 Lili Tang1,2,3,4
Lili Tang1,2,3,4 Fusheng Zhou1,2,3,4*
Fusheng Zhou1,2,3,4*- 1Department of Dermatology, The First Affiliated Hospital, Anhui Medical University, Hefei, China
- 2Institute of Dermatology, Anhui Medical University, Hefei, China
- 3Key Laboratory of Dermatology (Anhui Medical University), Ministry of Education, Hefei, China
- 4Inflammation and Immune Mediated Diseases Laboratory of Anhui Province, Hefei, China
The inflammasome is an important protein complex that cleaves the proinflammatory cytokines pro-IL-1β and pro-IL-18 into their active forms. Owing to its critical role in eliciting innate immune responses, IL-1β has been suggested to contribute to various skin diseases, including psoriasis, vitiligo, systemic lupus erythematosus (SLE), and atopic dermatitis (AD). Recently, several types of activators and inhibitors of different inflammasomes, as well as inflammasome-related genes and genetic susceptibility loci, have been identified in these immune-related common skin diseases. In particular, inflammasome activators and inhibitors presented highly cell-type-specific activity, suggesting that the inflammasome might perform different functions in different cell types. Moreover, most of these findings were based on experimental disease models, and the clinical features of the models partly resemble the typical symptoms of the diseases. In this review, from the perspective of activators and inhibitors, we collected evidence from the widely-studied inflammasomes, NLRP3, AIM2, and NLRP1, in psoriasis, vitiligo, SLE, and AD. Importantly, some small-molecule inhibitors hold therapeutic promise for the treatment of these diseases.
Introduction
The human skin is composed of epidermal and dermal layers that function as the first line of defense against various physical, chemical, and biological threats. The major structure of the epidermis can be divided into four tightly-connected and stratified parts, namely, the stratum basale, stratum spinosum, stratum granulosum, and stratum corneum (1). Skin homeostasis is maintained by various stem cells, which are responsible for organ renewal and injury repair. The strong self-renewal activity of these cells results in different cell lineages that comprise the mature adult tissue (2). Keratinocytes are the main cell type found in the epidermal layer; however, human skin also contains different types of immune cells, such as memory αβ-T cells, dendritic cells (DCs), macrophages, natural killer (NK) cells, δγ-T cells, innate lymphoid cells (ILCs), and melanocytes (3, 4). These cell types coordinate cutaneous immune responses against external stimuli. Aberrant immunological activation by microorganisms or autoantigens can induce inflammatory skin disorders, and even cancers.
The inflammasome is a high-molecular-weight protein complex found mainly in the cytosol of stimulated immune cells, and plays an important role in activating immune cascades by processing and generating the catalytically active protease caspase-1 (5). Caspase-1 further initiates downstream responses through its substrates gasdermin-D, interleukin (IL)-1β, and IL-18 and induces a type of cell death called pyroptosis (6). The inflammasome is a sensor for monitoring extra- and intracellular compartments for signs of infection or tissue injury, and is therefore regarded as a key component of the innate immune system. When the inflammasome was first described by Martinon et al. (5), it was implicated in almost every immune-related phenotype, including tissue healing, metabolism, infection, homeostasis, and tumorigenesis (7). The past 15 years have seen great progress in deciphering the role of the inflammasome in the pathophysiology of common skin diseases. In this review, we mainly focus on two well-studied types of inflammasomes, NLRP3, and AIM2. For activation and regulation information of the various inflammasomes, please see the relevant reviews (8, 9). We also discuss the main findings of inflammasome-related genes in common immune-related skin diseases, including psoriasis, vitiligo, systemic lupus erythematosus (SLE), and atopic dermatitis (AD).
Basic Concepts of the Inflammasome
The inflammasome can recognize damage-associated molecular patterns (DAMPs) or uncontrolled release of pathogen-associated molecular patterns (PAMPs) through pattern recognition receptors (PRRs). At least five canonical and several non-canonical inflammasomes have been identified based on the type of PRR involved (9). The five canonical PRRs comprise nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), RIG-like receptors, absent in melanoma 2 (AIM2)-like receptors (ALRs), Toll-like receptors (TLRs), and pyrin. The canonical inflammasomes mainly activate the protease caspase-1, while non-canonical inflammasomes target caspase-11 in mice and caspase-4 and/or−5 in human cells (10, 11). In addition to the recognition receptors, canonical inflammasome assembly requires the apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and effector protein pro-caspase-1 (12). The inflammasome must be tightly regulated through transcriptional, translational, and posttranslational mechanisms owing to its critical importance in innate immunity. First, the IL-1 receptor or other cytokines trigger the expression of inflammasome components (ASC, pattern receptors, pro-caspase-1, pro-IL-1β, and pro-IL-18). Second, DAMP- and PAMP-mediated signaling initiates the assembly of the multiprotein inflammasome, which entails pro-caspase-1 activation and cleavage of pro-IL-1β and pro-IL-18 into their active forms (13) (Figure 1). Secreted cytokines then induce effector cells, such as neutrophils, macrophages, and keratinocytes, to instigate inflammatory responses in damaged tissue (14).
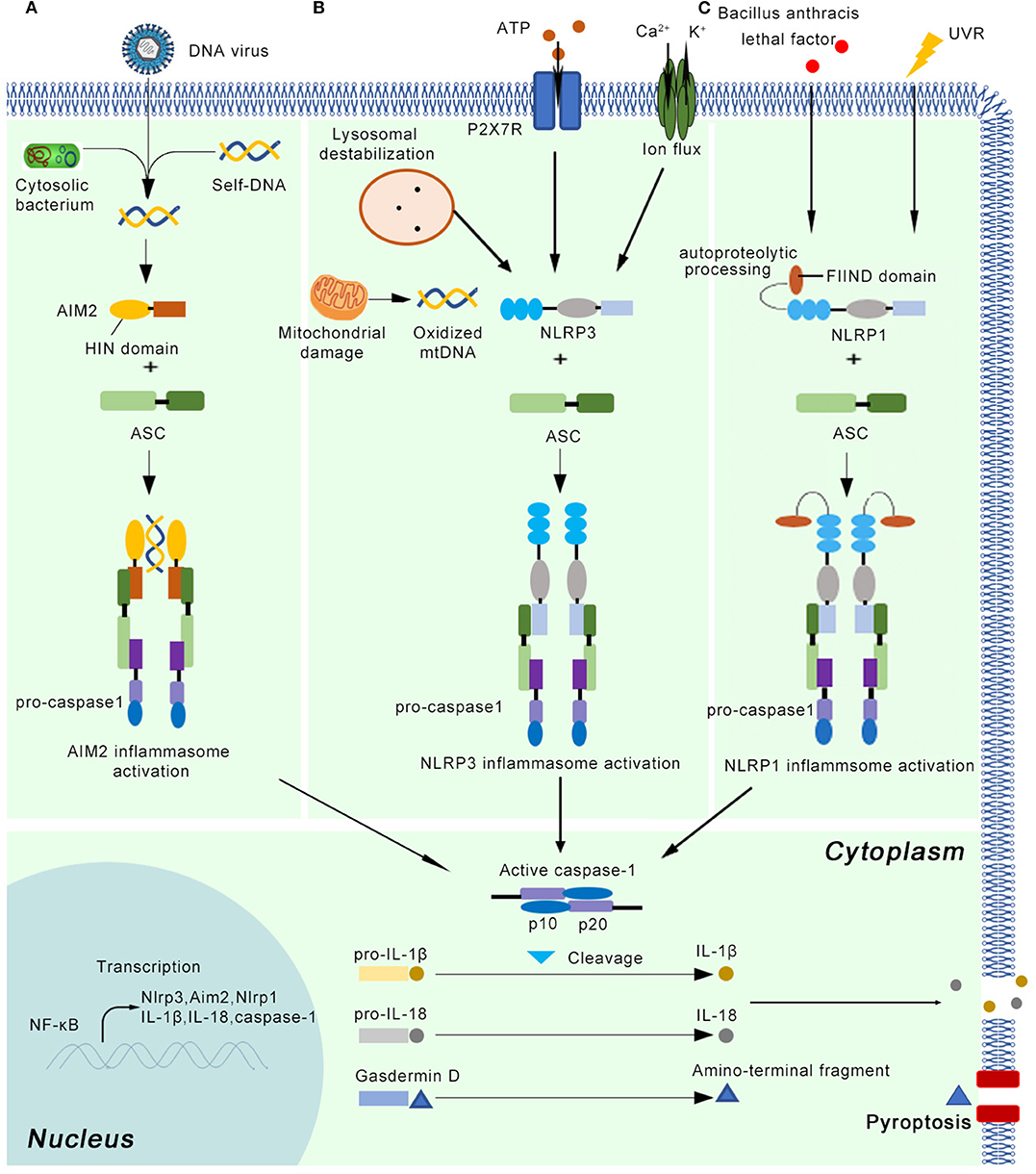
Figure 1. The basic concepts of the AIM2, NLRP3, and NLRP1 inflammasome. After priming by cytokine signals, the main components of the inflammasome (Nlrp3, Aim2, Nlrp1, pro-caspase-1, pro-IL-1β, and pro-IL-18) are transcribed in a NF-κB dependent manner. (A) The AIM2 inflammasome detects cytosolic dsDNA released from DNA viruses and cytosolic bacterium, as well as self-DNA. AIM2 senses and binds cytosolic dsDNA longer than 200 bp via its HIN domain. (B) The NLRP3 inflammasome can be activated by a variety of endogenous molecules, such as oxidized mitochondrial DNA, potassium efflux, extracellular ATP, lysosomal destabilization, intracellular calcium levels. (C) Autoproteolytic processing within the function-to-find domain (FIIND) is needed for the NLRP1 inflammasome activation. UV radiation and lethal factor of Bacillus anthracis can activate the NLRP1 inflammasome. Once the active inflammasome is formed, it directly recruits and cleaves pro-caspase1 into active caspase-1, which proteolytically activates the pro-inflammatory cytokines IL-1β and IL-18. In addition, the activated inflammasome cleaves gasdermin D into active N-terminal fragment, which drives a lytic type of cell death pyroptosis.
The NLRP3 Inflammasome
The NLRP3 inflammasome is the most complex and best-characterized member of the inflammasomes (5) (Figure 1). It can be primed by a wide range of extracellular inflammatory stimuli, such as bacteria, and viruses, as well as yeasts such as Candida albicans (15) and Malassezia spp. (16), in a NF-κB-independent manner (17). In addition, the NLRP3 inflammasome is activated in response to a variety of endogenous molecules indicative of tissue injury, such as oxidized mitochondrial DNA (18), potassium efflux (19), extracellular ATP (20), lysosomal destabilization (21), and intracellular calcium levels (22). The priming step results in the transcriptional induction of NLRP3 and activation of licensing receptors. Importantly, NLRP3 inflammasome activation can also be controlled by kinases such as Bruton's tyrosine kinase and JNK or Syk kinases through the recruitment of caspase-1 and regulation of ASC oligomerization, respectively (23–25). In human monocytes and macrophages, adenosine triphosphate (ATP) stimulation through P2X7R is also required to activate the NLRP3 inflammasome (26). After priming, NLRP3 oligomerization mediates the cleavage of pro-caspase-1, pro-IL-1β, and pro-IL-18 into their active forms (27). Although numerous regulators have been identified in both the priming and oligomerization stages, the exact mechanism by which NLRP3 is activated remains unclear (28).
