 Javier Lumbreras1*†
Javier Lumbreras1*† Marta Subias2*†Natalia Espinosa1
Marta Subias2*†Natalia Espinosa1 Juana María Ferrer3
Juana María Ferrer3 Emilia Arjona2
Emilia Arjona2 Santiago Rodríguez de Córdoba2
Santiago Rodríguez de Córdoba2- 1Unidad de Nefrología Infantil, Servicio de Pediatría, Hospital Universitari Son Espases–Instituto de Investigación Sanitaria Illes Balears (IdISBa), Palma de Mallorca, Spain
- 2Centro de Investigaciones Biológicas Margarita Salas and Ciber de Enfermedades Raras, Madrid, Spain
- 3Servicio de Inmunología, Hospital Universitari Son Espases–Instituto de Investigación Sanitaria Illes Balears (IdISBa), Palma de Mallorca, Spain
Thrombotic microangiopathy (TMA) has different etiological causes, and not all of them are well understood. In atypical hemolytic uremic syndrome (aHUS), the TMA is caused by the complement dysregulation associated with pathogenic mutations in complement components and its regulators. Here, we describe a pediatric patient with aHUS in whom the relatively benign course of the disease confused the initial diagnosis. A previously healthy 8-year-old boy developed jaundice, hematuria, hemolytic anemia, thrombopenia, and mild acute kidney injury (AKI) in the context of a diarrhea without hypertension nor oliguria. Spontaneous and complete recovery was observed from the third day of admission. Persistent low C3 plasma levels after recovery raised the suspicion for aHUS, which prompted clinicians to discard the initial diagnosis of Shigatoxin-associated HUS (STEC-HUS). A thorough genetic and molecular study of the complement revealed the presence of an isolated novel pathogenic C3 mutation. The relatively benign clinical course of the disease as well as the finding of a de novo pathogenic C3 mutation are remarkable aspects of this case. The data are discussed to illustrate the benefits of identifying the TMA etiological factor and the relevant contribution of the MCP aHUS risk polymorphism to the disease severity.
Introduction
Atypical hemolytic uremic syndrome (aHUS) is an ultra-rare disease characterized by acute kidney injury, thrombocytopenia, and microangiopathic hemolytic anemia, which results from an impaired protection of host endothelial cells from complement damage (1). The complement system is a key element of innate immunity with crucial roles in the elimination of pathogens, immune complexes, or cell remains. The complement activates by three pathways, classical (CP), lectin (LP), and alternative (AP), which generates protease complexes, named C3 convertases that cleave C3 to generate C3b. Convertase-generated C3b can form more AP C3 convertase, providing exponential amplification of the initial activation. Clustering of C3b around the surface-bound C3 convertase generates the C5 convertase, which cleaves C5 and initiates formation of the lytic membrane attack complex (MAC) (2). In health, the activation of C3 in plasma is kept at a very low level, and the deposition of C3b and further activation of complement are limited to the surface of pathogens by multiple regulatory proteins. The loss of complement regulation leads to the generation of proinflammatory components and/or tissue damage. Both situations have pathological consequences (3). Loss-of-function mutations in genes encoding the regulatory proteins factor H (FH), MCP, and factor I (FI), as well as gain-of-function mutations in the complement activating components factor B (FB) and C3, have been associated with aHUS (4–11). Criteria have been established to facilitate the clinical diagnosis of aHUS, but it is often difficult to exclude STEC-HUS and secondary HUS forms (12). Since 50–70% of aHUS patients have an underlying inherited and/or acquired complement abnormality (13, 14), genetic analyses are recommended to characterize the etiological factor, reinforce diagnosis, and assist patient management. We present a case that was initially classified as STEC-HUS but was reclassified to aHUS based on the complement follow-up and genetic analyses. We discuss the implications of the identification of a de novo gain-of-function C3 mutation in this case and the relevance of genotyping for the MCPggaac aHUS risk polymorphism.
