 Maria Stratigopoulou
Maria Stratigopoulou Tijmen P. van Dam
Tijmen P. van Dam Jeroen E. J. Guikema
Jeroen E. J. Guikema- Department of Pathology, Lymphoma and Myeloma Center Amsterdam (LYMMCARE), Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
The integrity of the genome is under constant threat of environmental and endogenous agents that cause DNA damage. Endogenous damage is particularly pervasive, occurring at an estimated rate of 10,000–30,000 per cell/per day, and mostly involves chemical DNA base lesions caused by oxidation, depurination, alkylation, and deamination. The base excision repair (BER) pathway is primary responsible for removing and repairing these small base lesions that would otherwise lead to mutations or DNA breaks during replication. Next to preventing DNA mutations and damage, the BER pathway is also involved in mutagenic processes in B cells during immunoglobulin (Ig) class switch recombination (CSR) and somatic hypermutation (SHM), which are instigated by uracil (U) lesions derived from activation-induced cytidine deaminase (AID) activity. BER is required for the processing of AID-induced lesions into DNA double strand breaks (DSB) that are required for CSR, and is of pivotal importance for determining the mutagenic outcome of uracil lesions during SHM. Although uracils are generally efficiently repaired by error-free BER, this process is surprisingly error-prone at the Ig loci in proliferating B cells. Breakdown of this high-fidelity process outside of the Ig loci has been linked to mutations observed in B-cell tumors and DNA breaks and chromosomal translocations in activated B cells. Next to its role in preventing cancer, BER has also been implicated in immune tolerance. Several defects in BER components have been associated with autoimmune diseases, and animal models have shown that BER defects can cause autoimmunity in a B-cell intrinsic and extrinsic fashion. In this review we discuss the contribution of BER to genomic integrity in the context of immune receptor diversification, cancer and autoimmune diseases.
Introduction
The adaptive immune response is of crucial importance for the elimination of pathogens, such as bacteria, viruses, and other foreign substances. Lymphocytes are the prime mediators of the adaptive immune response, recognizing antigens by their specific antigen receptors (AgR). DNA recombination and mutation processes ensure the generation of a vast array of AgRs. During lymphocyte development, the antigen-independent recombination of variable (V), diversity (D), and joining (J) gene segments assembles the genetic code for the T-cell receptor (TCR) and the B-cell receptor (BCR). This highly ordered process involves the generation of DNA double-strand breaks (DSB) followed by non-homologous end-joining (NHEJ) of the various gene segments. This process is unique for lymphocytes and requires the DNA nicking activity of recombination activating gene products 1 and 2 (RAG1, RAG2) (1).
B cells can undergo additional diversification processes that shape the immunoglobulin (Ig) repertoire in an antigen-dependent fashion. Class switch recombination (CSR) is an (predominantly) intrachromosomal looping process by which the constant region coding for the Ig isotype is exchanged, thereby altering the effector function of the expressed Ig. Switch regions that are located upstream of each Ig constant region are the targets for DSBs that are resolved by NHEJ, resulting in the looping out of DNA intervening the switch regions from upstream and downstream constant regions (2). Somatic hypermutation (SHM) is a crucial event for antibody affinity maturation. Point mutations are introduced in the recombined V(D)J and Ig switch regions. B cells with improved affinity for antigen as a result of these mutations are clonally selected to differentiate into memory B cells and plasma cells by competing for antibody-mediated antigen capture and subsequent acquisition of T-cell help within germinal centers (GC) in secondary lymphoid organs (3). CSR and SHM are initiated by the activation-induced cytidine deaminase (AID) (4, 5). AID instigates both events by provoking base damage directed at cytosines (C), generating deoxy-uracil (U) that triggers mutagenic processing by the base excision repair (BER) and mismatch repair (MMR) pathways, resulting in point mutations and DSBs.
Typically, BER is initiated by the recognition and removal of damaged bases by DNA glycosylases resulting in the formation of apurinic/apyrimidinic (AP) sites. These AP sites are highly mutagenic and require subsequent processing by AP endonucleases or by the AP lyase activity of bifunctional glycosylases, which nick the phosphodiester backbone of the AP site. The resulting DNA single-strand nicks can be processed into DSBs or be repaired by displacement synthesis (long-patch BER) or non-displacement synthesis (short-patch BER) (6, 7) (Figure 1). Interestingly, MMR is a primarily replication-linked repair pathway that acts on the same base lesions as BER. The three important steps that constitute the MMR pathway are: (i) mismatch recognition by MutS homolog (MSH) heterodimers (typically MSH2/MSH6; MutSα); (ii) recruitment of MutL homolog 1 (MLH1) and post-meiotic segregation-increased homolog 2 (PMS2) heterodimers (MutLα) and exonuclease 1 (EXO1), which are involved in the excision of a patch containing the damaged base(s); (iii) recruitment of DNA polymerases and fill-in synthesis (8). However, MMR can also act independently of DNA replication (9, 10). Importantly, in B cells undergoing CSR, AID-generated U:G mismatches give rise to MMR-dependent DSBs in the G1 phase of the cell cycle by patch excision of the mismatch-containing strand until a DNA nick on the opposite strand is reached (9). In addition, in B cells undergoing SHM, MMR displays a non-canonical (mutagenic) activity by the specific recruitment of the error-prone translesion polymerase POLH, which lacks proofreading activity. The error-prone activity of POLH is responsible for mutations at adenosine (A) and thymidine (T) bases during SHM, complementing a full spectrum of DNA mutations triggered by AID (11–13). The mechanistic basis for the switch to mutagenic non-canonical MMR (ncMMR) in B cells remains to be fully elucidated, and whether it is restricted to the G1 phase is currently unknown. However, in vitro and in vivo experiments indicate that the monoubiquitination of proliferating cell nuclear antigen (PCNA) is linked to ncMMR activity and is of crucial importance for mutations at A:T bases during SHM (10, 14, 15). Apparently, AID-dependent base lesions evade faithful DNA repair and elicit mutagenic repair, which critically involves BER and MMR (Figure 2).
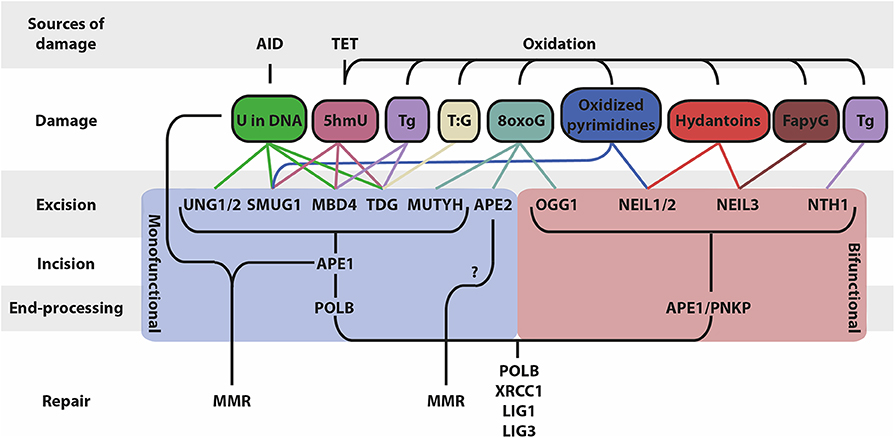
Figure 1. Schematic overview of BER and associated factors. BER functions on different types of DNA base lesions that are generated by AID, TET and through oxidation. BER occurs in four mains steps that differ based on the mono/bifunctionality of the glycosylase: (i) base excision, (ii) DNA backbone incision, (iii) DNA end processing, (iv) repair of the lesion (5hmU, 5-hydroxymethyluracil; Tg, thymine glycol; 5hmC, 5-hydroxymethylcytosine; 8oxoG, 8-oxoguanine; FapyG, 2,6-diamino-4-hydroxy-5-formamidopyrimidine).
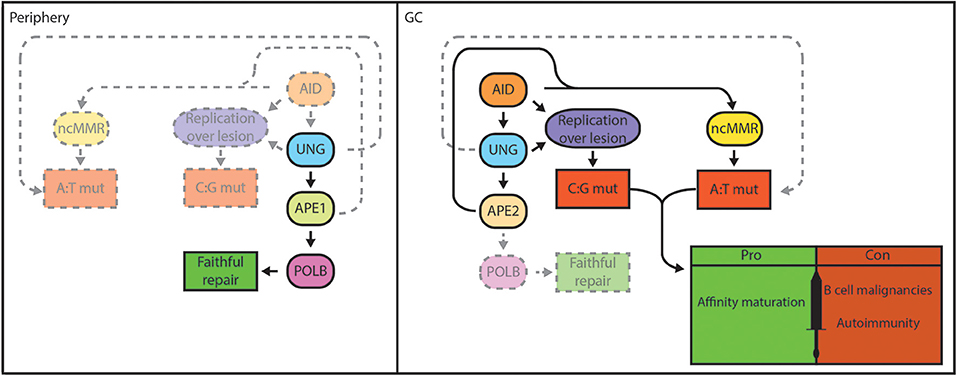
Figure 2. BER functions as a double-edged sword in B cells. Genomic integrity is safeguarded by the BER pathway in lymphocytes in the periphery. Damaged bases are faithfully repaired by UNG, APE1, and POLB in case of short-patch BER (left). In GC B cells, localized base damage is introduced in the Ig genes by AID (right). Some localized uracil lesions generated by AID overwhelm/escape BER and are replicated resulting in C:G transitions, but may also trigger A:T mutations. Others are engaged by UNG and are converted into AP sites that can escape processing by BER and are replicated by REV1, resulting in C:G transversions. Further processing of AP sites by BER generates nicks that serve to promote patch excision and mutagenic repair by ncMMR, which is responsible for the majority of A:T mutations. Mutagenic repair by ncMMR in GC B cells is stimulated by low POLB protein expression. AID-instigated mutations contribute to affinity maturation and immune function, but can also result in genomic instability that underlie B-cell malignancies or generate autoantibodies that cause autoimmune diseases.
Mutagenic repair can generate genomic alterations such as chromosomal translocations, and lead to the activation of oncogenes or inactivation of tumor suppressors. AgR diversification mechanisms are associated with hallmark genetic aberrations in B-cell lymphomas, mostly derived from illegitimate CSR events and off-target mutations caused by AID (16, 17). Mutagenic repair is coupled to a partial inactivation of the short-patch BER pathway in rapidly dividing GC B cells (Figure 2), which contributes to genomic instability (18) (preprint). Accordingly, the majority of human B-cell lymphomas are derived from the GC (19).
Autoimmune diseases are characterized by a breach of clonal tolerance that regulates the activation and survival of B cells independent of antigen specificity, often resulting in autoantibody production, which plays a pivotal role in the pathogenesis of these diseases (20). Isotype-switched autoantibodies are of crucial importance in several autoimmune disease models. Loss of AID function was shown to ameliorate disease manifestations in mouse models for multiple sclerosis (MS) (21, 22) and systemic lupus erythematosus (SLE) (23, 24), although from these studies it can not be concluded whether this is related to loss of CSR or SHM, as both require AID. However, in a recent study the effects of CSR versus SHM on the development of MS in a mouse model were assessed using an elegant approach, showing that CSR was of major importance for disease incidence and severity (25). Contrastingly, SHM activity was shown to be involved in the clonal redemption of anergic autoantibody-expressing B cells in humans and in autoantibody-dependent disease models in the mouse, by causing mutations that steer away from self-reactivity (26–28). Moreover, DNA repair pathways, including BER, have been linked to autoimmunity by suppressing the release of DNA, which is involved in the pathogenesis of autoimmune diseases (29, 30). DNA repair deficiencies may lead to the increased formation of endogenous DNA damage, which provokes apoptosis and the release of DNA fragments that can stimulate innate inflammatory responses. Upon endocytosis of extracellular DNA, engagement of endosomal and intracellular DNA sensors such as Toll-like receptor 9 (TLR9) and the cyclic-di-GMP-AMP synthetase (cGAS) results in the activation of the stimulator of interferon genes (STING) adaptor protein, which stimulates the release of proinflammatory cytokines that may contribute the pathogenesis of autoimmune diseases (31).
In this review we discuss the involvement of BER in lymphocyte development and function, with a particular focus on diseases such as lymphomas, leukemias, and autoimmune disorders.
DNA Glycosylases
DNA bases damaged by spontaneous and enzymatic deamination, depurination, oxidation or alkylation are repaired by the BER pathway. As an initial step, the damaged base is recognized and removed from the DNA backbone by one of the DNA glycosylases. Monofunctional glycosylases remove the damaged base and leave an AP site, which requires subsequent cleaving of the DNA backbone by an AP endonuclease, whereas bifunctional glycosylases possess both glycosylase and AP lyase activity. The various glycosylases recognize different types of damaged bases (Figure 1). Deamination of C to U is the most common base damage, which can be repaired by the monofunctional glycosylases UNG, TDG, MBD4, or SMUG1 (32). These enzymes have overlapping and distinct functions, mostly related to different substrate preferences (Table 1).
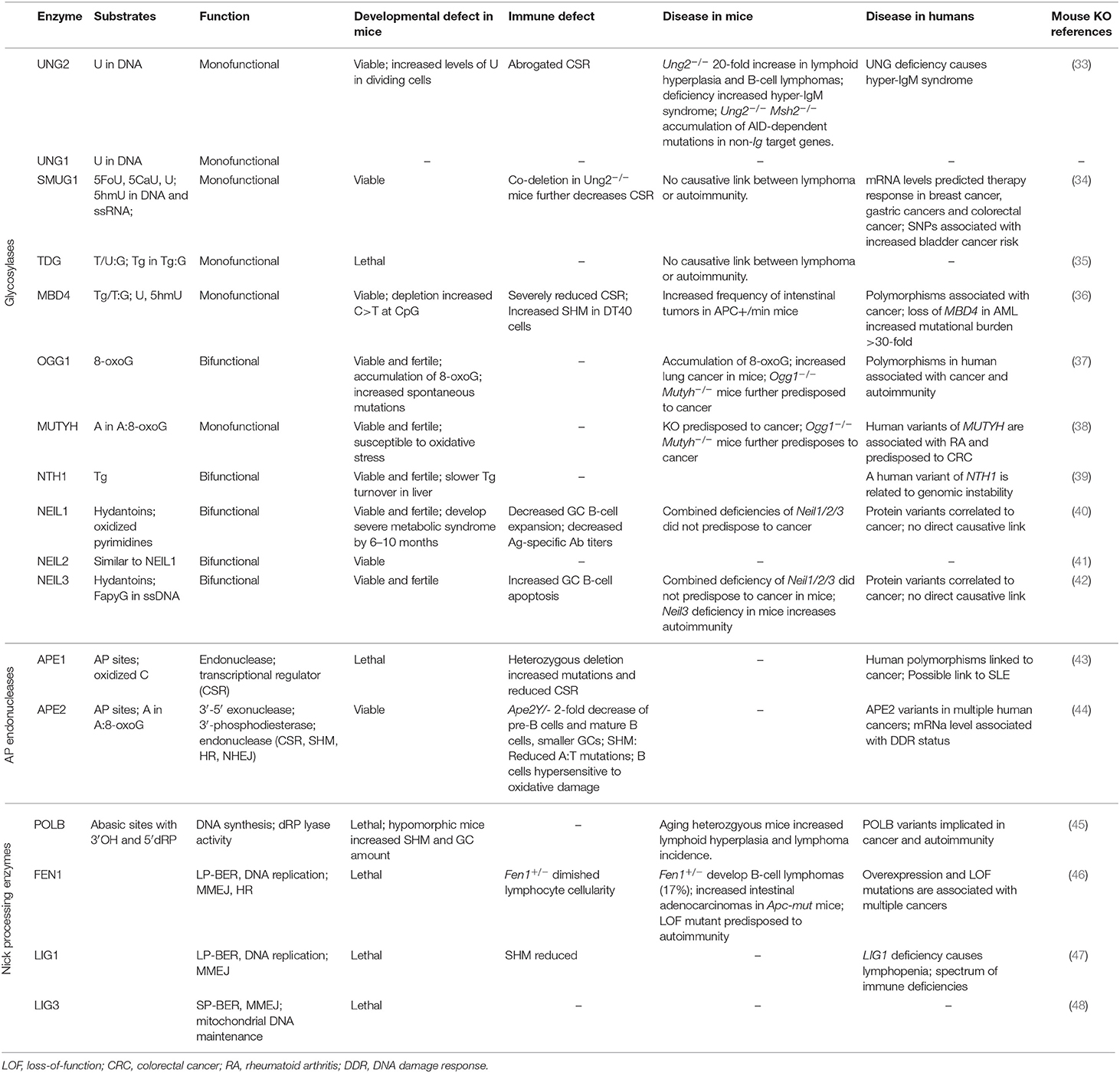
Table 1. An overview of biological and chemical attributes of BER proteins.
Uracil DNA Glycosylase (UNG)
The UNG gene encodes two isoforms that differ by their N-terminal sequence and localize to the mitochondria (UNG1) and the nucleus (UNG2) (49). Due to its high turnover rate UNG2 is the predominant glycosylase in U removal and is active on both single-stranded (ss) and double-stranded (ds) DNA, while TDG and MBD4 act on dsDNA (50–52), and SMUG1 preferentially acts on ssDNA (53). The expression of UNG peaks in S-phase, but there is sufficient evidence that UNG also acts at the G1-S transition of the cell cycle, which is especially relevant for U removal during CSR and SHM in B cells (9, 54).
UNG2 is the major DNA glycosylase involved in CSR and SHM, while SMUG1 has a backup function (55, 56). The homozygous deletion of UNG is not inherently lethal to embryonic development in mice. Ung−/− mice display a slow removal of U and increased steady-state levels of genomic U in dividing cells, whereas only a slight increase in the spontaneous mutation frequency was observed (57). Lymphocyte development appears to be unperturbed in Ung−/− mice, although a fraction (~20%) of aging Ung−/− mice develop lymphoid hyperplasia, which generally precedes the development of lymphomas in these animals. Ung−/− mice display an ~20-fold increased risk of developing B-cell lymphomas (58, 59). Importantly, ex vivo CSR to IgG3 and IgG1, induced by lipopolysaccharide (LPS) or LPS and interleukin-4 (IL4) is nearly abrogated (~10% of wildtype levels) in the absence of UNG (33). Interestingly, in mice UNG deficiency had minimal impact on the basal serum levels of Ig subclasses in vivo, whereas neutralizing switched Ig levels were severely (~100-fold) diminished in response to acute vesicular stomatitis virus (VSV) infection in mice. These results indicate that during chronic antigen exposure switched Ig originating from infrequent UNG-independent CSR can accumulate, whereas acute antigen exposure requires UNG for the timely and efficient generation of neutralizing switched Ig (60).
