 Kim R. van Daalen
Kim R. van Daalen Josephine F. Reijneveld
Josephine F. Reijneveld Niels Bovenschen
Niels Bovenschen- 1Cardiovascular Epidemiology Unit, Department of Public Health and Primary Care, University of Cambridge, Cambridge, United Kingdom
- 2Department of Pathology, University Medical Center Utrecht, Utrecht, Netherlands
- 3Center for Translational Immunology, University Medical Center Utrecht, Utrecht, Netherlands
Granzyme A (GrA) has long been recognized as one of the key players in the induction of cell death of neoplastic, foreign or infected cells after granule delivery by cytotoxic cells. While the cytotoxic potential of GrA is controversial in current literature, accumulating evidence now indicates roles for extracellular GrA in modulating inflammation and inflammatory diseases. This paper aims to explore the literature presenting current knowledge on GrA as an extracellular modulator of inflammation by summarizing (i) the presence and role of extracellular GrA in several inflammatory diseases, and (ii) the potential molecular mechanisms of extracellular GrA in augmenting inflammation.
Introduction
Granzymes are a family of homologous serine proteases primarily expressed by a collective of cytotoxic cells, i.e., cytotoxic T lymphocytes (CTLs), γδ T cells, natural killer (NK) cells, NK-T cells. Classically the role of cytotoxic cells is described as promoting cytotoxic lymphocyte-mediated eradication of neoplastic, foreign or infected cells via the induction of (apoptotic) cell death (1). Apoptosis mediated by cytotoxic cells is induced via engagement of the death receptor pathway or the granule secretory pathway (2). Whereas the death receptor pathway involves death cell surface receptor-ligand interaction and caspase recruitment, the granule secretory pathway delivers granzymes through a process involving the aid of perforin, a pore-forming protein, to target cells (3, 4). Upon recognition of a target cell, cytotoxic cells release the content of granules into the immunological synapse. Perforin provides granzymes access to the cytosol of the targets cell, where granzymes cleave their cohort of substrates to promote programmed cell death (5, 6). To date, five human granzymes are known (granzymes A, B, H, K, and M), while ten mouse granzymes have been identified (7). Granzyme A (GrA, a tryptase) and granzyme B (GrB, an aspartase) being best characterized (3, 8). Although, human granzymes are highly homologous in amino acid sequence (40%), they variate in their primary substrate specificity (amino acid after which the granzyme preferably cleaves) resulting in a unique granzyme degradome (9).
GrA is the most abundant protease present in cytotoxic granules and is reported as dominant mediator of toxicity in vitro (4). Among serine proteases GrA has an unique quaternary structure consisting of a disulphide-linked homodimer of 60kDa linked via Cys93 (10). Dimerization creates a high degree of specificity for GrA due an extended site for its substrates (11). GrA cleaves substrate after Arg or Lys like its closest homolog GrK, whilst GrB cleaves after Asp or Glu, GrM after Leu or Met and GrH after Tyr or Phe (12). One intracellular inhibitor has been identified for GrA (Serpinb6b) (13), whilst two extracellular inhibitors have been reported [Kazal-type pancreatic secretory trypsin inhibitor (14), serpin antithrombin III (SERPINC1) (15)]. With its tryptase-like activity GrA has been shown to activate caspase independent cell death pathways with the cleavage of the mitochondrial protein NDUFS3 resulting in reactive oxygen species (ROS) generation. Triggered by ROS, the SET complex translocates to the nucleus where GrA cleaves SET complex components which result in opening of the DNA (due to targeting of histones) and single stranded DNA nicks (16–20). However, both human studies and in vivo mouse studies indicate that GrA by itself is not cytotoxic in contrast with initial in vitro reports (21). Furthermore, new in vitro studies also indicate contrasting results with native human GrA showing a lack of cytotoxicity in vitro, whilst recombinant mouse GrA studies contrastingly demonstrate cytotoxicity in several in vitro assays (4, 22). This may suggest alternatives roles for GrA.
The functioning of GrB has recently been redefined by the discovery of multiple extracellular roles including the mediation of skin injury, inflammation and repair (23). Moreover, cumulative clinical and biochemical evidence indicate elevated levels of extracellular GrA in plasma, serum, synovial fluid and bronchoalveolar lavage (BAL) fluid. This includes patients with various viral infections, bacterial infections or other pro-inflammatory conditions (8, 24, 25). These elevated levels of extracellular GrA could potentially reflect spontaneous or inadvertent release of granzymes after elevated CTL/NK numbers in response to persistent inflammation, however extracellular biological effects are increasingly described (6, 8) (Figure 1). Furthermore, dendritic cells, mast cells and macrophages can express GrA but not perforin, suggesting perforin-independent (extracellular) roles for granzymes (26). Yet, little has so far been described on potential functioning of extracellular GrA.
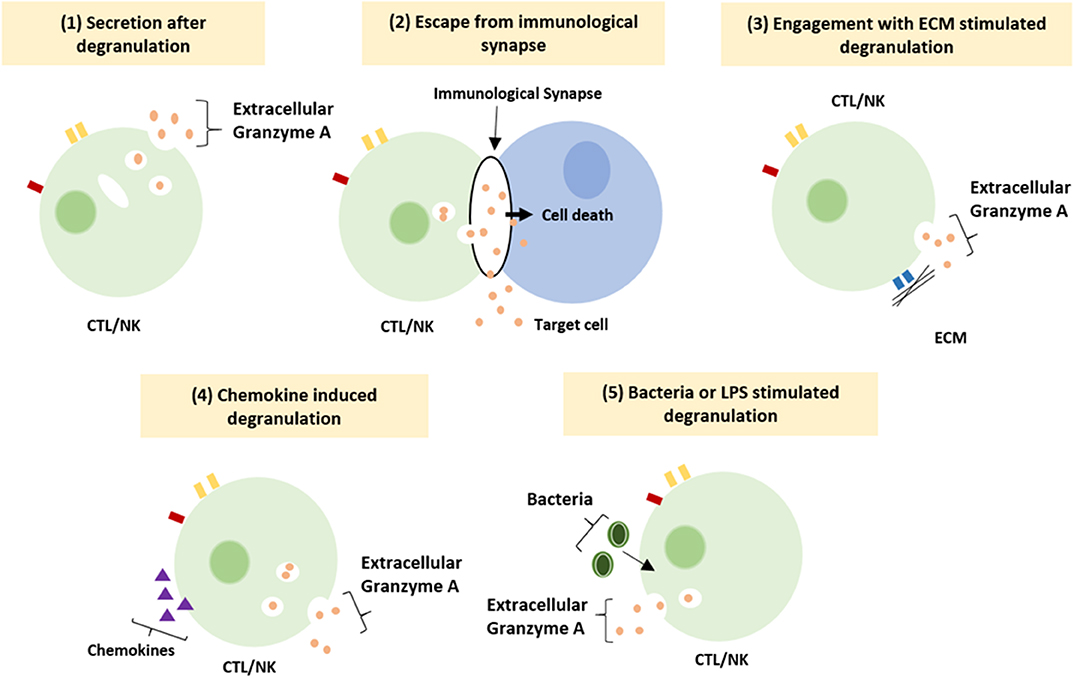
Figure 1. Suggested mechanism resulting in extracellular granzyme A release. Granzyme A can be release as a result of (1) constitutive granzyme A secretion after degranulation, (2) leakage of granzyme A from the immunological synapse, (3) degranulation after engagement of integrin with the ECM, (4) induction of degranulation by locally released chemokines (5) induction of degranulation by bacteria or LPS (8).
This paper aims to explore the literature presenting current knowledge on GrA as an extracellular modulator of inflammation by summarizing (i) the presence and suggested roles of extracellular GrA in several inflammatory diseases, and (ii) the molecular mechanisms of GrA in augmenting inflammation.
