 Rebecca A. Brown
Rebecca A. Brown Craig A. Byersdorfer
Craig A. Byersdorfer- Division of Blood and Marrow Transplant and Cellular Therapies, Department of Pediatrics, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
Allogeneic hematopoietic stem cell transplantation (aHSCT) is a curative therapy for a range of hematologic illnesses including aplastic anemia, sickle cell disease, immunodeficiency, and high-risk leukemia, but the efficacy of aHSCT is often undermined by graft-versus-host disease (GVHD), where T cells from the donor attack and destroy recipient tissues. Given the strong interconnection between T cell metabolism and cellular function, determining the metabolic pathways utilized by alloreactive T cells is fundamental to deepening our understanding of GVHD biology, including its initiation, propagation, and potential mitigation. This review summarizes the metabolic pathways available to alloreactive T cells and highlights key metabolic proteins and pathways linking T cell metabolism to effector function. Our current knowledge of alloreactive T cell metabolism is then explored, showing support for glycolysis, fat oxidation, and glutamine metabolism but also offering a potential explanation for how these presumably contradictory metabolic findings might be reconciled. Examples of additional ways in which metabolism impacts aHSCT are addressed, including the influence of butyrate metabolism on GVHD resolution. Finally, the caveats and challenges of assigning causality using our current metabolic toolbox is discussed, as well as likely future directions in immunometabolism, both to highlight the strengths of the current evidence as well as recognize some of its limitations.
Introduction
Allogeneic hematopoietic stem cell transplantation (aHSCT) is a potentially curative therapy for a wide range of hematologic maladies ranging from genetic diseases to aggressive leukemias and lymphomas (1–3). An unintended and potentially deadly consequence of aHSCT is graft-versus-host disease (GVHD), where donor T cells primed to react against alloantigens attack host tissues in the skin, gastrointestinal tract, and liver (4). While corticosteroid-induced immunosuppression can treat GVHD, corticosteroids are an imperfect therapy and durable remissions only occur in 50% of patients (5). Furthermore, the broad immunosuppression necessary to treat GVHD often limits physiologic immunity and impairs T cell mediated clearance of leukemia [aka the graft-versus-leukemia (GVL) effect (6, 7)]. Thus, common complications following the increased immunosuppression surrounding GVHD treatment include infection (8, 9) and cancer relapse (9), making development of GVHD especially dangerous. In fact, while absolute T cell depletion can minimize GVHD risk, the ensuing increase in infection, and disease relapse result in comparable overall survival (10). Thus, there is a need for novel approaches to treat and prevent GVHD while still preserving physiologic immunity and maintaining aHSCT efficacy.
It is broadly accepted that metabolism and immune cell function are linked, with immunologic differentiation influencing immune cell metabolism and metabolic pathways impacting immune responses (11, 12). It is therefore imperative to understand how metabolism influences T cell function in specific environmental contexts. This is particularly true during GVHD, where high levels of chronic antigen stimulation result in robustly activated T cells with a sharply increased metabolic demand. Understanding the unique metabolic profile of alloreactive T cells will enhance our ability to improve current therapeutic options and advance our contextual knowledge of in vivo T cell biology. This review will highlight our current understanding of alloreactive T cell metabolism in light of the major metabolic pathways, present evidence for involvement of various pathways at distinct stages of the process, define key metabolic regulators that influence substrate choice, and integrate multiple lines of evidence into a cohesive overarching hypothesis. We close by highlighting examples of additional ways in which metabolism can influence GVHD and discuss challenges to the interpretation of metabolic data.
Overview of Cellular Metabolism
Cellular metabolism is a complex interplay between multiple different enzymes, substrates, intermediates, and end products. Classically, glycolysis, and oxidative phosphorylation (OXPHOS) have been studied as the primary pathways that supply cellular energy. Glycolysis consists of a series of enzymatic steps that convert glucose into pyruvate. Depending on the intrinsic and extrinsic needs of the cell, pyruvate can then either be converted into lactate and excreted from the cell or channeled into acetyl-coA and further oxidized via OXPHOS. While lactate fermentation classically occurs in oxygen poor environments, T cells can perform glycolysis and lactate fermentation in oxygen replete environments, referred to as aerobic glycolysis. Although glycolysis might not be the best choice based solely on energy production, glycolytic intermediates can also act as substrates for anabolic pathways including amino acid synthesis, nucleotide synthesis, and the pentose phosphate pathway (PPP) (13), all processes necessary in actively proliferating cells.
Oxidative phosphorylation is a more efficient process used to generate cellular energy. Specifically, the tricarboxylic acid (TCA) cycle uses the end products of glycolysis, fatty acid oxidation, and glutamine metabolism to generate the reducing intermediates, NADH, and FADH2 (14). NADH and FADH2, in turn, fuel the electron transport chain (ETC) by donating electrons to Complex I and II (14), a process which results in ATP production and concurrent consumption of oxygen (14).
Metabolic Pathways Contributing to Alloreactive T Cell Effector Function
Classically, naïve T cells are considered largely quiescent, catabolically relying on OXPHOS to meet their modest energy demands. Upon activation, naïve T cells switch to anabolic metabolism (15) and despite the availability of oxygen, increase aerobic glycolysis in a process known as the Warburg effect (16, 17). Aerobic glycolysis produces less energy per molecule of substrate than oxidative pathways but has the advantage of maintaining redox balance (18) and allowing for the bulk of cellular machinery to be used in the production of biomolecules required for proliferation and T cell function, including cytokines (19, 20). In contrast to recently activated T cells, memory and regulatory T cells (Tregs) rely on oxidation of fatty acids and glucose to maintain their energetic balance (21–25). This classic view has recently been challenged, where effector T cells have been demonstrated to increase oxidative metabolism in vivo and be less reliant on glycolytic metabolism compared to in vitro activated cells (26). How alloreactive T cells meet their energetic demands during GVHD remains a work in progress, but evidence supports the adoption of both aerobic glycolysis and OXPHOS during early stages of T cell activation and disease initiation.
The studies highlighted in this review compare the profile of allogeneic T cells to either syngeneic or naïve T cell controls. While both syngeneic and naïve T cells are less activated than alloreactive cells, syngeneic T cells are the preferred negative control because they experience the inflammatory milieu of pre-transplant conditioning and most accurately reflect the lymphopenia-driven reconstitution of the immune system seen in human transplants. In contrast, naïve T cells are relatively inert. Thus, comparing alloreactive and naïve T cells risks identifying differences that are not unique to alloreactive T cells, but are instead characteristic of any proliferating T cell. Since the aim is to distinguish alloreactive from homeostatically proliferating T cells, syngeneic T cells remains the better negative control.
Since aerobic glycolysis is critical for physiologic T cell activation (16, 27, 28), it is natural to consider the role of glucose in alloreactive cells. However, discussions on glucose metabolism are challenging given that pyruvate, a critical glycolytic intermediate, can either be converted into lactate for fermentation or alternatively channeled through pyruvate dehydrogenase into the TCA cycle for oxidation. For the purposes of this review, we will consider both forms of glucose metabolism under one discussion. In two pre-clinical models of GVHD, glucose uptake increased in donor T cells 14 days post-transplant (allogeneic > syngeneic), accompanied by increased expression of glucose transporters Glut1 and Glut3 (29). Expression of the key glycolytic enzymes hexokinase (isoforms 1 and 2) and lactate dehydrogenase also increased in allogeneic cells, while donor T cell numbers decreased following treatment with the pan-glycolysis inhibitor 2-deoxyglucose (2DG) (29). In this same study, recipients experienced improved survival when treated with 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3-PO), an inhibitor of phosphofructokinase (30), the rate-limiting step in glycolytic metabolism (29, 31, 32). Consistent with glucose transport playing an important role in GVHD development, T cells lacking Glut1, which experienced a decreased glycolytic rate in vitro, were unable to induce GVHD in vivo. In this case, post-transplant weight loss and survival outcomes were similar between recipients of Glut1−/− T cells and those transplanted with bone marrow alone (33). In addition to its therapeutic potential, increased glucose uptake may also have diagnostic implications. Positron emission tomography using 18F-fluorodeoxyglucose (FDG-PET), a glucose analog, identifies increased tracer uptake in the gastrointestinal tract during GVHD in both mice and humans (34). Thus, inhibiting glycolytic metabolism, either genetically or pharmacologically, might constrain alloreactive T cell effector (Teff) function, while increased glucose trafficking may be of potential diagnostic value. The caveat is that glycolytic metabolism may also prove essential to the function of non-GVHD T cells.