Clinical Case
In October 2014, a previously healthy 8-year-old boy was evaluated at the pediatric emergency room (ER) in a tertiary care hospital for hematuria, asthenia, and mild jaundice observed in previous hours. Nonbloody diarrhea had been present for 3 days. Physical examination was unremarkable apart from mild jaundice. Nonfocal or generalized edema was found. He had no relevant personal or family past history. Initial blood test showed hemoglobin of 12.2 g/dl (>11.5 g/dl), platelets of 35,000/μl (>150,000/μl), creatinine of 84 μmol/L (<61 μmol/L), and normal transaminases, sodium, and potassium. Eight hours later, hemoglobin decreased to 10 g/dl and platelets to 28,900/μl; lactate dehydrogenase (LDH) was determined to be 1,657 U/L (<220 U/L) (Figure 1). Blood test was extended with haptoglobin (undetectable), and a blood smear showed 7–9 schistocytes per field. Creatinine increased to 93 μmol/L. Electrolytes, acid–base balance, and plasma proteins were in normal range. Urine protein to creatinine ratio (UPr/UCr) was 1,921 μg/μmol (<20 μg/μmol). Basic coagulation parameters were normal. Blood pressure remained spontaneously in normal range and diuresis preserved, without involvement of other organs or systems. Patient was admitted to the pediatric ward.
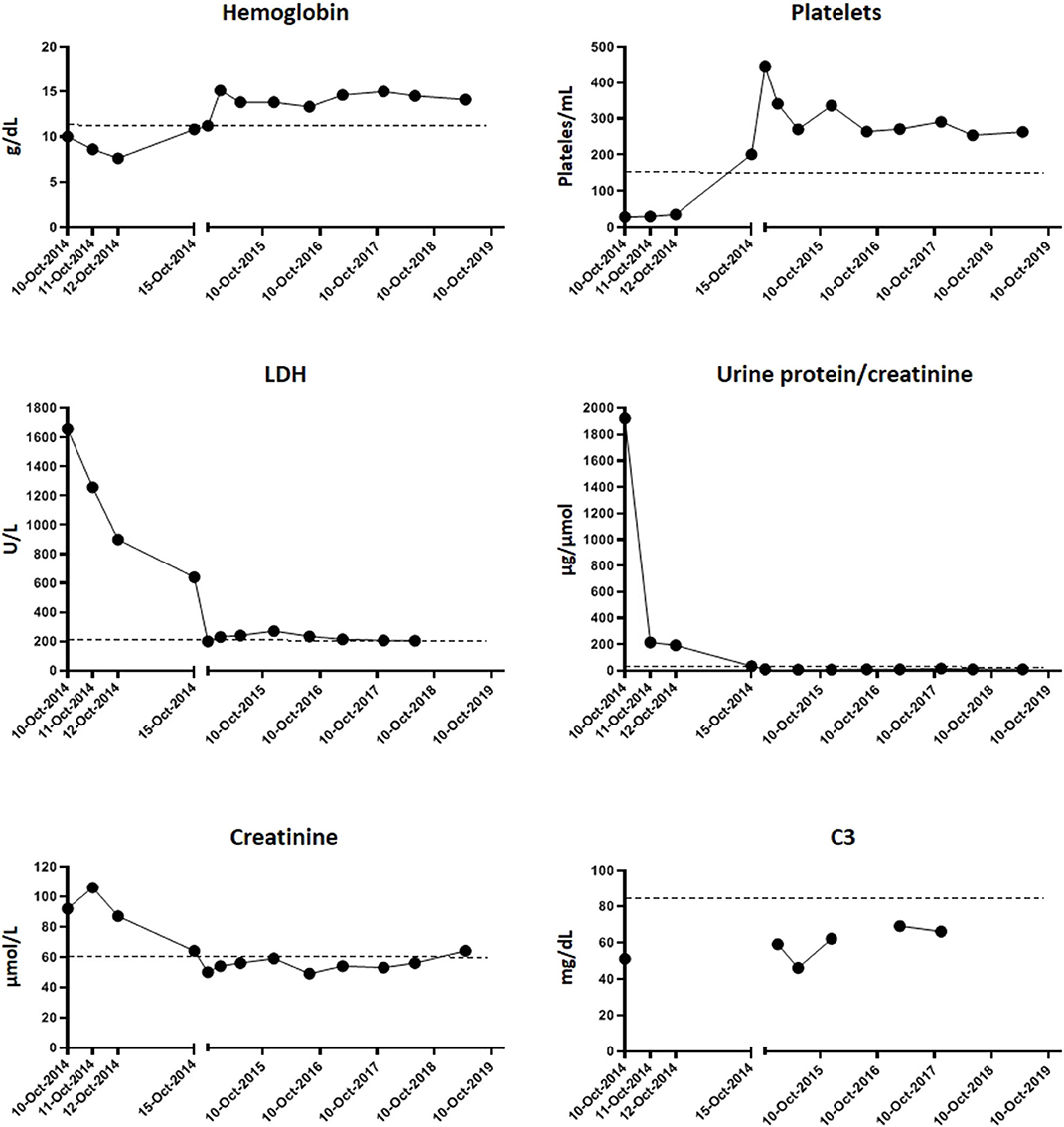
Figure 1. Evolution of hematological parameters and renal activity. Graphics show the blood test determinations since the day of admission (10-Oct-2014) to the day of discharge (15-Oct-2014) and then of the successive routine controls in the following years. Dotted lines represent the upper or minimum value of each parameter from which it is considered pathogenic (hemoglobin > 11.5 g/dl; platelets < 150/ml; LDH > 220 U/L; urine protein/creatinine > 20 μg/μmol; creatinine > 61 μmol/l; C3 < 85 mg/dl).