It was shown that UNG is required for the generation of DSBs in Ig S regions by processing AID-generated Us into nicks, which when in close vicinity on either DNA strand result in DSBs, or when further apart, require the MMR pathway to be converted into DSBs (61–63). During SHM, UNG plays a key role in the generation of C/G transversions, whereas C to T transition mutations are increased in Ung−/− mice (64). Ensuing work has shown that REV1 acts downstream of UNG to cause C/G transversions, bypassing AP sites by its translesion cytidyl transferase activity (65). In addition, UNG counteracts the accumulation of AID-generated Us that instruct the insertion of A on the other strand, resulting in C to T and G to A transitions. Mutations at A and T bases are also diminished in Ung−/− mice (33). The vast majority of A:T mutations during SHM depend on non-canonical MMR (ncMMR), where the U:G mismatch is recognized by MSH2/MSH6 heterodimers that recruits EXO1 to excise a patch of DNA that contains the U (66). However, EXO1 activity requires a pre-existing nick, which is provided by UNG and AP endonuclease activity. Of interest, the PMS2/MLH1 heterodimer also possesses endonuclease activity that nicks 5′ from U:G mismatches (67, 68). Perhaps this MMR-dependent nicking activity may partly explain the relatively unperturbed basal Ig levels in UNG deficient mice, by allowing infrequent UNG-independent CSR that accumulates over time due to chronic antigen exposure (60). PMS2 deficiency in mice had a negligible effect on A:T mutagenesis (68), but PMS2 may act as a backup in UNG deficient B cells, as A:T mutations were ~50% reduced in Ung−/− Pms2−/− mice (69). Despite the apparent role of UNG in the generation of the full spectrum of AID-induced mutations during SHM, antibody affinity maturation toward a complex antigen such as keyhole limpet hemocyanin (KLH) seems to be intact in UNG deficient animals (60). The mechanistic involvement of UNG in SHM and CSR has been largely corroborated in human subjects that lack UNG due to gene mutations (70–72). However, a hyper-IgM phenotype is frequently observed in human UNG deficient patients, whereas this was not apparent in Ung−/− mice. This may reflect a species-specific difference or perhaps a clinical bias caused by the mere fact that patients with severe immunodeficiency are more Likely to be identified due to clinical symptoms, while patients with mild symptoms remain largely undetected.
The role of UNG in lymphomagenesis has been addressed in several studies. Evidence for the promiscuous targeting of AID responsible for mutations in non-Ig loci was provided in Ung−/− Msh2−/− deficient mice (73). Due to the lack of repair or mutagenic processing, the accumulation of AID footprint mutations (74) was observed in various non-Ig genes, including B-cell lymphoma-associated oncogenes such as Bcl6, Pim1, and c-Myc (73). It was postulated that BER and MMR both protect the genome from AID off-target activity that may contribute to lymphomagenesis (75), but more recent data suggest that MMR has a dominant function in that regard (76). In addition, UNG was shown to be involved in the repair of AID-induced DNA damage at telomeres (77). In accordance, about 20% of aging Ung−/− mice develop lymphoid hyperplasia that may progress to B-cell lymphomas in about half of these mice when aged beyond 18 months. Based on histological features these lymphomas were classified as high-grade follicular lymphoma (FL) (58). Lymphoid hyperplasia was observed in some of the human subjects lacking UNG (2 out of 3), however, the rarity of this condition precludes any firm conclusions on whether UNG deficiency causes lymphomas in humans (71, 78). In contrast, UNG deficiency was shown to be protective for the development of BCL6-driven mouse diffuse large B-cell lymphomas (DLBCL), whereas MSH2 deficiency or the combined deficiency of UNG and MSH2 accelerated lymphomagenesis, accompanied by the accumulation of AID-dependent mutations in non-Ig target genes (76). These data suggest that MSH2 has a strong role in preventing mutations, whereas UNG is actually involved in the generation of mutations (downstream of AID) in BCL6-driven mouse lymphomas. In a c-Myc-driven lymphoma mouse model the deficiency of UNG had no impact on lymphomagenesis (79). These results underscore the dual character of BER in AID-instigated lesions, contributing to both faithful repair and mutagenic processing, depending on the context. How the balance between these contrasting outcomes is regulated is an important outstanding question in the field.
The involvement of UNG in autoimmune diseases is complex, as paradoxically, immunodeficiency (such as associated with the loss of UNG) is linked to autoimmune disease by multiple (indirect) means, for example by the loss of peripheral tolerance. Several excellent recent reviews discuss the potential mechanisms of immunodeficiency-related autoimmune diseases, which goes beyond the scope of this review (80–83). In a recent experimental model the connection between UNG and autoimmune disease was illustrated, showing that the loss of UNG resulted in a lower disease severity in the experimental autoimmune encephalomyelitis (EAE) mouse model for MS, which most likely is attributable to the reduction in isotype-switched autoantibodies (25). Interestingly, UNG deficiency in humans had no apparent effect on the frequency of autoreactive naïve B cells, whereas these were increased in AID-deficient subjects. These results indicate that AID is involved in a peripheral B-cell tolerance checkpoint that is related to SHM, but not CSR, as UNG deficient B cells can still undergo SHM but have severely impaired CSR (84).
Single-Strand Selective Monofunctional Uracil Glycosylase (SMUG1)
SMUG1 is monofunctional glycosylase that is involved in the removal of pyrimidine oxidation products such as 5-formyluracil (5FoU) and 5-carboxyuracil (5CaU) (85, 86), and is also involved in the removal of 5-hydroxymethyluracil (5hmU) from DNA, which is an oxidation product of thymine (34, 87). The ten eleven translocation (TET) dioxygenase enzymes can catalyze the conversion from T to 5hmU (88, 89) (Table 1; Figure 1). This lesion can also be present in ssRNA and be removed by SMUG1 (90). SMUG1 associates with a ribonucleoprotein complex in nucleoli and Cajal bodies and is involved in RNA quality control and co-transcriptional processing by removing 5hmU from ribosomal RNA and telomeric RNA, which is required for proper telomerase activity (90, 91). In addition, SMUG1 can be involved in the repair of U:G mismatches, and is the main means of U excision in Ung−/− mice (92). Strikingly, the expression of SMUG1 is much higher in mouse than in human cells and is not cell cycle-regulated (93). SMUG1 appears not to be directly involved in AgR diversifications. However, SMUG1 is capable of performing a backup function in the absence of UNG. For instance, the residual CSR in Ung−/− mice is significantly decreased by concomitant loss of SMUG1. Serum IgG3, IgG2b, and IgA levels were decreased ~4-, 10-, and 2-fold in Ung−/− Smug1−/− mice compared to Ung−/− mice at the age of 6 months, whereas IgG1 levels appeared to be unaffected. However, in 6 weeks-old Ung−/− Smug1−/− mice IgG1 levels showed an ~3-fold decrease compared to Ung−/− mice. In vitro IgG1 CSR was reduced 2–3-fold in Ung−/− Smug1−/− compared to Ung−/− splenic B-cell cultures, suggesting that the kinetics of SMUG1-dependent CSR is considerably slower in vivo (51). Moreover, ex vivo IgG1 CSR in Ung−/− B cells was restored by retroviral SMUG1 overexpression to a similar level as by retroviral UNG overexpression (22 vs. 17% IgG1 CSR, respectively) (94). Combined deficiencies for UNG and SMUG1 further reduced A:T mutagenesis (54% A:T mutations in the JH4 intron in wildtype mice; 55% in Smug1−/−, 46% in Ung−/−, 39% in Ung−/− Smug1−/−), indicating that SMUG1 can partially compensate for the loss of UNG by excision of U in the vicinity of U:G mismatches resulting in nicks required for patch excision (56). In agreement, SMUG1 overexpression in mice double deficient for MSH2 and UNG affected the SHM pattern, partly restoring the loss of C/G transversions and A:T mutations in Msh2−/− Ung−/− mice to about 50% of wildtype levels. Ex vivo IgG1 CSR is nearly ablated in Msh2−/− Ung−/− splenic B cells, but was restored to about 15–20% of wildtype levels when SMUG1 was overexpressed in these cells (55). It was shown that the endonuclease activity of PMS2 is responsible for the residual A:T mutagenesis in UNG and SMUG1 deficient B cells, but whether loss of PMS2 further diminishes A:T mutations in Ung−/− Smug1−/− mice remains to be tested (69). In conclusion, under normal conditions SMUG1 does not participate in CSR or SHM and is solely involved in these processes in cells that lack UNG activity.
As of yet, there are no studies available that implicate SMUG1 in lymphomagenesis or autoimmune diseases. However, the expression and the genetic variation of SMUG1 have been studied for different type of human cancers. SMUG1 mRNA expression negatively correlated with aggressive disease and survival in breast cancer, and predicted response to adjuvant therapy (95). Human gastric cancers with microsatellite instability are characterized by the downregulation of SMUG1 transcription, which could be related to the response to therapy (96). In addition, an association between bladder cancer and a SNP in the SMUG1 gene was reported (97). In mice deficient for SMUG1 and UNG there is accumulation of genomic U. Whole genome sequencing of UNG/SMUG1 deficient tumors revealed increased mutations, consisting primarily of C to T transitions within CpG sequences, which can be considered as a mutational signature for those tumors (98).
Thymine DNA Glycosylase (TDG)
TDG can repair T:G mismatches that arise from 5-methylcytosine (5mC) deamination (99). However, it has more pronounced activity toward U:G mismatch containing substrates in vitro (Table 1; Figure 1) (50, 51). However, whether TDG can excise U from U:G mismatches in vivo remains to be established. TDG is expressed at low levels in S-phase as it remains bound very tightly to the AP site after excision of U or T from the mismatch, which may stall the replication fork (100). In addition, TDG performs an important function in the transcriptional regulation of developmental genes by interacting with transcriptional cofactors at the promoters of these genes, and is essential for embryonic development (35). It was demonstrated that TDG is crucial for active demethylation and protection of CpG islands from hypermethylation. TDG associates with AID, and it was proposed that AID-dependent deamination of 5mC and 5hmC generates the substrates for TDG-mediated base removal and repair, thereby erasing the DNA methyl marks (101).
Despite its presumed association with AID, the exact role of TDG in AgR diversification mechanisms in B cells has not been studied extensively. It was shown that retroviral overexpression of TDG was not able to restore ex vivo CSR in UNG deficient B cells (94). TDG can only remove U from DNA when mispaired with G in dsDNA. We speculate that TDG perhaps is not be involved in CSR due to the fact that U excision from dsDNA is not efficient in triggering CSR. However, a possible role of TDG in AgR diversification in vivo remains unexplored. This would require a B-cell-specific conditional knockout model, which has not been reported (yet). Nonetheless, a mouse strain carrying a floxed Tdg allele is available, showing that the conditional knockout of Tdg in intestinal epithelial cells resulted in a 2-fold increase in adenomas in the small intestine of female tumor-predisposed APCmin/+ mice (102). This effect was attributed to the fact that TDG is a transcriptional coregulator of the estrogen receptor (103, 104), which has a protective effect on intestinal tumor formation (105).
It is conceivable that TDG is involved in aberrant DNA methylation in various cancers including lymphomas and leukemias, but conclusive data to support this is lacking. There are some indications that TDG may have a role in the malignant plasma cell neoplasia multiple myeloma (MM). In MM cell lines it was observed that the TDG gene was hypermethylated in comparison to normal human plasma cells, resulting in a lower TDG expression and less efficient DNA repair activity in response to hydrogen peroxide-induced DNA damage. The role of TDG in DNA repair was confirmed by compensation of repair capacity after exogenous expression of TDG in the KAS-6/1 MM cell line (106). Other in vitro and in vivo studies that support the involvement of TDG in cancer showed that TDG regulates the expression of tumor suppressor genes by interacting with several transcription factors, including the retinoic acid receptor (RARa), retinoid X receptor (RXR), estrogen receptor α (ERα), thyroid transcription factor 1 (TTF1), and histone acetyl-transferases p300 and CBP (103, 107–109).
Dysregulated epigenetic modifications have been implicated in autoimmune diseases. In the MRL/lpr mouse model for SLE it was shown that immune cells from lymph node and thymus show lower levels of DNA methylation compared to control mice. In human SLE, DNA hypomethylation was detected in B cells and in autoreactive T cells (110, 111). It was demonstrated that demethylating agents such as 5-azacytidine, procainamide and hydralazine induce autoreactivity in CD4+ T cells from healthy human donors, and provoke SLE-like manifestations in normal mice (111–113). These observations suggest an interesting potential link between BER-mediated DNA demethylation and autoimmune diseases (Figure 3). Mutations in BER genes have indeed been linked to SLE predisposition and lupus-like disease in mice (114–120), however, a direct association between TDG and autoimmune diseases has not been established.
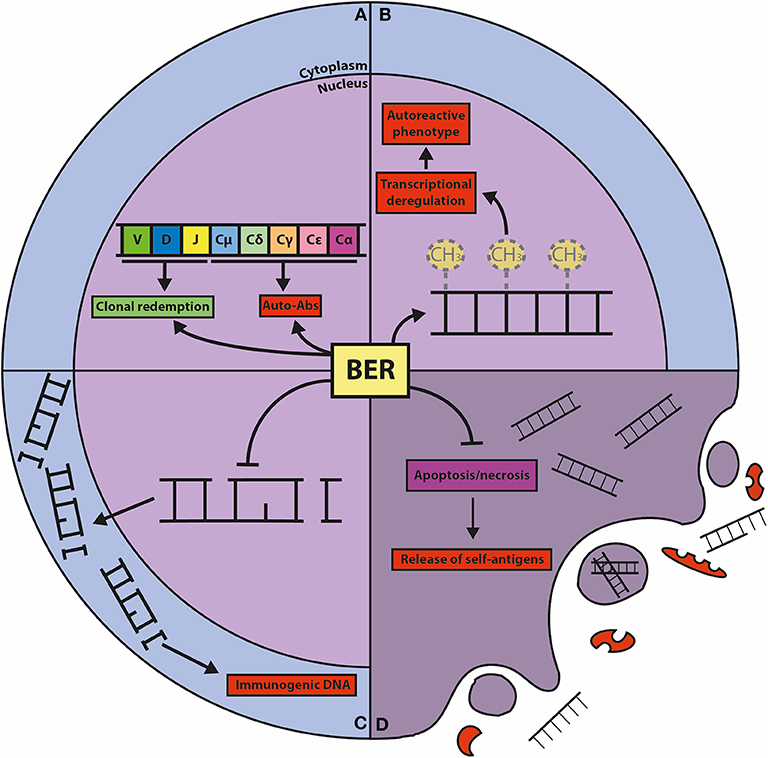
Figure 3. Graphic representation of the known connections between BER and autoimmunity. (A) BER is crucially involved in CSR and SHM, processes that shape the effector function and the repertoire of the humoral immune response. CSR is important for the generation of isotype-switched autoreactive antibodies (Auto-Abs), and SHM can result in the clonal redemption of autoreactive B cells by mutating away from self-reactivity. (B) The BER glycosylases TDG and MBD4 act as epigenetic regulators of autoimmunity through their DNA demethylating activities. Global and gene-specific DNA hypomethylation is associated with autoreactive features in lymphocytes. (C) The repair function of BER guards against damaged DNA in the cytoplasm, which is immunogenic and can elicit (chronic) inflammatory responses and provoke autoreactivity. (D) Cells with unrepaired DNA damage dedicated for apoptosis and necrosis release self-antigens that trigger autoimmunity, which is counteracted by the repair and transcriptional functions of BER components.
Methyl-CpG Binding Domain Protein 4 (MBD4)
MBD4 is structurally unrelated to UNG, SMUG1, and TDG, while it shares substrate specificity with these glycosylases. MBD4 can remove T from T:G mismatches at CpG sites, and U, 5hmU and thymine glycol (Tg) when mismatched with G (52) (Table 1; Figure 1). In contrast to Tdg knockout mice, Mbd4 knockout mice are viable and fertile (36), despite their apparent substrate overlap. Overexpression of MBD4 and AID was shown to cause bulk genome demethylation in zebrafish embryos whereas MBD4 and AID knockdown resulted in remethylation of certain genes, suggesting a role for MBD4 in active DNA demethylation, similar to TDG (121). The frequency of C to T transitions at CpG site was increased 3-fold in Mbd4−/− mice, indicating that MBD4 is important in preventing mutations at CpG sites (36).
MBD4 expression was induced in mouse splenic B cells activated to undergo CSR. However, targeted deletion of exon 3 or exons 2–5 of Mbd4 had no effect on CSR or SHM in mice (122, 123). Further analysis indicated that two isoforms of MBD4 are expressed in mouse B cells, a full length and a short form. Strikingly, expression of the short form is retained in the Mbd4 exon 3 and exon 2–5 deletion-mutant mouse strains, and importantly, glycosylase activity is potentially preserved in this short form (124). This prompted investigators to delete Mbd4 exons 6–8 in the CH12-F3 mouse cell line that can be induced to undergo CSR, showing that IgA isotype switching was ~4-fold reduced, which was associated with fewer Ig S region DSBs. Moreover, sequence analysis of S-S region junctions showed an increase of microhomology-mediated end-joining (MMEJ). The loss of intact MBD4 in these cells resulted in the decreased stability of the MSH2 and MLH1 MMR proteins (122, 124). Corroborating this, MBD4 was identified to interact with MLH1 in a yeast-two-hybrid screen (125). Importantly, splenic B cells from mice that lack MMR proteins show a 2- to 4-fold reduced capacity to undergo CSR due to fewer AID-instigated DSBs (62, 126, 127). Furthermore, splenic B cells from Mlh1−/− mice showed increased S-S region microhomology, perhaps because MLH1 diminishes the processivity of EXO1 during patch excision (128). These observations suggest that MBD4 acts in conjunction with MMR during CSR. It was hypothesized that MBD4 is involved in the recognition of U by the MLH1/PMS2 heterodimer and subsequent AP site generation, thereby triggering the recruitment of PCNA, which supports EXO1 recruitment, leading to patch excision and DSB generation (124). The extent to which MBD4 contributes to AP site generation in comparison to UNG has not been studied, but the rather severe reduction in CSR in Ung−/− mice suggests that this function of MBD4 is of minor importance. Rather, MBD4 may act as an accessory factor to MMR, facilitating the conversion of distal S region nicks to DSBs. This could be addressed in mice that lack the Sμ tandem repeats (TR), which are more dependent on MMR for CSR, due to loss of closely spaced palindromic AID hotspots (62). We hypothesize that loss of MBD4 has a much greater impact on CSR in these mice. The involvement of MBD4 in SHM was shown by CRISPR/Cas9-mediated deletion of MBD4 downstream from exon 5 in the chicken DT40 cell line (129). Loss of MBD4 resulted in a significant increase in SHM, mostly focused at C/G bases, but to a much lesser extent than in UNG deficient DT40 cells (130). These results suggest that MBD4 is involved in U removal during SHM and perhaps during CSR, but has a less prominent function than UNG, although it must be noted that these studies were conducted in DT40 cells overexpressing AID, which may have amplified the observed effects. These results await confirmation in a mouse model that lacks expression of the 3′ portion of the Mbd4 gene.