The AIM2 Inflammasome
The AIM2 inflammasome contains AIM2 as the recognition receptor. AIM2 mainly detects cytosolic dsDNA released from viruses and intracellular bacteria, as well as self-DNA (29) (Figure 1). By inducing the expression of IRF1, the host system controls the expression of GTPases known as guanylate-binding proteins (GBPs), which facilitates the sensing of cytosolic dsDNA. AIM2 senses and binds cytosolic dsDNA longer than 200 bp via its HIN domain, providing an oligomerization template (30). However, the mechanisms underlying how external DNA is sensed by PPRs are normally species-dependent. For example, Francisella novicida infections activate the AIM2 inflammasome through the interferon-inducible proteins GBP2, GBP5, and IRGB10 (31, 32), whereas GBP1 is required for AIM2 inflammasome-mediated detection of Salmonella (33).
In addition to its role in detecting exogenous bacterial DNA, the AIM2 inflammasome has been suggested to monitor self-DNA delivered by exosomes or damaged DNA within the nucleus (34, 35). AIM2-deficient mice are protected from ionizing radiation-induced cell death and severe tissue damage, suggesting that AIM2 mediates inflammasome activation through sensing dsDNA damage induced by exposure to ionizing radiation (34). Treatment with the cytotoxic agent irinotecan (CPT-11) leads to considerable intestinal release of dsDNA through exosome secretion, which then enters into innate immune cells and triggers the AIM2 inflammasome-mediated secretion of mature IL-1β and IL-18 (35).
The NLRP1 Inflammasome
NLRP1 is another member of NLR family that forms a new kind of inflammasome in human. NLRP1 inflammasome can mediate homotypic interactions through the PYD domain, using the same strategy as NLRP3 inflammasome. Interestingly, unlike NLRP3 protein, NLRP1 also has a function-to-find domain (FIIND) and a caspase activation and recruitment domain (CARD) (5) (Figure 1). NLRP1 inflammasome also interacts with ASC via CARD domain and activates their proteolytic function, which means this inflammasome can activate caspase-1 by CARD domain without recruiting ASC (36). Except for protein-protein interaction, additional event is needed for NLRP1 inflammasome activation. It has been showed that autoproteolytic processing within FIIND domain is necessary for the NLRP1 activity (37). UVB irradiation can activate NLRP1 inflammasome in human primary keratinocytes but not human fibroblasts (38). However, it is argued that NLRP3 inflammasome might also induce secretion of IL-1β and IL-18, indicating further investigation is needed (39). The lethal factor of Bacillus anthracis can activate NLRP1 inflammasome in rodents (40). The activation of NLRP1 inflammasome results in downstream response, including release of active caspase-1 cleavage enzyme, IL-1β and IL-18, as well as the activation of pyroptosis (41).
Inflammasomes in Systemic Lupus Erythematosus
SLE is a severe, devastating heterogeneous autoimmune disease characterized by loss of tolerance to self-antigens, chronic inflammation, and strong interindividual variation. The pathophysiology of SLE is highly complex and remains incompletely understood; however, there is substantial evidence indicating that the inflammasome might be involved in regulating cytokine secretion and inducing chronic inflammation in this condition, thereby partly contributing to disease development (Figure 2). Kahlenberg et al. were the first to observe that the expression of IL-1β/IL-18 was upregulated in the serum of SLE patients. They also found that inhibition of caspase-1 induced the aberrant differentiation of endothelial progenitor cells (EPCs) and circulating angiogenic cells (CACs), suggesting that the inflammasome machinery was involved in SLE etiology (42). In a clinical trial, IL-18 was found to be a predictive marker for long-term renal outcome. After 6 months of treatment, the serum IL-18 level in pediatric-onset SLE patients was significantly correlated with SLE global disease activity and the severity of lupus nephritis, suggesting that the NLRP3 inflammasome might modulate SLE treatment through IL-18 (43). A recent study revealed that Casp1−/− mice were strongly protected against pristane-induced autoantibody development and type I interferon responses, indicating that caspase-1 is an essential component in lupus development (44).
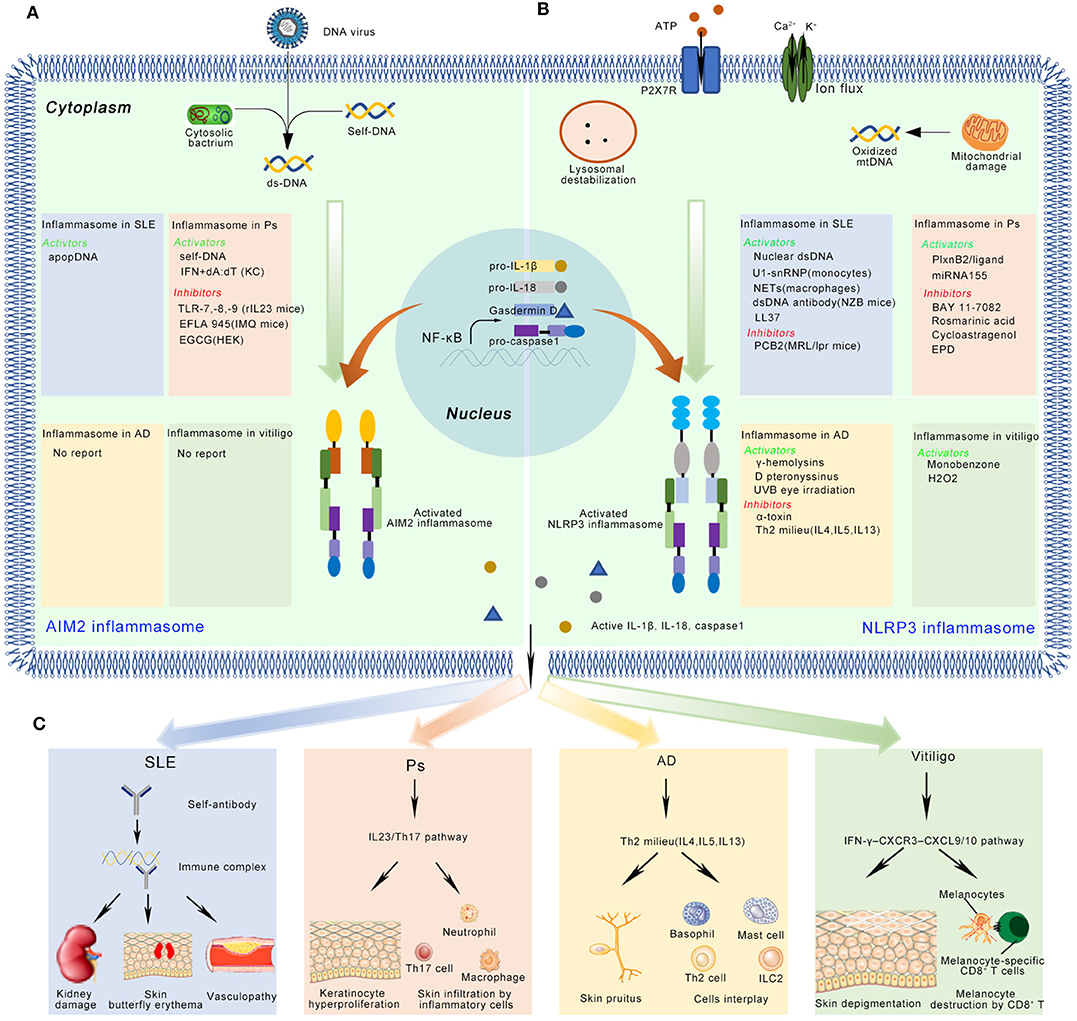
Figure 2. The activation and inhibition of the AIM2 and NLRP3 inflammasome in SLE, Ps, AD, and vitiligo. (A) In SLE, the AIM2 inflammasome can be activated by apopDNA in the patients. In psoriasis (Ps), the self-DNA in patients, dA:dT (after primed by IFN in keratinocyte) activate, while TLR-7/8/9, EFLA 945, and EGCG can inhibit the AIM2 inflammasome in Ps mice model or cultured cell lines. (B) In SLE, several NLRP3 activators (Nuclear dsDNA or dsDNA antibody, U1-snRNP, NETs, LL-37), and inhibitors (PCB2) have been identified in mice model or cell lines. In Ps, PlxnB2/ligand and miRNA155 activate the NLRP3 inflammasome, while BAY 11-7082, Rosmarinic acid, Cycloastragenol and EPD inhibit this inflammasome. In AD, activators (γ-hemolysins, D pteronyssinus, UVB eye irradiation) and inhibitors (α-toxin, Th2 milieu) of the NLRP3 inflammasome have been reported. In vitiligo, monobenzone, and H2O2 activate the NLRP3 inflammasome. (C) The active IL-1β and IL-18 might play multiple functions in immune skin disorders. Here, we showed their potential roles in the disease pathogenesis. In SLE, autoantibody binds with apopDNA, and generates immune complex. The immune complex might cause kidney damage, skin butterfly erythema and vasculopathy when it is deposited into different tissues. In Ps, IL-1β activates IL23/Th17 pathway, inducing a large number of inflammatory cytokines and chemokines. Neutrophils, Th17 cells, macrophages, and some other kind of immune cells infiltrate into skin. These immune cells crosstalk with keratinocytes and finally cause hyperproliferation in epidermis. In AD, the inflammasome regulates disease mainly through Th2 milieu. The typical symptoms include chronic, pruritic eczematous skin and the elevated serum concentrations of IgE. In vitiligo, the disease is regulated by IFN-γ-CXCR3–CXCL9/10 axis. The skin-resident melanocytes-specific CD8+ cells kill melanocytes, and finally cause depigmented skin patches.
Genetic Evidence for Inflammasome Involvement in SLE
SLE has a strong genetic background, and more than a 100 susceptibility loci have been identified (45). Some of these loci are located near or within genes coding primarily for inflammasome components (Table 1). Two genetic association studies revealed that variations in IL1B and NLRP1 are associated with SLE in Brazilian cohorts (47, 51). Several studies confirmed that genetic variation in the type I IFN signaling pathway increased the risk for developing SLE in humans and model mice (61, 62). Polymorphisms in two key receptors for NLRP3 priming, namely, P2X7R and TLR-9, are found to be associated with SLE in patients of different ethnicities (46, 48–50).