A key metabolic regulator that could connect glycolytic metabolism and alloreactive T cell function is the mammalian target of rapamycin (mTOR), a serine/threonine kinase belonging to the phosphoinositol 3-kinase (PI3K) family. mTOR is the catalytic subunit for either of two distinct protein complexes; mTORC1 (with scaffolding protein Raptor) and mTORC2 (with scaffolding protein RICTOR) (35). mTORC1 and mTORC2 each have unique roles within cells. mTORC1 promotes protein and lipid synthesis while mTORC2 promotes cytoskeletal rearrangement (36). In a variety of settings, mTOR integrates environmental signals into regulation of immune cell metabolism, differentiation, and effector function. In particular, mTOR and has been shown to play a key role in T cell activation and cell fate (37–39). Metabolically, T cell receptor stimulation activates mTOR via PI3K/Akt signaling, which then promotes glycolysis, glutaminolysis, and activation of the PPP (37, 40, 41). mTOR promotes glycolysis in part through the activation of HIF1α and c-Myc, transcription factors which drive expression of glycolytic proteins including pyruvate dehydrogenase kinase 1, hexokinase 2, and lactate dehydrogenase A (40, 42–44). Furthermore, the PI3K/Akt/mTORC1 pathway has been implicated in T cells as a key regulatory step for the expression and trafficking of the glucose transporter, Glut1, with mTORC inhibitors preventing Glut1 expression (33, 45). Thus, mTOR contributes to metabolic reprogramming following T cell activation by promoting both glycolysis and general anabolic pathways. mTOR signaling also influences T cell differentiation. T cells deficient in mTOR are unable to differentiate into T-helper type 1 (Th1), Th2, or Th17 cells under in vitro skewing conditions but still readily differentiate into FoxP3+ Treg cells (46). Furthermore, mTORC1, and mTORC2 promote differentiation of specific T cell subsets. mTORC2 promotes Th2 differentiation (47), while mTORC1 promotes development of Th17 cells (47, 48). The role played by mTORC1 in Th1 differentiation remains uncertain, with evidence both for (47), and against (48) mTORC1 involvement in Th1 responses.
mTOR has also been implicated in promoting the pathogenicity of alloreactive T cells. In pre-clinical models, mTOR activity increases in alloreactive T cells and both the pharmacologic inhibition of mTOR, and its genetic deletion, inhibit glycolysis without impacting OXPHOS (29, 49). These interventions also improve outcomes in animal models of GVHD (29). In the clinic, targeting mTOR using the inhibitors sirolimus (rapamycin) and everolimus is well-established and has been found to be efficacious for both GVHD prophylaxis and treatment (50, 51). Thus, inhibition of mTOR improves GVHD, in part through its inhibition of glycolysis.
In addition to glucose metabolism, there is ample evidence that alloreactive T cells rely on the energy and by-products of OXPHOS. This necessity of OXPHOS, in addition to an increase in lactate fermentation, is likely necessary due to the greater energetic demands experienced by T cells undergoing constant exposure to high levels of antigen in a near continuous manner. In allogeneic cells, oxygen consumption increased markedly compared to either naïve T cells or T cells recovered from syngeneic recipients (52). Mitochondrial activity also increased in allogeneic T cells with a corresponding increase in mitochondrial superoxide production (52, 53). Consistent with increased mitochondrial activity, alloreactive T cells upregulated expression of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc1α), a regulator of mitochondrial biogenesis (54). Finally, administration of BZ-423, an F1-F0 ATPase inhibitor that targets cells with increased mitochondrial respiration, improved survival, lowered clinical scores, and decreased lymphocytic infiltration into GVHD target organs in a murine model of GVHD (53). These results suggest that alloreactive T cells are preferentially susceptible to ETC inhibition, in part because of their increased reliance on mitochondrial respiration (53). Taken together these findings demonstrate an increased dependence on both OXPHOS and glycolytic metabolism in alloreactive T cells, making them metabolically distinct from other T cell populations.
Although the mechanisms that influence OXPHOS are complex, the cellular energy sensor AMP-activated protein kinase (AMPK) has been implicated as a driver of oxidative metabolism and could play a role. AMPK is heterotrimeric protein complex consisting of a serine/threonine kinase α subunit, a stabilizing β subunit, and a regulatory γ subunit. The γ subunit detects low intracellular energy levels by sensing the AMP/ATP ratio (increased during low energy states) and responds by facilitating activation of the AMPKα kinase domain (55). AMPK activation conserves energy by inhibiting anabolic pathways (e.g., fat and protein synthesis), while increasing energy production via catabolic pathways, including OXPHOS and autophagy (56, 57). In part, AMPK restricts anabolism by antagonizing mTORC1 via phosphorylation of Raptor as well as upstream regulator tuberous sclerosis complex 2 (28, 58). AMPK also directly promotes OXPHOS and fatty acid catabolism. In skeletal muscle, acetyl-coA carboxylase (ACC) produces malonyl-coA, which allosterically inhibits carnitine palmitoyl transferase 1 (CPT1a), a key enzyme in fat oxidation, and thereby blocks FAO (55, 59). AMPK inhibits ACC through phosphorylation, which decreases malonyl-CoA levels and thus increases FAO. In addition, ACC is a key enzyme in fatty acid synthesis (FAS) so that AMPK inhibition of ACC not only increases FAO but also reduces the anabolic process of FAS.
Given that AMPK is key to promoting both OXPHOS and FAO, it follows that AMPK would be integral to T cell homeostasis. However, the exact role for AMPK in T cells continues to evolve. Systemic ablation of AMPKα1 increased lymphocyte susceptibility to mitochondrial inhibition (i.e., treatment with oligomycin) but did not impact T cell development or differentiation (60). Later studies noted an increase in T cell glycolysis following global deletion of AMPKα1 and an increase in T cell production of the pro-inflammatory cytokines interferon gamma (IFN-γ) and interleukin 17a (IL-17a) (61). In a Listeria monocytogenes infection model, T cell-specific deletion of AMPKα1 impaired memory CD8 cell generation compared to wildtype T cells without impacting primary immune responses (62). More recently, AMPK was shown to be necessary for maximal Teff generation during both viral and bacterial challenges in vivo (63) and has been implicated in driving oxidative metabolism in T cell acute lymphoblastic leukemia (64). AMPK has also been suggested to be necessary for Treg development, with increased AMPK phosphorylation in cultured Treg cells and an increase in Treg percentages following in vivo administration of metformin, an indirect AMPK activator (24). Metformin also increases Treg number at the expense of Th17 cells in models of autoimmune arthritis (65, 66). However, whether AMPK is directly responsible for these changes, or they are driven by actions of metformin independent of AMPK, remains to be determined.
Despite characterization of AMPK in activated T cells, how AMPK contributes to GVHD pathogenicity is poorly understood. In the only paper to date investigating AMPK and GVHD, 5 days of metformin administration ameliorated GVHD severity and decreased disease lethality, with fewer Th17 and Th1 and increased Treg cells recovered from metformin-treated recipients (49). However, given the fact that metformin is a direct inhibitor of Complex I of the ETC (67–69), there is a high likelihood that metformin directly inhibits oxidative metabolism, a process necessary in alloreactive cells. In our hands, transplantation of donor T cells lacking both AMPK α1 and α2 improved GVHD-related lethality with decreased recovery of AMPK-deficient donor T cells, reduced T cell homing to target organs, and an improved Treg to Teff cell ratio (70). Indeed, these later results are in line with studies demonstrating an important role for AMPK in Teff cell survival and recovery (63). Thus, on balance, metformin treatment improves GVHD, but likely in an AMPK independent manner, as genetic elimination demonstrates that AMPK is necessary in donor T cells for maximal GVHD severity. In both cases, more work needs to be done to determine the exact mechanism behind the observed effects.
