 Rachel G. Scheraga
Rachel G. Scheraga Brian D. Southern
Brian D. Southern Lisa M. Grove
Lisa M. Grove Mitchell A. Olman
Mitchell A. Olman- 1Respiratory Institute, Cleveland Clinic, Cleveland, OH, United States
- 2Department of Inflammation and Immunity, Lerner Research Institute, Cleveland Clinic, Cleveland, OH, United States
Ion channels/pumps are essential regulators of innate immune cell function. Macrophages have been increasingly recognized to have phenotypic plasticity and location-specific functions in the lung. Transient receptor potential vanilloid 4 (TRPV4) function in lung injury has been shown to be stimulus- and cell-type specific. In the current review, we discuss the importance of TRPV4 in macrophages and its role in phagocytosis and cytokine secretion in acute lung injury/acute respiratory distress syndrome (ARDS). Furthermore, TRPV4 controls a MAPK molecular switch from predominately c-Jun N-terminal kinase, JNK activation, to that of p38 activation, that mediates phagocytosis and cytokine secretion in a matrix stiffness-dependent manner. Expanding knowledge regarding the downstream mechanisms by which TRPV4 acts to tailor macrophage function in pulmonary inflammatory diseases will allow for formulation of novel therapeutics.
Introduction
Ion channels and transporters are rapidly being recognized as essential for basic physiological functions of immune cells (1, 2). However, gaps in knowledge remain on the intracellular molecular mechanisms by which ion channels contribute to immune cell function. Calcium and other cations (such as sodium and potassium) have been shown to act as second messengers to regulate innate immune cell function and activation (3). For example, macrophage migration, polarization, phagocytosis, and cytokine secretion have been shown to be regulated through calcium (4–6). One such mechanism of calcium regulation in the cell is through the calcium permeable cation channel transient receptor potential vanilloid 4 (TRPV4). TRPV4 is a mechanosensitive cation channel that is essential for macrophage activation functions such as macrophage phagocytosis and cytokine secretion in a matrix stiffness-dependent manner (7, 8). The current review focuses on experimental data illustrating the importance of TRPV4 on immune cell function. We appreciate all the important contributions to the literature in this field, however given space considerations we have focused on what is perceived to be directly relevant to this review.
Macrophage Heterogeneity in the Lung
The lung is constantly exposed to inhaled particles and pathogens from the environment (9). Hence, lung innate immunity needs to be tightly regulated and phenotypically plastic in order to simultaneously maintain homeostasis and clear foreign invaders (10). Recently published data characterize macrophage phenotypic subsets based on their location in the lung [alveolar (AM) vs. interstitial (IM)] in naïve and injured (LPS/bleomycin treatment) conditions (11–13). The alveolar and interstitial subsets have been further divided based on site of origin into “resident” and “recruited” macrophage populations after injury or inflammation, by fate mapping, and lineage tracing models (11, 14–16). “Resident” macrophages populate the lung a few days after birth from fetal monocytes, and self-renew after injury (14, 15, 17). In contrast, recruited lung macrophages populate the lung only after injury and are derived from circulating monocytes that originated from the bone marrow (14, 15, 18). Despite this anatomic lineage and genomic classification, the in vivo biologic functions of both resident/recruited alveolar and interstitial macrophage populations are not fully understood. The microenvironment plays a key role in reprograming the monocyte/macrophage phenotypic response to lung injury (19, 20). Reprograming of the macrophage phenotype is not as simple as the classically-defined in vitro M1/M2 paradigm characterized by surface marker labeling (21–24). The molecular pathways in vivo by which the macrophage phenotype and function change in response to the microenvironment have yet to be fully described.
