 Ronit Mazor
Ronit Mazor Ira Pastan
Ira Pastan- 1Division of Cellular and Gene Therapies, Center for Biologics Evaluation and Research, Food and Drug Administration, Silver Spring, MD, United States
- 2Laboratory of Molecular Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, United States
Immunotoxins are cytolytic fusion proteins developed for cancer therapy, composed of an antibody fragment that binds to a cancer cell and a protein toxin fragment that kills the cell. Pseudomonas exotoxin A (PE) is a potent toxin that is used for the killing moiety in many immunotoxins. Moxetumomab Pasudotox (Lumoxiti) contains an anti-CD22 Fv and a 38 kDa portion of PE. Lumoxiti was discovered in the Laboratory of Molecular Biology at the U.S. National Cancer Institute and co-developed with Medimmune/AstraZeneca to treat hairy cell leukemia. In 2018 Lumoxiti was approved by the US Food and Drug Administration for the treatment of drug-resistant Hairy Cell Leukemia. Due to the bacterial origin of the killing moiety, immunotoxins containing PE are highly immunogenic in patients with normal immune systems, but less immunogenic in patients with hematologic malignancies, whose immune systems are often compromised. LMB-100 is a de-immunized variant of the toxin with a humanized antibody that targets mesothelin and a PE toxin that was rationally designed for diminished reactivity with antibodies and B cell receptors. It is now being evaluated in clinical trials for the treatment of mesothelioma and pancreatic cancer and is showing somewhat diminished immunogenicity compared to its un modified parental counterpart. Here we review the immunogenicity of the original and de-immunized PE immunotoxins in mice and patients, the development of anti-drug antibodies (ADAs), their impact on drug availability and their effect on clinical efficacy. Efforts to mitigate the immunogenicity of immunotoxins and its impact on immunogenicity will be described including rational design to identify, remove, or suppress B cell or T cell epitopes, and combination of immunotoxins with immune modulating drugs.
Introduction
Protein and cell based therapeutic agents have great potential to treat many human diseases. However, because many of these contain non-self sequences, they often elicit an immune response that blocks their efficacy. Clinical trials with chimeric antigen receptor-T cells (CAR-T) (1), enzyme replacement therapy (2), monoclonal antibodies (3), antibody drug conjugates (ADCs), immunotoxins (4), and viral based gene therapy vectors (5) have often failed to produce desired effects due to the formation of antibodies that neutralize the activity of the therapeutic agent.
Recombinant immunotoxins (RIT) are chimeric proteins that consist of a targeting element linked to a toxin. The targeting element is commonly an Fv portion of an antibody which targets a specific antigen on tumor or infected cells (6). RITs have been developed to treat a variety of indications, such as blood cancers (7, 8) solid tumors (9–11) graft-vs.-host disease (12), viral infections (13, 14), and autoimmune diseases (15). Pseudomonas exotoxin A (PE, also known as ETA) and diphtheria toxin are both favorable toxins for construction of RITs due to their high potency, expression and purification yields, ease of cloning, and relatively low non-specific toxicity compared to other toxins (16). Both toxins kill cells by catalyzing ADP ribosylation and inactivation of elongation factor 2, which results in arrest of protein translation, a fall in anti apoptotic proteins and apoptosis (11). Both toxins have been used as killing domains in antibody or cytokine targeted drugs and were approved for licensure by regulatory agencies. They represent “first in class” drugs for targeted toxins (17, 18).
Recently (September 2018), Moxetumomab pasudotox (Lumoxiti), whose pre-clinical and early clinical development took place in the Laboratory of Molecular Biology (LMB) at the U.S. National Cancer Institute and whose advanced clinical development took place at AstraZeneca, was approved by the U.S. Food and Drug Administration for the treatment of relapsed or refractory hairy cell leukemia. Lumoxiti is composed of an anti-CD22 Fv murine antibody fused to PE38, a 38 kDa truncated form of PE (Table 1) (26, 27). Encouraged by this success, major efforts are focused on developing PE based RITs against mesothelin and other proteins on solid tumors (20, 28–32).
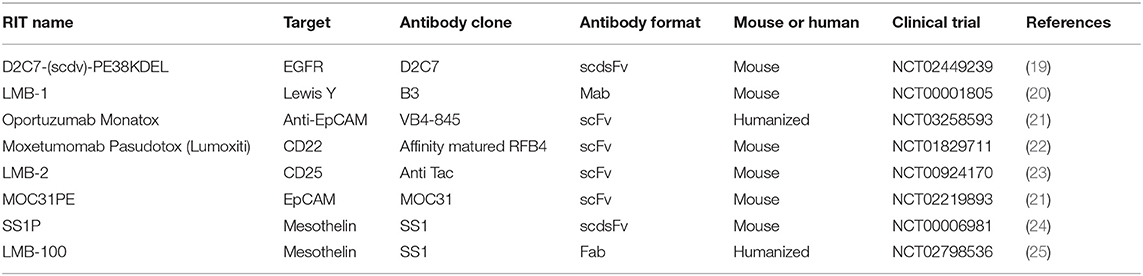
Table 1. Immunotoxins tested in patients.
Pseudomonas Exotoxin a (PE)
PE is the most toxic virulence factor of the opportunistic pathogen Pseudomonas aeruginosa (33), a Gram-negative bacterium (34). Pseudomonas aeruginosa is ubiquitous in soil and water and generally infects only immunocompromised and elderly populations (34). This indicates that immune competent patients can efficiently mount an immune response and maintain an immune memory against Pseudomonas aeruginosa toxins. Indeed, the immunogenicity of the PE based moiety is a major hurdle in immunotoxin clinical development. PE is composed of three structural domains. A binding domain (I), a processing domain (II) and the catalytic domain (III). For RIT construction, the binding domain was replaced with antibody fragments.
Methods to Assess Antibody Responses Against Recombinant Immunotoxins
Clinical development of RITs has been ongoing for about three decades. Immune monitoring of ADA by enzyme-linked immunosorbent assay (ELISA) or neutralizing antibodies (Nab) by neutralization assays (Nab assay) have changed in the past three decades as methods improved and as clinical development progressed. In early trials, ADAs were monitored using direct ELISA assays (28). A functional Nab assay was first reported in 1996 (20). The Nab assay entailed adding serum samples to two concentrations of immunotoxin and adding the mixture to sensitive cells. A sample was considered Nab positive if protein synthesis inhibition (20) or cytotoxic activity (7) was inhibited by 50 or 75%, respectively. Comparison of ADA positive patients and Nab positive assays revealed that all Nab positive samples are ADA positive but not all ADA positive samples are Nab positive (4). This indicates a higher sensitivity for the ADA assays and implies that some of the binding antibodies do not possess neutralization activity.
In the past decade, advancements in ADA monitoring methods and development of ultrasensitive assays have led to more specific and accurate monitoring approaches. Liang and colleague. improved the ELISA assay by minimizing the impact of PE38 immunodominance on the ability to detect ADA against the murine antibody fragment. They tested each patient's sample in a bridging ELISA with biotin-Lumoxiti coated plates in three conditions: with the CD22 fragment, with the PE38 fragment or with both. A signal was obtained using the addition of a ruthenium labeled Lumoxiti (35) and the fragments in the three conditions competed for binding with the ADAs.
To determine the immunogenicity cut-point for Lumoxiti (the OD or neutralization activity at which a sample is considered positive) samples from normal donors are commonly used. Because many naïve donors have been exposed to PE and have pre-existing antibodies, sample manipulation was necessary to obtain a sensitive cut-point for immunogenicity monitoring. To overcome this problem, an irrelevant PE-immunotoxin was added to serum samples to occupy the pre-existing antibodies prior to evaluating samples for cut point establishment (36).
Clinical Immunogenicity of Immunotoxins
Chemical Conjugates
The immunotoxin that was evaluated in a clinical trial (OVB3-PE) (Figure 1A) contained a full length murine antibody (clone OVB3) against an unknown antigen on ovarian cancer cells chemically conjugated to the PE protein (28). Formation of ADA against OVB3-PE was evaluated by ELISA, which showed that 16/16 of the patients developed anti PE ADA within 14 days. Human anti-mouse antibodies (HAMA) were also detected in 75% of the patients.
