 Nor Adzimah Johdi
Nor Adzimah Johdi Nur Fazilah Sukor
Nur Fazilah Sukor- UKM Medical Molecular Biology Institute (UMBI), National University of Malaysia, Bangi, Malaysia
Colorectal cancer is the third most common cancer in the world with increasing incidence and mortality rates globally. Standard treatments for colorectal cancer have always been surgery, chemotherapy and radiotherapy which may be used in combination to treat patients. However, these treatments have many side effects due to their non-specificity and cytotoxicity toward any cells including normal cells that are growing and dividing. Furthermore, many patients succumb to relapse even after a series of treatments. Thus, it is crucial to have more alternative and effective treatments to treat CRC patients. Immunotherapy is one of the new alternatives in cancer treatment. The strategy is to utilize patients’ own immune systems in combating the cancer cells. Cancer immunotherapy overcomes the issue of specificity which is the major problem in chemotherapy and radiotherapy. The normal cells with no cancer antigens are not affected. The outcomes of some cancer immunotherapy have been astonishing in some cases, but some which rely on the status of patients’ own immune systems are not. Those patients who responded well to cancer immunotherapy have a better prognostic and better quality of life.
Introduction
Colorectal cancer (CRC) is the third most common cancer in the world with an incidence of 10.2% and a mortality rate of 9.2% of all cancers (1, 2). Approximately 1.8 million new cases and mortality of 0.88 million were recorded until 2018. From 1975 to 2013, CRC incidence rate increased from 10–15 cases per 100,000 population of Americans between the age of 20–49 years old (3). In developing countries such as Argentina, Brazil, and China, the incidence and mortality rate due to CRC increased around 20% (4). Furthermore, the prognosis for patients with metastatic CRC remains poor with a median 5-year survival of only 18.5% in the United States and 27.7% in Europe (1, 2).
Standard conventional treatments for CRC are surgery, chemotherapy and radiotherapy. Depending on the localization and progression of the disease, these treatments can be used in combination (5–8). Total mesorectal excision (TME) through laparoscopic and transanal surgery approaches are often the options for localized cancer and whenever the tumor location is easy to access (9–11). However, complete removal of all cancer cells is often not possible. About 66 and 61% of stage II and III colon and rectal patients underwent further treatments with adjuvant chemotherapy and/or radiotherapy, respectively (12). These treatments have many side effects due to their unspecificity and cytotoxicity toward any cells that are growing and dividing (13, 14). Furthermore, 54% of patients relapse even after neoadjuvant treatment (15). Thus, it is crucial to have more alternative and effective treatments to treat CRC patients.
Cancer immunotherapy is one of the new alternatives in cancer treatment. In comparison to the standard treatment, this treatment manipulates and utilizes patients’ own immune system in combating the cancer cells. The innate and adaptive immune responses are alerted to recognize the cancer cells and potentially eradicate the tumor (16–18). Many successes have been reported especially in hematological malignancies and solid tumors (19, 20). Cancer immunotherapy overcomes the issue of specificity which is the major problem in chemotherapy and radiotherapy. Cancer immunotherapy targets cancer antigens on the malignant cells specifically, alerts the immune systems to the presence of foreign substances and eradicates cancer through the concert of immune responses. The normal cells with no cancer antigens are not affected. The outcomes of some cancer immunotherapies have been astonishing in some cases, but some which rely on the status of patients’ own immune systems are not. Those patients who responded well to cancer immunotherapy have a better prognostic and better quality of life.
Historically, cancer immunotherapy started in 1866 where notable tumor shrinkage was observed among cancer patients who were diagnosed with erysipelas caused by Streptococcus pyogenes (21, 22). Later in 1891, William Coley who is known as the ‘Father of Immunotherapy’ continued the discovery journey by introducing heat-inactivated Streptococcal bacteria (Coley’s toxin) into unresectable osteosarcoma patients with the hope that any side effects produced from the infection would shrink the tumor (23). The approach was successful for a time. The patients who developed erysipelas went into spontaneous remission (24, 25). Following this, Coley improved the formulation by combining live and attenuated Streptococcus erysipelas and Bacillus prodigiosus (26). Around 1000 patients were successfully treated using this method. After 8 years of hard work, Coley’s toxin was commercially available in 1899 (26). However, patients who underwent this treatment were exposed to extremely pathogenic bacteria. Furthermore, due to its unreproducible results, Coley’s toxin was opposed by most of the health practitioners. Surgery remained the most preferable way to treat cancer during that time (27).
After nearly two decades, immunotherapy once again captured scientists’ attention with the new concept of tumor-specific antigens which was found in a mouse model. This was followed by theories on acquired immunological tolerance and immunosurveillance (28–30). A year later in 1957, another cancer immunotherapy approach using interferon-α, a type of cytokine was introduced (31). The first cancer vaccine was also discovered during this era when 25 out of 114 (22%) gynecologic cancer patients went into remission upon treatment with adjuvant tumor lysate (32). In the subsequent years, novel findings on the importance of T cells in cancer immunity made cancer immunotherapy more exciting, thus lead to the discovery of dendritic cells and natural killer cells’ activities in mouse models (33–36). The first monoclonal antibody production using the hybridoma technique was also initiated in 1975 by Koehler and Milstein. They both were awarded a Nobel Prize in 1984 for this crucial finding which is widely used until today (37). Another significant finding in cancer immunotherapy was the discovery of the first immune checkpoint inhibitor namely CTLA-4 in 1988, which led to its first clinical trial in the year 2000 and approval by United States Food and Drugs Administration (FDA) to treat metastatic melanoma in 2011 (38). The emergence of cancer immunotherapy continued until the FDA-approved Interleukin-2 and the first monoclonal antibody (mAbs), Rituximab were used as anti-cancer therapies in 1992 and 1997, respectively (39, 40).
In the 20th century, the FDA has approved various types of immunotherapeutic drugs including Sipuleucel-T, a cancer vaccine to treat castration-resistant prostate cancer in 2010 (41, 42). Five years later, the first oncolytic virotherapy agent known as T-VEC was approved in treating metastatic melanoma (43). The chimeric antigen receptor (CAR) T-cell therapy was also introduced to relapsed B-cell acute lymphoblastic leukemia and diffuse large B-cell lymphoma patients in 2017 and 2018 after getting approval (44, 45). In the same year, Tasuku Honjo and James Allison received their Nobel Prize in Physiology due to their significant contributions in discovering the immune checkpoint inhibitors, PD-1 and CTLA-4, respectively (46). Currently, with an increasing number of FDA approved single and combinational immunotherapeutic drugs over the years, the cancer immunotherapy field is continuously showing potential in treating various types of malignancies.
Immune Classification
Cancer immunotherapies are classified based on the types of immune mechanisms that are involved either through passive or/and active mechanisms or based on antigen specificity (47). Passive immunotherapies are tumor-targeting mAbs, adoptive cell transfer (ACT) and oncolytic virotherapy while active immunotherapies are immunomodulatory mAbs, anti-cancer vaccines, immunostimulatory cytokines, inhibitor of immunosuppressive metabolism, pattern recognition receptor (PRR) agonists, immunogenic cell death inducer and other non-specific immunotherapeutic agents.
Monoclonal Antibodies (mAb)
Monoclonal antibodies (mAbs) are immunoglobulin molecules, which are made up of antigen-binding fragments that are connected to a constant region with two identical light and heavy chains. The light chains are made up of one variable and one constant domain while the heavy chains consist of one variable and three constant domains (48). There is also a special region within the variable domain with 3 loops known as the complementarity determining region (CDR) (48).
Initially, the hybridoma technique (49) and phage display (50) were used in producing murine mAbs in the laboratories. With advanced technologies, three types of antibody-engineered mAbs are produced using the same technique, namely chimeric, humanized and human monoclonal antibodies. Chimeric mAbs consist of cloned human amino acids at the constant domain whereas mouse amino acids are located at the variable domain. Chimeric mAbs can be produced by directly joining the variable region immunoglobulin of selected mouse hybridoma into the human constant region through in vitro cell-based technology (51). Examples of FDA approved chimeric mAbs are Rituximab that is used to treat Non-Hodgkin Lymphoma, Cetuximab to treat colorectal cancer and Dinutuximab that is used among neuroblastoma patients (52). On the other hand, humanized mAbs are generated through the addition of murine CDR into the human’s variable and constant domain through CDR grafting (53). Some of the FDA approved humanized mAbs are Trastuzumab used to treat breast cancer, Alemtuzumab to treat chronic myeloid leukemia and Bevacizumab to treat colorectal cancer (52). The third type of mAbs is the human mAbs which are produced when the whole mAbs are made up from human amino acids (54). This may be done through the preference of human antibody fragments from human hybridomas and in vitro libraries by transgenic mice (48). Human mAbs that are approved by FDA for cancer treatments are Panitumumab to treat colorectal cancer, Ofatumumab to treat chronic lymphocytic leukemia and Ramucirumab to treat gastric cancer (52).
Unlike polyclonal antibodies that bind to multiple epitopes, mAbs have a monovalent affinity which makes them bind to an epitope of antigens (55). The mAbs can recognize and bind specifically to tumor antigens of tumor-specific antigen (TSA) (56) or tumor-associated antigen (TAA) (57) that are present on cancer cells surface (Figure 1). TSAs are a group of mutated proteins due to somatic mutations and relatively are restricted to tumor cells (58). Their specificity in tumor cells makes them a good candidate for immunotherapy. One of the good examples of a TSA is the mutated p53 protein that is present in many cancer cells including colorectal cancer. As a result, the p53 synthetic long peptide vaccine was designed to treat metastatic CRC patients (59). The results show that around 90% of the respondents treated with this vaccine produced p53-specific-T cell response with low-grade toxicity suggesting that p53 is indeed one of the attractive TSAs in cancer immunotherapy (59).
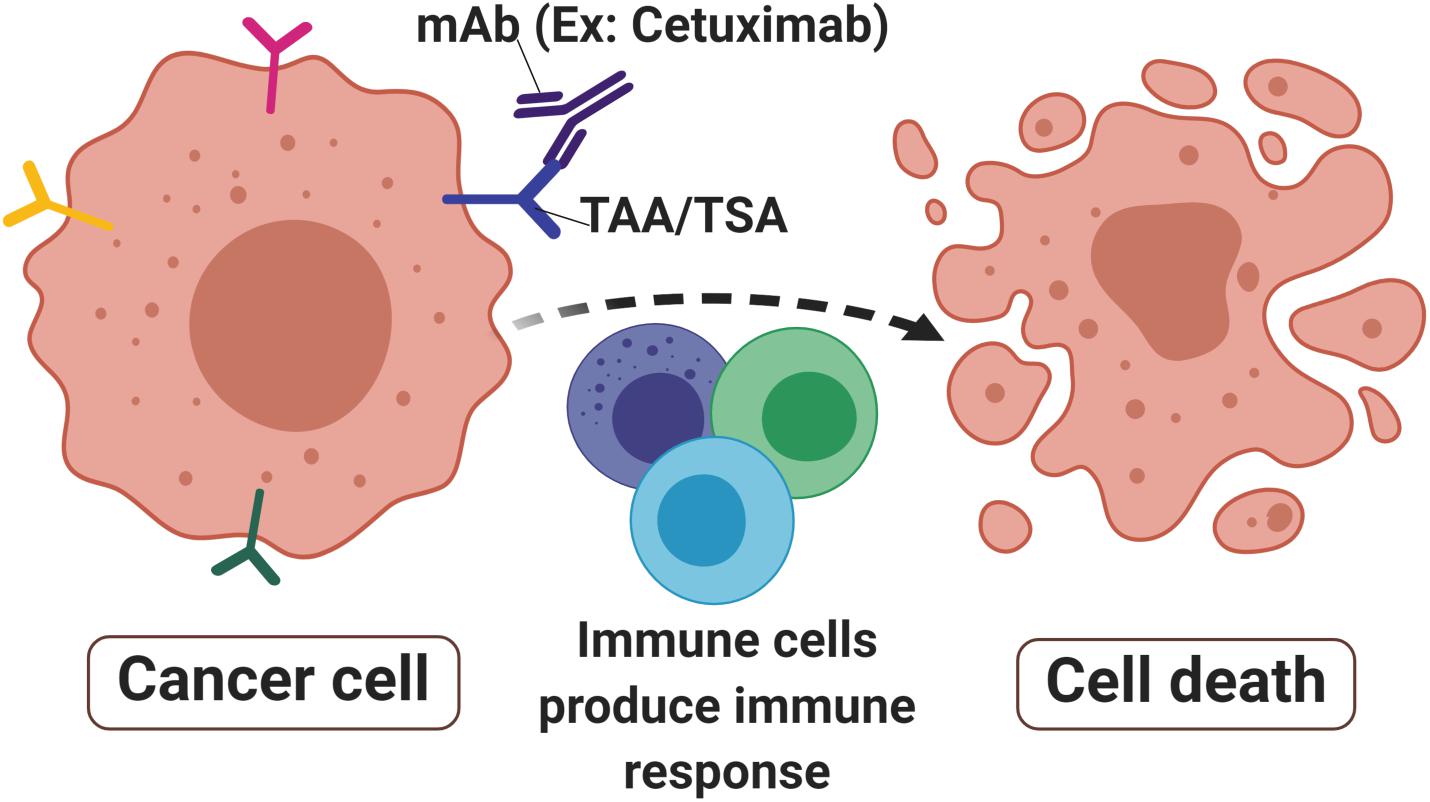
Figure 1. Monoclonal antibodies in cancer. Monoclonal antibodies (mAbs) such as Cetuximab are designed to target tumor-associated antigens (TAAs) or tumor-specific antigens (TSAs) that are abundant in cancer cells surface. The signals produced by receptor activities mediated immune cells toward malignant sites thus produced immune responses that lead to cell death to eradicate the tumor.
