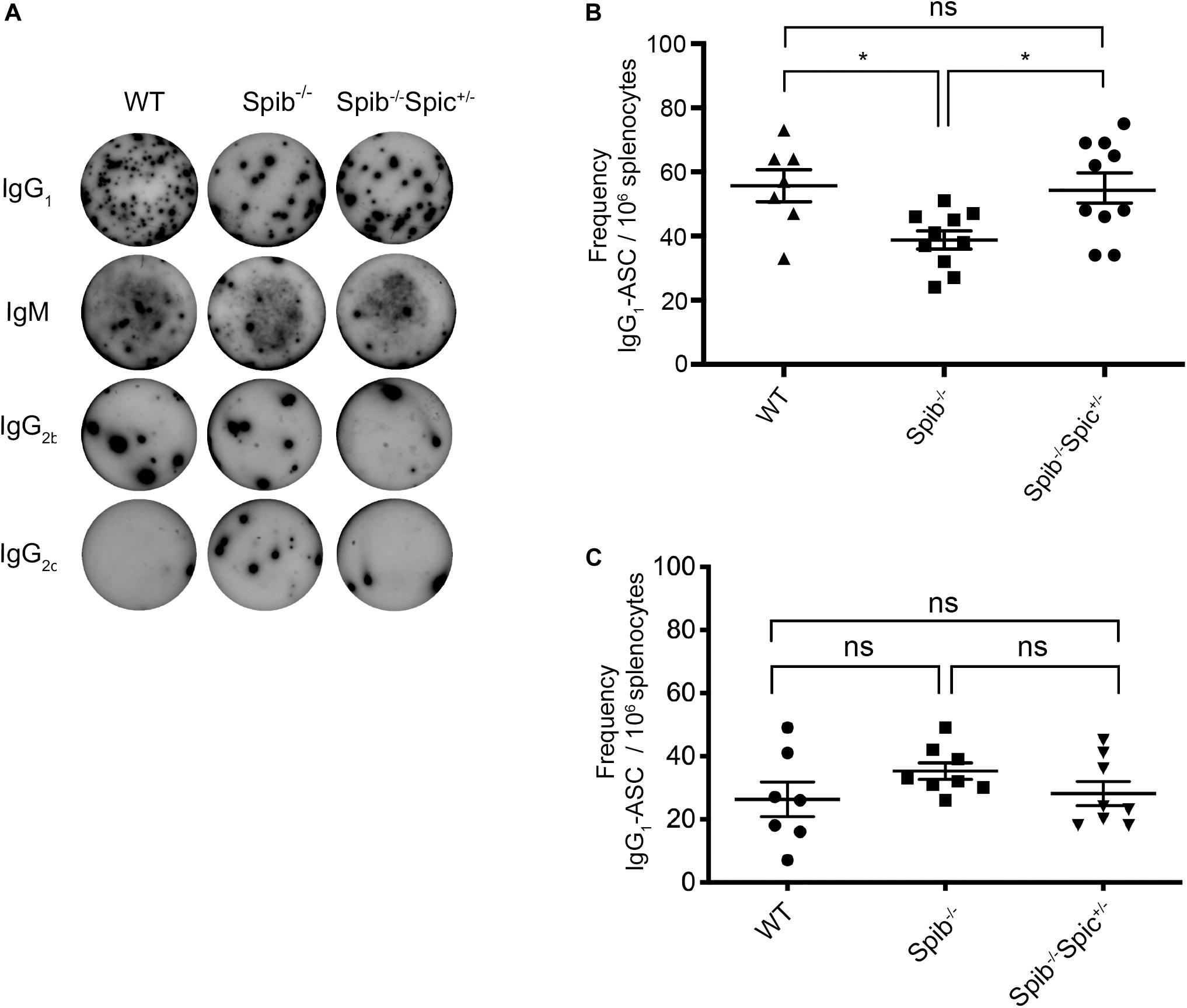
Figure 1. Differential regulation of secondary antibody responses by Spi-B and Spi-C. (A) Representative ELISpot wells for detection of IgG1, IgM, IgG2b, and IgG2c. (B) IgG1-ASC frequencies at day 37. Mice were immunized in 3 experiments of 3–4 mice of each genotype. Data are shown for n = 10 individual Spib–/– and Spib–/–Spic + /− mice, and n = 7 individual WT mice. (C) IgG1-ASC frequencies at day 7. Mice were immunized in 3 experiments of 2–3 mice of each genotype. Data are shown for n = 8 individual Spib–/– and Spib–/–Spic + /− mice, and n = 7 individual WT mice. Data are shown as mean ± SEM. Statistics were determined by one-way ANOVA with Tukey’s multiple comparisons test, *p < 0.05.
Since the day 37 response represents a combination of the 7-day primary response and the boosted 30-day response, we also measured the primary response of WT, Spib–/–, and Spib–/–Spic + /− mice using immunization with NP-OVA in alum followed by ELISPOT analysis at day 7 post-immunization. At this time point there were no significant differences between NP-reactive ASCs in responding Spib–/– and Spib–/–Spic + /− animals for IgG1 (Figure 1C), IgM, IgG2b, and IgG2c isotypes (Supplementary Figures S1D–F). We conclude that the significant difference observed in day 37 IgG1 or IgM responses represents a difference in the recall response to NP-OVA immunization, rather than in the primary response. Reduced secondary responses in Spib–/– mice, that were rescued in Spib–/–Spic + /− mice, suggest a role for Spi-B and Spi-C in promoting either GC B cell differentiation or memory B cell differentiation.
Accelerated Differentiation of Spib–/– B Cells Into Plasmablasts in Culture
PU.1 and Spi-B expression are down-regulated upon PC differentiation (40). Combined deficiency in PU.1 and Spi-B increases PC differentiation (19, 49). Immature PCs (plasmablasts) can be generated in culture using stimulation with CD40L, Interleukin-4 (IL-4), and Interleukin-5 (IL-5) (50). Culture of splenic B cells, enriched to ∼97% using magnetic beads, with CD40L + IL-4 + IL-5 promoted differentiation into CD138+ plasmablasts with increasing frequency in 3–5 days (Figure 2A). When WT or Spib–/– splenic B cells were enriched and cultured with CD40L+IL-4+IL-5 for 5 days, frequencies of CD138-expressing cells in Spib–/– cultures were significantly elevated compared to WT samples (Figures 2B,C). To further validate this culture system, enriched splenic B cells from WT, Spib–/– and Spib–/–Spic + /− mice were cultured with CD40L+IL-4+IL-5 for 4 days in order to generate equivalent numbers of CD138+ and CD138– cells (Figure 2A). Cultured cells were enriched for CD138+ and CD138– cells using magnetic beads. Total RNA was isolated and used to determine mRNA transcript levels of Prdm1, Irf4, Bcl6, and Pax5 using reverse transcription quantitative PCR (RT-qPCR). Transcript levels were normalized to TATA-binding protein (Tbp). We found mRNA transcript levels of Prdm1 (encoding Blimp-1) and Irf4 (encoding Interferon-Response Factor-4) to be increased in CD138+ enriched cells relative to that of the WT CD138-depleted fraction (Figures 2D,E). Differentiation of B cells into CD138+ cells was accompanied by downregulation of Bcl6, Pax-5 mRNA compared to CD138– cells (Figures 2F,G). These patterns of gene expression confirmed differentiation of B cells into plasmablasts under these culture conditions (51).