Maximum plasma creatinine was attained on the second day of admission (106 μmol/L). Hemolysis markers started to descend from the third day. He was discharged on the sixth day with hemoglobin of 10.8 g/dl, platelets of 201,000/μl, LDH of 639 U/L, creatinine of 65 μmol/L, and UPr/UCr of 34 μg/μmol (Figure 1).
Additional studies performed during admission revealed the following: plasma homocysteine was normal at 11.4 μmol/L (<15 μmol/L), autoantibodies [antinuclear antibody (ANA), antineutrophil cytoplasmic antibody (ANCA), and antiextractable nuclear antigen (anti-ENA)] were negative, C3 was 51 mg/dl (75–135 mg/dl) and C4 was 21 mg/dl (14–60 mg/dl), and plasma ADAMTS13 activity was 81% (>5%). A stool sample was only obtained after 4 days of admission. Because of its completely normal appearance and the satisfactory evolution of HUS at that moment, it was only tested for Shigatoxin. Blood and urine culture were sterile. The patient was under careful observation during admission without needing renal replacement therapy or any drug. The spontaneous and very favorable evolution, the previous history of diarrhea, and the justification of a negative Shigatoxin assay due to a late stool collection suggested STEC-HUS as the most likely etiology.
Successive controls showed a complete recovery of renal function and absence of hemolytic activity, anemia, and thrombopenia. No treatment was needed during follow-up. However, decreased C3 levels (46–62 mg/dl) (Figure 1) persisted, and subsequent analysis of factor B plasma levels revealed that they were in the lower part of the normal range (85–170 μg/ml) (Figure 2). Hypocomplementemia is not unusual during a STEC-SHU episode, but the complement normalizes afterwards in these patients (16). The persistent hypocomplementemia in our patients did not correlate with his favorable evolution, raising the suspicion of an underlying constitutive complement abnormality that prompted us to the realization of a complete complement molecular and genetic analysis.
