 Xiangru Li
Xiangru Li Zhenhua Hao
Zhenhua Hao Xiaorong Liu
Xiaorong Liu Wei Li
Wei Li- 1Beijing Key Laboratory for Genetics of Birth Defects, Beijing Pediatric Research Institute, Beijing Children's Hospital, Capital Medical University, Beijing, China
- 2MOE Key Laboratory of Major Diseases in Children, Beijing, China
- 3Genetics and Birth Defects Control Center, National Center for Children's Health, Beijing, China
- 4Department of Nephrology, Beijing Children's Hospital, Capital Medical University, National Center for Children's Health, Beijing, China
Alternative complement pathway (AP) plays an important role in the development of sepsis, which is life threatening. Deficiency of factor H-related protein 1 (FHR-1), which is a regulator of AP, has been considered as a susceptible factor for atypical hemolytic uremic syndrome (aHUS) and other types of nephropathy when an inducer such as infection exists. However, the underlying mechanism of the disease development is largely unknown. There is no report on CFHR1 gene knockout in any animal infection model and its function in vivo is still unclear. Here, a Cfhr1 knockout mouse was generated for investigating AP in sepsis and sepsis-induced acute kidney injury (AKI). We found that murine FHR-1 homolog (FHR-E) deficiency enhanced lipopolysaccharide (LPS)-induced AP activation both in vitro and in vivo and that Cfhr1 knockout mice exhibited more severe sepsis and AKI in response to LPS challenge. These results indicated that FHR-E deficiency promoted LPS-induced sepsis and AKI through AP over-activation, providing a mouse model for studying AP regulation and sepsis. This study revealed the function of FHR-E in vivo, which may further provide hints to the pathogenesis of FHR-1 deficiency-related diseases by enhancing LPS-induced AP activation.
Introduction
Sepsis is a critical health condition with high mortality rate (1, 2) and acute kidney injury (AKI) is a common severe complication of sepsis (3). Gram-negative bacteria infection is a predominant cause of severe infection-triggered sepsis (4). Sepsis is an intricate process during which complement system is activated and proved to be double-edged with benefits and harms (5). The complement system is crucial in immune surveillance (6) and has extensive cross-talk with coagulation system and inflammation for homeostasis (7). It is triggered through three pathways among which the alternative complement pathway (AP) activation is responsible for more than 80% of terminal complement activation (8, 9). AP plays an important role in endotoxin clearance during the process of sepsis (10). Lack of AP activation in individuals predisposes to infection (11–13). However, it is also life-threatening when complement is excessively activated, and inhibition of its over-activation prevents organ injuries (14). Therefore, uncovering the precise regulatory mechanism of AP during sepsis may shed light on sepsis intervention. However, the mechanism of AP regulation during sepsis remains elusive.
Factor H (FH) and its related proteins (FHRs) are regulators of AP (15) and responsible for determining complement activating surfaces (16). FH, the major regulatory factor of AP (17), has been widely investigated, while functions of FHRs are unclear or controversial. FH and FHRs consist of different number of complement control protein modules (CCPs). All FHRs contain homologous CCPs to CCP19, 20 of FH which are responsible for ligand recognition (18). In general, FH functions as a cofactor of Factor I (FI) to cleave C3b and accelerate the decay of C3 convertase (19, 20), while FHRs act as the competitor of FH (16). However, different FHRs may have different functions. FHR-2 inhibits C3 convertase (21). FHR-3 displays the cofactor activity for FI (22). FHR-5 inhibits C3 convertase in fluid phase and displays cofactor activity for FI (23). Nevertheless, it remains elusive for the physiological functions of FHRs.
Mutations in FH and FHRs are associated with various diseases (24). CFHR3-CFHR1 deletion increased the risk of atypical hemolytic uremic syndrome (aHUS) and systemic lupus erythematosus (SLE) (25–27). CFHR3-CFHR1 deletion was proved to protect against IgA nephropathy (IgAN) (28) and age-related macular degeneration (AMD) (29). The precise mechanism of this contradictory effect is unclear. In vitro studies of factor H-related protein 1 (FHR-1) have shown that FHR-1 interferes with the regulation of FH by competing with FH and inhibits the activity of C5 convertase and the formation of terminal complement complex (TCC) (30). No obvious C3 and C5 regulatory activity at physiological concentration has been found in FHR-1 (16), albeit it is the most abundant FHR protein (18, 31). Healthy individuals with CFHR3-CFHR1 deletion showed higher frequency of patrolling monocytes, plasmacytoid dendritic cells (DCs), and lower frequency of classical monocytes, myeloid DCs. Monocytes with CFHR3-CFHR1 deletion secreted higher level of IL-1β in response to LPS challenge (32). Necrotic cells bound to FHR-1 promotes the secretion of TNF-α, IL-1β, IL-18, and IL-6 by monocytes (33). These reports suggest multiple effects of CFHR1 deletion and highlight its complexity. The existence of relatively high frequency of healthy individuals with CFHR3-CFHR1 deletion (34) indicates that other triggers are essential to amplify the effect of FHR-1 deficiency, such as infections (35, 36). However, there is no report on FHR-1 deficiency in any animal infection model.
Three genes, Fhrb, Fhrc, and Fhre (alias Cfhr1), and two pseudogenes, Fhra and Fhrd, were identified in mouse (37, 38). Recombinant FHR-A, FHR-B, and FHR-C were reported to interact with human C3d, suggesting that murine FHRs function as homologs of human FHRs (39). None of FHR-A, FHR-B, and FHR-C contain the critical dimerization domain that exists in human FHR-1, FHR-2, and FHR-5 (39), while FHR-E is predicted to contain it (37). Thus, FHR-E is more likely the murine homolog of human FHR-1 or FHR-2 (40). Nomenclature of human and mouse FHRs differs and there exist discrepancies in literature and existing database (41). Inferred from the homology analysis (Figure 1), we use mouse FHR-1 homolog FHR-E (NP_056595) for the protein encoded by Cfhr1 (RefSeq# at NCBI: NM_015780, and a locus at MGI database: 2138169).
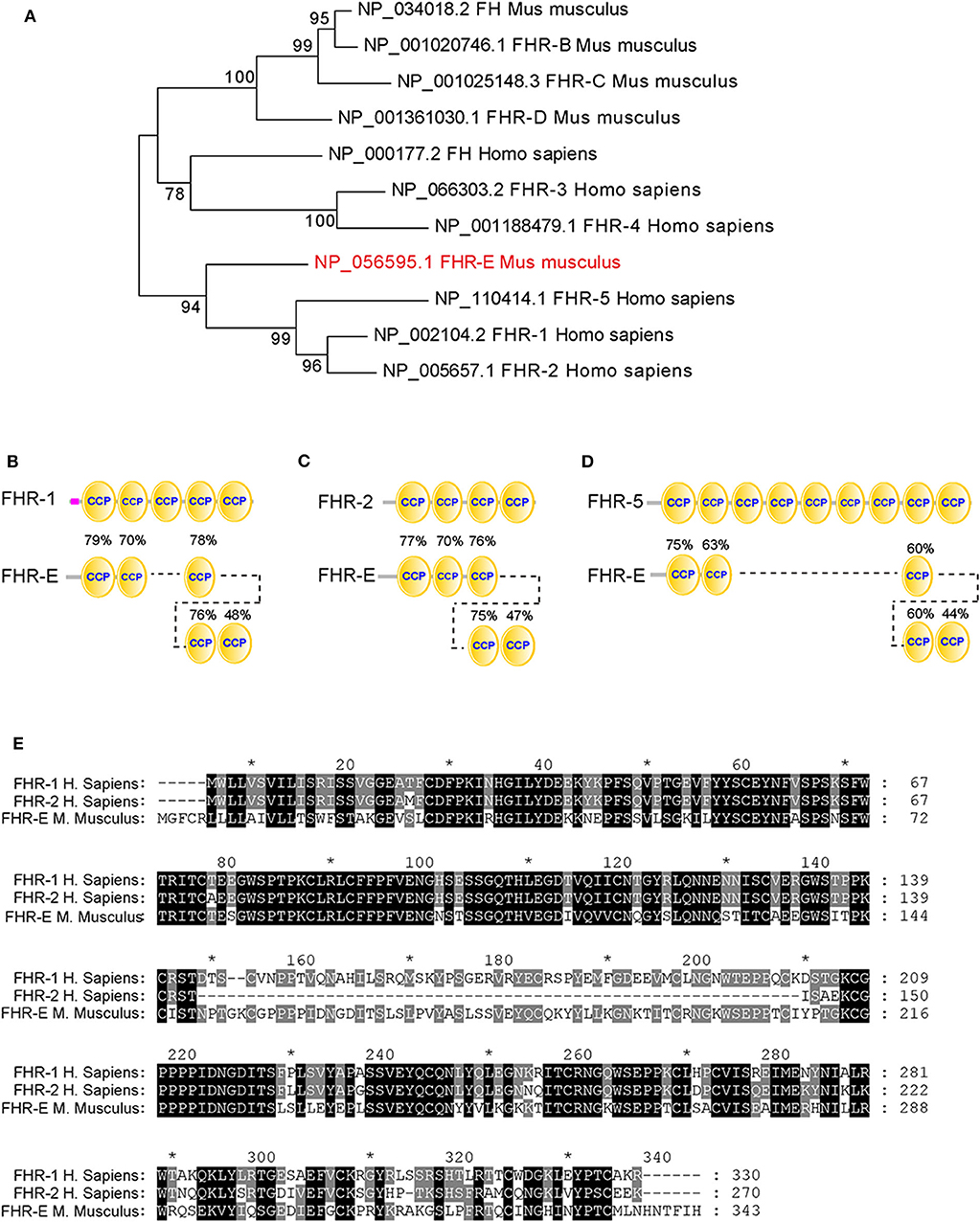
Figure 1. Homology analysis of different FHRs between human and mouse. (A) The evolutionary tree of different FHRs in human and mouse. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) were shown next to the branches. (B–D) The similarity analyses between FHR-E and human FHR-1, FHR-2, and FHR-5. The percentages marked on these figures denote the similarities between the CCP domains of FHR-E and human FHR-1, FHR-2, and FHR-5. (E) The sequence alignment of FHR-E and human FHR-1 and FHR-2. * Indicates the number of amino acid. It was marked every twenty amino acids.