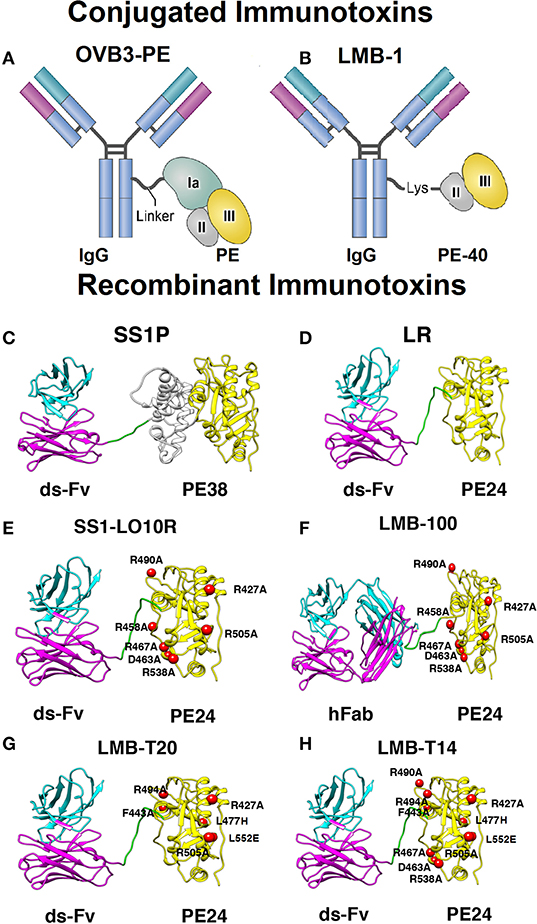
Figure 1. Models and structural models of conjugated and recombinant immunotoxins. (A) OVB3-PE is composed of a mouse IgG chemically conjugated to full length Pseudomonas exotoxin A (PE) using a linker. (B) LMB-1 consists of a mouse IgG chemically conjugated via a lysine residue to a 40 kDa fragment of PE that contains domain II (gray), domain III (yellow), and domain Ib (not shown). (C) SS1P consists of the disulfide-stabilized (ds) heavy chain Fv (VH; magenta) and light chain Fv (VL; cyan) of the antibody fragment SS1. The VH is linked to PE38 that contains domains II and III but Ia is deleted. (D) Lysosome resistance (LR) immunotoxin. The ds-Fv of SS1P is linked to a 24-kDa fragment consisting of domain III of PE38 (termed PE24) (E) SS1-LO10R. A 24-kDa fragment of PE24 with 6-point mutations in domain III designed to suppress binding to B cell receptors. Point mutations are marked with red balls. (F) LMB-100 consists of a humanized Fab linked to LO10R PE24 toxin fragment and 6 point mutations as in E. (G) LMB-T20. PE24 with 6-point mutations in domain III designed to diminish T cell epitopes. (H) LMB-T14. PE24 with 10-point mutations in domain III designed to diminish B and T cell epitopes. All images are based on the structures of native PE and IgG. Images (C–H) were adapted from Mazor et al. (37).
In a clinical trial evaluating LMB-1 (Figure 1B), in which domain I of PE was removed and the remaining 40 kDa protein attached to an antibody to Lewis-Y, 33/39 of the patients developed ADA responses against LMB-1 3 weeks after the first cycle of treatment. The remaining 10% who did not make neutralizing antibodies after the first cycle, were further treated and developed neutralizing antibodies after subsequent treatment cycles. ELISA assays indicated that eventually, 100% of the 38 patients made antibodies against the toxin moiety and 33/38 of the patients had HAMA against the antibody fragment (20).
Recombinant Immunotoxins
LMB-2 (structure similar to SS1P shown in Figure 1C), which is composed of a murine anti CD-25 Fv linked to PE38 (Table 1), was used to treat leukemia and lymphoma patients. Clinical evaluation showed several complete and partial responses; however, 10/35 of the patients developed Nabs, which prevented further treatment. Six of the patients developed Nabs after the first cycle of treatment. Three of the patients that developed Nabs also demonstrated immunogenicity related side effects including one anaphylactic reaction and other allergic grade 2–3 reactions (2/35) (7). These adverse events contra-indicated further treatments once Nabs are present.
The lowest immunogenicity rates were reported in early trials evaluating Lumoxiti for hematological malignancies; After the first treatment cycle, only 1/28 hairy cell leukemia (HCL) patients made Nabs and a total of 10/28 had Nabs throughout the entire phase 1 trial (38). Furthermore, of the 50 CLL patients that were treated with Lumoxiti, only two patients had a Nab response after four cycles of treatment (4, 39).
Phase II and III trials of Lumoxiti were monitored for presence of binding ADA rather than Nabs. In those trials, 65% of patients made ADA after two cycles (38). In a larger trial, 75% of patients had detectable ADA at the end of treatment (40). The difference in immunogenicity reports in the early trials is mostly explained by differences in monitoring methods; functional Nab assays are less sensitive than binding ADA assays.
The overall low rate of immunogenicity to Lumoxiti can be attributed to the immune status of the patients. Patients with HCL have usually been treated with Cladribine which kills immune cells in the bone marrow. Additionally, the leukemia cells infiltrate the marrow, causing immunosuppression. Furthermore, Lumoxiti targets the CD22 antigen which is highly expressed in the targeted cancer cells but also expressed in mature and immature B cells. It is likely that Lumoxiti kills some B cells that would mount an immune response against it.
A good example that exemplifies the importance of patients' immune status is that of LMB-2. Patients with hematological malignancies treated with LMB-2 had a relatively low rate of immunogenicity onset with 17% of the patients making neutralizing antibodies after the first cycle (7). In contrast, melanoma patients who received LMB-2 and had a normal immune system demonstrated a high level of immune response; 92% of patients made neutralizing antibodies after the first cycle (41).
Impact of Immunogenicity on Pharmacokinetics and Clinical Outcome
Generally, ADAs to therapeutic proteins have a risk of immune-related adverse events, including infusion-related reactions, allergic or anaphylactic reactions, delayed hypersensitivity, and autoimmunity (42). RITs show few of these responses. The only severe anaphylactic reaction reported occurred immediately after the first infusion of the RIT (7). Some patients reported grade 1, 2, or 3 skin reactions that were easily managed by a course of steroids [reviewed in (4)]. Neutralization and drug clearance are the main problems with RIT therapy, not immuno-toxicity. The low incidence of adverse side effects could be related to the relatively low doses administered and the small size (63 kDa) of the protein.
LMB-100 is a PE-based RIT engineered for decreased immunogenicity (Figure 1F). To study the impact of ADAs on LMB-100 levels, we analyzed immunogenicity and pharmacokinetic date from a clinical trial treating Pancreatic Ductal Adenocarcinoma with LMB-100 and nab-paclitaxel (25). Anti LMB-100 ADA were monitored using ultrasensitive methods to triage ADA positive and negative responses (screening assay). Patients with pre-existing antibodies were not excluded.
Using a cut point of O.D = 0.05, 9/20 had pre-existing antibodies. These low titers did not have much impact drug levels (Figure 2A). Cmax in 19/20 patients was well above 100 ng/ml. In cycle 2, only 6/13 patients were ADA negative and their Cmax well above 100 ng/ml. 7/13 of the patients were ADA positive still had effective blood levels (Figure 2B). Overall, more than half of patients receiving a second cycle of LMB-100 had detectable plasma drug concentrations. None of the patients received a third cycle of therapy due to toxicity of the nab-pactaxel. However, post treatment ADA monitoring showed that 9/10 of the patients evaluated were ADA positive; most of them with a very high OD signal (25).
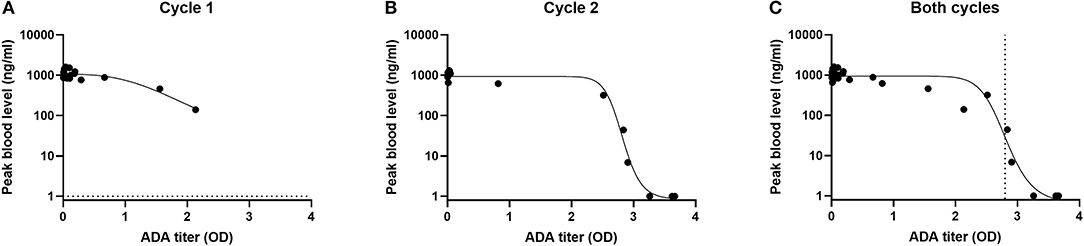
Figure 2. Impact of ADA on maximal concentration of LMB-100 (Cmax) in blood of patients. Cmax and ADA measurements were performed as described in (25). Cmax and ADA results were obtained from (25). Cmax values were log transformed and fitted to an asymmetric sigmodial, 5-parameter curve fit. Dotted line represents EC50.