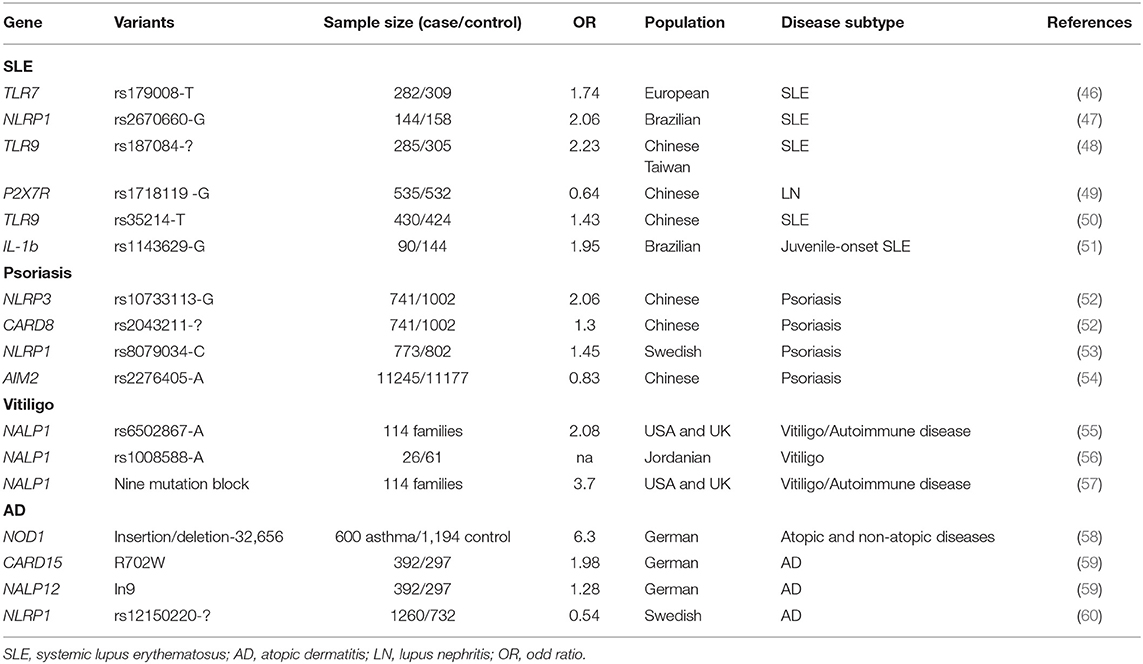
Table 1. Genetic evidence for inflammasome involvement in immune-related skin diseases.
The NLRP3 Inflammasome in SLE
Several studies have suggested that NLRP3 has an important role in SLE (Table 2). As the studies used more than one cell type and mouse model, various inflammasome aspects can be viewed from different directions. We subjectively divided these studies into three categories based on study design: studies mainly based on cultured cells; those mainly based on SLE-like mouse models; and those based on mice carrying specific gene deficiencies.
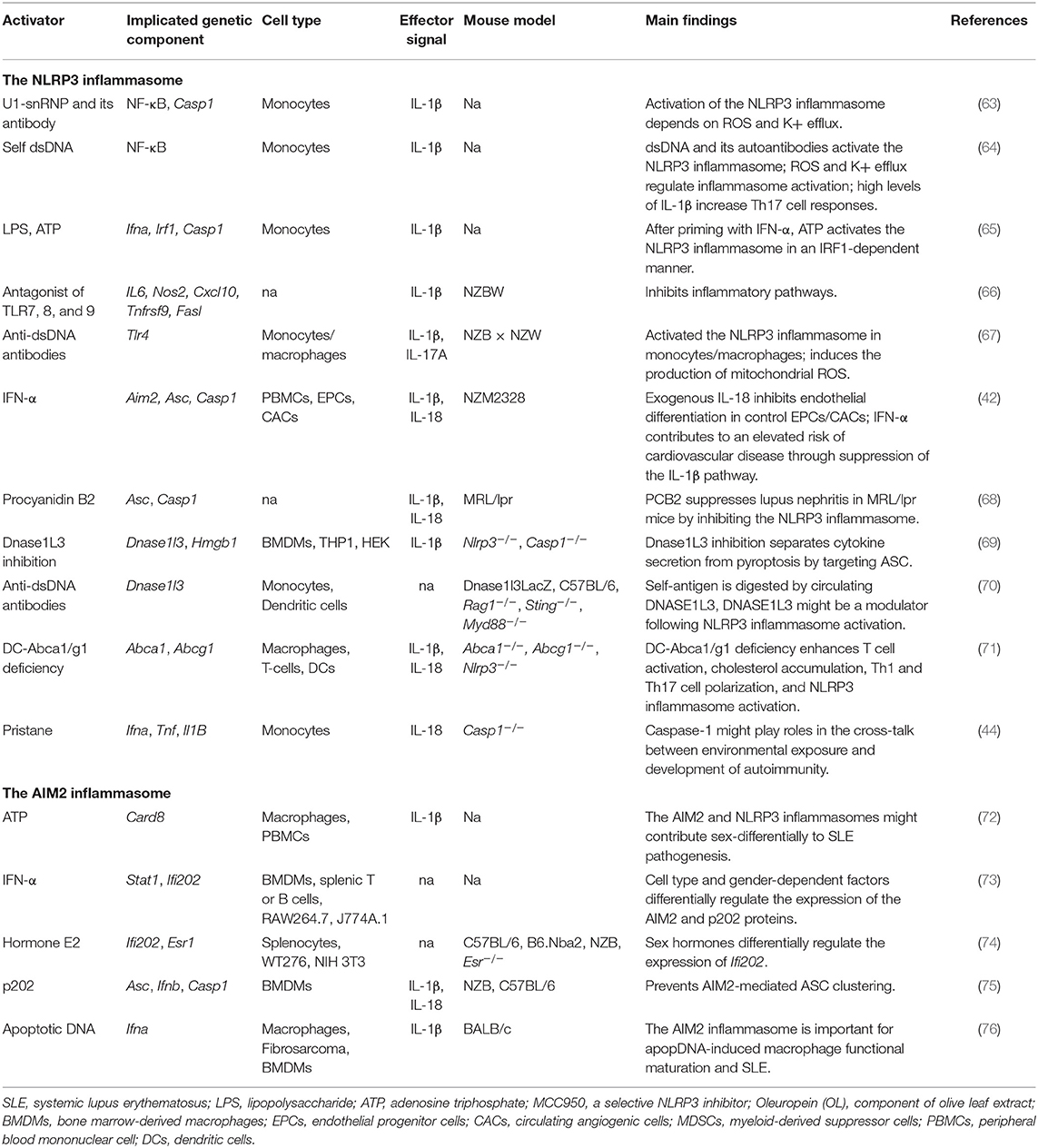
Table 2. The inflammasome in SLE.
Studies Based on Cultured Cells
Some monocyte-based studies have indicated the importance of the inflammasome in SLE. Nuclear dsDNA was shown to induce IL-1β secretion from human monocytes by activating the NLRP3 inflammasome, and this activation was modulated by reactive oxygen species (ROS) and K+ efflux. NLRP3 inflammasome activation led to increased IL-17 production from CD4+ T cells and triggered the downstream immune cascade (64). In addition to the endogenous DNA- and microbial nucleic acid-mediated inflammasome activation, endogenous U1-small nuclear ribonucleoprotein (U1-snRNP) was shown to activate the NLRP3 inflammasome in monocytes in the presence of anti-U1-snRNP antibodies (63). Type I interferons (IFNs) are important mediators of SLE. Two recent studies revealed that IFN-alpha levels are negatively correlated with the expression of NLRP3/NLRP1 inflammasomes. Exposure to IFN-alpha primed monocytes for inflammasome activation in an IFN regulatory factor 1 (IRF1)-dependent manner (65, 77).
Studies Based on SLE-Like Mouse Models
To date, several SLE-like mouse models have been developed, and while most partially mimic the clinical symptoms of this disease, none represent the entire spectra observed in SLE patients. Three types of spontaneous or induced models, NZB/NZM, MRL/lpr, and pristane-induced lupus, are widely used in inflammasome studies. Although these mice share some symptoms, they each present with specific clinical-like manifestations. NZB and NZM mice exhibit lymphadenopathy, anti-dsDNA IgG, and immune complex-mediated glomerulonephritis (78, 79), while MRL/lpr mice present with lymphadenopathy, DNA and RNA-directed autoantibodies, glomerulonephritis, and dermatitis (80). After pristane injection, the autoantibodies appeared in mice, along with the glomerulonephritis, arthritis, and anemia, and most of these phenotypes were type I interferon-mediated (81). Different types of inflammasome activators were identified using these models.
NZB and NZM mice
Antagonists of TLR-7,−8, and−9 inhibit NLRP3 inflammasome-related pathways in NZBW F1 lupus model mice, and therefore represent a potential therapeutic approach for lupus treatment (66). Furthermore, NZB mice exhibited an increase in IL-1β and IL-17A concentrations and the Th17/Treg cell ratio following injection of anti-dsDNA antibodies, suggesting that the NLRP3 inflammasome is involved in lupus pathogenesis in mice (67). Kahlenberg et al. found that neutrophil extracellular traps (NETs) can activate the NLRP3 inflammasome in lupus-affected macrophages, while LL-37 was shown to activate the NLRP3 inflammasome through P2X7 receptor-mediated potassium efflux (42).
MRL/lpr mice
Some SLE patients also displayed psychosis, seizures, and cognitive dysfunctions. MRL/lpr-derived strains have some advantages in the examination of these neuropsychiatric symptoms. MRL/lpr mice treated with procyanidin B2 (PCB2) showed reduced serum levels of IL-1β and IL-18 when compared with Nlrp3-deficient mice (68).
Studies Based on Mice Carrying Specific Gene Deficiencies
Dnase1L3 inhibition can block both NLRP3 and NLRC4 inflammasome-mediated secretion of IL-1β by targeting the ASC recruitment domain; however, it has little effect on NLRP3-dependent pyroptosis (69). Dnase1l3-deficient mice, a model of pediatric-onset SLE, showed the early presence of anti-dsDNA and anti-chromatin antibodies, suggesting that Dnase1L3 might function as a key modulator following NLRP3 inflammasome activation (70). Deletion of Abca1/Abcg1 induces enlarged lymph nodes and enhanced Th1 cell polarization. In this SLE-like model, the NLRP3 inflammasome was activated in dendritic cells but not in macrophages or T cells. NLRP3 deficiency significantly diminished the SLE-like symptoms, suggestive of the importance of regulating Abca1/g1 in dendritic cells (71). A recent study revealed that Casp1−/− mice are strongly protected against pristane-induced autoantibodies and type I IFN, indicating that caspase-1 is an essential component in lupus development (44).
The AIM2 Inflammasome in SLE
Studies Based on Cultured Cells
Accumulating evidence has suggested that the AIM2 inflammasome contributes to SLE pathogenesis (82) (Table 2). The levels of AIM2 mRNA are upregulated in the liver, PBMCs, and spleen of SLE patients when compared with healthy individuals (83). AIM2 mRNA expression is upregulated in macrophages derived from male, but not female, SLE patients after stimulation with 2 mM adenosine triphosphate (ATP), suggesting that the AIM2 inflammasome contributes to SLE in a gender-dependent manner (72). Similarly, Panchanathan et al. found that the expression of AIM2 can be induced in bone marrow-derived macrophages (BMDMs), but not splenic T or B cells, indicating that the AIM2 inflammasome is activated in a cell-specific manner (73).
