 Mimi Mu1,2,3†Peiyu Gao1,2,3†Qian Yang1,2,3Jing He1,2,3Fengjiao Wu1,2,3Xue Han1,2,3Shujun Guo1,2,3Zhongqing Qian1,2,3
Mimi Mu1,2,3†Peiyu Gao1,2,3†Qian Yang1,2,3Jing He1,2,3Fengjiao Wu1,2,3Xue Han1,2,3Shujun Guo1,2,3Zhongqing Qian1,2,3 Chuanwang Song1,2,3*
Chuanwang Song1,2,3*- 1Department of Immunology, School of Laboratory Medicine, Bengbu Medical College, Bengbu, China
- 2Anhui Provincial Key Laboratory of Infection and Immunity, Bengbu Medical College, Bengbu, China
- 3Anhui Province Key Laboratory of Immunology in Chronic Diseases, Bengbu Medical College, Bengbu, China
To maintain alveolar gas exchange, the alveolar surface has to limit unnecessary inflammatory responses. This involves crosstalk between alveolar epithelial cells (AECs) and alveolar macrophages (AMs) in response to damaging factors. We recently showed that insulin-like growth factor (IGF)-1 regulates the phagocytosis of AECs. AMs secrete IGF-1 into the bronchoalveolar lavage fluid (BALF) in response to inflammatory stimuli. However, whether AECs regulate the production of IGF-1 by AMs in response to inflammatory signals remains unclear, as well as the role of IGF-1 in controlling the alveolar balance in the crosstalk between AMs and AECs under inflammatory conditions. In this study, we demonstrated that IGF-1 was upregulated in BALF and lung tissues of acute lung injury (ALI) mice, and that the increased IGF-1 was mainly derived from AMs. In vitro experiments showed that the production and secretion of IGF-1 by AMs as well as the expression of TGF-β were increased in LPS-stimulated AEC-conditioned medium (AEC-CM). Pharmacological blocking of TGF-β in AECs and addition of TGF-β neutralizing antibody to AEC-CM suggested that this AEC-derived cytokine mediates the increased production and secretion of IGF-1 from AMs. Blocking TGF-β synthesis or treatment with TGF-β neutralizing antibody attenuated the increase of IGF-1 in BALF in ALI mice. TGF-β induced the production of IGF-1 by AMs through the PI3K/Akt signaling pathway. IGF-1 prevented LPS-induced p38 MAPK activation and the expression of the inflammatory factors MCP-1, TNF-α, and IL-1β in AECs. However, IGF-1 upregulated PPARγ to increase the phagocytosis of apoptotic cells by AECs. Intratracheal instillation of IGF-1 decreased the number of polymorphonuclear neutrophils in BALF of ALI model mice, reduced alveolar congestion and edema, and suppressed inflammatory cell infiltration in lung tissues. These results elucidated a mechanism by which AECs used TGF-β to regulate IGF-1 production from AMs to attenuate endogenous inflammatory signals during alveolar inflammation.
Introduction
Continuous and direct exposure of tissues to the outside environment require tight regulation of the alveolar immune response to effectively identify and eliminate invading microorganisms with minimal immunopathology to maintain optimal gas exchange (1). Alveolar macrophages (AMs) constitute the first line of defense in the respiratory tract and are key regulators of airway immune responses. AMs account for 95% of lower respiratory tract leukocytes in the stable state (2). AMs actively inhibit antigen-induced T cell proliferation and decrease dendritic cell (DC) antigen presentation in the lungs (3). These immunosuppressive properties as well as the capacity to suppress airway hyper responsiveness confer AMs the ability to control the immune response and help preserve the physiological functions of the lungs.
The mature alveolar epithelium is composed of type I and type II alveolar epithelial cells (AECs or alveolar parietal cells), which occupy 95 and 5% of the alveolar surface, respectively (4). Under normal conditions, AECs are a slow proliferating lung cell population with a renewal time of 4–6 weeks. Unlike type I AECs, type II AECs can re-enter the cell cycle and divide. Therefore, type II AECs are considered pluripotent cells that possess high plasticity, self-renewal ability, and the capacity to produce type I AECs through the process of transdifferentiation (5). In addition to acting as a physical barrier against pathogens and various inhaled environmental particles, AECs actively participate in the lung's immune response, which is the main regulator of alveolar balance (6).
AMs are attached to the surface of AECs, supporting the possible interaction between the two cells (7). Therefore, many studies have examined the role of AM-AEC association and communication in lung homeostasis and inflammation. AEC-conditioned medium (AEC-CM) is chemotactic for AM migration; however, RANTES, MCP-1, and GM-CSF neutralizing antibodies decrease the chemotactic activity of AEC-CM for AMs by 58, 29, and 47%, respectively (8). AM-mediated suppression of cysteinyl leukotriene pathway genes in the AEC promotes resistance to influenza A virus infection, thereby protecting the body from fatal infection (9). Bourdonnay et al. showed that AMs secrete SOCS1 and 3 in exosomes and vesicles, which are ingested by AECs and inhibit the activation of JAK/STAT inflammatory signals in AECs (10).
Insulin-like growth factor (IGF)-1 is a 7.5 KDa peptide composed of 70 amino acids and has 50% homology to insulin. IGF-1 acts as an important survival factor in different cells by preventing apoptosis and inducing cell proliferation. IGF-1 plays an important role in the development and balance of organs and disease states, including the lung (11). Low serum IGF-1 level is a high risk factor for bronchoalveolar dysplasia (12). Pulmonary fibrosis is associated with increased IGF-1 originated from AMs, and IGF-1 can stimulate the differentiation of fibroblasts into myofibroblast phenotypes, thus playing an important role in pulmonary fibrosis (13–15). Narasaraju et al. showed that IGF-1 promotes the proliferation and differentiation of AECs (16). Recent work from our group showed that IGF-1 inhibits inflammatory signaling in AECs (17). However, whether IGF-1 acts as a mediator between AMs and AECs and its role in regulating the balance of the alveolar microenvironment remain unclear.
In the present study, we showed that TGF-β produced by LPS-induced AECs stimulated the production of IGF-1 by AMs. This AM-derived IGF-1 attenuated pre-inflammatory signals in AECs. This two-way communication between AECs and AMs represents a novel means for fine tuning inflammatory responses in alveoli.
Materials and Methods
AEC Cell Culture
The mouse AEC line MLE-12 was suspended in high glucose DMEM medium containing 10% fetal bovine serum (FBS), inoculated into 24-well plates, and cultured in a 37°C, 5% CO2 incubator. After the cells reached 70–90% confluency, they were digested with 0.25% trypsin, collected, and passaged every other day.
Antibodies and Reagents
LPS (Escherichia coli O26:B6, L8274) was obtained from Sigma-Aldrich. TGF-β1 (CK33) was purchased from novoprotein company. TGF-β1 antibody (sc-146) was purchased from santa cruz biotechnology company. IGF-1 (MB-2917A) was purchased from Jiangsu Enzyme Biotech company. IGF-1 antibody (ab9572) was originated from abcam company. PPARγ antibody (16643-1-AP) was purchased from Proteintech. p-AKT antibody (AA329-1), phosphorylated P38MAPK antibody (AM063-1), GAPDH antibody (AG019-1), HRP-labeled goat anti-mouse antibody (A0216), HRP-labeled goat anti-rabbit antibody (A0208), PI3K inhibitor Wortmannin (S1952), Annexin V-FITC dye (C1062L), and TGF-β isotype antibody (A7007) were from Beyotime. Curcumin (458-37-7) was purchased from meilunbio company. Pirfenidone (53179-13-8) was from MedChemExpress. Lipo fectamine2000 (1854311) was purchased from Invitrogen.
Establishment of the Acute Lung Injury Model
BALB/c mice were randomly divided into a normal control group and an acute lung injury (ALI) group. ALI model mice were challenged with lipopolysaccharide (LPS, 4 mg/kg) through a single nasal instillation, and the normal control group was administered PBS instead of LPS. Mice were sacrificed 24 h after LPS challenge, and the bronchoalveolar lavage fluid (BALF) and lung tissues were collected and stored at −80°C until use.
BAL and Acquisition of BALF
Under anesthesia, the mice were intubated, and bronchoalveolar lavage was performed using 1 ml of sterile PBS six times. The BALF was centrifuged at 1,500 rpm for 5 min, and the supernatant was collected and stored at −80°C until use. Cells in BALF were washed twice with PBS, and part of the cell suspension was used for cell counting, whereas the other part was subjected to cell smear. Wright staining was performed, and the cells were classified and counted under a microscope.
Acquisition of Primary AMs
The BALF obtained as described above was centrifuged and resuspended in RPMI-1640 complete medium. The cell suspension was inoculated into a 6-well plate (1 × 106 cells/well) and cultured in a 37°C incubator for 2 h. The suspended cells were aspirated, and the adherent cells were kept as the AM fraction.
Induction and Labeling of Apoptotic Cells
The mouse alveolar epithelial cell line MLE-12 was resuspended in DMEM complete medium and seeded into 6-well plates (1 × 106 cells/well). After the cells became adherent, they were stimulated with 50 μM curcumin for 72 h. Cells were then collected and stained for 15 min with AnnexinV-FITC in the dark. The stained apoptotic cells were washed twice with cold sterile PBS for use.