Altogether, a correlation was observed between the ADA levels and drug blood levels (Figure 2C). Using a five-parameter asymmetric sigmoidal curve fit, EC50 has an OD = 2.8. Therefore, it can be estimated that samples with an OD lower than 2.8 will predict an effective Cmax and samples with OD >than EC50 will not. Previously, patients with positive signals on ADA or Nab assays were excluded from clinical trials. However, this data indicates that a positive call on the ADA assay does not predict a low blood level unless the titers are very high (Figure 2).
Kreitman et al. reported that a minimum of three to five cycles of treatment was required to obtain major responses including durable complete remissions (43). In earlier trials, patients were not allowed to complete the therapy once they developed Nabs. This was to avoid immunological side effects unnecessary ineffective RIT drug administration. Kreitman and colleagues were able to observe a correlation between the timing of antibody formation and the outcome of the treatment (43). In the phase 1 study, 65% of patients made ADAs after two cycles based on ELISA results (38). Most patients (80%) who did not achieve CR had a positive antidrug antibody ELISA test (38). In a larger trial, patients with favorable responses (complete or partial responses) had lower antibody titers (<10,000), which probably improved their drug blood levels for more treatment cycles compared to patients with stable or progressive disease (40). Furthermore, when SS1P (Figure 1C) was combined with pentostatin and cyclophosphamide to lower T and B cells and suppress anti-drug antibodies, more treatment cycles could be given to most of the patients and major tumor responses were observed in several patients with advanced refractory mesothelioma (44). Altogether, these findings indicate that patients with low or delayed immune responses are likely to respond better and justifies the efforts described below to mitigate the ADA response.
Strategies to Mitigate the Immunogenicity of RitS
Combination With Immune Modulating Drugs
Combination approaches to mitigate immunogenicity of RIT include targeting of the B cells that form the adaptive immune response, targeting the plasma cells that produce high titers of IgG, or targeting the T cells that support a neutralizing immune response. In addition, recent approaches have targeted regulatory factors of the immune system that suppress the immune responses (Figure 3).
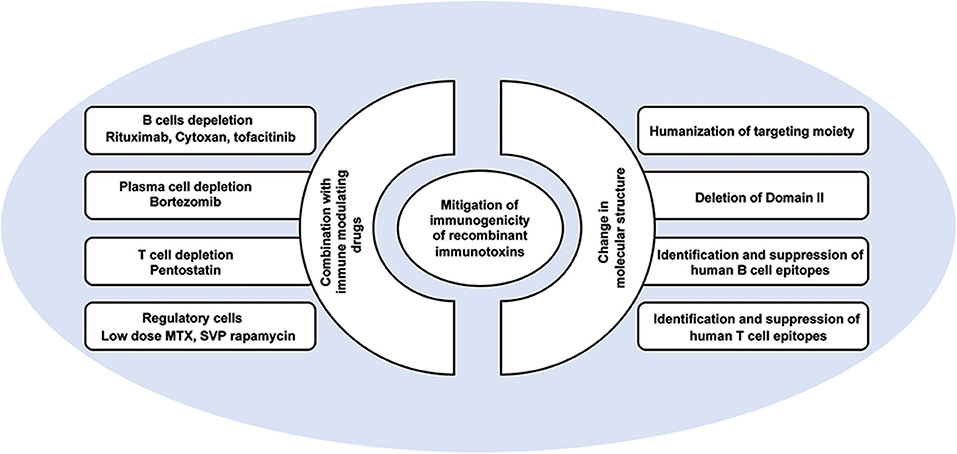
Figure 3. Approaches to mitigate immunogenicity of PE-based recombinant immunotoxins.
In 2004 five patients were pre-treated with rituximab to eliminate their B cells prior to LMB-1 administration. Binding ADA and Nab assay were used to monitor the development of human antibodies against LMB-1. Treatment with rituximab was effective in abrogating 99.9% of circulating CD20/CD19+ B cells in all patients (5/5). However, all these patients developed neutralizing anti-LMB-1 antibodies by day 21 of drug administration (45). This indicates that elimination of the peripheral B cells is not sufficient to eliminate the immune response.
To target both B and T cells, 10 refractory mesothelioma patients were treated with a combination of pentostatin and cyclophosphamide to kill B and T-cells. This combination delayed the formation of neutralizing antibodies to SS1P by several cycles. Out of the 10 patients treated, only two made Nabs after the first cycle, and 6 patients made Nabs after the second cycle. One patient did not make any Nabs throughout six treatment cycles (44). The toxicity observed in the trial described above was similar to the known side effects of pentostatin and cyclophosphamide.
In a T cell leukemia clinical trial evaluating the efficacy of LMB-2 after both cyclophosphamide and fludarabine, a great decrease in immunogenicity was observed. This delay translated to higher drug blood levels for multiple treatment cycles and very good anti-tumor responses (60% of the patients achieved complete remission) (23).
Elimination of pre-existing antibodies and plasma cells is a major goal for RITs due to a high prevalence of pre-existing antibodies from environmental exposure to PE, as well as the need to have more than one treatment cycles. Bortezomib is a reversible proteasome inhibitor that showed high efficacy in targeting long and short lived plasma cells that have high rates of Ig production (46). Manning et al. found that combination of SS1P with Bortezomib was able to reduce ADA formation by 50% compared to SS1P with no immune suppression in mice. Additional combination with pentostatin and cyclophosphamide reduced ADA formation by 88% (47).
Tofacitinib is a janus kinase 1 inhibitor that suppresses inflammatory responses. Treatment of mice with tofacitinib led to reduced numbers of CD127+ pro-B cells and reduction in B cell germinal center formation in mice spleens (48). Because normal Ig levels were still present during tofacitinib treatment, this agent specifically reduced ADAs.
Along with the immune depleting approaches, pre-clinical approaches to evaluate combinations with drug in low concentrations or encapsulated in nanoparticles have shown promising results. Low dose methotrexate (MTX) has been shown to reduce ADA formation against adalimumab [reviewed in (49, 50)] and against enzyme replacement therapy for infantile Pompe disease (51). Combination of low dose MTX with LMB-100 suppressed the formation of ADAs, maintained blood levels of LMB-100 and prevented its neutralization in immune competent mice. This did not compromise the immune response against a second antigen given after stopping MTX, suggesting contemporaneous immune tolerance (52).
To harness the immune modulatory properties of rapamycin, LMB collaborated with Selecta Bioscience that had encapsulated rapamycin in PLGA-PEG synthetic vaccine particles (SVP-R). Combination of SVP-R with LMB-100 produced a specific and transferable immune tolerance, which prevented ADA and Nab formation against the RIT in naïve mice and in mice that model pre-existing immunity (53). This approach was quickly translated to a clinical trial combining the two agents to treat mesothelioma patients. However, the combination resulted in an unforeseen lung toxicity in this patient population and the trial was discontinued (clinicaltrials.gov T03436732).
Change in Molecular Structure
The two-unit structure of RITs which includes a targeting antibody unit and a toxin unit, and the variable immunogenicity properties of those two units (i.e., preexisting antibodies to the toxin or the presence of a murine fragment in the antibody) allows tailored mitigation to each unit based on its properties and what is known in the art as de-immunization (Figure 3).
Mitigating the Immunogenicity of the Antibody Domain
Immunogenicity of therapeutic monoclonal antibodies can be mitigated by increasing the content of the human sequence. Such antibody engineering includes framework humanization, chimerization, and use of mice with humanized germlines. Such approaches can reduce the common immunogenicity rate from about 40% in chimeric antibodies to 9% in humanized antibodies (54, 55). However, in some cases, the immunogenicity against the variable complementarity determining region domains (CDRs) may still cause ADA formation (55).
Most RITs have a murine antibody fragment (Table 1). The lack of humanization of these agents can be explained by the fact that their development began before approaches to humanize antibodies were readily available. Furthermore, the immunogenicity of the bacterial PE is a much bigger barrier than HAMA (56). Recently, LMB-100 a “second generation RIT” containing a humanized Fab instead of a mouse Fv has entered clinical testing (Table 1) (25). The humanization was done by combining framework regions in the CDRs of the mouse anti-mesothelin antibody SS1 and human Fab. To improve the binding to mesothelin and to stabilize the CDRs tertiary structure, some back mutations within the mouse parent residues as well as the human sequences were introduced (as described in patent WO2015051199). The new humanized antibody had comparable binding affinity to mesothelin, and LMB-100 showed comparable thermal stability and technical developability to that of SS1P (57).