Extracellular Granzyme A During Inflammatory Disease
Microbial Infections, Bacteria
A plethora of pathogenic micro-organism exists (bacteria, viruses, parasites, and fungi) that can infect the human body. Exposure typically leads to acute infection that with appropriate immune response subsides in the elimination of the involved pathogen within days (27). With 1.6 million deaths and 10 million cases in 2017 alone, tuberculosis (TB) remains a global health problem with impaired control by the emergence of drug resistant forms of Mycobacterium tuberculosis (28, 29). Extracellular plasma GrA levels are increased in patients with TB in comparison with control patients, suggesting that extracellular GrA could be a potential new therapeutic target in the inflammatory response to mycobacteria (29–31). In TB, extracellular GrA has been found to induce inflammatory responses that lead to intracellular anti-microbial activity in vitro. Co-culture of mycobacteria infected macrophages with either mycobacteria specific Vγ9Vδ2 T cells or purified GrA result in the production of the pro-inflammatory cytokines TNFα and IL-1β by macrophages. This alone or in combination with unidentified factors results in the inhibition of intracellular mycobacterial growth. The inhibitory mechanism is independent of perforin, apoptosis, autophagy, nitric oxide production, type I interferons, and Fas/FasL. Necessity of TNFα is clear, as anti-TNF neutralizing antibodies prevent the inhibitory activity of Vγ9Vδ2 T cells and TNF knockdown prevents inhibition of intracellular mycobacterial growth. As high levels of TNFα is a marker of uncontrolled mycobacterial growth, it is currently believed that in vitro GrA mediated inhibition of mycobacterial growth includes other factors in addition to TNFα. The complete molecular signature responsible for GrA-mediated response and how TNFα influences mycobacterial growth therefore remains unknown (31). In an attempt to find a potential source of increased GrA in vivo, correlations between intracellular and extracellular granzymes were analyzed in active TB patients. However, this did not yield any significant results for the human lymphocyte populations studied (30). Furthermore, a recent mouse study on the in vivo role of GzmA in tuberculosis infection contradicts in vitro results. Although, GzmA is expressed by cytotoxic cells from mouse lungs during infection, GzmA knockout mice show no difference in lung bacterial burden compared to wildtype mice in long and short-term M. tuberculosis infection. This suggests extracellular GrA may not have a protective role in vivo in tuberculosis response (32).
Studies on sepsis and experimental endotoxemia found extracellular GrA plasma levels significantly higher in severe sepsis, septic shock and endotoxemia (33, 34). Experimental endotoxemia, a well-accepted model of systemic inflammation in humans, was induced by intravenous injection of the endotoxin LPS in volunteers. Extracellular plasma GrA increases peaking at 2 h post administration and associates with a decrease in the number of CTL and NK cells in the circulation. The latter suggests that LPS administration results in the activation of cytotoxic lymphocytes in vivo, which results in a quick granzyme release (33). Patients with melioidoses caused by Burkholderia pseudomallei, a gram-negative bacterium, also indicate increased serum levels of GrA in comparison with control subjects on admission and remained high during the 72 h study period. Serum GrA levels were not significantly elevated in patients with non-bacteremic melioidosis (33). Additionally, while patients infected with Salmonella enteric (1) in typhoid fever and Streptococcus Pneumoniae (35) in community-acquired pneumonia had elevated GrA levels in acute phase plasma and BAL, respectively, patients with Neisseria meningitides (36) infection did not, except for patients with shock. Extracellular plasma GrA levels in patients with typhoid fever correlate with IFN-γ, which is critical in the systemic control of S. enterica infections (1, 35). Combining these findings from several bacteria with the observation that stimulation with endotoxin strongly induces secretion of extracellular GrA, GrA release is most likely a general (acute-phase) immune response during bacterial infection and not specific for certain species (33).
Experimental results in mice suggest that GrA-induced release of pro-inflammatory cytokines contribute to the development of sepsis during infection with Brucella microti, without being essential for clearance of B. microti. GrA−/− mice injected with a sublethal dose of the bacterium clear the infection like wild-type (WT) mice, while GrB−/− mice, perforin−/− mice and mice depleted of cytotoxic CD8+ T cells do not. GrA−/− mice have a higher survival rate compared with WT mice and perforin or GrB depleted mice, which is correlated with a significant reduction in the levels of the cytokines IL-1α, IL-1β, IL-6. Transfer of WT NK cells into GrA−/− mice reverses susceptibility to sepsis, indicating that NK cells are the source of GrA during the infection (37). As both perforin−/− and perfxgzmAxB−/− mice are equally susceptible to sepsis as WT mice, this study was unable to determine whether the participation of GrA occurs intracellularly or extracellularly (7, 37). In agreement with this, similar studies reported that GrA−/− mice are more resistant to LPS induced toxicity compared to WT mice, GrA−/− mice show increased survival and lower bacterial load in BAL during S. Pneumoniae infection and GrA−/− mice have reduced levels of pro-inflammatory cytokines during S. Pneumoniae infection (21, 35, 38). These data support a role for this granzyme in pro-inflammatory cytokine signaling during infection. By targeting GrA, bacteria-mediated sepsis can therefore be ameliorated without compromising the ability of the immune system to control infection. Exact molecular mechanisms are to be determined (7).
Microbial Infections, Viruses
In addition to bacterial infections, viruses can infiltrate the human body and initiate an immune response mediated by cytotoxic lymphocytes (39). Longitudinal plasma samples from EBV-infected patients and one HIV-1 infected patient show increased levels of GrA in plasma during the acute phase of the infection and subsequent decline during its resolution. Increase in plasma GrA levels occur simultaneously with early markers of infection (40). Similarly, elevated plasma GrA is found in patients with dengue fever and cytomegalovirus infection after renal transplantation and in patients with respiratory syncytial virus infection (41–43). In the latter infection, plasma GrA clearly correlates with increased IL-8 levels and white blood cell counts after acute onset respiratory tract illness (43). In both non-human primates (44) and in chikungunya fever patients (44, 45), circulating GrA levels are elevated after chikungunya virus infection. Peak levels coincide with peaks of circulating IFNγ levels, increased viral load and disease scores in these patients (44, 45). It may seem conceivable that GrA contributes to the clearance of viral infections in humans by inducing a pro-inflammatory immune response in accordance with the correlated upregulation of cytokines and extracellular GrA in viral infections. Cytokines could directly inactivate the intracellular virus or they recruit and activate immune cells to remove infected cells (4). Yet, in vivo conflicting knockout mouse studies suggest these hypotheses may not be reliable. GrA deficiency does not seem to affect susceptibility to infections with e.g., chikungunya virus (44) and lymphocytic choriomeningitis virus (LMCV) (46). In contrast, GrA deficiency exhibited increased susceptibility to mousepox ectromelia (Ect) (47), herpes simplex virus (HSV) (48) and mouse cytomegalovirus (MCMV) (49). Control of the latter pathogens is often only delayed, without compromising mice survival, suggesting compensation mechanisms for absent GrA (50). Subcutaneous injection of enzymically active recombinant mouse GrA was able to mediate inflammation of chikungunya, zika and dengue virus (45).
Moreover, it has been found that several granzymes [e.g., GrH (51, 52), GrB (52), GrM (53)] can cleave viral [e.g., adenoviral DNA-binding protein (DBP) (52), phosphoprotein 71 (pp71) (53)] and host cell proteins involved in protein synthesis [e.g., La (51)] leading to restriction of viral replication (9, 54). In addition, the SET complex (a GrA/GrK substrate) has shown to be required for efficient HIV-1 infection by preventing autointegration (55) and for transcription of early genes during adenovirus infection (56). Whether extracellular GrA might also be able to cleave viral surface proteins, viral proteins expressed on the surface of infected cells, or host-surface proteins involved in the viral infection needs to be further explored (5, 9, 54). Interestingly, several viruses [e.g., cowpox virus (57), myxoma virus (58)] have been reported to encode proteins that inhibit GrB, suggesting immune evasive adaptations (59, 60), whilst no viral inhibitors have been described for GrA in the current literature.
Microbial Infections, Parasites
Parasites comprise diverse organisms with complex life cycles and often life-cycle specific interactions with the host immune systems. Many are long-term (chronic) persistent in the host due inadequate host immunity. Limited studies explored extracellular GrA in parasitic infections. A study in Cameroon showed that children presenting with clinical malaria (Plasmodium falciparum) have significantly increased concentrations of extracellular plasma GrA. Likewise, five healthy Dutch volunteers with no prior exposure to malaria that were experimentally infected by P. falciparum displayed extracellular plasma GrA increment 1-2 days prior to clinical symptoms and microscopically detectable parasitemia. This coincided with increases in IFNγ, IL-12p40, and IL-8. Although, granzymes are often considered as marker for CTL/NK involvement, additional roles in inflammation cannot be excluded and have to be determined (61). A study exploring the in vivo role of GzmA in mice showed that GzmA deficiency did not impact the outcome of Leishmania major infection (62). Other in vivo mice studies [e.g., on Trypanosoma cruzei (63)] used double knock-outs (e.g., GrA × B−/−) and therefore precludes drawing conclusions on GrA.