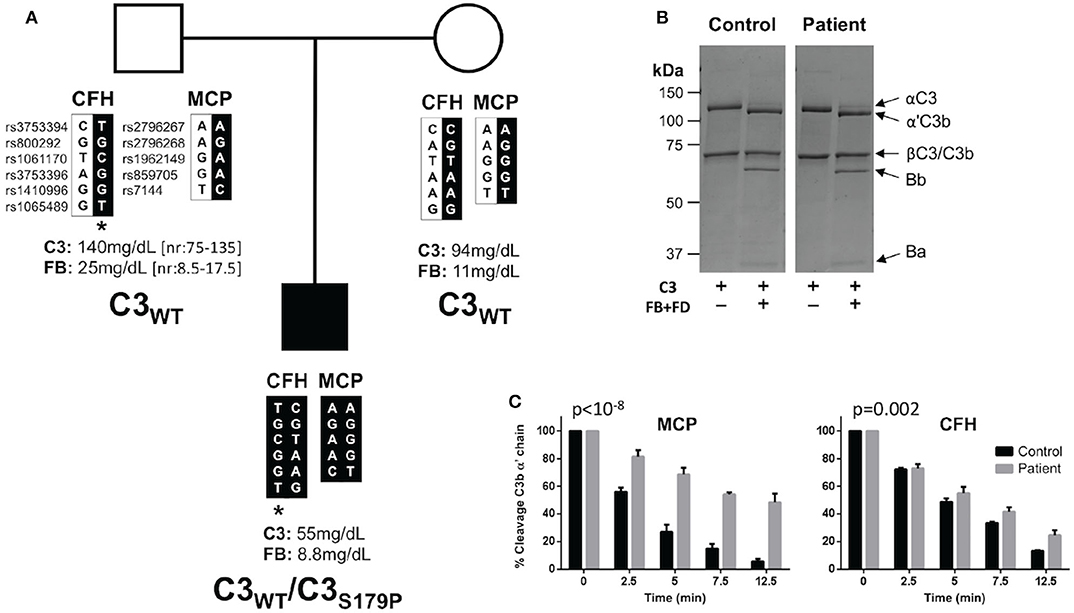
Figure 2. C3S179P is a novel de novo gain-of-function mutations that impairs regulation by MCP. (A) Pedigree depicting the segregation of CFH and MCP polymorphisms organized in haplotype blocks. Single nucleotide polymorphisms are identified by their “rs” numbers. In black are the haplotypes inherited by the patient, identified by a solid square. Asterisk identifies the CFH-H3 aHUS risk haplotype. Plasma levels of C3 and factor B (FB) are indicated for each individual. The patient is the only member of the pedigree carrying the C3S179P mutation. (B) C3 purified from the plasma-ethylenediaminetetraacetic acid (EDTA) of the patient activates normally to C3b by factor B (FB) and factor D (FD). Briefly, C3 was purified using a combination of sodium sulfate precipitation, lysine-sepharose chromatography, DEAE-Sepharose anion exchange chromatography, and Mono S HR 5/5 cation exchange chromatography as previously described (15). (C) C3b generated from the patient's C3 shows a marked resistant to inactivation by factor I in the presence of MCP. Differences between slopes were tested with a general linear model (GLM), with “time” as an integer variable and “strain” as a nominal one. The time × strain interaction was considered as the estimator of the differences between slopes. MCP (p < 10−8). FH (p = 0.002).
To search for mutations in complement genes, we used an in-house next generation sequencing (NGS) panel including all the complement genes relevant to aHUS (17). A complementary analysis of copy number variations was performed by multiplex ligation-dependent probe amplification (MLPA) with the P236 A1 ARMD mix 1 (MRC-Holland, Amsterdam, Netherlands). These analyses identified a C3 mutation in heterozygosis (c535T>C; p.S179P) that was confirmed by Sanger sequencing. This genetic variant has been found previously associated with aHUS (18). No other genetic alterations were found in this patient. Interestingly, none of his parents present this C3 variation (Figure 2A). Paternity was supported by the analysis of CFH and MCP polymorphisms. The patient carries the CFH-H3 aHUS risk polymorphism in heterozygosis, inherited from his father (Figure 2A). He does not carry the MCPggaac aHUS risk polymorphism.