Macrophage Function and Signaling in Acute Lung Injury
Other literature has extensively characterized the important macrophage mediated mechanisms of chronic lung injury. Herein, this review will focus on the role of macrophages in acute lung injury. Macrophages are the most abundant, and critical cells, that maintain homeostasis in the lung (9). Macrophages have also been shown to play an important role in orchestrating the acute lung injury and repair process (25). Acute lung injury, both from non-infectious and infectious inflammation is a complex process. Acute lung injury is a consequence of endothelial and/or alveolar epithelial injury, followed by recruitment, and accumulation of inflammatory cells in the injured/stiffened alveolus (26–28). Macrophages have surface receptors that recognize pathogen (PAMPs) and/or damage-associated molecular patterns (DAMPs) to recruit inflammatory cells (e.g., neutrophils, recruited alveolar/interstitial macrophages) and coordinate both activation and cessation of inflammation (29). Macrophages function to phagocytize invading organisms, apoptotic cells/neutrophils, or particles. In addition, macrophages secrete, and respond to pro- and anti-inflammatory cytokines and chemokines (e.g., IL-1β, TNF-α, IL-8, IL-6, IL-10) (30–34). Activation of macrophages in response to infection occurs in part through coordination of activation of key stress activated pathways including Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), Interferon regulatory factor 3 (IRF3), Stimulator of interferon genes (STING), and Mitogen activated protein kinases (MAPKs) (35). The MAPK family (i.e., p38, ERK, and JNK) exhibits functional cross-talk and redundant functions in inflammation (35). p38 and JNK have been shown to be activated in lung injury in response to infection and to be important in macrophage activation functions such as phagocytosis (36). Control of persistent activation of MAPKs is mainly regulated by the phosphatases, dual-specificity serine threonine phosphatases/MAPK phosphatases (DUSPs/MKPs) (37, 38). It has been described that increased lung stiffness regulates MAPK/phosphatase cross-talk, while others have shown that alveolar vessel wall stiffness increases >10-fold (from 3 to 45 kPa) after intratracheal LPS-induced lung injury in mice (39), thus providing mechanistic insight into how macrophages can respond to cues from the injured lung. However, distinct mechanisms whereby increased lung tissue stiffness/injury controls the MAPK and phosphatase cross-talk is poorly understood.
The TRPV4 Channel and Mechanobiology Of the Lung
Transient receptor potential (TRP) channels are a family of 6 transmembrane domain proteins that are permeable to multiple cations including calcium (40). TRP channels are widely expressed in multiple tissues and cell-types with varied physiologic functions (40). Specifically, the TRP family member, TRPV4, is a ubiquitously-expressed, plasma membrane-based, calcium-permeable channel that is sensitized and activated by both chemical [5,6-Epoxyeicosatrienoic acid (EET), 4 alpha-phorbol 12,13-didecanoate (4-αPDD)] and physical stimuli (temperature, stretch, and hypotonicity) (40–43). TRPV4 can initiate intracellular, celltype and context-specific signals that depend on local increases in intracellular calcium which could act as a second messenger, and/or induce heterodimerization with other channels, activate kinases, and/or directly interact with cytoskeletal proteins via intracellular amino-(NH2) and carboxy-(COOH) terminal tails (44–47).
It has been increasingly recognized that cellular responses depend on the biophysical properties of the surrounding lung tissue environment (48, 49). Thus, mechanical cues from lung tissue stretch/stiffness can alter cellular responses to soluble mediators (e.g., growth factors, cytokines, chemokines) resulting in cellular dysfunction and disease. The mechanosensitive channel, TRPV4 has been implicated in mouse models of lung injury/fibrosis, which include hydrochloric acid, pulmonary edema, ventilator-associated lung parenchymal overdistension, and from our group, pulmonary fibrosis (50–53). The recent mini-review by Michalick and Kuebler in Frontiers Immunology further supports the concept that TRPV4 may connect mechanosensation to immunity in the lung (54). Given TRPV4's published role on regulating activity and infectivity of RNA viruses such as Zika, it remains possible that TRPV4 plays a role in the profound lung injury observed in the current SARS-CoV-2 pandemic (55). Conflicting data exist on the role of TRPV4 in mouse models of lung inflammation/injury, which seem to depend on the inciting agent, mechanism of injury, and the effector cell type (50–53). In ventilator-induced lung injury, macrophage TRPV4 has shown to exacerbate the lung injury (51, 52). Similarly in acid-induced lung injury, TRPV4 also exacerbates the lung injury (53). Furthermore, a recent study using a single pharmacologic inhibitor of TRPV4 revealed decreased lung injury after intratracheal instillation of LPS for 24 h (56). Our data, in a clinically relevant infectious model of lung injury, support the hypothesis that TRPV4 is protective from injury (7). In support of our findings, epithelial cell TRPV4 similarly protects the lung, but in a somewhat distinct, rapid direct LPS-induced lung injury model (3 h) (57). Despite the conflicting data on the role of TRPV4 in mouse models of lung injury, some consensus exists on the importance of TRPV4 in macrophage signaling (7, 8, 52). Further understanding of the molecular mechanisms by which macrophage TRPV4 is involved in the pathogenesis of lung injury will allow for a therapeutic target to ameliorate lung injury.