Rheumatoid Arthritis
Rheumatoid Arthritis (RA) is a chronic autoimmune disease characterized by inflammation of the joints (synovitis), autoantibodies, systemic inflammation and cartilage and bone destruction (64, 65). The synovial membrane is infiltrated by multiple immune cells, including T cells, B cells, macrophages, and NK cells. Inflammation, predominantly mediated by IL-1 and TNF, leads to joint swelling and pain (64, 66). The final outcome of the disease is destruction of the joint (64, 65). Although external factors regulating TNF are well-known, endogenous factors may amplify TNF expression.
Increased levels of GrA in plasma, synovial fluid, and synovial tissue have been described in RA patients (40, 41, 66, 67). Markedly higher levels of GrA have been recorded in synovial fluid (up to 10-fold higher than patients with osteoarthritis or reactive arthritis), whereas GrA plasma levels are more similar to those of healthy controls. This suggest a local GrA release (40, 66). Mice with collagen induced arthritis (CIA) have significantly elevated extracellular levels of GrA in their joints and plasma and activated CLs at early and late RA stages. CIA was only slightly reduced in perforin−/− mice, therefore the function of GrA seems perforin independent (68). Together with the finding of activated cytolytic cells in RA synovium, these results strongly suggest that GrA is synthesized and secreted locally in the rheumatoid joint and plays a role in the pathogenesis of RA (24, 33). However, further studies are required to validate and explore these hypotheses.
The function of extracellular GrA in synovial inflammation and joint destruction remains unclear. As for bacterial sepsis, GrA promotes inflammation of the joint by stimulating the release of pro-inflammatory cytokines (7, 37, 66). IL-1β, TNF-α, IL-6, and IL-8, produced mainly by macrophages and fibroblasts, are abundantly present in the synovium of RA patients (66). CIA mice have increased levels of IL-6 and TNF-α during both early and late stages of RA compared with WT mice, whereas these cytokines are reduced in GrA−/− CI mice (68). Furthermore, GrA may contribute to rheumatoid arthritis partly by promoting mice osteoclast precursor differentiation via the stimulation of TNF-α secretion of monocytes and osteoclast precursors present in the inflammatory joint (68). Osteoclasts are cells that carry out bone resorption and have been shown to contribute to joint destruction in RA (69). Finally, ECM degradation induced by extracellular GrA might contribute to pathogenesis. Biologically active fibronectin fragments found in the synovium can induce neutrophil and monocyte chemotaxis, induce matrix metalloproteinase (MMP), induce chondrocyte aggrecanase expression and disrupt chondrocyte cell adhesion (8). ECM degradation may assist migration of activated cytotoxic T lymphocytes through the endothelial basement membrane and facilitate the influx of mononuclear cells contributing to hyperplasia and joint destruction (70).
Inflammatory Lung Disease
Numerous lung diseases are characterized by the presence of activated alveolar CTL and NK cells. Active GrA levels are locally increased in the BAL fluid, but not in blood, of patients with CD8+ T-cell-mediated hypersensitivity pneumonitis (HP) compared to control subjects. This is in contrast with increased plasmatic GrA concentrations in several viral, bacterial and parasitic infections (Table 1). Not surprisingly, TNFα, IL-6, and IL-8 are increased in BAL of HP patients coinciding with GrA levels (71). GrA positive cells and increased levels of GrA in sputum are also found in some smoking patients with asthma, smokers and nonsmoking patients with asthma (73, 74, 78). Both support a role for extracellular GrA in the lung inflammatory response. In allergic asthma there was no increase of GrA in BAL consistent with the absence of a lymphoid source (e.g., CTLs) (73, 74, 78). Moreover, GrA mRNA and IL-1β, IL-6, IL-8, and TNFα mRNA were upregulated in cells from BAL of patients with acute respiratory distress syndrome following sepsis. Though indirect, it has been suggested that this might indicate elevated extracellular GrA levels (79).
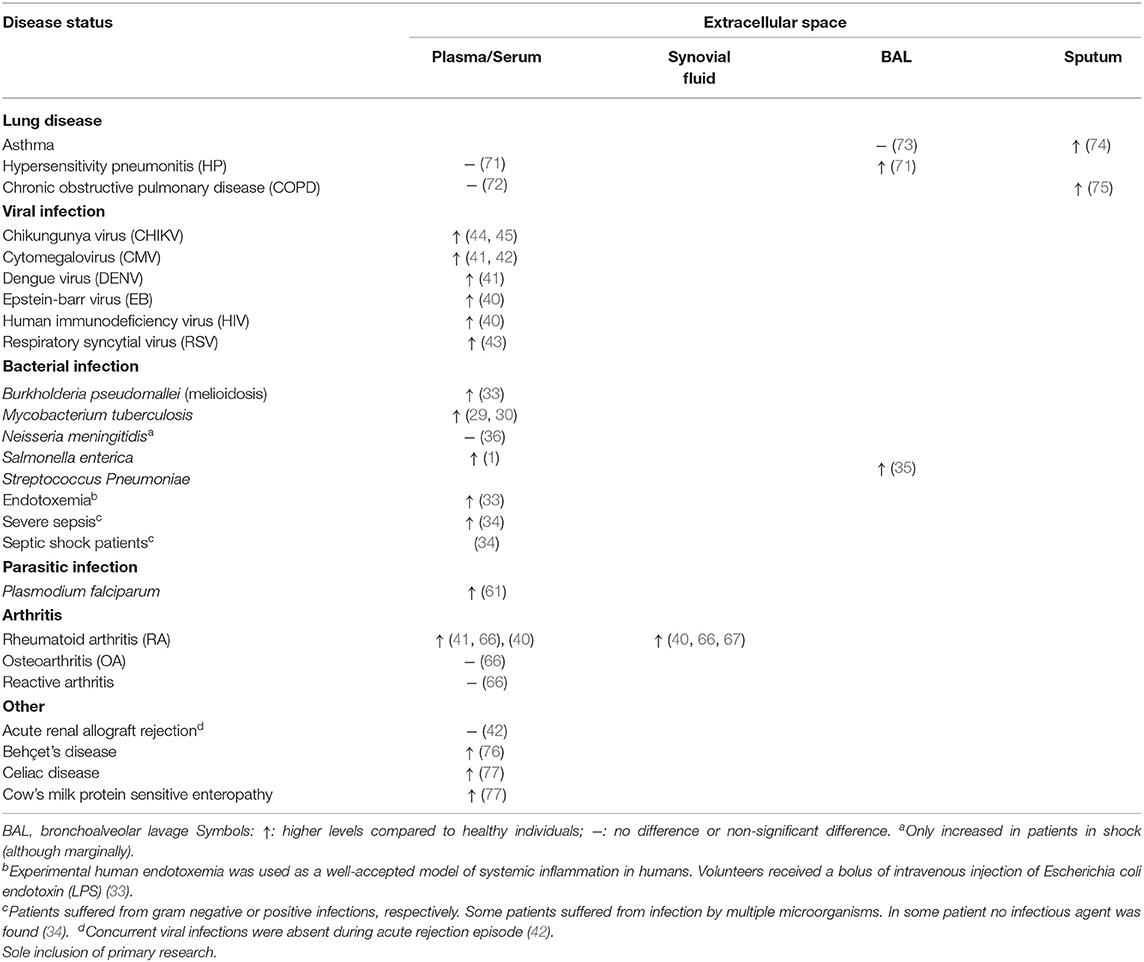
Table 1. Detection of extracellular granzyme A in patients with infections or pro-inflammatory disease.
Immunohistological studies indicate GrA expression by CTLs, NKs, alveolar macrophages, bronchiolar epithelium and type II pneumonocytes in both control subjects and chronic obstructive pulmonary disease (COPD) patients. GrA expression is significantly increased in sputum and/or lung specimens of patients with COPD in comparison with controls, but not in blood (72, 75, 80). Like in HP this suggest local lung GrA expression and tissue destruction (72). Several roles have been postulated in COPD. IL-6 and IL-8 show increased expression in COPD patients, indicating potential pro-inflammatory roles of GrA (80). Conjointly, recombinant rat GrA was found to cause rounding and detachment of an alveolar type II epithelial cell line (A549 cells), probably through its ability to cleave ECM, and was found to stimulate IL-8 via a mechanism involving microtubule disruption. This suggests that GrA might be involved in lung disease pathogenesis by loss of alveolar wall structures, chronic inflammation and neutrophil accumulation (81). It is noteworthy that the sulfated oligosaccharide k-carrageenan can inhibit the GrA promoted detachment of A549 cells in vitro, releasing IL-8. This might be of use for clinical purposes (82). Examination of BAL fluid and GrA−/− mouse models could provide further insights in the role of GrA in COPD patients.