Studies Based on Mouse Models
Recent studies have shown that p202 inhibits AIM2 inflammasome activation in response to cytosolic DNA (74, 75). IFN-inducible PYHIN and p202 are also associated with SLE (84). Macrophages derived from lupus nephritis-affected mice can be activated by apoptotic DNA (apopDNA). AIM2 expression is closely correlated with macrophage activation, and inhibition of AIM2 expression significantly ameliorates SLE syndrome in apopDNA-induced, lupus-affected mice (76).
The Inflammasome in Psoriasis
Psoriasis is a common inflammatory skin disease characterized by red scaly papules and plaques. The key features of psoriasis include aberrant proliferation and differentiation of keratinocytes; excessive infiltration of immune cells, such as T cells and DCs, into the skin; and production of various inflammatory cytokines and chemokines (1). TNF, IL-23, and IL-17 are key cytokines for disease development. The antimicrobial peptide (AMP) LL-37 can aggregate with dsDNA and initiate a cutaneous self-amplifying autoimmune reaction (85). Skin keratinocytes are key proinflammatory cells that respond to harmful insults through the coordinated production of cytokines, chemokines, and AMPs (86). The crosstalk between infiltrated immune cells and keratinocytes is central to the IL-17-mediated inflammatory response in psoriasis (87).
The levels of key inflammasome components, including NLRC4, NOD2, CARD6, and IFI16, are elevated in psoriatic epidermis (88, 90). Recently, several studies have reported that the AIM2 inflammasome is an important component of skin innate immunity. Kopfnagel found that human keratinocytes express AIM2 and respond to dsDNA with IL-1β secretion, indicating that the AIM2 inflammasome is a trigger for skin inflammation (91) (Figure 2).
Genetic Evidence for Inflammasome Involvement in Psoriasis
NLRP3 rs10733113 and CARD8 rs2043211 were found to increase the risk of psoriasis in a Swedish population, supporting the hypothesis that inflammasome variation predisposes individuals to psoriasis (52). Similarly, there is some evidence that NLRP1 rs8079034 also predisposes to psoriasis (53). Our group conducted a large-scale genome-wide association study in a Chinese population, and found that the rs2276405 AIM2 coding variant significantly increased the genetic risk for psoriasis in AA allele carriers (54) (Table 1).
The NLRP3 Inflammasome in Psoriasis
Although more than 40 psoriasis-like mouse models have been developed, imiquimod (IMQ)- and IL-23-induced models, or mice with specific gene deficiencies, are the most commonly used models in inflammasome studies (92). Various factors (microRNAs [miRNAs], genes, drugs) have been reported to activate or inhibit the NLRP3 inflammasome in cultured human keratinocytes or mice with IMQ-induced psoriasis (Table 3). A recent study reported that BAY 11-7082, an antagonist of NF-κB, can alleviate psoriasis-like dermatitis by inhibiting the NLRP3 inflammasome and the NF-κB pathway (97). PlxnB2 and its ligand were reported to activate inflammatory responses in keratinocytes through the NLRP3 inflammasome and the NF-κB pathway (94). In human primary keratinocytes, miRNA155 suppressor, rosmarinic acid (RA), cycloastragenol (CAG), and the effective part of Datura metel L. (EPD) have been shown to inhibit NLRP3-induced inflammatory cytokines such as IL-1β, IL-6, IL-8, CCL20, and TNF, indicating that they are therapeutic candidates for psoriasis treatment (89, 93, 95, 96). Different from that observed in the IMQ-induced psoriasis mouse model, recombinant IL (rIL)-23-induced psoriasiform dermatitis is largely dependent on the P2X7R signaling pathway. When Nlrp3-deficient mice were injected with rIL-23, psoriasiform phenotypes were considerably ameliorated, suggesting that the NLRP3 inflammasome contributes to this process. Diaz-Perez et al. further found that activation of the NLRP3 inflammasome occurred mainly through neutrophils and not keratinocytes or T cells (101). These findings supported that NLRP3 inflammasome activation might be primarily dependent on the source of activators and different mouse models used in these studies.
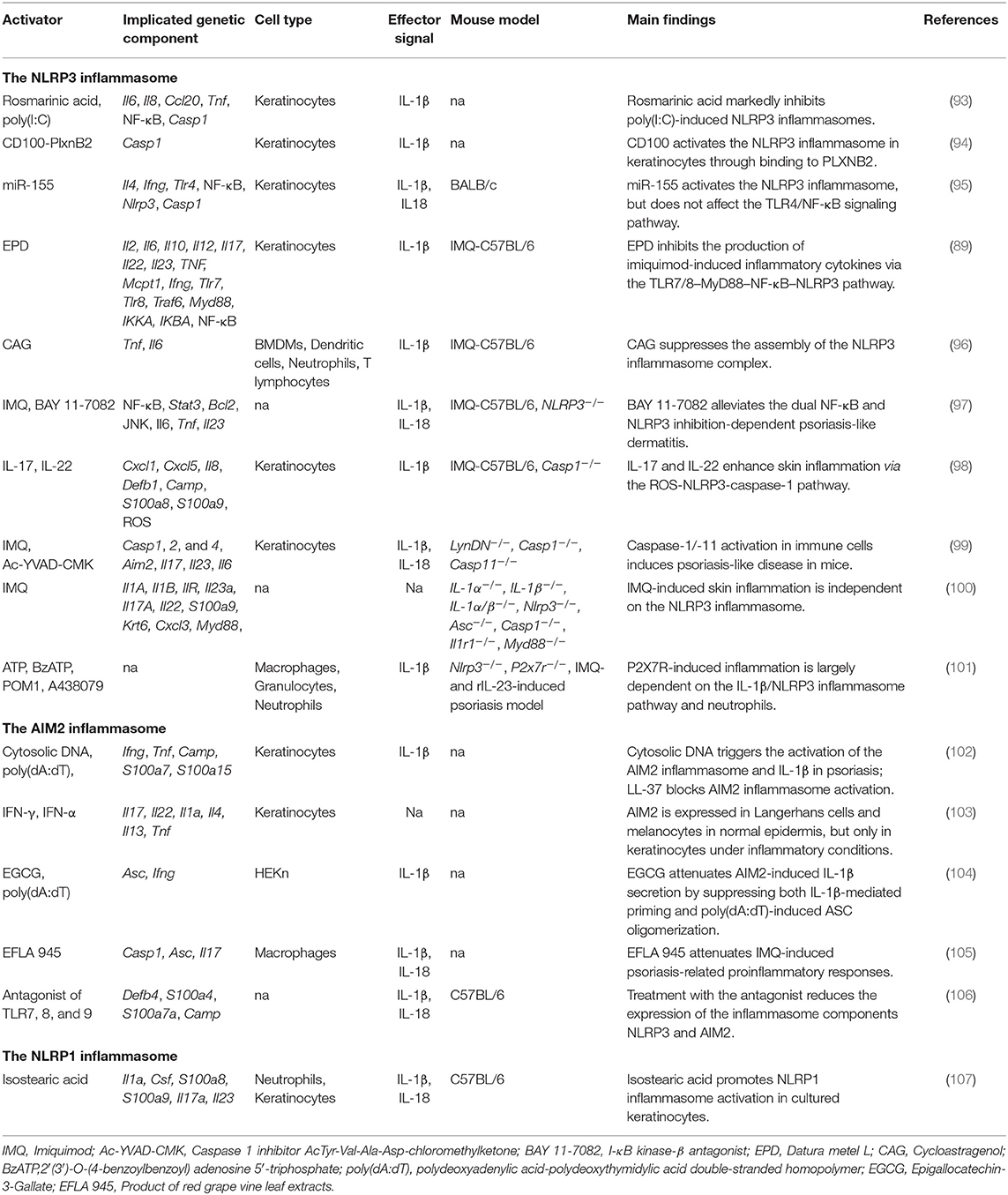
Table 3. The inflammasome in psoriasis.
Although it is known that the inflammasome contributes to the pathogenesis of psoriasis, the role of caspases in disease development remains controversial. In IMQ-treated Nlrp3−/− mice, psoriasiform lesions were comparable to those of normal controls; however, caspase-1 activity in the skin was markedly decreased, indicating that the NLRP3 inflammasome was required for caspase-1 activation, but dispensable for skin inflammation (100). However, Cho found that the NLRP3 inflammasome can be activated in IMQ-treated caspase-1-deficient mice, although the severity of psoriasis was much lower than in wild-type mice (98). Meanwhile, the functions of inflammation-related caspases were shown to be highly cell-type-specific. Activation of proinflammatory caspase-1 and caspase-11 in immune cells is sufficient to induce a psoriasis-like phenotype; however, in a psoriasis mouse model, these caspases are dispensable for inflammasome activation in keratinocytes/fibroblasts (99).
The AIM2 Inflammasome in Psoriasis
Cytosolic DNA of psoriatic skin triggers inflammation through activation of the AIM2 inflammasome and IL-1β (Table 3). In cultured keratinocytes, however, transfection of poly (dA:dT) induced IL-1β secretion only after priming by interferon gamma, suggesting that a proinflammatory cytokine microenvironment is essential for AIM2 inflammasome activation (102). Koning showed that AIM2 is expressed exclusively in Langerhans and melanocyte cells in normal epidermis, but is significantly upregulated in keratinocytes under inflammatory conditions such as psoriasis, AD, and allergic contact dermatitis (103). In an rIL-23-induced psoriasis mouse model, a TLR-7,−8, and−9 antagonist inhibited the dermal expression of Nlrp3 and Aim2 and reduced the secretion of Th1 and Th17 cytokines in skin and serum, suggesting that inflammasomes might be a therapeutic target for psoriasis treatment (106). Because murine keratinocytes do not express pro-IL-1β, IL-18 is the only cytokine that can be cleaved into its active form by inflammasomes in these cells (114). Recently, Chung reported that red vine leaf extract (EFLA 945) greatly attenuated IMQ-induced psoriasis phenotypes by inhibiting the activity of the AIM2 inflammasome (105). Epigallocatechin-3-gallate (EGCG) has been shown to inhibit AIM2-induced inflammatory cytokines, and attenuate caspase-1 activation in interferon gamma-primed HEKn cells (104).
IL-18 receptor knockout mice treated with Aldara exhibited thicker epidermis than that seen in normal controls. Aldara, a type of cream composed mainly of isostearic acid and IMQ, can be used to induce psoriasis-like lesions. Walter et al. found that isostearic acid was the key component in activating the NLRP1 inflammasome in a mouse model, indicating that Aldara might stimulate psoriasis-like phenotypes in different immune pathways requiring both inflammasome and IMQ-induced response (107).
The Inflammasome in Vitiligo
Vitiligo is an autoimmune skin disease characterized by the destruction of skin melanocytes and the presence of patchy white spots on the skin. Although the etiology of vitiligo has not been fully elucidated, evidence indicates that both genetic and environmental factors contribute to disease susceptibility (107). The IFN-γ-CXCR3–CXCL9/10 axis is suggested to be key for triggering skin inflammation by recruiting autoreactive CD8+ T cells (115) (Figure 2). Recently, Richmond et al. found that skin-resident memory CD8+ cells could not kill melanocytes by themselves, which should cooperate with recirculating memory CD8+ T cells so as to maintain the disease (116).