Annotation of C3S179P variant with six pathogenicity prediction algorithms (SIFT, POLYPHEN, Mutation Taster, MutAss, FATHMM and CADD) included in the ANNOVAR server (http://annovar.openbioinformatics.org/) indicated that it is most likely a benign C3 variant. However, because the C3 mutations associated with aHUS are gain-of-function mutations that normally are not predicted pathogenic, we purified the C3 protein from the patient's plasma and performed a complete functional characterization following standard procedures in our laboratory (15). These analyses demonstrated that the mutant C3S179P is present in the patient's plasma and shows an altered function with the characteristic of the C3 gain-of-function mutations that associate with aHUS (19–21). Briefly, when purified C3 from the patient was incubated with FB and FD, it completely activated to C3b, suggesting that C3S179P is normally activated by the AP C3 convertase (Figure 2B). When the patient C3b was tested for inactivation by FI in the presence of FH or MCP, we found that it was resistant to inactivation by FI in the presence of both cofactors, but much more resistant when MCP was the cofactor (Figure 2C).
The patient has remained completely asymptomatic without clinical or analytical data of disease activity or renal sequelae for 5 years, with an expectant attitude.
Discussion
aHUS is a rare, life-threatening renal pathology associated with complement dysregulation. Mutations in genes encoding the regulatory proteins factor H (CFH), factor H-related protein 1 (CFHR1), MCP (MCP), and factor I (CFI), as well as mutations in the complement components factor B (CFB) and C3 have been found in 50–70% of aHUS patients (13, 22). Importantly, while mutations in the complement regulators are loss-of-function, mutations in complement components like factor B and C3 are gain-of-function (13, 14). For C3, these aHUS-associated gain-of-function mutations result in C3b activated molecules being resistant to regulation by MCP, but not by factor H (19–21). The genetic and functional analyses performed in our patient concluded that he carries a C3 gain-of-function mutation that is prototypical of aHUS. This explains why our patient presents a constitutive complement alternative pathway activation with persistent consumption of C3. He has no familial history of aHUS because C3S179P is a de novo mutation, and he is the first in his pedigree carrying this genetic predisposition to aHUS. More interesting is the favorable disease outcome in our patient. Previous studies have shown that C3 mutations, like R161W, tend to be associated with severe aHUS presentations leading to end-stage renal disease. Others, like I1157T, associate with aHUS presentations characterized by multiple recurrences and prolonged favorable outcomes. Interestingly, the presence of the MCPggaac aHUS risk polymorphism influences the aHUS presentation in all carriers of C3 mutations (10, 19–21, 23), which may be justified because this polymorphism determines reduced expression of MCP on the cellular surface (8).
Our aHUS registry includes a total of 13 additional patients having a complete clinical record who carry a clearly pathogenic C3 mutation (Table 1). In total, this series comprises five different C3 mutations. Ten of these aHUS patients also carry the MCPggaac aHUS risk polymorphism (five in heterozygosis and five in homozygosis) (Table 1). This results in an allele frequency for the MCPggaac polymorphism in this group of patients (n = 14) of 0.54, which is significantly different (p < 0.0037) from that in the control Spanish population (AF = 0.28; n = 107). Notably, the only patient in this series who have had a relative favorable outcome is the only one who does not carry the MCPggaac risk polymorphism. Eculizumab treatment was initiated early after aHUS onset or to treat a bad evolution in three patients, and therefore, no conclusions can be reached in them regarding natural progression of aHUS. Notably, nine of the remaining 11 patients reached end-stage renal disease (ESRD) or had multiple recurrences (Table 1). Eight of these patients carry, in addition to the C3 gain-of-function mutation, the MCPggaac risk polymorphism or additional pathogenic mutations in the CFH, CFI, MCP, and THBD genes. The only patient who, like our current patient, does not carry the MCPggaac risk polymorphisms or additional pathogenic mutations had a very late onset (63 years old) without recurrences until she was 76 years old. Currently (80 years old), the patient presents chronic kidney disease but does not require hemodialysis. These registry data suggest that the likely explanation for the favorable disease outcome in our patient is that he does not carry additional genetic risk factors, in particular the MCPggaac risk polymorphism. A relevant question is why our patient had an aHUS episode. It is known that several viral pathogens interact with MCP and that viral infections may lead to a reduction in the pathogen's receptor. Therefore, one possibility could be that our patient underwent a transient decrease in the cell surface levels of MCP as a consequence of the infection that triggered the aHUS episode. However, this is just a speculation because, when MCP levels were tested, months after the aHUS episode, they were found normal.