In conclusion, locally elevated extracellular GrA is likely to play a role in general lymphocytic lung disease by enhancing inflammation in the lungs through release of pro-inflammatory cytokines and contributing to tissue damage. Nevertheless, additional research is required on the multifactorial roles of GrA in lung tissue destruction.
Other Inflammatory Diseases
Other diseases, such as Behçet's disease (BD), celiac disease (CD) and cow's milk protein sensitive enteropathy (CMSE), also show elevated GrA in serum and/or plasma (76, 77). Various micro-organisms, neutrophil hyperfunction, and autoimmune manifestation have been implicated as causative agents in BD. Using a BLT esterase assay, active BD patients show increased GrA levels in supernatants of lymphocytes correlated with the Vγ9Vδ2 expansion factor, suggesting active participation of CTLs and GrA in BD pathogenesis. The biological role of extracellular GrA in BD has not been studied. As GrA levels are elevated in both supernatant and serum, an extracellular role for GrA is speculated (76). Additionally, the intestinal immune response in both CMSE and CD is manifested by increased serum GrA, GrB and CD30. Elevated GrA correlates with the extent of CD villous atrophy. This offers new complementary means for diagnostic assessment of these diseases. More comprehensive studies on the detailed association and skewing of gut-associated lymphoid tissue and GrA functioning are needed (77).
Tumors
Questions are raised about the impact of pharmacological/biological inhibition of GrA in inflammatory carcinoma. Considering the variety of roles of extracellular GrA in immune response described, it has been speculated that extracellular GrA may contribute to the cancer response and/or tumor promotion. For example, ulcerative colitis (UC) patients have higher levels of infiltrating lymphocytes expressing GrA (83) and respond better to anti-inflammatory immunotherapy correlating with decrease in GrA (84). However, such observations could be circumstantial and not related to extracellular GrA functioning. Currently, there is a lack of (published) human, animal and in vitro studies in this context so far. Efforts to properly understand the potential role of GrA (intracellular and extracellular) in tumor progression can hold great promise for therapeutic approaches.
Pro-inflammatory Mechanisms of Extracellular Granzyme A
A central function of GrA in the modulation of pro-inflammatory cytokine expression, which is at least partly enhanced upon GrA intracellular delivery, is postulated (Figure 2) (95). Granzymes are localized both intra-and extracellular and thereby possess the potential to cleave on both sides of the membrane (96). Extracellular substrates for GrA are beginning to emerge. While direct in vivo evidence of granzyme-mediated cleavage of extracellular substrates is limited under physiological conditions, several substrates in vitro (Table 2) have formed the basis for hypothesis formulation linking multifactorial extracellular GrA activity to disease pathogenesis (Table 1).

Table 2. Extracellular substrates of GrA and suggested biological impact.
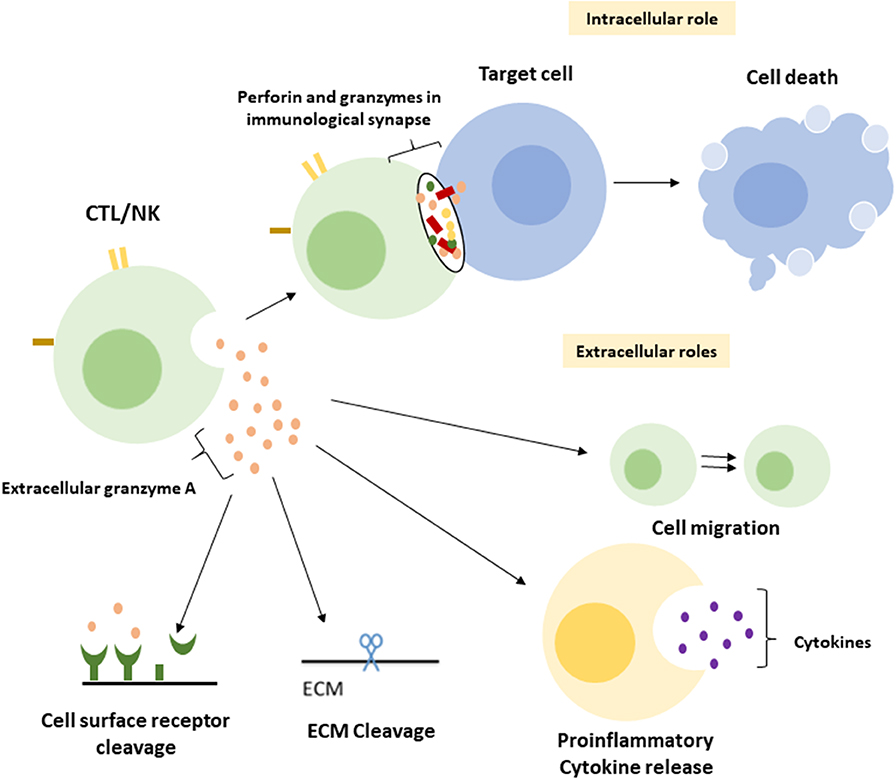
Figure 2. Intracellular and putative extracellular functions of Granzyme A. Classically GrA and other granzymes have been described as promoting cytotoxic lymphocyte mediated eradication of target cells via the induction of (apoptotic) cell death. Upon recognition of the target cell CTL release granule content into the immunological synapse, perforin provides access to the cytosol and granzymes promote cell death intracellularly (1). During a number of inflammatory disease statues GrA accumulates in extracellular space and is suggested to (i) induce release of pro-inflammatory cytokines in fibroblasts, epithelial cells, monocytes, and macrophages (3, 85, 86), (ii) remodel extracellular matrix (87–91), (iii) contribute to the migration of activated CTLs through tissue and extravasation of these cells from the vasculature (88), and (iv) cleavage of (cell surface) receptors as the Thrombin like receptor in neurite retraction (92–94).
Cytokine Release and Activation
First evidence suggesting a role of GrA as pro-inflammatory mediator in cytokine activity was proposed almost 25 years ago when it was found that GrA can function in vitro as an IL-1β-converting enzyme. GrA, but not GrB, cleaves the 31 kDa precursor form (pIL-1β) into the active 17 kDa cytokine in cultured macrophages expressing pIL-1β (101). Physiological relevance of this has never been shown (3). Although some have been able to show GrA alone can convert IL-1β to its mature form via direct cleavage, others were unsuccessful reproducing this maturation or suggest the aid of the inflammasome (3, 21, 85, 102).
In vitro human extracellular GrA stimulates release of various pro-inflammatory cytokines in multiple cell-types; IL-6 from fibroblast cell lines (lung, intestinal) and IL-8 from both fibroblasts (lung, skin, intestinal) and cultured (lung) epithelial cell lines (86). Additionally, GrA stimulates the production of IL-6, IL-8 and TNF-α in human peripheral blood mononuclear cells and purified monocytes without the presence of perforin (103), whilst inducing macrophages to produce TNF-α and IL-1 β (21, 85). Recently it has been shown that, although able to enhance the cytokine response induced by LPS, extracellular GrA is not enough to induce release of cytokines from human monocytes (26). At the same time, depletion of caspase-1 almost completely inhibits the release of IL-1β and IL-6 in monocytes, supporting arguments for involvement of an inflammasome. In contrast, GrA internalized by human blood derived-macrophages GrA can independently enhance cytokine expression (3, 102). A new model proposed that GrA in murine macrophages is induced by bacterial toxin, via the JAK-STAT pathway. GrA is subsequently released extracellularly via exocytosis and taken up by other macrophages, where it directly induces conversion of intracellular pro-IL-1β into mature IL-1β (102).
The molecular mechanism by which granzymes directly release pro-inflammatory cytokines remains unclear. Catalytic activity seems to be required for cytokine release and internalization of GrA partly enhances its release, suggesting activation of down-side effectors localized both internally and externally of cytokine producing cells (3, 21, 85, 86). Recently, we have examined the role of GrA in potentiating toll-like receptor (TLR) mediated cytokine response. Extracellular GrA potentiates a marked increase in TLR2/4 agonist-induced pro-inflammatory cytokines. Interestingly, inactive mutant GrA (Ser to Ala substitution in the GrA catalytic center) results in similar cytokine response as WT GrA, indicating an independence of the GrA catalytic activity. Thus, GrA can use its proteolytic activity to release cytokines in the absence of TLR stimulation or inactive GrA can make use of TLRs (95). Multiple studies suggest the presence of unidentified GrA-sensitive receptors on the cell-surface of reacting cells (21, 31, 85, 86, 95, 96).