MBD4 has been implicated in the onset and occurrence of cancer. An increased frequency of intestinal tumors was observed in Mbd4−/− Apcmin/+ mice, showing increased CpG to TpG mutations in the Apc gene (36, 131), although it must be noted that in the Wong et al. (131) study the Mbd4 exon 3 deletion mutant mouse strain was used, which potentially has retained glycosylase activity (124), raising some doubts on the involvement of MBD4 as a glycosylase in these studies. Nonetheless, MBD4 deficient human tumors carry more SNPs compared to MBD4 proficient tumors (132, 133). DNA repair capacity and cancer incidence were associated with MBD4 polymorphisms and frameshift mutations (134–137). Of interest, it was suggested that MBD4 exclusively acts as a tumor suppressor in MMR proficient mice as loss of MBD4 did not affect tumor onset or mutation frequency in Mlh1−/− or Msh2−/− mice (138), although the source of the MBD4 knockout mice in this study, and whether these may have retained glycosylase activity, has not been disclosed. These data suggest a potential epistatic interaction between MBD4 and MMR in cancer, similar to its role in CSR. The involvement of MBD4 in leukemia and lymphoma appears to be rather limited. Loss of MBD4 expression due to germline mutations was detected in a small number of acute myeloid leukemia (AML) patients. These patients displayed a >30-fold increased mutational burden compared to MBD4 proficient AML cases, with the vast majority of mutations being C to T transitions at CpG sites. Furthermore, it was found that hematopoietic stem cell clones with pathogenic mutations in the methyltransferase-coding gene DNMT3A repeatedly expanded in the course of the treatment in MBD4 deficient AML patients, suggesting that the loss of MBD4 drives clonal hematopoiesis (139). A fusion transcript that involves the MBD4 gene and the PTPRC gene was identified in a patient suffering from Sézary syndrome, which is an aggressive T-cell lymphoma of the skin (140). The functional consequences of this fusion transcript, and whether it involves aberrant MBD4 activity, remains unexplored.
There are some indications that MBD4 is involved in the pathogenesis of autoimmune diseases by provoking DNA demethylation of costimulatory genes that may lead to aberrant immune activation. In this scenario, MBD4 acts to remove T from T:G mismatches that arise by the deamination of 5mC, effectively erasing this methyl mark (52). In support, it was shown that to DNA hypomethylation in (autoreactive) lymphocyte populations is related to autoimmune diseases (141–143). Moreover, the expression of MBD4 mRNA showed an inverse correlation with DNA methylation in CD4+ T cells from SLE patients (144), and a positive correlation with the overexpression of costimulatory genes such as CD40LG, TNFSF7, ITGAL, PRF1, and KIR2DL4 (145). It was found that disease progression in MS was associated with elevated MBD4 gene expression in peripheral blood mononuclear cells (PBMNCs) (146), and an arthritis-linked genomic region that harbors the Mdb4 gene was identified as a quantitative trait locus in murine collagen-induced arthritis (147). However, a direct role for MBD4 in B cells of (autoantibody-mediated) autoimmune diseases has not been reported.
Bifunctional Oxidation Damage-Specific Glycosylases
In addition to base removal, bifunctional glycosylases also perform AP lyase activity, cleaving the DNA phosphodiester backbone 3′ to the AP site. As yet, five bifunctional glycosylases have been identified in mammals (OGG1, NTH1, NEIL1, NEIL2, NEIL3), which repair DNA bases damaged by oxidation (148) (Table 1; Figure 1). The AP lyase activity from these glycosylases differs from the AP endonucleases; OGG1, NTH1, and NEIL3 perform a β-elimination reaction on the AP site yielding a 3′ unsaturated aldehyde, whereas NEIL1 and NEIL2 perform an additional δ-elimination reaction converting the 3′ aldehyde to a 3′ phosphate. In contrast, AP endonucleases typically leave a 3′ hydroxyl and a 5′ deoxyribose phosphate (dRP) moiety upon AP site incision (149). These differences in DNA strand incision are important determinants for the ensuing DNA repair. The primary function of OGG1 is to remove 8-oxo-guanine (8-oxoG), an abundant mutagenic base damage caused by the reactive oxygen species (ROS) attack on guanine (150). Ogg1−/− mice are viable and fertile but accumulate 8-oxoG and show a moderate (2- to 3-fold) increase in mutations in liver cells (37). Escape from OGG1 excision results in the mispairing of the 8-oxoG lesion with A after replication. The adenine glycosylase MUTYH is responsible for a backup mechanism that excises the mispaired A, and subsequent repair of the AP site and incorporation of C provides another opportunity for OGG1 to excise the 8-oxoG, thereby repressing C to T and G to A transitions (32). Mutyh−/− mice are born at the expected Mendelian ratio and develop normally but are susceptible to oxidative stress and predisposed to develop tumors (38, 151). NTH1 is involved in the repair of thymine glycol (Tg), generated by ROS-mediated oxidation of thymine (39, 152). Homozygous Nth1 mutant mice show no gross developmental abnormalities but displayed a slower Tg turnover in the liver (39). Oxidized pyrimidines (C, T, U) are recognized and repaired by NEIL1 and NEIL2, but the preferred substrate for NEIL1 and NEIL2 are hydantoin lesions, which are derived from progressively oxidized 8-oxoG (153). NEIL1 and NEIL2 favor ssDNA structures such as bubbles and loops. NEIL1 associates with replication forks and is involved in pre-replicative repair in S-phase (154), whereas NEIL2 is mostly involved in repair at transcribed genes (155). Both NEIL1 and NEIL2 are involved in the maintenance of mitochondrial (mt) DNA, similar to OGG1 and NTH1 (156). Neil1−/− mice are born at the expected Mendelian rates and show no developmental defects. However, these mice show an accumulation of mtDNA damage and develop a severe metabolic syndrome by 6–10 months of age, characterized by obesity, fatty liver disease, and kidney vacuolization. It was hypothesized that mtDNA damage hampers replication and transcription of mitochondrial genes involved in metabolism, thereby impairing energy homeostasis (40). Neil2−/− mice are viable and show no overt phenotype, they accumulate oxidative DNA damage preferentially in transcribed regions, as expected. Neil2−/− mice display an increased responsiveness to inflammatory stimuli such as intranasal challenges with LPS, glucose oxidase (GOx), or tumor necrosis factor α (TNFα) (41). How these glycosylases are involved in innate inflammatory responses remains to be characterized. NEIL3 has an extended c-terminal tail compared to NEIL1 and NEIL2 and its properties were only quite recently characterized, showing that it recognizes hydantoins and 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG) in ssDNA in bubble structures, similar to NEIL1 and NEIL2, but uses a different AP lyase reaction mechanism (157). NEIL3 was found to be expressed predominantly in hematopoietic and lymphoid tissue, and in the brain (42, 158, 159). Neil3−/− mice are viable and fertile (42).
Typically, the bifunctional glycosylases have no major role in the AgR diversification mechanisms. OGG1 and NTH1 were demonstrated not to contribute to CSR (160). The role of MUTYH in CSR and SHM has not been investigated. Ogg1−/− mice displayed normal SHM, and OGG1 was shown not to be upregulated in mouse GC B cells (161), whereas it was increased in human GC B cells that undergo SHM (162). This discrepancy may be related to the chronic nature of GCs in human tonsil, accompanied by continuous apoptosis and release of oxidation products, while induced GCs in mouse tissues are transient. In contrast, Neil1−/− mice showed moderate (<2-fold) decrease in GC B-cell expansion after immunization with the model antigen nitrophenylactetyl-chicken γ-globulin (NP-CGG). Accordingly, the mutation frequency in the JH4 intron was slightly decreased (~80% of wildtype) in Neil1−/− mice (163). These results suggest that NEIL1 is required to curb endogenous oxidative damage related to the rapid expansion of GC B cells, but does not participate in the repair phase of AID-instigated DNA lesions. In agreement, NEIL1 was identified as a likely candidate gene for common variable immunodeficiency in a patient with 15q24 deletion (164). Neil3−/− mice showed a similar modest decrease in GC B-cell expansion (<2-fold), due to apoptosis. However, SHM was not significantly affected in these mice (116). Interestingly, 3 siblings from a consanguineous family were identified that carried a homozygous missense mutation in the NEIL3 gene, suffering from fatal infections and impaired B-cell function. Studies conducted on B cells from one of these patients showed a decreased capacity to undergo IgG and IgE CSR in vitro (116). These results underscore that the bifunctional glycosylases do not directly participate in the AgR diversification mechanisms, but are essential for the fitness of cells that undergo these processes.
Most of the bifunctional glycosylases are linked to cancer. Human tumor tissues were shown to accumulate clustered DNA lesions due to oxidative damage, suggesting impaired function of oxidative repair glycosylases and/or increased generation of oxidative agents (165). Moreover, knockout mouse models showed increased tumorigenesis with a differential tumor spectrum, likely reflecting tissue-specific dependencies. Aging Ogg1−/− mice accumulate genomic 8-oxoG and develop lung cancer (166), whereas most Mutyh−/− mice spontaneously develop intestinal tumors, which was increased by treatment with an oxidizing agent (151). In agreement, reduced activity and functional polymorphisms of OGG1 are associated with various types of human cancer, including lung cancer (167–169), and mutations in the MUTYH gene confer a heritable form of colorectal cancer predisposition (170). Of interest, in comparison to Ogg1−/− mice, Ogg1−/− Mutyh−/− mice were further predisposed to develop cancer, presenting with predominantly lung and ovarian tumors, and B-cell lymphomas (38). In contrast, B-cell lymphomas that arise in a Msh2−/− mouse strain depended on MUTYH, as lymphomagenesis was significantly delayed in Msh2−/− Mutyh−/− mice (171). These results underscore differential interdependencies of these BER components acting in a context-specific manner. Reduced expression of NTH1 mRNA was found in 36% of primary gastric carcinomas (172). It was shown that a functional germline variant of NTH1 that is expressed in a sizeable fraction of the human population (6%) causes genomic instability and cellular transformation in an experimental setting (173). It was reported that the expression of NEIL1 and NEIL2 inversely correlated with the number of somatic mutations in several cancers, whereas NEIL3 showed a positive correlation (174). Ectopic expression of a rare human NEIL1 germline variant devoid of glycosylase activity induced replication stress, DNA breaks and anchorage-independent growth, suggesting that it confers an increased risk for cancer (175). However, the (combined) deficiencies for the NEIL glycosylases (double and triple knockout mice) did not result in increased mutations or cancer predisposition in mice under normal conditions (176). These results suggest that there is limited overlap in the functions of the NEIL glycosylases. It remains to be addressed whether exposure to oxidative agents induces tumor formation in these mice. Several sporadic reports suggest the involvement of oxidative damage glycosylases in B-cell lymphomas. For instance, a deletion of the NEIL1 gene was found in a case of FL that transformed into a B-cell acute lymphoblastic leukemia (B-ALL) (177), and the expression of NEIL1 mRNA was part of a classifier that distinguished the germinal center B-cell-like (GCB) subtype from the activated B-cell-like (ABC) subgroup of DLBCL, and was associated with disease aggressiveness in the ABC subgroup (178). However, a clear role in B-cell lymphomas has not been reported, besides as a backup downstream of MMR defects (171). Most B-cell lymphomas are driven by genomic aberrations and mutations that are related to the GC response and Ig diversification mechanisms (179), the fact that the bifunctional glycosylases are not directly involved in Ig diversifications might explain the limited role of these enzymes in lymphomagenesis.
Oxidative DNA damage can provoke an inflammatory reaction by activation of innate immune receptors, which may ultimately trigger an immune response to self-antigens (180) (Figure 3). The glycosylases involved in mtDNA repair are especially relevant in this process, as these counteract the oxidation and release of mtDNA that activates the inflammasome, resulting in release of proinflammatory cytokines and mediators (181). In agreement, there is some evidence that SLE patients have increased levels of DNA damage compared to normal individuals, indicating that ROS-induced DNA damage and decreased OGG1 expression are involved in the development of SLE (182–184). In addition, there are several reports showing that OGG1 polymorphisms are associated with autoimmune diseases such as SLE, RA, and MS (115, 185–187). Also, two MUTYH polymorphisms were found to be associated with RA (188). Neil2−/− mice show no signs of autoimmune diseases, but develop severe lung inflammation upon intranasal challenge with LPS, which is associated with an accumulation of DNA damage and apoptosis that triggers local inflammation in the lung (41). The association with autoimmune disease appears especially strong in the case of NEIL3. Neil3−/− mice display elevated levels of autoantibody levels and develop nephritis when challenged with the immunostimulant poly(I:C). It was hypothesized that increased apoptosis of GC B cells from Peyer's patches and splenic B and T cells observed in Neil3−/− mice could stimulate autoreactivity due to release of self-antigens (Figure 3). Concordantly, NEIL3 deficient patients developed severe autoimmunity and suffered from fatal recurrent infections (116). These results indicate that NEIL3 is of crucial importance for the protection against autoimmunity. Importantly, the homozygous missense mutation found in the NEIL3 gene (D132V) in the consanguineous NEIL3 deficient patients is present in about 2% of healthy individuals from Middle Eastern descent (116), but whether this confers an increased risk to develop autoimmune disease remains to be studied.
AP Endonucleases
The AP endonucleases act after base removal by DNA glycosylases by nicking the DNA phosphodiester backbone. The AP endonucleases are grouped into two major groups according to their modes of action (Table 1; Figure 1). The abovementioned bifunctional glycosylases belong to class I, whereas the predominant AP endonucleases in most organisms belong to class II, which are AP endonucleases that cleave the DNA by a hydrolytic mechanism leaving a 3′ hydroxyl and a 5′ dRP group (189). In most cases, class I AP lyase requires the subsequent activity of class II AP endonucleases to remove 3′ blocking moieties and allow DNA polymerase-mediated repair synthesis (190). These non-redundant activities of class II AP endonucleases are of vital importance for cellular growth, preventing apoptosis by restoring (endogenous) DNA damage (191).
AP Endonuclease 1 (APE1)
In humans, two AP endonucleases are identified, APE1 and APE2, of which APE1 is predominantly active, accounting for >90% of AP site repair (192). APE1 is ubiquitously expressed, showing nuclear protein expression in all tissues. It appears that APE1 has a dual function, being involved in BER and acting as a transcriptional regulator by serving as a reducing donor for oxidized cysteines that hamper DNA binding of several transcription factors (193). Ape1 gene targeting in the mouse resulted in embryonic lethality, showing that APE1 is required for early embryonic development (43, 194). However, heterozygous Ape1+/− mice are viable but display haploinsufficiency, showing sensitivity to oxidative stress and increased mutations (195, 196). B-cell development is unaltered in Ape1+/− mice, and APE1 is expressed in resting and activated B cells. Of interest, ex vivo CSR was found to be modestly reduced (<2-fold) in Ape1+/− B cells compared to B cells from wildtype littermates. Moreover, a small molecule inhibitor of APE1 reduced CSR in vitro, independently of its effect on cell proliferation (44, 197). Consistent with the study from Guikema et al., further reduction to one copy of Ape1 in the CH12-F3 cell line, which normally expresses 3 copies, reduced the APE1 protein level by 60% and reduced IgA CSR by 50%. Importantly, deletion of all 3 copies of Ape1 reduced CSR efficiency to 20% of the wildtype level providing direct evidence that APE1 is required for efficient CSR (198). Fewer Sμ DSBs were observed in Ape1+/− B cells undergoing CSR, strongly suggesting that APE1 is responsible for the incisions at AP sites that are generated by AID and UNG during CSR (44). Interestingly, using a specific small molecule inhibitor it was suggested that the redox function of APE1 also plays a role in CSR by regulating interleukin-6 signaling and IgA expression in the CH12-F3 cell line (199). Recently, the dogma that CSR takes place within the GC was challenged by use of an elegant adoptive transfer mouse model. It was shown that CSR mostly occurs outside of the GC, prior to the onset of SHM (200). Having shown that CSR depends on AID, UNG and APE1 to generate S regions DSBs (4, 33, 44), it was demonstrated that APE1 expression is downregulated in GC B cells, thereby preventing CSR in the GC (200, 201). In agreement, APE1 does not seem to be required for SHM, which takes place in the GC (198, 201–203) (Figure 2).
The expression of APE1 and its subcellular localization are associated with various types of cancer and the response to therapy (204–209). Several polymorphic APE1 variants that show reduced DNA incision activity are linked to cancer risk and therapy sensitivity (210–213). There is no clear indication that APE1 is implicated in lymphomagenesis. However, the established relation between B-cell lymphomas and CSR-mediated chromosomal translocations (214), and the function of APE1 therein, suggest that APE1 functions as a crucial mediator of DSBs downstream of AID, which when repaired illegitimately lead to lymphoma-associated genomic lesions. In addition, the redox function of APE1 was shown to be a therapeutically amenable disease determinant in T-cell acute lymphoblastic leukemia (T-ALL) and AML (215–217).
A few reports indicate that APE1 may be involved in autoimmunity. The high mobility group box 1 protein (HMGB1) is released from necrotic cells, which triggers an inflammatory response in effector cells. Interestingly, the cytoplasmic expression of APE1 dampened this inflammatory response in monocytic cells, most likely mediated by its transcriptional regulatory function (218). In addition, APE1 is one of the nuclear target proteins for autoantibodies in SLE (219). It is conceivable that the role of APE1 in autoimmune disease is mainly related to its function in preventing DNA damage that may drive aberrant immune responses (220) (Figure 3).
AP Endonuclease 2 (APE2)
APE2 is a nuclear protein closely homologous to APE1 but with a weaker endonuclease activity (221). However, APE2 possesses strong 3′-5′ exonuclease and 3′-phosphodiesterase activity stimulated by the interaction with PCNA (222, 223). APE2 is involved in the repair of oxidative DNA damage by acting on damaged and mismatches DNA 3′ ends, removing A opposite of 8-oxoG (223). Additional evidence that APE2 participates in the response to oxidative DNA damage was provided, showing that the 3′-phosphodiesterase and 3′-5′ exonuclease activities are required for end resection, thereby stimulating homologous recombination-mediated repair (HR). Additionally, it was shown that APE2 participates in the ATR-CHK1 cell cycle checkpoint, facilitating CHK1 phosphorylation in response to oxidative DNA damage (224). Circumstantial evidence suggests that APE2 might also be involved in NHEJ, perhaps by serving as an end-cleaning/processing enzyme by virtue of its 3′-phosphodiesterase activity (225). Interestingly, the endonuclease activity of APE2 was shown to be crucial for the survival of cells with defective HR due to BRCA2 deficiency, whereas APE1 was not required (226).
APE2 deficient mice develop normally but show a 2-fold decrease in pre-B cell production in the bone marrow (227), and a similar reduction in newly formed and follicular B-cells in the spleen, but APE2 was shown not to be directly involved in V(D)J recombination. In addition, the expansion of early B-cell progenitors during the recovery after a chemotherapeutic challenge was hampered in APE2 deficient mice. Interestingly, B-cell cellularity was not altered in Ape2Y/− tp53−/− mice indicating that the loss of B cells was due to p53-dependent cell death triggered by DNA damage (228). It was found that APE2-deficient activated B cells are hypersensitive to oxidative damage, indicating that APE2 protects proliferating B cells from intracellular ROS (228). The expression of APE2 is upregulated in GC B cells, and APE2 deficient mice have ~2.5-fold fewer GC B cells due to the accumulation of AID-independent DNA damage. However, B-cell selection in the GC and affinity appeared not to be affected in APE2-deficient mice. The reduction in GC B-cell frequency was found to be related to AID-independent DNA damage resulting in the reduced expression of BCL6, which is required for the development of GC B cells (228). APE2 was shown to contribute to CSR in splenic B cells, participating in the formation of S region DSBs (44), however, its contribution seems to be of minor importance compared to APE1, as APE1 is sufficient for CSR, especially in cell lines that can be induced to undergo CSR (198, 202). However, in contrast to APE1, APE2 clearly participates in the SHM process, since APE2 deficient mice have a 2-fold reduced mutation frequency and altered mutation spectrum, with a significant reduction in mutations at A:T base pairs (59% of JH4 intron mutations wildtype mice versus 48% in APE2 deficient mice). It was suggested that APE2 contributes to DNA nicks that provide entry points for EXO1 that instigates ncMMR-mediated A:T mutations (201, 202). In addition, the 3′-5′ exonuclease activity of APE2 may also support SHM by the excision of short DNA patches that are targeted by AID and/or facilitate ncMMR (229) (Figure 2).