In contrast, TAAs have differentially expressed proteins that are present in both malignant and non-malignant cells. Although TAAs are expressed on normal cells, their expression on malignant cells has a unique characteristic resulting in specific immunogenicity (60). Nevertheless, because the antigens are also expressed on the normal cells, they may induce autoimmunity in the host (61). To overcome this, the self-antigen concept suggests that the self-reactive T cells are to be deactivated, therefore increasing their specificity in targeting the unique TAA on tumor cells (62). Furthermore, cancer vaccines must have a high tendency to specifically bind to tumor antigens and effectively kill them with minor adverse effects on normal cells. TAAs can be divided into four categories based on their expression pattern namely cancer-testis antigens (CTA), differentiation antigen, oncofoetal antigen and overexpressed antigens (62). CTA are aberrantly expressed protein in cancer and testis tissues with restricted expression in other normal tissues (63). A widely studied CTA in CRC is the melanoma-associated antigen (MAGE) group, particularly MAGE-A (64), MAGE-A-12 (65), and MAGE-A3 (66) variants. Differentiation antigens are expressed during cell differentiation stages such as Mucin 1 glycoprotein (MUC1) (61) and epithelial cell adhesion molecule (EpCAM) (67). These antigens may not create major side effects as they are tissue-specific and are only expressed during the cell differentiation stage (62). Oncofoetal antigens such as 5T4 (68, 69) and carcinoembryonic antigens (CEA) (70) are found in fetal tissues during its development as well as in several malignancies including ovarian, colorectal and breast cancers. Overexpressed antigens are huge, various groups of common proteins that can be found in both malignant and normal cells. However, they are highly expressed in malignant cells compared to the normal which thus have potential as an immunotherapeutic target (71). Some of the extensively studied overexpressed antigens in CRC are epidermal growth factor receptor (EGFR) (72), coiled-coil domain containing 34 (CCDC34) (73) and RAS-related protein (Rab-1A) (74). The binding of mAbs to TSA or TAA produces molecular signals to immune cells such as T cells, B cells and natural killer cells. This further initiates and activates receptors activities that lead to apoptosis and tumor-killing (75, 76).
The two major types of mAbs are tumor-targeting mAbs and immunomodulatory mAbs.
Tumor-Targeting mAbs
One of the passive immunotherapies is tumor-targeting mAbs and it is the most commonly mAbs used in treating hematological malignancies and other solid tumors. There are 76 mAbs that are approved by the European Medical Agency (EMA) and Food Drugs Administration (FDA) for therapeutic use (status 2017) (77). Table 1 listed the top 10 current EMA and/or FDA approved mAbs that are used for cancer treatments.
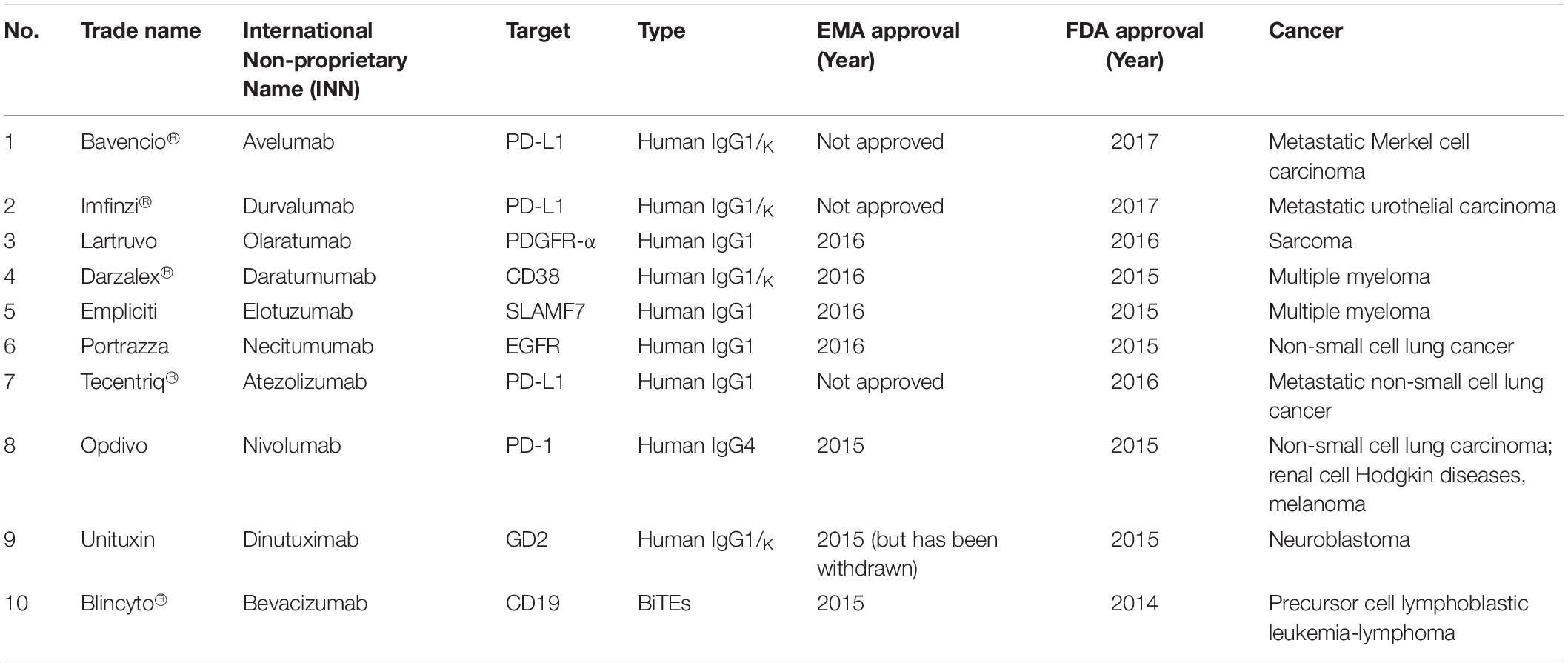
Table 1. The top 10 current EMA and/or FDA approved mAbs that are used for cancer treatments (78, 79).
Immunomodulatory mAbs
One of the common immunomodulatory mAbs is immune checkpoint inhibitors (ICIs). The ICIs are used to target and/or block immune checkpoints protein ligands on T cells surfaces or other immune cell subpopulations in restoring immune function. The immune checkpoints serve as key regulators that serve as an immune brake when there is sufficient immune response. However, in cancer, there is high activation and overexpression of immune checkpoints leading to suppression of anti-tumor immune response that favors malignant cell proliferation and spread (80, 81).
The most widely studied immune checkpoint targets are programmed cell death 1 (PD-1) and cytotoxic T lymphocyte antigen 4 (CTLA4) due to their overexpression and abundance in various solid tumors and hematological malignancies (82). Nevertheless, other checkpoints are currently being studied for their potential roles in tumor immunity regulation such as lymphocyte activation gene-3 (LAG-3), T cell immunoglobulin-3 (TIM-3), and T cell immunoglobulin and ITIM domain (TIGIT) (83–86).
In colorectal cancer, three FDA-approved ICIs drugs are targeting PD-1 and CTLA-4 (Table 2). However, ICI treatment efficiency is influenced by the microsatellite instability (MSI) status in each CRC patient (87, 88). MSI status is determined through immunohistochemistry staining and polymerase chain reaction targeting 5 MSI markers of BAT25, BAT26, D2S123, D5S346, and D17S250 (89). CRC patients are divided into three groups based on their mutation patterns of microsatellite instability-high (MSI-H), microsatellite instability-low (MSI-L), and microsatellite stable (MSS) (90, 91). ICI drugs targeting PD-1 and CTLA4 are more potent on metastatic CRC patients with MSI-H due to its higher tumor mutation burden (TMB). High TMB is positively correlated with high neo-antigen load thus increasing tumor immunogenicity (92). The majority of CRC patients with MSI-H benefited with this immunotherapy where the disease control rate was 80% using the combination of Nivolumab and Ipilimumab compared to patients having microsatellite stable (93).
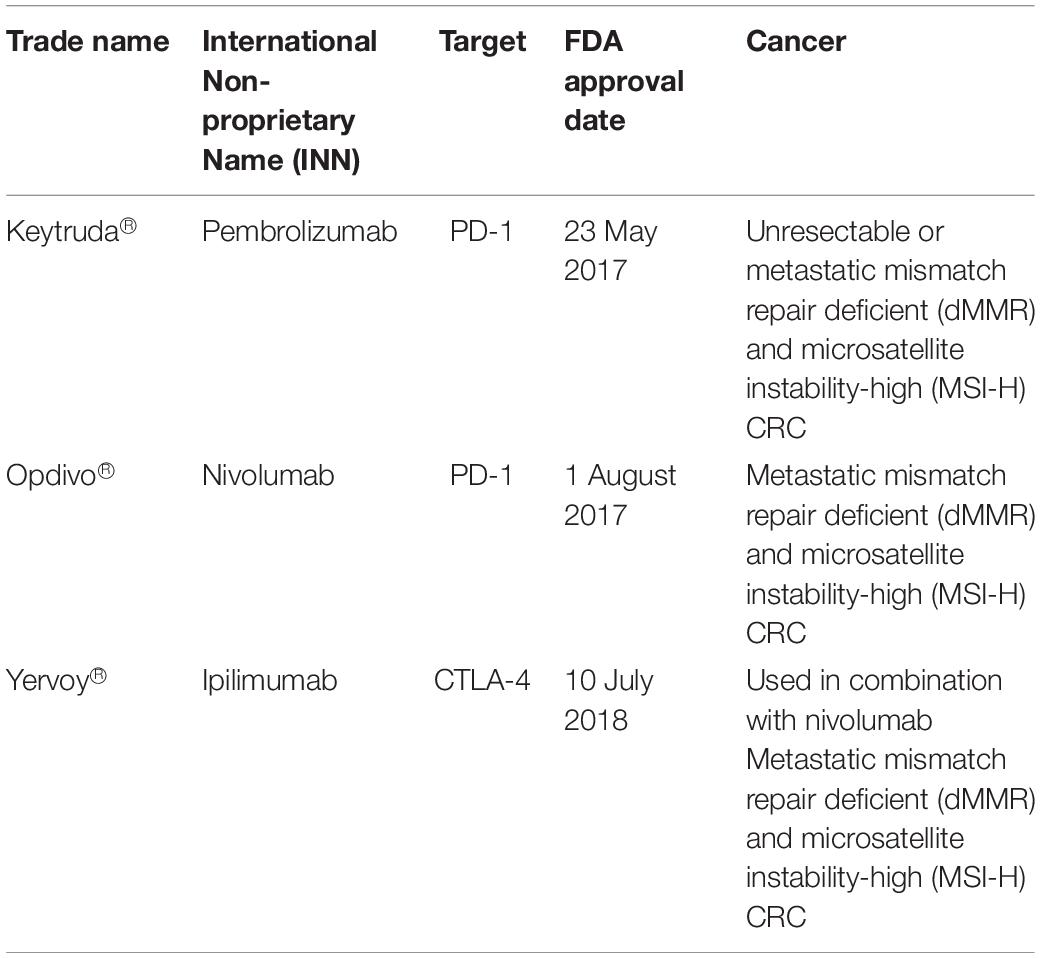
Table 2. FDA approved immune checkpoint inhibitors drugs for colorectal cancer treatments (78).
Cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4)
Cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) is one of the immune checkpoints that serve as co-inhibitory receptors on the T cell surface (38). The role of CTLA-4 in cancer immunotherapy was discovered through antibody generation that specifically targets the CTLA-4 glycoprotein (94). Under normal circumstances, T cell antigen receptor stimulation is regulated by CD28 co-stimulatory and CTLA-4 co-inhibitory signals (95). When there is an attack from a foreign substance, CD28 co-stimulatory signals increase to stimulate the T cell antigen receptors which further activate the downstream immune signaling (96). When the foreign substance has been cleared, CTLA-4 co-inhibitory signals are activated to stop immune signaling and thus prevent excessive immune responses or autoimmunity. Unlike CD28, CTLA-4 acts as negative regulatory feedback in T-cell stimulation. It switches off the T cell activity during priming and thus inhibits T cell activation and leads to antigenic tolerance. Hence, both CD28 co-stimulatory and CTLA-4 co-inhibitory signals are critical in maintaining T cell homeostasis and self-tolerance (97–99). However, there are some instances where CTLA-4 co-inhibitory signals are constitutively high and this can halt T cell activation (100). Such scenarios are seen in many cases of cancers and suppressor T cells such as T regulatory cells, suggesting the reason for lack of immune responses in cancer patients (101, 102). CTLA-4 protein is homologous to CD28 protein and has a higher binding affinity toward CD80 and CD86 on major histocompatibility complex (MHC) (Figure 2). This has been the avenue in immunotherapy in targeting CTLA-4 protein expression to stimulate the patient’s own T cell immune response. Many immune checkpoint inhibitor drugs are targeting CTLA-4 that is designed to bind and block this protein, thus allow CD28 binding to MHC and stimulate T cell immune response, activate downstream immune signaling and destroy the malignant cells (103, 104).
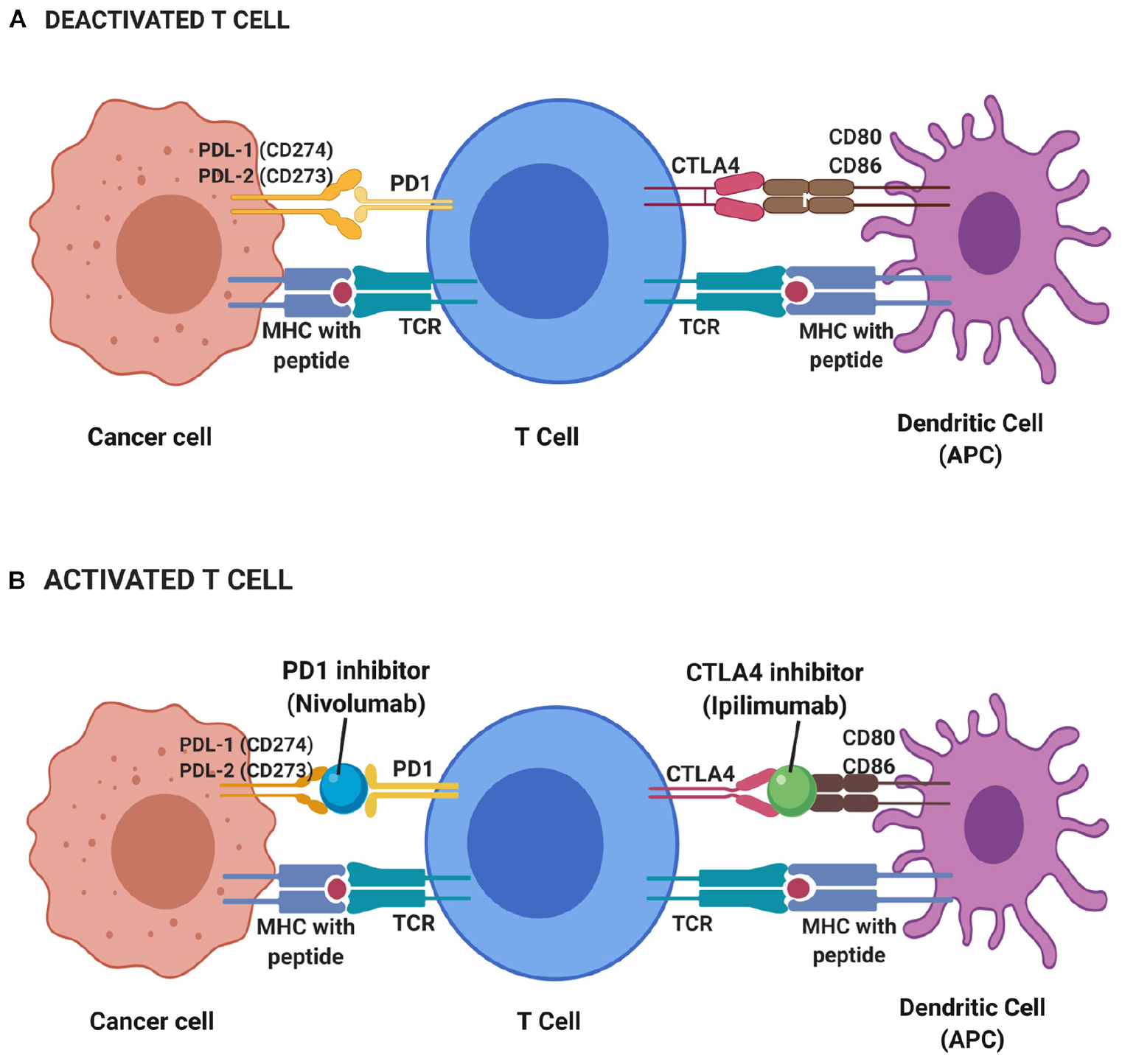
Figure 2. Deactivated (A) and activated (B) T cell-based on immune checkpoint inhibitors mechanism. During resting of T cell (deactivated T cell), CTLA4 and PD1 receptors on T cell surface binds to CD80 and CD86 on the antigen-presenting cell (APC) such as dendritic cell and PDL-1 and PDL-2 on cancer cell, respectively, while T cell receptor (TCR) binds to major histocompatibility complex (MHC) with the presence of peptide. Thus, no immune response triggered to kill cancer cells. T cells will only be activated with the presence of blockade or inhibitor on CTLA4 and PD1 receptors. Hence, a CTLA4 inhibitor known as Ipilimumab and PD1 inhibitor of Nivolumab functioned to block those receptors and elicit immune response thus leading to the apoptosis of cancer cells.
Programmed death 1 receptor and its ligand (PD-1/PD-L1)
Programmed death 1 (PD-1; CD279) is a novel member of the immunoglobulin gene superfamily (IgSF) with restricted expression in the thymus of mice (105). The PD-1 gene was first discovered in apoptosis gene screening and was shown to be involved in apoptosis of interleukin-3 (IL-3)-deprived LyD9 (a murine hematopoietic progenitor cell line) and stimulated 2B4.11 (a murine T-cell hybridoma) (105).
In humans, the PD-1 (PDCD1) gene is located at 2q37.3 which encodes for PD-1 proteins (106). This protein is a type I transmembrane glycoprotein with a size of 50–55 kDa (107). PD-1 is expressed in various immune cells such as CD4 and CD8 T cells, B cells, macrophages, dendritic cells and tumor-infiltrating lymphocytes (TILs) (108, 109). It functions as an immune checkpoint that balances the peripheral tolerance and regulates T-cell responses under normal conditions (110).
Programmed death 1 binds to two ligands, PD-L1 (CD274; B7-H1) and PD-L2 (CD273; B7-DC) with differential expression (111). This binding activates PD-1: PD-L1/L2 pathway which then mediates potent inhibitory signals to hinder the proliferation and function of T effector cells and have inimical effects on antiviral and antitumor immunity (112).
Similar to CTLA-4, the main role of PD-1/PDL1 interaction (PD-1 pathway) under normal conditions is as a brake for immune response in which the pathway can limit T-cell effector responses. One of the ways is through enhancing immunosuppressive regulatory cells (Tregs) development, thus prevent over-activation of the immune response in human peripheral tissues (113). This immune homeostasis is important in protecting us from autoimmune and severe inflammation.
However, this is not always the case in cancer. Overexpression of PD-1 is observed that leads to constant PD-1 (on the T cell surface) binding to its ligand, PDL1 (on the cancer cells). As a result, PD1/PDL1 signals are constitutively high, suppress the activation of T cells and cause antigenic tolerance. This allows immune cell evasion by cancer cells which then support the high tumor proliferation rate (114, 115). Therapeutic strategies targeting PD1/PDL1 pathways have resulted in many checkpoint inhibitors that function to interfere with PD-1/PDL1 binding through competitive binding and lead to restoring effector T cells activity in cancer patients. Drugs inhibitors were designed to bind and block PD1/PDL1 binding to restore effector T cells activation, proliferation, function and downstream immune signaling to destroy the malignant cells (104).
To date, there are five FDA approved PD-1 inhibitor drugs for various cancers (103, 116) (Table 3). The first FDA approved PD-1 inhibitor drug was Pembrolizumab (Keytruda®), a humanized monoclonal IgG4 antibody, used in treating melanoma (78). Phase I trial of the drug was tested in solid tumor and hematological malignancies patients. Results show that Pembrolizumab was well tolerated among multiple solid tumors patients (117). There was a significantly longer survival rate with minimum side effects among advanced non-small cell lung cancer (NSCLC) with high PDL1 expression compared to patients treated with platinum-based chemotherapy (118).