As granzyme activity is tightly regulated by serine protease inhibitors in vivo and extracellular GrA in complex with proteoglycans is resistant to serpins, regulation of granzyme activity might be essential to fine-tune pro-inflammatory cytokine response (95). GrA-inducible cytokines have been linked to diseases beyond those described above suggesting alternative roles to the traditionally proposed cytotoxic role for GrA in chronic inflammation (96).
Extracellular Matrix Remodeling
Early evidence of extracellular GrA disrupting basement membrane proteins was proposed in a study that showed that high GrA concentrations in vitro could cleave the α2(IV) chain of collagen IV in mice (88). Additionally, extracellular GrA cleaves fibronectin, myelin basic protein and heparan sulfate proteoglycans in vitro, suggesting a role in ECM remodeling (87–91, 97). Destruction and remodeling of the ECM could contribute to the migration of activated cytotoxic T lymphocytes through tissue and extravasation of these cells from the vasculature and to the pathogenesis of several viral, bacterial and parasitic diseases (88, 104). As GrA is elevated in COPD and identified extracellular GrA substrates are components of the alveolar ECM, GrA may contribute extracellularly to alveolar wall damage in COPD (75). Detachment of cells, such as long epithelial cells and small intestinal epithelial cells, due cleavage of ECM proteins, could lead to both cell death resulting in COPD in lung epithelial cells and maturation and exfoliation of cells in the intestine (81, 97). Furthermore, integrin interactions with ECM proteins (e.g., fibronectin and vitronectin) may induce T-cell activation and degranulation of GrA (8). Additional work is needed to establish the in vivo relevance of these findings.
Receptor Cleavage
Several receptors are identified as external substrates of GrA, including e.g., thrombin-like receptor on neurites and platelet thrombin receptors. Cleaving of these receptors by GrA may activate them and/or induce inflammation in disease (92, 93, 100). By cleavage and subsequent activation of the thrombin receptor on neurites, immediate neurite retraction and reversed stellation of astrocytes is induced. This may contribute to the etiology of autoimmune disorders of the nervous system (92). In contrast, GrA cleavage of the platelet thrombin receptor desensitizes response to thrombin-induced aggregation by platelets. This might be a result of competition of GrA with thrombin for ThR binding (93, 94). PAR-2 has also been suggested as a substrate of GrA, although available data is limited and conflicting. PAR-2 is a G-protein coupled receptor activated through the cleavage of the receptor's extracellular N-terminal domain. This yields the PAR-2 activating sequence that can subsequently induce the expression of e.g. pro-inflammatory cytokines (96, 100). One study found that treatment of P20 peptide (corresponding to the cleavage/activation site of the wt-rPAR-2 N terminus) with GrA yielded conversion to the PAR-2-activating peptide. However, calcium mobilization experiments did not show activation of PAR-2 by GrA (100) and another study notes that GrA fails to induce Ca influx to efficiently cleave various PAR-2 sub-types (21).
Concluding Remarks
Extracellular GrA is involved in several inflammatory diseases. After amplified debate on GrA's cytotoxic potential, novel functions challenge the traditional dogma of a protective role of GrA in immune homeostasis. The observations suggest that GrA might induce increased inflammation, (over)reaction of the immune system and pathogenesis (e.g., alveolar wall damage in COPD, sepsis, tumor promotion, rheumatoid arthritis). The elevated GrA levels in bodily fluids from patients with the described diseases and infections possibly arise from local release of GrA in the inflamed tissue. Considering its pro-inflammatory role, GrA is a potential target for anti-inflammatory interventions. Nevertheless, although implicated in cytokine release, ECM matrix remodeling and receptor cleavage, functional and pathological consequences of extracellular GrA release remain largely unknown. Further research is required to assess (i) mechanism and regulation of extracellular GrA release, (ii) extracellular and intracellular pathways activated for the promotion and/or release of cytokines, (iii) direct involvement of GrA in disease pathogenesis (e.g., virus infections, parasitic infections, cancer promotion, (iv) potential usage of GrA as target in therapies without ameliorating the immune systems' ability to control infections, e.g. by using the native GrA inhibitors, (v) the potential to use extracellular GrA expression for diagnostic tools, and (vi) whether known GrA inhibitors in the extracellular matrix (or other mechanism) are altered in favor of increased GrA activity in inflammatory disease.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. de Jong HK, Garcia-Laorden MI, Hoogendijk AJ, Parry CM, Maude RR, Dondorp AM, et al. Expression of intra- and extracellular granzymes in patients with typhoid fever. PLoS Negl Trop Dis. (2017) 11:e0005823. doi: 10.1371/journal.pntd.0005823
2. Anthony DA, Andrews DM, Watt SV, Trapani JA, Smyth MJ. Functional dissection of the granzyme family: cell death and inflammation. Immunol Rev. (2010) 235:73–92. doi: 10.1111/j.0105-2896.2010.00907.x
3. Wensink AC, Hack CE, Bovenschen N. Granzymes regulate pro-inflammatory cytokine responses. J Immunol. (2015) 194:491–7. doi: 10.4049/jimmunol.1401214
4. Froelich CJ, Pardo J, Simon MM. Granule-associated serine proteases: granzymes might not just be killer proteases. Trends Immunol. (2009) 30:117–23. doi: 10.1016/j.it.2009.01.002
5. Romero V, Andrade F. Non-apoptotic functions of granzymes. Tissue Antigens. (2008) 71:409–16. doi: 10.1111/j.1399-0039.2008.01013.x
6. Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ. (2010) 17:616–23. doi: 10.1038/cdd.2009.206
8. Buzza MS, Bird PI. Extracellular granzymes: current perspectives. Biol Chem. (2006) 387:827–37. doi: 10.1515/BC.2006.106
9. Domselaar R, Bovenschen N. Cell death-independent functions of granzymes: hit viruses where it hurts. Rev Med Virol. (2011) 21:301–14. doi: 10.1002/rmv.697
10. Bell JK, Goetz DH, Mahrus S, Harris JL, Fletterick RJ, Craik CS. The oligomeric structure of human granzyme A is a determinant of its extended substrate specificity. Nat Struct Biol. (2003) 10:527–34. doi: 10.1038/nsb944
11. Zhu P, Zhang D, Chowdhury D, Martinvalet D, Keefe D, Shi L, et al. Granzyme A, which causes single-stranded DNA damage, targets the double-strand break repair protein Ku70. EMBO Rep. (2006) 7:431–7. doi: 10.1038/sj.embor.7400622