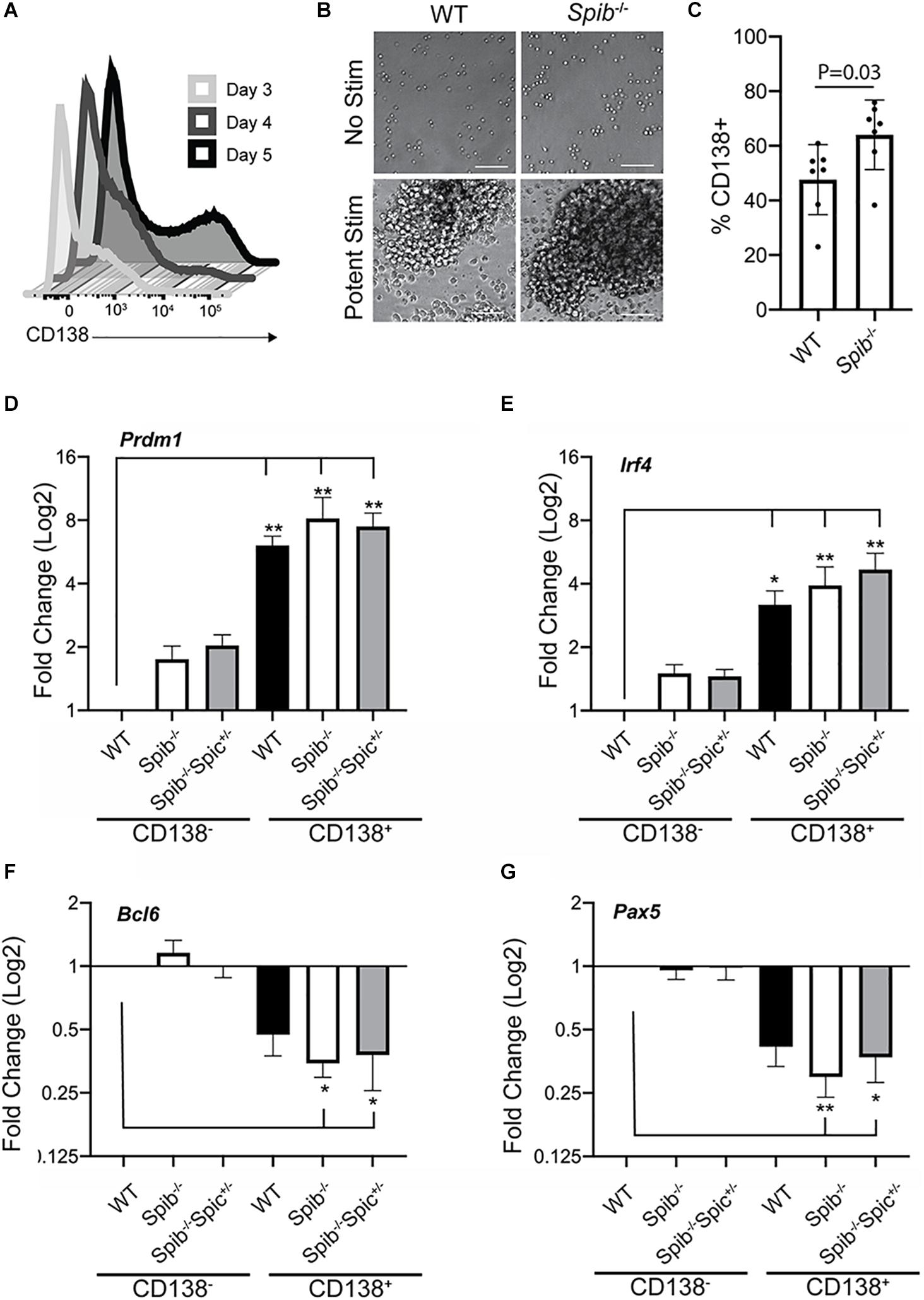
Figure 2. Plasmablast differentiation in cultures containing CD40L, IL-4, and IL-5. (A) Frequencies of CD138+ plasma cells following stimulation of enriched WT B cells for 3, 4, or 5 days with CD40L+IL-4+IL-5. One representative experiment is shown. (B) Photomicrographs of cell cultures, original magnification 20X. Scale bar indicates 50 μm. One representative experiment is shown. (C) Frequencies of CD138+ plasma cells following 5 days of culture. Means are shown ± SEM (n = 7 independent experiments with individual mice, unpaired t-test). (D–G) RT-qPCR analysis was performed for the indicated genes using total RNA prepared from CD138-enriched and -depleted samples, following 4 days of culture. Relative gene expression was normalized to Tbp. Fold change in expression was determined relative to WT CD138- cells, and is shown as mean ± SEM (n = 7 independent experiments with individual mice of each genotype, Kruskal-Wallis and Dunn’s multiple comparisons test), *p < 0.05; **p < 0.01.
To explore the kinetics of differentiation in successive rounds of cell division, WT, Spib–/–, or Spib–/–Spic + /− splenic B cells were loaded with CellTrace Violet and cultured with CD40L+IL-4+IL-5 for 5 days. Cells were divided into gates based on Celltrace violet staining, with cells in gate 1 indicating no divisions and cells in gate 7 having divided the greatest number of times (Figure 3A). This analysis revealed a marked increase in the proportion of Spib–/– CD138+ plasmablasts in the 3rd, 4th, and 5th gate, compared to WT cultures (Figures 3A,B). In contrast, Spib–/–Spic + /− B cells showed no significant differences from WT in any gate, suggesting a phenotype intermediate between WT and Spib–/– (Figure 3C). These results suggest that Spi-B, and to a lesser extent Spi-C, play roles in regulating plasmablast differentiation in response to CD40L+IL-4+IL-5.
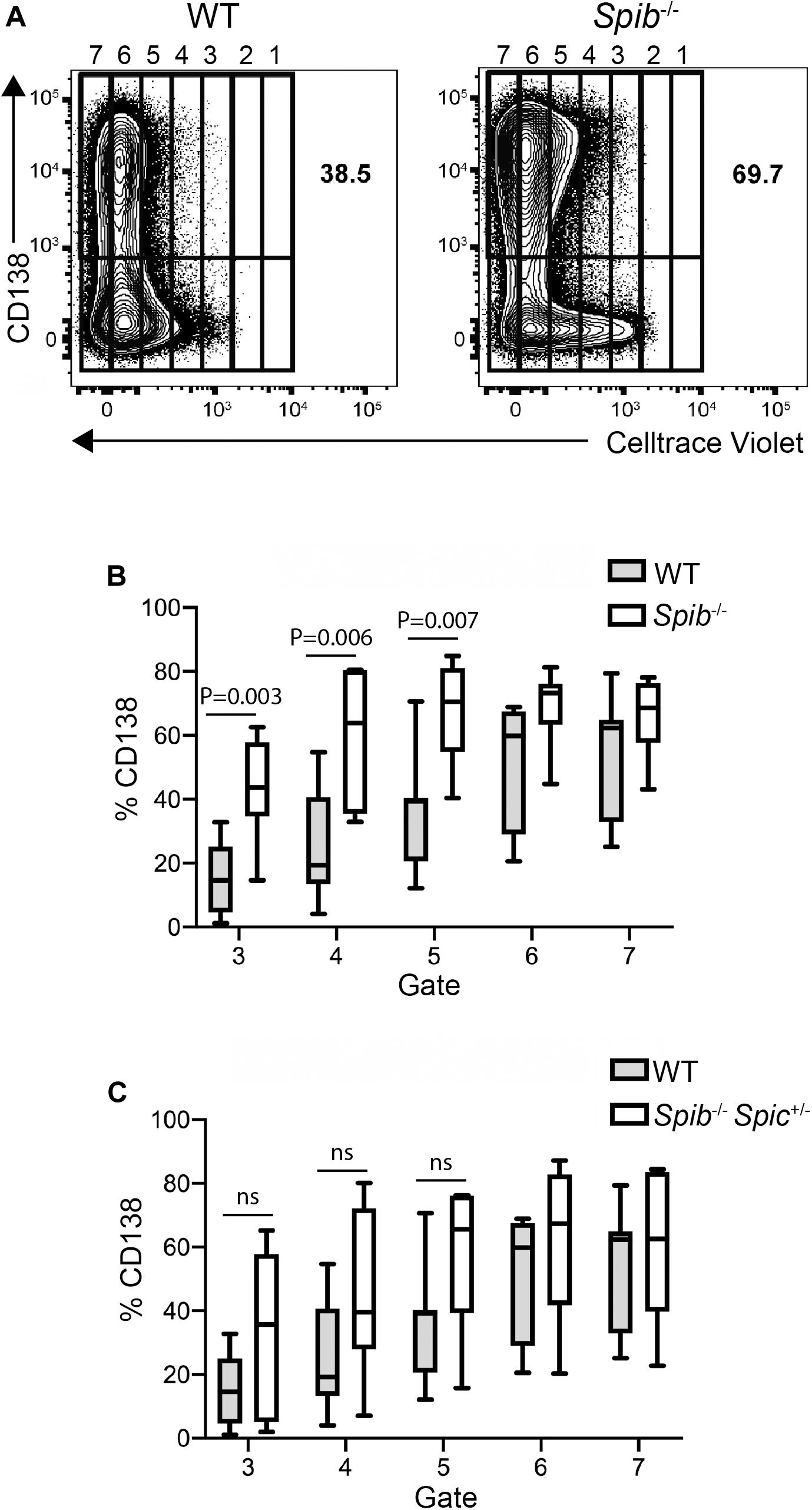
Figure 3. Accelerated plasmablast generation from Spib–/– B cells. (A) Increase in plasma cell frequency in cultured Spib–/– cells. Frequencies of CD138+ plasma cells were quantified in WT and Spib–/– enriched B cells, following 5 days of culture. Cells were assigned to arbitrary gates to track cell division. The rightmost gate denotes undivided cells. One representative experiment is shown. (B,C) Frequencies of CD138+ plasma cells were quantified in gates 3–7 for WT, Spib–/– and Spib–/–Spic + /− enriched B cells, following 5 days of culture. Data are shown as box-and-whisker plots in which the box represents the first and third quartile and the line represents the median (n = 7 independent experiments with individual mice of each genotype, one-way ANOVA and Holm–Sidak test).