Homology analysis has shown the difference between human and murine FHRs (41, 42), suggesting that murine FHRs may function differently from their human counterparts. FHR-B, FHR-C, and FHR-E are functional FHRs in mouse. FHR-B and FHR-C were detected in mouse plasma with anti-FH antibody (38). Mouse FHR-B interacts with C3b and pentraxin 3 to enhance complement activation and necrotic cells promoted this activation (43). Fhrc mRNA was found completely absent in the liver of three lupus-prone mouse strains and one diabetic-prone mouse strain (44). FHR-C may function on specific surfaces (39). It is unknown whether FHR-E exists in murine plasma and how it functions in AP. Its human homolog, FHR-1, was reported to function as a competitor of FH (22, 30, 45) and its deletion was associated with various autoimmune diseases. However, its function in vivo and whether it has a regulatory role alone in the activation of AP remain unclear.
In this study, Cfhr1 was deleted on C57BL/6 mouse to study the function of FHR-E on AP and the effect of FHR-E deficiency on LPS-induced sepsis. We found that FHR-E deficiency increased the mortality rate of LPS-induced sepsis and potentiated kidney injury through enhancing AP activation. Our data demonstrated a protective role of FHR-E during LPS-induced sepsis in vivo and highlighted its importance in AP regulation.
Materials and Methods
Mice
Cfhr1 was deleted on C57BL/6 mouse by using the CRISPR/Cas9 system developed by Biocytogen Co. (Beijing, China). The heterozygous mouse was backcrossed with wild-type C57BL/6 for more than six generations. All the mice were bred in the animal facility of the Institute of Genetics and Developmental Biology (IGDB), Chinese Academy of Science. All procedures were approved by the Institutional Animal Care and Use Committee of IGDB. The age-matched wild-type (WT) or heterozygous littermates were used as controls. All experiments were conducted on the offspring of mice backcrossed more than six generations.
Antibodies
The polyclonal antibody anti-FHR-E was generated in New Zealand white rabbit. The DNA fragment of the last three CCP domains (amino acids 146 to 343) of FHR-E was inserted into the expression vector pGEX-4T-1 and expressed in E. coli BL21 host cells. The recombinant protein was expressed as insoluble protein and separated by SDS-PAGE. The separated recombinant protein was used as an antigen to immunize the New Zealand white rabbit every other week. The rabbit was sacrificed 1 week after the fourth immunization and the sera was used as the anti-FHR-E polyclonal antibody. The rat anti-C3 (ab11862), rat anti-C5 (ab194637), sheep anti-FH (ab8842), and rabbit anti-fibrinogen (ab27913) were obtained from Abcam (Cambridge, UK). Purified rat anti-mouse C5a (558027) and biotin rat anti-mouse C5a (558028) were obtained from BD Pharmingen (California, USA).
DNA Extraction and Genotyping
Mice were marked by cutting different toes and the toes were lysed by 250 μl of lysis buffer (Tris–HCl pH 8.0 100 mM, EDTA pH 8.0 5 mM, NaCl 200 mM, 0.2% SDS, and 0.1 mg/ml Proteinase K) at 55°C for 3 h. After transient centrifugation, 150 μl of 5 M NaCl was added and mixed thoroughly. The mixture was centrifuged at 12,000 rpm for 15 min and the supernatant was collected by adding double volume of ethanol and mixed thoroughly. The mixture was centrifuged at 12,000 rpm for 15 min and then the supernatant was discarded. The pellet was washed with 70% ethanol. After the liquid was evaporated, 100 μl of ddH2O was added to dissolve the precipitated DNA. One forward primer located on the upstream of the deleted sequence, one reverse primer located on the deleted region, and the other reverse primer located on the downstream of the deleted sequence were used for genotyping PCR assays. The primer's sequences are 5′-CAGTAAGACTGCAAGAGACATATG-3′, 5′-CTAAGAGCAACAGGCGACAG-3′, and 5′-CATTTTAAAAGAAAAATAAGCCAGCCA-3′, respectively. The amplified products are depicted in Figure 2A.
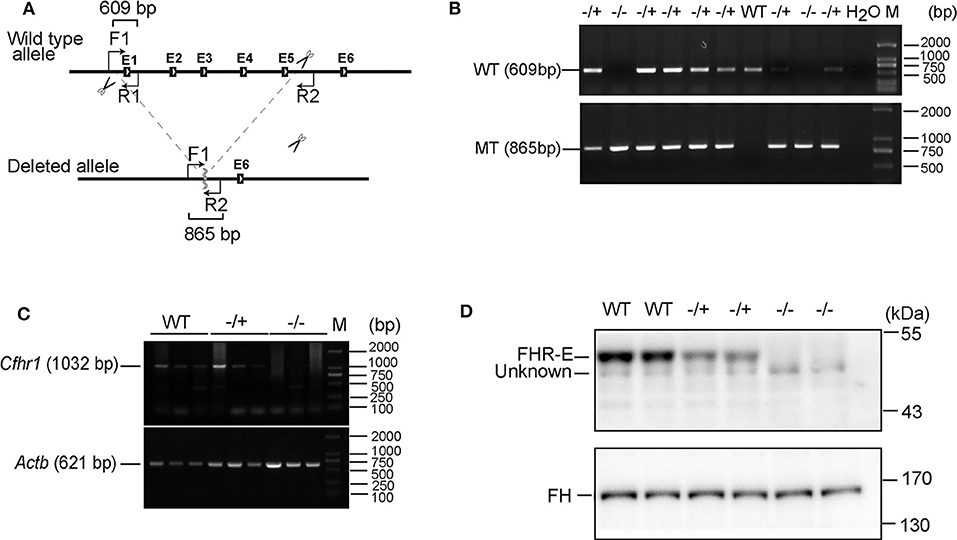
Figure 2. Verification of the absence of FHR-E in Cfhr1-KO mice. (A) The scheme of Cfhr1 knockout strategy. F1, R1, and R2 indicate the primers used for genotyping. The expected bands amplified by two pairs of primers were 609 and 865 bp, respectively. (B) Verification of Cfhr1 deletion from genome level. The upper bands were amplified with primers F1 and R1. These bands can only be amplified in wild-type and heterozygous mice and cannot be amplified in homozygous mice because of the absence of the sequence of primer R1. The lower bands were amplified with primers F1 and R2. These bands can only be amplified in heterozygous and homozygous mice. The sequence between primers F1 and R2 is too long to be amplified in wild-type mice. (C) Verification of Cfhr1 deletion from RNA level. Hepatic RNA of mice of different genotypes was extracted and reversely transcribed. Primers spanning the open reading frame (ORF, 1,032 bp, NM_015780) of Cfhr1 were used for RT-PCR. Actb was used as a semi-quantitative control. (D) Verification of FHR-E deficiency at the protein level. Plasma proteins of mice with different genotypes were separated by 8% SDS-PAGE gel. Proteins were transferred to the membrane and analyzed by Western blotting using anti-serum to mouse FHR-E, which was generated in rabbit immunized with a recombinant peptide of CCP 3–5 of FHR-E. An about 50 kDa specific FHR-E band was detected in WT and heterozygous mice, but not in homozygous mice. “Unknown” indicates an unknown protein recognized by anti-FHR-E antibody. Western blotting of FH was regarded as an internal control.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Mouse hepatic RNA was extracted using RNeasy Mini Kit (Qiagen, Germantown, Germany) and reversely transcribed with Bio-Rad transcript cDNA synthesis kit (Bio-Rad, California, USA). The cDNA was used as templates to amplify Cfhr1 and Actb (gene for β-actin). Primers of Cfhr1 were 5′-ATGGGGTTCTGTCGCCTGTTGC-3′ and 5′-TCAATGAATAAACGTATTGTGA-3′. Primers of Actb were 5′-TGATGGTGGGAATGGGTCAGA-3′ and 5′-CCGCTCGTTGCCAATAGTGAT-3′.
Immunoblotting
Equal volume of serum or plasma of mice was diluted 1:10 with PBS (Hyclone, Utah, USA). Equal volumes of diluted samples were separated with 8% SDS-PAGE gel and transferred onto polyvinylidene difluoride (PVDF) membrane (Millipore, Massachusetts, USA). The membrane was blocked with 5% defatted milk (BD, California, USA) for 1 h at room temperature and immunoblotted with specific antibody overnight at 4°C followed by incubation with HRP-conjugated secondary antibody (ZSGB-Bio, Beijing, China) for 1 h at room temperature. The membrane was visualized by ECL (Thermo Fisher Scientific, Massachusetts, USA or GE Healthcare, USA) and exposed with chemiluminescence apparatus (Beijing Sage Creation, Beijing, China). Protein bands' gray values were measured by NIH Image J.
LPS Treatment in vitro
Serum was diluted by AP buffer (2.5 mM barbital, 1.5 mM sodium barbital, 144 mM NaCl, 7 mM MgCl2, and 10 mM EGTA, pH 7.2–7.4). Ten-fold diluted serum was incubated with 0.125 mg/ml LPS O111:B4 (Sigma-Aldrich, Missouri, USA) or equal volume of PBS as control at 37°C for 30 min. The reactions were stopped by 50 mM EDTA. The samples were analyzed with 8% SDS-PAGE under reducing condition and protein bands' gray values were measured by NIH Image J.