Genetic Evidence for Inflammasome Involvement in Vitiligo
Relatively few studies have investigated the role of the inflammasome in melanocytes and/or vitiligo. Furthermore, most findings have come from genetic association studies (Table 1). In 2007, Jin et al. performed a genetic linkage, family-based association, target-region sequencing study on vitiligo-associated multiple autoimmune diseases, and found that single nucleotide polymorphisms (SNPs) near and within NALP1 were associated with vitiligo in Caucasian patients (55). This was the first evidence indicating that the inflammasome had a role in vitiligo pathogenesis. The same research group identified that peripheral blood monocytes expressing the NALP1 high-risk haplotype, covering L155H and M1184V substitutions, secreted a significantly greater amount of mature, bioactive IL-1β than those of other haplotype carriers. The high-risk haplotype did not lead to altered NALP1 mRNA or protein levels, indicating that this haplotype functions mainly through regulation of the NALP1 inflammasome (57). Consistent with their findings, NALP1 variants were also reported to be associated with Jordanian Arab vitiligo patients (56).
The NLRP3 Inflammasome in Vitiligo
In 2016, van den Boorn et al. found that monobenzone treatment resulted in melanocyte-specific skin inflammation characterized by macrophage infiltration and NK cell activation. Meanwhile, cutaneous lymph nodes showed an inflammasome-dependent influx of macrophages with a tissue-resident phenotype (108). However, recruitment of NK cells into the ear during monobenzone treatment was significantly inhibited in Nlrp3-deficient mice, suggesting that the NLRP3 inflammasome is key to monobenzone-induced inflammation in melanocytes. This indicates that the NLRP3 inflammasome and its downstream cytokines may be promising therapeutic targets for vitiligo treatment (108). Recently, Li et al. found that NLRP3 inflammasome activation was needed to promote innate immunity in keratinocytes. Deactivation of the NLRP3 inflammasome impaired CD8+ T cell recruitment and inhibited cytokine secretion in T cells derived from vitiligo patients (109) (Table 4).
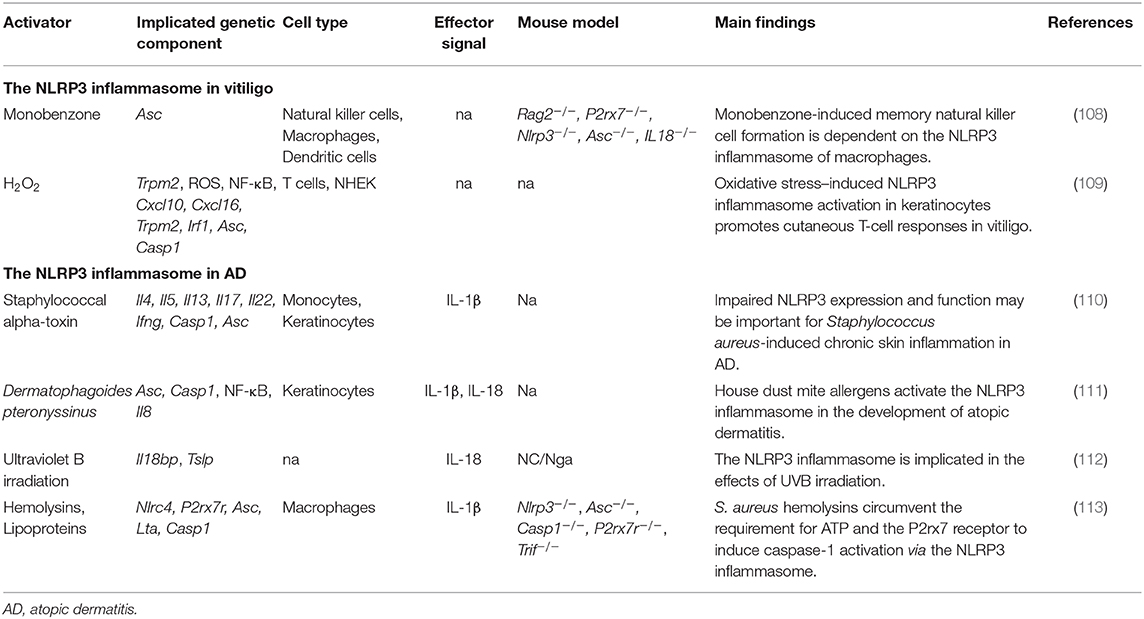
Table 4. The inflammasome in vitiligo and AD.
Other Types of Inflammasomes in Vitiligo
Two studies reported that NLRP1 levels were upregulated in both melanocytes and keratinocytes at the edge of progressing vitiligo lesions, which suggested that the NLRP1 inflammasome might drive the disease via two pathways (117, 118). In addition, NLRP1 and IL-1β levels in the skin may represent better markers than detection of lymphocyte infiltration to monitor vitiligo activity (117). There is some evidence to suggest that elevated IL-17 levels play important roles in stimulating inflammasome activation during vitiligo development (119, 120).
The Inflammasome in Atopic Dermatitis
AD is a chronic inflammatory dermatosis characterized by pruritic eczematous skin lesions and increased serum concentrations of immunoglobulin E (IgE) (121). The levels of AIM2 were found to be increased in keratinocytes derived from psoriasis and AD patients, which led to acute and chronic skin barrier disruption-related inflammation (103). Like most immune-related diseases, AD is affected by both genetic and environmental factors. The house dust mite and Staphylococcus aureus are common external agents that can trigger AD. Type 2 helper T cell-induced (Th-2 type) inflammation was reported to be essential for AD pathogenesis (122). IL-4, IL-13, and TSLP (thymic stromal lymphopoietin) are key players in Th-2 cell fate determination and in inducing the expression of IgE (1, 123). Meanwhile, skin barrier dysfunction is considered to be an important factor in AD etiology. For example, a nonsense mutation in the gene coding for the skin barrier protein filaggrin (FLG) can be observed in 20–40% of AD patients (124). Mutations in other genes such as SPINK5 and DSG1 that play roles in regulating stratum corneum formation or maintaining epithelial cell–cell tight junctions, also result in AD or AD-like dermatosis (125, 126).
Several studies have suggested that inflammasomes play key roles in disease development. In both human and mouse skin, the expression of IL-1A, IL-1B, IL-18, and IL-1RA was significantly higher in FLG mutant homozygous carriers than in either heterozygous carriers or wild-type subjects (127). AD-like dermatitis induced through FLG-deficiency was dependent on IL-1β and IL-1R1 signaling, but not NLRP3 inflammasome activation (128). Schuepbach-Mallepell found that the inflammasome inhibited the expression of TSLP and had a role in regulating Th1 and Th2 cell fate in the skin (129). In a chronic proliferative dermatitis animal model, Nlrp3- or Casp1- and -11-deficient mice showed reduced skin inflammation and delayed disease onset, suggesting that the inflammasome might be an important trigger for disease development (130).
Genetic Evidence for Inflammasome Involvement in AD
Several studies have revealed the inflammasome-related genetic variation involved in AD pathogenesis (Table 1). The NLRP1 coding variant rs12150220 showed a strong association with AD in a Swedish population. This SNP was located between the PYRIN and NACHT domains, potentially affecting inflammasome oligomerization (60). Polymorphisms in NOD1 and CARD15, two components involved in inflammasome assembly, increased the risk for AD in different populations (58, 131). Recently, NLRP3 polymorphisms have also been reported to be associated with AD (59, 132).
The NLRP3 Inflammasome in AD
The expression levels of NLRP3 and caspase-1 are lower in AD-affected skin than in healthy skin. Moreover, the gene expression of NLRP3 and ASC was significantly reduced in human keratinocytes stimulated with Th2 cytokines (IL-4, IL-5, and IL-13) (110). Several inflammasome activators have been found to be triggers for skin inflammation in AD (Figure 2). However, the reported effects have been contradictory. For example, Munoz-Planillo et al. found that S. aureus gamma-hemolysins alone could activate NLRP3 inflammasomes in macrophage cells. Alpha- and beta-hemolysins could also trigger inflammasome activation when coupled with bacterial lipoproteins. Interestingly, this activation was not dependent on the P2X7 receptor or the TLR adaptor MyD88, suggesting that S. aureus hemolysins might circumvent the requirement for the P2X7 receptor to activate the NLRP3 inflammasome (113). Inconsistent with this finding, Niebuhr et al. found that the S. aureus exotoxin, alpha-toxin, inhibited NLRP3 inflammasome activity by suppressing the expression of NLRP3, ASC, caspase-1, and IL1B in keratinocytes. The same trend was found in monocytes treated with Th2 milieu (LTA+alpha-toxin+IL-4, IL-5, and IL-13), suggesting that NLRP3 inflammasome impairment might contribute to skin inflammation in AD (110) (Figure 2). Dermatophagoides pteronyssinus activated the NLRP3 inflammasome in keratinocytes, and the released proinflammatory cytokines, IL-1β and IL-18, exacerbated the AD-associated symptoms (111). UVB eye irradiation was reported to aggravate AD symptoms through the NLRP3 inflammasome (112) (Table 4).
Conclusion
In the past 15 years, great progress has been made in discovering new inflammasome components and identifying new activators that trigger inflammation or stimulate the innate immune system in various organs, mouse models, and cell types. In this study, we reviewed the known canonical inflammasomes, especially the NLRP3 and AIM2 inflammasomes, and their roles in common immune-related skin diseases. After inflammasome-mediated cleavage, the proinflammatory cytokines IL-1β and IL-18 are processed into their active forms and further initiate downstream inflammation cascades, such as IL-23/Th17 signaling, in skin tissue.
For the important roles of IL-1β and IL-18 in immune-related skin disorders, several therapeutic drugs targeting of inflammasome components have been developed. For example, canakinumab, an anti-IL-1β monoclonal antibody, has been used to treat a generalized pustular psoriasis patient and resulted in complete remission of the lesions (133). P2X7 and EGCG have been shown to attenuate murine lupus symptoms by inhibiting the activation of NLRP3 inflammasome, thus can be viewed as a promising therapeutic agent in SLE treatment (134–136).
Several key points should be mentioned following this review. First, even though the inflammasome is essential for IL-1β and IL-18 cleavage, some inflammasome-independent mechanisms have been reported for IL-1β and IL-18 processing. For example, neutrophil- and macrophage-derived neutral serine proteinases, such as proteinase 3 (PR3) and cathepsin-G, can cleave pro-IL-1β into its bioactive form (137). Second, although at least 10 different types of inflammasome have been identified, most have not been extensively characterized (8, 9). It is not known whether they play roles in skin diseases similar to those played by AIM2 and NLRP3 inflammasomes, which requires further investigation. Third, activators and/or inflammasome types are highly cell-type specific. Inflammasome activators cannot initiate an immune response in all cell types, indicating that therapeutic inflammasome inhibitors, such as caspase-1/IL-1β inhibitors, may be effective in one cell type but not in others. This is very important in the clinical treatment of the relevant disease, because proper pharmacological inhibitors must be chosen that are likely to depend on the type of inflammatory infiltrate (138). Finally, because skin biology differs between humans and mice, caution must be exercised when translating experimental data from mouse models to humans. Although some inflammasome inhibitors show strong anti-inflammatory efficacy in mice, direct evidence for a similar effect in treating human skin diseases is still lacking.