Detection of Phagocytosis
MLE-12 cells were resuspended in DMEM complete medium and seeded into 6-well plates (105 cells/well). After adherence of the cells, FITC-labeled apoptotic cells (5 × 104 cells/well) were added and co-cultured for 4 h. The cells were collected, washed twice with PBS, and the cell suspension was fixed with 4% PFA. Flow cytometry was used to detect the phagocytosis of FITC-labeled apoptotic cells.
RNA Interference
MLE-12 cells (105 cells/well) were seeded into 6-well plates and allowed to adhere for 12 h before transfection. Cells were transfected using Lipofectamine® 2000 following the manufacturer's instructions. A 80 nM FAM-siRNA-Lipo2000 mixture was prepared according to the instructions. MLE-12 cells were co-cultured with this mixture in 400 μL FBS-free DMEM for 48 h, and protein was extracted from cells to verify the transfection efficacy.
The qRT-PCR
Total RNA was extracted using the TRIzol reagent. The first strand cDNA was synthesized using 2 μg of RNA as a template according to the kit instructions (RT TransScript First-Strand cDNA Synthesis SuperMix kit). Then, primers were designed and real-time PCR was performed according to the kit instructions (SYBRGreen TransStart Top Green qPCR SuperMix kit). The relative expression of each gene was analyzed using the 2−ΔΔCt method. The primer sequences (5′-3′) are listed below: PPARγ, Sense: ACTCATACATAAAGTCCTTCCCGC, Antisense: CTCTTGCACGGCTTCTACGG; LXRA, Sense: TCATCAAGGGAGCACGCTATGT, Antisense: CTTGAGCCTGTTCCTCTTCTTGC; LXRB, Sense: TCCGACCAGCCCAAAGTCAC, Antisense: GCTGTTTCTAGCAACATGATCTCAA; TNF-α, Sense: ACCCTCACACTCACAAACCA, Antisense: ATAGCAAATCGGCTGACGGT; IL-1β, Sense: AAAAGCCTCGTGCTGTCG, Antisense: TGCTTGTGAGGTGCTGATGTA; MCP-1, Sense: GTCCCTGTCATGCTTCTGG, Antisense: AAGTGCTTGAGGTGGTTGTG; GAPDH, Sense: CCTCGTCCCGTAGACAAAATG, Antisense: TGAGGTCAATGAAGGGGTCGT.
Western Blot Analysis
Protein was extracted from cells using NP-40 solution, and protein concentration was determined using the BAC method. Aliquots containing 30 μg of protein were separated by 6% SDS-polyacrylamide gel electrophoresis, followed by transfer to a nitrocellulose membrane. The membrane was blocked with 5% milk for 2 h, and then incubated with the following primary antibodies at 4°C overnight: IGF-1 (1: 500), p-Akt (1: 1000), PPARγ (1: 1000), p-p38 MAPK (1: 2000), and GAPDH (1: 1000). The membrane was washed with Tris-buffered saline containing 0.05% Tween-20 and incubated with HRP-labeled goat anti-mouse antibody (1: 2000) for 2 h. Bands on the membrane were visualized using a BeyoECL Plus kit and integrated optical density analysis was performed using Image J software.
Wet-Dry Weight Ratio of Lung Tissue
The lung tissues of mice in each group were collected, and PBS pulmonary arterial lavage was performed to remove residual blood. Lung tissues were placed on absorbent paper to eliminate surface moisture, and the weight (wet weight) was measured uniformly and recorded. Lung tissues were then placed in a 37°C incubator for 24 h until the weight became constant. Then, lung tissues were removed and weighed (dry weight). The wet/dry (W/D) weight ratio of lung tissues in each group of mice was calculated.
Determination of Protein Concentration in BALF
Mice were tracheally intubated, and the BALF was obtained as described above. The protein concentration in BALF was measured according to the kit instructions.
HE Staining of Lung Tissue
Mouse lung tissues were fixed for 24 h with 4% paraformaldehyde and then dehydrated for 12 h using a fully automatic tissue dehydrator. Lung tissues were embedded in paraffin, and paraffin blocks were cut into 5 μm thick slices on a microtome. The sections were dewaxed with different concentrations of xylene, and after immersion in a gradient of alcohol (high concentration to low concentration), tissues were stained with hematoxylin and eosin. The sections were transparent with xylene and then sealed with a neutral resin, and observed and photographed under a microscope.
Statistical Methods
Experimental data are expressed as the mean ± standard deviation. Data were analyzed using SPSS 16.0 software. Comparisons between multiple groups were performed using analysis of variance, and comparisons between two groups were performed using t-tests. P < 0.05 was considered statistically significant.
Results
Increased IGF-1 Production in Acute LPS Lung Injury Models
Recent studies show that IGF-1 is involved in the regulation of inflammation (18). We first examined the expression and secretion of IGF-1 in the lungs of mice with LPS-induced ALI. In these experiments, IGF-1 was quantitatively detected by ELISA in BALF and lung tissue homogenates. At 24 h after LPS administration, the content of IGF-1 was significantly higher in the BALF and lung tissue homogenates of treated mice than in those of control mice (Figures 1A,B), and the expression of IGF-1 in lung tissues was also increased (Figure 1C). The increased IGF-1 content in the lung tissue homogenate was reduced by 60% after BAL in LPS lung injury model mice (Figure 1D). This indicated that a large amount of IGF-1 in the lung tissue of ALI mice were removed by BAL.
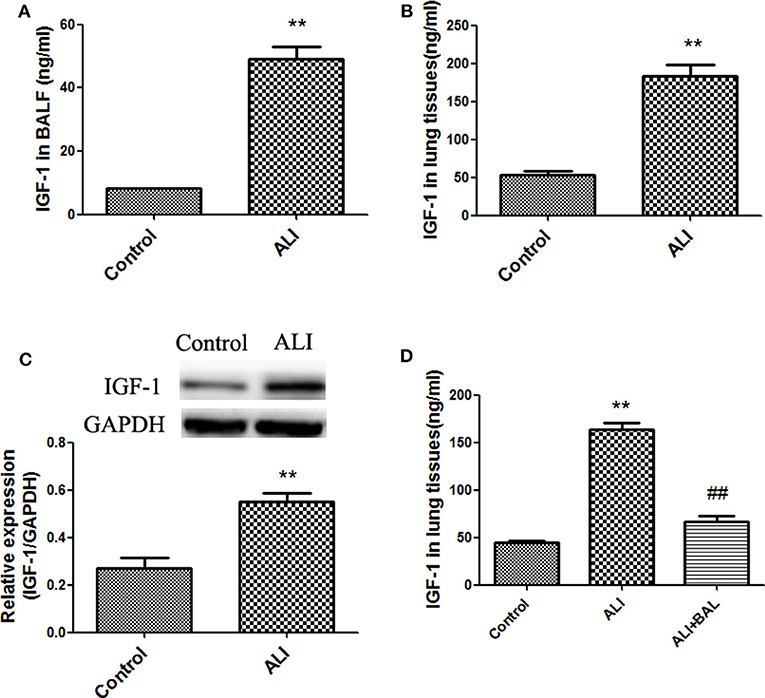
Figure 1. Increased IGF-1 production in acute LPS lung injury models. (A–D) BALB/c mice were instilled with LPS (4 mg/kg) into the nasal cavity to develop an ALI model. After 24 h of LPS instillation, IGF-1 content was detected in BALF (A) and lung tissue homogenates (B), and the expression of the IGF-1 protein in lung tissues (C) was detected by immunoblotting. **P < 0.01 vs. control group. (D) After ALI model mice were subjected to BAL (ALI + BAL), lung tissues were collected for homogenization, and the IGF-1 content in the homogenate was detected by ELISA. **P < 0.01 vs. the control group, ##P < 0.01 vs. the ALI group.
IGF-1 Is Derived From AMs in the ALI Model
IGF-1 is a polypeptide with a molecular weight of 7.6 KDa. Despite reports of IGF-1 produced after stimulation of epithelial cells and endothelial cells, studies show that the increased IGF-1 in the lungs of patients with allergic pneumonia is mainly derived from AMs (15). We therefore examined whether the elevated IGF-1 in the ALI model was derived from AMs. AMs were removed by 2-CA airway instillation, which showed that >77% of AMs were removed from ALI mouse airways (Figure 2A). After administration of 2-CA into the airway for 12 h, LPS was administered to generate the ALI model. The IGF-1 content in the BALF was significantly lower in 2-CA + ALI mice than in the LPS alone group (Figure 2B). In addition, the expression and secretion of IGF-1 in primary AMs of ALI model mice were significantly higher than those of normal control mice (Figures 2C,D). These results indicate that IGF-1 in the airway is mainly produced by AMs in mice exposed to ALI.