Given the increased oxidative metabolism of donor T cells during GVHD, an ongoing question becomes which substrate or substrates fuel this pathway. In fact, glucose utilization, fat oxidation, and glutamine metabolism have all been shown to be crucial for T cell proliferation and survival in various allogeneic contexts (15, 71, 72). T cells isolated on day 7 post-allogeneic transplant show elevated levels of fat import, higher acylcarnitine concentrations, and increased fatty acid oxidation (53, 54). These changes are supported by increased expression of CPT1a and CPT2, enzymes necessary for transport of long and very-long chain fatty acids into the mitochondria for β-oxidation. Furthermore, treatments with the FAO inhibitor etomoxir improved GVHD severity while simultaneously decreasing T cell proliferation and the number of donor T cells (54). This dependence on FAO may be most prevalent at early times post-transplant, as metabolic interrogation at later time points, in a distinct model of GVHD, demonstrated fat transport in allogeneic cells at an intermediate level between unstimulated and syngeneic T cells (29). Thus, the timing of FAO in alloreactive T cells, as well as its absolute necessity, remains unresolved.
In addition to lipids and glucose, glutamine is another common metabolic substrate for T cells. From the beginning, glutamine uptake and metabolism have been shown to increase following T cell activation and glutamine is required for both Th1 and Th17 differentiation (71, 73). Glick et al. demonstrated that glutamine can act as an anaplerotic nutrient source in alloreactive T cells to replenish TCA cycle intermediates and provide substrates for the PPP (74). However, in contrast to the glutamine dependence seen in alloreactive cells, which would suggest worsening disease with glutamine supplementation, there is strong evidence that systemic glutamine administration facilitates therapeutic recovery following aHSCT. In a murine model of GVHD, systemic glutamine administration increased Treg numbers and decreased serum levels of tumor necrosis factor α, limiting pro-inflammatory immune responses and improving recipient survival by 30% (75). In human patients, glutamine supplementation improved post-transplant survival with a trend toward decreased rates of GVHD (76). Thus, while there is much to be learned regarding glutamine metabolism in individual cell types, systemic glutamine administration appears to be protective.
Metabolism in Regulatory T Cells
Tregs provide crucial inhibitory signals to Teff and are key to dampening immune responses and promoting tolerance (77). In GVHD, Tregs are of immense interest because of their potential ability to correct the balance between inflammation and immunosuppression. In fact, the frequency of Treg, as marked by CD4+CD25+ expression, was lower in patients with chronic GVHD than in healthy controls or in patients post-transplant without chronic GVHD (78). Furthermore, enhancing Treg frequency, either through ultra-low dose IL-2 administration, or adoptive transfer of ex vivo expanded Treg, is an effective way to improve GVHD (79–81). Thus, increasing Treg frequency could be an essential component for GVHD prevention and treatment and metabolic interventions could play a large role in this effort.
In addition to filling a unique immunologic niche, Tregs have a metabolic profile distinct from Teff cells. Tregs generated in vitro increase their reliance on lipid and mitochondrial metabolism (24) while transgenic expression of the Glut1 receptor increases glycolysis, which impedes suppressive function. Opposing this signaling is the transcription factor Foxp3, which reprograms Treg metabolism toward OXPHOS and away from glycolysis (82, 83), driving up Treg suppressive capacity. Foxp3 is thought to achieve these results by inhibiting Myc (82), a transcription factor that promotes both glycolysis and glutamine metabolism (43). Thus, changes in suppressive function are metabolically dependent, with a loss of OXPHOS decreasing suppressor activity (84). This tuning of Treg suppressive capacity may be integral to the propagation and subsequent waning of an immune response. At times of high stimulation (e.g., infection) Treg increase in number but are minimally suppressive, allowing effector responses to proceed unabated. As the infection subsides, inflammatory signals decrease and FoxP3 levels stabilize, leading to the adoption of OXPHOS, which decreases Treg proliferation but increases suppressive function, limiting the effector response and restoring a state of tolerance (83).
Multiple studies have also demonstrated an integral link between mitochondrial metabolism and Treg mediated suppression both in vitro and in vivo (84–86). Deletion of complex III specifically in Treg led to development of a fatal early inflammatory disease (87) and transfer of Complex III deficient Treg was unable to protect recipients in a model of T-cell driven colitis (85). Treg also depend upon the mitochondrial transcription factor A (Tfam), which controls mitochondrial DNA copy number and is integral to ETC activity (88, 89). Loss of Tfam in Treg decreased mitochondrial respiration, blunted expression of inhibitory markers ICOS and CTLA4, and resulted in a severe inflammatory disorder (86). Interestingly, although mTOR signaling is known to drive T cell glycolysis (which is expected to decrease Treg function), mTOR is also required for proper Treg function and development. Compared to conventional T cells, Treg have higher mTORC1 activity (90, 91) and Treg-specific deletion of Raptor, an obligate component of the mTORC1 complex, resulted in a fatal inflammation and loss of Treg suppressor function (91). Similar results were found in mice lacking the mTOR protein, where Th2 responses increased significantly in the lungs and gastrointestinal tract of knock-out mice (86).
Despite our increased working knowledge of Treg metabolism, little is known about Treg metabolism in the alloreactive environment. Sirtuin-1 (Sirt1) is a class III histone deacetylase whose expression influences multiple metabolic pathways. Donor T cells lacking Sirt1 show increased FoxP3 stability in inducible Treg (iTreg) with a subsequent decreased conversion to pathogenic IFN-γ producing cells and a loss of follicular helper T cell development (92). In other studies, human iTregs propagated in vitro via pSTAT3 inhibition prevented xenogeneic GVHD yet spared donor antileukemia immunity. Metabolically, pSTAT3 inhibition shifted iTreg metabolism from OXPHOS to glycolysis, with a reduction in ETC activity. However, this metabolic impairment could be corrected by treating pSTAT3-inhibited Treg with coenzyme Q10, which restored OXPHOS and augmented their suppressive potency (93). In other work, adoptive transfer of Treg lacking vimentin, or pre-treated with the phosphokinase C inhibitor AEB071, improved GVHD survival, clinical scores, and weight loss to a greater degree than WT Treg. Mechanistically, absence or inhibition of vimentin enhanced oxidative metabolism within the Treg compartment and concomitantly increased their suppressive capacity (94). Finally, transplantation of adenosine producing CD150+ Treg into allogeneic animals decreased the severity of immune cell infiltration into the intestine (95). Thus, Treg function is a finely tuned process, accomplished through integration of multiple inputs including mTOR signaling, intracellular energy sensing, metabolic pathways, and the influence of local environmental cues including danger signals. Furthermore, Treg-associated metabolic changes found in other contexts appear to hold following aHSCT, in particular the association between increased OXPHOS and enhanced suppressive capacity.
Effects of Metabolic Inhibition on Graft-vs.-Tumor Responses
A major indication for allogeneic transplantation is relapsed or refractory leukemia and lymphoma. aHSCT's benefit in this setting derives from donor T cells reactivity against foreign tumor cells, the so-called Graft-versus-tumor (GVT) effect. It is expected that anything that interrupts T cell alloreactivity, or impairs allogeneic T cell number, might disrupt the therapeutic efficacy of allogeneic transplantation. And yet metabolic manipulation does not appear to be universally detrimental to anti-cancer responses. Donor T cells that lack AMPK induce less severe GVHD but continue to demonstrate preserved or even enhanced cytotoxic potential post-transplant (70). Similarly, mice treated with recombinant Thioredoxin at the time of transplantation exhibit decreased GVHD severity while simultaneously preserving GVT effects (96). In some cases, preserved cytotoxicity results from a preservation in cytokine responses in remaining T cells (70), coupled with a decrease in T cell dysfunction due to lower rates of GVHD (97). In other cases, better tumor control may result from the dual impact of metabolic modulation on both alloreactive T cells and the underlying malignancy (98, 99). It has also been argued that T cell activation, and hence metabolic demands, operate on a continuum, with GVHD-causing T cells at the far end of the activation and metabolic spectrum (100). In this case, highly active T cells would be more susceptible to metabolic or similar perturbations than anti-tumor T cells with more modest energy requirements. Indeed, Treg transfer experiments support this concept of differential sensitivity, as exogenous Treg administration sufficiently controls alloreactive T cell expansion without compromising GVT activity (101).