Serum AP Complement Activity Assays
LPS-stimulated AP activity was performed as previously published by ELISA (46). Briefly, LPS (2 μg/well) was coated on plates. Ten-fold diluted serum with AP buffer was incubated on plates at 37°C for 1 h. Serum diluted with EDTA buffer (2.5 mM barbital, 1.5 mM sodium barbital, 144 mM NaCl, and 50 mM EDTA, pH 7.2–7.4) was used as a negative control. After washing, the plates were incubated with rat anti-mouse C3 antibody and followed by incubation with HRP-conjugated anti-rat IgG. Tetramethylbenzidine (TMB, Solarbio, Beijing, China) was used as substrate and 1 M H2SO4 was used as a stop solution. The absorbance was measured with Multiskan GO microplate spectrophotometer (Thermo Scientific, Massachusetts, USA).
For the detection of LPS-induced C5a production, 10-fold diluted serum was incubated with 0.125 mg/ml LPS or equal volume of PBS as control at 37°C for 2 h and the reactions were stopped by 50 mM EDTA. The reaction mixtures were added to the plate, which was coated with purified rat anti-mouse C5a antibody (1 μg/ml) and incubated at 37°C for 2 h. The mixtures were discarded and biotin rat anti-mouse C5a (0.5 μg/ml) was added into the according plates as detection antibody. After washing, streptavidin HRP (1:1,000) (BD Pharmingen, California, USA) was added and incubated at 37°C for 1 h. TMB (Solarbio, Beijing, China) was used as substrate and 1 M H2SO4 was used as a stop solution. The absorbance was measured with Multiskan GO microplate spectrophotometer (Thermo Scientific, Massachusetts, USA).
LPS in vivo Challenge
Six- to eight-week-old male mice were injected intraperitoneally (i.p.) with 10 mg/kg LPS dissolved in PBS and the control group mice were injected i.p. with equal volume of PBS. The EDTA anticoagulant blood was extracted from the tail tip at different time points and the plasma was collected to analyze with Western blotting. Mice were sacrificed at different time points and the EDTA anticoagulant blood was harvested from heart puncture. Partial whole blood was used for complete blood counting using TEK-II Mini full-automatic animal blood analyzer (Tecom Science, Jiangxi, China). The plasma was collected for C3a, C5a, IL-1β and TNF-α concentration detection using ELISA kit as described below.
Plasma C3a, C5a, IL-1β, and TNF-α Measurements
LPS challenged mice were anesthetized and EDTA anticoagulant blood was collected by heart puncture. The whole blood was centrifuged at 4,000 rpm 4°C for 5 min and the supernatant was collected. Plasma levels of C3a and C5a were determined using ELISA kits (Mybiosource, California, USA) according to the manufacturer's instructions. The dilution factor of C3a and C5a was 1:4. Plasma IL-1β and TNF-α were measured with ELISA kits (R&D Systems, Minnesota, USA) according to the manufacturer's instructions.
Survival Curve
About 8-week-old mice were injected i.p. with 10 mg/kg LPS. Sixteen hours later, these mice were observed every half hour and the death of mice was recorded. The data were analyzed by GraphPad Prism version 5.0 using the Log-rank (Mantel–Cox) test.
Histological Analyses
Mice were sacrificed at 12 h after LPS challenge and the left kidneys were harvested. Half of the kidney was fixed in 4% paraformaldehyde for 24 h at 4°C, dehydrated using graded ethanol, cleared with xylene, and embedded in paraffin. Paraffin blocks were sliced into 3-μm sections. Hematoxylin and eosin (HE) staining was performed on the sections using hematoxylin and eosin staining kit (Solarbio, Beijing, China). Pictures were captured with Nicon Ci-L microscope (Nicon, Tokyo, Japan) at 200× magnification. Tubular damage was assessed by tubular dilation, necrosis, and apoptosis. Tubular dilations were counted in every non-overlap field and averaged. Cell necrosis and apoptosis were determined using in situ cell death detection kit (Roche, Basel, Switzerland). The fluorescent pictures were captured with an LSM 710 confocal fluorescence microscope (Zeiss, Oberkochen, Germany) at 100× magnification. Only dots overlapped with nuclei were counted and the whole cell number of each field was counted by Image J. Proportion of apoptotic and necrotic cells was computed.
Immunofluorescent Staining of Fibrin
Paraffin slides that were processed by xylene de-waxing and gradient ethanol hydrating were blocked by ready-to-use goat serum (Boster, Wuhan, China) at 37°C for 1 h and incubated with rabbit anti-fibrinogen (1:400) overnight at 4°C. After washing three times with PBS, the slides were incubated with Alexa Fluor 488 goat anti-rabbit IgG (1:1,000) (Invitrogen, California, USA) at 37°C for 1 h. The slides were washed three times and mounted with mounting medium with DAPI (ZSGB-Bio, Beijing, China). Images were captured using Leica TCS SP5 confocal laser scanning microscope (Leica Microsystems, Wetzlar, Germany) at 200× magnification. The fluorescence intensity and fluorescence positive area were analyzed by Image J.
Cluster Analysis and Homology Analysis
The protein sequences were downloaded from NCBI-Protein (https://www.ncbi.nlm.nih.gov/protein/). Amino acid sequences were aligned by ClustalW with MEGA 7.0 (47). The alignment diagram was drawn with GeneDoc. The evolutionary tree was constructed using the Neighbor-Joining method with Mega 7.0 (47, 48). The domain structures of FHRs were analyzed by SMART (http://smart.embl-heidelberg.de/). Domain similarities among different FHRs were computed with pBLAST.
Statistics
Data were presented as mean ± SEM. Student's t-test was used to compare the two groups. Time course of factors of different time points after LPS challenge was analyzed with one-way ANOVA by SPSS software. Comparison of survival curves was analyzed by Mantel–Cox test. Significant differences were considered when the p-value was less than 0.05 and extremely significant differences were considered when the p-value was less than 0.01.
Results
Homology Analysis of Human and Mouse FHR Proteins and Generation of Cfhr1 Knockout (KO) Mice
To ascertain the mouse FHR-E homolog of human FHR proteins, homology analysis between mouse and human FHRs was conducted. The evolutionary tree of FHRs showed that FHR-E converged with human FHR-1, FHR-2, and FHR-5 (Figure 1A). The overall homology between mouse FHR-E and human FHR-1/2/5 is about 70%, while FHRB and FHRC have about 40% homology to FHR-1/2/5 (40). Structure analyses of FHR-E and human FHR-1, FHR-2, and FHR-5 by SMART showed that they all consist of CCP modules. Amino acid sequence of each CCP in FHR-E was compared with human FHR-1, FHR-2, and FHR-5 by pBLAST and amino acid sequence alignment was analyzed by MEGA 7.0. Results showed that FHR-E shared more similarity with human FHR-1 (Figures 1B–E). With this regard, we selected the mouse Cfhr1 gene that encodes the FHR-E protein for the generation of a gene knockout colony.
The exons from 1 to 5 of Cfhr1 were deleted by using the CRISPR/Cas9 system on C57BL/6 mouse (Figure 2A). Verification of Cfhr1 deletion was conducted at DNA, mRNA, and protein levels (Figures 2B–D). The results demonstrated that FHR-E was detectable in murine plasma and was depleted in the knockout mice. Our homemade anti-FHR-E antibody recognized murine FH as well due to the high epitope identities to SCRs 19 and 20 of FH (data not shown). This is the first report of FHR-E in murine plasma. FHR-B and FHR-C has been detected in mouse plasma by using FH-specific antiserum (38). The unrecognized FHR-E in their experiment may be because of the antibody specificity. Both heterozygotes (Cfhr1−/+) and homozygotes (Cfhr1−/−) were viable and healthy bred in SPF (specific pathogen free) mouse facility.
FHR-E Deficiency Did Not Affect the Physiological Intrinsic AP Regulation
Under physiological condition, AP keeps at a low level of activation by spontaneous hydrolysis of C3 (49). To study whether Cfhr1 deletion can trigger complement dysregulation under physiological condition, the plasma levels of C3, C5, and FH, central molecules of complement and main soluble regulator of AP, were detected on 6- to 8-week-old male sibling mice. Western blotting results showed no obvious changes among mice of different genotypes (Figure 3A). The relative gray values of 18 mice were analyzed and results showed that there were no significant differences (Figures 3B–D). Furthermore, the concentrations of C3a and C5a, which are small cleaved fragments generated by complement activation, and the key mediators of inflammation were measured. Neither the level of C3a nor C5a had significant differences among different genotypes (Figures 3E,F). These results suggested that under physiological condition, the deletion of Cfhr1 was not sufficient to impact the AP regulation.