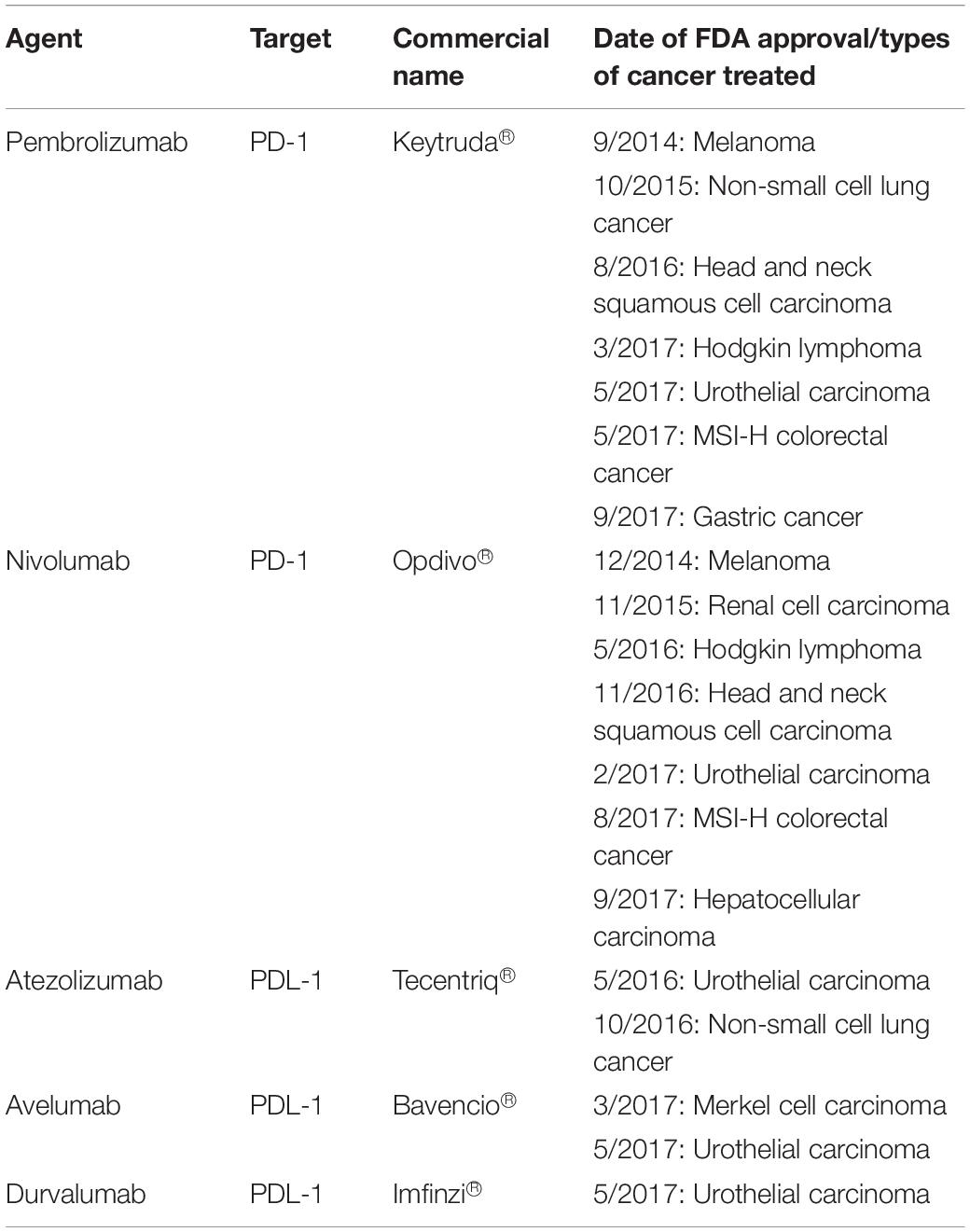
Table 3. FDA approved PD-1 and PDL-1 inhibitor drugs in various types of cancer immunotherapy (78, 116).
In CRC patients, Pembrolizumab shows significant benefit to the mismatch-repair deficient or microsatellite instability-high CRC patients (dMMR/MSI-H). Results show the progression-free survival rate up to 78% compared to mismatch-repair proficient, microsatellite stable (pMMR/MSS) patients of 11% (119). Another successful PD-1 inhibitor is Nivolumab (Opdivo®) that shows durable responses among the dMMR metastatic CRC (mCRC) cohort of patients. Approximately, 69% of these patients have 12 months of overall survival (OS) (120). Interestingly, a combination of Nivolumab with Ipilimumab (a CTLA4-targeting drug) demonstrates a higher response rate of up to 94% in these patients. This suggests that the combination of immune checkpoints therapy can greatly improve the efficacy of the treatment for dMMR/MSI-H mCRC patients (93).
Adoptive Cell Transfer (ACT)
Adoptive cell transfer is a cell-based therapy that uses cells either from the patient (autologous transfer) or other donors (allogeneic transfer) to improve immune function (121). There are 3 methods for the ACT; use of tumor-infiltrating lymphocytes (TILs), insertion of chimeric antigen receptor (CAR), and modification of T cell receptors (TCR) (122).
Adoptive cell transfer that uses TILs (ACT-TIL) was first successfully observed in 60% of metastatic melanoma patients who had not been treated with interleukin-2 (IL-2) and 40% of non-respondents IL-2 treatments which resulted in cancer regression (123). However, major drawbacks with ACT-TIL using patients’ own TIL was a limitation in generating tumor-specific T cells. This leads to the development of another approach using patients’ own genetically engineered-TCR. Approximately 13% (2 out of 15) metastatic melanoma patients benefited from this type of ACT with high sustained engineered T cells level were observed after a year of infusion (124). A few years later, ACT uses a better method of CAR insertion. This was shown to be safe to use among a cohort of clear cell renal cell carcinoma (ccRCC) and ovarian cancer patients with no toxicity observed although only two and none of these patients respond toward the treatment, respectively (125, 126).
CAR-T cells consist of antibody variable fragments specific to the antigen of interest that are fused to the isolated patient’s or donor’s T cells (127). In the CAR approach, T cells are isolated from a patient (autologous) or HLA-matched donor (allogeneic), cultured through ex vivo and genetically modified through the insertion of the chimeric antigen receptor (CAR) onto the T cells as CAR-T cells (128). The modified ex vivo CAR-T cells are re-infused back into the patient and monitored. The modifications are necessary for enhancing the T cells’ ability to recognize the antigen of interest and avoid the major histocompatibility complex restriction recognition. This leads to highly targeted antigen recognition and allows active trafficking to tumor sites, in vivo expansion and long-term persistence. Usually, CAR-T cells are engineered toward tumor-associated antigens (TAAs) such as CD19 in diffuse large B cell lymphoma (DLBCL) (129), interleukin 13 receptor alpha-2 (IL13Rα2) (130) and epidermal growth factor variant III (EGFRvIII) in glioblastoma (131) and carcinoembryonic antigen (CEA) in colorectal cancer (132) to promote cytotoxicity and apoptosis (Figure 3). The advantage of CAR-T cells is their specificity in targeting cell surface TAA in an MHC-independent manner. This allows more patients to be treated without the need for MHC-specific treatment. In addition, co-stimulatory domains such as CD28 or 4-1BB (CD137) can be added to improve CAR-T cells’ proliferation and survival rate in vivo, thus enhancing the anti-tumor activity of CAR-T cells (133, 134). Furthermore, the T cell responses produce memory cells that help to preserve the immunotherapeutic effect for several years even after treatments (135, 136).
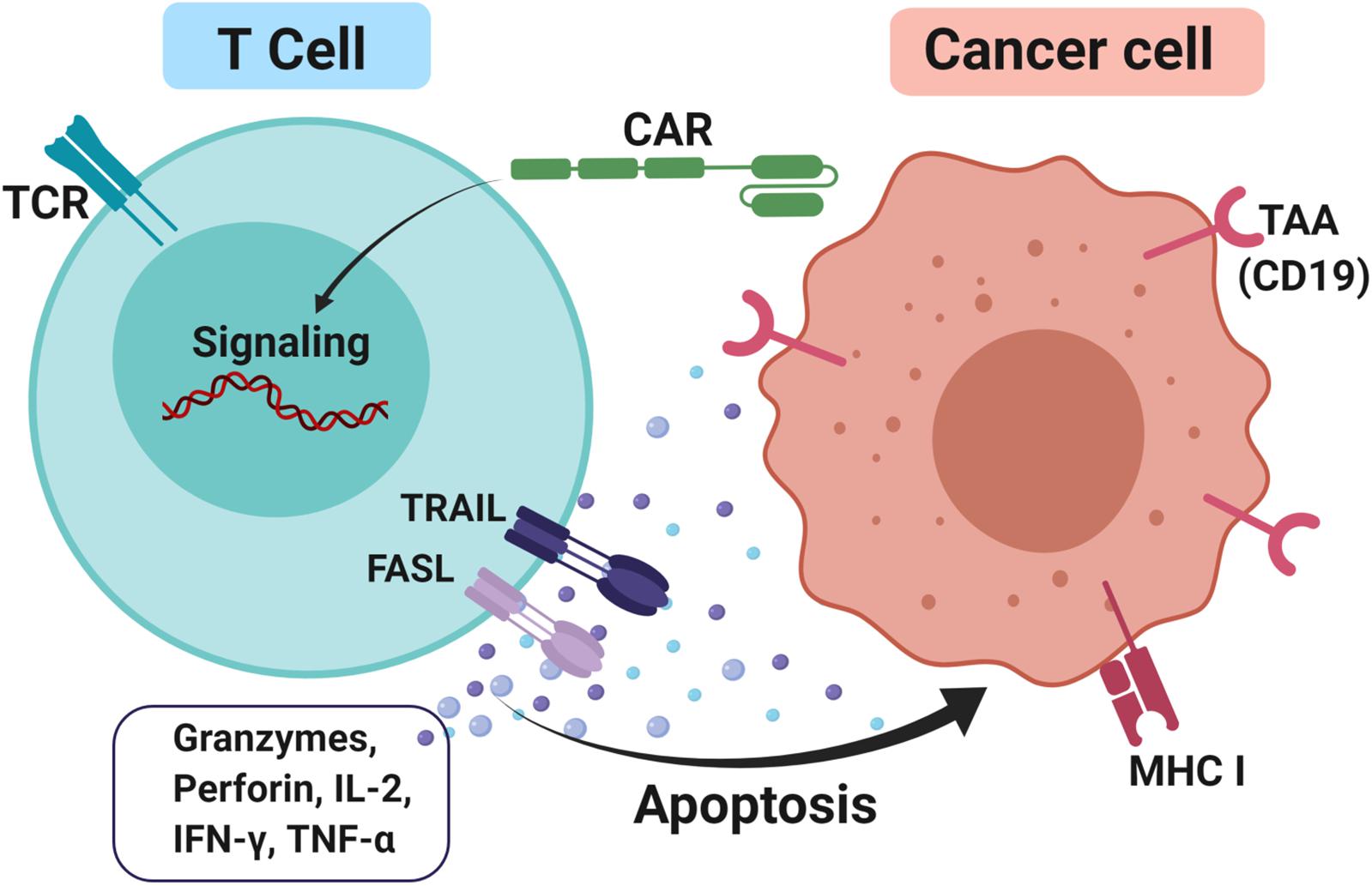
Figure 3. CAR-T cells approach to treating cancer. T cells were firstly isolated either from the patient itself or HLA matched donor through apheresis. Next, the cells were cultured and a genetically modified chimeric antigen receptor (CAR) was inserted into it and these T cells are now known as CAR-T cells. This modification is necessary to enhance T cells’ ability to recognize tumor-associated antigens (TAAs) such as CD19 and avoid the major histocompatibility complex class I (MHC I) restriction recognition on the cancer cell. Upon binding, FAS ligand (FASL) and TNF-related apoptosis-inducing ligand (TRAIL) promotes cytotoxicity by releasing effector cytokines and lead to cancer cell apoptosis.
Like many other methods, ACT comes with some limitations. This approach is highly technical and economically challenging for both the industry and patients due to the need to generate tumor-specific lymphocytes for each patient. Secondly, the patient who is receiving the allogeneic transfer is often exposed to the danger of graft-versus-host-disease (GvHD). Furthermore, toxicity is the main issue when targeting antigenic targets such as TAA which are also expressed on normal tissues but are overexpressed on the tumor. An example of these cross-reactivities was seen in CAR-T cells targeting HLA-A∗0201–restricted peptide in melanoma-associated antigen (MAGE)-A3. This approach had caused severe damage to gray matter in the brain as the TCR recognized a different but related epitope that was expressed at low levels in the normal brain cells (137). Another limitation is related to cytokine release syndrome (CRS), which is life-threatening toxicity that has been observed in the ACT. CRS refers to an elevated state of circulating level of cytokines, chemokines and other signaling proteins including interleukin 6 (IL-6) and interferon γ as a result of tumor lysis. This, in turn, activates more immune cells and other immune signalings, leading to excessive immune responses and toxicities (138). Other side effects including neurological problems such as problems remembering words, difficulty in speaking, being less alert, hallucinations, seizures and coma. In many patients, these problems fade on their own in a few days, but some have died from these problems. Nevertheless, FDA approved ACT was listed for many hematological malignancies (Table 4) but most of the solid tumors are still undergoing clinical trials.

Table 4. FDA approved CAR T cells treatments in cancers (78).