A recent study reported on the dysregulation of APE2 in a multitude of human cancers, showing that ~17% of cancer cases have genomic alterations involving APE2, mostly consisting of heterozygous deletions and gains. Liver, skin, and breast cancer showed the highest frequency (~24%), and somatic mutations in APE2 were found in uterine, skin, and lung cancers. Many of these mutations were speculated to affect PCNA and ssDNA binding, thereby reducing exonuclease activity. Moreover, APE2 mRNA levels were found to be upregulated in several cancer subtypes and showed a positive correlation with DNA damage response (DDR) genes (230). These data suggest that APE2 might be involved in cancer development, although direct functional evidence is not provided. These observations are in line with the notion that BER activity in tumors determines prognosis and response to therapy (231). A recent study reported on the upregulation of APE1 and APE2 expression in MM. Interestingly, knockdown of APE2 compromised HR proficiency of MM cells, consistent with the previously reported role of APE2 in HR (224, 229, 232).
Indications that APE2 is directly involved in other B-cell neoplasms or in autoimmune diseases have not been reported, but it's evident association with CSR and SHM indicate that APE2 has a role in genomic alterations in B-cell lymphomas and aberrant clonal selection in autoantibody-mediated diseases.
SINGLE Strand Nick Processing BER Enzymes
AP endonuclease- and AP lyase-generated nicks are processed by non-displacement DNA synthesis (short-patch BER) or displacement synthesis (long-patch BER). The mechanistic basis for the choice between these pathways involves various factors but is still poorly understood (233). Short-patch BER involves one nucleotide insertion by DNA polymerase β (POLB), whereas long-patch BER can be 2–16 nucleotides, executed by POLB or DNA polymerase δ/ε (POLD/E), stimulated by PCNA (234). The latter involves the trimming of overhanging ends by flap endonuclease 1 (FEN1) (Table 1; Figure 1).
DNA Polymerase β (POLB)
POLB has a dual catalytic function and is uniquely involved in BER. The C-terminal part of POLB is involved in DNA synthesis, while an independently folded N-terminal region is required for dRP lyase activity (235). POLB is the major lyase in the repair of oxidized and alkylated bases (236, 237). Although POLB lacks intrinsic 3′-5′ proofreading activity, it has a relatively low error rate in both short-patch as long-patch BER (238). Moreover, the ensuing DNA ligation step is inefficient when POLB inserts a mismatched or damaged base, providing an additional safety measure to prevent BER associated mutagenesis (239). POLB is an essential gene, mice with a heterozygous germline Polb deletion do not generate Polb−/− offspring. Embryos homozygous for the Polb deletion die at days 18–19 post-coitum (45). The structurally related DNA polymerase λ (POLL) contributes to BER as a backup for POLB but is apparently not sufficient for development (240). However, POLB deficient cell lines can be obtained by gene targeting and have indicated that POLB is required for BER (241).
Cell type-specific gene targeting by the Cre/Lox system and adoptive transfer of Polb−/− fetal liver cells have enabled the characterization of the role of POLB in lymphoid cells (45, 242). Using these approaches, it was shown that POLB is not required for T-cell or B-cell development, suggesting that POLB is not critically involved in V(D)J recombination (45, 242). It was shown that POLB is able to repair AID-instigated DNA nicks in S regions, thereby inhibiting CSR. However, this was only apparent for IgG2a, and to a lesser extent for IgG2b and IgG3 isotype switching. Of interest, the S regions located upstream of these specific constant regions contain the lowest density of AID hotspot motifs (especially for IgG2a), and it was hypothesized that AID-induced nicks may be limiting in these S regions. These results suggest that BER activity is overwhelmed by AID, and that POLB fails to repair all S region lesions during CSR (243). Furthermore, it was demonstrated that the GC response is normal in mice reconstituted with POLB deficient cells, showing unaltered mutation spectrum and levels, indicating that POLB is not involved in SHM, as was also the case for POLL (242, 244). When induced to undergo IgA CSR, the CH12-F3 cell lines accrue AID-dependent mutations in Sμ. However, the frequency of mutations at A:T base pairs is much lower in this cell line, as it true for other in vitro systems used to study AID-instigated mutagenesis, compared to in vivo B cells (0–20 vs. 50–60% of all mutations, respectively) (245–248). We have recently shown that knockdown of POLB in CH12-F3 cells, and in fibroblasts engineered to express AID, resulted in a 3-fold increase in A:T mutagenesis, which was associated with increased recruitment of MMR components (18) (preprint). Moreover, we have demonstrated that GC B cells in vivo lack POLB protein expression, in contrast to in vitro cultured cells, which functionally express POLB. This in part explains the lack of a GC phenotype in mice with POLB deficient lymphocytes. Furthermore, our studies suggest that POLB protein is destabilized in GC B cells due to the hypoxic microenvironment that marks the GC (18, 249, 250). Our data suggests that the specific loss of POLB in GC B cells is instrumental in the mutagenic repair of AID-dependent lesions (Figure 2). We speculate that the overall loss of POLB in combination with dUTP misincorporation related to DNA replication, and spontaneous base deaminations destabilize the genome in GC B cells, thereby potentially driving the development of GC B-cell derived lymphomas.
In line with this, the Sweasy group has identified and characterized various functional POLB variants that have been implicated in the development of cancer (251–255). It was reported that POLB variants can be detected in up to 30% of human cancers (256). These variants either have reduced repair activity or decreased fidelity. Importantly, expression of several of these variants resulted in cellular transformation, suggesting that these mutations are sufficient to drive cancer development (251). Moreover, Polb haploinsufficiency increased cancer incidence in aging mice. Of interest, ~40% of aging Polb+/− mice showed lymphoid hyperplasia, and aged Polb+/− mice had a 7-fold increase in lymphoma incidence compared to wildtype mice (257). These tumors develop without an obvious increase in mutation frequency, which would suggest that DSBs accumulate in these mice. For human B-cell neoplasia, a functional POLB P242R non-synonymous SNP, which bestows a slower catalytic activity, was identified as an independent prognostic marker in B-CLL patients (258, 259).
There is a potential link between POLB and autoimmune diseases. A genome-wide association study has identified an association between SLE and a functional POLB SNP that results in lowered POLB expression (260). In agreement, a mouse model with the hypomorphic Y265C POLB allele developed a lupus-like syndrome characterized by anti-nuclear antibodies, glomerular nephritis, and cervical lymphadenopathy (117). BCR repertoire analysis showed that the hypervariable complementarity determining region 3 (CDR3) is significantly shorter in bone marrow progenitor B cells and mature splenic B cells from POLB hypomorphic mice. No alterations in CSR were found in ex vivo activated splenic B cells from these mice. In addition, POLB hypomorphic mice displayed a significant increase in SHM frequency with increased transversions at G:C base pairs and increased A:T mutagenesis. The number of GCs were also increased in these mice. It was suggested that POLB participates in the V(D)J recombination by processing DNA ends and preventing nuclease activity prior to joining, thereby affecting CDR3 length. This role may have been overlooked in the adoptive transfer experiments that suggested POLB is not involved in V(D)J recombination (242). However, the relevance of this phenotype for the development of lupus remains unexplained. The effects of the hypomorphic POLB allele on SHM are difficult to reconcile with our findings showing that POLB is limiting in GC B cells due to low protein stability (18) (preprint). It might be that the hypomorphic mutant POLB has an increased stability in GC B cells, despite hampered catalytic activity. Perhaps mutagenic translesion (TLS) repair is favored in GC B cells expressing the mutant (stabile) POLB, as it is possible that the POLB mutant is recruited to AID-instigated nicks, but is not processive, thereby provoking the recruitment of TLS polymerases. Finally, it was hypothesized that the increased turnover and apoptosis in GCs in hypomorphic POLB mice increases the exposure to self-antigen, causing autoimmune disease (Figure 3).
Flap Endonuclease 1 (FEN1)
FEN1 is required for the removal of the displaced DNA strand in long-patch BER, and the processing of Okazaki fragments during lagging strand DNA synthesis (261). In addition, FEN1 is of crucial importance for MMEJ by cleaving the displaced 5′-flaps prior to joining (262), and can act as a 5′-3′ exonuclease to trim DNA ends during HR (263) (Table 1; Figure 1). Mice deficient for FEN1 could not obtained due to embryonic lethality, however, Fen1+/− mice develop relatively normal. Fen1+/− mice show diminished lymphocyte cellularity and premature thymic involution, likely related to the essential role of FEN1 in DNA synthesis and replication (46).
Interestingly, Fen1 haploinsufficiency accelerated cancer progression in Apc mutant mice, leading to an increased number of intestinal adenocarcinomas, characterized by microsatellite instability (MSI) (46). Knock-in of a cancer-related inactivating Fen1 mutation resulted in a high incidence of lung carcinomas (120). Overall, FEN1 overexpression as well as loss of function have been associated with human cancers, likely related to the dual nature of FEN1, being involved in DNA replication and in DNA repair (264). Moreover, functional defects in FEN1 were linked to increased risk in human cancer types (265–267). A considerable proportion (17%) of Fen1+/− mice developed B-cell lymphomas. In agreement, female mice with a compound Fen1 mutation in the nuclease domain developed B-cell lymphomas originating from GC B cells. The exact reason for this gender bias in lymphomagenesis is not understood, but might be related to the immunomodulatory effects of estrogen (268). B cells from these mice show normal ex vivo CSR and in vivo SHM, suggesting that FEN1 is not directly involved in Ig diversification-driven chromosomal aberrations linked to B-cell lymphomas, but rather, is involved in preventing DNA damage that is associated with the rapid proliferation of B cells in the GC.
A nuclease-deficient Fen1 mutant mouse model displayed a marked predisposition to chronic inflammation and autoimmunity (120). It was shown that the Fen1 E120D mutant mice accumulate undigested DNA in apoptotic cells, which may be at the basis of uncontrolled inflammatory responses by activation of stimulator of interferon genes (STING) signaling, which is linked to the development of autoimmune diseases (269, 270) (Figure 3). In agreement, Fen1 E120D mutant mice showed increased levels of anti-nuclear and anti-dsDNA antibodies (120). Moreover, the E120D mutation was found more frequently in lupus nephritis patients compared to healthy individuals (271).
DNA Ligase I and III
After nick processing, the DNA is sealed by DNA ligase, thereby completing BER. DNA ligase III (LIG3) is responsible for the nick-sealing step in short-patch BER (272), whereas DNA ligase I (LIG1) ligates the nick in long-patch BER (273) (Table 1; Figure 1). The accessory factor X-ray cross-complementing protein 1 (XRCC1) interacts with LIG3, serving as a scaffold protein that organizes repair by binding POLB. LIG1 plays an essential role in DNA replication by joining Okazaki fragments to extended DNA during lagging strand DNA synthesis (274). Both LIG1 and LIG3 were shown to be involved in MMEJ (275), whereas LIG3 and is the only DNA ligase that functions in mitochondria, where it acts to maintain mtDNA integrity independently of XRCC1 (276, 277). LIG1 and LIG3 are essential for embryonic development (47, 48). However, cell lines lacking LIG1 could be obtained due to the functional redundancy with LIG3 and LIG4, of which the latter mainly functions in NHEJ (278–280). Interestingly, biallelic LIG1 mutations were identified in 5 human subjects, these patients suffered from a spectrum of immune deficiencies characterized by lymphopenia, likely resulting from defective DNA repair in B cells and T cells (281).
Gene targeting in the CH12-F3 cell line has demonstrated that LIG1 and LIG3 are not required and act redundantly in CSR (278, 282), whereas loss of LIG4 decreased CSR, likely due to its central role in NHEJ (282). Interestingly, using an indirect approach it was shown that LIG1 is the major contributor to MMEJ-mediated CSR to IgG1, conditional deletion of Xrcc1 and Xrcc4 in mature B cells had no significant impact on CSR, whereas LIG3 expression was severely reduced in Xrcc1/4 deficient B cells (283). LIG1 mRNA is highly expressed in human tonsillar GC B cells compared to naïve B cells, whereas LIG3 mRNA expression was not found to be increased in GC B cells (284). The roles of LIG1 and LIG3 in SHM have not been directly assessed. However, a LIG1 deficient patient showed a much lower level of IGH somatic mutations in peripheral blood mononuclear cells, perhaps caused by blunted GC responses (281). Based on our findings we propose that AID/UNG/APE1-generated nicks are shunted toward error-prone repair by the ncMMR system, as POLB is limiting in GC B cells, thereby bypassing (short-patch) BER and nick-sealing by LIG3. LIG1 is responsible for sealing the nick in MMR-mediated patch repair and would be indirectly involved in SHM in this scenario (285). Of interest, SHM was increased in Peyer's patch GC B cells from Xrcc1+/− mice, which develop normally but are haploinsufficient. No obvious alteration in the SHM spectrum was observed in these mice (286). These data suggest that BER has limited access to AID-instigated lesions, causing uracils to escape faithful repair and become substrates for mutagenic repair.
Overexpression of each of the DNA ligases has been found in cancers, which is typically related to increased proliferation and reliance on DNA end-joining due to (therapy-induced) DNA damage (287). LIG3 was shown to promote chromosomal translocations in cell lines upon induction of distal DSBs (288). Increased expression of LIG1 was linked to genomic instability by causing trinucleotide repeat instability due to reduced slipped DNA repair (289). On the other hand, loss of LIG1 function has also been associated with cancer predisposition (290). Moreover, DNA ligases are under consideration as targets for anticancer therapy (291, 292). LIG1 and LIG3 have not been directly associated with lymphomagenesis, but it was demonstrated that C-MYC, which is frequently deregulated in B-cell lymphomas and MM, drives the expression LIG3 (293, 294). Given their role in MMEJ, which is responsible for illegitimate recombinations, LIG1 and LIG3 are likely to be of pivotal importance for genomic instability and clonal evolution in B-cell malignancies. In agreement, it was shown that MMEJ repair activity was increased in B-CLL cells compared to normal B cells (295).
The involvement of LIG1 and LIG3 in autoimmune diseases has not been reported, however, it is conceivable that LIG1 and LIG3 may prevent autoimmunity as they are crucial for the maintenance of mitochondrial and nuclear DNA, which can have immunostimulatory effects that may underlie a breach of clonal tolerance (Figure 3).
Concluding Remarks
In lymphocytes, strategies have evolved that both utilize the faithful nature of BER to ensure genetic integrity, as well as subvert it to allow (localized) mutagenic repair for AgR diversifications. Many of the key players of the ubiquitous BER pathway are of essential importance for AgR diversification mechanisms that shape the adaptive immune response, and are at the basis of B-cell neoplasia and autoimmune diseases. In that sense, the BER pathway cannot be regarded in isolation, as it acts in concert with other repair pathways. For instance, the specific loss of the late gap-filling step of short-patch BER in GC B cells favors mutagenic repair by ncMMR, thereby contributing to diversification. However, mutagenic repair is also responsible for oncogenic mutations and rearrangements that drive the development and progression of B-cell malignancies. BER can thus be regarded as a double-edged sword that on the one hand protects the organism by ensuring highly specific adaptive immune responses and on the other hand jeopardizes it by provoking genomic instability. In that regard, the BER pathway is also of pivotal importance to minimize the release of immunogenic DNA that can instigate autoimmune diseases. Here too, the double-edged nature of the BER pathway is apparent (Figure 2). BER is instrumental in the development of autoantibodies, but at the same time prevents the release of (extracellular) DNA and DNA-protein complexes that (hyper) activate cytosolic and endosomal DNA sensors leading to auto-inflammatory and autoimmune diseases (Figure 3). From that point of view, the development of B-cell neoplasia and autoantibody-driven autoimmunity can have a common etiology as in both cases it relies on the evasion of checkpoints that either safeguards genomic integrity or prevents autoimmunity. Recent experimental and clinical data has suggested that these checkpoints may overlap, since patients with autoimmune diseases have an increased risk to develop B-cell lymphomas (296–298), and several B-cell lymphoma subsets are characterized by the expression of autoreactive BCRs (299–303). In addition, lymphoma driver mutations have been detected in rare B cells that produce pathogenic autoantibodies from primary Sjögren's syndrome patients, underscoring a potential common origin for B-cell lymphoma and autoimmune disease (304). BER has been linked to these conditions, acting to both prevent and to provoke disease, depending on the context and timing. These mechanisms will remain the subject of intense research in the coming years, which will provide important new insight into the complexity of biological systems that drive adaptive immunity and related diseases.
Author Contributions
MS, TD, and JG wrote and edited the manuscript and generated figures.
Funding
Research in the Guikema laboratory is part of a project (COSMIC; www.cosmic-h2020.eu) that has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 765158. TD was a recipient of the Amsterdam Academic Medical Center PhD scholarship 2019.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the members of the Guikema laboratory for helpful discussions.