Metabolic Influence Beyond T Cells
It was been known for some time that Treg induction in the gastrointestinal tract is influenced by production of short chain fatty acids (SCFAs), commonly produced by commensal bacteria, and primarily in the forms of butyrate and propionate. SCFAs induce Foxp3 expression (102, 103) by either inhibiting histone deacetylases or by activating G-protein receptor 43 (GPR43) (103). Butyrate levels were found to be excessively low in intestinal tissues following allogeneic transplantation and administration of exogenous butyrate increased these levels back to normal while decreasing GVHD severity and improving weight loss and clinical scores (104). However, these beneficial effects were found to be independent of Treg and instead resulted from direct salutary effects of butyrate on the intestinal epithelial cells (IECs), essentially improving the host response to injury. In a follow-up study, GPR43 expression was found to be necessary to realize the GVHD-protective effects of butyrate and because of this necessity GVHD severity increased in mice lacking GPR43 (105). Ultimately, GPR43 signaling increased inflammasome activation in IECs and enhanced IEC integrity and epithelial repair secondary to increases in local cytokine secretion including IL-18. Thus, metabolites and metabolic pathways beyond those utilized directly by T cells can have a profound effect on GVHD pathobiology, in part by influencing host cell responses.
Toward a Unifying Theory of Alloreactive T Cell Metabolism
As highlighted thus far, alloreactive T cell metabolism is complex, with studies implicating a role for multiple and sometimes opposing metabolic pathways and substrates. While some contradictory findings might relate to minor differences in animal models, or the time point tested, an additional possibility is that the metabolic pathways being considered are not mutually exclusive and alloreactive T cells might upregulate multiple pathways at the same time. To this point, data from our lab demonstrates that CD8 T cells isolated from allogeneic recipients on day 7 post-transplant simultaneously increased both OXPHOS, as measured by oxygen consumption rates, and glycolysis, as measured by extracellular acidification (Figure 1). These data argue that OXPHOS and glycolysis are not mutually exclusive pathways within T cells and instead hint that alloreactive T cells might increase both aerobic glycolysis and OXPHOS simultaneously to meet their increased energy needs.
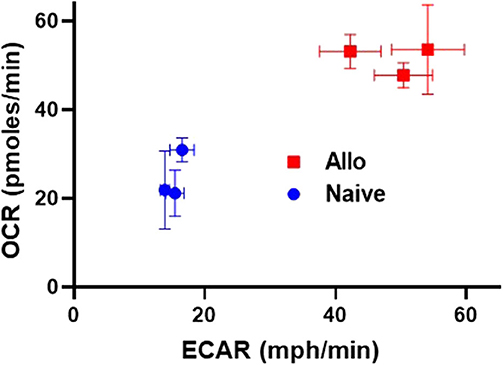
Figure 1. Alloreactive CD8+ T cells simultaneously increase both OXPHOS and glycolysis. 2 × 106 CD45.1+ B6 T cells and 5 × 106 B6 bone marrow (BM) cells were transplanted into irradiated, allogeneic (B6 × DBA F1) recipients. On day 7 post-transplant, donor CD8+ T cells (CD8+CD45.1+TCR-β+) were flow-sorted and 2 × 105 cells placed into a Seahorse metabolic analyzer. Values for both oxygen consumption rate (OCR) and extracellular acidification of the media (ECAR), a proxy for glycolysis, were simultaneously increased in day 7 CD8+ donor T cells.
Current Challenges
Inherently, our understanding of T cell metabolism is limited by the availability of current methods—namely pharmacologic manipulation and genetic deletion. While both tools can determine the role of a particular target in a specific pathway or disease process, pharmacologic manipulation and genetic deletion both come with limitations. For pharmacologic activators and inhibitors, this limitation is often an unintended effect of the drug, whereas genetic knockouts are often compromised by timing of the deletion in relation to when the effects are being measured.
Looking more closely at pharmacologic manipulation, activators and inhibitors can work indirectly, have off-target effects, or mediate on-target effects in off-target cells/tissues. The use of metformin as an AMPK activator is a perfect example of this challenge (106–108). Because metformin inhibits complex I of the ETC, it activates AMPK indirectly by increasing the AMP:ATP ratio (109–111). Thus, it is often difficult to determine if metformin-related changes are due to AMPK activation or instead to metformin's direct effects on the ETC. Inhibitors can also cause off target effects. Many foundational studies used the CPT1a inhibitor etomoxir to study FAO (112, 113) and connect FAO with specific changes in Treg and/or memory T cell (Tmem) populations (24, 25, 114). Recently, however, it was shown that the higher concentrations of etomoxir used in most studies not only inhibited long-chain FAO, but also broadly inhibited OXPHOS secondary to a decreased abundance of TCA cycle intermediates (115). Thus, given the potential off-target effects of etomoxir, interpretation of many seminal findings have come under increased scrutiny. Lastly, pharmacologic administration can cause unwanted effects in off-target tissues, resulting in toxic side effects and limiting the utility of the drug. For example, the glutamine antagonist, 6-Diazo-5-oxo-L-norleucine (DON) (116), promotes tolerance in combination with glycolytic inhibitors (117). However, its use has been limited because of severe on target, off-tissue toxicity, particularly to the gastrointestinal tract (118, 119). This challenge has led to the development of a DON pro-drug to more specifically deliver glutamine inhibition directly to tissues of interest (120).
Regarding genetic knockouts, while enhanced technologies can target a specific gene of interest within a given tissue type, timing of the deletion, particularly vis-à-vis measurement of effect, becomes extraordinarily important. In most cases of T cell specific-deletion, Cre recombinase is expressed under control of either the CD4 or lymphocyte protein kinase (Lck) promoter. In both cases, genetic deletion occurs during T cell development, leaving a long time for T cells to utilize alternative compensatory pathways, akin to taking the back-roads into work when the highway is unavailable. This point is important to keep in mind, as experiments using genetic knockouts might easily yield results confounded by the adoption of compensatory pathways.
In the example cited earlier, Raud et al. (115) used CD4 promoter driven Cre expression to delete CPT1a specifically in T cells, allowing them to conclude that Tmem and Treg cells developed in vitro and in vivo in the absence of CPT1a (presumably lacking FAO). While these findings appear to contradict a necessity for FAO in Treg and Tmem cells (24, 25, 114), alternative explanations exist. A distinct possibility is that an extended loss of CPT1a allowed T cells to become dependent on other metabolic pathways (121). In fact, Raud et al. (115) concede that their experiments did not limit medium or short-chain FAO, leaving open the possibility of this alternative pathway. In this one example, acute inhibition of FAO (as would be the intent with pharmacologic inhibitors) might give a very different outcome than prolonged absence of fat oxidation using genetic models, highlighting that timing of genetic deletion vis-à-vis measurement of effect must be strongly considered in every situation.
The potential for metabolic flexibility also influences our approach in treating T cell-driven pathogenesis. For example, treatment efficacy could improve by targeting multiple metabolic pathways simultaneously, in essence restricting T cells from upregulating compensatory pathways. Akin to retroviral therapy for human immunodeficiency virus (HIV) (122, 123), where a combination of drugs targets different viral components, targeting multiple metabolic pathways concurrently may be necessary to overcome the metabolic adaptations of pathogenic cells. In the context of solid organ transplantation, the combination DON (glutamine inhibition), 2-DG (glycolytic inhibition), and metformin (targeting OXPHOS) effectively promoted tolerance in fully mismatched skin and heart allograft models (117). Alternatively, inhibiting a mediator central to the metabolic reprogramming of multiple pathways might also be feasible. During GVHD, PD-L1 was shown to be central to reprogramming multiple T cell pathways and PD-L1 deficient T cells reduced glycolysis, OXPHOS, and FAO, improving GVHD outcomes in the process (124).