In CRC, CAR-T cells target carcinoembryonic antigens (CEA) and guanylyl cyclase C (GUCY2C) (142, 143) tumor-associated glycoprotein (TAG72) (144), epithelial cell adhesion molecule (EpCAM) (145), NK cell surface receptor ligands (NKG2DLs) such as major histocompatibility complex (MHC) class I-related chain A and B (MICA and MICB, respectively) and six unique long 16 binding protein (UL-BP1-6) (146). The CAR-T cell therapy can work effectively only if these targets are highly expressed in colorectal carcinoma tissues with low expression in other normal tissues. Normally, these molecules are present at low or undetectable levels on normal cells but rapidly appear on the surface of stressed, malignant transformed and infected cells. For example, therapeutic potential targeting CEA was observed with tumor regression and antigen specificity but with some CRS toxicity and severe colitis (143, 147, 148). These effects were due to the overexpression of CEA in both colorectal adenocarcinomas and several normal colonic mucosae of the gastrointestinal tract (149). Despite some of the drawbacks of CEA targeted CAR-T cell approaches, a phase I trial was performed among CEA + mCRC patients to evaluate its efficacy. Interestingly, there were no CAR-T related severe adverse events observed among all the 10 patients involved in this study (150). Furthermore, combination treatment between CEA-CAR-T cells in addition to recombinant human interleukin 12 (rh-IL12) in mouce models show effective and elevated anti-tumor activity of CAR-T cells among various types of solid tumors including CRC (151).
However, some limitations are encountered in effective targeting of solid tumors by CAR-T cells. This explains why CAR-T cells have not proceeded into commercialization and approval for solid tumors. One of the challenges is the efficient trafficking and infiltrating of solid tumors. The microenvironment of solid tumors contains an abundant fibrous matrix and immunosuppressive cells, which protects the tumor tissue and resists immune cell attack. This includes certain chemokines such as CXCL1, CXCL12, and CXCL5 that are secreted by the tumor cells to inhibit the effective delivery of the CAR-T cells (152, 153). Therefore, overcoming this hindrance is through engineering chemokine receptor (CXCL1 receptor) – T cells. This has greatly drive CAR-T cells to migrate toward chemokines secreted-tumor cells (154). However, having the CXCL1 receptor- engineered T cells is not an ultimate solution. Even if the CAR-T cells successfully traffick and infiltrate the cancer cells, the nature of the tumor and environment themselves further inhibit the actions of the CAR-T cell. This refers to the architecture of the cancer cells such as extensive vascular leakage, poor integrity of tissue structure, hypoxia and low pH. In hypoxic conditions, the acidic tumor microenvironment lacks the necessary essential amino acids. Thus, T cells are likely to experience anergy, exhaustion, senescence and stemness, making it a challenge to achieve the desired CAR-T cells tumor killing (155). Also, other immune suppressor cells such as T cells (Tregs), myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages are present in the tumor microenvironment that further inhibit the CAR-T cells from being activated and producing a response toward cancer cells. Furthermore, immune checkpoint receptors on tumor cells or immunosuppressive cells are able to inhibit T cells by binding to negative regulatory ligands on T cells. As an example, PD-L1 on the tumor cell surface binds to PD1 on T cells, which will inhibit CAR-T cell activation. Lastly, the tumor-derived inhibitory cytokines such as transforming growth factor-β (TGF-β) are able to deter the function of CAR-T cells in killing the cancer cells. TGF-β plays a major role in alleviating the antitumor response where it can downregulate CD8 + effector T cell function and upregulates Treg maturation (156).
Modification of T cell receptors (TCR) is another approach for ACT and it is known as TCR transduced therapy. It is quite similar to CAR-T cells but their mechanisms for recognizing antigens are quite different. In CAR-T cells, antibody fragments are employed to bind the specific antigens on the surface of cancer cells. In contrast, TCRs use heterodimers consisting of alpha and beta-peptide chains to recognize polypeptide fragments presented by MHC molecules (157). This allows recognition of an intracellular, cell surface antigen or a neo-antigen produced by tumor cells after mutation. The TCR-T cell therapy directly modifies TCR binding to tumor antigens with high-affinity by genetic engineering technology (158). Therefore, it requires the identification of specific targets on cancer cells to ensure minimal off-target effects and cross-reactivity in other cells.
In TCR-T cell therapy, the MHC-dependent allows more antigen targeting compared to CARs. These antigens include MART-1, Gp100, CEA, NY-ESO-1, MAGE-A3, MAGE-A4, and others, which are suitable for TCR-T cell therapy (Table 5). In addition, TCR can also be targeted toward neoantigens generated by random mutations in tumor DNA and cancer-testis antigens (159).
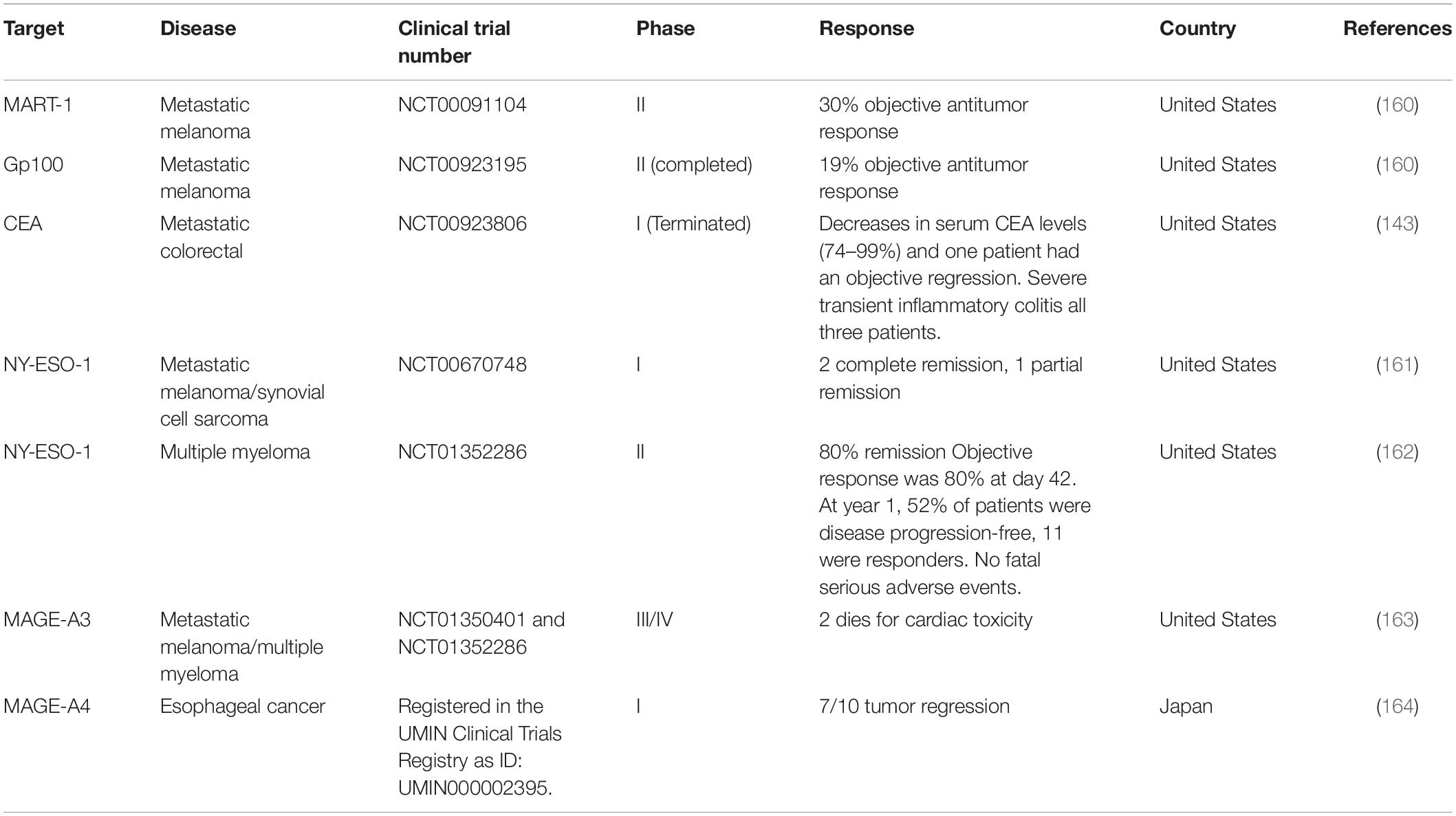
Table 5. Current clinical trials of TCR-T therapy for solid tumors with reported outcomes.
Normally, the affinity of human TCRs toward cancer antigens is relatively low, which makes it impossible to recognize and kill tumors effectively. With the advance of engineering technology, genetically-modified TCR is encoded in T cells that give it better specificity and affinity toward cancer antigen. For example in the multiple myeloma patients, TCR was engineered through modifications of several key amino acids in order to have a higher affinity for cancer antigens, NY-ESO-1 (165). The clinical trial showed that 80% of these patients had a good clinical response while 70% of them had a complete or near-complete response (165). Another example is TCR-T cell therapy toward MART-1 for the treatment of metastatic melanoma patients. Results in clinical trials showed higher antitumor reactivity with objective cancer regressions were seen in 30 and 19% of patients who received the human or mouse TCR, respectively. However, patients suffered some acceptable side effects without CRS (160). These findings show that T cells expressing highly reactive TCRs could mediate cancer regression in humans and target rare cognate–antigen-containing cells throughout the body. This is an important implication for the gene therapy of cancer. The reported outcomes of clinical TCR-T cell therapies are listed in Table 5. The first report on TCR-T cell therapy in colon cancer was targeting CEA antigens where some evidence of clinical response was seen but with severe colitis due to the presence of CEA in normal cells in the colon (143). This demonstrates the feasibility of T-cell therapy in metastatic colon cancer, but also the limitations of targeting CEA as an antigen. Following the promising results, another attempt was developed for treating advanced metastatic colon cancer patients. This was using mRNA-engineered T Cells targeting transforming growth factor β- receptor type II (TGFβII) frameshift antigen which is expressed in microsatellite instability positive (MSI+) colon cancer (166). However, the clinical trial was terminated due to severe adverse effects. Other ongoing TCR therapies are against KRAS G12V + tumor (NCT03190941) and KRAS G12D + tumor (NCT03745326), both are at clinical trials phase I/II (166).
T cell receptors therapy although it is a very promising approach comes with many challenges including good targets selection, specific TCRs search, optimal TCR affinity screening, safety evaluation, time and cost (159). In addition, since TCR therapy is highly dependent on MHC for peptide presentation, it may escape immune surveillance due to the downregulation or mutation of MHC molecules in the tumor environment, resulting in clinical limitations. Furthermore, hybridization (mismatch) between exogenous and endogenous chains may occur and induce harmful recognition of self-antigens, leading to graft-versus-host disease (167). Although higher TCR affinity offers a great benefit, there is a possible risk of false targeting. In a nutshell, TCR-T cell therapy has shown some therapeutic potential but there are still many limitations that should be considered carefully.
Cancer Vaccines
Cancer cells express altered self-antigens that induce weaker responses compared to foreign antigens such as infectious agents. Often, immune stimulants and adjuvant are incorporated together with the cancer vaccines to enhance the effects. Cancer vaccine includes autologous patient-derived immune cell vaccines, tumor antigen-expressing recombinant virus vaccines, peptide vaccines, DNA vaccines and heterologous whole-cell vaccines derived from established human tumor cell lines (168).
Preventive (prophylactic) and therapeutic are the approaches of cancer vaccines treatments in which preventive cancer vaccines intended to minimize cancer incidence, morbidity and mortality while therapeutic vaccines aimed to treat current malignancies and may prevent (169, 170). To date, there are three FDA approved therapeutic cancer vaccines (Table 6).