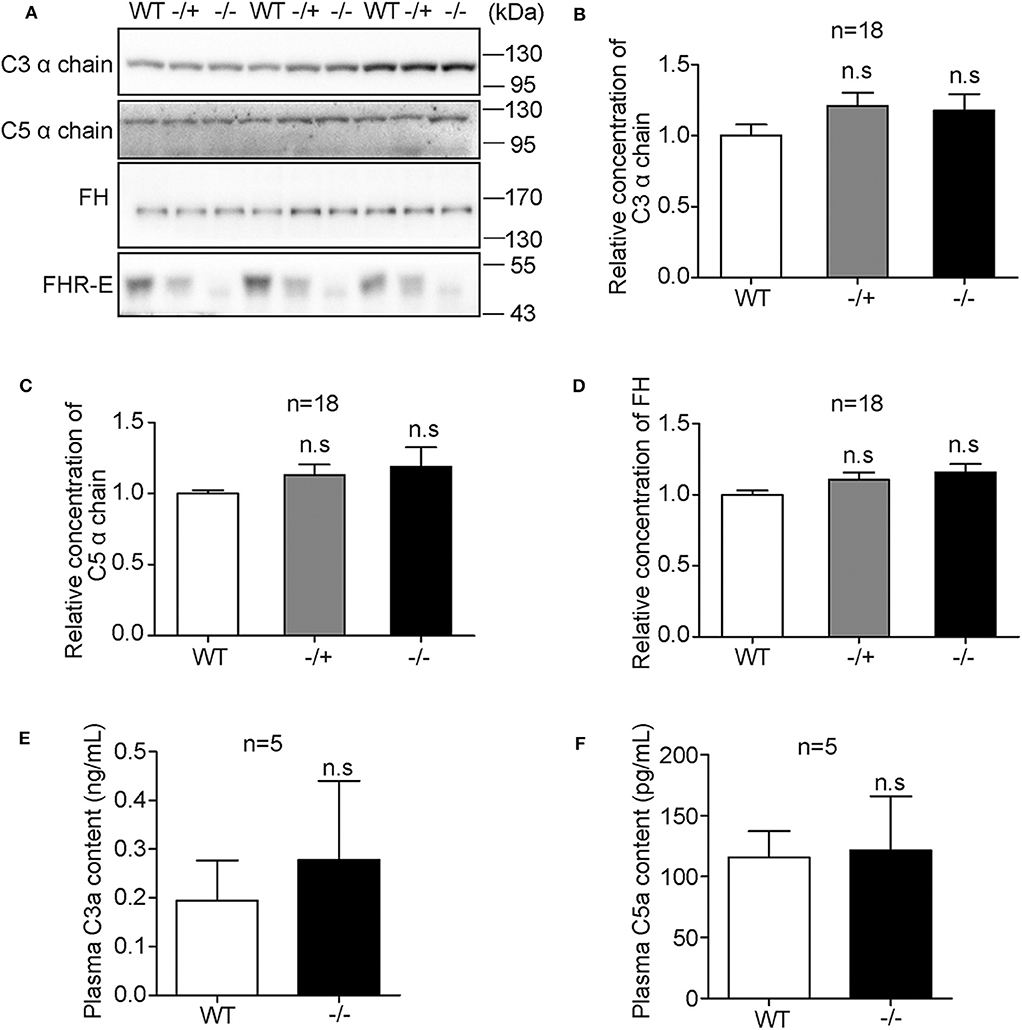
Figure 3. Detection of plasma levels of major complement factors among mice of different genotypes under physiological conditions. (A) Representative Western blotting analyses of plasma C3, C5, FH, and FHR-E. Equal volume of plasma was loaded. Eighteen mice in each group were included. (B–D) The gray values of C3, C5, and FH were measured with Image J. The calculated gray values of each blot were divided by the average gray value of wild type in the blot. (E,F) Measurements of plasma concentration of C3a and C5a with ELISA kit. EDTA anti-coagulant blood was obtained from heart puncture. Five mice in each group were used. Very low background absorption was detected in different genotype mice. n.s., not significant.
AP Activation Is Enhanced in Cfhr1−/− Mice in Response to LPS Treatment in vitro
LPS, the principal component of gram-negative bacteria cell wall, is an activator of AP (50) and responsible for gram-negative bacteria induced sepsis (51). To test the effect of FHR-E deficiency on LPS-induced AP, in vitro LPS-induced AP activation experiments were performed. LPS was incubated with serum from mice of different genotypes and the mixtures were analyzed by Western blotting. A higher amount of C3-activated fragment in Cfhr1−/− mice was observed compared to the wild-type group (Figures 4A,B). AP activity measured by ELISA assay (46, 52) was also conducted to verify this effect. As expected, Cfhr1−/− mice showed more deposited C3b on LPS (Figure 4C). Furthermore, LPS-induced C5a production, an ensuing event of AP activation, was determined and significantly higher concentration of C5a was detected in Cfhr1−/− mice compared to wild-type mice (Figure 4D). An increasing tendency was observed in the heterozygotes, but there was no significant difference between WT and heterozygotes, suggesting that dose effects of FHR-E in heterozygotes may exist. Thus, FHR-E deficiency promoted LPS-induced AP activation in vitro.
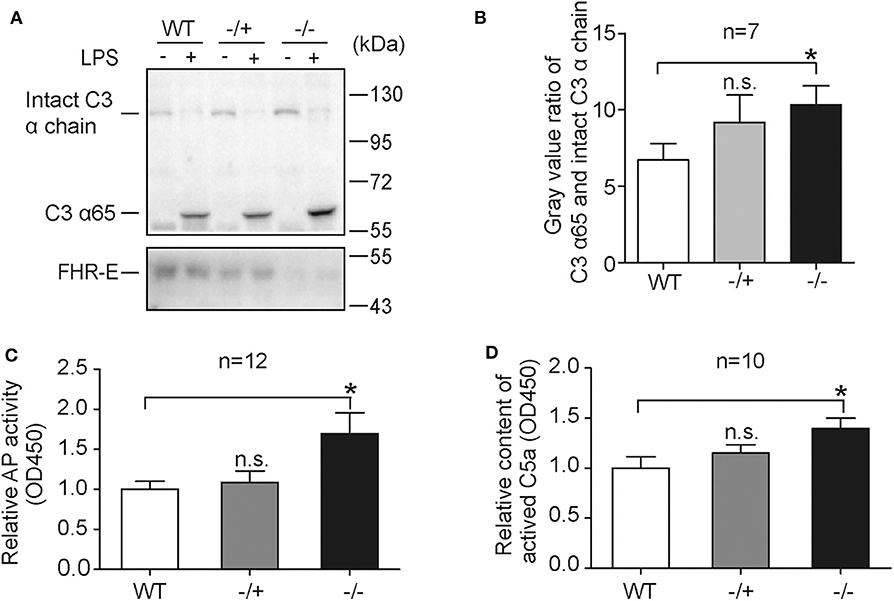
Figure 4. Serological measurements of in vitro LPS stimulation. (A) Representative Western blotting analysis of C3 activation by LPS treatment among mice of different genotypes. Western blotting analysis of FHR-E was used for genotyping. Equal volumes of PBS were used as negative controls. Equal volume of protein was loaded. Seven mice in each group were used. (B) The gray values of C3 α65 fragment and intact C3 α chain in (A) were measured with Image J. The gray value of C3 α65 was divided by the gray value of intact C3 α chain in the same lane. The ratio of C3 α65 and intact C3 α chain was used to represent cleavage status of C3 α chain. (C) Detection of AP activation by plate bounded LPS in diluted serum with ELISA method. Equivalent LPS was coated on the plate. AP buffer and EDTA buffer diluted serum was added into the plate and incubated for 1 h at 37°C. EDTA buffer diluted serum was used as a negative control for background subtraction. After washing, the LPS-bound C3b was detected with C3 antibody. Twelve mice in each group were used. (D) Measurement of the concentration of C5a produced at 2 h after LPS in vitro stimulation. AP buffer diluted serum was incubated with LPS or PBS for 2 h at 37°C. Equal volume of PBS was used as a negative control for background subtraction. The quantity of C5a was detected with sandwich enzyme-linked immunosorbent assay. Ten pairs of mice were used. n.s., not significant. *p < 0.05.
FHR-E Deficiency Resulted in Enhanced Activation of Complement Pathway and Inflammation in vivo
To further study the regulatory function of FHR-E, in vivo challenge of LPS was applied. EDTA anticoagulant blood of different time points was collected from the tail tip to monitor the content changes of main complement or coagulation factors using Western blotting. Our results showed that the level of C3 decreased over time and that the level of C5 increased at first and decreased from 1 h after LPS challenge. No significant difference was observed among different genotypes at different points. However, more significant decrease of C5 content over time was observed in Cfhr1−/− mice compared with wild-type mice (Figures 5A–C). The levels of FH and von Willebrand factor (VWF), which is involved in hemostasis, were also determined. The content of VWF increased at first and decreased from 3 h post LPS challenge (Figures 5A,E) while the content of FH did not have any observable change over time (Figures 5A,D). This demonstrated that the hemostasis was promptly triggered and gradually vanished as the exhaustion of clotting factor, and that FHR-E deficiency and LPS stimulation did not affect the level of FH. The contents of FHR-E in wild-type and heterozygous mice were also measured. Interestingly, we found that FHR-E increased significantly at 3 h post-LPS challenge (Figures 5A,F). The data in Figure 5 were obtained from the same batches of mice.