Evidence for Genetic Interaction of Spi-B, Spi-C, and Bach2
Bach1 and Bach2 are related basic region leucine zipper proteins that de-repress target genes upon interaction with heme (30). Bach1 and Bach2 transcriptionally repress target genes including Spic in common lymphoid progenitors (36, 37). In macrophages, heme-induced Bach1 degradation promotes differentiation into red pulp macrophages in a Spi-C-dependent manner (38). B cells express Bach2, that been shown to be a key regulator of the MBC versus PC fate decision (35). Whether Bach2 represses Spic in the B cell lineage to regulate B cell differentiation has not been investigated. We examined patterns of expression of Spib, Spic, and Bach2, using published RNA-seq data from enriched murine B cell populations including splenic PCs or cultured plasmablasts (40). Spib was highly expressed in all B cell subsets, and was downregulated upon PC differentiation (Figure 4A, left panel). Spic was expressed in Fo and MZ B cells, was expressed at low levels in peritoneal B1 and GC B cells, and was upregulated during PC differentiation with highest expression in PCs (Figure 4A, left panel). Bach2 was expressed in all B cell subsets, with maximal expression in GC B cells, and was downregulated upon PC differentiation (Figure 4A, right panels). Tbp was stably expressed across all B cell subsets (Figure 4A, right panel). Bach2 expression correlated with Spib expression (r = 0.89, p = 0.03 by Spearman’s test) but did not correlate with Spic expression.
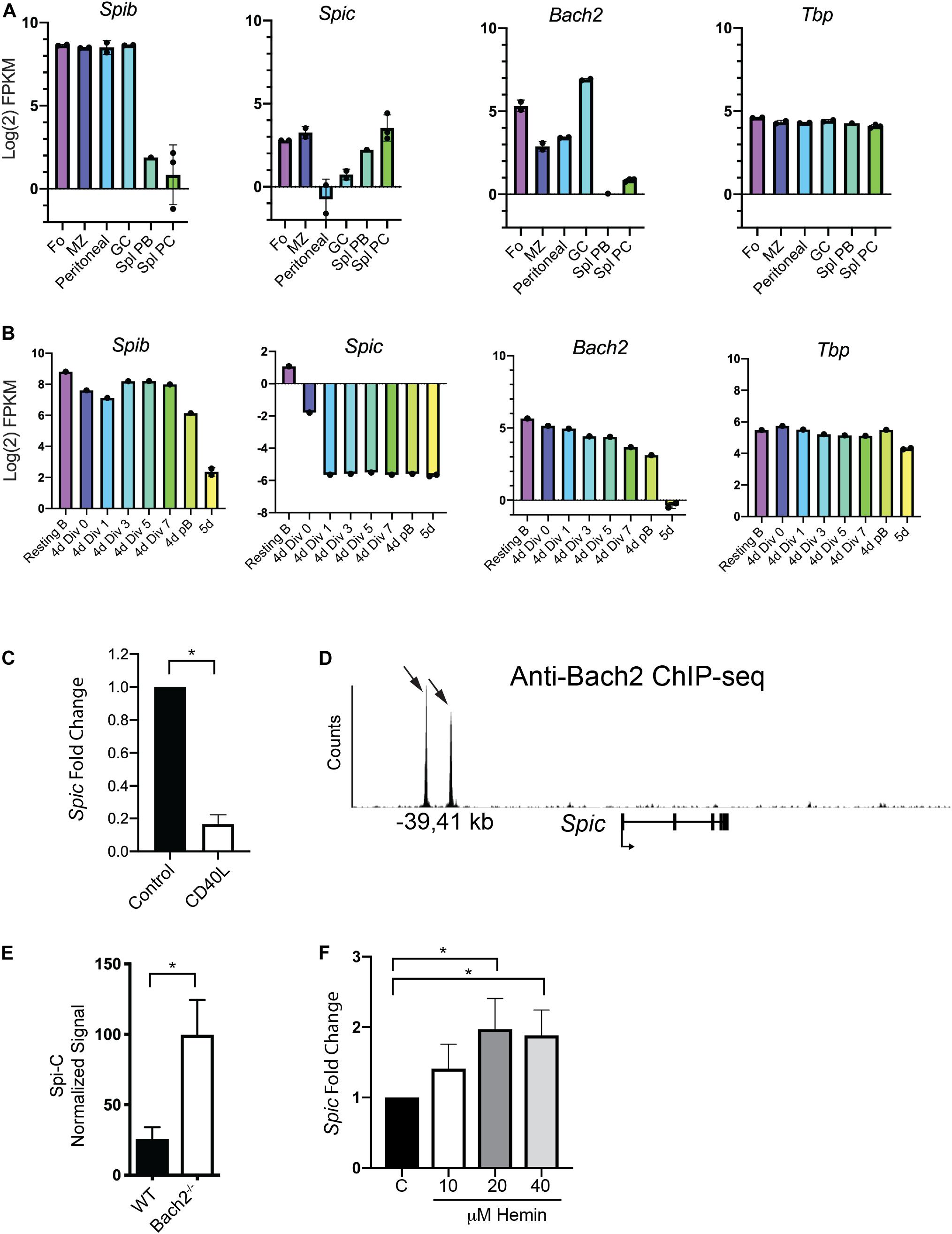
Figure 4. Evidence for repression of Spic by Bach2. (A) Analysis of gene expression in sorted mouse B cell populations. RNA-seq data from (40) was re-analyzed for enriched B cell populations including Follicular (Fo), Marginal Zone (MZ), Peritoneal, Germinal Center (GC), Splenic Plasmablast (Spl PB), and Splenic Plasma Cell (Spl PC). Y-axis shows gene expression as Log2 Fragments per Kilobase of transcript per Million mapped reads (FPKM). Dots indicate biological replicates. (B) Analysis of gene expression in cultured plasmablasts enriched based on cell division number. RNA-seq data from (40) was re-analyzed to compare gene expression in Resting B cells; B cells cultured for 4 days in CD40L+IL-4+IL-5 and sorted for 0, 1, 3, 5, or 7 cell divisions; CD138+ plasmablasts from 4 day cultures, and CD138+ plasmablasts from 5 day cultures (5 days). Dots indicate biological replicates. (C) Reduced Spic mRNA expression upon 24 h culture with CD40L. Control represents Spic mRNA expression in freshly enriched splenic B cells. Result is shown as mean ± SEM (n = 6 independent experiments with individual mice of each genotype, one sample t and Wilcoxon test) *p < 0.05. (D) Interaction of Bach2 with regulatory regions in the Spic locus. ChIP-seq data from (37) was re-analyzed to show interaction of Bach2 with a putative regulatory element located at –39 and –41 kb upstream of the Spic transcription start site. Black arrows indicate locations of Bach2 binding sites. (E) Increased Spic mRNA expression in anti-IgM-stimulated splenic B cells lacking Bach2. Agilent microarray data from (52) was re-analyzed; y-axis shows Spic normalized signal. Result is shown as mean ± SEM (n = 3 biological replicates, unpaired t-test), *p < 0.05. (F) Increased Spic mRNA transcript levels upon culture of B cells with hemin. Enriched splenic B cells were cultured with CD40L+IL-4+IL-5 and concentrations of hemin indicated on the x-axis. Spic mRNA transcript levels were determined using RT-qPCR of RNA prepared after 4 days of culture. Result is shown as mean ± SEM (n = 6 independent experiments with individual mice, Kruskal-Wallis and Dunn’s Multiple Comparisons test), *p < 0.05.