TRPV4 in Macrophages and Lung Injury
The calcium ion channel, TRPV4 is an essential mechanosensor that is required for effective phagocytosis in vitro and protects against infection-associated lung injury in vivo (7, 8). TRPV4 in macrophages have been shown to exacerbate ventilator-associated lung injury, and macrophages are key effector cells in the lung injury process (51, 52). Calcium has long been described as a mediator of many discrete steps in the phagocytic process (58). In addition, effective phagocytosis requires cytoskeletal rearrangements and direct interaction with the biophysical properties of the matrix (19, 20, 59). However, the key regulatory ion channels/pumps by which calcium influx into the cell is controlled during phagocytosis is not fully elucidated.
Published work first revealed that differentiated murine bone marrow-derived macrophages (BMDMs) express equal amounts of TRPV4 that was functionally active with or without LPS. TRPV4 in macrophages functions to effectively phagocytize both non-opsonized, E. coli bacteria, and opsonized (FcR-dependent), IgG-coated latex beads, in vitro in response to LPS (8). The LPS-stimulated phagocytic response was induced, in our hands, under conditions of pathophysiologic-range extracellular matrix stiffnesses, in the range noted in inflamed or injured lung (≥8–25 kPa) (16). TRPV4 had no effect on basal phagocytosis. In addition, TRPV4 downregulated LPS-induced IL-1β secretion and upregulated IL-10 secretion. Further, this TRPV4 mediated anti-inflammatory cytokine profile (↓ IL-1β, ↑ IL-10) was dependent on pathophysiologic-range matrix stiffness. To apply in vivo relevance, TRPV4 was found to be required for effective alveolar macrophage phagocytosis of IgG-coated latex beads in live mice in vivo (8). Taken together, TRPV4 is necessary for effective opsonized and non-opsonized macrophage phagocytosis and an anti-inflammatory cytokine profile, in a stiffness-dependent manner in vitro and in vivo (8).
To expand on this work, TRPV4's in vivo relevance to human disease, and molecular mechanism by which TRPV4 mediates its phagocytic and cytokine secretory effects was determined. TRPV4 was found to function to protect the lung from injury in an experimental model of Pseudomonas aeruginosa pneumonia in intact mice (7). Lung injury was measured by (i) inflammatory cell infiltration, (ii) total protein in whole lung lavage, (iii) cytokine secretion, and (iv) lung parenchymal consolidation. In addition, TRPV4 was required for effective clearance of the P. aeruginosa bacteria as measured by colony forming units retained in the lung in WT, as compared to global TRPV4 KO mice. Next, macrophages were identified as the critical cell type required to clear the P. aeruginosa infection by flow cytometric techniques (7).
To determine the molecular mechanism by which TRPV4 protects the lung from injury and clears bacteria, putative intracellular signaling pathways were investigated that are known to regulate LPS signals in macrophages (60). TRPV4 controlled molecular switching from predominate JNK activation to that of p38, in a stiffness-dependent manner. Since MAPK phosphorylation occurs commonly through the phosphatase family DUSPs/MKPs, it was postulated that DUSPs controlled the TRPV4-mediated MAPK molecular switch. TRPV4 acted to increase DUSP1 and then functioned to selectively dephosphorylate/deactivate JNK. Hence, the TRPV4-mediated MAPK molecular switch was found to be controlled through DUSP1 in a stiffness-dependent manner. TRPV4 additionally enhanced p38 activation thereby driving effective phagocytosis, while inhibiting JNK thereby decreasing pro-inflammatory cytokine secretion (IL-6, CCL2, and CXCL1). Finally, TRPV4 is also required for macrophage phagocytosis and p38 activation in healthy human monocyte-derived macrophages. Taken together, published work shows that TRPV4 in macrophages protected the lung from infection-associated lung injury through regulation of MAPK activation switching via DUSP1 (7). TRPV4 provides a novel mechanistic link between the mechanoenvironmental properties of the lung and innate immune cell function (Figure 1).