12. Mahrus S, Craik CS. Selective chemical functional probes of granzymes A and B reveal granzyme B is a major effector of natural killer cell-mediated lysis of target cells. Chem Biol. (2005) 12:567–77. doi: 10.1016/j.chembiol.2005.03.006
13. Kaiserman D, Stewart SE, Plasman K, Gevaert K, Van Damme P, Bird PI. Identification of serpinb6b as a species-specific mouse granzyme A inhibitor suggests functional divergence between human and mouse granzyme A. J Biol Chem. (2014) 289:9408–17. doi: 10.1074/jbc.M113.525808
14. Tsuzuki S, Kokado Y, Satomi S, Yamasaki Y, Hirayasu H, Iwanaga T, et al. Purification and identification of a binding protein for pancreatic secretory trypsin inhibitor: A novel role of the inhibitor as an anti-granzyme A. Biochem J. (2003) 372:227–33. doi: 10.1042/bj20021891
15. Masson D, Tschopp J. Inhibition of lymphocyte protease granzyme A by antithrombin III. Mol Immunol. (1988) 25:1283–9. doi: 10.1016/0161-5890(88)90043-0
16. Martinvalet D, Dykxhoorn DM, Ferrini R, Lieberman J. Granzyme A cleaves a mitochondrial complex i protein to initiate caspase-independent cell death. Cell. (2008) 133:681–92. doi: 10.1016/j.cell.2008.03.032
17. Fan Z, Beresford PJ, Zhang D, Xu Z, Novina CD, Yoshida A, et al. Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat Immunol. (2003) 4:145–53. doi: 10.1038/ni885
18. Beresford PJ, Xia Z, Greenberg AH, Lieberman J. Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity. (1999) 10:585–94. doi: 10.1016/S1074-7613(00)80058-8
19. Zhang D, Beresford PJ, Greenberg AH, Lieberman J. Granzymes A and B directly cleave lamins and disrupt the nuclear lamina during granule-mediated cytolysis. Proc Natl Acad Sci USA. (2001) 98:5746–51. doi: 10.1073/pnas.101329598
20. Vidyarani M, Selvaraj P, Raghavan S, Narayanan PR. Regulatory role of 1, 25-dihydroxyvitamin D3 and vitamin D receptor gene variants on intracellular granzyme A expression in pulmonary tuberculosis. Exp Mol Pathol. (2009) 86:69–73. doi: 10.1016/j.yexmp.2008.10.002
21. Metkar SS, Menaa C, Pardo J, Wang B, Wallich R, Freudenberg M, et al. Human and mouse granzyme a induce a pro-inflammatory cytokine response. Immunity. (2008) 29:720–33. doi: 10.1016/j.immuni.2008.08.014
22. Kaiserman D, Bird CH, Sun J, Matthews A, Ung K, Whisstock JC, et al. The major human and mouse granzymes are structurally and functionally divergent. J Cell Biol. (2006) 175:619–30. doi: 10.1083/jcb.200606073
23. Turner CT, Lim D, Granville DJ. Granzyme B in skin inflammation and disease. Matrix Biol. (2019) 75-76:126–40. doi: 10.1016/j.matbio.2017.12.005
24. Boivin WA, Cooper DM, Hiebert PR, Granville DJ. Intracellular versus extracellular granzyme B in immunity and disease: challenging the dogma. Lab Investig. (2009) 89:1195–220. doi: 10.1038/labinvest.2009.91
25. Bovenschen N, Spijkers SNM, Wensink AC, Schellens IMM, van Domselaar R, van Baarle D. Elevated granzyme M-expressing lymphocytes during cytomegalovirus latency and reactivation after allogeneic stem cell transplantation. Clin Immunol. (2014) 150:1–11. doi: 10.1016/j.clim.2013.11.005
26. Wensink AC, Kok HM, Meeldijk J, Fermie J, Froelich CJ, Hack CE, et al. Granzymes A and K differentially potentiate LPS-induced cytokine response. Cell Death Discov. (2016) 2:1–9. doi: 10.1038/cddiscovery.2016.84
27. Thakur A, Mikkelsen H, Jungersen G. Intracellular Pathogens: Host Immunity and Microbial Persistence Strategies. J Immunol Res. (2019) 2019:1356540. doi: 10.1155/2019/1356540
28. Teo AKJ, Singh SR, Prem K, Hsu LY, Yi S. Delayed diagnosis and treatment of pulmonary tuberculosis in high-burden countries: A systematic review protocol. BMJ Open. (2019) 9:807. doi: 10.1136/bmjopen-2019-029807
29. Guggino G, Orlando V, Cutrera S, La Manna MP, Di Liberto D, Vanini V, et al. Granzyme A as a potential biomarker of Mycobacterium tuberculosis infection and disease. Immunol Lett. (2015) 166:87–91. doi: 10.1016/j.imlet.2015.05.019
30. Garcia-Laorden MI, Blok DC, Kager LM, Hoogendijk AJ, van Mierlo GJ, Lede IO, et al. Increased intra- and extracellular granzyme expression in patients with tuberculosis. Tuberculosis. (2015) 95:575–80. doi: 10.1016/j.tube.2015.05.016
31. Spencer CT, Abate G, Sakala IG, Xia M, Truscott SM, Eickhoff CS, et al. Granzyme A produced by γ9δ2 T cells induces human macrophages to inhibit growth of an intracellular pathogen. PLoS Pathog. (2013) 9:e1003119. doi: 10.1371/journal.ppat.1003119
32. Uranga S, Marinova D, Martin C, Pardo J, Aguilo N. Granzyme A is expressed in mouse lungs during Mycobacterium tuberculosis infection but does not contribute to protection in vivo. PLoS ONE. (2016) 11:e0153028. doi: 10.1371/journal.pone.0153028
33. Lauw FN, Simpson AJH, Hack CE, Prins JM, Wolbink AM, van Deventer SJH, et al. Soluble granzymes are released during human endotoxemia and in patients with severe infection due to gram-negative bacteria. J Infect Dis. (2000) 182:206–13. doi: 10.1086/315642
34. Zeerleder S, Hack CE, Caliezi C, van Mierlo G, Eerenberg-Belmer A, Wolbink A, et al. Activated cytotoxic T cells and NK cells in severe sepsis and septic shock and their role in multiple organ dysfunction. Clin Immunol. (2005) 116:158–65. doi: 10.1016/j.clim.2005.03.006
35. van den Boogaard FE, van Gisbergen KPJM, Vernooy JH, Medema JP, Roelofs JJTH, van Zoelen MAD, et al. Granzyme A impairs host defense during Streptococcus pneumoniae pneumonia. Am J Physiol - Lung Cell Mol Physiol. (2016) 311:L507–L16. doi: 10.1152/ajplung.00116.2016
36. van Woensel JBM, Biezeveld MH, Hack CE, Bos AP, Kuijpers TW. Elastase and granzymes during meningococcal disease in children: correlation to disease severity. Intensive Care Med. (2005) 31:1239–47. doi: 10.1007/s00134-005-2720-3
37. Arias MA, Jiménez de Bagües MP, Aguiló N, Menao S, Hervás-Stubbs S, de Martino A, et al. Elucidating sources and roles of granzymes A and B during bacterial infection and sepsis. Cell Rep. (2014) 8:420–9. doi: 10.1016/j.celrep.2014.06.012
38. Anthony DA, Andrews DM, Chow M, Watt SV, House C, Akira S, et al. A Role for Granzyme M in TLR4-driven inflammation and endotoxicosis. J Immunol. (2010) 185:1794–803. doi: 10.4049/jimmunol.1000430
39. Mabbott NA. The influence of parasite infections on host immunity to co-infection with other pathogens. Front Immunol. (2018) 9:2579. doi: 10.3389/fimmu.2018.02579
40. Spaeny-Dekking EH, Hanna WL, Wolbink AM, Wever PC, Kummer JA, Kummer AJ, et al. Extracellular granzymes A and B in humans: detection of native species during CTL responses in vitro and in vivo. J Immunol. (1998) 160:3610–6.