Finally, careful study of T cell metabolism as well as secondary/tertiary compensatory pathways will improve the timing and specificity of our inhibition. In this regard, aHSCT offers the distinct advantage of having a period in which donor cells are manipulated ex vivo prior to transplantation into recipients. Leveraging this advantage, metabolic inhibitors or activators could be applied exclusively to donor cells, sparing tissues from systemic administration of the compound. Furthermore, it may be possible to inhibit a first pathway through ex vivo manipulation (e.g., FAO), followed by subsequent inhibition of a compensatory pathway in vivo through systemic administration of a second inhibitor (e.g., glutamine metabolism). Cells lesioned in the first pathway would be expected to be more sensitive to secondary inhibition, while T cells arising de novo (and not having experienced the primary inhibition) would be spared. Ultimately, precise metabolic modulation and the potential for simultaneous inhibition of multiple metabolic pathways, will enhance the efficacy of metabolism-based therapies.
Future Directions
The breadth of research highlighted thus far lays a strong foundation for an increased understanding of T cell metabolism during aHSCT. However, outstanding questions remain regarding the heterogeneity of T cells recovered post-transplant and whether murine findings are translatable to humans. Like many complex disease processes, T cells collected and analyzed post-aHSCT represent a heterogenous population of cells including those driving the alloreactive response and bystanders simply responding to the inflammatory milieu. Studying metabolism in whole T cells during GVHD thus glosses over differences between individual T cell subsets and complicates the understanding of metabolism in the most highly alloreactive T cells. One approach to clarify this issue has been to use GVHD models in which donor T cells respond to a specific antigen. For example, transgenic CD8 T cells that recognize the SIINFEKL peptide of ovalbumin (i.e., OT-1 T cells) can be injected into CAG-OVA recipients which express ovalbumin as a self-antigen. Using this model, T cells transplanted into CAG-OVA recipients expressed increased levels of ROS and PD-1 compared to OT-1 T cells responding to immunization with CAG-OVA expressing dendritic cells (125). In a different study, OT-1 T cells isolated during GVHD increased fat transport while bystander (non-OVA reactive) T cells did not (54). However, the use of transgenic systems for the study of metabolism in alloreactive T cells has been limited.
Another approach is to use single-cell metabolomics to determine the individual metabolic profiles of donor T cells isolated from allogeneic recipients, a breakthrough that has the potential to revolutionize the study of immunometabolism during GVHD. While there are many groups working to develop single-cell metabolic technologies, the approach remains relatively new and faces numerous challenges, including the technical hurdle of how to collect cells without altering their metabolite abundance (126–129). To circumvent these issues, Miller et al. (130) developed a system that measures the activity of five key metabolic enzymes in conjunction with cell marker analysis to measure metabolic activity with single-cell resolution. Although this alternative approach is promising, there is no measure of global metabolic activity and the process is not high throughput. Thus, measuring metabolic activity at the single cell level, although highly promising for the study of GVHD, is currently limited by available technology.
Finally, the studies highlighted in this review have primarily utilized models using murine T cells and known strain combinations. While animal models have been integral to building foundational knowledge, their inherent limitations, including differences in murine and human immunology, pathogen free housing conditions, and genetic homogeneity can limit the translatability of the findings. Humanized murine models of xenogeneic GVHD (xGVHD) improve upon existing animal models by injecting human peripheral blood mononuclear cells (hPBMCs) into immunodeficient mice (131), allowing for the expansion and activation of human T cells in vivo. Increased use of xGVHD models will undoubtedly increase the likelihood that experimental findings will apply to human patients. However, xGVHD models themselves are imperfect and do not improve entirely upon deficiencies of classical animal models. Thus, patient samples may prove to be the best way to validate laboratory findings and ensure that observations are applicable to real world scenarios. In recent years, the repertoire of available technologies for in vivo human work has expanded, making the in vivo study of T cell metabolism during GVHD more feasible than ever. For example, recent publications have used administration of non-radioactive isotope tracers to study tissue metabolism in human patients in real time (132, 133). It could be envisioned that using non-radioactive isotope tracing in GVHD patients at the time of diagnosis could help to determine the dynamics of T cell metabolism during an active alloreactive immunologic response in vivo.
Conclusion
This review has investigated recent developments in our understanding of alloreactive T cell metabolism. While evidence suggests that many metabolic pathways are active in alloreactive T cells (Figure 2), including glycolysis, OXPHOS, FAO, and glutamine metabolism, there is no current consensus on the relative importance of each pathway or their temporal necessity. Furthermore, while studies have examined the significance of glycolysis and OXPHOS independent from each other, it is likely that glycolysis and OXPHOS increase simultaneously in alloreactive cells to meet enhanced energetic demands. In addition, the inherent ability of T cells to exhibit metabolic flexibility, adopting a compensatory metabolism when an initial pathway is lesioned (as occurs with genetic deletions), coupled with the indirect and somewhat pleiotropic nature of metabolic activators and inhibitors, makes assignment of causality difficult in many cases. Finally, Treg exhibit a distinct metabolic profile linking oxidative metabolism to T cell suppressive function, a phenomenon that appears to hold during allogeneic transplantation, yet remains the focus of intense investigation. In the end, studying T cell metabolism in the context of GVHD will help to deepen our understanding of in vivo T cell biology and identify novel therapies for the treatment of T cell-mediated pathologies.
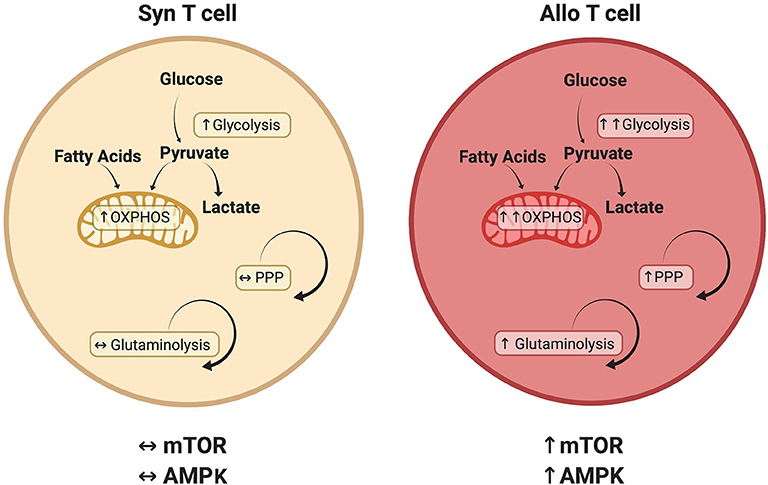
Figure 2. Metabolic pathways that distinguish alloreactive and syngeneic T cells. T cells isolated from allogeneic recipients have a unique metabolic profile including a greater increase in both glycolysis and oxidative phosphorylation (OXPHOS) (29, 53, 124). While various carbon sources contribute to this increased oxidative metabolism, including glucose (29, 52) and fatty acids (54), the relative contribution of each substrate remains to be determined. Glutaminolysis and the PPP are also preferentially upregulated in alloreactive T cells, as is activation of the AMPK and mTOR pathways (29, 70, 74). Because T cell metabolism is a dynamic process, this figure represents known or suspected metabolic activity in donor T cells on day 7 post-transplant. The relative contribution of each pathway is likely to change over time as discussed in the text.