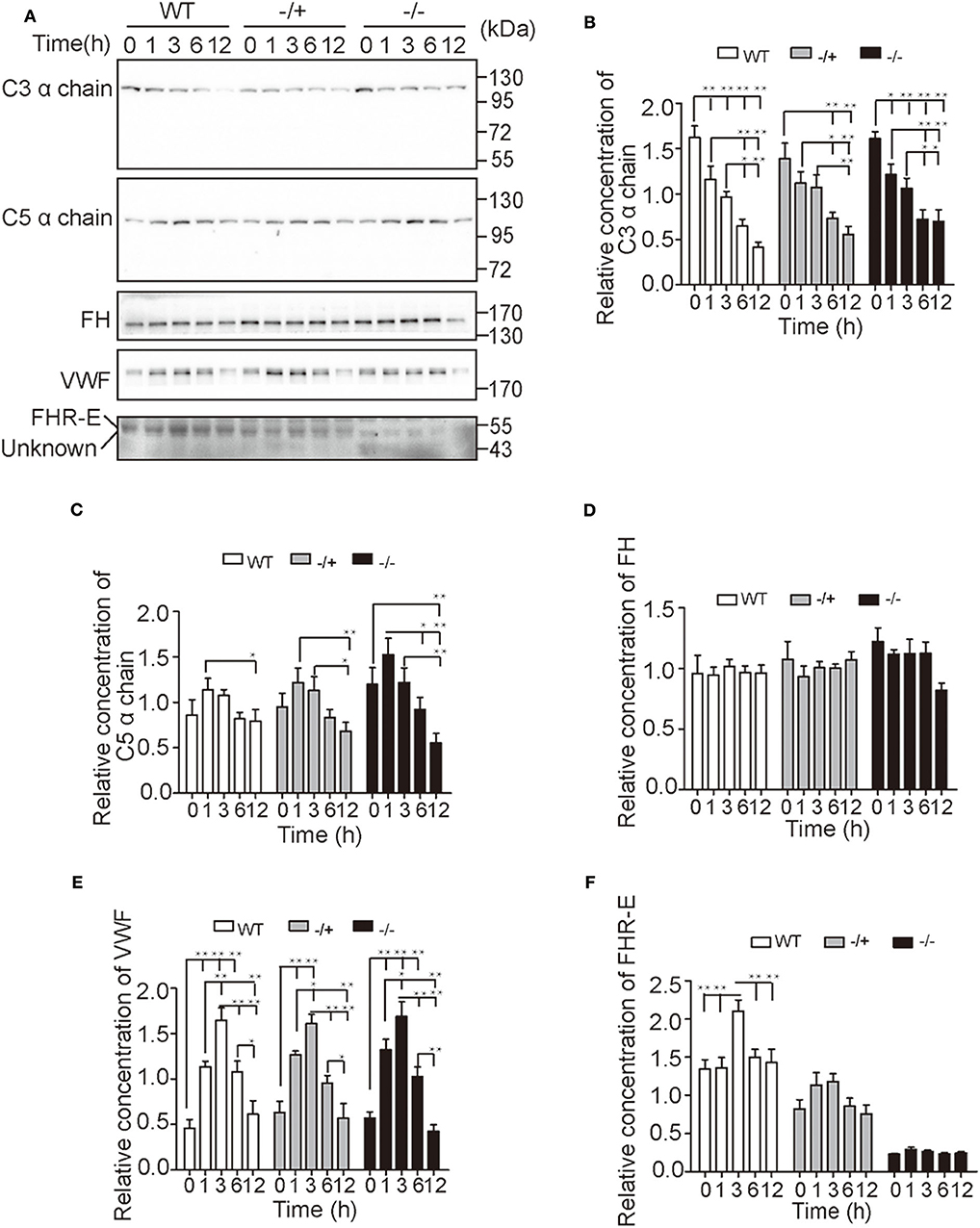
Figure 5. Detection of major complement and coagulation factors after in vivo LPS challenge. (A) Time-course changes of C3, C5, FH, VWF, and FHR-E expression levels analyzed by Western blotting. Equal volume of plasma was loaded. Five mice in each group were used. “Unknown” means unknown proteins recognized by the anti-FHR-E antibody. (B–F) The gray value quantification of C3, C5, FH, VWF, and FHR-E in (A) measured with Image J. The gray values of each membrane were divided by the average gray value of all samples on the membrane. The data were analyzed with one-way ANOVA. *p < 0.05, **p < 0.01.
Furthermore, another group of mice were challenged with LPS and EDTA anticoagulant blood of different time points was harvested from heart puncture. The contents of plasma C3a and C5a of Cfhr1−/− mice were significantly higher than wild-type controls at 3 h post-challenge (Figures 6A,B). These results demonstrated that FHR-E deficiency accelerated LPS-induced AP. The plasma IL-1β and TNF-α contents at 3 h post-challenge were detected and elevated concentrations of IL-1β and TNF-α were found in Cfhr1−/− mice (Figures 6C,D). Subsequently, we observed that the proportion of granulocytes significantly increased and lymphocytes mildly decreased at 6 h after challenge, which suggests that Cfhr1−/− mice had more severe inflammation (Figures 6E,F). The quantity of red blood cells in Cfhr1−/− mice at 12 h after LPS challenge was dramatically lower than the wild-type mice, which indicates that much more congestion could happen in Cfhr1 deletion mice (Figure 6G). In summary, the Cfhr1−/− mice had features of enhanced complement activation and inflammation, which may lead to organ injury.
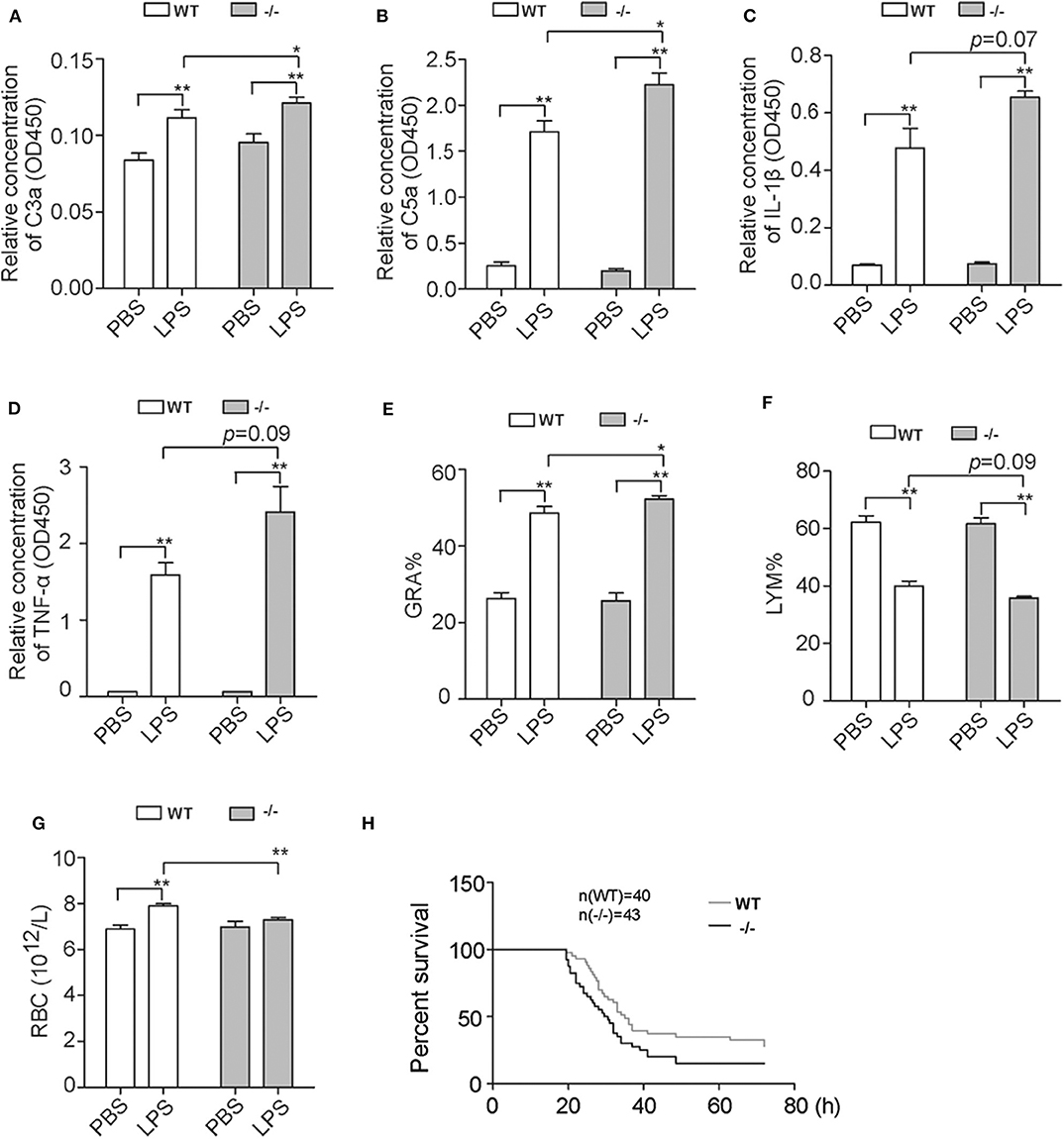
Figure 6. Detection of complement-activated products and blood cells after in vivo LPS challenge. (A,B) C3a and C5a concentration measurements with ELISA kit at 3 h after in vivo LPS challenge. Four mice in each group were included. (C,D) IL-1β and TNF-α concentration measurements with ELISA kit at 3 h after in vivo LPS challenge. Three mice in each group were included. (E–G) Percentages of granulocytes and lymphocytes at 6 h after LPS challenge and red cells at 12 h after LPS challenge measured by animal blood analyzer. GRA, granulocytes. LYM, lymphocytes. RBC, red blood cells. Four mice in each group were included. (H) The survival data of two groups with i.p. injection of 10 mg/kg LPS. The data were analyzed by Gehan–Breslow–Wilcoxon test. The number of wild-type mice and Cfhr1−/− mice was 40 and 43, respectively. *p < 0.05, **p < 0.01.
To explore the endpoint effect of FHR-E deficiency on LPS-induced sepsis, the mortality assay was performed. Cfhr1−/− and wild-type mice were administrated i.p. injection of 10 mg/kg LPS. The survival data were recorded and analyzed. Compared to the wild-type mice, the average survival time of Cfhr1−/− mice was significantly shorter and the mortality rate of Cfhr1−/− mice was significantly higher (Figure 6H; p = 0.0425, n = 43 in the wild-type group, n = 40 in the Cfhr1−/− mice group). Median survival time of wild-type and Cfhr1−/− mice was 35 and 30 h, respectively. At 26 h post-LPS challenge, 14% of wild-type mice died while 35% of Cfhr1−/− mice died. Thus, FHR-E plays a protective role on LPS-induced sepsis.
FHR-E Deficiency Promoted LPS-Induced AKI
Human CFHR1 deletion was associated with nephropathy (27, 53) and kidney injury can be induced by complement overactivation (54). Urea and creatinine, which are indicators of renal function, were tested at 12 h post-challenge and significant increases were observed in both WT and KO groups (Figures 7A,B). However, we did not observe an apparent increase in the KO group compared with the WT group after LPS treatment. This suggests that renal function was not worsened dramatically in FHR-E deficiency at the time of measurement. We then performed histological examination of kidney. Renal tissue sections of 12 h after LPS challenge were prepared and HE staining was conducted. Kidney injury degree was assessed by the tubular dilations, apoptosis, and necrosis. Tubular dilations were quantified, and cell apoptosis and necrosis were determined through TUNEL assay. More tubular dilations (Figures 7C,D) and more cell apoptosis and necrosis (Figures 7E,F) were exhibited in Cfhr1−/− mice. Fibrin, which is the marker of vascular injury, was also stained to testify tissue injury. We found that Cfhr1−/− mice had significantly more fibrin deposition than wild-type mice (Figures 8A–C). Plasma fibrin was also analyzed by Western blotting and higher concentration of plasma fibrin was found in Cfhr1−/− mice (Figure 8D).