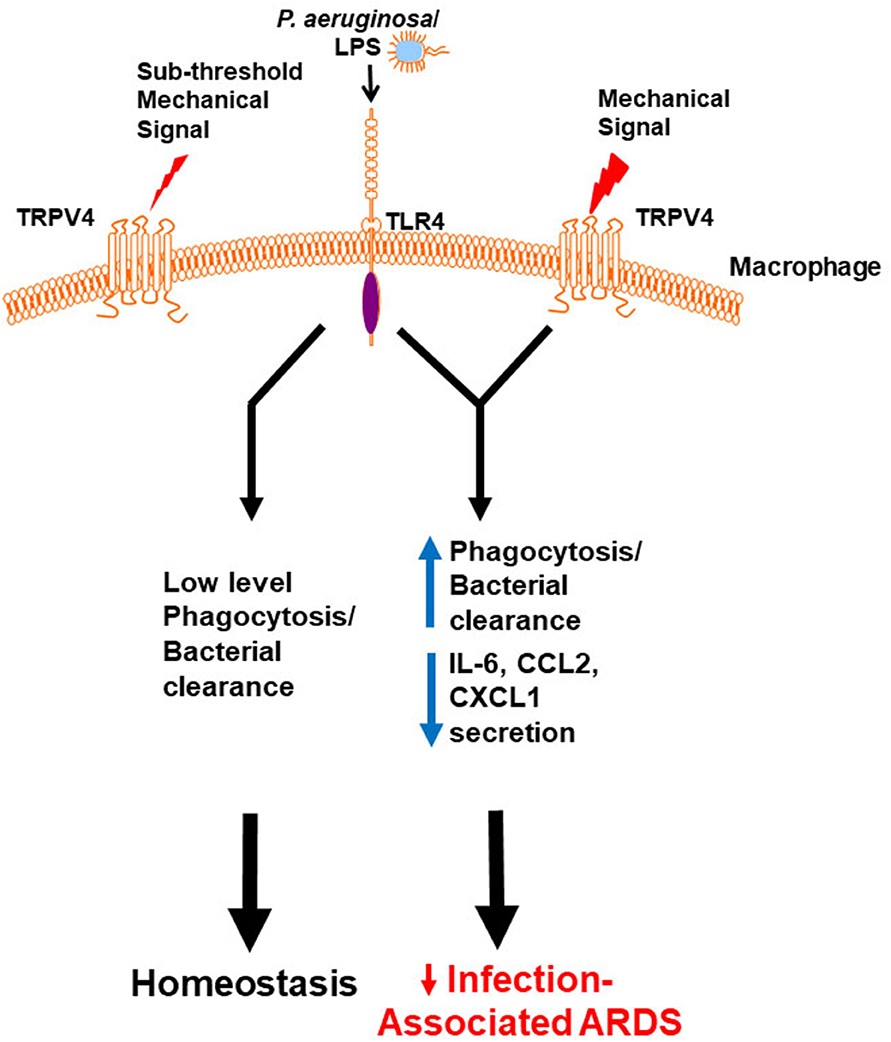
Figure 1. Working model demonstrates that a mechanical signal through TRPV4 regulates the LPS response. Our data shows in the absence of an above threshold mechanical signal, TRPV4 does not influence the minimal LPS/TLR4 response, leading to low level phagocytosis/bacterial clearance, and resultant lung homeostasis. In the presence of an above threshold mechanical signal, TRPV4 modulates with the LPS/TLR4 response to increase phagocytosis and decrease pro-inflammatory cytokine secretion, thereby protecting the lung from infection-associated injury/ARDS (7). Adapted from original publication in The Journal of Immunology. Copyright© 2020 The American Association of Immunologists, Inc.