41. Bade B, Lohrmann J, Brinke A, Wolbink AM, Wolbink G-J, ten Berge IJM, et al. Detection of soluble human granzyme K in vitro and in vivo. Eur J Immunol. (2005) 35:2940–948. doi: 10.1002/eji.200526249
42. Berge IJ, Wever P, Wolbink A, Surachno J, Wertheim PM, Spaeny LH, et al. Increased systemic levels of soluble granzymes A and B during primary cytomegalovirus infection after renal transplantation. Transplant Proc. (1998) 30:3972–4. doi: 10.1016/S0041-1345(98)01308-6
43. Bem RA, Bos AP, Bots M, Wolbink AM, van Ham SM, Medema JP, et al. Activation of the granzyme pathway in children with severe respiratory syncytial virus infection. Pediatr Res. (2008) 63:650–5. doi: 10.1203/PDR.0b013e31816fdc32
44. Wilson JAC, Prow NA, Schroder WA, Ellis JJ, Cumming HE, Gearing LJ, et al. RNA-Seq analysis of chikungunya virus infection and identification of granzyme A as a major promoter of arthritic inflammation. PLOS Pathog. (2017) 13:e1006155. doi: 10.1371/journal.ppat.1006155
45. Schanoski AS, Le TT, Kaiserman D, Rowe C, Prow NA, Barboza DD, et al. Granzyme A in chikungunya and other arboviral infections. Front Immunol. (2020) 10:3083. doi: 10.3389/fimmu.2019.03083
46. Ebnet K, Hausmann M, Lehmann-Grube F, Müllbacher A, Kopf M, Lamers M, et al. Granzyme A-deficient mice retain potent cell-mediated cytotoxicity. EMBO J. (1995) 14:4230–9. doi: 10.1002/j.1460-2075.1995.tb00097.x
47. Müllbacher A, Waring P, Hla RT, Tran T, Chin S, Stehle T, et al. Granzymes are the essential downstream effector molecules for the control of primary virus infections by cytolytic leukocytes. Proc Natl Acad Sci USA. (1999) 96:13950–5. doi: 10.1073/pnas.96.24.13950
48. Pereira RA, Simon MM, Simmons A. Granzyme A, a noncytolytic component of CD8+ cell granules, restricts the spread of herpes simplex virus in the peripheral nervous systems of experimentally infected mice. J Virol. (2000) 74:1029–32. doi: 10.1128/JVI.74.2.1029-1032.2000
49. Riera L, Gariglio M, Valente G, Müllbacher A, Museteanu C, Landolfo S, et al. Murine cytomegalovirus replication in salivary glands is controlled by both perforin and granzymes during acute infection. Eur J Immunol. (2000) 30:1350–5. doi: 10.1002/(SICI)1521-4141(200005)30:5<1350::AID-IMMU1350>3.0.CO;2-J
50. Arias M, Martínez-Lostao L, Santiago L, Ferrandez A, Granville DJ, Pardo J. The untold story of granzymes in oncoimmunology: novel opportunities with old acquaintances. Trends in Cancer. (2017) 3:407–22. doi: 10.1016/j.trecan.2017.04.001
51. Romero V, Fellows E, Jenne DE, Andrade F. Cleavage of La protein by granzyme H induces cytoplasmic translocation and interferes with La-mediated HCV-IRES translational activity. Cell Death Differ. (2009) 16:340–48. doi: 10.1038/cdd.2008.165
52. Andrade F, Fellows E, Jenne DE, Rosen A, Young CSH. Granzyme H destroys the function of critical adenoviral proteins required for viral DNA replication and granzyme B inhibition. EMBO J. (2007) 26:2148–57. doi: 10.1038/sj.emboj.7601650
53. van Domselaar R, Philippen LE, Quadir R, Wiertz EJHJ, Kummer JA, Bovenschen N. Noncytotoxic inhibition of cytomegalovirus replication through NK cell protease granzyme m-mediated cleavage of viral phosphoprotein 71. J Immunol. (2010) 185:7605–13. doi: 10.4049/jimmunol.1001503
54. Andrade F. Non-cytotoxic antiviral activities of granzymes in the context of the immune antiviral state. Immunol Rev. (2010) 235:128–46. doi: 10.1111/j.0105-2896.2010.00909.x
55. Yan N, Cherepanov P, Daigle JE, Engelman A, Lieberman J. The SET complex acts as a barrier to autointegration of HIV-1. PLoS Pathog. (2009) 5:e1000327 doi: 10.1371/journal.ppat.1000327
56. Haruki H, Okuwaki M, Miyagishi M, Taira K, Nagata K. Involvement of template-activating factor I/SET in transcription of adenovirus early genes as a positive-acting factor. J Virol. (2006) 80:794–801. doi: 10.1128/JVI.80.2.794-801.2006
57. Quan LT, Caputo A, Bleackley RC, Pickup DJ, Salvesen GS. Granzyme B is inhibited by the cowpox virus serpin cytokine response modifier A. J Biol Chem. (1995) 270:10377–9. doi: 10.1074/jbc.270.18.10377
58. Turner PC, Sancho MC, Thoennes SR, Caputo A, Bleackley RC, Moyer RW. Myxoma virus Serp2 is a weak inhibitor of granzyme B and interleukin-1β-converting enzyme in vitro and unlike crma cannot block apoptosis in cowpox virus-infected cells. J Virol. (1999) 73:6394–404. doi: 10.1128/JVI.73.8.6394-6404.1999
59. Andrade F, Bull HG, Thornberry NA, Ketner GW, Casciola-Rosen LA, Rosen A. Adenovirus L4-100K assembly protein is a granzyme B substrate that potently inhibits granzyme B-mediated cell death. Immunity. (2001) 14:751–61. doi: 10.1016/S1074-7613(01)00149-2
60. Jerome KR, Chen Z, Lang R, Torres MR, Hofmeister J, Smith S, et al. HSV and glycoprotein J inhibit caspase activation and apoptosis induced by granzyme B or Fas. J Immunol. (2001) 167:3928–35. doi: 10.4049/jimmunol.167.7.3928
61. Hermsen CC, Konijnenberg Y, Mulder L, Loe C, Van Deurem M, Van der Meer JWM, et al. Circulating concentrations of soluble granzyme A and B increase during natural and experimental Plasmodium falciparum infections. Clin Exp Immunol. (2003) 132:467–72. doi: 10.1046/j.1365-2249.2003.02160.x
62. Eisert V, Münster U, Simon MM, Moll H. The course of leishmania major infection in mice lacking granzyme-mediated mechanisms. Immunobiology. (2002) 205:314–20. doi: 10.1078/0171-2985-00134
63. Müller U, Sobek V, Balkow S, Hölscher C, Müllbacher A, Museteanu C, et al. Concerted action of perforin and granzymes is critical for the elimination of Trypanosoma cruzi from mouse tissues, but prevention of early host death is in addition dependent onthe FasL/Fas pathway. Eur J Immunol. (2003) 33:70–8. doi: 10.1002/immu.200390009
64. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. (2010) 376:1094–108. doi: 10.1016/S0140-6736(10)60826-4
65. Sardar S, Andersson Å. Old and new therapeutics for Rheumatoid Arthritis: in vivo models and drug development. Immunopharmacol Immunotoxicol. (2016) 38:2–13. doi: 10.3109/08923973.2015.1125917
66. Tak PP, Spaeny-dekking L, Kraan MC, Breedveld FC, Froelich CJ, Hack CE. The levels of soluble granzyme A and B are elevated in plasma and synovial fluid of patients with rheumatoid arthritis (RA). Clin Exp Immunol. (1999) 116:366–70. doi: 10.1046/j.1365-2249.1999.00881.x
67. Kummer JA, Tak PP, Brinkman BM, van Tilborg AA, Kamp AM, Verweij CL, et al. Expression of granzymes A and B in synovial tissue from patients with rheumatoid arthritis and osteoarthritis. Clin Immunol Immunopathol. (1994) 73:88–95. doi: 10.1006/clin.1994.1173
68. Santiago L, Menaa C, Arias M, Martin P, Jaime-S Anchez P, Metkar S, et al. Granzyme A Contributes to Inflammatory Arthritis in Mice Through Stimulation of Osteoclastogenesis. ARTHRITIS Rheumatol. (2017) 69:320–34. doi: 10.1002/art.39857
69. Hirayama T, Danks L, Sabokbar A, Athanasou NA. Osteoclast formation and activity in the pathogenesis of osteoporosis in rheumatoid arthritis. Rheumatology (Oxford). (2002) 41:1232–9. doi: 10.1093/rheumatology/41.11.1232
70. Müller-Ladner U, Kriegsmann Jör, Tschopp Jür, Gay RE, Gay S. Demonstration of granzyme A and perforin messenger RNA in the synovium of patients with rheumatoid arthritis. Arthritis Rheum. (1995) 38:477–84. doi: 10.1002/art.1780380404
71. Tremblay GM, Wolbink AM, Cormier Y, Hack CE. Granzyme activity in the inflamed lung is not controlled by endogenous serine proteinase inhibitors. J Immunol. (2000) 165:3966–969. doi: 10.4049/jimmunol.165.7.3966
72. Hodge G, Mukaro V, Holmes M, Reynolds PN, Hodge S. Enhanced cytotoxic function of natural killer and natural killer T-like cells associated with decreased CD94 (Kp43) in the chronic obstructive pulmonary disease airway. Respirology. (2013) 18:369–76. doi: 10.1111/j.1440-1843.2012.02287.x
73. Tschopp CM, Spiegl N, Didichenko S, Lutmann W, Julius P, Virchow JC, et al. Granzyme B, a novel mediator of allergic inflammation: its induction and release in blood basophils and human asthma. Blood. (2006) 108:2290–9. doi: 10.1182/blood-2006-03-010348
74. Proklou A, Soulitzis N, Neofytou E, Rovina N, Zervas E, Gaga M, et al. Granule cytotoxic activity and oxidative DNA damage in smoking and nonsmoking patients with asthma. Chest. (2013) 144:1230–7. doi: 10.1378/chest.13-0367
75. Vernooy JHJ, Möller GM, van Suylen RJ, van Spijk MP, Cloots RHE, Hoet PH, et al. Increased granzyme A expression in Type II pneumocytes of patients with severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2007) 175:464–72. doi: 10.1164/rccm.200602-169OC
76. Accardo-Palumbo A, Ferrante A, Cadelo M, Ciccia F, Parrinello G, Lipari L, et al. The level of soluble Granzyme A is elevated in the plasma and in the Vgamma9/Vdelta2 T cell culture supernatants of patients with active Behçet's disease. Clin Exp Rheumatol. (2004) 22:S45–9.