Next, we examined published RNA-seq data from splenic B cells stimulated with CD40L+IL-4+IL-5, then sorted based on cell division (40). Spib and Bach2 were downregulated over the course of PC differentiation (Figure 4B, first and third panel). In contrast to its pattern of expression in freshly enriched cells, Spic was sharply downregulated in culture with CD40L+IL-4+IL-5 (Figure 4B, second panel), while Tbp was stably expressed across all cell divisions (Figure 4B, fourth panel). Spic therefore showed a different pattern of expression in culture compared to freshly isolated populations, as it was expressed at high levels in splenic plasmablasts and PCs (Figure 4A) but was nearly undetectable in cultured plasmablasts (Figure 4B). To determine if Spic mRNA levels are regulated by CD40L, RNA was prepared from freshly enriched splenic B cells, or enriched B cells cultured 24 h with CD40L. RT-qPCR analysis showed that Spic was downregulated upon culture with CD40L (Figure 4C). This result suggests that CD40 signaling may explain differences in Spic expression in mice compared to cell culture, and may explain the low impact of Spic heterozygosity on plasma cell differentiation in cultures containing CD40L+IL-4+IL-5.
Next, we investigated whether Bach2 can repress Spic in B cells. Examination of Bach2 ChIP-seq analysis (36) demonstrated that Bach2 interacts directly with two sites located within upstream regulatory elements within the Spic locus (Figure 4D). To determine if Spic is repressed by Bach2 in B cells, we examined published microarray data from WT and Bach2–/– B cells activated by anti-IgM stimulation (52). This analysis showed that Spic is up-regulated by approximately 4-fold in Bach2–/– B cells compared to WT B cells (Figure 4E). Finally, Bach2 is a heme-binding protein, and its repressor activity is decreased by interaction with heme (30). Culture of splenic B cells with hemin in addition to CD40L+IL-4+IL-5 increased Spic mRNA transcript levels (Figure 4F). These results suggest that Bach2 is a transcriptional repressor of Spic in B cells.
Chromatin Immunoprecipitation-Sequencing Analysis of Spi-C Binding in B Cells
Genome-wide analysis of Spi-C interaction in the genome of B cells has not previously been reported because chromatin immunoprecipitation (ChIP)-grade antibodies for this transcription factor are not available. To determine Spi-C binding sites in the genome of B cells, we generated Abelson-transformed 38B9 pre-B cell lines infected with a retroviral vector encoding 3XFLAG-tagged murine Spi-C (12). Anti-FLAG ChIP-seq was performed, and reads were mapped to mouse genome Mm9 to identify binding sites. There were 1037 Spi-C binding sites identified using a e value cutoff of ≤10–5 (Figure 5A). These sites were then intersected with those previously identified for PU.1 in pro-B cells (13) and Spi-B in WEHI-279 cells (14). 554 peaks were unique to Spi-C only, 239 peaks were shared with PU.1; 57 peaks were shared with Spi-B; and 187 peaks were shared with both PU.1 and Spi-B (Figure 5A). For Spi-C peaks shared with either PU.1 or Spi-B, the most frequently occurring motif was a purine-rich sequence closely resembling the motif previously published for these transcription factors (Figures 5B,C) (14).
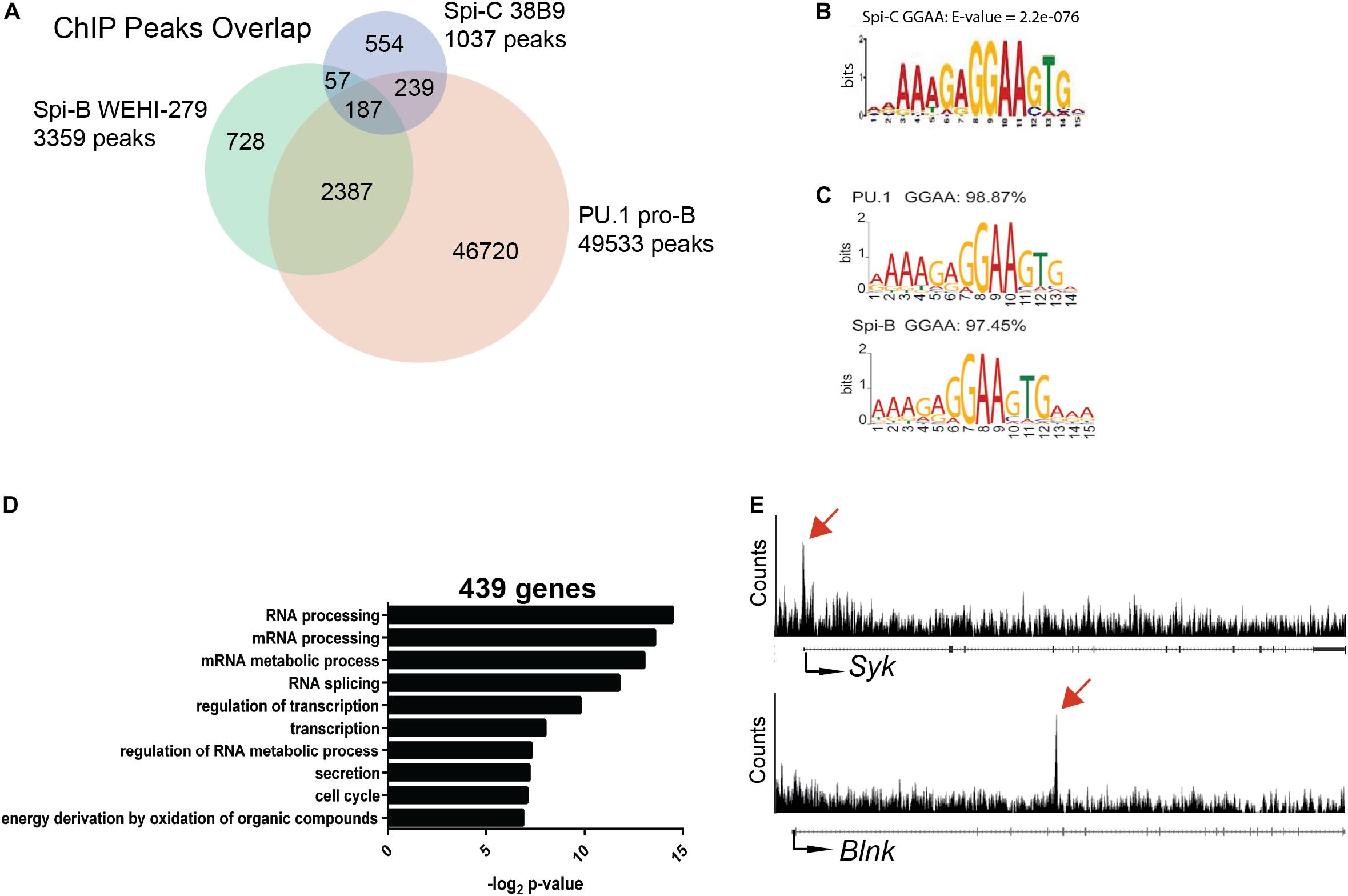
Figure 5. ChIP-seq analysis of Spi-C interaction in 38B9 cells. (A) Venn diagram showing overlap in binding sites. Genomic regions from ChIP analysis (peaks) were compared for Spi-C in 38B9 cells (blue), PU.1 in pro-B cells (red) and Spi-B in WEHI-279 B lymphoma cells (green). Numbers indicate the number of unique or overlapping peaks. (B) DNA interaction motifs recovered from Spi-C ChIP-seq analysis. (C) DNA interaction motifs recovered from PU.1 (top) and Spi-B (bottom) ChIP-seq analysis. (D) Biological pathways were identified from 439 genes interacting with Spi-C using DAVID analysis. (E) Representative Spi-C peaks in 38B9 cells for genes encoding Syk (top panel) and B cell linker protein (Blnk, lower panel). Red arrows indicate location of Spi-C peaks.