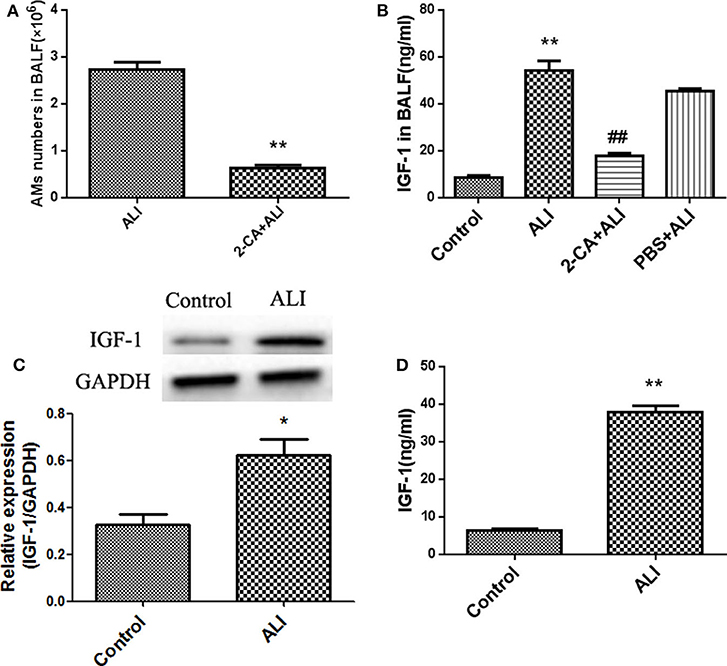
Figure 2. IGF-1 is derived from AMs in the ALI model. (A–D) ALI model mice were generated as described in Materials and Methods (Antibodies and Reagents). The 2-CA (10 μg 2-CA in 20 μL PBS) was dripped into the nasal cavity for 12 h before the generation of the ALI model, and AMs were removed from ALI model mice (2-CA + ALI). Mice in the PBS + ALI group were ALI model mice treated with PBS instead of 2-CA. (A) After 24 h of LPS instillation, primary AMs were obtained and counted as described in Materials and Methods (BAL and Acquisition of BALF). **P < 0.01 vs. the ALI group. (B) BALF was obtained as described in Materials and Methods (Establishment of the Acute Lung Injury Model), and the IGF-1 content was detected by ELISA. **P < 0.01 vs. the control group, ##P < 0.01 vs. the ALI group. (C,D) The expressions of AM band IGF-1 in normal control and ALI model mice were detected by immunoblotting (C) and ELISA (D), respectively. *P < 0.05, **P < 0.01 vs. the control group.
IGF-1 Production Is Enhanced in AMs by LPS-Stimulated AEC-CM
LPS induced IGF-1 production in vivo, and airway IGF-1 was mainly produced by AMs in ALI. This suggested that IGF-1 was produced by LPS-stimulated AMs or alternatively, a factor derived from LPS-stimulated AECs induces AMs to produce IGF-1. To test these possibilities, we examined the indirect effects of LPS-activated AEC-derived secretion factors and the direct effects of LPS on IGF-1 secretion by AMs. Mouse-derived MLE-12 AECs were treated with different doses of LPS for 4 h, and the AEC-CM produced was added to mouse primary AMs for 2 h. IGF-1 production was measured by western blotting and ELISA (Figure 3A). LPS increased the ability of AMs to secrete and express IGF-1 in the AEC-CM in a dose-dependent manner, and the greatest effect was observed at a dose of 10 ng/mL LPS. Direct treatment of primary AMs with LPS for 12 h also increased the production of IGF-1, although the increase did not reach statistical significance (Figure 3B). These results indicated that the production of IGF-1 by AMs was induced by a factor secreted by LPS-stimulated AECs. Therefore, in subsequent experiments, we focused on identifying the AEC-derived factor responsible for this effect.
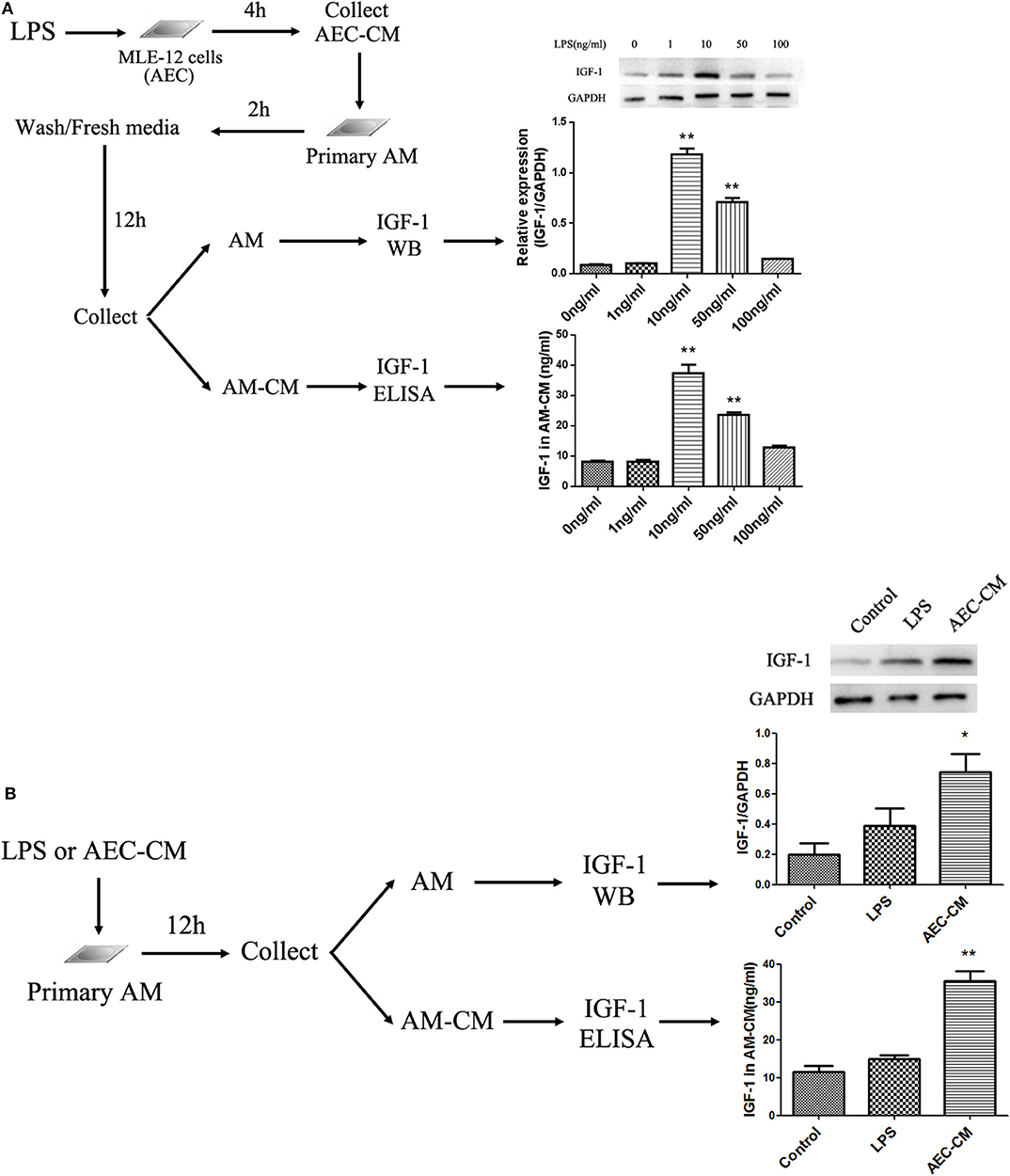
Figure 3. IGF-1 production is enhanced in AMs by LPS-stimulated AEC-CM. (A) AECs (1 ×106 cells/well) were inoculated into a 6-well plate and allowed to adhere before treatment with different doses of LPS (1, 10, 50, and 100 ng/mL) for 4 h and collection of AEC-CM. Primary AMs were extracted as described in Materials and Methods and cultured (1 ×106 cells/well). After the cells adhered to the wall, AEC-CM was added for 2 h. After several washes, AMs were resuspended in RPMI-1640 complete medium and cultured for an additional 12 h. AM-CM was collected and the IGF-1 content was detected by ELISA. Immunoblotting was used to detect IGF-1 protein expression in AMs. **P < 0.01 vs. the 0 ng/mL group. (B) Primary AMs (1 ×106 cells/well) were inoculated into a 6-well plate and stimulated with LPS (10 ng/mL) or AEC-CM (10 ng/mL, LPS stimulation group) for 12 h. The expression of AM IGF-1 was detected by immunoblotting, and the content of IGF-1 in AM-CM was detected by ELISA. *P < 0.05, **P < 0.01 vs. the control group.
Identification of Factors Produced by LPS-Treated AECs That Induce IGF-1 Secretion by AMs
AECs secrete a variety of mediators to regulate AM activity. Among them, MCP-1, IL-6, TNF-α, IL-1β, GM-CSF, and TGF-β are induced by LPS (19–22). Therefore, the levels of these factors in AECs stimulated with LPS (10 ng/mL) for 2, 8, and 24 h were measured by ELISA to determine the effect of LPS in our experimental system. As shown in Figure 4A, LPS strongly upregulated TNF-α and moderately upregulated MCP-1, IL-1β, and TGF-β, whereas IL-6 and GM-CSF did not increase significantly. To determine whether these factors could induce IGF-1 secretion from AMs, primary AMs were directly exposed to each mediator for 1 h in vitro, and the IGF-1 content in the AM-CM produced was analyzed by ELISA. As shown in Figure 4B, TGF-β was the only factor that significantly increased IGF-1 levels in AM-CM. In addition, TGF-β stimulation upregulated the expression of IGF-1 in AMs (Figure 4C). These results suggested that TGF-β was a likely AEC-derived candidate factor inducing IGF-1 secretion by AMs in the context of LPS stimulation.