Author Contributions
RB reviewed the literature, drafted the manuscript, and reviewed the final product. CB reviewed and interpreted the primary literature, edited the manuscript draft, and critically revised the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants to CB from the National Institute of Health—NHBLI (K08 HL123631 and R01 HL144556), the Hyundai Motor Company (Hope on Wheels Scholar grant), the American Society of Hematology (Scholar Award), and the Be the Match Foundation (Amy Strelzer Manasevit Award) and to RB from the American Society of Hematology (Physician Scientist Career Development Award).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med. (2006) 354:1813–26. doi: 10.1056/NEJMra052638
2. Li HW, Sykes M. Emerging concepts in haematopoietic cell transplantation. Nat Rev Immunol. (2012) 12:403–16. doi: 10.1038/nri3226
3. Ljungman P, Bregni M, Brune M, Cornelissen J, de Witte T, Dini G, et al. Allogeneic and autologous transplantation for haematological diseases, solid tumours and immune disorders: current practice in Europe 2009. Bone Marrow Transplant. (2010) 45:219–34. doi: 10.1038/bmt.2009.141
4. Zeiser R, Blazar BR. Acute graft-versus-host disease - biologic process, prevention, and therapy. N Engl J Med. (2017) 377:2167–79. doi: 10.1056/NEJMra1609337
5. Saliba RM, de Lima M, Giralt S, Andersson B, Khouri IF, Hosing C, et al. Hyperacute GVHD: risk factors, outcomes, and clinical implications. Blood. (2007) 109:2751–8. doi: 10.1182/blood-2006-07-034348
6. Marmont AM, Horowitz MM, Gale RP, Sobocinski K, Ash RC, van Bekkum DW, et al. T-cell depletion of HLA-identical transplants in leukemia. Blood. (1991) 78:2120–30. doi: 10.1182/blood.V78.8.2120.2120
7. Singh AK, McGuirk JP. Allogeneic stem cell transplantation: a historical and scientific overview. Cancer Res. (2016) 76:6445–51. doi: 10.1158/0008-5472.CAN-16-1311
8. Lum LG. The kinetics of immune reconstitution after human marrow transplantation. Blood. (1987) 69:369–80. doi: 10.1182/blood.V69.2.369.bloodjournal692369
9. D'Souza A, Lee S, Zhu X, Pasquini M. Current use and trends in hematopoietic cell transplantation in the united states. Biol Blood Marrow Transplant. (2017) 23:1417–21. doi: 10.1016/j.bbmt.2017.05.035
10. Ho VT, Soiffer RJ. The history and future of T-cell depletion as graft-versus-host disease prophylaxis for allogeneic hematopoietic stem cell transplantation. Blood. (2001) 98:3192–204. doi: 10.1182/blood.V98.12.3192
11. Mathis D, Shoelson SE. Immunometabolism: an emerging' ' frontier. Nat Rev Immunol. (2011) 11:81. doi: 10.1038/nri2922
12. O'Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
13. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
14. Mills EL, Kelly B, O'Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol. (2017) 18:488–98. doi: 10.1038/ni.3704
15. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
16. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. (2002) 16:769–77. doi: 10.1016/S1074-7613(02)00323-0
17. Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol. (2012) 33:168–73. doi: 10.1016/j.it.2012.01.010
18. Geltink RIK, Kyle RL, Pearce EL. Unraveling the complex interplay between T cell metabolism and function. Annu Rev Immunol. (2018) 36:461–88. doi: 10.1146/annurev-immunol-042617-053019
19. Chang CH, Curtis JD, Maggi LB, Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
20. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science. (2016) 354:481–4. doi: 10.1126/science.aaf6284
21. Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. (2005) 5:844–52. doi: 10.1038/nri1710
22. Yusuf I, Fruman DA. Regulation of quiescence in lymphocytes. Trends Immunol. (2003) 24:380–6. doi: 10.1016/S1471-4906(03)00141-8
23. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. (2011) 208:1367–76. doi: 10.1084/jem.20110278
24. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
25. van der Windt GJW, O'Sullivan D, Everts B, Huang SCC, Buck MD, Curtis JD, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci USA. (2013) 110:14336–41. doi: 10.1073/pnas.1221740110
26. Ma EH, Verway MJ, Johnson RM, Roy DG, Steadman M, Hayes S, et al. Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated CD8+ T cells. Immunity. (2019) 51:856–70.e5. doi: 10.1016/j.immuni.2019.09.003
27. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. (2008) 180:4476–86. doi: 10.4049/jimmunol.180.7.4476
28. Almeida L, Lochner M, Berod L, Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol. (2016) 28:514–24. doi: 10.1016/j.smim.2016.10.009
29. Nguyen HD, Chatterjee S, Haarberg KMK, Wu Y, Bastian D, Heinrichs J, et al. Metabolic reprogramming of alloantigen-activated T cells after hematopoietic cell transplantation. J Clin Invest. (2016) 126:1337–52. doi: 10.1172/JCI82587
30. Schoors S, De Bock K, Cantelmo AR, Georgiadou M, Ghesquière B, Cauwenberghs S, et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. (2014) 19:37–48. doi: 10.1016/j.cmet.2013.11.008
31. Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. (2012) 337:975–80. doi: 10.1126/science.1222278
32. Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther. (2008) 7:110–20. doi: 10.1158/1535-7163.MCT-07-0482
33. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. (2014) 20:61–72. doi: 10.1016/j.cmet.2014.05.004
34. Stelljes M, Hermann S, Albring J, Köhler G, Löffler M, Franzius C, et al. Clinical molecular imaging in intestinal graft-versus-host disease: mapping of disease activity, prediction, and monitoring of treatment efficiency by positron emission tomography. Blood. (2008) 111:2909–18. doi: 10.1182/blood-2007-10-119164
35. Liu Y, Zhang D, Liu X. mTOR signaling in T cell immunity and autoimmunity. Int Rev Immunol. (2015) 34:50–66. doi: 10.3109/08830185.2014.933957
36. Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. (2009) 122:3589–94. doi: 10.1242/jcs.051011
37. Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. (2012) 12:325–38. doi: 10.1038/nri3198
38. Myers DR, Wheeler B, Roose JP. mTOR and other effector kinase signals that impact T cell function and activity. Immunol Rev. (2019) 291:134–53. doi: 10.1111/imr.12796
39. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
40. Linke M, Fritsch SD, Sukhbaatar N, Hengstschläger M, Weichhart T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. (2017) 591:3089–103. doi: 10.1002/1873-3468.12711
41. Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. (2010) 39:171–83. doi: 10.1016/j.molcel.2010.06.022
42. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. (2006) 3:187–97. doi: 10.1016/j.cmet.2006.01.012
43. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
44. Kim J, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
45. Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. (2007) 18:1437–46. doi: 10.1091/mbc.e06-07-0593
46. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. (2009) 30:832–44. doi: 10.1016/j.immuni.2009.04.014
47. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. (2011) 12:295–303. doi: 10.1038/ni.2005
48. Kurebayashi Y, Nagai S, Ikejiri A, Ohtani M, Ichiyama K, Baba Y, et al. PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORγ. Cell Rep. (2012) 1:360–73. doi: 10.1016/j.celrep.2012.02.007
49. Park MJ, Lee SY, Moon SJ, Son HJ, Lee SH, Kim EK, et al. Metformin attenuates graft-versus-host disease via restricting mammalian target of rapamycin/signal transducer and activator of transcription 3 and promoting adenosine monophosphate-activated protein kinase-autophagy for the balance between T helper 17 and Tregs. Transl Res. (2016) 173:115–30. doi: 10.1016/j.trsl.2016.03.006
50. Abouelnasr A, Roy J, Cohen S, Kiss T, Lachance S. Defining the role of sirolimus in the management of graft-versus-host disease: from prophylaxis to treatment. Biol Blood Marrow Transplant. (2013) 19:12–21. doi: 10.1016/j.bbmt.2012.06.020
51. Lutz M, Mielke S. New perspectives on the use of mTOR inhibitors in allogeneic haematopoietic stem cell transplantation and graft-versus-host disease. Br J Clin Pharmacol. (2016) 82:1171–9. doi: 10.1111/bcp.13022