To identify genes regulated by Spi-C, Cistrome Beta Minus (53) was used to identify genes with transcription start sites located within 15 kb of Spi-C binding peaks. 439 genes were identified to be associated with Spi-C interaction. Among these, 56 genes were unique to Spi-C, while 383 were shared with either PU.1 or Spi-B. Gene ontology analysis was performed to identify biological pathways associated with genes uniquely occupied by Spi-C. The top three pathways included RNA processing, mRNA processing, and mRNA metabolic process (Figure 5D). Genes with Spi-C/Spi-B/PU.1 shared binding sites included Syk and Blnk, genes for which these transcription factors have been demonstrated to have opposing functions (15, 26) (Figure 5E, upper panels). These results suggested that half of Spi-C binding sites are coincident with PU.1 and/or Spi-B binding sites, but Spi-C also has unique binding sites and therefore may exert unique functions in B cells.
Regulation of Bach2 by Spi-B and Spi-C
Next, our goal was to determine if Spi-B and Spi-C interact with regions in the Bach2 locus. Four regions of interest (ROI 1-4) were identified in the Bach2 locus based on regions of open chromatin marked by IMMGEN ATAC-seq analysis in enriched follicular B cells (54) (Figure 6A). ROI 1-4 were found to interact to differing extents with PU.1, Spi-B, and Spi-C based on the ChIP-seq analysis described above (Figure 6A). Multiple species conservation analysis (PhastCons46wayPlacental) of ROIs revealed a high degree of conservation in ROIs 1 and 3, found in Bach2 introns 1 and 2, respectively (Figure 6B). To determine if Spi-B and Spi-C could interact with ROI 1 and 3 in primary B cells, splenic B cells from WT mice were enriched and stimulated overnight with CD40L+IL-4+IL-5, then transduced with MIGR1 (control), MIG-3XFLAG-SpiB, or MIG-3XFLAG-SpiC retroviral vectors (Figure 6C). Mean transduction efficiency, as determined by percentage of GFP+ cells, was 56% in MIGR1-transduced cells, 16% in MIG-3XFLAG-Spi-B-infected cells, and 15% in MIG-3XFLAG-Spi-C-infected cells (Figure 6D). Chromatin from transduced and stimulated B cells was immunoprecipitated using anti-FLAG antibodies and used as input for qPCR targeting ROIs 1 and 3, and on a negative control region (NCR) selected from intron 2 of Bach2. QPCR Analysis revealed significant enrichment of Spi-C at ROI 1 (Figure 6F) and ROI 3 (Figure 6G), relative to the NCR (Figure 5E) and also compared to that of MIGR1-transduced cells (Figures 6E–G). Spi-B interaction was likewise observed at ROI 1 and 3, compared to basal MIGR1 and NCR enrichment (Figures 6E–G). Therefore Spi-B and Spi-C can interact with ROI 1 and ROI 3 in B cells.
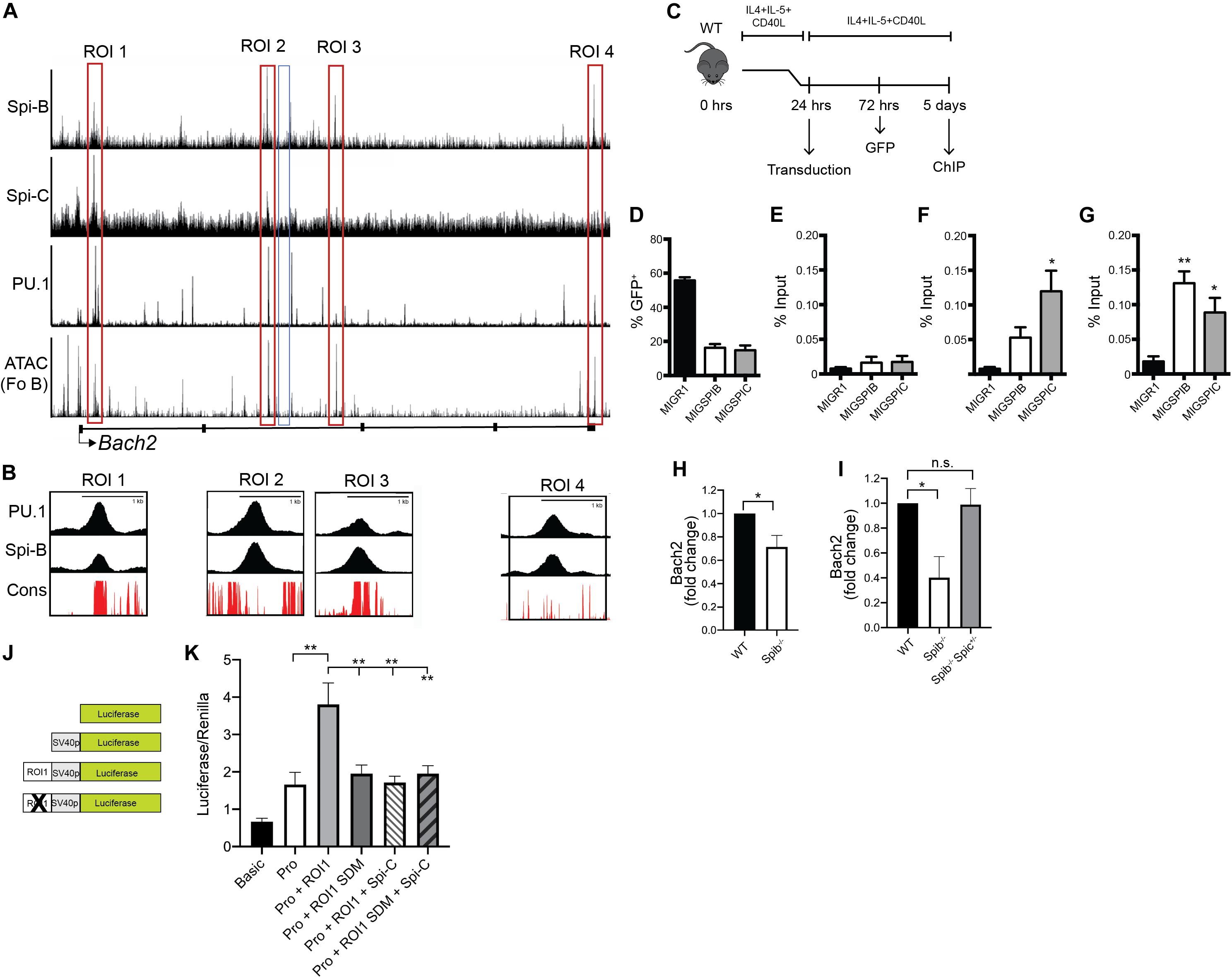
Figure 6. Regulation of Bach2 by Spi-B and Spi-C. (A) UCSC genome browser tracks of ChIP-seq analysis of 3XFLAG-tagged Spi-B in WEHI-279 cells (top panel) and Spi-C in 38B9 cells (second panel). Also shown is ChIP-seq analysis of 3X-FLAG-tagged PU.1 binding in pro-B cells (third panel) and IMMGEN ATAC-seq analysis performed in enriched follicular B cells (Fo B) (Fourth panel). Bach2 gene structure is shown below, with black boxes denoting exons and lines representing introns. Red boxes indicate regions-of-interest (ROIs 1-4), while the blue box denotes the negative control region (NCR). (B) Multiple placental species conservation analysis (in red) visualized as superimposed UCSC tracks of ChIP-seq (ROI 1-4, in black) within the Bach2 locus for PU.1 and Spi-B. (C) Schematic depicting the workflow for primary B cell transduction, followed by chromatin immunoprecipitation (ChIP). (D) Frequency of GFP+ cells, 48 h following transduction of WT enriched B cells with the indicated retroviral vectors. Bars represent the mean transduction efficiency ± SEM. (E–G) Enrichment of FLAG-tagged Spi-B and Spi-C in cultured WT B cells at the NCR (E), ROI 1 (F), and ROI 3 (G). Results are shown as percentage of input DNA, with bars representing mean% input ± SEM (n = 3 independent experiments using cells from individual mice, one-way ANOVA with Tukey’s multiple comparisons test), *p < 0.05; **p < 0.01. (H) Reduced Bach2 mRNA transcript levels in Spib–/– B cells. RT-qPCR analysis was performed on RNA prepared from splenic B cells enriched from WT and Spib–/– mice (n = 5 independent experiments using individual mice of each genotype, one sample t and Wilcoxon test), *p < 0.05. (I) Reduced Bach2 mRNA transcript levels in Spib–/– GC B cells. RT-qPCR analysis was performed on RNA prepared from splenic GC B cells enriched from WT, Spib–/–, and Spib–/– Spic + /− mice by cell sorting, 10 days after immunization with NP-KLH. Result is shown for representative experiment with triplicate technical replicates, Kruskal-Wallis with Dunn’s multiple comparisons test, *p < 0.05. (J) Schematic of luciferase reporters pGL3-basic (first panel), pGL3-promoter (second panel), pGL3-promoter-ROI 1 (third panel), and pGL3-promoter-RO1 1 mutant (fourth panel). (K) Bach2 ROI 1 shows enhancer activity in WEHI-279 cells that is reduced by mutation of the ETS binding site (n = 4 independent experiments using triplicate technical replicates, mixed-effects ANOVA with Tukey’s multiple comparisons test), **p < 0.01.