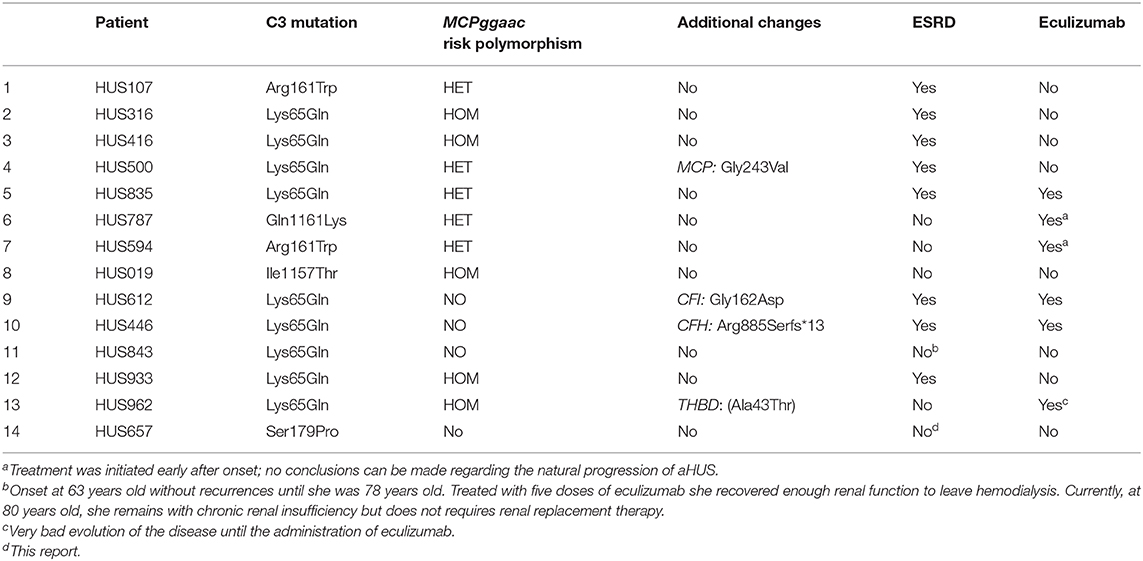
Table 1. Atypical hemolytic uremic syndrome (aHUS) patients carrying C3 mutations in our aHUS registry.
Although our patient is currently asymptomatic and presents normal renal function, our functional characterization of the C3S179P variant indicates that it is an important aHUS genetic risk factor. This has important implications. In fact, we cannot exclude that under exposure to a strong environmental trigger (e.g., an infection), our patient will experience a more severe aHUS recurrence. While we strongly recommend normal life to avoid unnecessary anxiety in the patient and its family, we also suggest active surveillance of the patient with specific recommendations. Regular determination of blood pressure, blood count, and measurement of biochemical markers for hemolysis (bilirubin, LDH, haptoglobin), as well as plasma creatinine, proteinuria, and albuminuria are performed. The patient and his parents are instructed to go the ER in case of presenting symptoms suggesting activity of his disease, such as hematuria, oliguria, edema, and significant general malaise in the context of some intercurrent process that can act as a trigger. Ultimately, it is reassuring to know that we have the “magic bullet” of eculizumab, if this patient should need it (24). In conclusion, complete understanding of the etiological factor in the TMA patient is critical to strengthen diagnosis and assist patient management.