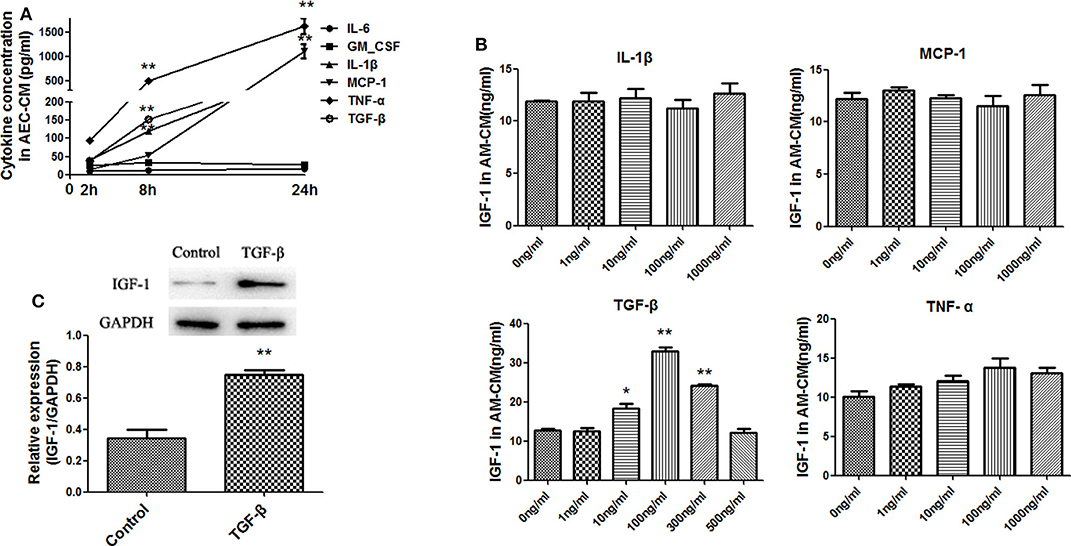
Figure 4. Identification of factors produced by LPS-treated AECs that induce IGF-1 secretion by AMs. (A) AECs (2 ×105 cells/well) were inoculated into a 24-well plate and stimulated with LPS (10 ng/mL) for 2, 8, and 24 h. The supernatants were analyzed by ELISA for IL-6, GM-CSF, IL-1β, MCP- 1, TNF-α, and TGF-β content. **P < 0.01 vs. the 2 h group. (B) Primary AMs (1 ×106 cells/well) were inoculated into 6-well plates and stimulated h with different concentrations (1, 10, 100, and 1,000 ng/mL) of IL-1β, TNF-α, MCP-1, and TGF-β for 24 h. The IGF-1 content in AM-CM was measured by ELISA. *P < 0.05 and **P < 0.01 vs. the 0 ng/mL group. (C) Primary AMs (1 ×106 cells/well) were inoculated into 6-well plates and stimulated with TGF-β (100 ng/mL) for 24 h. The expression level of IGF-1 in AMs was detected by western blotting. **P < 0.01 vs. the control group.
TGF-β Is Necessary for IGF-1 Production in AMs Induced by LPS-Stimulated AEC-CM
Antibody neutralization experiments were used to assess the effect of AEC-derived TGF-β on AMs. Addition of a neutralizing antibody against TGF-β to AEC-CM blocked the ability of AEC-CM to induce the expression and secretion of IGF-1 by AMs, whereas a neutralizing antibody against MCP-1 had no effect (Figure 5A). To confirm these results, AECs were treated with a TGF-β synthesis inhibitor before LPS stimulation to block TGF-β production from AECs. This treatment partially blocked the ability of AEC-CM to induce IGF-1 secretion from AMs (Figures 5B,C). Taken together, these results suggested that TGF-β was an important mediator originated from LPS-stimulated AECs that induces IGF-1 secretion from AMs.
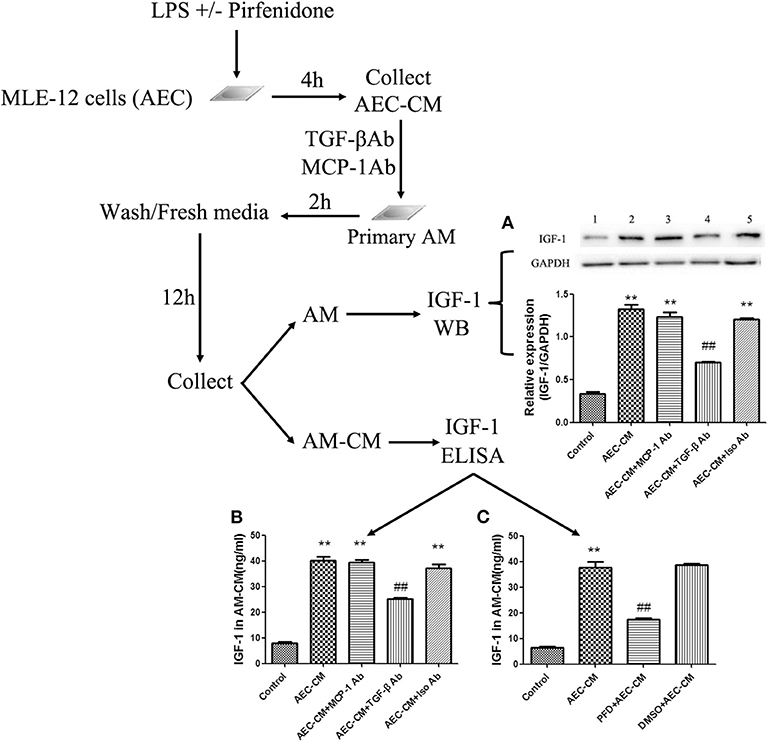
Figure 5. TGF-β is necessary for IGF-1 production in AMs induced by LPS-stimulated AEC-CM. (A–C) AECs (1 ×106 cells/well) were inoculated into a 6-well plate, stimulated with LPS (10 ng/mL) for 4 h, and AEC-CM was collected. AEC-CM was incubated for 30 min at 4°C with anti-MCP-1 antibody (MCP-Ab) or anti-TGF-β antibody (TGF-β Ab) or isotype control antibody (Iso Ab). Primary AMs were stimulated with the treated AEC-CM, washed, and resuspended in RPMI-1640 complete medium for 12 h. The content of IGF-1 in AM-CM was detected by ELISA (A), and the expression of IGF-1 in AM was detected by western blotting (B). In addition, AECs were treated with pirfenidone (50 ng/μL) for 1 h before stimulation with LPS (10 ng/mL), and the AEC-CM was collected and added to AMs for 2 h. After washing and resuspending AMs in culture medium for 12 h, the IGF-1 content in AM-CM was detected by ELISA (C). **P < 0.01 vs. the control group, ##P < 0.01 vs. the AEC-CM group.
IGF-1 Elevation in the ALI Model Is TGF-β Dependent
Instillation of LPS into the airways of mice upregulated TGF-β in BALF (Figure 6A). To determine whether TGF-β was involved in the LPS-induced increase in IGF-1 production in vivo, mice were treated with the TGF-β synthesis inhibitor pirfenidone 24 h before LPS stimulation. Pirfenidone partially blocked the LPS-induced increase in IGF-1 in the lungs (Figure 6A). In addition, treatment with anti-TGF-β antibody by intratracheal instillation before LPS stimulation significantly reduced IGF-1 content in BALF in ALI model mice (Figure 6B). These results indicated that TGF-β acted as a mediator that promotes the secretion of IGF-1 by AMs in response to LPS stimulation.
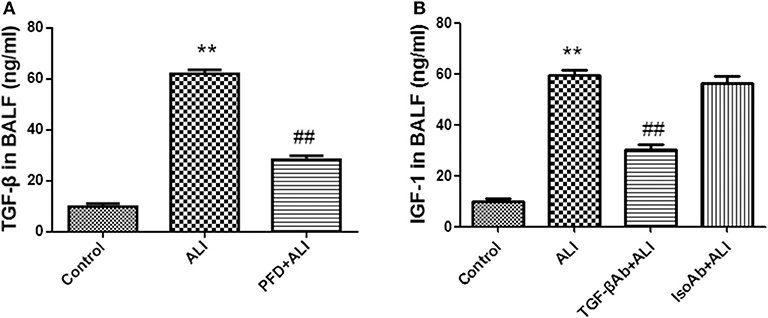
Figure 6. IGF-1 elevation in the ALI model is TGF-β dependent. (A,B) ALI model mice were generated as described in Materials and Methods (Antibodies and Reagents). At 24 h before LPS instillation, pirfenidone (400 μg/kg) (A) or TGF-β antibody (400 μg/kg) (B) was instilled into the nasal cavity once every 12 h. At 24 h after LPS instillation, the content of TGF-β in the BALF of each group of mice was measured by ELISA. **P < 0.01 vs. the control group, ##P < 0.01 vs. the ALI group.
TGF-β Induces IGF-1 Production in AMs via the PI3K/Akt Signaling Pathway
Not only does TGF-β activate Smad signaling, but also it can activate the PI3K/Akt signaling pathway (23). We showed that TGF-β activated Akt in AMs in a dose-dependent manner (Figure 7A), which suggested TGF-β activated the PI3K/Akt signaling pathway in AMs. Blocking the PI3K signaling pathway with Wortmannin partially suppressed the expression and secretion of IGF-1 in AMs induced by TGF-β (Figures 7B,C). These results indicated that TGF-β induced the production of IGF-1 in AMs through the PI3K/Akt signaling pathway.