Important Future Directions
Ongoing questions remain regarding the molecular mechanism by which TRPV4 activity is regulated or how TRPV4 is directly activated. In addition, the molecular signals by which TRPV4 regulates the MAPK molecular switch remain unknown. Since TRPV4 can directly interact with signaling molecules via its amino (NH2) and carboxy (COOH) terminal intracellular tails, the key signaling molecules that interact with TRPV4 to enhance macrophage phagocytosis and limit cytokine secretion is an active area of investigation. It remains possible that the TRPV4 interacting partners are not TLR4-dependent, as other data suggests that TRPV4 interacts with PI3K to mediate pulmonary fibrosis (61). The role of TRPV4 in different macrophage populations after infection remains an important question. Other cation/calcium channels (e.g., Piezo) have been shown to have an effect on immune cells in a mechanosensitive manner and interestingly recent work demonstrates TRPV4 is required for Piezo1-induced pancreatitis (62, 63). The molecular pathways in vivo by which the macrophage phenotype and function change in response to the microenvironment have yet to be fully described. For example, it will also be interesting in the future to explore TRPV4's action in response to (a) other types of infectious stimuli (e.g., Gram positive organisms, viral infections such as SARS-CoV-2), (b) sex differences, and (c) transcriptional/epigenetic mechanisms. An important goal of future work is to integrate mouse and human macrophage experiments, however there is limitation to this approach. It is well-known that mouse models do not fully recapitulate human disease, and the mouse immune system is programmed differently than that of humans (64). In order to circumvent this limitation, investigators utilize human diseased tissue which may provide insight into disease mechanisms, however it is usually in an in vitro setting. Hence, data obtained from mouse models and human disease tissue have their own independent strengths. Therefore, it is important to interpret the findings in a contextual nature to determine the relevance to mechanisms of human disease and design targeted pharmacologic therapies.
Conclusion
In summary, macrophages function in the lung to maintain homeostasis and clear environmental particles and pathogens. Extensive macrophage heterogeneity and plasticity allows for fine-tuning of the inflammatory response upon inflammation or infection. Ion channels have been shown to play a key role in regulating innate immune function and contribute to the pathogenesis of inflammatory/infectious lung diseases. The cation channel TRPV4 has been implicated in lung diseases associated with parenchymal stretch and inflammation or infection. The data outlined in this review show the importance of macrophage TRPV4 in response to infection and lung injury. The data shows that TRPV4 is (a) functionally active in macrophages, (b) required for effective non-opsonized and opsonized phagocytosis in vitro and in vivo, and (c) required for secretion of an anti-inflammatory cytokine profile by macrophages. These phagocytic and cytokine effects in macrophages were both dependent on matrix stiffness in the range of injured or fibrotic lung (8). In addition, TRPV4 (a) protects the lung from injury after P. aeruginosa pneumonia, (b) mediates the lung injury effects through MAPK molecular switching, and (c) is required for effective macrophage phagocytosis in human macrophages. This MAPK switching effect in macrophages was also dependent on matrix stiffness in the range of injured or fibrotic lung (7). Collectively, TRPV4 is shown to play a novel role in protecting the lung from infection-associated lung injury by regulating the phagocytic and cytokine secretory response to infection, and therefore may be a potential therapeutic target in the pathogenesis of acute lung injury.
Author Contributions
RS, BS, LG, and MO reviewed the literature and wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH grants (HL-133380) to RS, (HL-132079) to BS, and (HL-133721) to MO.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Jentsch TJ, Hubner CA, Fuhrmann JC. Ion channels: function unravelled by dysfunction. Nat Cell Biol. (2004) 6:1039–47. doi: 10.1038/ncb1104-1039
2. Hubner CA, Jentsch TJ. Ion channel diseases. Hum Mol Gene. (2002) 11:2435–45. doi: 10.1093/hmg/11.20.2435