Mitigating the Immunogenicity of the Toxin
Identification of B Cell Epitopes
Antibodies and B-cell receptors bind to regions on the surface of a protein called B cell epitopes. These epitopes often cluster on the surface of the antigen and can control most immune responses (58). Roscoe et al. used synthetic peptides from PE38 to map the B cell epitopes in serum samples from monkeys and humans treated with immunotoxins (59, 60). This approach identified linear epitopes but not discontinuous conformational B cell epitopes on the toxin. Onda and Nagata immunized mice with RIT and used a capture assay to isolate monoclonal antibodies that reacted with native PE38 in solution (61). They discovered seven murine conformational epitopes in PE38 and identified single point alanine substitutions that abolished binding to those antibodies. They constructed and characterized a novel de-immunized mouse RIT named 8M. This RIT retained excellent cytotoxic and anti-tumor activity and importantly, had a low immunogenicity response after injection into mice. These experiments established the first proof that removing B cell epitopes could greatly diminish immunogenicity (61, 62).
Human B-cell epitopes in domain III were mapped using phage display. These studies focused on domain III of PE, because it was found that most of domain II was not needed to make active immunotoxins and could be removed (63). B cells were isolated from 7 patients receiving immunotoxin therapy and phage Fv libraries was prepared from B cells that contained Fvs reacting with domain III of PE. This selected library should represent the antibody repertoire that can bind and neutralize RITs with domain III. Then an immunotoxin library was constructed. This library contained 36 mutant PE immunotoxin constructs, each with a single point mutation replacing large amino acids like arginine, glutamine and glutamic acid with alanine. Then, the phage library was panned against each mutant RIT in the mutant library, identifying point mutations that abolish binding (64). Seven major B cell epitopes were identified and subsequently silenced by converting a key residue in the epitope to alanine. The modified toxin was named LO10 (Figure 1E) (representing the initials of the last name of the two scientists developing it). The LO10 toxin (Table 2) has been used to make immunotoxins targeting both CD22 and mesothelin. LMB-100 contains the LO10 mutations and is the first “de-immunized” PE based toxin that has advanced to clinical development.
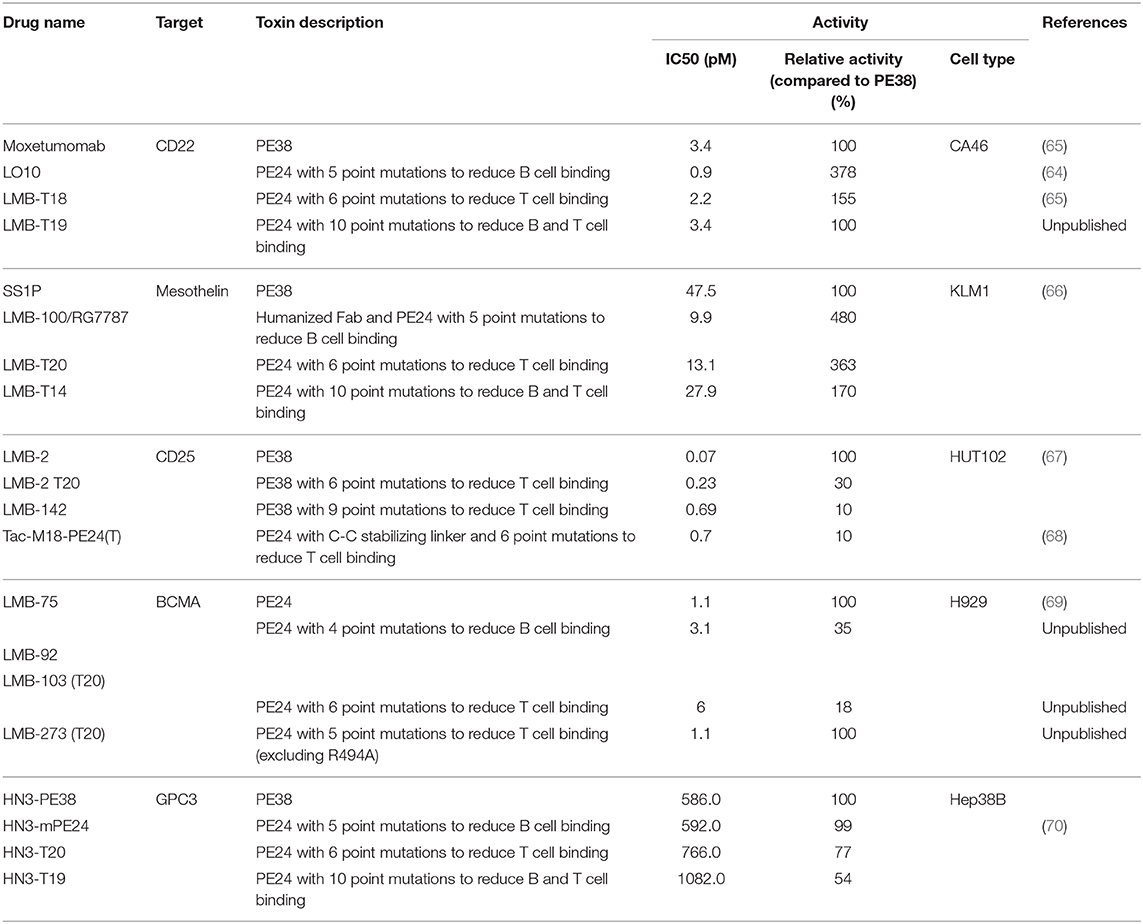
Table 2. Recombinant Immunotoxins that were mutated to decrease immunogenicity.
A similar approach was used to identify B cell epitopes in diphtheria toxin (71). Highly hydrophilic amino acids on the surface of the toxin were mutated, and the mutant constructs were injected into mice for screening. Constructs that did not activate the mouse immune system are speculated to be of low immunogenicity in humans as well (71). This approach, while simpler than the strategy used to generate LO10, suffers from the fact that mice and human have different self and non-self-selection, and immunogenic regions that activate a human immune system may not activate a mouse immune system.
Deletion of Domain II of PE38
Protease evasion can reduce processing of the protein in the endosome and late endosome and therefore, reduce peptide presentation by MHC II molecules and T-cell activation. Weldon et al. found that domain II of PE38 was very sensitive to lysosomal protease digestion and furthermore that 102/113 amino acids in domain II can be removed without loss of activity as long as the furin cleavage site (in amino acids 274–284) remained (72) (Figure 1D). Deletion of the majority of domain II had the additional benefit of deletion of the immunogenic B and T cell epitopes in that domain. RITs with the resulting mutant toxin (designated LR for lysosome protease resistance) or PE24 (Table 2) were tested in three strains of mice and showed a greatly decreased antibody response (73).
T Cell Epitopes
Elimination of B cell epitopes as described above should be effective in evading pre-existing antibodies. However, deletion of the immunodominant B cell epitopes cannot prevent B cells with low affinity B-cell-receptors from undergoing affinity maturation and class switching. These processes are supported by professional antigen presenting cells and helper T cells (58, 74). Unlike B cells, T-cell receptor specificity, does not change on antigen encounter. Once T-cell epitopes are eliminated, formation of new specificities is not expected (75). In a proof of concept study, the murine T cell epitopes in PE38 were mapped using a peptide library and IL2 ELISpot of immunized mice spleens. Alanine scanning of each amino acid within 15 mer epitopes revealed single point mutations that can prevent the T cell response. A new RIT was constructed with several point mutations in PE38 that were effective on preventing anti PE antibodies and Nabs (76). Additional studies in BALB/c mice reinforced the identification of a subdominant murine T cell epitope in domain III (77). This study also showed that a slightly modified version of the de-immunized PE (A505H) using a different mode of administration and adjuvant has a significantly lower immunogenicity compared to PE24.
The human T cell epitopes in PE38 mapped using PBMCs from 50 donors that share similar HLA to the typical patient population in the western world. The PBMC were expanded with PE38 to allow antigen processing and presentation and enrich the T cells that recognize PE38 epitopes (78). The enriched T cells were re-stimulated with over-lapping peptides that span the sequence of PE38. T cell activation was monitored d using IL-2 ELISpot (79). IL-2 supports T-cell activation, differentiation, and memory and is a less specialized cytokine than IL-4 or IFN-γ (80). Twenty-three peptides whose sequence overlap had positive responses and made up eight T cell epitopes (65). One of these epitopes, located in domain II, was present in 21/50 donors (79). The eight T cell epitopes identified in naïve donor PBMC were also identified using samples from 16 cancer patients previously treated with PE38 containing RITs and who had mounted an immune response to the protein. This supports the conclusion that PE38 has eight T cell epitopes and other regions of the protein are less immunogenic. Interestingly, similar assays using PBMCs from immunized HCL patients show several epitopes missing (65). Further work will address the absence of some epitopes in HCL patients and why cells recognizing these epitopes are absent in these patients.