Data Availability Statement
All datasets presented in this study are included in the article/supplementary files.
Ethics Statement
Written informed consent was obtained from the participant's legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
JL, MS, and SR designed the study. JL, MS, EA, and SR performed the experiments, collected, and analyzed the data. JL and SR drafted the manuscript, which was revised and approved by all coauthors. All authors contributed to the article and approved the submitted version.
Funding
SR was supported by the Spanish Ministerio de Economía y Competitividad-FEDER (SAF2015-66287R) and the Autonomous Region of Madrid (S2017/BMD-3673).
Conflict of Interest
SR has received honoraria from Alexion Pharmaceuticals for giving lectures and participating in advisory boards. JL has received honoraria from Alexion Pharmaceuticals for giving lectures. None of these activities has had any influence on the results or interpretation in this article.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
All the members of the Multidisciplinary Working Group on Thrombotic Microangiopathies from the Hospital Universitari Son Espases, in Palma de Mallorca, Spain are gratefully acknowledged. We are in debt to all patients and relatives participating in this study. This work was developed under the supervision of the Spanish Registry of atypical Hemolytic Uremic Syndrome and C3-Glomerulopathy (aHUS/C3G).
References
1. Moake JL. Thrombotic microangiopathies. N Engl J Med. (2002) 347:589–600. doi: 10.1056/NEJMra020528
2. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
3. de Cordoba SR, Tortajada A, Harris CL, Morgan BP. Complement dysregulation and disease: from genes and proteins to diagnostics and drugs. Immunobiology. (2012) 217:1034–46. doi: 10.1016/j.imbio.2012.07.021
4. Noris M, Brioschi S, Caprioli J, Todeschini M, Bresin E, Porrati F, et al. Familial haemolytic uraemic syndrome and an MCP mutation. Lancet. (2003) 362:1542–7. doi: 10.1016/S0140-6736(03)14742-3
5. Perez-Caballero D, Gonzalez-Rubio C, Gallardo ME, Vera M, Lopez-Trascasa M, Rodriguez de Cordoba S, et al. Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome. Am J Hum Genet. (2001) 68:478–84. doi: 10.1086/318201
6. Richards A, Kemp EJ, Liszewski MK, Goodship JA, Lampe AK, Decorte R, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci USA. (2003) 100:12966–71. doi: 10.1073/pnas.2135497100
7. Warwicker P, Goodship TH, Donne RL, Pirson Y, Nicholls A, Ward RM, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. (1998) 53:836–44. doi: 10.1111/j.1523-1755.1998.00824.x
8. Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, Carreras Berges L, Lopez-Trascasa M, Sanchez-Corral P, et al. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet. (2005) 14:703–12. doi: 10.1093/hmg/ddi066
9. Fremeaux-Bacchi V, Dragon-Durey MA, Blouin J, Vigneau C, Kuypers D, Boudailliez B, et al. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet. (2004) 41:e84. doi: 10.1136/jmg.2004.019083
10. Fremeaux-Bacchi V, Miller EC, Liszewski MK, Strain L, Blouin J, Brown AL, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. (2008) 112:4948–52. doi: 10.1182/blood-2008-01-133702
11. Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, Carreras L, Arranz EA, Garrido CA, et al. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA. (2007) 104:240–5. doi: 10.1016/j.molimm.2006.07.076
12. Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Fremeaux-Bacchi V, Kavanagh D, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “kidney disease: improving global outcomes” (KDIGO) controversies conference. Kidney Int. (2017) 91:539–51. doi: 10.1016/j.kint.2016.10.005