Author Contributions
FZ contributed to the conception and design of the paper, provided approval for publication of the content, and agreed to be accountable for all aspects of the work. LT drafted the work and revised it critically for the content.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Dainichi T, Kitoh A, Otsuka A, Nakajima S, Nomura T, Kaplan DH, et al. The epithelial immune microenvironment (EIME) in atopic dermatitis and psoriasis. Nat Immunol. (2018) 19:1286–98. doi: 10.1038/s41590–018–0256–2
2. Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. (2009) 10:207–17. doi: 10.1038/nrm2636
3. Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol. (2014) 14:289–301. doi: 10.1038/nri3646
4. Kupper TS, Fuhlbrigge RC. Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev Immunol. (2004) 4:211–22. doi: 10.1038/nri1310
5. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL–beta. Molecular cell. (2002) 10:417–26. doi: 10.1016/s1097–276500599–3
6. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase−11 cleaves gasdermin D for non–canonical inflammasome signalling. Nature. (2015) 526:666–71. doi: 10.1038/nature15541
7. Rathinam VAK, Chan FK. Inflammasome, inflammation, and tissue homeostasis. Trends Mol Med. (2018) 24:304–18. doi: 10.1016/j.molmed.2018.01.004
8. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. (2013) 13:397–411. doi: 10.1038/nri3452
9. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
10. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non–canonical inflammasome activation targets caspase-11. Nature. (2011) 479:117–21. doi: 10.1038/nature10558
11. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. (2013) 341:1246–9. doi: 10.1126/science.1240248
12. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577–019–0165–0
13. Wree A, Marra F. The inflammasome in liver disease. J Hepatol. (2016) 65:1055–6. doi: 10.1016/j.jhep.2016.07.002
14. Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
15. Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, et al. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe. (2009) 5:487–97. doi: 10.1016/j.chom.2009.05.002
16. Kistowska M, Fenini G, Jankovic D, Feldmeyer L, Kerl K, Bosshard P, et al. Malassezia yeasts activate the NLRP3 inflammasome in antigen–presenting cells via Syk–kinase signalling. Exp Dermatol. (2014) 23:884–9. doi: 10.1111/exd.12552
17. Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol. (2012) 13:333–42. doi: 10.1038/ni.2237
18. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
19. Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. (2007) 14:1583–9. doi: 10.1038/sj.cdd.4402195
20. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. (2006) 440:228–32. doi: 10.1038/nature04515
21. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. (2008) 9:847–56. doi: 10.1038/ni.1631
22. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci USA. (2012) 109:11282–7. doi: 10.1073/pnas.1117765109
23. Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, et al. Bruton's tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun. (2015) 6:7360. doi: 10.1038/ncomms8360
24. Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez-Cuellar E, Shen Y, et al. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck–like aggregates and inflammasome activity. Nat Immunol. (2013) 14:1247–55. doi: 10.1038/ni.2749
25. Okada M, Matsuzawa A, Yoshimura A, Ichijo H. The lysosome rupture–activated TAK1–JNK pathway regulates NLRP3 inflammasome activation. J Biol Chem. (2014) 289:32926–36. doi: 10.1074/jbc.M114.579961
26. Netea MG, Nold–Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, et al. Differential requirement for the activation of the inflammasome for processing and release of IL−1beta in monocytes and macrophages. Blood. (2009) 113:2324–35. doi: 10.1182/blood−2008–03–146720
27. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
28. He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. (2016) 530:354–7. doi: 10.1038/nature16959
29. Hornung V, Ablasser A, Charrel–Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase−1–activating inflammasome with ASC. Nature. (2009) 458:514–8. doi: 10.1038/nature07725
30. Morrone SR, Matyszewski M, Yu X, Delannoy M, Egelman EH, Sohn J. Assembly–driven activation of the AIM2 foreign–dsDNA sensor provides a polymerization template for downstream ASC. Nat Commun. (2015) 6:7827. doi: 10.1038/ncomms8827
31. Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Ruhl S, et al. Guanylate–binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol. (2015) 16:476–84. doi: 10.1038/ni.3119
32. Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, et al. IRGB10 liberates bacterial ligands for sensing by the aim2 and caspase−11–NLRP3 Inflammasomes. Cell. (2016) 167:382–96. doi: 10.1016/j.cell.2016.09.012
33. Fisch D, Bando H, Clough B, Hornung V, Yamamoto M, Shenoy AR, et al. Human GBP1 is a microbe–specific gatekeeper of macrophage apoptosis and pyroptosis. EMBO J. (2019) 38:e100926. doi: 10.15252/embj.2018100926
34. Hu B, Jin C, Li HB, Tong J, Ouyang X, Cetinbas NM, et al. The DNA–sensing AIM2 inflammasome controls radiation–induced cell death and tissue injury. Science. (2016) 354:765–8. doi: 10.1126/science.aaf7532
35. Lian TF, Xu YP, Li LF, Su XD. Crystal structure of tetrameric arabidopsis MYC2 reveals the mechanism of enhanced interaction with DNA. Cell Rep. (2017) 19:1334–42. doi: 10.1016/j.celrep.2017.04.057
36. Guey B, Bodnar M, Manie SN, Tardivel A, Petrilli V. Caspase−1 autoproteolysis is differentially required for NLRP1b and NLRP3 inflammasome function. Proc Natl Acad Sci USA. (2014) 111:17254–9. doi: 10.1073/pnas.1415756111
37. Finger JN, Lich JD, Dare LC, Cook MN, Brown KK, Duraiswami C, et al. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. The J Biol Chem. (2012) 287:25030–7. doi: 10.1074/jbc.M112.378323
38. Sand J, Haertel E, Biedermann T, Contassot E, Reichmann E, French LE, et al. Expression of inflammasome proteins and inflammasome activation occurs in human, but not in murine keratinocytes. Cell Death Dis. (2018) 9:24. doi: 10.1038/s41419–017–0009–4
39. Hasegawa T, Nakashima M, Suzuki Y. Nuclear DNA damage–triggered NLRP3 inflammasome activation promotes UVB–induced inflammatory responses in human keratinocytes. Biochem Biophys Res Commun. (2016) 477:329–35. doi: 10.1016/j.bbrc.2016.06.106
40. Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathogens. (2012) 8:e1002638. doi: 10.1371/journal.ppat.1002638
41. Zhao D, Wu Y, Zhuang J, Xu C, Zhang F. Activation of NLRP1 and NLRP3 inflammasomes contributed to cyclic stretch–induced pyroptosis and release of IL−1beta in human periodontal ligament cells. Oncotarget. (2016) 7:68292–302. doi: 10.18632/oncotarget.11944
42. Kahlenberg JM, Thacker SG, Berthier CC, Cohen CD, Kretzler M, Kaplan MJ. Inflammasome activation of IL−18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J Immunol. (2011) 187:6143–56. doi: 10.4049/jimmunol.1101284
43. Wu CY, Yang HY, Yao TC, Liu SH, Huang JL. Serum IL−18 as biomarker in predicting long–term renal outcome among pediatric–onset systemic lupus erythematosus patients. Medicine (Baltimore). (2016) 95:e5037. doi: 10.1097/MD.0000000000005037
44. Kahlenberg JM, Yalavarthi S, Zhao W, Hodgin JB, Reed TJ, Tsuji NM, et al. An essential role of caspase 1 in the induction of murine lupus and its associated vascular damage. Arthritis Rheumatol. (2014) 66:152–62. doi: 10.1002/art.38225
45. Deng Y, Tsao BP. Updates in lupus genetics. Curr Rheumatol Rep. (2017) 19:68. doi: 10.1007/s11926–017–0695–z
46. dos Santos BP, Valverde JV, Rohr P, Monticielo OA, Brenol JC, Xavier RM, et al. TLR7/8/9 polymorphisms and their associations in systemic lupus erythematosus patients from southern Brazil. Lupus. (2012) 21:302–9. doi: 10.1177/0961203311425522
47. Pontillo A, Girardelli M, Kamada AJ, Pancotto JA, Donadi EA, Crovella S, et al. Polimorphisms in inflammasome genes are involved in the predisposition to systemic lupus erythematosus. Autoimmunity. (2012) 45:271–8. doi: 10.3109/08916934.2011.637532
48. Huang LH, Kuo HC, Pan CT, Lin YS, Huang YH, Li SC. Multiomics analyses identified epigenetic modulation of the S100A gene family in Kawasaki disease and their significant involvement in neutrophil transendothelial migration. Clin Epigenetics. (2018) 10:135. doi: 10.1186/s13148–018–0557–1
49. Chen GM, Feng CC, Ye QL, Tao JH, Li R, Peng H, et al. Association of P2X7R gene polymorphisms with systemic lupus erythematosus in a Chinese population. Mutagenesis. (2013) 28:351–5. doi: 10.1093/mutage/get007
50. Zhang J, Zhu Q, Meng F, Lei H, Zhao Y. Association study of TLR−9 polymorphisms and systemic lupus erythematosus in northern Chinese Han population. Gene. (2014) 533:385–8. doi: 10.1016/j.gene.2013.08.051
51. Pontillo A, Reis EC, Liphaus BL, Silva CA, Carneiro–Sampaio M. Inflammasome polymorphisms in juvenile systemic lupus erythematosus. Autoimmunity. (2015) 48:434–7. doi: 10.3109/08916934.2015.1064399
52. Carlstrom M, Ekman AK, Petersson S, Soderkvist P, Enerback C. Genetic support for the role of the NLRP3 inflammasome in psoriasis susceptibility. Exp Dermatol. (2012) 21:932–7. doi: 10.1111/exd.12049
53. Ekman AK, Verma D, Fredrikson M, Bivik C, Enerback C. Genetic variations of NLRP1: susceptibility in psoriasis. Br J Dermatol. (2014) 171:1517–20. doi: 10.1111/bjd.13178