52. Saha A, Aoyama K, Taylor PA, Koehn BH, Veenstra RG, Panoskaltsis-Mortari A, et al. Host programmed death ligand 1 is dominant over programmed death ligand 2 expression in regulating graft-versus-host disease lethality. Blood. (2013) 122:3062–73. doi: 10.1182/blood-2013-05-500801
53. Gatza E, Wahl DR, Opipari AW, Sundberg TB, Reddy P, Liu C, et al. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci Transl Med. (2011) 3:67ra8. doi: 10.1126/scitranslmed.3001975
54. Byersdorfer CA, Tkachev V, Opipari AW, Goodell S, Swanson J, Sandquist S, et al. Effector T cells require fatty acid metabolism during murine graft-versus-host disease. Blood. (2013) 122:3230–7. doi: 10.1182/blood-2013-04-495515
55. Ma EH, Poffenberger MC, Wong AHT, Jones RG. The role of AMPK in T cell metabolism and function. Curr Opin Immunol. (2017) 46:45–52. doi: 10.1016/j.coi.2017.04.004
56. Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. (2003) 546:113–20. doi: 10.1016/S0014-5793(03)00560-X
57. Adams WC, Chen YH, Kratchmarov R, Yen B, Nish SA, Lin WHW, et al. Anabolism-associated mitochondrial stasis driving lymphocyte differentiation over self-renewal. Cell Rep. (2016) 17:3142–52. doi: 10.1016/j.celrep.2016.11.065
58. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. (2008) 30:214–26. doi: 10.1016/j.molcel.2008.03.003
59. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. (2018) 19:121–35. doi: 10.1038/nrm.2017.95
60. Mayer A, Denanglaire S, Viollet B, Leo O, Andris F. AMP-activated protein kinase regulates lymphocyte responses to metabolic stress but is largely dispensable for immune cell development and function. Eur J Immunol. (2008) 38:948–56. doi: 10.1002/eji.200738045
61. MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, et al. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol. (2011) 187:4187–98. doi: 10.4049/jimmunol.1100367
62. Rolf J, Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA. AMPKα1: a glucose sensor that controls CD8 T-cell memory. Eur J Immunol. (2013) 43:889–96. doi: 10.1002/eji.201243008
63. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. (2015) 42:41–54. doi: 10.1016/j.immuni.2014.12.030
64. Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ, et al. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-all cell stress and survival. Cell Metab. (2016) 23:649–62. doi: 10.1016/j.cmet.2016.03.008
65. Kang KY, Kim YK, Yi H, Kim J, Jung HR, Kim IJ, et al. Metformin downregulates Th17 cells differentiation and attenuates murine autoimmune arthritis. Int Immunopharmacol. (2013) 16:85–92. doi: 10.1016/j.intimp.2013.03.020
66. Son HJ, Lee J, Lee SY, Kim EK, Park MJ, Kim KW, et al. Metformin attenuates experimental autoimmune arthritis through reciprocal regulation of Th17/Treg balance and osteoclastogenesis. Mediators Inflamm. (2014) 2014:973986. doi: 10.1155/2014/973986
67. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. (2000) 348(Pt 3):607–14. doi: 10.1042/bj3480607
68. El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. (2000) 275:223–8. doi: 10.1074/jbc.275.1.223
69. Fontaine E. Metformin-Induced mitochondrial complex I inhibition: facts, uncertainties, and consequences. Front Endocrinol. (2018) 9:753. doi: 10.3389/fendo.2018.00753
70. Beezhold K, Moore N, Chiaranunt P, Brown R, Byersdorfer CA. Deletion of AMP-activated protein kinase (AMPK) in donor T cells protects against graft-verus-host disease through control of regulatory T cell expansion and target organ infiltration. Blood. (2016) 128:806–6. doi: 10.1182/blood.V128.22.806.806
71. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. (2010) 185:1037–44. doi: 10.4049/jimmunol.0903586
72. Pollizzi KN, Sun IH, Patel CH, Lo YC, Oh MH, Waickman AT, et al. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8(+) T cell differentiation. Nat Immunol. (2016) 17:704–11. doi: 10.1038/ni.3438
73. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. (2014) 40:692–705. doi: 10.1016/j.immuni.2014.04.007
74. Glick GD, Rossignol R, Lyssiotis CA, Wahl D, Lesch C, Sanchez B, et al. Anaplerotic metabolism of alloreactive T cells provides a metabolic approach to treat graft-versus-host disease. J Pharmacol Exp Ther. (2014) 351:298–307. doi: 10.1124/jpet.114.218099
75. Song EK, Yim JM, Yim JY, Song MY, Rho HW, Yim SK, et al. Glutamine protects mice from acute graft-versus-host disease (aGVHD). Biochem Biophys Res Commun. (2013) 435:94–9. doi: 10.1016/j.bbrc.2013.04.047
76. da Gama Torres HO, Vilela EG, da Cunha AS, Goulart EMA, Souza MHC, Aguirre ACC, et al. Efficacy of glutamine-supplemented parenteral nutrition on short-term survival following allo-SCT: a randomized study. Bone Marrow Transplant. (2008) 41:1021–7. doi: 10.1038/bmt.2008.27
77. Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
78. Zorn E, Kim HT, Lee SJ, Floyd BH, Litsa D, Arumugarajah S, et al. Reduced frequency of FOXP3+ CD4+CD25+ regulatory T cells in patients with chronic graft-versus-host disease. Blood. (2005) 106:2903–11. doi: 10.1182/blood-2005-03-1257
79. Kennedy-Nasser AA, Ku S, Castillo-Caro P, Hazrat Y, Wu MF, Liu H, et al. Ultra low-dose IL-2 for GVHD prophylaxis after allogeneic hematopoietic stem cell transplantation mediates expansion of regulatory T cells without diminishing antiviral and antileukemic activity. Clin Cancer Res. (2014) 20:2215–25. doi: 10.1158/1078-0432.CCR-13-3205
80. Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. (2011) 365:2055–66. doi: 10.1056/NEJMoa1108188
81. Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, et al. Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Sci Transl Med. (2011) 3:83ra41. doi: 10.1126/scitranslmed.3001809
82. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. (2017) 25:1282–93.e7. doi: 10.1016/j.cmet.2016.12.018
83. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat Immunol. (2016) 17:1459–66. doi: 10.1038/ni.3577
84. Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, et al. Essential role of mitochondrial energy metabolism in Foxp3+ T-regulatory cell function and allograft survival. FASEB J. (2015) 29:2315–26. doi: 10.1096/fj.14-268409
85. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martínez-Reyes I, et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature. (2019) 565:495–9. doi: 10.1038/s41586-018-0846-z
86. Chapman NM, Zeng H, Nguyen TLM, Wang Y, Vogel P, Dhungana Y, et al. mTOR coordinates transcriptional programs and mitochondrial metabolism of activated Treg subsets to protect tissue homeostasis. Nat Commun. (2018) 9:2095. doi: 10.1038/s41467-018-04392-5
87. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. (2013) 38:225–36. doi: 10.1016/j.immuni.2012.10.020
88. Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, et al. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. (2004) 13:935–44. doi: 10.1093/hmg/ddh109
89. Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. (1998) 18:231–6. doi: 10.1038/ng0398-231
90. Procaccini C, De Rosa V, Galgani M, Abanni L, Calì G, Porcellini A, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. (2010) 33:929–41. doi: 10.1016/j.immuni.2010.11.024
91. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature. (2013) 499:485–90. doi: 10.1038/nature12297
92. Daenthanasanmak A, Iamsawat S, Chakraborty P, Nguyen HD, Bastian D, Liu C, et al. Targeting Sirt-1 controls GVHD by inhibiting T-cell allo-response and promoting Treg stability in mice. Blood. (2019) 133:266–79. doi: 10.1182/blood-2018-07-863233
93. Walton K, Fernandez MR, Sagatys EM, Reff J, Kim J, Lee MC, et al. Metabolic reprogramming augments potency of human pSTAT3-inhibited iTregs to suppress alloreactivity. JCI Insight. (2020) 5:e136437. doi: 10.1172/jci.insight.136437
94. McDonald-Hyman C, Muller JT, Loschi M, Thangavelu G, Saha A, Kumari S, et al. The vimentin intermediate filament network restrains regulatory T cell suppression of graft-versus-host disease. J Clin Invest. (2018) 128:4604–21. doi: 10.1172/JCI95713
95. Yang S, Sheng X, Xiang D, Wei X, Chen T, Yang Z, et al. CD150highTreg cells may attenuate graft versus host disease and intestinal cell apoptosis after hematopoietic stem cell transplantation. Am J Transl Res. (2019) 11:1299–310.