77. Augustin MT, Kokkonen J, Karttunen R, Karttunen TJ. Serum granzymes and CD30 are increased in children's milk protein sensitive enteropathy and celiac disease. J Allergy Clin Immunol. (2005) 115:157–62. doi: 10.1016/j.jaci.2004.10.009
78. Annoni R, Silva LFF, Nussbaumer-Ochsner Y, Van Schadewijk A, Mauad T, Hiemstra PS, et al. Increased expression of granzymes A and B in fatal asthma. Eur Respir J. (2015) 45:1485–8. doi: 10.1183/09031936.00213814
79. Hashimoto S, Kobayashi A, Kooguchi K, Kitamura Y, Onodera H, Nakajima H. Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med. (2000) 161:237–43. doi: 10.1164/ajrccm.161.1.9810007
80. Zhao CZ, Fang XC, Wang D, Tang F Di, Wang XD. Involvement of type II pneumocytes in the pathogenesis of chronic obstructive pulmonary disease. Respir Med. (2010) 104:1391–5. doi: 10.1016/j.rmed.2010.06.018
81. Yoshikawa Y, Hirasyasu H, Tsuzuki S, Fushiki T. Granzyme A causes detachment of alveolar epithelial A549 cells accompanied by promotion of interleukin-8 release. Biosci Biotechnol Biochem. (2008) 72:2481–4. doi: 10.1271/bbb.80362
82. Yoshikawa Y, Hirayasu H, Tsuzuki S, Fushiki T. Carrageenan inhibits granzyme A-induced detachment of and interleukin-8 release from alveolar epithelial A549 cells. Cytotechnology. (2008) 58:63–7. doi: 10.1007/s10616-008-9175-7
83. Müller S, Lory J, Corazza N, Griffiths GM, Z'graggen K, Mazzucchelli L, et al. Activated CD4+ and CD8+ cytotoxic cells are present in increased numbers in the intestinal mucosa from patients with active inflammatory bowel disease. Am J Pathol. (1998) 152:261–8.
84. Tew GW, Hackney JA, Gibbons D, Lamb CA, Luca D, Egen JG, et al. Association between response to etrolizumab and expression of integrin αe and granzyme A in colon biopsies of patients with ulcerative colitis. Gastroenterology. (2016) 150:477–87.e9. doi: 10.1053/j.gastro.2015.10.041
85. Sower LE, Froelich CJ, Allegretto N, Rose PM, Hanna WD, Klimpel GR. Extracellular activities of human granzyme A. Monocyte activation by granzyme A versus alpha-thrombin. J Immunol. (1996) 156:2585–90.
86. Sower LE, Klimpel GR, Hanna W, Froelich CJ. Extracellular Activities of Human Granzymes. Cell Immunol. (1996) 171:159–63. doi: 10.1006/cimm.1996.0187
87. Vettel U, Brunner G, Bar-Shavit R, Vlodavsky I, Kramer MD. Charge-dependent binding of granzyme A (MTSP-1) to basement membranes. Eur J Immunol. (1993) 23:279–82. doi: 10.1002/eji.1830230144
88. Simon MM, Kramer MD, Prester M, Gay S. Mouse T-cell associated serine proteinase 1 degrades collagen type IV: a structural basis for the migration of lymphocytes through vascular basement membranes. Immunology. (1991) 73:117–9.
89. Simon MM, Prester M, Nerz G, Kramer MD, Fruth U. Release of biologically active fragments from human plasma-fibronectin by murine T cell-specific proteinase 1 (TSP-1). Biol Chem Hoppe Seyler. (1988) 369 (Suppl):107–12.
90. Vettel U, Bar-Shavit R, Simon MM, Brunner G, Vlodavsky I, Kramer MD. Coordinate secretion and functional synergism of T cell-associated serine proteinase-1 (MTSP-1) and endoglycosidase(s) of activated T cells. Eur J Immunol. (1991) 21:2247–51. doi: 10.1002/eji.1830210936
91. Simon MM, Simon HG, Fruth U, Epplen J, Müller-Hermelink HK, Kramer MD. Cloned cytolytic T-effector cells and their malignant variants produce an extracellular matrix degrading trypsin-like serine proteinase. Immunology. (1987) 60:219.
92. Suidan HS, Bouvier J, Schaerer E, Stone SR, Monard D, Tschopp J. Granzyme A released upon stimulation of cytotoxic T lymphocytes activates the thrombin receptor on neuronal cells and astrocytes. Proc Natl Acad Sci USA. (1994) 91:8112–6. doi: 10.1073/pnas.91.17.8112
93. Parry MAA, Myles T, Rg Tschopp J, Stone SR. Cleavage of the thrombin receptor: identification of potential activators and inactivators. Biochem J. (1996) 320:335–41.
94. Suidan HS, Clemetson KJ, Brown-Luedi M, Niclou SP, Clementson JM, Tschopp J, et al. The serine protease granzyme A does not induce platelet aggregation but inhibits responses triggered by thrombin. Biochem J. (1996) 315:939–45. doi: 10.1042/bj3150939
95. Eck JA, van Shan L, Meeldijk J, Hack CE, Bovenschen N. A novel pro-inflammatory role for granzyme A. Cell Death Dis. (2017) 2017:82. doi: 10.1038/cddis.2017.56
96. Hendel A, Hiebert PR, Boivin WA, Williams SJ, Granville DJ. Granzymes in age-related cardiovascular and pulmonary diseases. Cell Death Differ. (2010) 17:596–606. doi: 10.1038/cdd.2010.5
97. Hirayasu H, Yoshikawa Y, Tsuzuki S, Fushiki T. A lymphocyte serine protease granzyme A causes detachment of a small-intestinal epithelial cell line (IEC-6). Biosci Biotechnol Biochem. (2008) 72:2294–302. doi: 10.1271/bbb.80140
98. Vanguri P, Lee E, Henkart P, Shin ML. Hydrolysis of myelin basic protein in myelin membranes by granzymes of large granular lymphocytes. J Immunol. (1993) 150:2431–9.
99. Brunner G, Simon MM, Kramer MD. Activation of pro-urokinase by the human T cell-associated serine proteinase HuTSP-1. FEBS Lett. (1990) 260:141–4. doi: 10.1016/0014-5793(90)80087-Y
100. Hansen KK, Sherman PM, Cellars L, Andrade-Gordon P, Pan Z, Baruch A, et al. A major role for proteolytic activity and proteinase-activated receptor-2 in the pathogenesis of infectious colitis. Proc Natl Acad Sci. (2005) 102:8363–8. doi: 10.1073/pnas.0409535102
101. Irmler M, Hertig S, MacDonald HR, Sadoul R, Becherer JD, Proudfoot A, et al. Granzyme A is an interleukin 1 beta-converting enzyme. J Exp Med. (1995) 181:1917–22. doi: 10.1084/jem.181.5.1917
102. Hildebrand D, Bode KA, Rieß D, Cerny D, Waldhuber A, Römmler F, et al. Granzyme A Produces Bioactive IL-1β through a Nonapoptotic Inflammasome-Independent Pathway. Cell Rep. (2014) 9:910–7. doi: 10.1016/j.celrep.2014.10.003
103. Campbell RA, Franks Z, Bhatnagar A, Rowley JW, Manne BK, Supiano MA, et al. Granzyme A in human platelets regulates the synthesis of pro-inflammatory cytokines by monocytes in aging. J Immunol. (2018) 200:295–304. doi: 10.4049/jimmunol.1700885
Keywords: Granzyme A, granzymes, inflammation, extracellular, inflammatory disease
Citation: van Daalen KR, Reijneveld JF and Bovenschen N (2020) Modulation of Inflammation by Extracellular Granzyme A. Front. Immunol. 11:931. doi: 10.3389/fimmu.2020.00931
Received: 06 February 2020; Accepted: 21 April 2020;
Published: 19 May 2020.
Edited by:
Fabrice Cognasse, Groupe Sur L'immunité Des Muqueuses Et Agents Pathogènes (GIMAP), FranceReviewed by:
Julian Pardo, Fundacion Agencia Aragonesa para la Investigacion y el Desarrollo, SpainDavid J. Granville, University of British Columbia, Canada
Copyright © 2020 van Daalen, Reijneveld and Bovenschen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Niels Bovenschen, bi5ib3ZlbnNjaGVuQHVtY3V0cmVjaHQubmw=
†These authors share first authorship