3. Vig M, Kinet J-P. Calcium signaling in immune cells. Nat Immunol. (2009) 10:21–7. doi: 10.1038/ni.f.220
4. Nunes P, Demaurex N. The role of calcium signaling in phagocytosis. J Leukocyte Biol. (2010) 88:57–68. doi: 10.1189/jlb.0110028
5. Desai BN, Leitinger N. Purinergic and calcium signaling in macrophage function and plasticity. Front Immunol. (2014) 5:580. doi: 10.3389/fimmu.2014.00580
6. Chauhan A, Sun Y, Sukumaran P, Quenum Zangbede FO, Jondle CN, Sharma A, et al. M1 macrophage polarization is dependent on TRPC1-mediated calcium entry. iScience. (2018) 8:85–102. doi: 10.1016/j.isci.2018.09.014
7. Scheraga RG, Abraham S, Grove LM, Southern BD, Crish JF, Perelas A, et al. TRPV4 protects the lung from bacterial pneumonia via MAPK molecular pathway switching. J Immunol. (2020) 204:1310–21. doi: 10.4049/jimmunol.1901033
8. Scheraga RG, Abraham S, Niese KA, Southern BD, Grove LM, Hite RD, et al. TRPV4 mechanosensitive ion channel regulates lipopolysaccharide-stimulated macrophage phagocytosis. J Immunol. (2016) 196:428–36. doi: 10.4049/jimmunol.1501688
9. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. (2014) 14:81–93. doi: 10.1038/nri3600
10. Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, et al. Three unique interstitial macrophages in the murine lung at steady state. Am J Resp Cell Mol Biol. (2017) 57:66–76. doi: 10.1165/rcmb.2016-0361OC
11. Mould KJ, Barthel L, Mohning MP, Thomas SM, McCubbrey AL, Danhorn T, et al. Cell origin dictates programming of resident versus recruited macrophages during acute lung injury. Am J Resp Cell Mol Biol. (2017) 57:294–306. doi: 10.1165/rcmb.2017-0061OC
12. Mould KJ, Jackson ND, Henson PM, Seibold M, Janssen WJ. Single cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI insight. (2019) 4:e126556. doi: 10.1172/jci.insight.126556
13. McCubbrey AL, Barthel L, Mohning MP, Redente EF, Mould KJ, Thomas SM, et al. Deletion of c-FLIP from CD11b(hi) macrophages prevents development of bleomycin-induced lung fibrosis. Am J Resp Cell Mol Biol. (2018) 58:66–78. doi: 10.1165/rcmb.2017-0154OC
14. Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
15. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
16. Morales-Nebreda L, Misharin AV, Perlman H, Budinger GRS. The heterogeneity of lung macrophages in the susceptibility to disease. Eur Resp Rev. (2015) 24:505–9. doi: 10.1183/16000617.0031-2015
17. Hashimoto D, Chow A, Noizat C, Teo P, Beasley Mary B, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
18. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. (2017) 214:2387–404. doi: 10.1084/jem.20162152
19. Van Goethem E, Poincloux R, Gauffre F, Maridonneau-Parini I, Le Cabec V. Matrix architecture dictates three-dimensional migration modes of human macrophages: differential involvement of proteases and podosome-like structures. J Immunol. (2010) 184:1049–61. doi: 10.4049/jimmunol.0902223
20. Blakney AK, Swartzlander MD, Bryant SJ. The effects of substrate stiffness on the in vitro activation of macrophages and in vivo host response to poly(ethylene glycol)-based hydrogels. J Biomed Mater Res A. (2012) 100A:1375–86. doi: 10.1002/jbm.a.34104
21. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
22. Gordon S, Plüddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. (2014) 262:36–55. doi: 10.1111/imr.12223
23. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
24. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
25. Aggarwal NR, King LS, D'Alessio FR. Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol. (2014) 306:L709–25. doi: 10.1152/ajplung.00341.2013
26. Han S, Mallampalli RK. The acute respiratory distress syndrome: from mechanism to translation. J Immunol. (2015) 194:855–60. doi: 10.4049/jimmunol.1402513
27. Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD. Evolution of bronchoalveolar cell populations in adult respiratory distress syndrome. Am J Resp Crit Care Med. (1994) 150:113–22. doi: 10.1164/ajrccm.150.1.8025736
28. Suzuki T, Arumugam P, Sakagami T, Lachmann N, Chalk C, Sallese A, et al. Pulmonary macrophage transplantation therapy. Nature. (2014) 514:450–4. doi: 10.1038/nature13807
29. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
30. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. (2000) 342:1334–49. doi: 10.1056/NEJM200005043421806
31. Martin TR, Ruzinski JT, Steinberg KP. Cytokine balance in the lungs of patients with acute respiratory distress syndrome (ARDS). Am J Resp Crit Care Med. (1998). 157:A679.