54. Zuo X, Sun L, Yin X, Gao J, Sheng Y, Xu J, et al. Whole–exome SNP array identifies 15 new susceptibility loci for psoriasis. Nat Commun. (2015) 6:6793. doi: 10.1038/ncomms7793
55. Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, et al. NALP1 in vitiligo–associated multiple autoimmune disease. N Engl J Med. (2007) 356:1216–25. doi: 10.1056/NEJMoa061592
56. Alkhateeb A, Qarqaz F. Genetic association of NALP1 with generalized vitiligo in Jordanian Arabs. Arch Dermatol Res. (2010) 302:631–4. doi: 10.1007/s00403–010–1064–1
57. Levandowski CB, Mailloux CM, Ferrara TM, Gowan K, Ben S, Jin Y, et al. NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin−1beta processing via the NLRP1 inflammasome. Proc Natl Acad Sci USA. (2013) 110:2952–6. doi: 10.1073/pnas.1222808110
58. Hysi P, Kabesch M, Moffatt MF, Schedel M, Carr D, Zhang Y, et al. NOD1 variation, immunoglobulin E and asthma. Hum Mol Genet. (2005) 14:935–41. doi: 10.1093/hmg/ddi087
59. Macaluso F, Nothnagel M, Parwez Q, Petrasch–Parwez E, Bechara FG, Epplen JT, et al. Polymorphisms in NACHT–LRR (NLR) genes in atopic dermatitis. Exp Dermatol. (2007) 16:692–8. doi: 10.1111/j.1600–0625.2007.00589.x
60. Bivik C, Verma D, Winge MC, Lieden A, Bradley M, Rosdahl I, et al. Genetic variation in the inflammasome and atopic dermatitis susceptibility. J Invest Dermatol. (2013) 133:2486–9. doi: 10.1038/jid.2013.168
61. Crow MK. Type I interferon in the pathogenesis of lupus. J Immunol. (2014) 192:5459–68. doi: 10.4049/jimmunol.1002795
62. Niewold TB. Interferon alpha as a primary pathogenic factor in human lupus. J Interferon Cytokine Res. (2011) 31:887–92. doi: 10.1089/jir.2011.0071
63. Shin MS, Kang Y, Lee N, Kim SH, Kang KS, Lazova R, et al. U1–small nuclear ribonucleoprotein activates the NLRP3 inflammasome in human monocytes. J Immunol. (2012) 188:4769–75. doi: 10.4049/jimmunol.1103355
64. Shin MS, Kang Y, Lee N, Wahl ER, Kim SH, Kang KS, et al. Self double–stranded (ds)DNA induces IL−1beta production from human monocytes by activating NLRP3 inflammasome in the presence of anti–dsDNA antibodies. J Immunol. (2013) 190:1407–15. doi: 10.4049/jimmunol.1201195
65. Liu J, Berthier CC, Kahlenberg JM. Enhanced inflammasome activity in systemic lupus erythematosus is mediated via type i interferon–induced up–regulation of interferon regulatory factor 1. Arthritis Rheumatol. (2017) 69:1840–9. doi: 10.1002/art.40166
66. Zhu FG, Jiang W, Bhagat L, Wang D, Yu D, Tang JX, et al. A novel antagonist of Toll–like receptors 7, 8 and 9 suppresses lupus disease–associated parameters in NZBW/F1 mice. Autoimmunity. (2013) 46:419–28. doi: 10.3109/08916934.2013.798651
67. Zhang H, Fu R, Guo C, Huang Y, Wang H, Wang S, et al. Anti–dsDNA antibodies bind to TLR4 and activate NLRP3 inflammasome in lupus monocytes/macrophages. J Transl Med. (2016) 14:156. doi: 10.1186/s12967–016–0911–z
68. He J, Sun M, Tian S. Procyanidin B2 prevents lupus nephritis development in mice by inhibiting NLRP3 inflammasome activation. Innate Immun. (2018) 24:307–15. doi: 10.1177/1753425918780985
69. Shi G, Abbott KN, Wu W, Salter RD, Keyel PA. Dnase1L3 Regulates Inflammasome–Dependent Cytokine Secretion. Front Immunol. (2017) 8:522. doi: 10.3389/fimmu.2017.00522
70. Sisirak V, Sally B, D'Agati V, Martinez–Ortiz W, Ozcakar ZB, David J, et al. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell. (2016) 166:88–101. doi: 10.1016/j.cell.2016.05.034
71. Westerterp M, Gautier EL, Ganda A, Molusky MM, Wang W, Fotakis P, et al. Cholesterol accumulation in dendritic cells links the inflammasome to acquired immunity. Cell Metab. (2017) 25:1294–304. doi: 10.1016/j.cmet.2017.04.005
72. Yang CA, Huang ST, Chiang BL. Sex–dependent differential activation of NLRP3 and AIM2 inflammasomes in SLE macrophages. Rheumatology. (2015) 54:324–31. doi: 10.1093/rheumatology/keu318
73. Panchanathan R, Duan X, Arumugam M, Shen H, Liu H, Choubey D. Cell type and gender–dependent differential regulation of the p202 and Aim2 proteins: implications for the regulation of innate immune responses in SLE. Mol Immunol. (2011) 49:273–80. doi: 10.1016/j.molimm.2011.08.022
74. Panchanathan R, Shen H, Bupp MG, Gould KA, Choubey D. Female and male sex hormones differentially regulate expression of Ifi202, an interferon–inducible lupus susceptibility gene within the Nba2 interval. J Immunol. (2009) 183:7031–8. doi: 10.4049/jimmunol.0802665
75. Yin Q, Sester DP, Tian Y, Hsiao YS, Lu A, Cridland JA, et al. Molecular mechanism for p202–mediated specific inhibition of AIM2 inflammasome activation. Cell Rep. (2013) 4:327–39. doi: 10.1016/j.celrep.2013.06.024
76. Zhang W, Cai Y, Xu W, Yin Z, Gao X, Xiong S. AIM2 facilitates the apoptotic DNA–induced systemic lupus erythematosus via arbitrating macrophage functional maturation. J Clin Immunol. (2013) 33:925–37. doi: 10.1007/s10875–013–9881–6
77. Yang Q, Yu C, Yang Z, Wei Q, Mu K, Zhang Y, et al. Deregulated NLRP3 and NLRP1 inflammasomes and their correlations with disease activity in systemic lupus erythematosus. J Rheumatol. (2014) 41:444–52. doi: 10.3899/jrheum.130310
78. Helyer BJ, Howie JB. Renal disease associated with positive lupus erythematosus tests in a cross–bred strain of mice. Nature. (1963) 197:197. doi: 10.1038/197197a0
79. Waters ST, Fu SM, Gaskin F, Deshmukh US, Sung SS, Kannapell CC, et al. NZM2328: a new mouse model of systemic lupus erythematosus with unique genetic susceptibility loci. Clin Immunol. (2001) 100:372–83. doi: 10.1006/clim.2001.5079
80. Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, McConahey PJ, et al. Spontaneous murine lupus–like syndromes. clinical and immunopathological manifestations in several strains. J Exp Med. (1978) 148:1198–215. doi: 10.1084/jem.148.5.1198
81. Satoh M, Reeves WH. Induction of lupus–associated autoantibodies in BALB/c mice by intraperitoneal injection of pristane. J Exp Med. (1994) 180:2341–6. doi: 10.1084/jem.180.6.2341
82. Choubey D, Panchanathan R. Interferon (IFN)–inducible absent in melanoma 2 proteins in the negative regulation of the type I IFN response: implications for lupus nephritis. Cytokine. (2019) doi: 10.1016/j.cyto.2019.03.008
83. Ding L, Dong G, Zhang D, Ni Y, Hou Y. The regional function of cGAS/STING signal in multiple organs: one of culprit behind systemic lupus erythematosus? Med Hypotheses. (2015) 85:846–9. doi: 10.1016/j.mehy.2015.09.026
84. Choubey D, Panchanathan R. Absent in Melanoma 2 proteins in SLE. Clin Immunol. (2017) 176:42–8. doi: 10.1016/j.clim.2016.12.011
85. Dombrowski Y, Schauber J. Cathelicidin LL−37: a defense molecule with a potential role in psoriasis pathogenesis. Exp Dermatol. (2012) 21:327–30. doi: 10.1111/j.1600–0625.2012.01459.x
86. Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nat Rev Immunol. (2009) 9:679–91. doi: 10.1038/nri2622
87. Wagner EF, Schonthaler HB, Guinea–Viniegra J, Tschachler E. Psoriasis: what we have learned from mouse models. Nat Rev Rheumatol. (2010) 6:704–14. doi: 10.1038/nrrheum.2010.157
88. Hiruma J, Harada K, Motoyama A, Okubo Y, Maeda T, Yamamoto M, et al. Key component of inflammasome, NLRC4, was identified in the lesional epidermis of psoriatic patients. J Dermatol. (2018) 45:971–7. doi: 10.1111/1346–8138.14478
89. Yang BY, Cheng YG, Liu Y, Liu Y, Tan JY, Guan W, et al. Ameliorates imiquimod–induced psoriasis–like dermatitis and inhibits inflammatory cytokines production through TLR7/8–MyD88–NF–kappaB–NLRP3 inflammasome pathway. Molecules. (2019) 24. doi: 10.3390/molecules24112157
90. Tervaniemi MH, Katayama S, Skoog T, Siitonen HA, Vuola J, Nuutila K, et al. NOD–like receptor signaling and inflammasome–related pathways are highlighted in psoriatic epidermis. Sci Rep. (2016) 6:22745. doi: 10.1038/srep22745
91. Kopfnagel V, Wittmann M, Werfel T. Human keratinocytes express AIM2 and respond to dsDNA with IL−1beta secretion. Exp Dermatol. (2011) 20:1027–9. doi: 10.1111/j.1600–0625.2011.01382.x
92. Hawkes JE, Adalsteinsson JA, Gudjonsson JE, Ward NL. Research techniques made simple: murine models of human psoriasis. J Invest Dermatol. (2018) 138:e1–8. doi: 10.1016/j.jid.2017.10.013
93. Zhou MW, Jiang RH, Kim KD, Lee JH, Kim CD, Yin WT, et al. Rosmarinic acid inhibits poly(I:C)–induced inflammatory reaction of epidermal keratinocytes. Life Sci. (2016) 155:189–94. doi: 10.1016/j.lfs.2016.05.023
94. Zhang C, Xiao C, Dang E, Cao J, Zhu Z, Fu M, et al. CD100–Plexin–B2 promotes the inflammation in psoriasis by activating NF–kappaB and the inflammasome in keratinocytes. J Invest Dermatol. (2018) 138:375–83. doi: 10.1016/j.jid.2017.09.005
95. Luo Q, Zeng J, Li W, Lin L, Zhou X, Tian X, et al. Silencing of miR155 suppresses inflammatory responses in psoriasis through inflammasome NLRP3 regulation. Int J Mol Med. (2018) 42:1086–95. doi: 10.3892/ijmm.2018.3677
96. Deng G, Chen W, Wang P, Zhan T, Zheng W, Gu Z, et al. Inhibition of NLRP3 inflammasome–mediated pyroptosis in macrophage by cycloastragenol contributes to amelioration of imiquimod–induced psoriasis–like skin inflammation in mice. Int Immunopharmacol. (2019) 74:105682. doi: 10.1016/j.intimp.2019.105682
97. Irrera N, Vaccaro M, Bitto A, Pallio G, Pizzino G, Lentini M, et al. BAY 11–7082 inhibits the NF–kappaB and NLRP3 inflammasome pathways and protects against IMQ–induced psoriasis. Clin Sci. (2017) 131:487–98. doi: 10.1042/CS20160645
98. Cho KA, Suh JW, Lee KH, Kang JL, Woo SY. IL−17 and IL−22 enhance skin inflammation by stimulating the secretion of IL−1beta by keratinocytes via the ROS–NLRP3–caspase−1 pathway. Int Immunol. (2012) 24:147–58. doi: 10.1093/intimm/dxr110