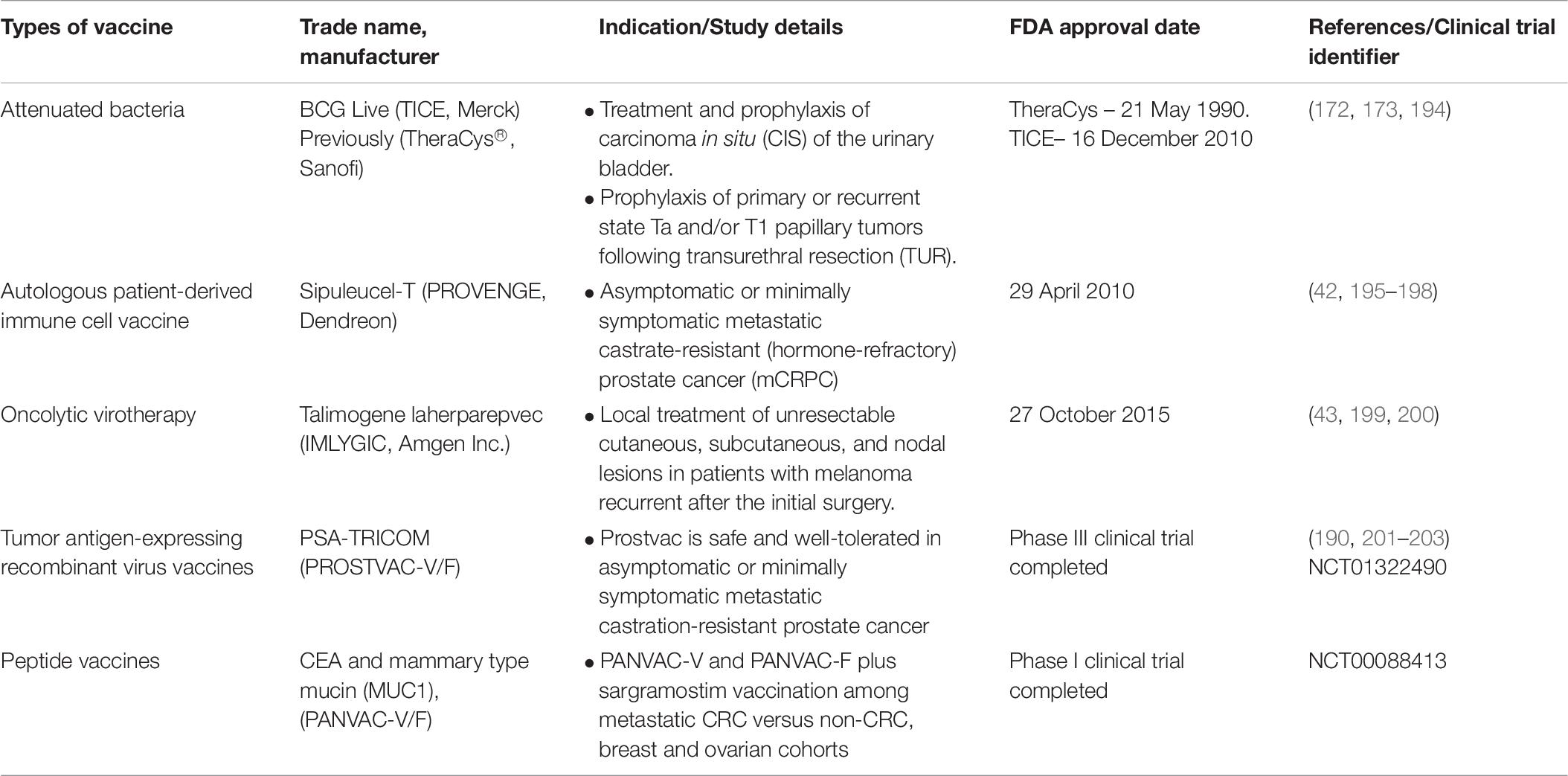
Table 6. FDA approved and clinical trial cancer vaccines (78).
The first FDA approved cancer vaccine was TheraCys® (Sanofi) (171). It is an intravesical, attenuated Connaught strain of Bacillus Calmette-Guarin (BCG) derived from Mycobacterium Bovis. TheraCys® is used in the treatment and prophylaxis of urothelial carcinoma-in situ (CIS), particularly non-muscle invasive bladder cancer (NMIBC) subtype. Results showed that approximately 74% of the patients showed a complete response with BCG compared to Doxorubicin (172). This vaccine also improves the protection against superficial bladder cancer recurrence. A randomized trial demonstrated that 70% of the CIS patients have a complete response to BCG therapy instead of doxorubicin suggesting the effectiveness of this vaccine compare to chemotherapy (173). At the start, two BCG strains were available; Connaught (TheraCys®, Sanofi) and Tice (TICE®, Merck). However, due to the supply shortage of TheraCys®, it was discontinued and replaced by TICE®. Nevertheless, a study conducted previously shows that treatments using different BCG strains among NMIBC patients have a significant role as BCG Connaught treatment was more effective than BCG Tice in terms of 5 years recurrence-free survival rate (174). Furthermore, a cohort study among 2099 NMIBC patients using both strains indicates that Connaught was more effective than Tice among patients without BCG maintenance (175).
Another FDA approved therapeutic vaccine is Sipuleucel-T (Provenge; Dendreon) which used to treat prostate cancer patients who are asymptomatic or minimally symptomatic metastatic castration-resistant. This vaccine uses the patient’s own immune cells (dendritic cells, T cells, B cells and natural killer cells) isolated through leukapheresis. The immune cells were cultured and incubated with PA2024, a fusion protein made up of prostatic acid phosphatase (PAP) which is linked to granulocyte-macrophage colony-stimulating factor (GM-CSF) as the adjuvant (41, 176). Antigen-MHC complex was presented on activated DCs surface during incubation thus inducing CD4+ and CD8+ cells to act against PAP while GM-CSF improves DCs maturation upon being introduced into patients (177). This vaccine also shows significant survival benefit (50% higher than in control) for this population of asymptomatic patients who have not been treated with chemotherapy, except for docetaxel (whose inherent toxicities often lead patients and physicians to delay administration until symptoms develop) (42). Currently approaches to enhance efficacy are considered to increase the efficiency of Sipuleucel-T in a wider range of patients. A study to look at the adverse effects related to the usage of this vaccine was performed and an adverse event spectrum was consistent with no new safety concerns observed from 2010 to 2017 (178).
Talimogene laherparepvec (T-VEC) (IMLYGIC®, Amgen Inc.) is the latest FDA approved vaccine to treat unresectable cutaneous, subcutaneous, and nodal lesions melanoma recurrent before initial surgery (179). T-VEC is a genetically modified oncolytic virotherapy made up of herpes simplex virus type I (HSV-1) (180). Significantly higher (16.3%) durable response rate (DRR) was demonstrated among unresected stage IIIB to IV melanoma patients injected with T-VEC compared to GM-CSF (2.1%) (43). A randomized trial among advance and unresectable melanoma patients revealed that combination therapy of T-VEC and ipilimumab have a significantly higher (39%) objective response rate compared to ipilimumab only (18%) due to their substantial activity (181, 182).
CRC Cancer Vaccines
To date, there are no CRC cancer vaccines approved by the FDA. Most of them are still undergoing clinical trials. Many studies are looking at various attractive, overexpressed single and combination uses of TAAs such as CEA (183–185), MAGE (186, 187), and MUC1 (188, 189). One of the cancer vaccine examples is the Poxviral vaccine regimen which targets TAAs such as CEA and MUC-1 using TRICOM vaccination strategy. In this approach, poxviruses are used as a viral vector for targeting CEA and MUC-1 in combination with T-cell co-stimulatory molecules (B7-1, ICAM-1, and LFA-3). The addition of the co-stimulatory molecules has been shown to greatly enhance the immune response to the antigen (170, 184, 190). The recombinant poxviruses were engineered to express both signal 1 (antigen) and signal 2 (co-stimulatory molecules), with each transgene being driven by a different poxvirus promoter. Preclinical studies have demonstrated that when a TAA transgene is placed into a poxvirus genome, its expression leads to a more vigorous host T-cell immune response to the TAA than would be achieved otherwise (191). Approximately 56% of the metastatic carcinoma patients showed a significant immune response toward MUC-1 and/or CEA following this vaccination (184, 190). In a phase I clinical trial of PANVAC-V, PANVAC-F and sargramostim (GM-CSF immunostimulator), this type of vaccine is associated with enhancing immune responses and has shown evidence of clinical activity among advanced CRC versus non-CRC, breast and ovarian carcinoma cohort.
Another peptide-based cancer vaccine targets CEA only. The study which was conducted using transgenic mice has shown that immunization with CEA peptides can elicit an immune response to kill cancer cells and hence may improve survival (192). Vaccination with an anti-CEA can break immune tolerance to TAA CEA and induce anti-CEA antibodies as well as CEA-specific CD4+ T-helper responses in colon cancer patients and mice transgenic for human CEA (193). Furthermore, combination strategies with the CTL peptides of CEA to get both CDT-helper and CTL responses in the transgenic CEA/HLA-A2 mouse model have shown overall significant immune responses and survival (192). The combined vaccination strategy results in the potential use of this vaccination strategy for future clinical applications.
Cytokines
Cytokines are a group of small cell-signaling polypeptides with a molecular weight of less than 30 kDa (204). They are secreted by various cells mainly the immune cells (T cells, neutrophils and macrophages), endothelial cells, fibroblasts, and other stromal cells (205). There are more than 130 cytokines with various roles. However, their main function is similar which is to stimulate and modulate robust immune responses toward inflammations and infections (206). Besides, these glycoproteins can act on the cells that produce them (autocrine action), in adjacent cells (paracrine action) or in distant cells (endocrine action) (207).
Cytokines are one of the potential polypeptides used in immunotherapy since they can enhance patients’ immune responses well. Three recombinant cytokines such as recombinant interferon-alpha and interleukin-2 drugs have been approved by the FDA and EMA for the treatment of several malignancies (Table 7) (208). A renewed interest in the anti-tumor properties of cytokines has led to an exponential increase in the number of clinical trials that explore the safety and efficacy of cytokine-based drugs, not only as single agents but also in combination with other immunomodulatory drugs. These second-generation drugs under clinical development include known molecules with novel mechanisms of action, new targets, and fusion proteins that increase the half-life and target cytokine activity to the tumor microenvironment or the desired effector immune cells. In addition, the detrimental activity of immunosuppressive cytokines can be blocked by antagonistic antibodies, small molecules, cytokine traps or siRNAs.
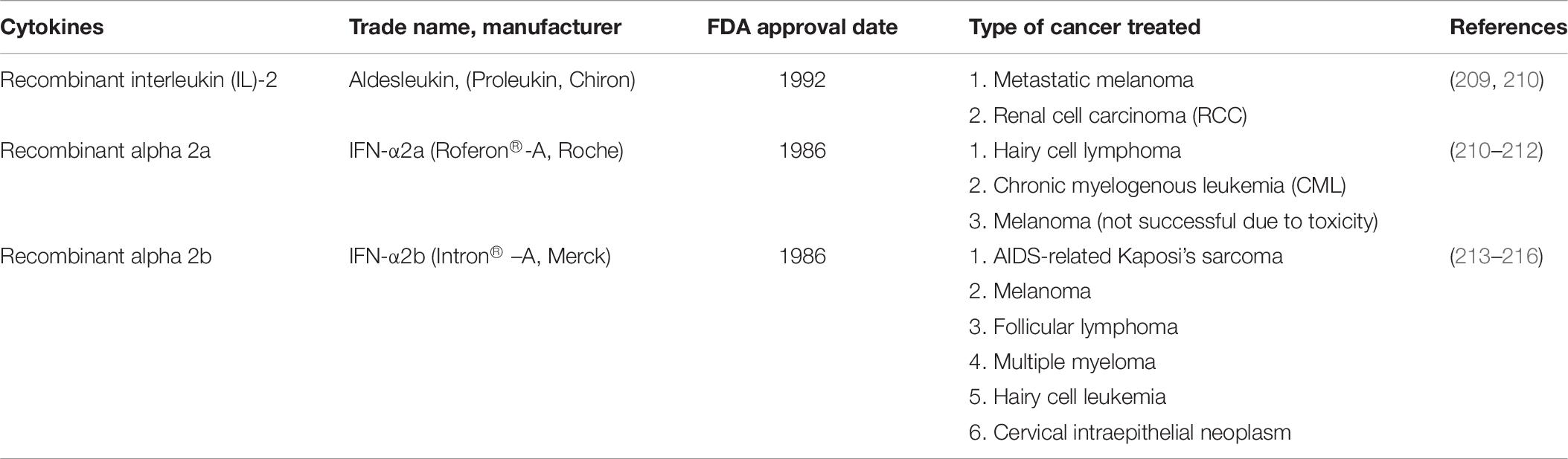
Table 7. FDA and EMA approved cytokines drugs for various cancer treatments.