32. Martin TR. Lung cytokines and ARDS: roger S. Mitchell lecture. Chest. (1999) 116:2S−8S. doi: 10.1378/chest.116.suppl_1.2S
33. Meduri GU, Kohler G, Headley S, Tolley EA, Stentz F, Postlethwaite A. Inflammatory cytokines in the BAL of patients with ARDS. Chest. (1995) 108:1303–14. doi: 10.1378/chest.108.5.1303
34. Parsons PE, Moss M, Vannice JL, Moore EE, Moore FA, Repine JE. Circulating IL-1ra and IL-10 levels are increased but do not predict the development of acute respiratory distress syndrome in at-risk patients. Am J Respir Crit Care Med. (1997) 155:1469–73. doi: 10.1164/ajrccm.155.4.9105096
35. Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. (2002) 12:9–18. doi: 10.1038/sj.cr.7290105
36. Arcaroli J, Yum H-K, Kupfner J, Park JS, Yang K-Y, Abraham E. Role of p38 MAP kinase in the development of acute lung injury. Clin Immunol. (2001) 101:211–9. doi: 10.1006/clim.2001.5108
37. Kim C, Sano Y, Todorova K, Carlson BA, Arpa L, Celada A, et al. The kinase p38 alpha serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression. Nat Immunol. (2008) 9:1019–27. doi: 10.1038/ni.1640
38. Hammer M, Mages J, Dietrich H, Servatius A, Howells N, Cato ACB, et al. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J Exp Med. (2006) 203:15–20. doi: 10.1084/jem.20051753
39. Meng F, Mambetsariev I, Tian Y, Beckham Y, Meliton A, Leff A, et al. Attenuation of lipopolysaccharide-induced lung vascular stiffening by lipoxin reduces lung inflammation. Am J Resp Cell Mol Biol. (2014) 52:152–61. doi: 10.1165/rcmb.2013-0468OC
40. Montell C, Birnbaumer L, Flockerzi V. The TRP channels, a remarkably functional family. Cell. (2002) 108:595–8. doi: 10.1016/S0092-8674(02)00670-0
41. Everaerts W, Nilius B, Owsianik G. The vanilloid transient receptor potential channel TRPV4: from structure to disease. Prog Biophys Mol Biol. (2010) 103:2–17. doi: 10.1016/j.pbiomolbio.2009.10.002
42. Gao X, Wu L, O'Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J Biol Chem. (2003) 278:27129–37. doi: 10.1074/jbc.M302517200
43. Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci USA. (2003) 101:396–401. doi: 10.1073/pnas.0303329101
44. Kanugula AK, Adapala RK, Midha P, Cappelli HC, Meszaros JG, Paruchuri S, et al. Novel non-canonical regulation of soluble VEGF/VEGFR2 signaling by mechanosensitive ion channel TRPV4. FASEB J. (2018) 33:195–203. doi: 10.1096/fj.201800509R
45. Jie P, Hong Z, Tian Y, Li Y, Lin L, Zhou L, et al. Activation of transient receptor potential vanilloid 4 induces apoptosis in hippocampus through downregulating PI3K/Akt and upregulating p38 MAPK signaling pathways. Cell Death Dis. (2015) 6:e1775. doi: 10.1038/cddis.2015.146
46. Sharma S, Goswami R, Merth M, Cohen J, Lei KY, Zhang DX, et al. TRPV4 ion channel is a novel regulator of dermal myofibroblast differentiation. Am J Physiol Cell Physiol. (2017) 312:C562–72. doi: 10.1152/ajpcell.00187.2016
47. Goswami C, Kuhn J, Heppenstall PA, Hucho T. Importance of non-selective cation channel TRPV4 interaction with cytoskeleton and their reciprocal regulations in cultured cells. PLoS ONE. (2010) 5:e11654. doi: 10.1371/journal.pone.0011654
48. Tschumperlin DJ, Boudreault F, Liu F. Recent advances and new opportunities in lung mechanobiology. J Biomechan. (2010) 43:99–107. doi: 10.1016/j.jbiomech.2009.09.015
49. Southern BD, Grove LM, Rahaman SO, Abraham S, Scheraga RG, Niese KA, et al. Matrix-driven myosin II mediates the pro-fibrotic fibroblast phenotype. J Biol Chem. (2016) 291:6083–95. doi: 10.1074/jbc.M115.712380
50. Rahaman SO, Grove LM, Paruchuri S, Southern BD, Abraham S, Niese KA, et al. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J Clin Invest. (2014) 124:5225–38. doi: 10.1172/JCI75331