HLA binding algorithms that predict the binding affinity of peptides to polymorphic HLA II molecules can be used to predict or narrow down peptides for potential T-cell epitopes. Overpredictions are expected for such algorithm predicted epitopes due to various factors involved in T-cell activation that cannot be predicted by the HLA binding, including antigen processing in the endosome, T-cell receptor binding, and T-cell activation. To compare the experimentally identified epitopes with in silico predicted epitopes, two primary HLA binding algorithms: Propred (81) and IEDB Consensus (82) were used to predict promiscuous binding to 15 common HLA DR alleles (83). Venn diagrams showing comparison of the predicted peptides using the in-silico analysis and the experimental approach is shown in Figure 4. The top 30 stringently predicted peptides had an overlap of 15 peptides with the 23 experimental peptides. This left 8 peptides (representing four epitopes) mis-identified by the analysis as negative. A less stringent threshold of 56 peptides (choosing 50% of the peptides as positive) had a much better precision and predicted 21 of the 23 peptides. However, the epitope in peptides 8 and 9 was overlooked by the algorithm. While overpredictions are expected, underpredictions are not, and they such reduce the effectiveness of these computational tools for prediction of T-cell epitopes. HLA binding inhibition assays revealed that the missed epitope in peptides 8 and 9 was solely presented by HLA DP presentation molecules and not DR (84). The algorithm could not have predicted binding to this peptide, because the query was limited to DR alleles. Interestingly, re-analysis of the HLA binding prediction by adding 8 DP alleles still failed to recognize this epitope as a strong binder (84). This indicates that HLA binding algorithms cannot accurately predict all T-cell epitopes and should always be validated with experimental work.
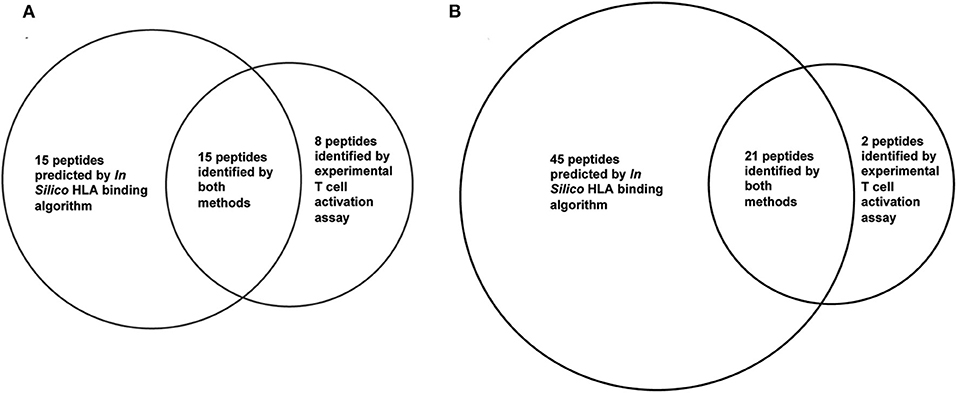
Figure 4. Overlap of experimental T cell epitopes and in silico HLA binding prediction. Twenty three T cell epitope were identified in PE38 by experimental T cell activation assays using 50 PBMC donors. In silico predicted binders in PE38 were predicted using the IEDB consensus HLA class II binding algorithm and 15 HLA-DR alleles. A peptide was considered a potential epitope using a threshold of (A) top 30 predicted binders and (B) top 56 predicted binders (50%). Peptides 8 and 9 were false negatively predicted using both thresholds.
The epitopes in domain II of PE38 were eliminated by deletion of the whole domain, except for the 11 amino acid furin cleavage site, which does not contain an epitope. To modify the epitopes in domain III, alanine scanning was used to identify amino acids that impact the T-cell epitopes. To ensure that the point mutation did not introduce a new T cell epitope PBMCs were stimulated with the mutant RIT and re-stimulated with the mutant peptides. Two of the epitopes (epitopes 2 and 6) were difficult to solve by alanine scanning, because the mutations caused loss in activity. To aid with that, Rosetta computational protein design methods was combined with an HLA binding algorithm to identify mutations that disrupt the binding to HLA II molecule and as the same time still maintained cytotoxic activity (85). Epitope 2 (in domain III) was not resolved using a single point mutation and required a combination of two-point mutations to diminish the T cell responses significantly (R494A and R505A). However, the cytotoxic activity was reduced 2- to 3-fold by one of these mutations (Table 2).
The six point mutations designed to remove of suppress T cell epitopes were combined into new RITs. LMB-T18 targets CD22 (65), LMB-T20 (Figure 1G), targets mesothelin, HN3-T20 targets GP3 (70), and LMB-273 targets BCMA (69). Each protein contains the mutated toxin as shown in Table 2. Re-analysis of LMB-T20 for T cell activation showed that cryptic or new epitopes did not emerge as a result of altered antigen processing in LMB-T20 (86).
Interestingly, when the de-immunized toxin used to make a RIT that targets human CD25 to kill human T-cell malignancies, the deletion of domain II significantly impaired the cytotoxic activity (67, 68). The dependency on domain II for cytotoxic activity is receptor specific and probably attributable to a variable internalization pathway. To improve the cytotoxic activity of CD25-targeting immunotoxin, PE38 was de-immunized with three more mutations in domain II (Table 2).
B and T Cell De-immunized Immunotoxin
Intriguingly, two of the mutations intended to eliminate T cell epitopes are the same mutations that diminished binding to B cell epitopes. Both (R505A and R427A) have very high accessible surface area (150 and 142 Å, respectively) indicating these arginines are located on the surface of the molecule. Since B cell epitopes are known to contain bulky hydrophilic amino acids like arginine (87–89), it is not surprising that mutations that diminish T cell epitopes also diminish B cell epitopes. Other reports have shown that important epitopes may be shared by B and T cells (90–92), and a functional link between B and T cells that recognize overlapping peptides has been suggested (93).
To reduce reactivity with both B and T-cells, the mutations that eliminated T- and B-cell epitopes were incorporated into a single RIT that targets mesothelin (66). The final RIT (LMB-T14) (Figure 1H) has good cytotoxic and anti-tumor activity vs. human cell lines, patient-derived cells, and mouse tumor models. LMB-14 has reduced binding to serum from patients who developed antibodies compared to its unmutated parental immunotoxin. Unexpectedly, remapping of T-cell epitopes of LMB-T14 revealed that two mutations, that were introduced to eliminate conformational B-cell epitope, created a new T-cell epitope. This demonstrates the challenging balance between cytotoxic activity, B-cell and T-cell reactivity during de-immunization (66).
Translation of De-immunization Effort (Immunogenicity of LMB-100)
The effectivity of T cell de-immunization efforts has not yet been tested in clinical settings. However, LMB-100, a B cell de-immunized RIT, has been tested in a recent trial.
It is difficult to compare the immunogenicity rate in this study to previous ones due to significant variation in the immunogenicity monitoring assays. While the immunogenicity response against SS1P was mostly monitored using a functional Nab assay, immunogenicity response against LMB-100 was monitored using an ADA bridge ELISA. Furthermore, blood half time concentration cannot be compared due to differences in dose, size and structure that can impact half time regardless of immunogenicity. Lastly, the clinical design of the SS1P study excluded patients who had elevated pre-existing antibodies to SS1P, presumably due to prior exposure to Pseudomonas aeruginosa, while 9/20 patients in the LMB-100 trial had pre-existing ADA.
To try and compare SS1P and its de-immunized counterpart, Alewine et al. compared the number of patients with effective RIT Cmax levels (>100 ng/ml). They noted that more than half of patients receiving a second cycle of LMB-100 had detectable plasma drug concentrations. These results compare favorably with SS1P, for which more than 90% of the patients had undetectable drug levels by the start of cycle 2, after excluding patients with high preexisting antibodies (94, 95). This clearly indicates that LMB-100 de-immunization decreased the impact of immunogenicity. However, this improvement was only enough to allow one additional dose on the second cycle for most patients. Only a single patient was ADA negative after the completion of the therapy. We conclude that humanization of the antibody and silencing of the B cell epitopes (and some of the T cell epitopes) is helpful, but not sufficient to completely prevent an immune response. Future work is required to evaluate if the complete T cell de-immunized molecules (LMB-T20 and LMB-T14) are more effective in diminishing the immune response.
Summary and Conclusion
In this review, we described various methods to monitor the immune response against RITs and efforts made to minimize the immunogenicity response in patients by combination therapy or rational design. LMB-100 is the first humanized and de-immunized RIT that was rationally designed for reduced B cell epitopes and evaluated in patient. Although it showed lower rates of immunogenicity compared to its parental RIT (SSIP), formation of ADA and Nab was delayed but not eradicated. Future work will require evaluation of novel approaches like elimination of both the B and T cell epitopes or combination therapy of immune suppressive agents and the de-immunized RIT.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This research was supported by the Intramural Research Program of the NIH, NCI, CCR.