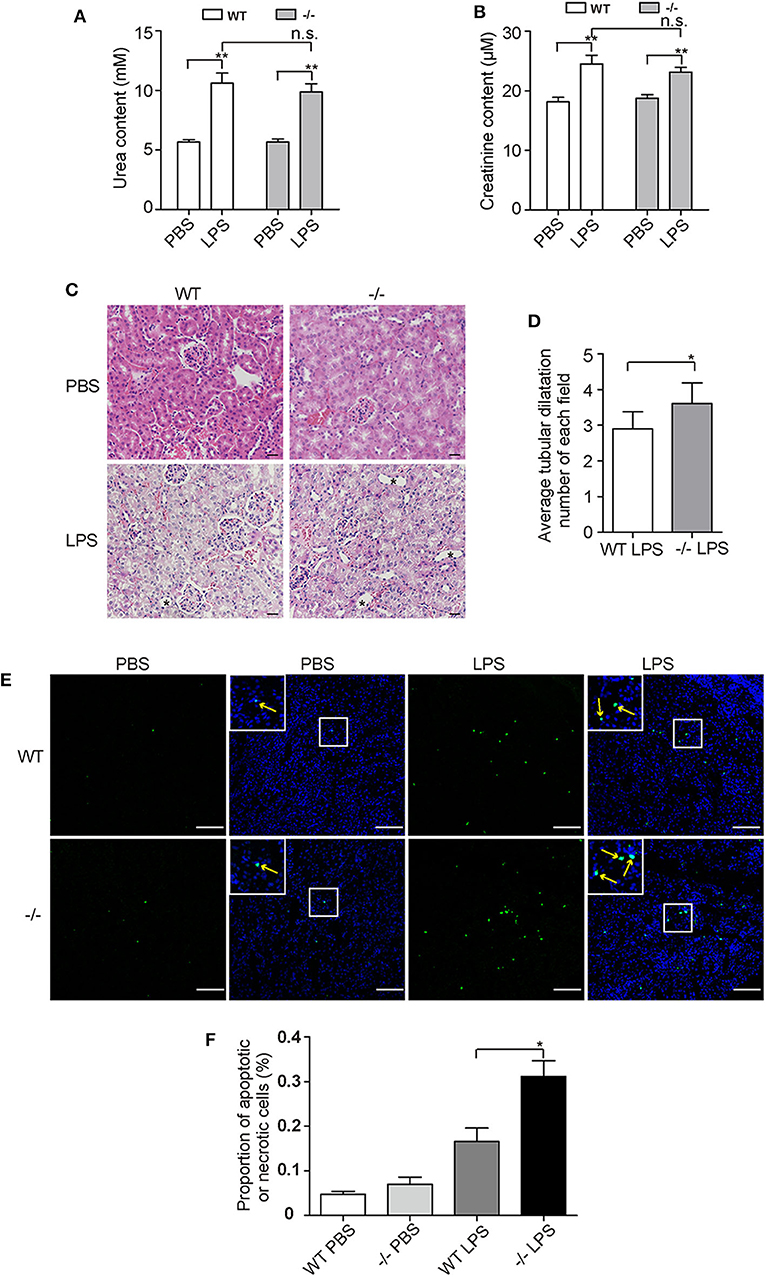
Figure 7. Tests of kidney injury at 12 h after LPS challenge. Three mice in each group were used. (A,B) Measurements of urea and creatinine content. (C) Hematoxylin and eosin (HE) staining of kidney sections. Pictures were taken at a magnification of 200×. The slice thickness was 3 μM. The bar is 20 μM. Tubular dilations were marked by asterisks. (D) Average tubular dilation number of each field. Non-overlapped fields of one section were counted and averaged. (E) TUNEL staining of kidney sections. Pictures were taken at a magnification of 100×. The green signals indicate nuclei of apoptotic or necrotic cell whose DNA strand breaks in nuclei were labeled by fluorescein-dUTP. The blue signals indicate total nuclei, which were stained with DAPI. The field in the upper left corner is the zoom picture in the white square box. Arrows point to the fluorescence positive points. The bar is 100 μM. (F) Proportion of apoptotic and necrotic cells. Green fluorescence positive points that were overlapped with blue points of 10 non-overlapped fields were counted. Total nuclei numbers were recognized as total cell numbers counted by Image J. The ratio of fluorescence nuclei among total nuclei was calculated. *p < 0.05, **p < 0.01, n.s., not significant.
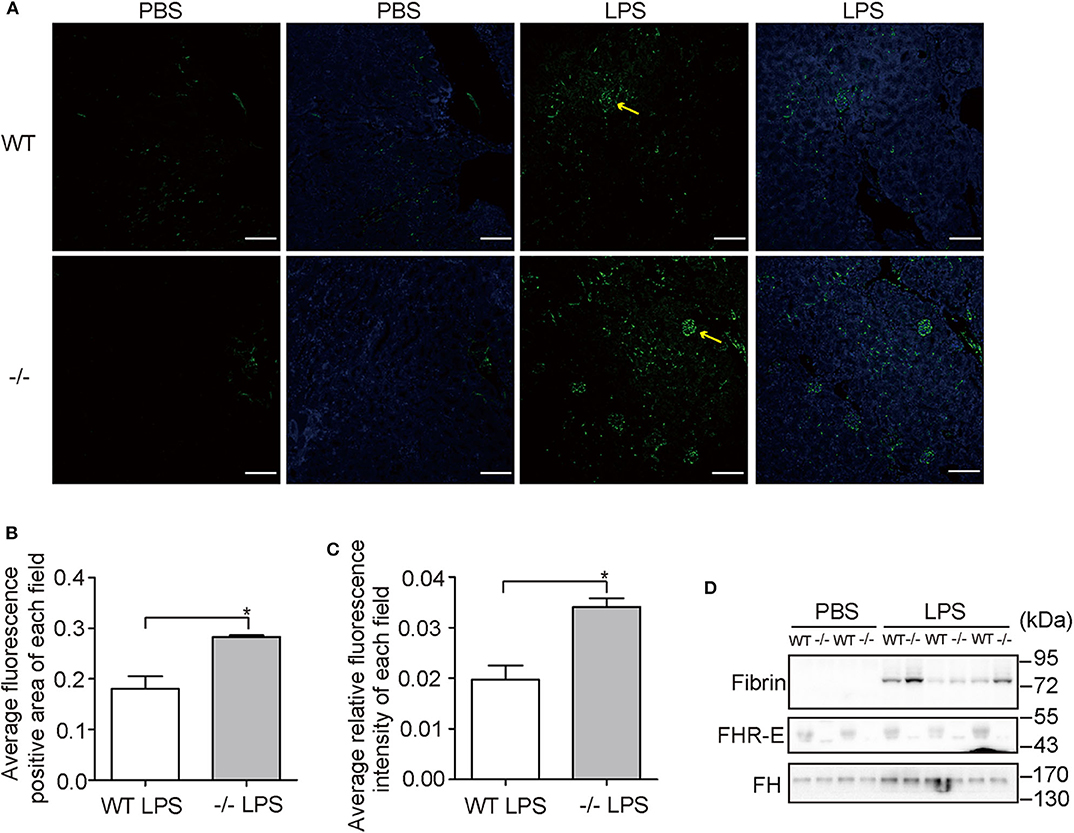
Figure 8. Vascular damage examinations. Three mice in each group were used. (A) Immunostaining of fibrin on kidney sections. Pictures were taken at a magnification of 200×. The green signals indicate fibrin. The blue signals indicate total nuclei stained with DAPI. Arrows point to the fluorescence positive area. The bar is 50 μM. (B) Comparison of fluorescence positive area between WT and Cfhr1−/− mice. Fluorescence positive area of each field was calculated by Image J. Twenty non-overlapped fields were counted and averaged. (C) Comparison of fluorescence intensity between wild-type and Cfhr1−/− mice. Fluorescence intensity of each field was calculated by Image J. Twenty non-overlapped fields were counted and averaged. (D) Western blotting analysis of plasma fibrin of 12 h after LPS challenge. Western blotting of FHR-E was performed to determine the genotypes. Equal volumes of plasma were loaded. The Western blotting of FH on the same blot was regarded as an internal control. *p < 0.05.
Discussion
In this study, Cfhr1 knockout mice were generated to further investigate its role in AP regulation. No obvious changes of C3a and C5a concentration in plasma were found in Cfhr1 knockout mice. These results suggest that the basal level of FHR-E plays negligible roles in spontaneously activated AP pathway. This may explain why many individuals with CFHR1 homozygous deletion are healthy (34). Surprisingly, with FHR-E deficiency, increased C3 and C5 cleavage were found after LPS stimulation in vitro, suggesting that FHR-E itself inhibits LPS-induced AP activation. This may be explained by the conformation switch model in which soluble FH exists in a low-affinity latent conformation and transits to high-affinity activated conformation by interacting with self-surface targets (55). Based on this model, FH may play negligible inhibition on LPS-induced AP and FHR-E may compete C3b with positive regulators of AP, like properdin, which plays an essential role in LPS induced AP activation (46). When FHR-E is absent, AP activity is enhanced by the positive regulators. The inhibitory effect of FHR-1 on C5 convertase has been reported (30) and was regarded as a therapeutic target (56). The result that more C5a production after LPS challenge in Cfhr1−/− mice was observed supports the idea that FHR-E has functional homology to human FHR-1.