51. Hamanaka K, Jian MY, Weber DS, Alvarez DF, Townsley MI, Al-Mehdi AB, et al. TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am J Physiol Lung Cell Mol Physiol. (2007) 293:L923–32. doi: 10.1152/ajplung.00221.2007
52. Hamanaka K, Jian MY, Townsley MI, King JA, Liedtke W, Weber DS, et al. TRPV4 channels augment macrophage activation and ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. (2010) 299:L353–L62. doi: 10.1152/ajplung.00315.2009
53. Balakrishna S, Song W, Achanta S, Doran SF, Liu B, Kaelberer MM, et al. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. (2014) 307:L158–72. doi: 10.1152/ajplung.00065.2014
54. Michalick L, Kuebler WM. TRPV4-A missing link between mechanosensation and immunity. Front Immunol. (2020) 11:413. doi: 10.3389/fimmu.2020.00413
55. Doñate-Macián P, Jungfleisch J, Pérez-Vilaró G, Rubio-Moscardo F, Perálvarez-Marín A, Diez J, et al. The TRPV4 channel links calcium influx to DDX3X activity and viral infectivity. Nat Commun. (2018) 9:2307. doi: 10.1038/s41467-018-04776-7
56. Rayees S, Joshi JC, Tauseef M, Anwar M, Baweja S, Rochford I, et al. PAR2-mediated cAMP generation suppresses TRPV4-dependent Ca2+ signaling in alveolar macrophages to resolve TLR4-induced inflammation. Cell Rep. (2019) 27:793–805.e4. doi: 10.1016/j.celrep.2019.03.053
57. Alpizar YA, Boonen B, Sanchez A, Jung C, López-Requena A, Naert R, et al. TRPV4 activation triggers protective responses to bacterial lipopolysaccharides in airway epithelial cells. Na Commun. (2017) 8:1059. doi: 10.1038/s41467-017-01201-3
58. Gronski MA, Kinchen JM, Juncadella IJ, Franc NC, Ravichandran KS. An essential role for calcium flux in phagocytes for apoptotic cell engulfment and the anti-inflammatory response. Cell Death Diff. (2009) 16:1323–31. doi: 10.1038/cdd.2009.55
59. Fereol S, Fodil R, Labat B, Galiacy S, Laurent VM, Louis B, et al. Sensitivity of alveolar macrophages to substrate mechanical and adhesive properties. Cell Motility Cytoskel. (2006) 63:321–40. doi: 10.1002/cm.20130
60. Yang Y, Kim SC, Yu T, Yi Y-S, Rhee MH, Sung G-H, et al. Functional roles of p38 mitogen-activated protein kinase in macrophage-mediated inflammatory responses. Mediat inflam. (2014) 2014:352371. doi: 10.1155/2014/352371
61. Grove LM, Mohan ML, Abraham S, Scheraga RG, Southern BD, Crish JF, et al. Translocation of TRPV4-PI3Kγ complexes to the plasma membrane drives myofibroblast transdifferentiation. Sci Signal. (2019) 12:eaau1533. doi: 10.1126/scisignal.aau1533
62. Solis AG, Bielecki P, Steach HR, Sharma L, Harman CCD, Yun S, et al. Mechanosensation of cyclical force by PIEZO1 is essential for innate immunity. Nature. (2019) 573:69–74. doi: 10.1038/s41586-019-1485-8
63. Swain SM, Romac JMJ, Shahid RA, Pandol SJ, Liedtke W, Vigna SR, et al. TRPV4 channel opening mediates pressure-induced pancreatitis initiated by Piezo1 activation. J Clin Invest. (2020) 130:2527–41. doi: 10.1172/JCI134111
64. Weinberg JB, Misukonis MA, Shami PJ, Mason SN, Sauls DL, Dittman WA, et al. Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood. (1995) 86:1184–95. doi: 10.1182/blood.V86.3.1184.1184
Keywords: TRPV4 (transient receptor potential vanilloid-4), macrophage, innate immunity, lung inflammation and injury, MAPK
Citation: Scheraga RG, Southern BD, Grove LM and Olman MA (2020) The Role of TRPV4 in Regulating Innate Immune Cell Function in Lung Inflammation. Front. Immunol. 11:1211. doi: 10.3389/fimmu.2020.01211
Received: 24 February 2020; Accepted: 15 May 2020;
Published: 26 June 2020.
Edited by:
Susanna Zierler, Ludwig Maximilian University of Munich, GermanyReviewed by:
Anthony George Tsolaki, Brunel University London, United KingdomWolfgang Liedtke, Duke University, United States
Copyright © 2020 Scheraga, Southern, Grove and Olman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mitchell A. Olman, b2xtYW5tQGNjZi5vcmc=