References
1. Schatz DG, Spanopoulou E. Biochemistry of V(D)J recombination. Curr Top Microbiol Immunol. (2005) 290:49–85. doi: 10.1007/3-540-26363-2_4
2. Stavnezer J, Guikema JEJ, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. (2008) 26:261–92. doi: 10.1146/annurev.immunol.26.021607.090248
3. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
4. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. (2000) 102:553–63. doi: 10.1016/s0092-8674(00)00078-7
5. Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2). Cell. (2000) 102:565–75. doi: 10.1016/s0092-8674(00)00079-9
6. Krokan HE, Bjoras M. Base excision repair. Cold Spring Harb Perspect Biol. (2013) 5:a012583. doi: 10.1101/cshperspect.a012583
7. Matsumoto Y, Kim K, Bogenhagen DF. Proliferating cell nuclear antigen-dependent abasic site repair in Xenopus laevis oocytes: an alternative pathway of base excision DNA repair. Mol Cell Biol. (1994) 14:6187–97. doi: 10.1128/mcb.14.9.6187
8. Bak ST, Sakellariou D, Pena-Diaz J. The dual nature of mismatch repair as antimutator and mutator: for better or for worse. Front Genet. (2014) 5:287. doi: 10.3389/fgene.2014.00287
9. Schrader CE, Guikema JEJ, Linehan EK, Selsing E, Stavnezer J. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J Immunol. (2007) 179:6064–71. doi: 10.4049/jimmunol.179.9.6064
10. Pena-Diaz J, Bregenhorn S, Ghodgaonkar M, Follonier C, Artola-Boran M, Castor D, et al. Noncanonical mismatch repair as a source of genomic instability in human cells. Mol Cell. (2012) 47:669–80. doi: 10.1016/j.molcel.2012.07.006
11. Wilson TM, Vaisman A, Martomo SA, Sullivan P, Lan L, Hanaoka F, et al. MSH2-MSH6 stimulates DNA polymerase eta, suggesting a role for A:T mutations in antibody genes. J Exp Med. (2005) 201:637–45. doi: 10.1084/jem.20042066
12. Neuberger MS, Rada C. Somatic hypermutation: activation-induced deaminase for C/G followed by polymerase eta for A/T. J Exp Med. (2007) 204:7–10. doi: 10.1084/jem.20062409
13. Reynaud C-A, Delbos F, Faili A, Gueranger Q, Aoufouchi S, Weill J-C. Competitive repair pathways in immunoglobulin gene hypermutation. Philos Trans R Soc Lond B Biol Sci. (2009) 364:613–9. doi: 10.1098/rstb.2008.0206
14. Langerak P, Nygren AOH, Krijger PHL, van den Berk PCM, Jacobs H. A/T mutagenesis in hypermutated immunoglobulin genes strongly depends on PCNAK164 modification. J Exp Med. (2007) 204:1989–98. doi: 10.1084/jem.20070902
15. Roa S, Avdievich E, Peled JU, Maccarthy T, Werling U, Kuang FL, et al. Ubiquitylated PCNA plays a role in somatic hypermutation and class-switch recombination and is required for meiotic progression. Proc Natl Acad Sci USA. (2008) 105:16248–53. doi: 10.1073/pnas.0808182105
16. Bahjat M, Guikema JEJ. The complex interplay between DNA injury and repair in enzymatically induced mutagenesis and DNA damage in B lymphocytes. Int J Mol Sci. (2017) 18:1876. doi: 10.3390/ijms18091876
17. Perez-Duran P, de Yebenes VG, Ramiro AR. Oncogenic events triggered by AID, the adverse effect of antibody diversification. Carcinogenesis. (2007) 28:2427–33. doi: 10.1093/carcin/bgm201
18. Bahjat M, Stratigopoulou M, Pilzecker B, van Dam TP, Mobach S, Bende RJ, et al. DNA polymerase β prevents AID-instigated mutagenic non-canonical mismatch DNA repair. bioRxiv [preprint]. (2020). doi: 10.1101/2020.01.30.926964
19. Mlynarczyk C, Fontan L, Melnick A. Germinal center-derived lymphomas: the darkest side of humoral immunity. Immunol Rev. (2019) 288:214–39. doi: 10.1111/imr.12755
20. Martin F, Chan AC. B cell immunobiology in disease: evolving concepts from the clinic. Annu Rev Immunol. (2006) 24:467–96. doi: 10.1146/annurev.immunol.24.021605.090517
21. Sun Y, Peng I, Senger K, Hamidzadeh K, Reichelt M, Baca M, et al. Critical role of activation induced cytidine deaminase in experimental autoimmune encephalomyelitis. Autoimmunity. (2013) 46:157–67. doi: 10.3109/08916934.2012.750301
22. Galicia G, Boulianne B, Pikor N, Martin A, Gommerman JL. Secondary B cell receptor diversification is necessary for T cell mediated neuro-inflammation during experimental autoimmune encephalomyelitis. PLoS ONE. (2013) 8:e61478. doi: 10.1371/journal.pone.0061478
23. Umiker BR, McDonald G, Larbi A, Medina CO, Hobeika E, Reth M, et al. Production of IgG autoantibody requires expression of activation-induced deaminase in early-developing B cells in a mouse model of SLE. Eur J Immunol. (2014) 44:3093–108. doi: 10.1002/eji.201344282
24. Zhu J, Hay AN, Potter AA, Richwine MW, Sproule T, LeRoith T, et al. Abrogated AID function prolongs survival and diminishes renal pathology in the BXSB mouse model of systemic lupus erythematosus. J Immunol. (2020) 204:1091–100. doi: 10.4049/jimmunol.1900501
25. Galicia G, Lee DSW, Ramaglia V, Ward LA, Yam JY, Leung LYT, et al. Isotype-switched autoantibodies are necessary to facilitate central nervous system autoimmune disease in Aicda(-/-) and Ung(-/-) mice. J Immunol. (2018) 201:1119–30. doi: 10.4049/jimmunol.1700729
26. McDonald G, Medina CO, Pilichowska M, Kearney JF, Shinkura R, Selsing E, et al. Accelerated systemic autoimmunity in the absence of somatic hypermutation in 564Igi: a mouse model of systemic lupus with knocked-in heavy and light chain genes. Front Immunol. (2017) 8:1094. doi: 10.3389/fimmu.2017.01094
27. Reed JH, Jackson J, Christ D, Goodnow CC. Clonal redemption of autoantibodies by somatic hypermutation away from self-reactivity during human immunization. J Exp Med. (2016) 213:1255–65. doi: 10.1084/jem.20151978
28. Burnett DL, Langley DB, Schofield P, Hermes JR, Chan TD, Jackson J, et al. Germinal center antibody mutation trajectories are determined by rapid self/foreign discrimination. Science. (2018) 360:223–6. doi: 10.1126/science.aao3859
29. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. (2002) 416:603–7. doi: 10.1038/416603a
30. Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. (2008) 134:587–98. doi: 10.1016/j.cell.2008.06.032
31. Lou H, Pickering MC. Extracellular DNA and autoimmune diseases. Cell Mol Immunol. (2018) 15:746–55. doi: 10.1038/cmi.2017.136
32. Svilar D, Goellner EM, Almeida KH, Sobol RW. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid Redox Signal. (2011) 14:2491–507. doi: 10.1089/ars.2010.3466
33. Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol. (2002) 12:1748–55. doi: 10.1016/s0960-9822(02)01215-0
34. Kemmerich K, Dingler FA, Rada C, Neuberger MS. Germline ablation of SMUG1 DNA glycosylase causes loss of 5-hydroxymethyluracil- and UNG-backup uracil-excision activities and increases cancer predisposition of Ung-/-Msh2-/- mice. Nucleic Acids Res. (2012) 40:6016–25. doi: 10.1093/nar/gks259
35. Cortazar D, Kunz C, Selfridge J, Lettieri T, Saito Y, MacDougall E, et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. (2011) 470:419–23. doi: 10.1038/nature09672
36. Millar CB, Guy J, Sansom OJ, Selfridge J, MacDougall E, Hendrich B, et al. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science. (2002) 297:403–5. doi: 10.1126/science.1073354
37. Klungland A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe B, et al. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci USA. (1999) 96:13300–5. doi: 10.1073/pnas.96.23.13300
38. Xie Y, Yang H, Cunanan C, Okamoto K, Shibata D, Pan J, et al. Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-ras oncogene in lung tumors. Cancer Res. (2004) 64:3096–102. doi: 10.1158/0008-5472.can-03-3834
39. Takao M, Kanno S, Shiromoto T, Hasegawa R, Ide H, Ikeda S, et al. Novel nuclear and mitochondrial glycosylases revealed by disruption of the mouse Nth1 gene encoding an endonuclease III homolog for repair of thymine glycols. EMBO J. (2002) 21:3486–93. doi: 10.1093/emboj/cdf350
40. Vartanian V, Lowell B, Minko IG, Wood TG, Ceci JD, George S, et al. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc Natl Acad Sci USA. (2006) 103:1864–9. doi: 10.1073/pnas.0507444103
41. Chakraborty A, Wakamiya M, Venkova-Canova T, Pandita RK, Aguilera-Aguirre L, Sarker AH, et al. Neil2-null mice accumulate oxidized DNA bases in the transcriptionally active sequences of the genome and are susceptible to innate inflammation. J Biol Chem. (2015) 290:24636–48. doi: 10.1074/jbc.M115.658146
42. Torisu K, Tsuchimoto D, Ohnishi Y, Nakabeppu Y. Hematopoietic tissue-specific expression of mouse Neil3 for endonuclease. J Biochem. (2005) 138:763–72. doi: 10.1093/jb/mvi168
43. Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci USA. (1996) 93:8919–23. doi: 10.1073/pnas.93.17.8919
44. Guikema JEJ, Linehan EK, Tsuchimoto D, Nakabeppu Y, Strauss PR, Stavnezer J, et al. APE1- and APE2-dependent DNA breaks in immunoglobulin class switch recombination. J Exp Med. (2007) 204:3017–26. doi: 10.1084/jem.20071289
45. Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. (1994) 265:103–6. doi: 10.1126/science.8016642
46. Kucherlapati M, Yang K, Kuraguchi M, Zhao J, Lia M, Heyer J, et al. Haploinsufficiency of Flap endonuclease (Fen1) leads to rapid tumor progression. Proc Natl Acad Sci USA. (2002) 99:9924–9. doi: 10.1073/pnas.152321699
47. Petrini JH, Xiao Y, Weaver DT. DNA ligase I mediates essential functions in mammalian cells. Mol Cell Biol. (1995) 15:4303–8. doi: 10.1128/mcb.15.8.4303
48. Puebla-Osorio N, Lacey DB, Alt FW, Zhu C. Early embryonic lethality due to targeted inactivation of DNA ligase III. Mol Cell Biol. (2006) 26:3935–41. doi: 10.1128/MCB.26.10.3935-3941.2006
49. Nilsen H, Otterlei M, Haug T, Solum K, Nagelhus TA, Skorpen F, et al. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. (1997) 25:750–5. doi: 10.1093/nar/25.4.750
50. Waters TR, Swann PF. Kinetics of the action of thymine DNA glycosylase. J Biol Chem. (1998) 273:20007–14. doi: 10.1074/jbc.273.32.20007
51. Neddermann P, Jiricny J. Efficient removal of uracil from G.U mispairs by the mismatch-specific thymine DNA glycosylase from HeLa cells. Proc Natl Acad Sci USA. (1994) 91:1642–6. doi: 10.1073/pnas.91.5.1642
52. Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. (1999) 401:301–4. doi: 10.1038/45843
53. Haushalter KA, Todd Stukenberg MW, Kirschner MW, Verdine GL. Identification of a new uracil-DNA glycosylase family by expression cloning using synthetic inhibitors. Curr Biol. (1999) 9:174–85. doi: 10.1016/s0960-9822(99)80087-6
54. Sharbeen G, Yee CWY, Smith AL, Jolly CJ. Ectopic restriction of DNA repair reveals that UNG2 excises AID-induced uracils predominantly or exclusively during G1 phase. J Exp Med. (2012) 209:965–74. doi: 10.1084/jem.20112379
55. Di Noia JM, Rada C, Neuberger MS. SMUG1 is able to excise uracil from immunoglobulin genes: insight into mutation versus repair. EMBO J. (2006) 25:585–95. doi: 10.1038/sj.emboj.7600939
56. Dingler FA, Kemmerich K, Neuberger MS, Rada C. Uracil excision by endogenous SMUG1 glycosylase promotes efficient Ig class switching and impacts on A:T substitutions during somatic mutation. Eur J Immunol. (2014) 44:1925–35. doi: 10.1002/eji.201444482
57. Nilsen H, Rosewell I, Robins P, Skjelbred CF, Andersen S, Slupphaug G, et al. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol Cell. (2000) 5:1059–65. doi: 10.1016/s1097-2765(00)80271-3
58. Nilsen H, Stamp G, Andersen S, Hrivnak G, Krokan HE, Lindahl T, et al. Gene-targeted mice lacking the Ung uracil-DNA glycosylase develop B-cell lymphomas. Oncogene. (2003) 22:5381–6. doi: 10.1038/sj.onc.1206860
59. Andersen S, Ericsson M, Dai HY, Pena-Diaz J, Slupphaug G, Nilsen H, et al. Monoclonal B-cell hyperplasia and leukocyte imbalance precede development of B-cell malignancies in uracil-DNA glycosylase deficient mice. DNA Repair. (2005) 4:1432–41. doi: 10.1016/j.dnarep.2005.08.004
60. Zahn A, Daugan M, Safavi S, Godin D, Cheong C, Lamarre A, et al. Separation of function between isotype switching and affinity maturation in vivo during acute immune responses and circulating autoantibodies in UNG-deficient mice. J Immunol. (2013) 190:5949–60. doi: 10.4049/jimmunol.1202711
61. Stavnezer J, Schrader CE. Mismatch repair converts AID-instigated nicks to double-strand breaks for antibody class-switch recombination. Trends Genet. (2006) 22:23–8. doi: 10.1016/j.tig.2005.11.002
62. Min IM, Schrader CE, Vardo J, Luby TM, D'Avirro N, Stavnezer J, et al. The Smu tandem repeat region is critical for Ig isotype switching in the absence of Msh2. Immunity. (2003) 19:515–24. doi: 10.1016/s1074-7613(03)00262-0
63. Eccleston J, Yan C, Yuan K, Alt FW, Selsing E. Mismatch repair proteins MSH2, MLH1, and EXO1 are important for class-switch recombination events occurring in B cells that lack nonhomologous end joining. J Immunol. (2011) 186:2336–43. doi: 10.4049/jimmunol.1003104
64. Krijger PHL, Langerak P, van den Berk PCM, Jacobs H. Dependence of nucleotide substitutions on Ung2, Msh2, and PCNA-Ub during somatic hypermutation. J Exp Med. (2009) 206:2603–11. doi: 10.1084/jem.20091707
65. Krijger PHL, Tsaalbi-Shtylik A, Wit N, van den Berk PCM, de Wind N, Jacobs H. Rev1 is essential in generating G to C transversions downstream of the Ung2 pathway but not the Msh2+Ung2 hybrid pathway. Eur J Immunol. (2013) 43:2765–70. doi: 10.1002/eji.201243191
66. Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF, et al. The biochemistry of somatic hypermutation. Annu Rev Immunol. (2008) 26:481–511. doi: 10.1146/annurev.immunol.26.021607.090236
67. Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. (2006) 126:297–308. doi: 10.1016/j.cell.2006.05.039
68. van Oers JMM, Roa S, Werling U, Liu Y, Genschel J, Hou HJ, et al. PMS2 endonuclease activity has distinct biological functions and is essential for genome maintenance. Proc Natl Acad Sci USA. (2010) 107:13384–9. doi: 10.1073/pnas.1008589107
69. Girelli Zubani G, Zivojnovic M, De Smet A, Albagli-Curiel O, Huetz F, Weill J-C, et al. Pms2 and uracil-DNA glycosylases act jointly in the mismatch repair pathway to generate Ig gene mutations at A-T base pairs. J Exp Med. (2017) 214:1169–80. doi: 10.1084/jem.20161576
70. IJspeert H, van Schouwenburg PA, Pico-Knijnenburg I, Loeffen J, Brugieres L, Driessen GJ, et al. Repertoire sequencing of B cells elucidates the role of UNG and mismatch repair proteins in somatic hypermutation in humans. Front Immunol. (2019) 10:1913. doi: 10.3389/fimmu.2019.01913
71. Imai K, Slupphaug G, Lee W-I, Revy P, Nonoyama S, Catalan N, et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat Immunol. (2003) 4:1023–8. doi: 10.1038/ni974
72. Kavli B, Andersen S, Otterlei M, Liabakk NB, Imai K, Fischer A, et al. B cells from hyper-IgM patients carrying UNG mutations lack ability to remove uracil from ssDNA and have elevated genomic uracil. J Exp Med. (2005) 201:2011–21. doi: 10.1084/jem.20050042
73. Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, et al. Two levels of protection for the B cell genome during somatic hypermutation. Nature. (2008) 451:841–5. doi: 10.1038/nature06547
74. Xue K, Rada C, Neuberger MS. The in vivo pattern of AID targeting to immunoglobulin switch regions deduced from mutation spectra in msh2-/- ung-/- mice. J Exp Med. (2006) 203:2085–94. doi: 10.1084/jem.20061067
75. Liu M, Schatz DG. Balancing AID and DNA repair during somatic hypermutation. Trends Immunol. (2009) 30:173–81. doi: 10.1016/j.it.2009.01.007
76. Gu X, Booth CJ, Liu Z, Strout MP. AID-associated DNA repair pathways regulate malignant transformation in a murine model of BCL6-driven diffuse large B-cell lymphoma. Blood. (2016) 127:102–12. doi: 10.1182/blood-2015-02-628164
77. Cortizas EM, Zahn A, Safavi S, Reed JA, Vega F, Di Noia JM, et al. UNG protects B cells from AID-induced telomere loss. J Exp Med. (2016) 213:2459–72. doi: 10.1084/jem.20160635
78. Sousa MML, Krokan HE, Slupphaug G. DNA-uracil and human pathology. Mol Aspects Med. (2007) 28:276–306. doi: 10.1016/j.mam.2007.04.006
79. Green B, Martin A, Belcheva A. Deficiency in the DNA glycosylases UNG1 and OGG1 does not potentiate c-Myc-induced B-cell lymphomagenesis. Exp Hematol. (2018) 61:52–8. doi: 10.1016/j.exphem.2018.02.006
80. Schmidt RE, Grimbacher B, Witte T. Autoimmunity and primary immunodeficiency: two sides of the same coin? Nat Rev Rheumatol. (2017) 14:7–18. doi: 10.1038/nrrheum.2017.198
81. Todoric K, Koontz JB, Mattox D, Tarrant TK. Autoimmunity in immunodeficiency. Curr Allergy Asthma Rep. (2013) 13:361–70. doi: 10.1007/s11882-013-0350-3
82. Goyal R, Bulua AC, Nikolov NP, Schwartzberg PL, Siegel RM. Rheumatologic and autoimmune manifestations of primary immunodeficiency disorders. Curr Opin Rheumatol. (2009) 21:78–84. doi: 10.1097/BOR.0b013e32831cb939
83. Grammatikos AP, Tsokos GC. Immunodeficiency and autoimmunity: lessons from systemic lupus erythematosus. Trends Mol Med. (2012) 18:101–8. doi: 10.1016/j.molmed.2011.10.005
84. Cantaert T, Schickel J-N, Bannock JM, Ng Y-S, Massad C, Delmotte FR, et al. Decreased somatic hypermutation induces an impaired peripheral B cell tolerance checkpoint. J Clin Invest. (2016) 126:4289–302. doi: 10.1172/JCI84645
85. Masaoka A, Matsubara M, Hasegawa R, Tanaka T, Kurisu S, Terato H, et al. Mammalian 5-formyluracil-DNA glycosylase. 2. Role of SMUG1 uracil-DNA glycosylase in repair of 5-formyluracil and other oxidized and deaminated base lesions. Biochemistry. (2003) 42:5003–12. doi: 10.1021/bi0273213
86. Darwanto A, Theruvathu JA, Sowers JL, Rogstad DK, Pascal T, Goddard W 3rd, et al. Mechanisms of base selection by human single-stranded selective monofunctional uracil-DNA glycosylase. J Biol Chem. (2009) 284:15835–46. doi: 10.1074/jbc.M807846200
87. Kuznetsova AA, Iakovlev DA, Misovets IV, Ishchenko AA, Saparbaev MK, Kuznetsov NA, et al. Pre-steady-state kinetic analysis of damage recognition by human single-strand selective monofunctional uracil-DNA glycosylase SMUG1. Mol Biosyst. (2017) 13:2638–49. doi: 10.1039/c7mb00457e
88. Pfaffeneder T, Spada F, Wagner M, Brandmayr C, Laube SK, Eisen D, et al. Tet oxidizes thymine to 5-hydroxymethyluracil in mouse embryonic stem cell DNA. Nat Chem Biol. (2014) 10:574–81. doi: 10.1038/nchembio.1532
89. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. (2009) 324:930–5. doi: 10.1126/science.1170116
90. Jobert L, Skjeldam HK, Dalhus B, Galashevskaya A, Vagbo CB, Bjoras M, et al. The human base excision repair enzyme SMUG1 directly interacts with DKC1 and contributes to RNA quality control. Mol Cell. (2013) 49:339–45. doi: 10.1016/j.molcel.2012.11.010
91. Kroustallaki P, Lirussi L, Carracedo S, You P, Esbensen QY, Gotz A, et al. SMUG1 promotes telomere maintenance through telomerase RNA processing. Cell Rep. (2019) 28:1690–702.e10. doi: 10.1016/j.celrep.2019.07.040
92. Nilsen H, Haushalter KA, Robins P, Barnes DE, Verdine GL, Lindahl T. Excision of deaminated cytosine from the vertebrate genome: role of the SMUG1 uracil-DNA glycosylase. EMBO J. (2001) 20:4278–86. doi: 10.1093/emboj/20.15.4278
93. Doseth B, Visnes T, Wallenius A, Ericsson I, Sarno A, Pettersen HS, et al. Uracil-DNA glycosylase in base excision repair and adaptive immunity: species differences between man and mouse. J Biol Chem. (2011) 286:16669–80. doi: 10.1074/jbc.M111.230052
94. Di Noia JM, Williams GT, Chan DTY, Buerstedde J-M, Baldwin GS, Neuberger MS. Dependence of antibody gene diversification on uracil excision. J Exp Med. (2007) 204:3209–19. doi: 10.1084/jem.20071768
95. Abdel-Fatah TMA, Albarakati N, Bowell L, Agarwal D, Moseley P, Hawkes C, et al. Single-strand selective monofunctional uracil-DNA glycosylase (SMUG1) deficiency is linked to aggressive breast cancer and predicts response to adjuvant therapy. Breast Cancer Res Treat. (2013) 142:515–27. doi: 10.1007/s10549-013-2769-6
96. D'Errico M, de Rinaldis E, Blasi MF, Viti V, Falchetti M, Calcagnile A, et al. Genome-wide expression profile of sporadic gastric cancers with microsatellite instability. Eur J Cancer. (2009) 45:461–9. doi: 10.1016/j.ejca.2008.10.032
97. Xie H, Gong Y, Dai J, Wu X, Gu J. Genetic variations in base excision repair pathway and risk of bladder cancer: a case-control study in the United States. Mol Carcinog. (2015) 54:50–7. doi: 10.1002/mc.22073
98. Alsoe L, Sarno A, Carracedo S, Domanska D, Dingler F, Lirussi L, et al. Uracil accumulation and mutagenesis dominated by cytosine deamination in CpG dinucleotides in mice lacking UNG and SMUG1. Sci Rep. (2017) 7:7199. doi: 10.1038/s41598-017-07314-5
99. Wiebauer K, Jiricny J. Mismatch-specific thymine DNA glycosylase and DNA polymerase beta mediate the correction of G.T mispairs in nuclear extracts from human cells. Proc Natl Acad Sci USA. (1990) 87:5842–5. doi: 10.1073/pnas.87.15.5842
100. Hardeland U, Kunz C, Focke F, Szadkowski M, Schar P. Cell cycle regulation as a mechanism for functional separation of the apparently redundant uracil DNA glycosylases TDG and UNG2. Nucleic Acids Res. (2007) 35:3859–67. doi: 10.1093/nar/gkm337
101. Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. (2011) 146:67–79. doi: 10.1016/j.cell.2011.06.020
102. Xu J, Cortellino S, Tricarico R, Chang W-C, Scher G, Devarajan K, et al. Thymine DNA glycosylase (TDG) is involved in the pathogenesis of intestinal tumors with reduced APC expression. Oncotarget. (2017) 8:89988–97. doi: 10.18632/oncotarget.21219
103. Chen D, Lucey MJ, Phoenix F, Lopez-Garcia J, Hart SM, Losson R, et al. T:G mismatch-specific thymine-DNA glycosylase potentiates transcription of estrogen-regulated genes through direct interaction with estrogen receptor alpha. J Biol Chem. (2003) 278:38586–92. doi: 10.1074/jbc.M304286200
104. Liu Y, Duong W, Krawczyk C, Bretschneider N, Borbely G, Varshney M, et al. Oestrogen receptor beta regulates epigenetic patterns at specific genomic loci through interaction with thymine DNA glycosylase. Epigenetics Chromatin. (2016) 9:7. doi: 10.1186/s13072-016-0055-7
105. Weyant MJ, Carothers AM, Mahmoud NN, Bradlow HL, Remotti H, Bilinski RT, et al. Reciprocal expression of ERalpha and ERbeta is associated with estrogen-mediated modulation of intestinal tumorigenesis. Cancer Res. (2001) 61:2547–51.