To determine if Spi-B is an activator of Bach2, we measured Bach2 mRNA levels in freshly isolated Spib–/– splenic B cells. We observed reduced Bach2 mRNA levels in B cells enriched from the spleen of Spib–/– mice (Figure 6H). Based on the observation that both Spi-B and Bach2 are expressed at high levels in GC B cells (Figure 4A), we performed the following experiment. WT, Spib–/–, and Spib–/– Spic + /− mice were immunized with alum-precipitated NP-KLH. Ten days after immunization, a time point reported to have high frequencies of GC B cells following immunization (17), CD38– CD95+ CD19+ CD138– splenic GC B cells (55) were enriched from the spleen of immunized mice by cell sorting. RT-qPCR analysis showed that Bach2 mRNA transcripts were reduced in Spib–/– GC B cells relative to WT GC B cells (Figure 6I). In contrast, Bach2 mRNA transcripts were not reduced in Spib–/– Spic + /− GC B cells relative to WT GC B cells (Figure 6I). These data suggest that Spi-B and Spi-C regulate Bach2 expression in naïve and GC B cells.
Bach2 ROI 1 was cloned and tested for enhancer activity in combination with a minimal SV40 promoter in WEHI-279 B lymphoma cells that express high levels of Spi-B, but do not express Spi-C (14) (Figure 6J). ROI 1 increased activity of the luciferase reporter (Figure 6K). Mutation of the ETS site (GGAA → GGCC) reduced activity of ROI 1 (Figure 6K). Co-transfection with a Spi-C expression vector repressed activity of the wild-type ROI 1 reporter, but did not repress activity of the mutant ROI 1 reporter plasmid (Figure 6K). Taken together, these data suggest that Spi-B is a transcriptional activator of Bach2 through a binding site in ROI 1, while Spi-C can function as a transcriptional repressor at this site.
Discussion
The goal of this study was to determine the roles of Spi-B and Spi-C in regulation of B cell antibody responses. Our results demonstrate that Spi-B and Spi-C play opposing roles in secondary antibody responses and plasmablast differentiation. Spic heterozygosity rescued frequencies of IgG1 antibody-secreting cells following secondary antigenic challenge in Spib–/– mice. Culture of enriched splenic B cells with CD40L+IL-4+IL-5 revealed that Spib–/– B cells differentiated into CD138-expressing plasmablasts with accelerated kinetics relative to WT cells. Using ChIP-seq, ChIP, and luciferase assays, we showed that Spi-B and Spi-C interact with intronic regions of the Bach2 locus to regulate transcription. Experiments indicated that Spi-B activated Bach2, Spi-C repressed Bach2, and Spi-C and Bach2 were mutually cross-antagonistic. Together, our results suggest that Spi-B and Spi-C are involved in shaping antibody-forming responses by influencing the differentiation of activated B cells.
Our results showed that secondary IgG1 antibody responses and GC B cell frequencies are reduced in Spib–/– mice relative to WT mice, and are partially rescued by deletion of one Spic allele in Spib–/–Spic + /− mice. In contrast, plasmablast differentiation in culture was accelerated in Spib–/– B cells relative to WT B cells. These results suggest that Spi-B promotes GC and/or memory B cell differentiation, but inhibits plasmablast differentiation. These observations fit well with previous observations that Spi-B is required for sustaining germinal centers (17), while in contrast Spi-B is normally downregulated upon plasmablast differentiation (Figure 4A), and that ectopic expression of Spi-B impairs plasmablast differentiation (20). We expect that the explanation for reduced secondary antibody responses in Spib–/– mice might be a combination of reduced GC and/or memory B cell generation in Spib–/– mice, and a tendency for naïve Spib–/– B cells to differentiate into plasmablasts instead of GC B cells. We speculate that Spi-B and Spi-C may regulate cell fate decisions of naïve or GC B cells to differentiate into GC, memory or plasmablast cells. However, answering this question will require further experiments performed at single cell resolution.
ChIP-seq studies for Spi-C have not been previously performed because commercially available antibodies recognizing Spi-C do not work in ChIP. Therefore, we performed anti-FLAG ChIP-seq of 3XFLAG-tagged Spi-C in 38B9 pre-B cells. Interestingly, we found that ectopic expression of Spi-C could not be sustained in WEHI-279 lymphoma cells because it induced high rates of apoptosis (data not shown). ChIP-seq analysis for Spi-C revealed a 5′-GGAA-3′ motif that was consistent with that described for PU.1 and Spi-B (14). Examples of genes bound by PU.1, Spi-B, and Spi-C were Syk encoding the SYK tyrosine kinase and Blnk encoding B cell linker protein, both of which are key mediators of BCR signaling (15, 26). Thus, our Spi-C ChIP analysis revealed unique and shared sites at which Spi-C can compete with PU.1 and/or Spi-B binding in the genome of B cells in order to function as a regulator of PU.1 and/or Spi-B function.
Spi-C function in B cell antibody-forming responses has not been previously studied. We found that Spic heterozygosity rescued frequencies of IgG1 antibody-secreting cells following secondary antigenic challenge in Spib–/– mice. These results are consistent with previous observations suggesting that Spi-C functions to oppose activity of Spi-B (28). Recently, Spi-C was found to bind DNA cooperatively with BCLAF to displace PU.1 or Spi-B from binding sites in developing B cells (27). Interestingly, Spic mRNA levels were discovered to be expressed differently in vivo than in culture (Figures 4A,B). This difference was found to be due to downregulation of Spic mRNA expression by CD40L (Figures 4B,C). Conversely, we found that Spic mRNA levels were increased in stimulated B cells from Bach2–/– mice (Figure 4E) or in cultures treated with heme (Figure 4G). These experiments suggest that Spi-C is dynamically regulated in response to external signals such as CD40L and heme. We speculate that Spi-C responsiveness to heme may play a role in generation of plasmablasts in response to hemolytic bacterial infections (56).
The results presented in this study suggest the following model. Spi-B may function as an activator of Bach2 in B cells to promote efficient GC and memory B cell differentiation (33, 35, 57). Upon induction of the plasmablast differentiation program, Spib is downregulated, leading to downregulated Bach2 and induction of the plasmablast differentiation program by de-repression of Blimp-1 and Spi-C (58). Increased Spi-C expression would help enforce the plasmablast differentiation program in part by repression of Bach2. CD40/CD40L engagement of activated B cells by T follicular helper cells might down-regulate Spic to promote the GC/memory B cell fate. This model provides a framework for further study.
In summary, B cell developmental decisions are governed by mutually cross-antagonistic transcription factor networks including IRF4 versus IRF8 (11) or Bach2 versus Blimp-1 (35, 58). Our results suggest that Spi-B versus Spi-C may represent nodes in an additional opposing transcription factor network governing B cell differentiation. Understanding the molecular circuitry that governs B cell fate decisions during immune responses may ultimately have important implications for the design of vaccination strategies.