96. Sofi MH, Wu Y, Schutt SD, Dai M, Daenthanasanmak A, Heinrichs Voss J, et al. Thioredoxin-1 confines T cell alloresponse and pathogenicity in graft-versus-host disease. J Clin Invest. (2019) 129:2760–74. doi: 10.1172/JCI122899
97. Kornblau SM, Aycox PG, Stephens C, McCue LD, Champlin RE, Marini FC. Control of graft-versus-host disease with maintenance of the graft-versus-leukemia effect in a murine allogeneic transplant model using retrovirally transduced murine suicidal lymphocytes. Exp Hematol. (2007) 35:842–53. doi: 10.1016/j.exphem.2007.02.008
98. Beezhold K, Byersdorfer CA. Targeting immuno-metabolism to improve anti-cancer therapies. Cancer Lett. (2018) 414:127–35. doi: 10.1016/j.canlet.2017.11.005
99. Gerner RR, Macheiner S, Reider S, Siegmund K, Grabherr F, Mayr L, et al. Targeting NAD immunometabolism limits severe graft-versus-host disease and has potent antileukemic activity. Leukemia. (2020) 34:1885–97. doi: 10.1038/s41375-020-0709-0
100. Byersdorfer CA. The role of Fatty Acid oxidation in the metabolic reprograming of activated t-cells. Front Immunol. (2014) 5:641. doi: 10.3389/fimmu.2014.00641
101. Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. (2003) 9:1144–50. doi: 10.1038/nm915
102. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. (2013) 504:446–50. doi: 10.1038/nature12721
103. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. (2013) 341:569–73. doi: 10.1126/science.1241165
104. Mathewson ND, Jenq R, Mathew AV, Koenigsknecht M, Hanash A, Toubai T, et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol. (2016) 17:505–13. doi: 10.1038/ni.3400
105. Fujiwara H, Docampo MD, Riwes M, Peltier D, Toubai T, Henig I, et al. Microbial metabolite sensor GPR43 controls severity of experimental GVHD. Nat Commun. (2018) 9:3674. doi: 10.1038/s41467-018-06048-w
106. Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. (2002) 51:2074–81. doi: 10.2337/diabetes.51.7.2074
107. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. (2001) 108:1167–74. doi: 10.1172/JCI13505
108. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. (2005) 310:1642–6. doi: 10.1126/science.1120781
109. Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. (2010) 120:2355–69. doi: 10.1172/JCI40671
110. Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. (2010) 11:554–65. doi: 10.1016/j.cmet.2010.04.001
111. Stephenne X, Foretz M, Taleux N, van der Zon GC, Sokal E, Hue L, et al. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia. (2011) 54:3101–10. doi: 10.1007/s00125-011-2311-5
112. Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem. (2006) 281:37372–80. doi: 10.1074/jbc.M608372200
113. Lopaschuk GD. Treating ischemic heart disease by pharmacologically improving cardiac energy metabolism. Am J Cardiol. (1998) 82:14K−7K. doi: 10.1016/S0002-9149(98)00532-3
114. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. (2009) 460:103–7. doi: 10.1038/nature08097
115. Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, et al. Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab. (2018) 28:504–15.e7. doi: 10.1016/j.cmet.2018.06.002
116. Thomas AG, O'Driscoll CM, Bressler J, Kaufmann W, Rojas CJ, Slusher BS. Small molecule glutaminase inhibitors block glutamate release from stimulated microglia. Biochem Biophys Res Commun. (2014) 443:32–6. doi: 10.1016/j.bbrc.2013.11.043
117. Lee CF, Lo YC, Cheng CH, Furtmüller GJ, Oh B, Andrade-Oliveira V, et al. Preventing allograft rejection by targeting immune metabolism. Cell Rep. (2015) 13:760–70. doi: 10.1016/j.celrep.2015.09.036
118. Lemberg KM, Vornov JJ, Rais R, Slusher BS. We're Not “DON” Yet: optimal dosing and prodrug delivery of 6-Diazo-5-oxo-L-norleucine. Mol Cancer Ther. (2018) 17:1824–32. doi: 10.1158/1535-7163.MCT-17-1148
119. Potter MC, Baxter VK, Mathey RW, Alt J, Rojas C, Griffin DE, et al. sequelae induced by alphavirus infection of the CNS are attenuated by treatment with the glutamine antagonist 6-diazo-5-oxo-l-norleucine. J Neurovirol. (2015) 21:159–73. doi: 10.1007/s13365-015-0314-6
120. Rais R, Jančarík A, Tenora L, Nedelcovych M, Alt J, Englert J, et al. Discovery of 6-Diazo-5-oxo-l-norleucine (DON) prodrugs with enhanced CSF delivery in monkeys: a potential treatment for glioblastoma. J Med Chem. (2016) 59:8621–33. doi: 10.1021/acs.jmedchem.6b01069
121. Van den Bossche J, van der Windt GJW. Fatty acid oxidation in macrophages and T cells: time for reassessment? Cell Metab. (2018) 28:538–40. doi: 10.1016/j.cmet.2018.09.018
122. Balzarini J, Pelemans H, Karlsson A, De ClercQ E, Kleim JP. Concomitant combination therapy for HIV infection preferable over sequential therapy with 3TC and non-nucleoside reverse transcriptase inhibitors. Proc Natl Acad Sci USA. (1996) 93:13152–7. doi: 10.1073/pnas.93.23.13152
123. Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med. (1997) 337:734–9. doi: 10.1056/NEJM199709113371102
124. Saha A, O'Connor RS, Thangavelu G, Lovitch SB, Dandamudi DB, Wilson CB, et al. Programmed death ligand-1 expression on donor T cells drives graft-versus-host disease lethality. J Clin Invest. (2016) 126:2642–60. doi: 10.1172/JCI85796
125. Tkachev V, Goodell S, Opipari AW, Hao LY, Franchi L, Glick GD, et al. Programmed death-1 controls T cell survival by regulating oxidative metabolism. J Immunol. (2015) 194:5789–800. doi: 10.4049/jimmunol.1402180
126. Zenobi R. Single-cell metabolomics: analytical and biological perspectives. Science. (2013) 342:1243259. doi: 10.1126/science.1243259
127. Amantonico A, Urban PL, Zenobi R. Analytical techniques for single-cell metabolomics: state of the art and trends. Anal Bioanal Chem. (2010) 398:2493–504. doi: 10.1007/s00216-010-3850-1
128. Couvillion SP, Zhu Y, Nagy G, Adkins JN, Ansong C, Renslow RS, et al. New mass spectrometry technologies contributing towards comprehensive and high throughput omics analyses of single cells. Analyst. (2019) 144:794–807. doi: 10.1039/C8AN01574K
129. Zhang L, Vertes A. Single-cell mass spectrometry approaches to explore cellular heterogeneity. Angew Chem Int Ed. (2018) 57:4466–77. doi: 10.1002/anie.201709719
130. Miller A, Nagy C, Knapp B, Laengle J, Ponweiser E, Groeger M, et al. Exploring metabolic configurations of single cells within complex tissue microenvironments. Cell Metab. (2017) 26:788–800.e6. doi: 10.1016/j.cmet.2017.08.014
131. Walsh NC, Kenney LL, Jangalwe S, Aryee KE, Greiner DL, Brehm MA, et al. Humanized mouse models of clinical disease. Annu Rev Pathol. (2017) 12:187–215. doi: 10.1146/annurev-pathol-052016-100332
132. Kim IY, Suh SH, Lee IK, Wolfe RR. Applications of stable, nonradioactive isotope tracers in in vivo human metabolic research. Exp Mol Med. (2016) 48:e203. doi: 10.1038/emm.2015.97
Keywords: alloreactive T cells, GVHD biology, immunometabolism, glycolysis, fatty acid oxidation (FAO), mammalian target of rapamycin (mTOR), AMPK
Citation: Brown RA and Byersdorfer CA (2020) Metabolic Pathways in Alloreactive T Cells. Front. Immunol. 11:1517. doi: 10.3389/fimmu.2020.01517
Received: 03 April 2020; Accepted: 09 June 2020;
Published: 24 July 2020.
Edited by:
Michael Uhlin, Karolinska Institutet (KI), SwedenReviewed by:
Yongxia Wu, Medical University of South Carolina, United StatesYi Zhang, Temple University, United States
Copyright © 2020 Brown and Byersdorfer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Craig A. Byersdorfer, Y3JhaWcuYnllcnNkb3JmZXJAY2hwLmVkdQ==