106. Peng B, Hurt EM, Hodge DR, Thomas SB, Farrar WL. DNA hypermethylation and partial gene silencing of human thymine- DNA glycosylase in multiple myeloma cell lines. Epigenetics. (2006) 1:138–45. doi: 10.4161/epi.1.3.2938
107. Um S, Harbers M, Benecke A, Pierrat B, Losson R, Chambon P. Retinoic acid receptors interact physically and functionally with the T:G mismatch-specific thymine-DNA glycosylase. J Biol Chem. (1998) 273:20728–36. doi: 10.1074/jbc.273.33.20728
108. Missero C, Pirro MT, Simeone S, Pischetola M, Di Lauro R. The DNA glycosylase T:G mismatch-specific thymine DNA glycosylase represses thyroid transcription factor-1-activated transcription. J Biol Chem. (2001) 276:33569–75. doi: 10.1074/jbc.M104963200
109. Tini M, Benecke A, Um S-J, Torchia J, Evans RM, Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol Cell. (2002) 9:265–77. doi: 10.1016/s1097-2765(02)00453-7
110. Garaud S, Le Dantec C, Jousse-Joulin S, Hanrotel-Saliou C, Saraux A, Mageed RA, et al. IL-6 modulates CD5 expression in B cells from patients with lupus by regulating DNA methylation. J Immunol. (2009) 182:5623–32. doi: 10.4049/jimmunol.0802412
111. Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. (1990) 33:1665–73. doi: 10.1002/art.1780331109
112. Mizugaki M, Yamaguchi T, Ishiwata S, Shindo H, Hishinuma T, Nozaki S, et al. Alteration of DNA methylation levels in MRL lupus mice. Clin Exp Immunol. (1997) 110:265–9. doi: 10.1111/j.1365-2249.1997.tb08326.x
113. Cornacchia E, Golbus J, Maybaum J, Strahler J, Hanash S, Richardson B. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J Immunol. (1988) 140:2197–200.
114. Meas R, Burak MJ, Sweasy JB. DNA repair and systemic lupus erythematosus. DNA Repair. (2017) 56:174–82. doi: 10.1016/j.dnarep.2017.06.020
115. Lee H-T, Lin C-S, Lee C-S, Tsai C-Y, Wei Y-H. The role of hOGG1 C1245G polymorphism in the susceptibility to lupus nephritis and modulation of the plasma 8-OHdG in patients with systemic lupus erythematosus. Int J Mol Sci. (2015) 16:3757–68. doi: 10.3390/ijms16023757
116. Massaad MJ, Zhou J, Tsuchimoto D, Chou J, Jabara H, Janssen E, et al. Deficiency of base excision repair enzyme NEIL3 drives increased predisposition to autoimmunity. J Clin Invest. (2016) 126:4219–36. doi: 10.1172/JCI85647
117. Senejani AG, Liu Y, Kidane D, Maher SE, Zeiss CJ, Park H-J, et al. Mutation of POLB causes lupus in mice. Cell Rep. (2014) 6:1–8. doi: 10.1016/j.celrep.2013.12.017
118. Han J-W, Zheng H-F, Cui Y, Sun L-D, Ye D-Q, Hu Z, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. (2009) 41:1234–7. doi: 10.1038/ng.472
119. Lin Y-J, Wan L, Huang C-M, Chen S-Y, Huang Y-C, Lai C-H, et al. Polymorphisms in the DNA repair gene XRCC1 and associations with systemic lupus erythematosus risk in the Taiwanese Han Chinese population. Lupus. (2009) 18:1246–51. doi: 10.1177/0961203309345777
120. Zheng L, Dai H, Zhou M, Li M, Singh P, Qiu J, et al. Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nat Med. (2007) 13:812–9. doi: 10.1038/nm1599
121. Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. (2008) 135:1201–12. doi: 10.1016/j.cell.2008.11.042
122. Grigera F, Bellacosa A, Kenter AL. Complex relationship between mismatch repair proteins and MBD4 during immunoglobulin class switch recombination. PLoS ONE. (2013) 8:e78370. doi: 10.1371/journal.pone.0078370
123. Bardwell PD, Martin A, Wong E, Li Z, Edelmann W, Scharff MD. Cutting edge: the G-U mismatch glycosylase methyl-CpG binding domain 4 is dispensable for somatic hypermutation and class switch recombination. J Immunol. (2003) 170:1620–4. doi: 10.4049/jimmunol.170.4.1620
124. Grigera F, Wuerffel R, Kenter AL. MBD4 facilitates immunoglobulin class switch recombination. Mol Cell Biol. (2017) 37:e00316–16. doi: 10.1128/MCB.00316-16
125. Bellacosa A, Cicchillitti L, Schepis F, Riccio A, Yeung AT, Matsumoto Y, et al. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc Natl Acad Sci USA. (1999) 96:3969–74. doi: 10.1073/pnas.96.7.3969
126. Schrader CE, Vardo J, Stavnezer J. Role for mismatch repair proteins Msh2, Mlh1, and Pms2 in immunoglobulin class switching shown by sequence analysis of recombination junctions. J Exp Med. (2002) 195:367–73. doi: 10.1084/jem.20011877
127. Min IM, Rothlein LR, Schrader CE, Stavnezer J, Selsing E. Shifts in targeting of class switch recombination sites in mice that lack mu switch region tandem repeats or Msh2. J Exp Med. (2005) 201:1885–90. doi: 10.1084/jem.20042491
128. Schrader CE, Vardo J, Stavnezer J. Mlh1 can function in antibody class switch recombination independently of Msh2. J Exp Med. (2003) 197:1377–83. doi: 10.1084/jem.20022190
129. Arakawa H, Saribasak H, Buerstedde J-M. Activation-induced cytidine deaminase initiates immunoglobulin gene conversion and hypermutation by a common intermediate. PLoS Biol. (2004) 2:E179. doi: 10.1371/journal.pbio.0020179
130. Saribasak H, Saribasak NN, Ipek FM, Ellwart JW, Arakawa H, Buerstedde J-M. Uracil DNA glycosylase disruption blocks Ig gene conversion and induces transition mutations. J Immunol. (2006) 176:365–71. doi: 10.4049/jimmunol.176.1.365
131. Wong E, Yang K, Kuraguchi M, Werling U, Avdievich E, Fan K, et al. Mbd4 inactivation increases cright-arrowT transition mutations and promotes gastrointestinal tumor formation. Proc Natl Acad Sci USA. (2002) 99:14937–42. doi: 10.1073/pnas.232579299
132. Rodrigues M, Mobuchon L, Houy A, Alsafadi S, Baulande S, Mariani O, et al. Evolutionary routes in metastatic uveal melanomas depend on MBD4 alterations. Clin Cancer Res. (2019) 25:5513–24. doi: 10.1158/1078-0432.CCR-19-1215
133. Tricarico R, Cortellino S, Riccio A, Jagmohan-Changur S, Van der Klift H, Wijnen J, et al. Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis. Oncotarget. (2015) 6:42892–904. doi: 10.18632/oncotarget.5740
134. Allione A, Guarrera S, Russo A, Ricceri F, Purohit R, Pagnani A, et al. Inter-individual variation in nucleotide excision repair pathway is modulated by non-synonymous polymorphisms in ERCC4 and MBD4 genes. Mutat Res. (2013) 751–2:49–54. doi: 10.1016/j.mrfmmm.2013.08.005
135. Xiong X-D, Luo X-P, Liu X, Jing X, Zeng L-Q, Lei M, et al. The MBD4 Glu346Lys polymorphism is associated with the risk of cervical cancer in a Chinese population. Int J Gynecol Cancer. (2012) 22:1552–6. doi: 10.1097/IGC.0b013e31826e22e4
136. Yamada T, Koyama T, Ohwada S, Tago K, Sakamoto I, Yoshimura S, et al. Frameshift mutations in the MBD4/MED1 gene in primary gastric cancer with high-frequency microsatellite instability. Cancer Lett. (2002) 181:115–20. doi: 10.1016/s0304-3835(02)00043-5
137. Riccio A, Aaltonen LA, Godwin AK, Loukola A, Percesepe A, Salovaara R, et al. The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat Genet. (1999) 23:266–8. doi: 10.1038/15443
138. Sansom OJ, Bishop SM, Bird A, Clarke AR. MBD4 deficiency does not increase mutation or accelerate tumorigenesis in mice lacking MMR. Oncogene. (2004) 23:5693–6. doi: 10.1038/sj.onc.1207767
139. Sanders MA, Chew E, Flensburg C, Zeilemaker A, Miller SE, Al Hinai AS, et al. MBD4 guards against methylation damage and germ line deficiency predisposes to clonal hematopoiesis and early-onset AML. Blood. (2018) 132:1526–34. doi: 10.1182/blood-2018-05-852566
140. Izykowska K, Przybylski GK, Gand C, Braun FC, Grabarczyk P, Kuss AW, et al. Genetic rearrangements result in altered gene expression and novel fusion transcripts in sezary syndrome. Oncotarget. (2017) 8:39627–39. doi: 10.18632/oncotarget.17383
141. Brandt B, Rashidiani S, Ban A, Rauch TA. DNA methylation-governed gene expression in autoimmune arthritis. Int J Mol Sci. (2019) 20:5646. doi: 10.3390/ijms20225646
142. Celarain N, Tomas-Roig J. Changes in deoxyribonucleic acid methylation contribute to the pathophysiology of multiple sclerosis. Front Genet. (2019) 10:1138. doi: 10.3389/fgene.2019.01138
143. Andlauer TFM, Buck D, Antony G, Bayas A, Bechmann L, Berthele A, et al. Novel multiple sclerosis susceptibility loci implicated in epigenetic regulation. Sci Adv. (2016) 2:e1501678. doi: 10.1126/sciadv.1501678
144. Balada E, Ordi-Ros J, Serrano-Acedo S, Martinez-Lostao L, Vilardell-Tarres M. Transcript overexpression of the MBD2 and MBD4 genes in CD4+ T cells from systemic lupus erythematosus patients. J Leukoc Biol. (2007) 81:1609–16. doi: 10.1189/jlb.0107064
145. Balada E, Castro-Marrero J, Felip L, Ordi-Ros J, Vilardell-Tarres M. Associations between the expression of epigenetically regulated genes and the expression of DNMTs and MBDs in systemic lupus erythematosus. PLoS ONE. (2012) 7:e45897. doi: 10.1371/journal.pone.0045897
146. Fagone P, Mangano K, Di Marco R, Touil-Boukoffa C, Chikovan T, Signorelli S, et al. Expression of DNA methylation genes in secondary progressive multiple sclerosis. J Neuroimmunol. (2016) 290:66–9. doi: 10.1016/j.jneuroim.2015.11.018
147. Yu X, Teng H, Marques A, Ashgari F, Ibrahim SM. High resolution mapping of Cia3: a common arthritis quantitative trait loci in different species. J Immunol. (2009) 182:3016–23. doi: 10.4049/jimmunol.0803005
148. Wallace SS. DNA glycosylases search for and remove oxidized DNA bases. Environ Mol Mutagen. (2013) 54:691–704. doi: 10.1002/em.21820
149. Fromme JC, Banerjee A, Verdine GL. DNA glycosylase recognition and catalysis. Curr Opin Struct Biol. (2004) 14:43–9. doi: 10.1016/j.sbi.2004.01.003
150. Kanvah S, Joseph J, Schuster GB, Barnett RN, Cleveland CL, Landman U. Oxidation of DNA: damage to nucleobases. Acc Chem Res. (2010) 43:280–7. doi: 10.1021/ar900175a
151. Sakamoto K, Tominaga Y, Yamauchi K, Nakatsu Y, Sakumi K, Yoshiyama K, et al. MUTYH-null mice are susceptible to spontaneous and oxidative stress induced intestinal tumorigenesis. Cancer Res. (2007) 67:6599–604. doi: 10.1158/0008-5472.CAN-06-4802
152. Dizdaroglu M, Karahalil B, Senturker S, Buckley TJ, Roldan-Arjona T. Excision of products of oxidative DNA base damage by human NTH1 protein. Biochemistry. (1999) 38:243–6. doi: 10.1021/bi9819071
153. Leipold MD, Muller JG, Burrows CJ, David SS. Removal of hydantoin products of 8-oxoguanine oxidation by the Escherichia coli DNA repair enzyme, FPG. Biochemistry. (2000) 39:14984–92. doi: 10.1021/bi0017982
154. Hegde ML, Hegde PM, Bellot LJ, Mandal SM, Hazra TK, Li G-M, et al. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc Natl Acad Sci USA. (2013) 110:E3090–9. doi: 10.1073/pnas.1304231110
155. Banerjee D, Mandal SM, Das A, Hegde ML, Das S, Bhakat KK, et al. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J Biol Chem. (2011) 286:6006–16. doi: 10.1074/jbc.M110.198796
156. Han D, Schomacher L, Schule KM, Mallick M, Musheev MU, Karaulanov E, et al. NEIL1 and NEIL2 DNA glycosylases protect neural crest development against mitochondrial oxidative stress. Elife. (2019) 8:e49044. doi: 10.7554/eLife.49044
157. Liu M, Bandaru V, Bond JP, Jaruga P, Zhao X, Christov PP, et al. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc Natl Acad Sci USA. (2010) 107:4925–30. doi: 10.1073/pnas.0908307107
158. Rolseth V, Runden-Pran E, Luna L, McMurray C, Bjoras M, Ottersen OP. Widespread distribution of DNA glycosylases removing oxidative DNA lesions in human and rodent brains. DNA Repair. (2008) 7:1578–88. doi: 10.1016/j.dnarep.2008.06.007
159. Hildrestrand GA, Neurauter CG, Diep DB, Castellanos CG, Krauss S, Bjoras M, et al. Expression patterns of Neil3 during embryonic brain development and neoplasia. BMC Neurosci. (2009) 10:45. doi: 10.1186/1471-2202-10-45
160. Ucher AJ, Linehan EK, Teebor GW, Schrader CE, Stavnezer J. The DNA glycosylases Ogg1 and Nth1 do not contribute to Ig class switching in activated mouse splenic B cells. PLoS ONE. (2012) 7:e36061. doi: 10.1371/journal.pone.0036061
161. Winter DB, Phung QH, Zeng X, Seeberg E, Barnes DE, Lindahl T, et al. Normal somatic hypermutation of Ig genes in the absence of 8-hydroxyguanine-DNA glycosylase. J Immunol. (2003) 170:5558–62. doi: 10.4049/jimmunol.170.11.5558
162. Kuo FC, Sklar J. Augmented expression of a human gene for 8-oxoguanine DNA glycosylase (MutM) in B lymphocytes of the dark zone in lymph node germinal centers. J Exp Med. (1997) 186:1547–56. doi: 10.1084/jem.186.9.1547
163. Mori H, Ouchida R, Hijikata A, Kitamura H, Ohara O, Li Y, et al. Deficiency of the oxidative damage-specific DNA glycosylase NEIL1 leads to reduced germinal center B cell expansion. DNA Repair (Amst). (2009) 8:1328–32. doi: 10.1016/j.dnarep.2009.08.007
164. Romano R, Zaravinos A, Liadaki K, Caridha R, Lundin J, Carlsson G, et al. NEIL1 is a candidate gene associated with common variable immunodeficiency in a patient with a chromosome 15q24 deletion. Clin Immunol. (2017) 176:71–6. doi: 10.1016/j.clim.2017.01.006
165. Nowsheen S, Wukovich RL, Aziz K, Kalogerinis PT, Richardson CC, Panayiotidis MI, et al. Accumulation of oxidatively induced clustered DNA lesions in human tumor tissues. Mutat Res. (2009) 674:131–6. doi: 10.1016/j.mrgentox.2008.09.010
166. Sakumi K, Tominaga Y, Furuichi M, Xu P, Tsuzuki T, Sekiguchi M, et al. Ogg1 knockout-associated lung tumorigenesis and its suppression by Mth1 gene disruption. Cancer Res. (2003) 63:902–5.