Data Availability Statement
The datasets generated for this study can be found in the ChIP-seq data is available from the Gene Expression Omnibus accession GSE115593.
Ethics Statement
The animal study was reviewed and approved by the Western University Council on Animal Care Committee.
Author Contributions
RD conceived the study. A-SL, HR, PS, CB, DS, LX, and YT performed the experiments and interpreted the data. SH, SK, and RD interpreted the data. A-SL and RD wrote the manuscript.
Funding
This work was supported by CIHR grants MOP-106581 and MOP-142258 and NSERC grant 04749-2016 to RD.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Rajon Hossain, and Michelle Ho, and LX (Western University) in genotyping the mice used in the present study, Rajiv Jain (Western University) for his contribution of Bcl-6, Blimp-1, and Bach2 primer sequences, and Dr. Kenneth M. Murphy (Washington University School of Medicine) for providing the Spic + /− mice. We also thank Heather Craig (Western University) and Kristin Chadwick (London Regional Flow Cytometry Facility) for their help with flow cytometry.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00841/full#supplementary-material
References
1. Nutt, SL, Hodgkin, PD, Tarlinton, DM, and Corcoran, LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. (2015) 15:160–71. doi: 10.1038/nri3795
2. De Silva, NS, and Klein, U. Dynamics of B cells in germinal centres. Nat Rev Immunol. (2015) 15:137–48. doi: 10.1038/nri3804
3. Kerfoot, SM, Yaari, G, Patel, JR, Johnson, KL, Gonzalez, DG, Kleinstein, SH, et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity. (2011) 34:947–60. doi: 10.1016/j.immuni.2011.03.024
4. Victora, GD, and Nussenzweig, MC. Germinal centers. Annu Rev Immunol. (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
5. Allen, CDC, Ansel, KM, Low, C, Lesley, R, Tamamura, H, Fujii, N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. (2004) 5:943–52. doi: 10.1038/ni1100
6. Gitlin, AD, Shulman, Z, and Nussenzweig, MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature. (2014) 509:637–40. doi: 10.1038/nature13300
7. Shaffer, AL, Lin, K-I, Kuo, TC, Yu, X, Hurt, EM, Rosenwald, A, et al. Blimp-1 Orchestrates Plasma Cell Differentiation by Extinguishing the Mature B Cell Gene Expression Program. Immunity. (2002) 17:51–62. doi: 10.1016/S1074-7613(02)00335-7
8. Lin, K-I, Angelin-Duclos, C, Kuo, TC, and Calame, K. Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Mol Cell Biol. (2002) 22:4771–80. doi: 10.1128/mcb.22.13.4771-4780.2002
9. Crotty, S, Johnston, RJ, and Schoenberger, SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol. (2010) 11:114–20. doi: 10.1038/ni.1837
10. Ochiai, K, Muto, A, Tanaka, H, Takahashi, S, and Igarashi, K. Regulation of the plasma cell transcription factor Blimp-1 gene by Bach2 and Bcl6. Int Immunol. (2008) 20:453–60. doi: 10.1093/intimm/dxn005
11. Xu, H, Chaudhri, VK, Wu, Z, Biliouris, K, Dienger-Stambaugh, K, Rochman, Y, et al. Regulation of bifurcating B cell trajectories by mutual antagonism between transcription factors IRF4 and IRF8. Nat Immunol. (2015) 16:1274–81. doi: 10.1038/ni.3287
12. DeKoter, RP, Geadah, M, Khoosal, S, Xu, LS, Thillainadesan, G, Torchia, J, et al. Regulation of follicular B cell differentiation by the related E26 transformation-specific transcription factors PU.1, Spi-B, and Spi-C. J Immunol. (2010) 185:7374–84. doi: 10.4049/jimmunol.1001413
13. Batista, CR, Li, SKH, Xu, LS, Solomon, LA, and DeKoter, RP. PU.1 Regulates Ig light chain transcription and rearrangement in Pre-B cells during B cell development. J Immunol. (2017) 198:1565–74. doi: 10.4049/jimmunol.1601709
14. Solomon, LA, Li, SKH, Piskorz, J, Xu, LS, and DeKoter, RP. Genome-wide comparison of PU.1 and Spi-B binding sites in a mouse B lymphoma cell line. BMC Genomics. (2015) 16:76. doi: 10.1186/s12864-015-1303-0
15. Xu, LS, Sokalski, KM, Hotke, K, Christie, DA, Zarnett, O, Piskorz, J, et al. Regulation of B cell linker protein transcription by PU.1 and Spi-B in murine B cell acute lymphoblastic leukemia. J Immunol. (2012) 189:3347–54. doi: 10.4049/jimmunol.1201267
16. Christie, DA, Xu, LS, Turkistany, SA, Solomon, LA, Li, SK, Yim, E, et al. PU.1 opposes IL-7-dependent proliferation of developing B cells with involvement of the direct target gene bruton tyrosine kinase. J Immunol. (2015) 194:595–605. doi: 10.4049/jimmunol.1401569
17. Su, GH, Chen, HM, Muthusamy, N, Garrett-Sinha, LA, Baunoch, D, Tenen, DG, et al. Defective B cell receptor-mediated responses in mice lacking the Ets protein. Spi-B. EMBO J. (1997) 16:7118–29. doi: 10.1093/emboj/16.23.7118
18. Garrett-Sinha, LA, Su, GH, Rao, S, Kabak, S, Hao, Z, Clark, MR, et al. PU.1 and Spi-B are required for normal B cell receptor-mediated signal transduction. Immunity. (1999) 10:399–408. doi: 10.1016/s1074-7613(00)80040-0
19. Willis, SN, Tellier, J, Liao, Y, Trezise, S, Light, A, O’Donnell, K, et al. Environmental sensing by mature B cells is controlled by the transcription factors PU.1 and SpiB. Nat Commun. (2017) 8:1426. doi: 10.1038/s41467-017-01605-1
20. Schmidlin, H, Diehl, SA, Nagasawa, M, Scheeren, FA, Schotte, R, Uittenbogaart, CH, et al. Spi-B inhibits human plasma cell differentiation by repressing BLIMP1 and XBP-1 expression. Blood. (2008) 112:1804–12. doi: 10.1182/blood-2008-01-136440
21. Kohyama, M, Ise, W, Edelson, BT, Wilker, PR, Hildner, K, Mejia, C, et al. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature. (2009) 457:318–21. doi: 10.1038/nature07472
22. Bemark, M, Martensson, A, Liberg, D, and Leanderson, T. Spi-C, a novel Ets protein that is temporally regulated during B lymphocyte development. J Biol Chem. (1999) 274:10259–67. doi: 10.1074/jbc.274.15.10259
23. Hashimoto, S, Nishizumi, H, Hayashi, R, Tsuboi, A, Nagawa, F, Takemori, T, et al. Prf, a novel Ets family protein that binds to the PU.1 binding motif, is specifically expressed in restricted stages of B cell development. Int Immunol. (1999) 11:1423–9. doi: 10.1093/intimm/11.9.1423
24. Schweitzer, BL, Huang, KJ, Kamath, MB, Emelyanov, AV, Birshtein, BK, and DeKoter, RP. Spi-C has opposing effects to PU.1 on gene expression in progenitor B cells. J Immunol. (2006) 177:2195–207. doi: 10.4049/jimmunol.177.4.2195
25. Zhu, X, Schweitzer, BL, Romer, EJ, Sulentic, CEW, and DeKoter, RP. Transgenic expression of Spi-C impairs B-cell development and function by affecting genes associated with BCR signaling. Eur J Immunol. (2008) 38:2587–99. doi: 10.1002/eji.200838323
26. Bednarski, JJ, Pandey, R, Schulte, E, White, LS, Chen, BR, Sandoval, GJ, et al. RAG-mediated DNA double-strand breaks activate a cell type-specific checkpoint to inhibit pre-B cell receptor signals. J Exp Med. (2016) 213:209–23. doi: 10.1084/jem.20151048