CFHR1 deletion has been reported to be associated with renal disease in an incomplete penetrance and a second hit such as infection is considered to be essential for disease development (57). It is unknown whether CFHR1 homozygous deletion alone would trigger the onset of these conditions upon infection. One reported case showed that shiga toxin was a potential trigger of CFHR1 deletion-related thrombotic microangiopathy (58). Here, we chose LPS, which is the main constituent of the wall of gram-negative bacteria as a stimulator to study the effect of Cfhr1 deletion in mice for possible pathological changes. Both in vitro and in vivo assays showed that FHR-E deficiency significantly promoted LPS-induced AP activation. These results suggested that FHR-E may play a coordinated role with FH in determining complement activation. However, the mechanism of selective activation of AP remains enigmatic. In many aHUS patients with CFHR1 deletion, FH autoantibody was detectable (27). It was believed that aHUS may result from the blocking of FH function by FH auto-antibody. However, this cannot explain those aHUS patients with CFHR1 deletion without FH auto-antibody (59). Our results suggest that FHR-E deficiency promotes infection-induced damage through enhancing AP activation. This may provide a hint for FHR-1 deficiency-related nephropathy. Nevertheless, how FHR-E functions in this complex process warrants further investigation. In addition, we did not see much difference of FH level in FHR-E deficiency mice.
To the best of our knowledge, there has been no report of FHRs on any inflammatory disease mouse models. Our results revealed that Cfhr1 knockout mice challenged with LPS served as a good model to study the intricate network of complement, inflammation, and coagulation in sepsis and AKI. We found that Cfhr1−/− mice showed higher content of C3a and C5a, more severe inflammation, more blood coagulation, and more severe renal injury after LPS challenge compared with the wild-type group. However, we did not see significantly higher levels of urea and creatinine in Cfhr1−/− mice compared to the wild-type mice upon LPS challenge. C3a and C5a, which are two main products of complement activation, have been shown to have anti-inflammatory (60, 61) and pro-inflammatory function (62), respectively. Administration of C5a receptor antagonist peptide in thrombotic glomerulonephritis model significantly reduced leucocyte accumulation and thrombus formation in glomeruli (63) and improved survival of mice with sepsis (64), while administration of C3a receptor antagonist exacerbated mortality of mice with sepsis (64). The regulation mechanism of C3a and C5a on sepsis development, which seemed to have converse effect, is still unclear. In our study, the deficiency of FHR-E promoted complement activation with more C3a and C5a production, which in turn induced more inflammation and eventually accelerated coagulation and tissue injury. These results suggested that C5a may play a more potent role on sepsis development. Our Cfhr1−/− mouse model provides a unique tool to study the underlying mechanism in which AP is activated upon LPS challenge to induce tissue injury. Beyond the scope of this study, it is speculative that patients with FHR deficiency may be susceptible to severe conditions under SARS-CoV-2 infection when sepsis is induced in COVID-19 patients.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee of IGDB (Institute of Genetics and Developmental Biology, Chinese Academy of Science).
Author Contributions
XLi, ZH, and WL designed the experiments, analyzed the data, and supervised the project. XLiu contributed to the initiation and design of the project. XLi and WL wrote the manuscript. XLi and ZH performed the assays.
Funding
This study was partially supported by grants from the Ministry of Science and Technology of China (2016YFC1000306) and from National Natural Science Foundation of China (31830054; 91854110).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Seymour CW, Rea TD, Kahn JM, Walkey AJ, Yealy DM, Angus DC. Severe sepsis in pre-hospital emergency care: analysis of incidence, care, and outcome. Am J Respir Crit Care Med. (2012) 186:1264–71. doi: 10.1164/rccm.201204-0713OC
2. Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, et al. Incidence and trends of sepsis in US Hospitals using clinical vs claims data, 2009-2014. JAMA. (2017) 318:1241–9. doi: 10.1001/jama.2017.13836
3. Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. (2004) 351:159–69. doi: 10.1056/NEJMra032401
4. Alberti C, Brun-Buisson C, Burchardi H, Martin C, Goodman S, Artigas A, et al. Epidemiology of sepsis and infection in ICU patients from an international multicentre cohort study. Intensive Care Med. (2002) 28:108–21. doi: 10.1007/s00134-001-1143-z
5. Markiewski MM, DeAngelis RA, Lambris JD. Complexity of complement activation in sepsis. J Cell Mol Med. (2008) 12:2245–54. doi: 10.1111/j.1582-4934.2008.00504.x
6. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
7. Keragala CB, Draxler DF, McQuilten ZK, Medcalf RL. Haemostasis and innate immunity - a complementary relationship: A review of the intricate relationship between coagulation and complement pathways. Br J Haematol. (2018) 180:782–98. doi: 10.1111/bjh.15062
8. Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol. (2004) 138:439–46. doi: 10.1111/j.1365-2249.2004.02627.x
9. Harboe M, Mollnes TE. The alternative complement pathway revisited. J Cell Mol Med. (2008) 12:1074–84. doi: 10.1111/j.1582-4934.2008.00350.x
10. Charchaflieh J, Wei J, Labaze G, Hou YJ, Babarsh B, Stutz H, et al. The role of complement system in septic shock. Clin Dev Immunol. (2012) 2012:407324. doi: 10.1155/2012/407324
11. Sprong T, Roos D, Weemaes C, Neeleman C, Geesing CL, Mollnes TE, et al. Deficient alternative complement pathway activation due to factor D deficiency by 2 novel mutations in the complement factor D gene in a family with meningococcal infections. Blood. (2006) 107:4865–70. doi: 10.1182/blood-2005-07-2820
12. Genel F, Atlihan F, Gulez N, Sjoholm AG, Skattum L, Truedsson L. Properdin deficiency in a boy with fulminant meningococcal septic shock. Acta Paediatr. (2006) 95:1498–500. doi: 10.1080/08035250600603008
13. Slade C, Bosco J, Unglik G, Bleasel K, Nagel M, Winship I. Deficiency in complement factor B. N Engl J Med. (2013) 369:1667–9. doi: 10.1056/NEJMc1306326
14. Silasi-Mansat R, Zhu H, Popescu NI, Peer G, Sfyroera G, Magotti P, et al. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood. (2010) 116:1002–10. doi: 10.1182/blood-2010-02-269746
15. Medjeral-Thomas N, Pickering MC. The complement factor H-related proteins. Immunol Rev. (2016) 274:191–201. doi: 10.1111/imr.12477
16. Jozsi M, Tortajada A, Uzonyi B, Goicoechea de Jorge E, Rodriguez de Cordoba S. Factor H-related proteins determine complement-activating surfaces. Trends Immunol. (2015) 36:374–84. doi: 10.1016/j.it.2015.04.008
17. Rodriguez de Cordoba S, Esparza-Gordillo JE, Goicoechea de Jorge, Lopez-Trascasa M, Sanchez-Corral P. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol. (2004) 41:355–67. doi: 10.1016/j.molimm.2004.02.005
18. Sanchez-Corral P, Pouw RB, Lopez-Trascasa M, Jozsi M. Self-damage caused by dysregulation of the complement alternative pathway: relevance of the factor H protein family. Front Immunol. (2018) 9:1607. doi: 10.3389/fimmu.2018.01607
19. Pangburn MK, Schreiber RD, Muller-Eberhard HJ. Human complement C3b inactivator: isolation, characterization, and demonstration of an absolute requirement for the serum protein beta1H for cleavage of C3b and C4b in solution. J Exp Med. (1977) 146:257–70. doi: 10.1084/jem.146.1.257
20. Weiler JM, Daha MR, Austen KF, Fearon DT. Control of the amplification convertase of complement by the plasma protein beta1H. Proc Natl Acad Sci USA. (1976) 73:3268–72. doi: 10.1073/pnas.73.9.3268
21. Eberhardt HU, Buhlmann D, Hortschansky P, Chen Q, Bohm S, Kemper MJ, et al. Human factor H-related protein 2 (CFHR2) regulates complement activation. PLoS ONE. (2013) 8:e78617. doi: 10.1371/journal.pone.0078617
22. Fritsche LG, Lauer N, Hartmann A, Stippa S, Keilhauer CN, Oppermann M, et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Hum Mol Genet. (2010) 19:4694–704. doi: 10.1093/hmg/ddq399
23. McRae JL, Duthy TG, Griggs KM, Ormsby RJ, Cowan PJ, Cromer BA, et al. Human factor H-related protein 5 has cofactor activity, inhibits C3 convertase activity, binds heparin and C-reactive protein, and associates with lipoprotein. J Immunol. (2005) 174:6250–6. doi: 10.4049/jimmunol.174.10.6250
24. Skerka C, Chen Q, Fremeaux-Bacchi V, Roumenina LT. Complement factor H related proteins (CFHRs). Mol Immunol. (2013) 56:170–80. doi: 10.1016/j.molimm.2013.06.001
25. Zhao J, Wu H, Khosravi M, Cui H, Qian X, Kelly JA, et al. Association of genetic variants in complement factor H and factor H-related genes with systemic lupus erythematosus susceptibility. PLoS Genet. (2011) 7:e1002079. doi: 10.1371/journal.pgen.1002079
26. Zipfel PF, Edey M, Heinen S, Jozsi M, Richter H, Misselwitz J, et al. Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet. (2007) 3:e41. doi: 10.1371/journal.pgen.0030041
27. Jiang H, Fan MN, Yang M, Lu C, Zhang M, Liu XH, et al. Association among complement factor H autoantibodies, deletions of CFHR, and the risk of atypical hemolytic uremic syndrome. Int J Environ Res Public Health. (2016) 13:E1209. doi: 10.3390/ijerph13121209
28. Xie J, Kiryluk K, Li Y, Mladkova N, Zhu L, Hou P, et al. Fine mapping implicates a deletion of CFHR1 and CFHR3 in protection from IgA nephropathy in Han Chinese. J Am Soc Nephrol. (2016) 27:3187–94. doi: 10.1681/ASN.2015111210
29. Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. (2006) 38:1173–7. doi: 10.1038/ng1890
30. Heinen S, Hartmann A, Lauer N, Wiehl U, Dahse HM, Schirmer S, et al. Factor H-related protein 1 (CFHR-1) inhibits complement C5 convertase activity and terminal complex formation. Blood. (2009) 114:2439–47. doi: 10.1182/blood-2009-02-205641
31. van Beek AE, Pouw RB, Brouwer MC, van Mierlo G, Geissler J, Ooijevaar-de Heer P, et al. Factor H-related (FHR)-1 and FHR-2 form homo- and heterodimers, while FHR-5 circulates only as homodimer in human plasma. Front Immunol. (2017) 8:1328. doi: 10.3389/fimmu.2017.01328
32. Bhasym A, Gurjar BS, Prabhu S, Puraswani M, Khandelwal P, Saini H, et al. Altered peripheral blood leucocyte phenotype and responses in healthy individuals with homozygous deletion of FHR1 and FHR3 genes. J Clin Immunol. (2019) 39:336–45. doi: 10.1007/s10875-019-00619-2
33. Irmscher S, Brix SR, Zipfel SLH, Halder LD, Mutluturk S, Wulf S, et al. Serum FHR1 binding to necrotic-type cells activates monocytic inflammasome and marks necrotic sites in vasculopathies. Nat Commun. (2019) 10:2961. doi: 10.1038/s41467-019-10766-0
34. Holmes LV, Strain L, Staniforth SJ, Moore I, Marchbank K, Kavanagh D, et al. Determining the population frequency of the CFHR3/CFHR1 deletion at 1q32. PLoS ONE. (2013) 8:e60352. doi: 10.1371/journal.pone.0060352
35. Dragon-Durey MA, Blanc C, Marliot F, Loirat C, Blouin J, Sautes-Fridman C, et al. The high frequency of complement factor H related CFHR1 gene deletion is restricted to specific subgroups of patients with atypical haemolytic uraemic syndrome. J Med Genet. (2009) 46:447–50. doi: 10.1136/jmg.2008.064766
36. Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, et al. Atypical aHUS: state of the art. Mol Immunol. (2015) 67:31–42. doi: 10.1016/j.molimm.2015.03.246
37. Vik DP, Munoz-Canoves P, Kozono H, Martin LG, Tack BF, Chaplin DD. Identification and sequence analysis of four complement factor H-related transcripts in mouse liver. J Biol Chem. (1990) 265:3193–201.