13. de Cordoba SR. Complement genetics and susceptibility to inflammatory disease. Lessons from genotype-phenotype correlations. Immunobiology. (2016) 221:709–14. doi: 10.1016/j.imbio.2015.05.015
14. Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, et al. Atypical aHUS: state of the art. Mol Immunol. (2015) 67:31–42. doi: 10.1016/j.molimm.2015.03.246
15. Martinez-Barricarte R, Heurich M, Valdes-Canedo F, Vazquez-Martul E, Torreira E, Montes T, et al. Human C3 mutation reveals a mechanism of dense deposit disease pathogenesis and provides insights into complement activation and regulation. J Clin Invest. (2010) 120:3702–12. doi: 10.1172/JCI43343
16. Exeni RA, Fernandez-Brando RJ, Santiago AP, Fiorentino GA, Exeni AM, Ramos MV, et al. Pathogenic role of inflammatory response during Shiga toxin-associated hemolytic uremic syndrome (HUS). Pediatr Nephrol. (2018) 33:2057–71. doi: 10.1007/s00467-017-3876-0
17. Cavero T, Arjona E, Soto K, Caravaca-Fontan F, Rabasco C, Bravo L, et al. Severe and malignant hypertension are common in primary atypical hemolytic uremic syndrome. Kidney Int. (2019) 96:995–1004. doi: 10.1016/j.kint.2019.05.014
18. Osborne AJ, Breno M, Borsa NG, Bu F, Fremeaux-Bacchi V, Gale DP, et al. Statistical validation of rare complement variants provides insights into the molecular basis of atypical hemolytic uremic syndrome and C3 glomerulopathy. J Immunol. (2018) 200:2464–78. doi: 10.4049/jimmunol.1701695
19. Martinez-Barricarte R, Heurich M, Lopez-Perrote A, Tortajada A, Pinto S, Lopez-Trascasa M, et al. The molecular and structural bases for the association of complement C3 mutations with atypical hemolytic uremic syndrome. Mol Immunol. (2015) 66:263–73. doi: 10.1016/j.molimm.2015.03.248
20. Roumenina LT, Frimat M, Miller EC, Provot F, Dragon-Durey MA, Bordereau P, et al. A prevalent C3 mutation in aHUS patients causes a direct C3 convertase gain of function. Blood. (2012) 119:4182–91. doi: 10.1182/blood-2011-10-383281
21. Schramm EC, Roumenina LT, Rybkine T, Chauvet S, Vieira-Martins P, Hue C, et al. Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood. (2015) 125:2359–69. doi: 10.1182/blood-2014-10-609073
22. Vieira-Martins P, El Sissy C, Bordereau P, Gruber A, Rosain J, Fremeaux-Bacchi V. Defining the genetics of thrombotic microangiopathies. Transfus Apher Sci. (2016) 54:212–9. doi: 10.1016/j.transci.2016.04.011
23. Siomou E, Gkoutsias A, Serbis A, Kollios K, Chaliasos N, Fremeaux-Bacchi V. aHUS associated with C3 gene mutation: a case with numerous relapses and favorable 20-year outcome. Pediatr Nephrol. (2016) 31:513–7. doi: 10.1007/s00467-015-3267-3
Keywords: C3, MCP risk polymorphism, atypical hemolytic uremic syndrome, de novo mutation, case report
Citation: Lumbreras J, Subias M, Espinosa N, Ferrer JM, Arjona E and Rodríguez de Córdoba S (2020) The Relevance of the MCP Risk Polymorphism to the Outcome of aHUS Associated With C3 Mutations. A Case Report. Front. Immunol. 11:1348. doi: 10.3389/fimmu.2020.01348
Received: 05 April 2020; Accepted: 27 May 2020;
Published: 16 July 2020.
Edited by:
Paolo Macor, University of Trieste, ItalyReviewed by:
Viviana P. Ferreira, University of Toledo, United StatesZoltan Prohaszka, Semmelweis University, Hungary
Copyright © 2020 Lumbreras, Subias, Espinosa, Ferrer, Arjona and Rodríguez de Córdoba. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Javier Lumbreras, amF2aWVyLmx1bWJyZXJhc0Bzc2liLmVz; Marta Subias, bS5zdWJpYXNAY2liLmNzaWMuZXM=
†These authors share first authorship