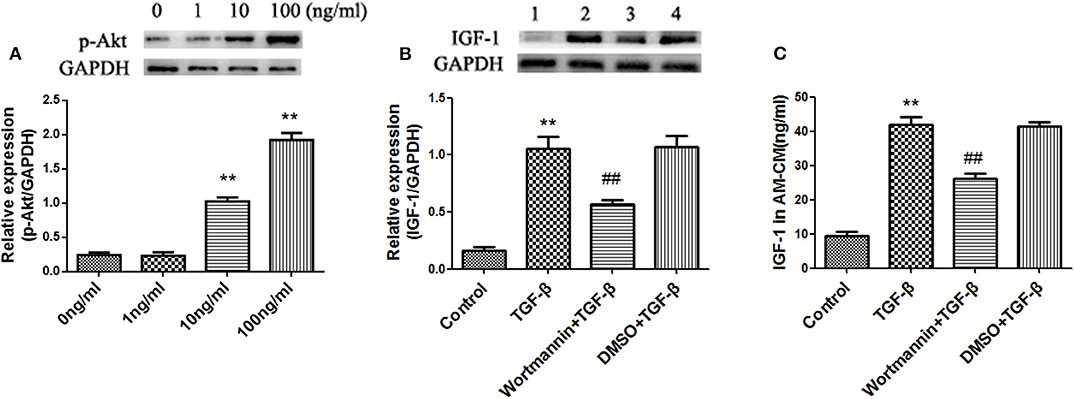
Figure 7. TGF-β induces IGF-1 production in AMs via the PI3K/Akt signaling pathway. (A) Primary AMs (1 ×106 cells/well) were inoculated into 6-well plates and stimulated with different concentration of TGF-β (0, 1, 10, and 100 ng/mL) for 24 h. The expression of p-Akt in AMs was detected by western blotting. **P < 0.01 vs. the 0 ng/ml group. (B,C) Primary AMs (1 ×106 cells/well) were inoculated into a 6-well plate and stimulated with Wortmannin (1 mM) for 2 h, followed by TGF-β (100 ng/mL) treatment for 24 h. IGF-1 protein expression in AMs was detected by immunoblotting, and the IGF-1 content in AM-CM was detected by ELISA. **P < 0.01 vs. the control group, ##P < 0.01 vs. the TGF-β group.
IGF-1 Prevents LPS-Induced p38 MAPK Activation and MCP-1, TNF-α, and IL-1β Production in AECs
We showed that LPS-treated AECs induced IGF-1 secretion by AMs through the release of TGF-β. Therefore, we investigated the functional effects of IGF-1 on AEC inflammation-related signaling in AECs. For this purpose, AECs were pretreated with IGF-1 for 2 h and then stimulated with LPS before analysis of p38 MAPK activation and expression of inflammatory factors. Compared with the LPS single stimulation group, pretreatment with IGF-1 significantly prevented LPS-induced p38 MAPK activation (Figure 8A) and MCP-1, TNF-α, and IL-1β expression in AECs (Figure 8B). Similarly, before stimulation of AECs with LPS, pretreatment with IGF-1 for 1 h significantly reduced LPS-induced secretion of MCP-1, TNF-α, and IL-1β in AECs (Figure 8C).
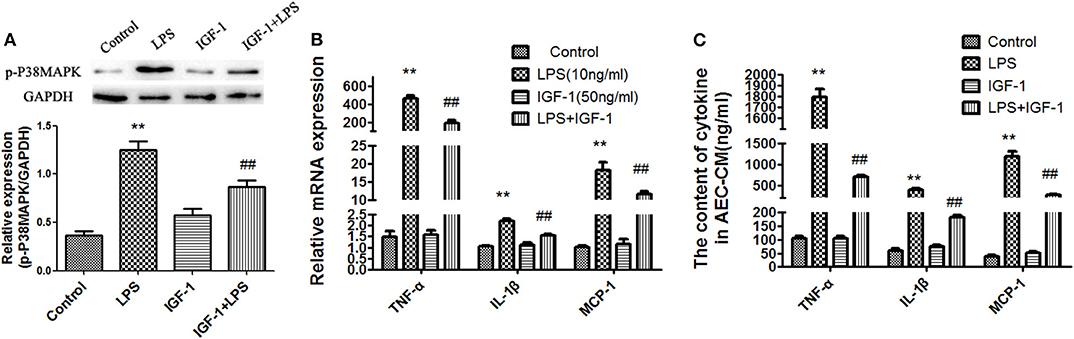
Figure 8. IGF-1 prevents LPS-induced p38 MAPK activation and MCP-1, TNF-α, and IL-1β production in AECs. (A–C) AECs (1 ×106 cells/well) were inoculated into a 6-well plate and pre-stimulated with IGF-1 (50 ng/mL) for 1 h, followed by LPS (10 ng/mL) for a total of 24 h. (A) The expression of p-p38 MAPK in AECs was detected by immunoblotting. (B) The mRNA expression levels of the cytokines TNF-α, IL-1β, and MCP-1 in AECs was detected by qRT-PCR. (C) The cytokines TNF-α, IL-1β, and MCP-1 in AEC-CM were detected by ELISA. **P < 0.01 vs. the control group, ##P < 0.01 vs. the LPS group.
IGF-1 Promotes Apoptotic Cell Phagocytosis by AECs Through PPARγ
Phagocytosis is the process by which phagocytes take up particles larger than 0.5 μm in diameter. Phagocytosis of apoptotic cells plays an important role in the resolution of inflammation and the maintenance of tissue balance (24). We therefore examined the effect of IGF-1 on AEC phagocytosis for apoptotic cells. As shown in Figures 9A,B, IGF-1 promoted phagocytosis of apoptotic cells by AECs in a dose-dependent manner, and 50 ng/ml IGF-1 had the strongest effect. Because PPARγ and LXR are involved in phagocytosis, we investigated the role of these transcription factors in IGF-1-induced phagocytosis in AECs. IGF-1 had no effect on LXR expression in AECs (Figure 9C); therefore, we focused on PPARγ in subsequent experiments. IGF-1 upregulated PPARγ in AECs in a time- and dose-dependent manner (Figures 9D,E), indicating that IGF-1 signaling induced the expression of PPARγ in AECs. PPARγ gene interference inhibited the effect of IGF-1 on the phagocytosis of apoptotic cells by AECs (Figure 9F). This indicated that IGF-1 promoted the phagocytosis of apoptotic cells by AECs by inducing the expression of PPARγ.
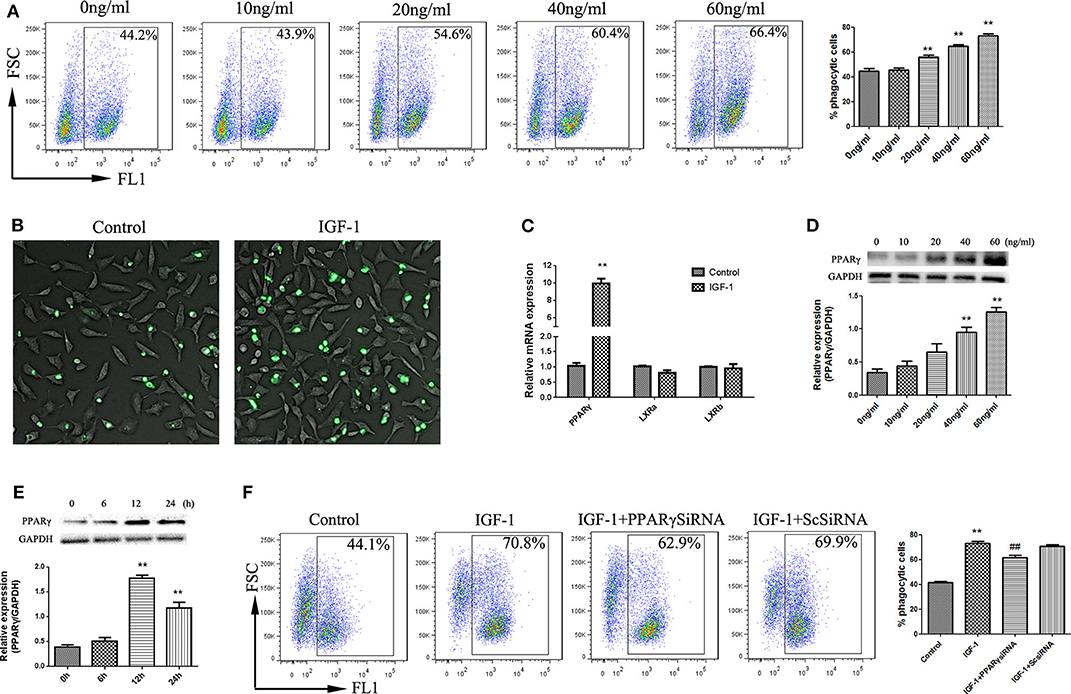
Figure 9. IGF-1 promotes apoptotic cell phagocytosis by AECs through PPARγ. (A,B) AECs (1 ×105 cells/well) were inoculated into a 6-well plate, stimulated with different concentrations of IGF-1 (0, 10, 20, 40, and 60 ng/mL) for 12 h, and co-cultured with FITC-labeled apoptotic cells for a total of 4 h. Phagocytosis of apoptotic cells was detected by flow cytometry (A) and fluorescence microscopy (B). **P < 0.01 vs. the 0 ng/mL group. (C) AECs (1 ×106 cells/well) were inoculated into a 6-well plate, stimulated with IGF-1 (60 ng/mL) for 24 h, and qRT-PCR was used to detect PPARγ, LxRA, and LxRB mRNA expression in AECs. **P < 0.01 vs. the control group. (D) AECs (1 ×105 cells/well) were inoculated into a 6-well plate and treated with IGF-1 at different concentrations (0, 10, 20, 40, and 60 ng/mL) for 24 h. The protein expression of PPARγ was detected by western blotting. **P < 0.01 vs. the 0 ng/ml group. (E) AECs were stimulated with IGF-1 (60 ng/mL) for 6, 12, and 24 h, and the protein expression of PPARγ in AECs was detected by western blotting. **P < 0.01 vs. the 0 h group. (F) AECs (1 ×105 cells/well) were inoculated into a 6-well plate, transfected with PPARγ siRNA or scrambled (Sc) siRNA for 36 h, and stimulated with IGF-1 (60 ng/mL) for 12 h. FITC-labeled apoptotic cells were added to the culture for 4 h. AEC phagocytosis of apoptotic cells was detected by flow cytometry. **P < 0.01 vs. the control group, ##P < 0.01 vs. the IGF-1 group.