99. Aira LE, Goncalves D, Bossowski JP, Rubio–Patino C, Chiche J, Paul–Bellon R, et al. Caspase 1/11 deficiency or pharmacological inhibition mitigates psoriasis–like phenotype in mice. J Invest Dermatol. (2019) 139:1306–17. doi: 10.1016/j.jid.2018.11.031
100. Rabeony H, Pohin M, Vasseur P, Petit–Paris I, Jegou JF, Favot L, et al. IMQ–induced skin inflammation in mice is dependent on IL−1R1 and MyD88 signaling but independent of the NLRP3 inflammasome. Eur J Immunol. (2015) 45:2847–57. doi: 10.1002/eji.201445215
101. Diaz–Perez JA, Killeen ME, Yang Y, Carey CD, Falo LD Jr, et al. Extracellular ATP and IL−23 form a local inflammatory circuit leading to the development of a neutrophil–dependent psoriasiform dermatitis. J Invest Dermatol. (2018) 138:2595–605. doi: 10.1016/j.jid.2018.05.018
102. Dombrowski Y, Peric M, Koglin S, Kammerbauer C, Goss C, Anz D, et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med. (2011) 3:82ra38. doi: 10.1126/scitranslmed.3002001
103. de Koning HD, Bergboer JG, van den Bogaard EH, van Vlijmen–Willems IM, Rodijk–Olthuis D, Simon A, et al. Strong induction of AIM2 expression in human epidermis in acute and chronic inflammatory skin conditions. Exp Dermatol. (2012) 21:961–4. doi: 10.1111/exd.12037
104. Yun M, Seo G, Lee JY, Chae GT, Lee SB. Epigallocatechin−3–gallate attenuates the AIM2–induced secretion of IL−1beta in human epidermal keratinocytes. Biochem Biophys Res Commun. (2015) 467:723–9. doi: 10.1016/j.bbrc.2015.10.075
105. Chung IC, Yuan SN, OuYang CN, Hu SI, Lin HC, Huang KY, et al. EFLA 945 restricts AIM2 inflammasome activation by preventing DNA entry for psoriasis treatment. Cytokine. (2019) 127:154951. doi: 10.1016/j.cyto.2019.154951
106. Jiang W, Zhu FG, Bhagat L, Yu D, Tang JX, Kandimalla ER, et al. A Toll–like receptor 7, 8, and 9 antagonist inhibits Th1 and Th17 responses and inflammasome activation in a model of IL−23–induced psoriasis. J Invest Dermatol. (2013) 133:1777–84. doi: 10.1038/jid.2013.57
107. Walter A, Schafer M, Cecconi V, Matter C, Urosevic–Maiwald M, Belloni B, et al. Aldara activates TLR7–independent immune defence. Nat Commun. (2013) 4:1560. doi: 10.1038/ncomms2566
108. van den Boorn JG, Jakobs C, Hagen C, Renn M, Luiten RM, Melief CJ, et al. Inflammasome–Dependent induction ofadaptive NK cell memory. Immunity. (2016) 44:1406–21. doi: 10.1016/j.immuni.2016.05.008
109. Li S, Kang P, Zhang W, Jian Z, Zhang Q, Yi X, et al. Activated NLR family pyrin domain containing 3 (NLRP3) inflammasome in keratinocytes promotes cutaneous T–cell response in patients with vitiligo. J Allergy Clin Immunol. (2019) doi: 10.1016/j.jaci.2019.10.036
110. Niebuhr M, Baumert K, Heratizadeh A, Satzger I, Werfel T. Impaired NLRP3 inflammasome expression and function in atopic dermatitis due to Th2 milieu. Allergy. (2014) 69:1058–67. doi: 10.1111/all.12428
111. Dai X, Sayama K, Tohyama M, Shirakata Y, Hanakawa Y, Tokumaru S, et al. Mite allergen is a danger signal for the skin via activation of inflammasome in keratinocytes. J Allergy Clin Immunol. (2011) 127:806–14 e1–4. doi: 10.1016/j.jaci.2010.12.006
112. Hiramoto K, Yamate Y, Yokoyama S. Ultraviolet B eye irradiation aggravates atopic dermatitis via adrenocorticotropic hormone and NLRP3 inflammasome in NC/Nga mice. Photodermatol Photoimmunol Photomed. (2018) 34:200–10. doi: 10.1111/phpp.12372
113. Munoz–Planillo R, Franchi L, Miller LS, Nunez G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus–induced activation of the Nlrp3 inflammasome. J Immunol. (2009) 183:3942–8. doi: 10.4049/jimmunol.0900729
114. Feldmeyer L, Werner S, French LE, Beer HD. Interleukin−1, inflammasomes and the skin. Eur J Cell Biol. (2010) 89:638–44. doi: 10.1016/j.ejcb.2010.04.008
115. Rashighi M, Agarwal P, Richmond JM, Harris TH, Dresser K, Su MW, et al. CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci Transl Med. (2014) 6:223ra23. doi: 10.1126/scitranslmed.3007811
116. Riding RL, Harris JE. The Role of Memory CD8(+) T Cells in Vitiligo. J Immunol. (2019) 203:11–9. doi: 10.4049/jimmunol.1900027
117. Marie J, Kovacs D, Pain C, Jouary T, Cota C, Vergier B, et al. Inflammasome activation and vitiligo/nonsegmental vitiligo progression. Br J Dermatol. (2014) 170:816–23. doi: 10.1111/bjd.12691
118. Petrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol. (2007) 19:615–22. doi: 10.1016/j.coi.2007.09.002
119. Bhardwaj S, Rani S, Srivastava N, Kumar R, Parsad D. Increased systemic and epidermal levels of IL−17A and IL−1beta promotes progression of non–segmental vitiligo. Cytokine. (2017) 91:153–61. doi: 10.1016/j.cyto.2016.12.014
120. Zhou L, Shi YL, Li K, Hamzavi I, Gao TW, Huggins RH, et al. Increased circulating Th17 cells and elevated serum levels of TGF–beta and IL−21 are correlated with human non–segmental vitiligo development. Pigment Cell Melanoma Res. (2015) 28:324–9. doi: 10.1111/pcmr.12355
121. Eichenfield LF, Tom WL, Chamlin SL, Feldman SR, Hanifin JM, Simpson EL, et al. Guidelines of care for the management of atopic dermatitis: section 1. diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. (2014) 70:338–51. doi: 10.1016/j.jaad.2013.10.010
122. Novak N, Bieber T, Leung DY. Immune mechanisms leading to atopic dermatitis. J Allergy Clin Immunol. (2003) 112(Suppl.6):S128–39. doi: 10.1016/j.jaci.2003.09.032
123. Yoo J, Omori M, Gyarmati D, Zhou B, Aye T, Brewer A, et al. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J Exp Med. (2005) 202:541–9. doi: 10.1084/jem.20041503
124. Palmer CN, Irvine AD, Terron–Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss–of–function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. (2006) 38:441–6. doi: 10.1038/ng1767
125. Chavanas S, Bodemer C, Rochat A, Hamel–Teillac D, Ali M, Irvine AD, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. (2000) 25:141–2. doi: 10.1038/75977
126. Samuelov L, Sarig O, Harmon RM, Rapaport D, Ishida–Yamamoto A, Isakov O, et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet. (2013) 45:1244–8. doi: 10.1038/ng.2739
127. Kezic S, O'Regan GM, Lutter R, Jakasa I, Koster ES, Saunders S, et al. Filaggrin loss–of–function mutations are associated with enhanced expression of IL−1 cytokines in the stratum corneum of patients with atopic dermatitis and in a murine model of filaggrin deficiency. J Allergy Clin Immunol. (2012) 129:1031–9. doi: 10.1016/j.jaci.2011.12.989
128. Schwartz C, Moran T, Saunders SP, Kaszlikowska A, Floudas A, Bom J, et al. Spontaneous atopic dermatitis in mice with a defective skin barrier is independent of ILC2 and mediated by IL−1beta. Allergy. (2019) 74:1920–33. doi: 10.1111/all.13801
129. Schuepbach–Mallepell S, Philippe V, Bruggen MC, Watanabe H, Roques S, Baldeschi C, et al. Antagonistic effect of the inflammasome on thymic stromal lymphopoietin expression in the skin. J Allergy Clin Immunol. (2013) 132:1348–57. doi: 10.1016/j.jaci.2013.06.033
130. Douglas T, Champagne C, Morizot A, Lapointe JM, Saleh M. The inflammatory caspases−1 and −11 mediate the pathogenesis of dermatitis in sharpin–deficient mice. J Immunol. (2015) 195:2365–73. doi: 10.4049/jimmunol.1500542
131. Kabesch M, Peters W, Carr D, Leupold W, Weiland SK, von Mutius E. Association between polymorphisms in caspase recruitment domain containing protein 15 and allergy in two German populations. J Allergy Clin Immunol. (2003) 111:813–7. doi: 10.1067/mai.2003.1336
132. Zhang Q, Fan HW, Zhang JZ, Wang YM, Xing HJ. NLRP3 rs35829419 polymorphism is associated with increased susceptibility to multiple diseases in humans. Genet Mol Res. (2015) 14:13968–80. doi: 10.4238/2015.October.29.17
133. Skendros P, Papagoras C, Lefaki I, Giatromanolaki A, Kotsianidis I, Speletas M, et al. Successful response in a case of severe pustular psoriasis after interleukin-1beta inhibition. Br J Dermatol. (2017) 176:212–5. doi: 10.1111/bjd.14685
134. Zhao J, Wang H, Huang Y, Zhang H, Wang S, Gaskin F, et al. Lupus nephritis: glycogen synthase kinase 3beta promotion of renal damage through activation of the NLRP3 inflammasome in lupus–prone mice. Arthritis Rheumatol. (2015) 67:1036–44. doi: 10.1002/art.38993
135. Zhao J, Wang J, Zhou M, Li M, Li M, Tan H. Curcumin attenuates murine lupus via inhibiting NLRP3 inflammasome. Int Immunopharmacol. (2019) 69:213–6. doi: 10.1016/j.intimp.2019.01.046
136. Zhao R, Zhou H, Su SB. A critical role for interleukin−1beta in the progression of autoimmune diseases. Int Immunopharmacol. (2013) 17:658–69. doi: 10.1016/j.intimp.2013.08.012
137. Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, et al. Converting enzyme–independent release of tumor necrosis factor alpha and IL−1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci USA. (1999) 96:6261–6. doi: 10.1073/pnas.96.11.6261
Keywords: inflammasome, immune-related, psoriasis, vitiligo, systemic lupus erythematosus, atopic dermatitis, mouse model
Citation: Tang L and Zhou F (2020) Inflammasomes in Common Immune-Related Skin Diseases. Front. Immunol. 11:882. doi: 10.3389/fimmu.2020.00882
Received: 12 February 2020; Accepted: 16 April 2020;
Published: 12 May 2020.
Edited by:
Guochang Hu, University of Illinois at Chicago, United StatesReviewed by:
Stefan W. Ryter, Harvard Medical School, United StatesGuangxun Meng, Institut Pasteur of Shanghai (CAS), China
Copyright © 2020 Tang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fusheng Zhou, YmlvemhvdWZzQDE2My5jb20=