Conflict of Interest
Drs. IP and RM are inventors on patents describing how to make less immunogenic immunotoxins. These patents have all been assigned to NIH.
Acknowledgments
We thank Dr. John Weldon for providing helpful comments.
References
1. Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. (2013) 1:26–31. doi: 10.1158/2326-6066.CIR-13-0006
2. Harmatz P. Enzyme replacement therapies and immunogenicity in lysosomal storage diseases: is there a pattern? Clin Ther. (2015) 37:2130–4. doi: 10.1016/j.clinthera.2015.06.004
3. Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med. (2008) 358:1109–17. doi: 10.1056/NEJMoa074943
4. Mazor R, Onda M, Pastan I. Immunogenicity of therapeutic recombinant immunotoxins. Immunol Rev. (2016) 270:152–64. doi: 10.1111/imr.12390
5. Sun JY, Chatterjee S, Wong KKJr. Immunogenic issues concerning recombinant adeno-associated virus vectors for gene therapy. Curr Gene Ther. (2002) 2:485–500. doi: 10.2174/1566523023347616
6. Wilkins DK, Mayer A. Development of antibodies for cancer therapy. Expert Opin Biol Ther. (2006) 6:787–96. doi: 10.1517/14712598.6.8.787
7. Kreitman RJ, Wilson WH, White JD, Stetler-Stevenson M, Jaffe ES, Giardina S, et al. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J Clin Oncol. (2000) 18:1622–36. doi: 10.1200/JCO.2000.18.8.1622
8. Wayne AS, Fitzgerald DJ, Kreitman RJ, Pastan I. Immunotoxins for leukemia. Blood. (2014) 123:2470–7. doi: 10.1182/blood-2014-01-492256
9. Sausville EA, Headlee D, Stetler-Stevenson M, Jaffe ES, Solomon D, Figg WD, et al. Continuous infusion of the anti-CD22 immunotoxin IgG-RFB4-SMPT-dgA in patients with B-cell lymphoma: a phase I study. Blood. (1995) 85:3457–65. doi: 10.1182/blood.V85.12.3457.bloodjournal85123457
10. Azemar M, Djahansouzi S, Jager E, Solbach C, Schmidt M, Maurer AB, et al. Regression of cutaneous tumor lesions in patients intratumorally injected with a recombinant single-chain antibody-toxin targeted to ErbB2/HER2. Breast Cancer Res Treat. (2003) 82:155–64. doi: 10.1023/B:BREA.0000004371.48757.19
11. Shafiee F, Aucoin MG, Jahanian-Najafabadi A. Targeted diphtheria toxin-based therapy: a review article. Front Microbiol. (2019) 10:2340. doi: 10.3389/fmicb.2019.02340
12. Groth C, van Groningen LFJ, Matos TR, Bremmers ME, Preijers F, Dolstra H, et al. Phase I/II trial of a combination of anti-CD3/CD7 immunotoxins for steroid-refractory acute graft-versus-host disease. Biol Blood Marrow Transplant. (2019) 25:712–9. doi: 10.1016/j.bbmt.2018.10.020
13. Berger EA, Pastan I. Immunotoxin complementation of HAART to deplete persisting HIV-infected cell reservoirs. PLoS Pathog. (2010) 6:e1000803. doi: 10.1371/journal.ppat.1000803
14. Krishna BA, Spiess K, Poole EL, Lau B, Voigt S, Kledal TN, et al. Targeting the latent cytomegalovirus reservoir with an antiviral fusion toxin protein. Nat Commun. (2017) 8:14321. doi: 10.1038/ncomms14321
15. Zhao P, Wang P, Dong S, Zhou Z, Cao Y, Yagita H, et al. Depletion of PD-1-positive cells ameliorates autoimmune disease. Nat Biomed Eng. (2019) 3:292–305. doi: 10.1038/s41551-019-0360-0
16. Antignani A, Fitzgerald D. Immunotoxins: the role of the toxin. Toxins (Basel). (2013) 5:1486–502. doi: 10.3390/toxins5081486
17. Eklund JW, Kuzel TM. Denileukin diftitox: a concise clinical review. Expert Rev Anticancer Ther. (2005) 5:33–8. doi: 10.1586/14737140.5.1.33
18. Fancher KM, Lally-Montgomery ZC. Moxetumomab pasudotox: a first-in-class treatment for hairy cell leukemia. J Oncol Pharm Pract. (2019) 25:1467–72. doi: 10.1177/1078155219838041
19. Chandramohan V, Bao X, Keir ST, Pegram CN, Szafranski SE, Piao H, et al. Construction of an immunotoxin, D2C7-(scdsFv)-PE38KDEL, targeting EGFRwt and EGFRvIII for brain tumor therapy. Clin Cancer Res. (2013) 19:4717–27. doi: 10.1158/1078-0432.CCR-12-3891
20. Pai LH, Wittes R, Setser A, Willingham MC, Pastan I. Treatment of advanced solid tumors with immunotoxin LMB-1: an antibody linked to Pseudomonas exotoxin. Nat Med. (1996) 2:350–3. doi: 10.1038/nm0396-350
21. Andersson Y, Engebraaten O, Juell S, Aamdal S, Brunsvig P, Fodstad O, et al. Phase I trial of EpCAM-targeting immunotoxin MOC31PE, alone and in combination with cyclosporin. Br J Cancer. (2015) 113:1548–55. doi: 10.1038/bjc.2015.380
22. Kreitman RJ, Pastan I. Contextualizing the use of moxetumomab pasudotox in the treatment of relapsed or refractory hairy cell leukemia. Oncologist. (2020) 25:e170–7. doi: 10.1634/theoncologist.2019-0370
23. Kreitman RJ, Stetler-Stevenson M, Jaffe ES, Conlon KC, Steinberg SM, Wilson W, et al. Complete remissions of adult T-cell leukemia with anti-CD25 recombinant immunotoxin LMB-2 and chemotherapy to block immunogenicity. Clin Cancer Res. (2016) 22:310–8. doi: 10.1158/1078-0432.CCR-15-1412
24. Hassan R, Thomas A, Alewine C, Le DT, Jaffee EM, Pastan I. Mesothelin immunotherapy for cancer: ready for prime time? J Clin Oncol. (2016) 34:4171–9. doi: 10.1200/JCO.2016.68.3672
25. Alewine C, Ahmad M, Peer CJ, Hu ZI, Lee MJ, Yuno A, et al. Phase I/II study of the mesothelin-targeted immunotoxin LMB-100 with nab-paclitaxel for patients with advanced pancreatic adenocarcinoma. Clin Cancer Res. (2019) 26:828–36. doi: 10.1158/1538-7445.PANCA19-PR07
26. Dhillon S. Moxetumomab pasudotox: first global approval. Drugs. (2018) 78:1763–7. doi: 10.1007/s40265-018-1000-9
27. Lin AY, Dinner SN. Moxetumomab pasudotox for hairy cell leukemia: preclinical development to FDA approval. Blood Adv. (2019) 3:2905–10. doi: 10.1182/bloodadvances.2019000507
28. Pai LH, Bookman MA, Ozols RF, Young RC, Smith JW II, Longo DL, et al. Clinical evaluation of intraperitoneal Pseudomonas exotoxin immunoconjugate OVB3-PE in patients with ovarian cancer. J Clin Oncol. (1991) 9:2095–103. doi: 10.1200/JCO.1991.9.12.2095
29. Hassan R, Kreitman RJ, Pastan I, Willingham MC. Localization of mesothelin in epithelial ovarian cancer. Appl Immunohistochem Mol Morphol. (2005) 13:243–7. doi: 10.1097/01.pai.00000141545.36485.d6
30. Tang Z, Qian M, Ho M. The role of mesothelin in tumor progression and targeted therapy. Anticancer Agents Med Chem. (2013) 13:276–80. doi: 10.2174/1871520611313020014
31. Alewine C, Xiang L, Yamori T, Niederfellner G, Bosslet K, Pastan I. Efficacy of RG7787, a next-generation mesothelin-targeted immunotoxin, against triple-negative breast and gastric cancers. Mol Cancer Ther. (2014) 13:2653–61. doi: 10.1158/1535-7163.MCT-14-0132
32. Hollevoet K, Mason-Osann E, Liu XF, Imhof-Jung S, Niederfellner G, Pastan I. In vitro and in vivo activity of the low-immunogenic antimesothelin immunotoxin RG7787 in pancreatic cancer. Mol Cancer Ther. (2014) 13:2040–9. doi: 10.1158/1535-7163.MCT-14-0089-T
33. Michalska M, Wolf P. Pseudomonas exotoxin A: optimized by evolution for effective killing. Front Microbiol. (2015) 6:963. doi: 10.3389/fmicb.2015.00963
34. Wolf P, Elsasser-Beile U. Pseudomonas exotoxin A: from virulence factor to anti-cancer agent. Int J Med Microbiol. (2009) 299:161–76. doi: 10.1016/j.ijmm.2008.08.003