IGF-1 Reduces Airway Inflammation in ALI Mice
Elimination of apoptotic cells contributes to the resolution of inflammation. In vitro experiments demonstrated that IGF-1 promotes apoptotic cell phagocytosis by AECs and inhibits LPS-induced inflammatory signals. Therefore, we next investigated the potential effect of IGF-1 on LPS-induced airway inflammation in vivo. At 24 h after LPS challenge, mice showed significantly increased polymorphonuclear neutrophils (PMNs) in BALF, marked alveolar congestion and exudation, and increased inflammatory cell infiltration compared with that in normal control mice. Mice pretreated with IGF-1 directly into the airway 24 h before the establishment of the ALI model showed significantly reduced infiltration of inflammatory cells and a significantly lower number of PMNs and protein content in BALF than LPS treated mice (Figure 10). These data support the view that IGF-1 secretion by AMs is enhanced in response to AEC-derived TGF-β, which can serve as an important functional brake for the inflammatory response in alveoli during infection.
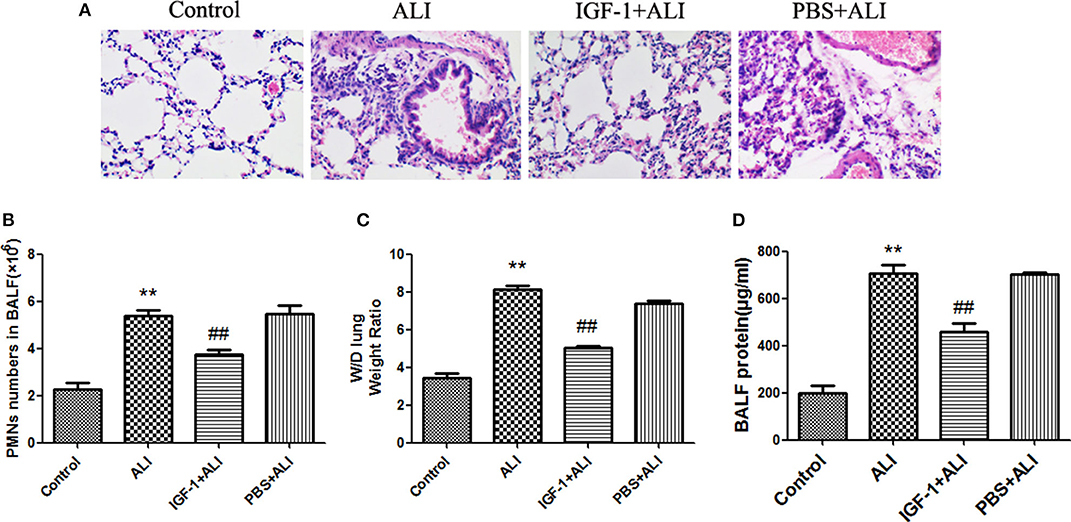
Figure 10. IGF-1 reduces airway inflammation in ALI mice. (A–D) ALI model mice were prepared as described in Materials and Methods (Antibodies and Reagents). At 24 h before LPS nasal drops, BALB/c mice received nasal drops of IGF-1 (0.8 mg/kg) once every 12 h. At 24 h after LPS treatment, lung tissues and BALF were collected from mice. (A) The lung tissues of each group were stained with HE, and the pathological damage was observed under a microscope. (B) The BALF of each group of mice was centrifuged, cell pellets were smeared and stained with Wright's staining solution, and the number of neutrophils was counted. (C) Wet-dry weight ratios of lung tissues in each group of mice were calculated as described in Materials and Methods (Western Blot Analysis). (D) The total protein content of BALF in each group of mice was measured according to Materials and Methods (Wet-Dry Weight Ratio of Lung Tissue). **P < 0.01 vs. the control group, ##P < 0.01 vs. the ALI group.
Discussion
In this study, we showed that TGF-β produced by LPS-induced AECs stimulated the production of IGF-1 by AMs, and IGF-1 prevented the generation of pre-inflammatory signals in AECs and promoted the phagocytosis of apoptotic cells by AECs. IGF-1 is a polypeptide growth factor that plays a key role in regulating cell proliferation, differentiation, metabolism, and survival. It also demonstrated pleiotropy in some organs and was involved in the development of different disease states (25). For example, IGF-1 is closely related to the pathological processes of obesity, cardiovascular disease, central nervous system disease, tumor development, and other diseases. In addition, current research indicates that IGF-1 is associated with lung disease. IGF-1 is upregulated in patients with idiopathic and secondary pulmonary fibrosis (26). The level of IGF-1 mRNA is significantly increased in asthmatic patients (27). The serum IGF-1 level is decreased in chronic obstructive pulmonary disease patients in association with the severity of the disease (28). However, serum IGF-1 levels are elevated in ALI/ARDS patients (29). In the present study, we showed that IGF-1 levels were increased in the lung tissues of ALI model mice, and the IGF-1 in the airways of ALI model mice was derived from AMs. IGF-1 is produced in a variety of cells, including dental pulp stem cells, nucleus pulposus cells, macrophages, endothelial cells, and epithelial cells (17). Wang et al. recently reported that epidermal T cells can also produce IGF-1 (30). Han et al. showed that IGF-1 in Th2 type cytokine-stimulated mice is mainly derived from AMs, which is consistent with the present findings (31).
Stimulation of gastric vascular endothelial cells with nerve growth factor leads to the production of IGF-1 to prevent cells from damage caused by indomethacin (32). IL-15 promotes the repair of diabetic skin wounds by increasing the production of IGF-1 by epidermal T cells (30). In this study, we demonstrated that AEC-derived TGF-β induced IGF-1 production by AMs. TGF-β is a key factor for immune balance and tolerance that functions by preventing the expansion and activity of certain components of the immune system. It not only suppresses the function of adaptive immune cells such as T-cell DC cells, but also regulates the behavior of innate immune cells such as natural killer cells, macrophages, and neutrophils, which form a negative regulatory network in the immune system (33, 34).
TGF-β1 can both initiate the classic Smad signaling pathway and induce non-Smad signaling pathways such as the MAPK, Rho, and PI3K pathways (35, 36). In this study, we showed that TGF-β1 induced IGF-1 production in lung epithelial cells through the PI3K/Akt pathway. Wygrecka et al. demonstrated that TGF-β1 induces IGF-1 production in human lung fibroblasts through the PI3K/Akt pathway (37). The PI3K/Akt pathway also mediates the expression of type I collagen induced by TGF-β1 in retinal pigment epithelial cells and human mesangial cells (38). In vascular smooth muscle cells, TGF-β activates the PI3K/Akt pathway through Smad3 signaling, and p38 MAPK is the most important bridge connecting Smad3 and Akt (39). Both, the PI3K and Smad3 signaling pathways are involved in TGF-β1-induced cardiac fibroblast type I and type III collagen expression, and PI3K blockade reduces the phosphorylation of Smad3 initiated by TGF-β1. These findings indicate that the PI3K and Smad3 pathways may be integrated in the TGF-β1 signal (40).
Common signaling pathways between LPS and IGF-1 signals, such as PI3K/Akt and MAPK, may be involved in crosstalk between immune signals and endocrine signals. Exposure to IGF-1 prevents LPS-induced sickness behavior and the expression of pro-inflammatory factors in mice and rats (41, 42). IGF-1 prevents LPS-induced NOS2 expression in INS-1 cells by activating the PI3K/Akt signaling pathway (43). In buffalo granulocytes, IGF-1 downregulates LPS-induced expression of IL-1β, IL-6, and TNF-α, and delays LPS-induced phosphorylation of ERK1/2 (44). In this study, IGF-1 inhibited the increase of IL-1β, TNF-α, MCP-1 induced by LPS and the activation of p38 MAPK in AECs, whereas it had no effect on IL-6 production in AECs.
IGF-1 plays an important role in cell growth, migration, and differentiation, in addition to regulating the function of phagocytic cells. Exposure of mouse macrophages to IGF-1 significantly increases their phagocytic activity (45), whereas it does not affect the phagocytic function of rat macrophages (46). Jakn et al. showed that IGF-1 binds to IGF-1R on airway epithelial cells of non-professional phagocytic cells, which can promote the phagocytosis of microparticles by airway epithelial cells (31). In previous studies from our group, we showed that IGF-1 promotes the phagocytosis of fluorescent microspheres by alveolar epithelial cells (17). In this study, we demonstrated that IGF-1 promoted the phagocytosis of apoptotic cells by the alveolar epithelium. The phagocytosis of apoptotic cells by AECs is an important mechanism for suppressing airway inflammation. Our findings suggested that IGF-1 promoted the resolution of airway inflammation and accelerated the repair of inflammatory injury.