Summary
In this review, we provide an overview of the novel trends in the immunotherapy field that are yielding therapeutic benefits in CRC patients. We summarize the advantages and drawbacks of each type of immunotherapy in Table 8. The future for immunotherapy is vast with different approaches. What it takes is smart strategies in overcoming the evasion of cancer cells from immune recognition.
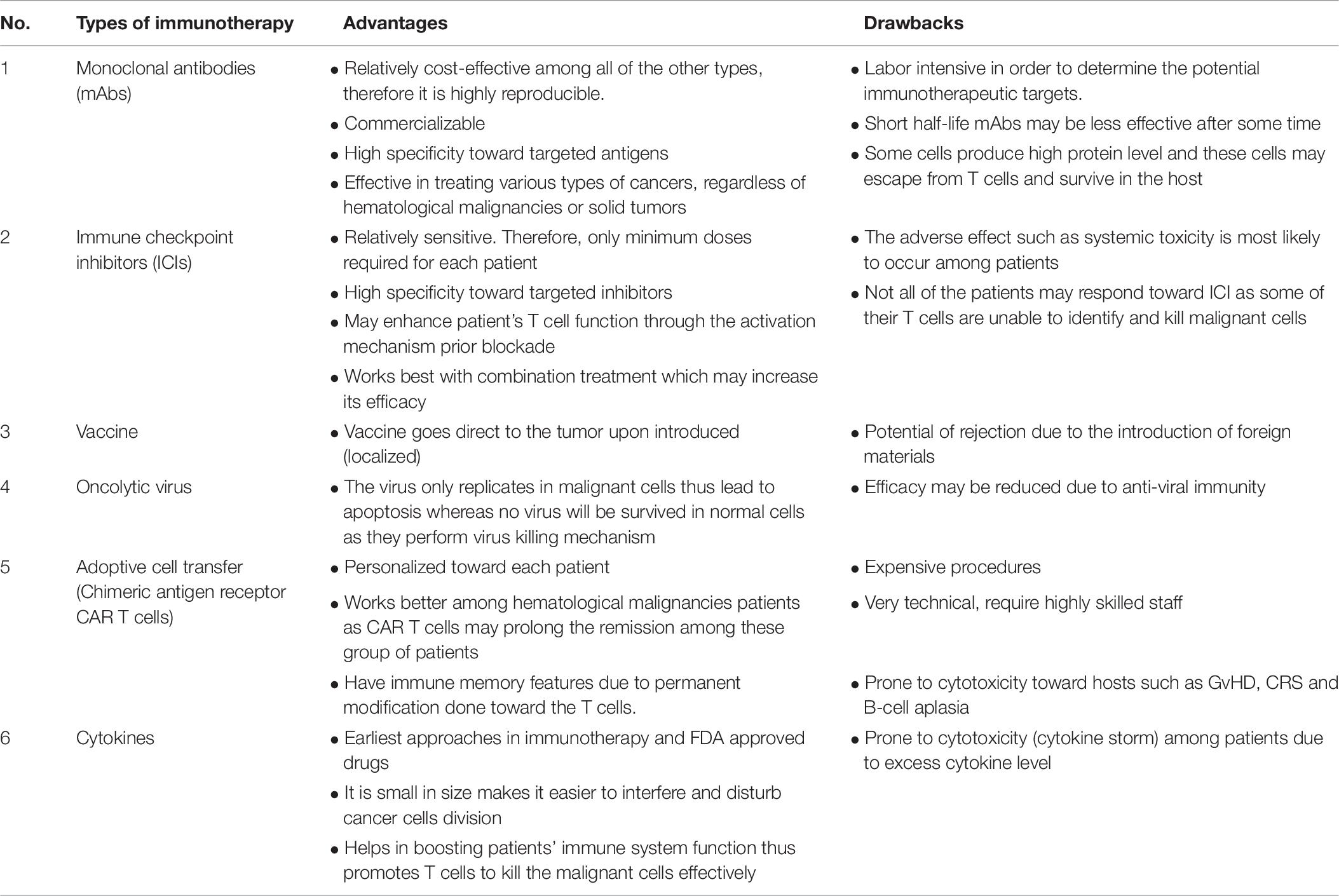
Table 8. The type of novel trends in the immunotherapy with their advantages and drawbacks.
Author Contributions
NJ and NS contributed to the conception and design of the study. NS wrote the first draft of the manuscript, looked for the raw material and summarize the findings, and constructed the diagrams and tables. NJ did the final editing and final input of ideas. Both authors contributed to manuscript revision, read and approved the submitted version.
Funding
This work was supported by the Ministry of Energy, Science, Technology, Environment and Climate Change (MESTECC) Malaysia, under Science Fund, grant number: 02-01-02-SF1329.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Ferlay J, Ervik M, Lam F, Colombet M, Mery L, Piñeros M, et al. Global Cancer Observatory: Cancer Today. (2019). Available online at: https://gco.iarc.fr/today/data/factsheets/cancers/10_8_9-Colorectum-fact-sheet.pdf (accessed September 21, 2019).
3. Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. (2017) 67:177–93. doi: 10.3322/caac.21395
4. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. (2016) 66:683–91. doi: 10.1136/gutjnl-2015-310912
5. Schmoll HJ, Van Cutsem E, Stein A, Valentini V, Glimelius B, Haustermans K, et al. ESMO consensus guidelines for management of patients with colon and rectal cancer. A personalized approach to clinical decision making. Ann Oncol. (2012) 23:2479–516. doi: 10.1093/annonc/mds236
6. Van Cutsem E, Cervantes A, Nordlinger B, Arnold D. Metastatic colorectal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2014) 25(Suppl. 3):iii1–9. doi: 10.1093/annonc/mdu260
7. Yoshino T, Arnold D, Taniguchi H, Pentheroudakis G, Yamazaki K, Xu RH, et al. Pan-Asian adapted ESMO consensus guidelines for the management of patients with metastatic colorectal cancer: A JSMO-ESMO initiative endorsed by CSCO, KACO, MOS, SSO and TOS. Ann Oncol. (2018) 29:44–70. doi: 10.1093/annonc/mdx738
8. Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. (2016) 27:1386–422. doi: 10.1093/annonc/mdw235
9. Bonjer HJ, Deijen CL, Abis GA, Cuesta MA, Van der Pas MHGM, De Lange-de Klerk ESM, et al. A randomized trial of laparoscopic versus open surgery for rectal cancer. N Engl J Med. (2015) 372:1324–32. doi: 10.1056/NEJMoa1414882
10. MacFarlane JK, Ryall RDH, Heald RJ. Mesorectal excision for rectal cancer. Lancet. (1993) 341:457–60. doi: 10.1016/0140-6736(93)90207-W
11. Park SC, Sohn DK, Kim MJ, Chang HJ, Han KS, Hyun JH, et al. Phase II clinical trial to evaluate the efficacy of transanal endoscopic total mesorectal excision for rectal cancer. Dis Colon Rectum. (2018) 61:554–60. doi: 10.1097/DCR.0000000000001058
12. Miller KD, Nogueira L, Mariotto AB, Rowland JH, Yabroff KR, Alfano CM, et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. (2019) 0:1–23. doi: 10.3322/caac.21565
13. Garg MB, Lincz LF, Adler K, Scorgie FE, Ackland SP, Sakoff JA. Predicting 5-fluorouracil toxicity in colorectal cancer patients from peripheral blood cell telomere length: a multivariate analysis. Br J Cancer. (2012) 107:1525–33. doi: 10.1038/bjc.2012.421
14. González-Perera I, Gutiérrez-Nicolás F, Nazco-Casariego GJ, Ramos-Diaz R, Hernández-San GR, Pérez-Pérez JA, et al. 5-fluorouracil toxicity in the treatment of colon cancer associated with the genetic polymorphism 2846 A>G (rs67376798). J Oncol Pharm Pract. (2017) 23:396–8. doi: 10.1177/1078155216647202
15. Ogura A, Konishi T, Cunningham C, Garcia-Aguilar J, Iversen H, Toda S, et al. Neoadjuvant (Chemo)radiotherapy with total mesorectal excision only is not sufficient to prevent lateral local recurrence in enlarged nodes: results of the multicenter lateral node study of patients with low cT3/4 rectal cancer. J Clin Oncol. (2018) 37:33–43. doi: 10.1200/JCO
16. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. (2013) 14:1014–22. doi: 10.1038/ni.2703
17. Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, et al. Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol. (2014) 122:91–128. doi: 10.1016/B978-0-12-800267-4.00003-1
18. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. (2011) 29:235–71. doi: 10.1146/annurev-immunol-031210-101324
19. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. (2017) 10:1–10. doi: 10.1186/s13045-017-0453-8
20. Nixon NA, Blais N, Ernst S, Kollmannsberger C, Bebb G, Butler M, et al. Current landscape of immunotherapy in the treatment of solid tumours, with future opportunities and challenges. Curr Oncol. (2018) 25:e373–84. doi: 10.3747/co.25.3840
21. Fehleisen F. Ueber die züchtung der erysipelkokken auf künstlichem nåhrboden und ihre uebertragbarkeit auf den menschen. Dtsch Med Wochenschr. (1882) 8:553–4. doi: 10.1055/s-0029-1196806
22. Busch W. Aus der sitzung der medicinischen section vom 13 November 1867. Berlin Klin Wochenschr. (1868) 5:137.
23. Coley WB. II. Contribution to the knowledge of sarcoma. Ann Surg. (1891) 14:199–220. doi: 10.1097/00000658-189112000-00015
24. McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. (2006) 26:154–8.
25. Nikolaou M, Nikolaou G, Digklia A, Pontas C, Tsoukalas N, Kyrgias G, et al. Immunotherapy of cancer: Developments and reference points, an unorthodox approach. Integr Cancer Ther. (2019) 18:1–10. doi: 10.1177/1534735419827090
26. Coley WB. The treatment of inoperable sarcoma by bacterial toxins (The mixed toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc R Soc Med. (1910) 3:1–48. doi: 10.1177/003591571000301601
27. Dobosz P, Dzieciątkowski T. The intriguing history of cancer immunotherapy. Front Immunol. (2019) 10:2965. doi: 10.3389/fimmu.2019.02965
28. Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res. (1970) 13:1–27. doi: 10.1159/000386035
29. Burnet M. Immunological factors in the process of carcinogenesis. Br Med Bull. (1964) 20:154–8. doi: 10.1093/oxfordjournals.bmb.a070310
30. Klein G, Sjogren HO, Klein E, Hellstrom KE. Demonstration of resistance against methylcholanthrene-induced sarcomas in the primary autochthonous host. Cancer Res. (1960) 20:1561–72.
31. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc London Ser B Biol Sci. (1957) 147:258–67. doi: 10.1098/rspb.1957.0048
32. Graham JB, Graham RM. The effect of vaccine on cancer patients. Surg Gynecol Obstet. (1959) 109:131–8. doi: 10.1097/00006534-195911000-00016
33. Miller JFAP, Mitchell GF, Weiss NS. Cellular basis of the immunological defects in thymectomized mice. Nature. (1967) 214:992–7. doi: 10.1038/214992a0
34. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice: I. Morphology, quantitation, tissue distribution. J Exp Med. (1973) 137:1142–62. doi: 10.1084/jem.137.5.1142
35. Kiessling R, Klein E, Wigzell H. Natural killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol. (1975) 5:112–7. doi: 10.1002/eji.1830050208
36. Kiessling R, Klein E, Pross H, Wigzell H. Natural killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol. (1975) 5:117–21. doi: 10.1002/eji.1830050209
37. Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. (1975) 256:495–7. doi: 10.1038/256495a0
38. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, et al. A new member of the immunoglobulin superfamily-CTLA-4. Nature. (1988) 328:267–70. doi: 10.1038/328267a0
39. Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood. (1997) 90:2188–95.