38. Hellwage J, Eberle F, Babuke T, Seeberger H, Richter H, Kunert A, et al. Two factor H-related proteins from the mouse: expression analysis and functional characterization. Immunogenetics. (2006) 58:883–93. doi: 10.1007/s00251-006-0153-y
39. Antonioli AH, White J, Crawford F, Renner B, Marchbank KJ, Hannan JP, et al. Modulation of the alternative pathway of complement by murine factor H-related proteins. J Immunol. (2018) 200:316–26. doi: 10.4049/jimmunol.1602017
40. Pouw RB, Vredevoogd DW, Kuijpers TW, Wouters D. Of mice and men: the factor H protein family and complement regulation. Mol Immunol. (2015) 67:12–20. doi: 10.1016/j.molimm.2015.03.011
41. Bohlson SS, Garred P, Kemper C, Tenner AJ. Complement nomenclature-deconvoluted. Front Immunol. (2019) 10:1308. doi: 10.3389/fimmu.2019.01308
42. Vik DP, Keeney JB, Munoz-Canoves P, Chaplin DD, Tack BF. Structure of the murine complement factor H gene. J Biol Chem. (1988) 263:16720–4.
43. Cserhalmi M, Csincsi AI, Mezei Z, Kopp A, Hebecker M, Uzonyi B, et al. The murine factor H-related protein FHR-B promotes complement activation. Front Immunol. (2017) 8:1145. doi: 10.3389/fimmu.2017.01145
44. Mehta G, Ferreira VP, Skerka C, Zipfel PF, Banda NK. New insights into disease-specific absence of complement factor H related protein C in mouse models of spontaneous autoimmune diseases. Mol Immunol. (2014) 62:235–48. doi: 10.1016/j.molimm.2014.06.028
45. Goicoechea de Jorge E, Caesar JJ, Malik TH, Patel M, Colledge M, Johnson S, et al. Dimerization of complement factor H-related proteins modulates complement activation in vivo. Proc Natl Acad Sci USA. (2013) 110:4685–90. doi: 10.1073/pnas.1219260110
46. Kimura Y, Miwa T, Zhou L, Song WC. Activator-specific requirement of properdin in the initiation and amplification of the alternative pathway complement. Blood. (2008) 111:732–40. doi: 10.1182/blood-2007-05-089821
47. Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. (2016) 33:1870–4. doi: 10.1093/molbev/msw054
48. Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. (1987) 4:406–25.
49. Pangburn MK, Schreiber RD, Muller-Eberhard HJ. Formation of the initial C3 convertase of the alternative complement pathway. Acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med. (1981) 154:856–67. doi: 10.1084/jem.154.3.856
50. Mey A, Ponard D, Colomb M, Normier G, Binz H, Revillard JP. Acylation of the lipid A region of a Klebsiella pneumoniae LPS controls the alternative pathway activation of human complement. Mol Immunol. (1994) 31:1239–46. doi: 10.1016/0161-5890(94)90074-4
51. Opal SM. Endotoxins and other sepsis triggers. Contrib Nephrol. (2010) 167:14–24. doi: 10.1159/000315915
52. Ueda Y, Mohammed I, Song D, Gullipalli D, Zhou L, Sato S, et al. Murine systemic thrombophilia and hemolytic uremic syndrome from a factor H point mutation. Blood. (2017) 129:1184–96. doi: 10.1182/blood-2016-07-728253
53. Palamuthusingam D, Mantha M, Oliver K, Bavishi K, Dheda S. Mini review: a unique case of crescentic C3 glomerulonephritis. Nephrology. (2017) 22:261–4. doi: 10.1111/nep.12925
54. Huber-Lang M, Sarma VJ, Lu KT, McGuire SR, Padgaonkar VA, Guo RF, et al. Role of C5a in multiorgan failure during sepsis. J Immunol. (2001) 166:1193–9. doi: 10.4049/jimmunol.166.2.1193
55. Makou E, Herbert AP, Barlow PN. Functional anatomy of complement factor H. Biochemistry. (2013) 52:3949–62. doi: 10.1021/bi4003452
56. Michelfelder S, Fischer F, Waldin A, Horle KV, Pohl M, Parsons J, et al. The MFHR1 fusion protein is a novel synthetic multitarget complement inhibitor with therapeutic potential. J Am Soc Nephrol. (2018) 29:1141–53. doi: 10.1681/ASN.2017070738
57. Valoti E, Alberti M, Iatropoulos P, Piras R, Mele C, Breno M, et al. Rare functional variants in complement genes and anti-FH autoantibodies-associated aHUS. Front Immunol. (2019) 10:853. doi: 10.3389/fimmu.2019.00853
58. Nalluru SS, Sridharan M, Go RS, Said S, Marshall AL. Shiga toxin as a potential trigger of CFHR1 deletion-associated thrombotic microangiopathy. Am J Med Sci. (2018) 356:492–8. doi: 10.1016/j.amjms.2018.05.012
59. Gurjar BS, Manikanta Sriharsha T, Bhasym A, Prabhu S, Puraswani M, Khandelwal P, et al. Characterization of genetic predisposition and autoantibody profile in atypical haemolytic-uraemic syndrome. Immunology. (2018) 154:663–72. doi: 10.1111/imm.12916
60. Kildsgaard J, Hollmann TJ, Matthews KW, Bian K, Murad F, Wetsel RA. Cutting edge: targeted disruption of the C3a receptor gene demonstrates a novel protective anti-inflammatory role for C3a in endotoxin-shock. J Immunol. (2000) 165:5406–9. doi: 10.4049/jimmunol.165.10.5406
61. Coulthard LG, Woodruff TM. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J Immunol. (2015) 194:3542–8. doi: 10.4049/jimmunol.1403068
62. Czermak BJ, Sarma V, Bless NM, Schmal H, Friedl HP, Ward PA. In vitro and in vivo dependency of chemokine generation on C5a and TNF-alpha. J Immunol. (1999) 162:2321–5.
63. Kondo C, Mizuno M, Nishikawa K, Yuzawa Y, Hotta N, Matsuo S. The role of C5a in the development of thrombotic glomerulonephritis in rats. Clin Exp Immunol. (2001) 124:323–9. doi: 10.1046/j.1365-2249.2001.01513.x
Keywords: sepsis, FHR-E, alternative complement pathway, acute kidney injury, LPS
Citation: Li X, Hao Z, Liu X and Li W (2020) Deficiency of Mouse FHR-1 Homolog, FHR-E, Accelerates Sepsis, and Acute Kidney Injury Through Enhancing the LPS-Induced Alternative Complement Pathway. Front. Immunol. 11:1123. doi: 10.3389/fimmu.2020.01123
Received: 28 January 2020; Accepted: 07 May 2020;
Published: 19 June 2020.
Edited by:
Philippe Saas, INSERM U1098 Interactions Hôte-Greffon-Tumeur & Ingénierie Cellulaire et Génique, FranceReviewed by:
Pilar Sánchez-Corral, University Hospital La Paz Research Institute (IdiPAZ), SpainChristine Skerka, Leibniz Institute for Natural Product Research and Infection Biology, Germany
Copyright © 2020 Li, Hao, Liu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaorong Liu, bHhyYmNoQHNpbmEuY29t; Wei Li, bGl3ZWlAYmNoLmNvbS5jbg==
†These authors have contributed equally to this work