In this study, we showed that IGF-1 promoted the phagocytosis of apoptotic cells by lung epithelial cells by inducing the expression of PPARγ in lung epithelial cells. PPARγ and LXR are important transcription factors that regulate the expression of phagocytic receptors and bridge molecules to mediate phagocytosis (47). In this study, IGF-1 had no effect on the expression of LXR in AECs; therefore, related experiments focused on PPARγ.
Lee et al. showed that IGF-1 participates in the adipogenesis of mesenchymal stem cells by regulating the expression of PPARγ (48). In addition, IGF-1 induces the proliferation and invasion of UM cells by inhibiting Foxo3a (49). In hippocampal neural stem cells, IGF-1 upregulation activates the PI3K/Akt pathway, which upregulates the Brn4 transcription factor, thereby causing the differentiation of stem cells into neuronal phenotypes (50). NF-AT, CREB and other transcription factors are also activated by IGF-1 (51). These studies indicated that IGF-1 can activate multiple transcription factors to mediate its biological activity in vivo.
In summary, the results of this study indicated that TGF-β1 derived from AECs activated AMs to secrete IGF-1 into the alveolar fluid in response to stimulation of the airway by inflammatory signals. This AM-derived IGF-1 attenuated the p38 MAPK inflammatory signal in AECs and promoted the phagocytosis of apoptotic cells by AECs. This two-way communication between AECs and AMs represents a well-tuned system for the regulation of the inflammatory response in alveoli (Figure 11).
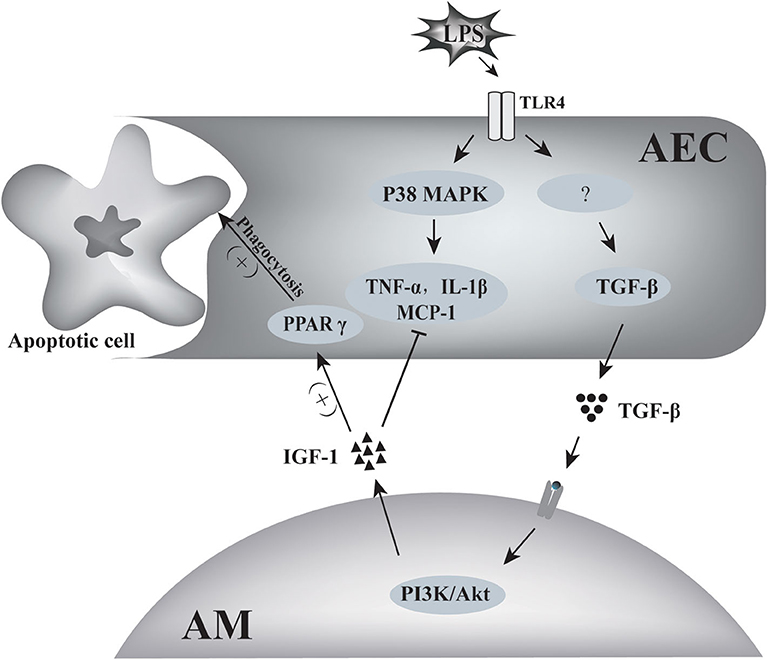
Figure 11. Schematic of the two-way communication between AMs and AECs in LPS-induced airway inflammation. LPS induced the production of inflammatory factors such as TNF-α, MCP-1, IL-1β by AECs through p38 MAPK signaling. AECs also produced TGF-β in response to LPS stimulation. TGF-β induced IGF-1 production by AMs through the PI3K/Akt signaling pathway. IGF-1 prevented LPS-induced p38 MAPK activation and MCP-1, TNF-α, and IL-1 β production in AECs, and it also promoted AEC phagocytosis of apoptotic cells through PPARγ.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics Statement
The animal study was reviewed and approved by the Ethics Committee of Bengbu Medical College, Anhui, China.
Author Contributions
MM and PG conceived and designed the experiments and performed the experiments. QY, JH, FW, XH, and SG conceived and designed the experiments, analyzed the data, and contributed reagents, materials, analysis tools. ZQ and CS analyzed the data, contributed reagents, materials, analysis tools, prepared figures and/or tables, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Science Foundation of China (Nos. 81273273 and 81801573), Anhui Provincial Natural Science Foundation (Nos. 1708085MH218, 1808085QH253, and 2008085QH410), Scientific Research Innovation Team Project of Bengbu Medical College (No. BYKC201902), Natural Science Research Program of Bengbu Medical College: (Nos. BYKY1716ZD, BYKY1804ZD, and BYKY2019013ZD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Holt PG, Strickland DH, Wikström ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol. (2008) 8:142–52. doi: 10.1038/nri2236
2. Zasłona Z, Przybranowski S, Wilke C, van Rooijen N, Teitz-Tennenbaum S, Osterholzer JJ, et al. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol. (2014) 193:4245–53. doi: 10.4049/jimmunol.1400580
3. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. (2014) 14:81–93. doi: 10.1038/nri3600
4. Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. (2015) 16:27–35. doi: 10.1038/ni.3045
5. Fehrenbach H. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir Res. (2001) 2:33–46. doi: 10.1186/rr36
6. Guillot L, Nathan N, Tabary O, Thouvenin G, Le Rouzic P, Corvol H, et al. Alveolar epithelial cells: master regulators of lung homeostasis. Int J Biochem Cell Biol. (2013) 45:2568–73. doi: 10.1016/j.biocel.2013.08.009
7. Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature. (2014) 506:503–6. doi: 10.1038/nature12902
8. O'Brien AD, Standiford TJ, Christensen PJ, Wilcoxen SE, Paine R III. Chemotaxis of alveolar macrophages in response to signals derived from alveolar epithelial cells. J Lab Clin Med. (1998) 131:417–24. doi: 10.1016/S0022-2143(98)90142-1
9. Cardani A, Boulton A, Kim TS, Braciale TJ. Alveolar macrophages prevent lethal influenza pneumonia by inhibiting infection of type-1 alveolar epithelial cells. PLoS Pathog. (2017) 13:e1006140. doi: 10.1371/journal.ppat.1006140
10. Bourdonnay E, Zasłona Z, Penke LRK, Speth JM, Schneider DJ, Przybranowski S, et al. Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. J Exp Med. (2015) 212:729–42. doi: 10.1084/jem.20141675
11. Hung CF, Rohani MG, Lee S-S, Chen P, Schnapp LM. Role of IGF-1 pathway in lung fibroblast activation. Respir Res. (2013) 14:102–102. doi: 10.1186/1465-9921-14-102
12. Löfqvist C, Hellgren G, Niklasson A, Engström E, Ley D, Hansen-Pupp I, et al. Low postnatal serum IGF-I levels are associated with bronchopulmonary dysplasia (BPD). Acta Paediatr. (2012) 101:1211–6. doi: 10.1111/j.1651-2227.2012.02826.x
13. Chetty A, Nielsen HC. Regulation of cell proliferation by insulin-like growth factor 1 in hyperoxia-exposed neonatal rat lung. Mol Genet Metab. (2002) 75:265–75. doi: 10.1006/mgme.2002.3295
14. Bessich JL, Nymon AB, Moulton LA, Dorman D, Ashare A. Low levels of insulin-like growth factor-1 contribute to alveolar macrophage dysfunction in cystic fibrosis. J Immunol. (2013) 191:378–85. doi: 10.4049/jimmunol.1300221
15. He J, Mu M, Wang H, Ma H, Tang X, Fang Q, et al. Upregulated IGF-1 in the lungs of asthmatic mice originates from alveolar macrophages. Mol Med Rep. (2019) 19:1266–71. doi: 10.3892/mmr.2018.9726
16. Narasaraju TA, Chen H, Weng T, Bhaskaran M, Jin N, Chen J, et al. Expression profile of IGF system during lung injury and recovery in rats exposed to hyperoxia: a possible role of IGF-1 in alveolar epithelial cell proliferation and differentiation. J Cell Biochem. (2006) 97:984–98. doi: 10.1002/jcb.20653
17. Wang H, He J, Luo Y, Mu M, Guo S, Shen L, et al. IGF-1 Promotes endocytosis of alveolar epithelial cells through PI3K signaling. Ann Clin Lab Sci. (2019) 49:3–8.
18. Arroba AI, Campos-Caro A, Aguilar-Diosdado M, Valverde ÁM. IGF-1, inflammation and retinal degeneration: a close network. Front Aging Neurosci. (2018) 10:203. doi: 10.3389/fnagi.2018.00203
19. Andrzejczak D, Górska D, Czarnecka E. Influence of enalapril, quinapril and losartan on lipopolysaccharide (LPS)-induced serum concentrations of TNF-alpha, IL-1 beta, IL-6 in spontaneously hypertensive rats (SHR). Pharmacol Rep. (2007) 59:437–46.