167. Goode EL, Ulrich CM, Potter JD. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol Biomarkers Prev. (2002) 11:1513–30.
168. Hung RJ, Hall J, Brennan P, Boffetta P. Genetic polymorphisms in the base excision repair pathway and cancer risk: a HuGE review. Am J Epidemiol. (2005) 162:925–42. doi: 10.1093/aje/kwi318
169. Paz-Elizur T, Ben-Yosef R, Elinger D, Vexler A, Krupsky M, Berrebi A, et al. Reduced repair of the oxidative 8-oxoguanine DNA damage and risk of head and neck cancer. Cancer Res. (2006) 66:11683–9. doi: 10.1158/0008-5472.CAN-06-2294
170. Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, et al. Inherited variants of MYH associated with somatic G:C–>T:A mutations in colorectal tumors. Nat Genet. (2002) 30:227–32. doi: 10.1038/ng828
171. Russo MT, De Luca G, Casorelli I, Degan P, Molatore S, Barone F, et al. Role of MUTYH and MSH2 in the control of oxidative DNA damage, genetic instability, and tumorigenesis. Cancer Res. (2009) 69:4372–9. doi: 10.1158/0008-5472.CAN-08-3292
172. Goto M, Shinmura K, Igarashi H, Kobayashi M, Konno H, Yamada H, et al. Altered expression of the human base excision repair gene NTH1 in gastric cancer. Carcinogenesis. (2009) 30:1345–52. doi: 10.1093/carcin/bgp108
173. Galick HA, Kathe S, Liu M, Robey-Bond S, Kidane D, Wallace SS, et al. Germ-line variant of human NTH1 DNA glycosylase induces genomic instability and cellular transformation. Proc Natl Acad Sci USA. (2013) 110:14314–9. doi: 10.1073/pnas.1306752110
174. Shinmura K, Kato H, Kawanishi Y, Igarashi H, Goto M, Tao H, et al. Abnormal expressions of DNA glycosylase genes NEIL1, NEIL2, and NEIL3 are associated with somatic mutation loads in human cancer. Oxid Med Cell Longev. (2016) 2016:1546392. doi: 10.1155/2016/1546392
175. Galick HA, Marsden CG, Kathe S, Dragon JA, Volk L, Nemec AA, et al. The NEIL1 G83D germline DNA glycosylase variant induces genomic instability and cellular transformation. Oncotarget. (2017) 8:85883–95. doi: 10.18632/oncotarget.20716
176. Rolseth V, Luna L, Olsen AK, Suganthan R, Scheffler K, Neurauter CG, et al. No cancer predisposition or increased spontaneous mutation frequencies in NEIL DNA glycosylases-deficient mice. Sci Rep. (2017) 7:4384. doi: 10.1038/s41598-017-04472-4
177. Ning Y, Foss A, Kimball AS, Neill N, Matz T, Schultz R. Characterization of a case of follicular lymphoma transformed into. Mol Cytogenet. (2013) 6:34. doi: 10.1186/1755-8166-6-34
178. Blenk S, Engelmann J, Weniger M, Schultz J, Dittrich M, Rosenwald A, et al. Germinal center B cell-like (GCB) and activated B cell-like (ABC) type of diffuse large B cell lymphoma (DLBCL): analysis of molecular predictors, signatures, cell cycle state and patient survival. Cancer Inform. (2007) 3:399–420.
179. Basso K, Dalla-Favera R. Germinal centres and B cell lymphomagenesis. Nat Rev Immunol. (2015) 15:172–84. doi: 10.1038/nri3814
180. Chen Y, Zhou Z, Min W. Mitochondria, oxidative stress and innate immunity. Front Physiol. (2018) 9:1487. doi: 10.3389/fphys.2018.01487
181. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. (2015) 520:553–7. doi: 10.1038/nature14156
182. Lee H-T, Lin C-S, Lee C-S, Tsai C-Y, Wei Y-H. Increased 8-hydroxy-2'-deoxyguanosine in plasma and decreased mRNA expression of human 8-oxoguanine DNA glycosylase 1, anti-oxidant enzymes, mitochondrial biogenesis-related proteins and glycolytic enzymes in leucocytes in patients with systemic lupus erythematosus. Clin Exp Immunol. (2014) 176:66–77. doi: 10.1111/cei.12256
183. Evans MD, Cooke MS, Akil M, Samanta A, Lunec J. Aberrant processing of oxidative DNA damage in systemic lupus erythematosus. Biochem Biophys Res Commun. (2000) 273:894–8. doi: 10.1006/bbrc.2000.3078
184. Arneth B. Systemic lupus erythematosus and DNA degradation and elimination defects. Front Immunol. (2019) 10:1697. doi: 10.3389/fimmu.2019.01697
185. Chen S-Y, Wan L, Huang C-M, Huang Y-C, Sheu JJ-C, Lin Y-J, et al. Association of the C-285T and A5954G polymorphisms in the DNA repair gene OGG1 with the susceptibility of rheumatoid arthritis. Rheumatol Int. (2012) 32:1165–9. doi: 10.1007/s00296-010-1738-1
186. Mohamed RH, El-Shal AS, El-Shahawy EE, Abdel Galil SM. Association of XRCC1 and OGG1 DNA repair gene polymorphisms with rheumatoid arthritis in Egyptian patients. Gene. (2016) 578:112–6. doi: 10.1016/j.gene.2015.12.021
187. Karahalil B, Orhan G, Ak F. The impact of detoxifying and repair gene polymorphisms and the levels of serum ROS in the susceptibility to multiple sclerosis. Clin Neurol Neurosurg. (2015) 139:288–94. doi: 10.1016/j.clineuro.2015.10.028
188. Kung Y-J, Tsai KS, Huang C-M, Lin H-J, Chen T-H, Hsu Y-A, et al. MUTYH gene polymorphisms as risk factors for rheumatoid arthritis. Biomed Res Int. (2015) 2015:893796. doi: 10.1155/2015/893796
189. Levin JD, Demple B. Analysis of class II (hydrolytic) and class I (beta-lyase) apurinic/apyrimidinic endonucleases with a synthetic DNA substrate. Nucleic Acids Res. (1990) 18:5069–75. doi: 10.1093/nar/18.17.5069
190. Warner HR, Demple BF, Deutsch WA, Kane CM, Linn S. Apurinic/apyrimidinic endonucleases in repair of pyrimidine dimers and other lesions in DNA. Proc Natl Acad Sci USA. (1980) 77:4602–6. doi: 10.1073/pnas.77.8.4602
191. Fung H, Demple B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol Cell. (2005) 17:463–70. doi: 10.1016/j.molcel.2004.12.029
192. Chen DS, Herman T, Demple B. Two distinct human DNA diesterases that hydrolyze 3'-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res. (1991) 19:5907–14. doi: 10.1093/nar/19.21.5907
193. Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. (1992) 11:3323–35.
194. Ludwig DL, MacInnes MA, Takiguchi Y, Purtymun PE, Henrie M, Flannery M, et al. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutat Res. (1998) 409:17–29. doi: 10.1016/s0921-8777(98)00039-1
195. Meira LB, Devaraj S, Kisby GE, Burns DK, Daniel RL, Hammer RE, et al. Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress. Cancer Res. (2001) 61:5552–7.
196. Huamani J, McMahan CA, Herbert DC, Reddick R, McCarrey JR, MacInnes MI, et al. Spontaneous mutagenesis is enhanced in apex heterozygous mice. Mol Cell Biol. (2004) 24:8145–53. doi: 10.1128/MCB.24.18.8145-8153.2004
197. Madhusudan S, Smart F, Shrimpton P, Parsons JL, Gardiner L, Houlbrook S, et al. Isolation of a small molecule inhibitor of DNA base excision repair. Nucleic Acids Res. (2005) 33:4711–24. doi: 10.1093/nar/gki781
198. Masani S, Han L, Yu K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Mol Cell Biol. (2013) 33:1468–73. doi: 10.1128/MCB.00026-13
199. Frossi B, Antoniali G, Yu K, Akhtar N, Kaplan MH, Kelley MR, et al. Endonuclease and redox activities of human apurinic/apyrimidinic endonuclease 1 have distinctive and essential functions in IgA class switch recombination. J Biol Chem. (2019) 294:5198–207. doi: 10.1074/jbc.RA118.006601
200. Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, et al. Class-switch recombination occurs infrequently in germinal centers. Immunity. (2019) 51:337–50.e7. doi: 10.1016/j.immuni.2019.07.001
201. Stavnezer J, Linehan EK, Thompson MR, Habboub G, Ucher AJ, Kadungure T, et al. Differential expression of APE1 and APE2 in germinal centers promotes error-prone repair and A:T mutations during somatic hypermutation. Proc Natl Acad Sci USA. (2014) 111:9217–22. doi: 10.1073/pnas.1405590111
202. Sabouri Z, Okazaki I-M, Shinkura R, Begum N, Nagaoka H, Tsuchimoto D, et al. Apex2 is required for efficient somatic hypermutation but not for class switch recombination of immunoglobulin genes. Int Immunol. (2009) 21:947–55. doi: 10.1093/intimm/dxp061
203. Islam H, Kobayashi M, Honjo T. Apurinic/apyrimidinic endonuclease 1 (APE1) is dispensable for activation-induced cytidine deaminase (AID)-dependent somatic hypermutation in the immunoglobulin gene. Int Immunol. (2019) 31:543–54. doi: 10.1093/intimm/dxz028
204. Herring CJ, West CM, Wilks DP, Davidson SE, Hunter RD, Berry P, et al. Levels of the DNA repair enzyme human apurinic/apyrimidinic endonuclease (APE1, APEX, Ref-1) are associated with the intrinsic radiosensitivity of cervical cancers. Br J Cancer. (1998) 78:1128–33. doi: 10.1038/bjc.1998.641
205. Robertson KA, Bullock HA, Xu Y, Tritt R, Zimmerman E, Ulbright TM, et al. Altered expression of Ape1/ref-1 in germ cell tumors and overexpression in NT2 cells confers resistance to bleomycin and radiation. Cancer Res. (2001) 61:2220–5.
206. Kelley MR, Cheng L, Foster R, Tritt R, Jiang J, Broshears J, et al. Elevated and altered expression of the multifunctional DNA base excision repair and redox enzyme Ape1/ref-1 in prostate cancer. Clin Cancer Res. (2001) 7:824–30.
207. Bobola MS, Blank A, Berger MS, Stevens BA, Silber JR. Apurinic/apyrimidinic endonuclease activity is elevated in human adult gliomas. Clin Cancer Res. (2001) 7:3510–8.
208. Di Maso V, Avellini C, Croce LS, Rosso N, Quadrifoglio F, Cesaratto L, et al. Subcellular localization of APE1/Ref-1 in human hepatocellular carcinoma: possible prognostic significance. Mol Med. (2007) 13:89–96. doi: 10.2119/2006-00084.dimaso
209. Sheng Q, Zhang Y, Wang R, Zhang J, Chen B, Wang J, et al. Prognostic significance of APE1 cytoplasmic localization in human epithelial ovarian cancer. Med Oncol. (2012) 29:1265–71. doi: 10.1007/s12032-011-9931-y
210. Hadi MZ, Coleman MA, Fidelis K, Mohrenweiser HW, Wilson DM 3rd. Functional characterization of ape1 variants identified in the human population. Nucleic Acids Res. (2000) 28:3871–9. doi: 10.1093/nar/28.20.3871
211. Hu JJ, Smith TR, Miller MS, Mohrenweiser HW, Golden A, Case LD. Amino acid substitution variants of APE1 and XRCC1 genes associated with ionizing radiation sensitivity. Carcinogenesis. (2001) 22:917–22. doi: 10.1093/carcin/22.6.917
212. Gu D, Wang M, Wang M, Zhang Z, Chen J. The DNA repair gene APE1 T1349G polymorphism and cancer risk: a meta-analysis of 27 case-control studies. Mutagenesis. (2009) 24:507–12. doi: 10.1093/mutage/gep036
213. Karahalil B, Bohr VA, Wilson DM 3rd. Impact of DNA polymorphisms in key DNA base excision repair proteins on cancer risk. Hum Exp Toxicol. (2012) 31:981–1005. doi: 10.1177/0960327112444476
214. Kuppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. (2001) 20:5580–94. doi: 10.1038/sj.onc.1204640
215. Ding J, Fishel ML, Reed AM, McAdams E, Czader MB, Cardoso AA, et al. Ref-1/APE1 as a transcriptional regulator and novel therapeutic target in pediatric T-cell leukemia. Mol Cancer Ther. (2017) 16:1401–11. doi: 10.1158/1535-7163.MCT-17-0099
216. Fishel ML, Colvin ES, Luo M, Kelley MR, Robertson KA. Inhibition of the redox function of APE1/Ref-1 in myeloid leukemia cell lines results in a hypersensitive response to retinoic acid-induced differentiation and apoptosis. Exp Hematol. (2010) 38:1178–88. doi: 10.1016/j.exphem.2010.08.011
217. Kohl V, Flach J, Naumann N, Brendel S, Kleiner H, Weiss C, et al. Antileukemic efficacy in vitro of talazoparib and APE1 inhibitor III combined with decitabine in myeloid malignancies. Cancers (Basel). (2019) 11:1493. doi: 10.3390/cancers11101493
218. Yuk J-M, Yang C-S, Shin D-M, Kim K-K, Lee SK, Song YJ, et al. A dual regulatory role of apurinic/apyrimidinic endonuclease 1/redox factor-1 in. Antioxid Redox Signal. (2009) 11:575–88. doi: 10.1089/ars.2008.2196
219. Luo H, Wang L, Bao D, Wang L, Zhao H, Lian Y, et al. Novel autoantibodies related to cell death and DNA repair pathways in systemic lupus erythematosus. Genomics Proteomics Bioinformatics. (2019) 17:248–59. doi: 10.1016/j.gpb.2018.11.004
220. Souliotis VL, Vlachogiannis NI, Pappa M, Argyriou A, Ntouros PA, Sfikakis PP. DNA damage response and oxidative stress in systemic autoimmunity. Int J Mol Sci. (2019) 21:55. doi: 10.3390/ijms21010055
221. Tsuchimoto D, Sakai Y, Sakumi K, Nishioka K, Sasaki M, Fujiwara T, et al. Human APE2 protein is mostly localized in the nuclei and to some extent in the mitochondria, while nuclear APE2 is partly associated with proliferating cell nuclear antigen. Nucleic Acids Res. (2001) 29:2349–60. doi: 10.1093/nar/29.11.2349
222. Burkovics P, Szukacsov V, Unk I, Haracska L. Human Ape2 protein has a 3'-5' exonuclease activity that acts preferentially on mismatched base pairs. Nucleic Acids Res. (2006) 34:2508–15. doi: 10.1093/nar/gkl259
223. Burkovics P, Hajdu I, Szukacsov V, Unk I, Haracska L. Role of PCNA-dependent stimulation of 3'-phosphodiesterase and 3'-5' exonuclease activities of human Ape2 in repair of oxidative DNA damage. Nucleic Acids Res. (2009) 37:4247–55. doi: 10.1093/nar/gkp357
224. Willis J, Patel Y, Lentz BL, Yan S. APE2 is required for ATR-Chk1 checkpoint activation in response to oxidative stress. Proc Natl Acad Sci USA. (2013) 110:10592–7. doi: 10.1073/pnas.1301445110
225. Menon V, Povirk LF. End-processing nucleases and phosphodiesterases: an elite supporting cast for the non-homologous end joining pathway of DNA double-strand break repair. DNA Repair. (2016) 43:57–68. doi: 10.1016/j.dnarep.2016.05.011
226. Mengwasser KE, Adeyemi RO, Leng Y, Choi MY, Clairmont C, D'Andrea AD, et al. Genetic screens reveal FEN1 and APEX2 as BRCA2 synthetic lethal targets. Mol Cell. (2019) 73:885–99.e6. doi: 10.1016/j.molcel.2018.12.008
227. Guikema JEJ, Gerstein RM, Linehan EK, Cloherty EK, Evan-Browning E, Tsuchimoto D, et al. Apurinic/apyrimidinic endonuclease 2 is necessary for normal B cell development and recovery of lymphoid progenitors after chemotherapeutic challenge. J Immunol. (2011) 186:1943–50. doi: 10.4049/jimmunol.1002422
228. Guikema JEJ, Linehan EK, Esa N, Tsuchimoto D, Nakabeppu Y, Woodland RT, et al. Apurinic/apyrimidinic endonuclease 2 regulates the expansion of germinal centers by protecting against activation-induced cytidine deaminase-independent DNA damage in B cells. J Immunol. (2014) 193:931–9. doi: 10.4049/jimmunol.1400002
229. Hossain MA, Lin Y, Yan S. Single-strand break end resection in genome integrity: mechanism and regulation by APE2. Int J Mol Sci. (2018) 19:2389. doi: 10.3390/ijms19082389
230. Jensen KA, Shi X, Yan S. Genomic alterations and abnormal expression of APE2 in multiple cancers. Sci Rep. (2020) 10:3758. doi: 10.1038/s41598-020-60656-5
231. Malfatti MC, Antoniali G, Codrich M, Burra S, Mangiapane G, Dalla E, et al. New perspectives in cancer biology from a study of canonical and non-canonical functions of base excision repair proteins with a focus on early steps. Mutagenesis. (2020) 35:129–49. doi: 10.1093/mutage/gez051
232. Kumar S, Talluri S, Pal J, Yuan X, Lu R, Nanjappa P, et al. Role of apurinic/apyrimidinic nucleases in the regulation of homologous recombination in myeloma: mechanisms and translational significance. Blood Cancer J. (2018) 8:92. doi: 10.1038/s41408-018-0129-9
233. Fortini P, Dogliotti E. Base damage and single-strand break repair: mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair. (2007) 6:398–409. doi: 10.1016/j.dnarep.2006.10.008
234. Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. (1997) 16:3341–8. doi: 10.1093/emboj/16.11.3341
235. Prasad R, Beard WA, Strauss PR, Wilson SH. Human DNA polymerase beta deoxyribose phosphate lyase. Substrate specificity and catalytic mechanism. J Biol Chem. (1998) 273:15263–70. doi: 10.1074/jbc.273.24.15263
236. Sobol RW, Prasad R, Evenski A, Baker A, Yang XP, Horton JK, et al. The lyase activity of the DNA repair protein beta-polymerase protects from. Nature. (2000) 405:807–10. doi: 10.1038/35015598
237. Allinson SL, Dianova II, Dianov GL. DNA polymerase beta is the major dRP lyase involved in repair of oxidative base lesions in DNA by mammalian cell extracts. EMBO J. (2001) 20:6919–26. doi: 10.1093/emboj/20.23.6919
238. Osheroff WP, Jung HK, Beard WA, Wilson SH, Kunkel TA. The fidelity of DNA polymerase beta during distributive and processive DNA synthesis. J Biol Chem. (1999) 274:3642–50. doi: 10.1074/jbc.274.6.3642
239. Caglayan M. The ligation of pol beta mismatch insertion products governs the formation of promutagenic base excision DNA repair intermediates. Nucleic Acids Res. (2020) 48:3708–21. doi: 10.1093/nar/gkaa151
240. Braithwaite EK, Prasad R, Shock DD, Hou EW, Beard WA, Wilson SH. DNA polymerase lambda mediates a back-up base excision repair activity in extracts of mouse embryonic fibroblasts. J Biol Chem. (2005) 280:18469–75. doi: 10.1074/jbc.M411864200
241. Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, et al. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature. (1996) 379:183–6. doi: 10.1038/379183a0
242. Esposito G, Texido G, Betz UA, Gu H, Muller W, Klein U, et al. Mice reconstituted with DNA polymerase beta-deficient fetal liver cells are able to mount a T cell-dependent immune response and mutate their Ig genes normally. Proc Natl Acad Sci USA. (2000) 97:1166–71. doi: 10.1073/pnas.97.3.1166
243. Wu X, Stavnezer J. DNA polymerase beta is able to repair breaks in switch regions and plays an inhibitory role during immunoglobulin class switch recombination. J Exp Med. (2007) 204:1677–89. doi: 10.1084/jem.20070756
244. Schrader CE, Linehan EK, Ucher AJ, Bertocci B, Stavnezer J. DNA polymerases beta and lambda do not directly affect Ig variable region somatic hypermutation although their absence reduces the frequency of mutations. DNA Repair. (2013) 12:1087–93. doi: 10.1016/j.dnarep.2013.09.002
245. Martin A, Bardwell PD, Woo CJ, Fan M, Shulman MJ, Scharff MD. Activation-induced cytidine deaminase turns on somatic hypermutation in hybridomas. Nature. (2002) 415:802–6. doi: 10.1038/nature714
246. Matthews AJ, Husain S, Chaudhuri J. Binding of AID to DNA does not correlate with mutator activity. J Immunol. (2014) 193:252–7. doi: 10.4049/jimmunol.1400433
247. Xiao Z, Ray M, Jiang C, Clark AB, Rogozin IB, Diaz M. Known components of the immunoglobulin A:T mutational machinery are intact in burkitt lymphoma cell lines with G:C bias. Mol Immunol. (2007) 44:2659–66. doi: 10.1016/j.molimm.2006.12.006
248. Yoshikawa K, Okazaki I-M, Eto T, Kinoshita K, Muramatsu M, Nagaoka H, et al. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science. (2002) 296:2033–6. doi: 10.1126/science.1071556
249. Cho SH, Raybuck AL, Stengel K, Wei M, Beck TC, Volanakis E, et al. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature. (2016) 537:234–8. doi: 10.1038/nature19334
250. Abbott RK, Thayer M, Labuda J, Silva M, Philbrook P, Cain DW, et al. Germinal center hypoxia potentiates immunoglobulin class switch recombination. J Immunol. (2016) 197:4014–20. doi: 10.4049/jimmunol.1601401
251. Sweasy JB, Lang T, Starcevic D, Sun K-W, Lai C-C, Dimaio D, et al. Expression of DNA polymerase {beta} cancer-associated variants in mouse cells results in cellular transformation. Proc Natl Acad Sci USA. (2005) 102:14350–5. doi: 10.1073/pnas.0505166102
252. Alnajjar KS, Garcia-Barboza B, Negahbani A, Nakhjiri M, Kashemirov B, McKenna C, et al. A Change in the rate-determining step of polymerization by the K289M DNA polymerase beta cancer-associated variant. Biochemistry. (2017) 56:2096–105. doi: 10.1021/acs.biochem.6b01230
253. Nemec AA, Abriola L, Merkel JS, de Stanchina E, DeVeaux M, Zelterman D, et al. DNA Polymerase beta germline variant confers cellular response to cisplatin therapy. Mol Cancer Res. (2017) 15:269–80. doi: 10.1158/1541-7786.MCR-16-0227-T
254. Nemec AA, Murphy DL, Donigan KA, Sweasy JB. The S229L colon tumor-associated variant of DNA polymerase beta induces cellular transformation as a result of decreased polymerization efficiency. J Biol Chem. (2014) 289:13708–16. doi: 10.1074/jbc.M114.550400
255. Donigan KA, Sun K, Nemec AA, Murphy DL, Cong X, Northrup V, et al. Human POLB gene is mutated in high percentage of colorectal tumors. J Biol Chem. (2012) 287:23830–9. doi: 10.1074/jbc.M111.324947
256. Starcevic D, Dalal S, Sweasy JB. Is there a link between DNA polymerase beta and cancer? Cell Cycle. (2004) 3:998–1001.