27. Soodgupta, D, White, LS, Yang, W, Johnston, R, Andrews, JM, Kohyama, M, et al. RAG-mediated DNA breaks attenuate PU.1 activity in early B cells through activation of a SPIC-BCLAF1 complex. Cell Rep. (2019) 29:829.e–43.e. doi: 10.1016/j.celrep.2019.09.026
28. Li, SK, Solomon, LA, Fulkerson, PC, and DeKoter, RP. Identification of a negative regulatory role for Spi-C in the murine B cell lineage. J Immunol. (2015) 194:3978–3807. doi: 10.4049/jimmunol.1402432
29. Oyake, T, Itoh, K, Motohashi, H, Hayashi, N, Hoshino, H, Nishizawa, M, et al. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol Cell Biol. (1996) 16:6083–95. doi: 10.1128/mcb.16.11.6083
30. Watanabe-Matsui, M, Muto, A, Matsui, T, Itoh-Nakadai, A, Nakajima, O, Murayama, K, et al. Heme regulates B-cell differentiation, antibody class switch, and heme oxygenase-1 expression in B cells as a ligand of Bach2. Blood. (2011) 117:5438–48. doi: 10.1182/blood-2010-07-296483
31. Igarashi, K, and Itoh-Nakadai, A. Orchestration of B lymphoid cells and their inner myeloid by Bach. Curr Opin Immunol. (2016) 39:136–42. doi: 10.1016/j.coi.2016.01.012
32. Muto, A, Tashiro, S, Nakajima, O, Hoshino, H, Takahashi, S, Sakoda, E, et al. The transcriptional programme of antibody class switching involves the repressor Bach2. Nature. (2004) 429:566–71. doi: 10.1038/nature02596
33. Shinnakasu, R, Inoue, T, Kometani, K, Moriyama, S, Adachi, Y, Nakayama, M, et al. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol. (2016) 17:861–9. doi: 10.1038/ni.3460
34. Muto, A, Ochiai, K, Kimura, Y, Itoh-Nakadai, A, Calame, KL, Ikebe, D, et al. Bach2 represses plasma cell gene regulatory network in B cells to promote antibody class switch. EMBO J. (2010) 29:4048–61. doi: 10.1038/emboj.2010.257
35. Kometani, K, Nakagawa, R, Shinnakasu, R, Kaji, T, Rybouchkin, A, Moriyama, S, et al. Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity. (2013) 39:136–47. doi: 10.1016/j.immuni.2013.06.011
36. Itoh-Nakadai, A, Hikota, R, Muto, A, Kometani, K, Watanabe-Matsui, M, Sato, Y, et al. The transcription repressors Bach2 and Bach1 promote B cell development by repressing the myeloid program. Nat Immunol. (2014) 15:1171–80. doi: 10.1038/ni.3024
37. Itoh-Nakadai, A, Matsumoto, M, Kato, H, Sasaki, J, Uehara, Y, Sato, Y, et al. A Bach2-cebp gene regulatory network for the commitment of multipotent hematopoietic progenitors. Cell Rep. (2017) 18:2401–14. doi: 10.1016/j.celrep.2017.02.029
38. Haldar, M, Kohyama, M, So, AYL, Kc, W, Wu, X, Briseño, CG, et al. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell. (2014) 156:1223–34. doi: 10.1016/j.cell.2014.01.069
39. Pfaffl, M. Quantification strategies in real-time PCR Michael W. Pfaffl. In: SA Bustin editor. A-Z Quant PCR. La Jolla, CA: International University Line (2004). p. 87–112.
40. Shi, W, Liao, Y, Willis, SN, Taubenheim, N, Inouye, M, Tarlinton, DM, et al. Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat Immunol. (2015) 16:663–73. doi: 10.1038/ni.3154
41. Morita, S, Kojima, T, and Kitamura, T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. (2000) 7:1063–6. doi: 10.1038/sj.gt.3301206
42. Afgan, E, Baker, D, van den Beek, M, Blankenberg, D, Bouvier, D, Cech, M, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. (2016) 44:W3–10. doi: 10.1093/nar/gkw343
43. Langmead, B, Trapnell, C, Pop, M, and Salzberg, SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. (2009) 10:R25. doi: 10.1186/gb-2009-10-3-r25
44. Zhang, Y, Liu, T, Meyer, CA, Eeckhoute, J, Johnson, DS, Bernstein, BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. (2008) 9:R137. doi: 10.1186/gb-2008-9-9-r137
45. Shin, H, Liu, T, Manrai, AK, and Liu, XS. CEAS: cis-regulatory element annotation system. Bioinformatics. (2009) 25:2605–6. doi: 10.1093/bioinformatics/btp479
46. Huang da, W, and Sherman, BT. Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. (2009) 4:44–57. doi: 10.1038/nprot.2008.211
47. Ma, W, Noble, WS, and Bailey, TL. Motif-based analysis of large nucleotide data sets using MEME-ChIP. Nat Protoc. (2014) 9:1428–50. doi: 10.1038/nprot.2014.083
48. Imanishi, T, and Makela, O. Strain differences in the fine specificity of mouse anti-hapten antibodies. Eur J Immunol. (1973) 3:323–30. doi: 10.1002/eji.1830030602
49. Carotta, S, Willis, SN, Hasbold, J, Inouye, M, Pang, SHM, Emslie, D, et al. The transcription factors IRF8 and PU.1 negatively regulate plasma cell differentiation. J Exp Med. (2014) 211:2169–81. doi: 10.1084/jem.20140425
50. Hasbold, J, Corcoran, LM, Tarlinton, DM, Tangye, SG, and Hodgkin, PD. Evidence from the generation of immunoglobulin G-secreting cells that stochastic mechanisms regulate lymphocyte differentiation. Nat Immunol. (2004) 5:55–63. doi: 10.1038/ni1016
51. Sciammas, R, Shaffer, AL, Schatz, JH, Zhao, H, Staudt, LM, and Singh, H. Graded Expression of Interferon Regulatory Factor-4 Coordinates Isotype Switching with Plasma Cell Differentiation. Immunity. (2006) 25:225–36. doi: 10.1016/j.immuni.2006.07.009
52. Miura, Y, Morooka, M, Sax, N, Roychoudhuri, R, Itoh-Nakadai, A, Brydun, A, et al. Bach2 promotes B cell receptor-induced proliferation of B lymphocytes and represses cyclin-dependent kinase inhibitors. J Immunol. (2018) 200:2882–93. doi: 10.4049/jimmunol.1601863
53. Wang, S, Sun, H, Ma, J, Zang, C, Wang, C, Wang, J, et al. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc. (2013) 8:2502–15. doi: 10.1038/nprot.2013.150
54. Yoshida, H, Lareau, CA, Ramirez, RN, Rose, SA, Maier, B, Wroblewska, A, et al. The cis-regulatory atlas of the mouse immune system. Cell. (2019) 176:897.e–912.e. doi: 10.1016/j.cell.2018.12.036
55. Oliver, AM, Martin, F, and Kearney, JF. Mouse CD38 is down-regulated on germinal center B cells and mature plasma cells. J Immunol. (1997) 158:1108–15.
56. Dutra, FF, and Bozza, MT. Heme on innate immunity and inflammation. Front Pharmacol. (2014) 5:115. doi: 10.3389/fphar.2014.00115
57. Shinnakasu, R, and Kurosaki, T. Regulation of memory B and plasma cell differentiation. Curr Opin Immunol. (2017) 45:126–31. doi: 10.1016/j.coi.2017.03.003
Keywords: Spi-B, Spi-C, plasma cell, antibody, Bach2
Citation: Laramée A-S, Raczkowski H, Shao P, Batista C, Shukla D, Xu L, Haeryfar SMM, Tesfagiorgis Y, Kerfoot S and DeKoter R (2020) Opposing Roles for the Related ETS-Family Transcription Factors Spi-B and Spi-C in Regulating B Cell Differentiation and Function. Front. Immunol. 11:841. doi: 10.3389/fimmu.2020.00841
Received: 18 December 2019; Accepted: 14 April 2020;
Published: 08 May 2020.
Edited by:
Harry W. Schroeder, University of Alabama at Birmingham, United StatesReviewed by:
Stephen Nutt, Walter and Eliza Hall Institute of Medical Research, AustraliaPrasad Srikakulapu, University of Virginia, United States
Copyright © 2020 Laramée, Raczkowski, Shao, Batista, Shukla, Xu, Haeryfar, Tesfagiorgis, Kerfoot and DeKoter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rodney DeKoter, cmRla290ZXJAdXdvLmNh