20. Li Y, Lu M, Alvarez-Lugo L, Chen G, Chai TC. Granulocyte-macrophage colony-stimulating factor (GM-CSF) is released by female mouse bladder urothelial cells and expressed by the urothelium as an early response to lipopolysaccharides (LPS). Neurourol Urodyn. (2017) 36:1020–5. doi: 10.1002/nau.23057
21. Aidoukovitch A, Anders E, Dahl S, Nebel D, Svensson D, Nilsson B-O. The host defense peptide LL-37 is internalized by human periodontal ligament cells and prevents LPS-induced MCP-1 production. J Periodont Res. (2019) 54:662–70. doi: 10.1111/jre.12667
22. Vasanthkumar T, Hanumanthappa M, Lakshminarayana R. Curcumin and capsaicin modulates LPS induced expression of COX-2, IL-6 and TGF-β in human peripheral blood mononuclear cells. Cytotechnology. (2019) 71:963–76. doi: 10.1007/s10616-019-00338-x
23. Hamidi A, Song J, Thakur N, Itoh S, Marcusson A, Bergh A, et al. TGF-β promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85α. Sci Signal. (2017) 10:eaal4186. doi: 10.1126/scisignal.aal4186
24. Yoon KW. Dead cell phagocytosis and innate immune checkpoint. BMB Rep. (2017) 50:496–503. doi: 10.5483/BMBRep.2017.50.10.147
25. Troncoso R, Ibarra C, Vicencio JM, Jaimovich E, Lavandero S. New insights into IGF-1 signaling in the heart. Trends Endocrinol Metab. (2014) 25:128–37. doi: 10.1016/j.tem.2013.12.002
26. Krein PM, Winston BW. Roles for insulin-like growth factor I and transforming growth factor-beta in fibrotic lung disease. Chest. (2002) 122(Suppl. 6):289S−93S. doi: 10.1378/chest.122.6_suppl.289S
27. Hoshino M, Nakamura Y, Sim JJ, Yamashiro Y, Uchida K, Hosaka K, et al. Inhaled corticosteroid reduced lamina reticularis of the basement membrane by modulation of insulin-like growth factor (IGF)-I expression in bronchial asthma. Clin Exp Allergy. (1998) 28:568–77. doi: 10.1046/j.1365-2222.1998.00277.x
28. Ye M, Yu H, Yu W, Zhang G, Xiao L, Zheng X, et al. Evaluation of the significance of circulating insulin-like growth factor-1 and C-reactive protein in patients with chronic obstructive pulmonary disease. J Int Med Res. (2012) 40:1025–35. doi: 10.1177/147323001204000321
29. Krein PM, Sabatini PJB, Tinmouth W, Green FHY, Winston BW. Localization of insulin-like growth factor-I in lung tissues of patients with fibroproliferative acute respiratory distress syndrome. Am J Respir Crit Care Med. (2003) 167:83–90. doi: 10.1164/rccm.2201012
30. Wang Y, Bai Y, Li Y, Liang G, Jiang Y, Liu Z, et al. IL-15 enhances activation and IGF-1 production of dendritic epidermal T cells to promote wound healing in diabetic mice. Front Immunol. (2017) 8:1557. doi: 10.3389/fimmu.2017.01557
31. Han CZ, Juncadella IJ, Kinchen JM, Buckley MW, Klibanov AL, Dryden K, et al. Macrophages redirect phagocytosis by non-professional phagocytes and influence inflammation. Nature. (2016) 539:570–4. doi: 10.1038/nature20141
32. Ahluwalia A, Jones MK, Hoa N, Tarnawski AS. NGF protects endothelial cells from indomethacin-induced injury through activation of mitochondria and upregulation of IGF-1. Cell Signal. (2017) 40:22–9. doi: 10.1016/j.cellsig.2017.08.006
33. Stewart AG, Thomas B, Koff J. TGF-β: master regulator of inflammation and fibrosis. Respirology. (2018) 23:1096–7. doi: 10.1111/resp.13415
34. Chen J, Gingold JA, Su X. Immunomodulatory TGF-β signaling in hepatocellular carcinoma. Trends Mol Med. (2019) 25:1010–23. doi: 10.1016/j.molmed.2019.06.007
35. Zhang YE. Non-smad signaling pathways of the TGF-β family. Cold Spring Harb Perspect Biol. (2017) 9:a022129. doi: 10.1101/cshperspect.a022129
36. Hanna A, Frangogiannis NG. The role of the TGF-β superfamily in myocardial infarction. Front Cardiovasc Med. (2019) 6:140. doi: 10.3389/fcvm.2019.00140
37. Wygrecka M, Zakrzewicz D, Taborski B, Didiasova M, Kwapiszewska G, Preissner KT, et al. TGF-β1 induces tissue factor expression in human lung fibroblasts in a PI3K/JNK/Akt-dependent and AP-1-dependent manner. Am J Respir Cell Mol Biol. (2012) 47:614–27. doi: 10.1165/rcmb.2012-0097OC
38. Yokoyama K, Kimoto K, Itoh Y, Nakatsuka K, Matsuo N, Yoshioka H, et al. The PI3K/Akt pathway mediates the expression of type I collagen induced by TGF-β2 in human retinal pigment epithelial cells. Graefe's Arch Clin Exp Ophthalmol. (2012) 250:15–23. doi: 10.1007/s00417-011-1766-x
39. Suwanabol PA, Seedial SM, Zhang F, Shi X, Si Y, Liu B, et al. TGF-β and Smad3 modulate PI3K/Akt signaling pathway in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. (2012) 302:H2211–19. doi: 10.1152/ajpheart.00966.2011
40. Voloshenyuk TG, Landesman ES, Khoutorova E, Hart AD, Gardner JD. Induction of cardiac fibroblast lysyl oxidase by TGF-β1 requires PI3K/Akt, Smad3, and MAPK signaling. Cytokine. (2011) 55:90–7. doi: 10.1016/j.cyto.2011.03.024
41. Dantzer R, Gheusi G, Johnson RW, Kelley KW. Central administration of insulin-like growth factor-1 inhibits lipopolysaccharide-induced sickness behavior in mice. Neuroreport. (1999) 10:289–92. doi: 10.1097/00001756-199902050-00015
42. Pang Y, Campbell L, Zheng B, Fan L, Cai Z, Rhodes P. Lipopolysaccharide-activated microglia induce death of oligodendrocyte progenitor cells and impede their development. Neuroscience. (2010) 166:464–75. doi: 10.1016/j.neuroscience.2009.12.040
43. Castrillo A, Bodelón OG, Boscá L. Inhibitory effect of IGF-I on type 2 nitric oxide synthase expression in Ins-1 cells and protection against activation-dependent apoptosis: involvement of phosphatidylinositol 3-kinase. Diabetes. (2000) 49:209–17. doi: 10.2337/diabetes.49.2.209
44. Onnureddy K, Ravinder Onteru SK, Singh D. IGF-1 attenuates LPS induced pro-inflammatory cytokines expression in buffalo (Bubalus bubalis) granulosa cells. Mol Immunol. (2015) 64:136–43. doi: 10.1016/j.molimm.2014.11.008
45. Dos Santos Reis MD, Dos Santos YMO, de Menezes CA, Borbely KSC, Smaniotto S. Resident murine macrophage migration and phagocytosis are modulated by growth hormone. Cell Biol Int. (2018) 42:615–23. doi: 10.1002/cbin.10939
46. Xiao W, Chen P, Wang R, Dong J. Overload training inhibits phagocytosis and ROS generation of peritoneal macrophages: role of IGF-1 and MGF. Eur J Appl Physiol. (2013) 113:117–25. doi: 10.1007/s00421-012-2418-5
47. Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y, et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARγ and LXRα pathways. Circ Res. (2011) 108:985–95. doi: 10.1161/CIRCRESAHA.110.233775
48. Lee JH, Lee SH, Lee HS, Ji ST, Jung SY, Kim JH, et al. Lnk is an important modulator of insulin-like growth factor-1/Akt/peroxisome proliferator-activated receptor-gamma axis during adipogenesis of mesenchymal stem cells. Korean J Physiol Pharmacol. (2016) 20:459–66. doi: 10.4196/kjpp.2016.20.5.459
49. Yan F, Liao R, Farhan M, Wang T, Chen J, Wang Z, et al. Elucidating the role of the FoxO3a transcription factor in the IGF-1-induced migration and invasion of uveal melanoma cancer cells. Biomed Pharmacother. (2016) 84:1538–50. doi: 10.1016/j.biopha.2016.11.027
50. Zhang X, Zhang L, Cheng X, Guo Y, Sun X, Chen G, et al. IGF-1 promotes Brn-4 expression and neuronal differentiation of neural stem cells via the PI3K/Akt pathway. PLoS ONE. (2014) 9:e113801. doi: 10.1371/journal.pone.0113801
Keywords: AEC, AMs, IGF-1, TGF-β, inflammation
Citation: Mu M, Gao P, Yang Q, He J, Wu F, Han X, Guo S, Qian Z and Song C (2020) Alveolar Epithelial Cells Promote IGF-1 Production by Alveolar Macrophages Through TGF-β to Suppress Endogenous Inflammatory Signals. Front. Immunol. 11:1585. doi: 10.3389/fimmu.2020.01585
Received: 01 May 2020; Accepted: 15 June 2020;
Published: 21 July 2020.
Edited by:
Detlef Neumann, Hannover Medical School, GermanyReviewed by:
Nico Lachmann, Hannover Medical School, GermanyAnagha Malur, East Carolina University, United States
Copyright © 2020 Mu, Gao, Yang, He, Wu, Han, Guo, Qian and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chuanwang Song, Y2h1YW53YW5nc29uZ0Bob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work