35. Vainshtein I, Sun B, Roskos LK, Liang M. A novel approach to assess domain specificity of anti-drug antibodies to moxetumomab pasudotox, an immunotoxin with two functional domains. J Immunol Methods. (2019) 477:112688. doi: 10.1016/j.jim.2019.112688
36. Schneider AK, Vainshtein I, Roskos LK, Chavez C, Sun B, Liang M. An immunoinhibition approach to overcome the impact of pre-existing antibodies on cut point establishment for immunogenicity assessment of moxetumomab pasudotox. J Immunol Methods. (2016) 435:68–76. doi: 10.1016/j.jim.2016.05.007
37. Mazor R, King EM, Pastan I. Strategies to reduce the immunogenicity of recombinant immunotoxins. Am J Pathol. (2018) 188:1736–43. doi: 10.1016/j.ajpath.2018.04.016
38. Kreitman RJ, Tallman MS, Robak T, Coutre S, Wilson WH, Stetler-Stevenson M, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol. (2012) 30:1822–8. doi: 10.1200/JCO.2011.38.1756
39. Kreitman RJ, Squires DR, Stetler-Stevenson M, Noel P, FitzGerald DJ, Wilson WH, et al. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J Clin Oncol. (2005) 23:6719–29. doi: 10.1200/JCO.2005.11.437
40. Kreitman RJ, Dearden C, Zinzani PL, Delgado J, Karlin L, Robak T, et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia. (2018) 32:1768–77. doi: 10.1038/s41375-018-0210-1
41. Powell DJ Jr, Felipe-Silva A, Merino MJ, Ahmadzadeh M, Allen T, Levy C, et al. Administration of a CD25-directed immunotoxin, LMB-2, to patients with metastatic melanoma induces a selective partial reduction in regulatory T cells in vivo. J Immunol. (2007) 179:4919–28. doi: 10.4049/jimmunol.179.7.4919
42. Khan DA. Hypersensitivity and immunologic reactions to biologics: opportunities for the allergist. Ann Allergy Asthma Immunol. (2016) 117:115–20. doi: 10.1016/j.anai.2016.05.013
43. Kreitman RJ. Immunoconjugates and new molecular targets in hairy cell leukemia. Hematology Am Soc Hematol Educ Program. (2012) 2012:660–6. doi: 10.1182/asheducation.V2012.1.660.3798659
44. Hassan R, Miller AC, Sharon E, Thomas A, Reynolds JC, Ling A, et al. Major cancer regressions in mesothelioma after treatment with an anti-mesothelin immunotoxin and immune suppression. Sci Transl Med. (2013) 5:208ra147. doi: 10.1126/scitranslmed.3006941
45. Hassan R, Williams-Gould J, Watson T, Pai-Scherf L, Pastan I. Pretreatment with rituximab does not inhibit the human immune response against the immunogenic protein LMB-1. Clin Cancer Res. (2004) 10:16–8. doi: 10.1158/1078-0432.CCR-1160-3
46. Meister S, Schubert U, Neubert K, Herrmann K, Burger R, Gramatzki M, et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. (2007) 67:1783–92. doi: 10.1158/0008-5472.CAN-06-2258
47. Manning ML, Mason-Osann E, Onda M, Pastan I. Bortezomib reduces pre-existing antibodies to recombinant immunotoxins in mice. J Immunol. (2015) 194:1695–701. doi: 10.4049/jimmunol.1402324
48. Onda M, Ghoreschi K, Steward-Tharp S, Thomas C, O'Shea JJ, Pastan IH, et al. Tofacitinib suppresses antibody responses to protein therapeutics in murine hosts. J Immunol. (2014) 193:48–55. doi: 10.4049/jimmunol.1400063
49. Jani M, Barton A, Warren RB, Griffiths CEM, Chinoy H. The role of DMARDs in reducing the immunogenicity of TNF inhibitors in chronic inflammatory diseases. Rheumatology (Oxford). (2014) 53:213–22. doi: 10.1093/rheumatology/ket260
50. Strik AS, van den Brink GR, Ponsioen C, Mathot R, Löwenberg M, D'Haens GR. Suppression of anti-drug antibodies to infliximab or adalimumab with the addition of an immunomodulator in patients with inflammatory bowel disease. Aliment Pharmacol Ther. (2017) 45:1128–34. doi: 10.1111/apt.13994
51. Messinger YH, Mendelsohn NJ, Rhead W, Dimmock D, Hershkovitz E, Champion M, et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet Med. (2012) 14:135–42. doi: 10.1038/gim.2011.4
52. King EM, Mazor R, Cuburu N, Pastan I. Low-dose methotrexate prevents primary and secondary humoral immune responses and induces immune tolerance to a recombinant immunotoxin. J Immunol. (2018) 200:2038–45. doi: 10.4049/jimmunol.1701430
53. Mazor R, King EM, Onda M, Cuburu N, Addissie S, Crown D, et al. Tolerogenic nanoparticles restore the antitumor activity of recombinant immunotoxins by mitigating immunogenicity. Proc Natl Acad Sci USA. (2018) 115:E733–42. doi: 10.1073/pnas.1717063115
54. Hwang WY, Foote J. Immunogenicity of engineered antibodies. Methods. (2005) 36:3–10. doi: 10.1016/j.ymeth.2005.01.001
55. Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. (2010) 2:256–65. doi: 10.4161/mabs.2.3.11641
56. Nagata S, Pastan I. Removal of B cell epitopes as a practical approach for reducing the immunogenicity of foreign protein-based therapeutics. Adv Drug Deliv Rev. (2009) 61:977–85. doi: 10.1016/j.addr.2009.07.014
57. Bauss F, Lechmann M, Krippendorff BF, Staack R, Herting F, Festag M, et al. Characterization of a re-engineered, mesothelin-targeted Pseudomonas exotoxin fusion protein for lung cancer therapy. Mol Oncol. (2016) 10:1317–29. doi: 10.1016/j.molonc.2016.07.003
58. Berzofsky JA. Intrinsic and extrinsic factors in protein antigenic structure. Science. (1985) 229:932–40. doi: 10.1126/science.2410982
59. Roscoe DM, Jung SH, Benhar I, Pai L, Lee BK, Pastan I. Primate antibody response to immunotoxin: serological and computer-aided analysis of epitopes on a truncated form of Pseudomonas exotoxin. Infect Immun. (1994) 62:5055–65. doi: 10.1128/IAI.62.11.5055-5065.1994
60. Roscoe DM, Pai LH, Pastan I. Identification of epitopes on a mutant form of Pseudomonas exotoxin using serum from humans treated with Pseudomonas exotoxin containing immunotoxins. Eur J Immunol. (1997) 27:1459–68. doi: 10.1002/eji.1830270624
61. Onda M, Nagata S, FitzGerald DJ, Beers R, Fisher RJ, Vincent JJ, et al. Characterization of the B cell epitopes associated with a truncated form of Pseudomonas exotoxin (PE38) used to make immunotoxins for the treatment of cancer patients. J Immunol. (2006) 177:8822–34. doi: 10.4049/jimmunol.177.12.8822
62. Onda M, Beers R, Xiang L, Nagata S, Wang QC, Pastan I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc Natl Acad Sci USA. (2008) 105:11311–6. doi: 10.1073/pnas.0804851105
63. Weldon JE, Pastan I. A guide to taming a toxin–recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. (2011) 278:4683–700. doi: 10.1111/j.1742-4658.2011.08182.x
64. Liu W, Onda M, Lee B, Kreitman RJ, Hassan R, Xiang L, et al. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc Natl Acad Sci USA. (2012) 109:11782–7. doi: 10.1073/pnas.1209292109
65. Mazor R, Eberle JA, Hu X, Vassall AN, Onda M, Beers R, et al. Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc Natl Acad Sci USA. (2014) 111:8571–6. doi: 10.1073/pnas.1405153111
66. Mazor R, Onda M, Park D, Addissie S, Xiang L, Zhang J, et al. Dual B- and T-cell de-immunization of recombinant immunotoxin targeting mesothelin with high cytotoxic activity. Oncotarget. (2016) 7:29916–26. doi: 10.18632/oncotarget.9171