257. Cabelof DC, Ikeno Y, Nyska A, Busuttil RA, Anyangwe N, Vijg J, et al. Haploinsufficiency in DNA polymerase beta increases cancer risk with age and alters mortality rate. Cancer Res. (2006) 66:7460–5. doi: 10.1158/0008-5472.CAN-06-1177
258. Sellick GS, Wade R, Richards S, Oscier DG, Catovsky D, Houlston RS. Scan of 977 nonsynonymous SNPs in CLL4 trial patients for the identification of genetic variants influencing prognosis. Blood. (2008) 111:1625–33. doi: 10.1182/blood-2007-08-110130
259. Yamtich J, Nemec AA, Keh A, Sweasy JB. A germline polymorphism of DNA polymerase beta induces genomic instability and cellular transformation. PLoS Genet. (2012) 8:e1003052. doi: 10.1371/journal.pgen.1003052
260. Sheng Y-J, Gao J-P, Li J, Han J-W, Xu Q, Hu W-L, et al. Follow-up study identifies two novel susceptibility loci PRKCB and 8p11.21 for systemic lupus erythematosus. Rheumatology. (2011) 50:682–8. doi: 10.1093/rheumatology/keq313
261. Li X, Li J, Harrington J, Lieber MR, Burgers PM. Lagging strand DNA synthesis at the eukaryotic replication fork involves binding and stimulation of FEN-1 by proliferating cell nuclear antigen. J Biol Chem. (1995) 270:22109–12. doi: 10.1074/jbc.270.38.22109
262. Liang L, Deng L, Chen Y, Li GC, Shao C, Tischfield JA. Modulation of DNA end joining by nuclear proteins. J Biol Chem. (2005) 280:31442–9. doi: 10.1074/jbc.M503776200
263. Kikuchi K, Taniguchi Y, Hatanaka A, Sonoda E, Hochegger H, Adachi N, et al. Fen-1 facilitates homologous recombination by removing divergent sequences at DNA break ends. Mol Cell Biol. (2005) 25:6948–55. doi: 10.1128/MCB.25.16.6948-6955.2005
264. Illuzzi JL, Wilson DM 3rd. Base excision repair: contribution to tumorigenesis and target in anticancer treatment paradigms. Curr Med Chem. (2012) 19:3922–36. doi: 10.2174/092986712802002581
265. Figueroa JD, Malats N, Real FX, Silverman D, Kogevinas M, Chanock S, et al. Genetic variation in the base excision repair pathway and bladder cancer risk. Hum Genet. (2007) 121:233–42. doi: 10.1007/s00439-006-0294-y
266. Yang M, Guo H, Wu C, He Y, Yu D, Zhou L, et al. Functional FEN1 polymorphisms are associated with DNA damage levels and lung cancer risk. Hum Mutat. (2009) 30:1320–8. doi: 10.1002/humu.21060
267. Chang JS, Wrensch MR, Hansen HM, Sison JD, Aldrich MC, Quesenberry CPJ, et al. Base excision repair genes and risk of lung cancer among San Francisco Bay Area latinos and African-Americans. Carcinogenesis. (2009) 30:78–87. doi: 10.1093/carcin/bgn261
268. Larsen E, Kleppa L, Meza TJ, Meza-Zepeda LA, Rada C, Castellanos CG, et al. Early-onset lymphoma and extensive embryonic apoptosis in two domain-specific Fen1 mice mutants. Cancer Res. (2008) 68:4571–9. doi: 10.1158/0008-5472.CAN-08-0168
269. Kumar V. A STING to inflammation and autoimmunity. J Leukoc Biol. (2019) 106:171–85. doi: 10.1002/JLB.4MIR1018-397RR
270. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. (2015) 15:760–70. doi: 10.1038/nri3921
271. Wang J, Cao P, Qi Y-Y, Chen X-P, Ma L, Deng R-R, et al. The relationship between cell apoptosis dysfunction and FEN1 E160D mutation in lupus nephritis patients. Autoimmunity. (2017) 50:476–80. doi: 10.1080/08916934.2017.1402302
272. Caldecott KW, McKeown CK, Tucker JD, Ljungquist S, Thompson LH. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol Cell Biol. (1994) 14:68–76. doi: 10.1128/mcb.14.1.68
273. Pascucci B, Stucki M, Jonsson ZO, Dogliotti E, Hubscher U. Long patch base excision repair with purified human proteins. DNA ligase I as patch size mediator for DNA polymerases delta and epsilon. J Biol Chem. (1999) 274:33696–702. doi: 10.1074/jbc.274.47.33696
274. Levin DS, Bai W, Yao N, O'Donnell M, Tomkinson AE. An interaction between DNA ligase I and proliferating cell nuclear antigen: implications for okazaki fragment synthesis and joining. Proc Natl Acad Sci USA. (1997) 94:12863–8. doi: 10.1073/pnas.94.24.12863
275. Liang L, Deng L, Nguyen SC, Zhao X, Maulion CD, Shao C, et al. Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res. (2008) 36:3297–310. doi: 10.1093/nar/gkn184
276. Simsek D, Furda A, Gao Y, Artus J, Brunet E, Hadjantonakis A-K, et al. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature. (2011) 471:245–8. doi: 10.1038/nature09794
277. Gao Y, Katyal S, Lee Y, Zhao J, Rehg JE, Russell HR, et al. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature. (2011) 471:240–4. doi: 10.1038/nature09773
278. Han L, Masani S, Hsieh C-L, Yu K. DNA ligase I is not essential for mammalian cell viability. Cell Rep. (2014) 7:316–20. doi: 10.1016/j.celrep.2014.03.024
279. Arakawa H, Bednar T, Wang M, Paul K, Mladenov E, Bencsik-Theilen AA, et al. Functional redundancy between DNA ligases I and III in DNA replication in vertebrate cells. Nucleic Acids Res. (2012) 40:2599–610. doi: 10.1093/nar/gkr1024
280. Grawunder U, Zimmer D, Fugmann S, Schwarz K, Lieber MR. DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol Cell. (1998) 2:477–84. doi: 10.1016/s1097-2765(00)80147-1
281. Maffucci P, Chavez J, Jurkiw TJ, O'Brien PJ, Abbott JK, Reynolds PR, et al. Biallelic mutations in DNA ligase 1 underlie a spectrum of immune deficiencies. J Clin Invest. (2018) 128:5489–504. doi: 10.1172/JCI99629
282. Masani S, Han L, Meek K, Yu K. Redundant function of DNA ligase 1 and 3 in alternative end-joining during immunoglobulin class switch recombination. Proc Natl Acad Sci USA. (2016) 113:1261–6. doi: 10.1073/pnas.1521630113
283. Boboila C, Oksenych V, Gostissa M, Wang JH, Zha S, Zhang Y, et al. Robust chromosomal DNA repair via alternative end-joining in the absence of X-ray repair cross-complementing protein 1 (XRCC1). Proc Natl Acad Sci USA. (2012) 109:2473–8. doi: 10.1073/pnas.1121470109
284. Hogerkorp C-M, Borrebaeck CAK. The human CD77- B cell population represents a heterogeneous subset of cells comprising centroblasts, centrocytes, and plasmablasts, prompting phenotypical revision. J Immunol. (2006) 177:4341–9. doi: 10.4049/jimmunol.177.7.4341
285. Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, et al. Reconstitution of 5'-directed human mismatch repair in a purified system. Cell. (2005) 122:693–705. doi: 10.1016/j.cell.2005.06.027
286. Saribasak H, Maul RW, Cao Z, McClure RL, Yang W, McNeill DR, et al. XRCC1 suppresses somatic hypermutation and promotes alternative nonhomologous end joining in Igh genes. J Exp Med. (2011) 208:2209–16. doi: 10.1084/jem.20111135
287. Tomkinson AE, Naila T, Khattri Bhandari S. Altered DNA ligase activity in human disease. Mutagenesis. (2020) 35:51–60. doi: 10.1093/mutage/gez026
288. Simsek D, Brunet E, Wong SY-W, Katyal S, Gao Y, McKinnon PJ, et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet. (2011) 7:e1002080. doi: 10.1371/journal.pgen.1002080
289. Lopez Castel A, Tomkinson AE, Pearson CE. CTG/CAG repeat instability is modulated by the levels of human DNA ligase I and its interaction with proliferating cell nuclear antigen: a distinction between replication and slipped-DNA repair. J Biol Chem. (2009) 284:26631–45. doi: 10.1074/jbc.M109.034405
290. Harrison C, Ketchen A-M, Redhead NJ, O'Sullivan MJ, Melton DW. Replication failure, genome instability, and increased cancer susceptibility in mice with a point mutation in the DNA ligase I gene. Cancer Res. (2002) 62:4065–74.
291. Saquib M, Ansari MI, Johnson CR, Khatoon S, Kamil Hussain M, Coop A. Recent advances in the targeting of human DNA ligase I as a potential new strategy for cancer treatment. Eur J Med Chem. (2019) 182:111657. doi: 10.1016/j.ejmech.2019.111657
292. Singh DK, Krishna S, Chandra S, Shameem M, Deshmukh AL, Banerjee D. Human DNA ligases: a comprehensive new look for cancer therapy. Med Res Rev. (2014) 34:567–95. doi: 10.1002/med.21298
293. Muvarak N, Kelley S, Robert C, Baer MR, Perrotti D, Gambacorti-Passerini C, et al. c-MYC Generates repair errors via increased transcription of alternative-NHEJ factors, LIG3 and PARP1, in tyrosine kinase-activated leukemias. Mol Cancer Res. (2015) 13:699–712. doi: 10.1158/1541-7786.MCR-14-0422
294. Caracciolo D, Di Martino MT, Amodio N, Morelli E, Montesano M, Botta C, et al. miR-22 suppresses DNA ligase III addiction in multiple myeloma. Leukemia. (2019) 33:487–98. doi: 10.1038/s41375-018-0238-2
295. Gassner FJ, Schubert M, Rebhandl S, Spandl K, Zaborsky N, Catakovic K, et al. Imprecision and DNA break repair biased towards incompatible end joining in leukemia. Mol Cancer Res. (2018) 16:428–38. doi: 10.1158/1541-7786.MCR-17-0373
296. Zintzaras E, Voulgarelis M, Moutsopoulos HM. The risk of lymphoma development in autoimmune diseases: a meta-analysis. Arch Intern Med. (2005) 165:2337–44. doi: 10.1001/archinte.165.20.2337
297. Anderson LA, Gadalla S, Morton LM, Landgren O, Pfeiffer R, Warren JL, et al. Population-based study of autoimmune conditions and the risk of specific lymphoid malignancies. Int J Cancer. (2009) 125:398–405. doi: 10.1002/ijc.24287
298. Morton LM, Slager SL, Cerhan JR, Wang SS, Vajdic CM, Skibola CF, et al. Etiologic heterogeneity among non-Hodgkin lymphoma subtypes: the interlymph non-hodgkin lymphoma subtypes project. J Natl Cancer Inst Monogr. (2014) 2014:130–44. doi: 10.1093/jncimonographs/lgu013
299. Bende RJ, Janssen J, Beentjes A, Wormhoudt TAM, Wagner K, Haacke EA, et al. Salivary gland MALT lymphomas of Sjgren's syndrome patients in majority express rheumatoid factors affinity-selected for IgG. Arthritis Rheumatol. (2020). doi: 10.1002/art.41263. [Epub ahead of print].
300. Bende RJ, Janssen J, Wormhoudt TAM, Wagner K, Guikema JEJ, van Noesel CJM. Identification of a novel stereotypic IGHV4-59/IGHJ5-encoded B-cell receptor subset expressed by various B-cell lymphomas with high affinity rheumatoid factor activity. Haematologica. (2016) 101:e200–3. doi: 10.3324/haematol.2015.139626
301. Hoogeboom R, Wormhoudt TA, Schipperus MR, Langerak AW, Dunn-Walters DK, Guikema JEJ, et al. A novel chronic lymphocytic leukemia subset expressing mutated IGHV3-7-encoded rheumatoid factor B-cell receptors that are functionally proficient. Leukemia. (2013) 27:738–40. doi: 10.1038/leu.2012.238
302. Stamatopoulos K, Belessi C, Moreno C, Boudjograh M, Guida G, Smilevska T, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: pathogenetic implications and clinical correlations. Blood. (2007) 109:259–70. doi: 10.1182/blood-2006-03-012948
303. Gemenetzi K, Agathangelidis A, Zaragoza-Infante L, Sofou E, Papaioannou M, Chatzidimitriou A, et al. B cell receptor immunogenetics in B cell lymphomas: immunoglobulin genes as key to ontogeny and clinical decision making. Front Oncol. (2020) 10:67. doi: 10.3389/fonc.2020.00067
Keywords: base excision repair (BER), germinal center (GC), lymphoma, autoimmune diseases, class switch recombination (CSR), somatic hypermutation (SHM)
Citation: Stratigopoulou M, van Dam TP and Guikema JEJ (2020) Base Excision Repair in the Immune System: Small DNA Lesions With Big Consequences. Front. Immunol. 11:1084. doi: 10.3389/fimmu.2020.01084
Received: 27 March 2020; Accepted: 05 May 2020;
Published: 29 May 2020.
Edited by:
Amy L. Kenter, University of Illinois at Chicago, United StatesReviewed by:
Cristina Rada, University of Cambridge, United KingdomJavier Marcelo Di Noia, Institute of Clinical Research De Montreal (IRCM), Canada
Copyright © 2020 Stratigopoulou, van Dam and Guikema. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeroen E. J. Guikema, ai5lLmd1aWtlbWFAYW1zdGVyZGFtdW1jLm5s
†These authors have contributed equally to this work