67. Mazor R, Kaplan G, Park D, Jang Y, Lee F, Kreitman R, et al. Rational design of low immunogenic anti CD25 recombinant immunotoxin for T cell malignancies by elimination of T cell epitopes in PE38. Cell Immunol. (2017) 313:59–66. doi: 10.1016/j.cellimm.2017.01.003
68. Kaplan G, Mazor R, Lee F, Jang Y, Leshem Y, Pastan I. Improving the in vivo efficacy of an anti-Tac (CD25) immunotoxin by Pseudomonas exotoxin A domain II engineering. Mol Cancer Ther. (2018) 17:1486–93. doi: 10.1158/1535-7163.MCT-17-1041
69. Shancer Z, Williams M, Igelman A, Nagata S, Ise T, Pastan I, et al. Preclinical development of anti-BCMA immunotoxins targeting multiple myeloma. Antib Ther. (2018) 1:19–25. doi: 10.1093/abt/tby004
70. Fleming BD, Urban DJ, Hall MD, Longerich T, Greten TF, Pastan I, et al. Engineered anti-GPC3 immunotoxin, HN3-ABD-T20, produces regression in mouse liver cancer xenografts through prolonged serum retention. Hepatology. (2019) 71:1696–711. doi: 10.1002/hep.30949
71. Schmohl JU, Todhunter D, Oh S, Vallera DA. Mutagenic deimmunization of diphtheria toxin for use in biologic drug development. Toxins (Basel). (2015) 7:4067–82. doi: 10.3390/toxins7104067
72. Weldon JE, Xiang L, Chertov O, Margulies I, Kreitman RJ, FitzGerald DJ, et al. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood. (2009) 113:3792–800. doi: 10.1182/blood-2008-08-173195
73. Hansen JK, Weldon JE, Xiang L, Beers R, Onda M, Pastan I. A recombinant immunotoxin targeting CD22 with low immunogenicity, low nonspecific toxicity, and high antitumor activity in mice. J Immunother. (2010) 33:297–304. doi: 10.1097/CJI.0b013e3181cd1164
74. White TJ, Ibrahimi IM, Wilson AC. Evolutionary substitutions and the antigenic structure of globular proteins. Nature. (1978) 274:92–4. doi: 10.1038/274092a0
75. Yeung VP, Chang J, Miller J, Barnett C, Stickler M, Harding FA. Elimination of an immunodominant CD4+ T cell epitope in human IFN-beta does not result in an in vivo response directed at the subdominant epitope. J Immunol. (2004) 172:6658–65. doi: 10.4049/jimmunol.172.11.6658
76. Mazor R, Crown D, Addissie S, Jang Y, Kaplan G, Pastan I. Elimination of murine and human T-cell epitopes in recombinant immunotoxin eliminates neutralizing and anti-drug antibodies in vivo. Cell Mol Immunol. (2017) 14:432–42. doi: 10.1038/cmi.2015.91
77. Moss DL, Park HW, Mettu RR, Landry SJ. Deimmunizing substitutions in Pseudomonas exotoxin domain III perturb antigen processing without eliminating T-cell epitopes. J Biol Chem. (2019) 294:4667–81. doi: 10.1074/jbc.RA118.006704
78. Oseroff C, Sidney J, Kotturi MF, Kolla R, Alam R, Broide DH, et al. Molecular determinants of T cell epitope recognition to the common Timothy grass allergen. J Immunol. (2010) 185:943–55. doi: 10.4049/jimmunol.1000405
79. Mazor R, Vassall AN, Eberle JA, Beers R, Weldon JE, Venzon DJ, et al. Identification and elimination of an immunodominant T-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin A. Proc Natl Acad Sci USA. (2012) 109:E3597–603. doi: 10.1073/pnas.1218138109
80. Tassignon J, Burny W, Dahmani S, Zhou L, Stordeur P, Byl B, et al. Monitoring of cellular responses after vaccination against tetanus toxoid: comparison of the measurement of IFN-gamma production by ELISA, ELISPOT, flow cytometry and real-time PCR. J Immunol Methods. (2005) 305:188–98. doi: 10.1016/j.jim.2005.07.014
81. Singh H, Raghava GP. ProPred: prediction of HLA-DR binding sites. Bioinformatics. (2001) 17:1236–7. doi: 10.1093/bioinformatics/17.12.1236
82. Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinformatics. (2010) 11:568. doi: 10.1186/1471-2105-11-568
83. Mazor R, Tai CH, Lee B, Pastan I. Poor correlation between T-cell activation assays and HLA-DR binding prediction algorithms in an immunogenic fragment of Pseudomonas exotoxin A. J Immunol Methods. (2015) 425:10–20. doi: 10.1016/j.jim.2015.06.003
84. Mazor R, Addissie S, Jang Y, Tai CH, Rose J, Hakim F, et al. Role of HLA-DP in the presentation of epitopes from the truncated bacterial PE38 immunotoxin. AAPS J. (2017) 19:117–29. doi: 10.1208/s12248-016-9986-y
85. King C, Garza EN, Mazor R, Linehan JL, Pastan I, Pepper M, et al. Removing T-cell epitopes with computational protein design. Proc Natl Acad Sci USA. (2014) 111:8577–82. doi: 10.1073/pnas.1321126111
86. Mazor R, Zhang J, Xiang L, Addissie S, Awuah P, Beers R, et al. Recombinant immunotoxin with T-cell epitope mutations that greatly reduce immunogenicity for treatment of mesothelin-expressing tumors. Mol Cancer Ther. (2015) 14:2789–96. doi: 10.1158/1535-7163.MCT-15-0532
87. Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. (1998) 280:1–9. doi: 10.1006/jmbi.1998.1843
88. Ansari HR, Raghava GP. Identification of conformational B-cell epitopes in an antigen from its primary sequence. Immunome Res. (2010) 6:6. doi: 10.1186/1745-7580-6-6
89. Onda M, Beers R, Xiang L, Lee B, Weldon JE, Kreitman RJ, et al. Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc Natl Acad Sci USA. (2011) 108:5742–7. doi: 10.1073/pnas.1102746108
90. Barnett BC, Burt DS, Graham CM, Warren AP, Skehel JJ, Thomas DB. I-Ad restricted T cell recognition of influenza hemagglutinin. Synthetic peptides identify multiple epitopes corresponding to antibody-binding regions of the HA1 subunit. J Immunol. (1989) 143:2663–9.
91. Kaliyaperumal A, Michaels MA, Datta SK. Naturally processed chromatin peptides reveal a major autoepitope that primes pathogenic T and B cells of lupus. J Immunol. (2002) 168:2530–7. doi: 10.4049/jimmunol.168.5.2530
92. Ratto-Kim S, de Souza MS, Currier JR, Karasavvas N, Sidney J, Rolland M, et al. Identification of immunodominant CD4-restricted epitopes co-located with antibody binding sites in individuals vaccinated with ALVAC-HIV and AIDSVAX B/E. PLoS ONE. (2015) 10:e0115582. doi: 10.1371/journal.pone.0115582
93. Steede NK, Rust BJ, Hossain MM, Freytag LC, Robinson JE, Landry SJ. Shaping T cell–B cell collaboration in the response to human immunodeficiency virus type 1 envelope glycoprotein gp120 by peptide priming. PLoS ONE. (2013) 8:e65748. doi: 10.1371/journal.pone.0065748
94. Hassan R, Bullock S, Premkumar A, Kreitman RJ, Kindler H, Willingham MC, et al. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a Bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. (2007) 13:5144–9. doi: 10.1158/1078-0432.CCR-07-0869
Keywords: recombinant immunotoxins, neutralizing antibodies, anti-drug antibodies (ADA), B cell epitopes, T cell epitopes, moxetumomab pasudotox, LMB-100
Citation: Mazor R and Pastan I (2020) Immunogenicity of Immunotoxins Containing Pseudomonas Exotoxin A: Causes, Consequences, and Mitigation. Front. Immunol. 11:1261. doi: 10.3389/fimmu.2020.01261
Received: 30 March 2020; Accepted: 18 May 2020;
Published: 26 June 2020.
Edited by:
Bernard Maillere, Commissariat à l'Energie Atomique et aux Energies Alternatives (CEA), FranceReviewed by:
Theo Thepen, Otto von Guericke University Magdeburg, GermanyStefan Barth, University of Cape Town, South Africa
Copyright © 2020 Mazor and Pastan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ronit Mazor, cm9uaXQubWF6b3JAZmRhLmhocy5nb3Y=