40. Rosenberg SA, Lotze MT, Yang JC, Topalian SL, Chang AE, Schwartzentruber DJ, et al. Prospective randomized trial of high-dose interleukin-2 alone or in conjunction with lymphokine-activated killer cells for the treatment of patients with advanced cancer. JNCI J Natl Cancer Inst. (1993) 85:622–32. doi: 10.1093/jnci/85.8.622
41. Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. (2011) 17:3520–6. doi: 10.1158/1078-0432.CCR-10-3126
42. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. (2010) 363:411–22. doi: 10.1056/NEJMoa1001294
43. Andtbacka RHI, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. (2015) 33:2780–8. doi: 10.1200/JCO.2014.58.3377
44. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-Cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
45. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-Cell lymphoma. N Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
46. Guo ZS. The 2018 Nobel Prize in medicine goes to cancer immunotherapy (editorial for BMC cancer). BMC Cancer. (2018) 18:1086. doi: 10.1186/s12885-018-5020-3
47. Galluzzi L, Vacchelli E, Bravo-San Pedro JM, Buque A, Senovilla L, Baracco EE, et al. Classification of current anticancer immunotherapies. Oncotarget. (2014) 5:12472–508. doi: 10.18632/oncotarget.2998
48. Brekke OH, Sandlie I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat Rev Drug Discov. (2003) 2:52–62. doi: 10.1038/nrd984
49. Köhler G, Milstein C. Derivation of specific antibody-producing tissue culture and tumor lines by cell fusion. Eur J Immunol. (1976) 6:511–9. doi: 10.1002/eji.1830060713
50. Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. (1985) 228:1315–7. doi: 10.1126/science.4001944
51. Kurosawa K, Lin W, Ohta K. Chimeric antibodies. Methods Mol Biol. (2014) 1060:139–48. doi: 10.1007/978-1-62703-586-6_8
52. Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, et al. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. (2020) 27:1–30. doi: 10.1186/s12929-019-0592-z
53. Kettleborough CA, Saldanha J, Heath VJ, Morrison CJ, Bendig MM. Humanization of a mouse monoclonal antibody by CDR-grafting: the importance of framework residues on loop conformation. Protein Eng. (1991) 4:773–83. doi: 10.1093/protein/4.7.773
54. Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJT. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov. (2010) 9:325–38. doi: 10.1038/nrd3003
55. Lipman NS, Jackson LR, Trudel LJ, Weis-Garcia F. Monoclonal versus polyclonal antibodies: distinguishing characteristics, applications, and information resources. ILAR Journal. (2005) 46:258–68. doi: 10.1093/ilar.46.3.258
56. Bubeník J, Barešová M, Viklický V, Jakoubková J, Sainerová H, Donner J. Established cell line of urinary bladder carcinoma (T24) containing tumour-specific antigen. Int J Cancer. (1973) 11:765–73. doi: 10.1002/ijc.2910110327
57. Hollinshead A, Elias EG, Arlen M, Buda B, Mosley M, Scherrer J. Specific active immunotherapy in patients with adenocarcinoma of the colon utilizing tumor-associated antigens (TAA). A phase I clinical trial. Cancer. (1985) 56:480–9. doi: 10.1002/1097-0142(19850801)56:33.0.co;2-2
58. Dickinson JP, Caspary EA, Field EJ. A common tumour specific antigen. Br J Cancer. (1973) 27:99–105. doi: 10.1038/bjc.1973.13
59. Speetjens FM, Kuppen PJK, Welters MJP, Essahsah F, Van Den Brink AMEGV, Lantrua MGK, et al. Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clin Cancer Res. (2009) 15:1086–95. doi: 10.1158/1078-0432.CCR-08-2227
60. Alatrash G, Crain AK, Molldrem JJ. Tumor-Associated Antigens. Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation. Amsterdam: Elsevier (2019). p. 107–25. doi: 10.1016/B978-0-12-812630-1.00007-4
61. Cloosen S, Arnold J, Thio M, Bos GMJ, Kyewski B, Germeraad WTV. Expression of tumor-associated differentiation antigens, MUC1 glycoforms and CEA, in human thymic epithelial cells: implications for self-tolerance and tumor therapy. Cancer Res. (2007) 67:3919–26. doi: 10.1158/0008-5472.CAN-06-2112
62. Even-Desrumeaux K, Baty D, Chames P. State of the art in tumor antigen and biomarker discovery. Cancers (Basel). (2011) 3:2554–96. doi: 10.3390/cancers3022554
63. Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev. (2002) 188:22–32. doi: 10.1034/j.1600-065X.2002.18803.x
64. Kerkar SP, Wang ZF, Lasota J, Park T, Patel K, Groh E, et al. MAGE-A is more highly expressed than NY-ESO-1 in a systematic immunohistochemical analysis of 3668 cases. J Immunother. (2016) 39:181–7. doi: 10.1097/CJI.0000000000000119
65. Yanagi T, Nagai K, Shimizu H, Matsuzawa SI. Melanoma antigen A12 regulates cell cycle via tumor suppressor p21 expression. Oncotarget. (2017) 8:68448–59. doi: 10.18632/oncotarget.19497
66. Santha Kumara HMC, Grieco MJ, Caballero OL, Su T, Ahmed A, Ritter E, et al. MAGE-A3 is highly expressed in a subset of colorectal cancer patients. Cancer Immun. (2012) 12:1–9.
67. Nicolazzo C, Raimondi C, Francescangeli F, Ceccarelli S, Trenta P, Magri V, et al. EpCAM-expressing circulating tumor cells in colorectal cancer. Int J Biol Markers. (2017) 32:e415–20. doi: 10.5301/ijbm.5000284
68. Starzynska T, Marsh PJ, Schofield PF, Roberts SA, Myers KA, Stern PL. Prognostic significance of 5T4 oncofetal antigen expression in colorectal carcinoma. Br J Cancer. (1994) 69:899–902. doi: 10.1038/bjc.1994.173
69. Besneux M, Greenshields-Watson A, Scurr MJ, MacLachlan BJ, Christian A, Davies MM, et al. The nature of the human T cell response to the cancer antigen 5T4 is determined by the balance of regulatory and inflammatory T cells of the same antigen-specificity: Implications for vaccine design. Cancer Immunol Immunother. (2019) 68:247–56. doi: 10.1007/s00262-018-2266-1
70. Wang W, Li Y, Zhang X, Jing J, Zhao X, Wang Y, et al. Evaluating the significance of expression of CEA mRNA and levels of CEA and its related proteins in colorectal cancer patients. J Surg Oncol. (2014) 109:440–4. doi: 10.1002/jso.23503
71. Bright RK, Bright JD, Byrne JA. Overexpressed oncogenic tumor-self antigens. Hum Vaccines Immunother. (2014) 10:3297–305. doi: 10.4161/hv.29475
72. Liu J, Zhou Q, Xu J, Wang J, Zhang Y. Detection of EGFR expression in patients with colorectal cancer and the therapeutic effect of cetuximab. J BUON. (2016) 21:95–100.
73. Geng W, Liang W, Fan Y, Ye Z, Zhang L. Overexpression of CCDC34 in colorectal cancer and its involvement in tumor growth, apoptosis and invasion. Mol Med Rep. (2018) 17:465–73. doi: 10.3892/mmr.2017.7860
74. Wang ZK, Cheng ZW, Chen SJ, Zhu XG, Gu YP, Yang XD, et al. Aberrant expression of Rab1A and its prognostic significance in human colorectal cancer. Eur Rev Med Pharmacol Sci. (2018) 22:4509–17. doi: 10.26355/eurrev_201807_15505
75. Michaud HA, Eliaou JF, Lafont V, Bonnefoy N, Gros L. Tumor antigen-targeting monoclonal antibody-based immunotherapy: orchestrating combined strategies for the development of long-term antitumor immunity. Oncoimmunology. (2014) 3:e955684. doi: 10.4161/21624011.2014.955684
76. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. (2012) 12:278–87. doi: 10.1038/nrc3236
77. Biopharma LLC.BIOPHARMA®: Biopharmaceutical Products in the U.S. and European Markets. (2019). Available online at: www.biopharma.com (accessed June 26, 2019).
78. Food and Drug Administration.Resources for Information on Approved Drugs. (2019). Available online at: www.fda.gov/drugs/informationondrugs/approveddrugs (accessed June 28, 2019).
79. European Medicines Agency. (2019). Available online at: www.ema.europa.eu/ema (accessed June 28, 2019).
80. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. (2018) 32:1267–84. doi: 10.1101/gad.314617.118
81. Pardoll D. Cancer and immune system: basic concepts and targets for intervention. Semin Oncol. (2015) 42:523–38. doi: 10.1053/j.seminoncol.2015.05.003
82. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. (2018) 8:86. doi: 10.3389/fonc.2018.00086
83. Joller N, Kuchroo VK. Tim-3, Lag-3, and TIGIT. Curr Top Microbiol Immunol. (2017) 410:127–56. doi: 10.1007/82_2017_62
84. Kisielow M, Kisielow J, Capoferri-Sollami G, Karjalainen K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur J Immunol. (2005) 35:2081–8. doi: 10.1002/eji.200526090
85. Kong Y, Zhu L, Schell TD, Zhang J, Claxton DF, Ehmann WC, et al. T-cell immunoglobulin and ITIM domain (TIGIT) associates with CD8+ T-cell exhaustion and poor clinical outcome in AML patients. Clin Cancer Res. (2016) 22:3057–66. doi: 10.1158/1078-0432.CCR-15-2626
86. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. (2002) 415:536–41. doi: 10.1038/415536a
87. Menter DG, Davis JS, Broom BM, Overman MJ, Morris J, Kopetz S. Back to the colorectal cancer consensus molecular subtype future. Curr Gastroenterol Rep. (2019) 21:1–12. doi: 10.1007/s11894-019-0674-9
88. Rodriguez-Salas N, Dominguez G, Barderas R, Mendiola M, García-Albéniz X, Maurel J, et al. Clinical relevance of colorectal cancer molecular subtypes. Crit Rev Oncol Hematol. (2017) 109:9–19. doi: 10.1016/j.critrevonc.2016.11.007
89. Shemirani AI, Haghighi MM, Zadeh SM, Fatemi SR, Taleghani MY, Zali N, et al. Simplified MSI marker panel for diagnosis of colorectal cancer. Asian Pac J Cancer Prev. (2011) 12:2101–4. doi: 10.17179/excli2018-1455
90. Kawakami H, Zaanan A, Sinicrope FA. Microsatellite instability testing and its role in the management of colorectal cancer. Curr Treat Options Oncol. (2015) 16:1–15. doi: 10.1007/s11864-015-0348-2
91. Nojadeh JN, Behrouz Sharif S, Sakhinia E. Microsatellite instability in colorectal cancer. EXCLI J. (2018) 17:159–68. doi: 10.17179/excli2017-948
92. Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. (2016) 15:857–65. doi: 10.1016/j.celrep.2016.03.075
93. Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. (2018) 36:773–9. doi: 10.1200/JCO.2017.76.9901
94. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. (1995) 182:459–65. doi: 10.1084/jem.182.2.459
95. Sanchez-Lockhart M, Rojas AV, Fettis MM, Bauserman R, Higa TR, Miao H, et al. T cell receptor signaling can directly enhance the avidity of CD28 ligand binding. PLoS One (2014) 9:e89263. doi: 10.1371/journal.pone.0089263
97. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways similarities, differences, and implications of their inhibition. Am J Clin Oncol Cancer Clin Trials. (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
98. Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. (2001) 19:565–94. doi: 10.1146/annurev.immunol.19.1.565
99. Egen JG, Kuhns MS, Allison JP. CTLA-4: New insights into its biological function and use in tumor immunotherapy. Nat Immunol. (2002) 3:611–8. doi: 10.1038/ni0702-611
100. Intlekofer AM, Thompson CB. At the bench: Preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J Leukoc Biol. (2013) 94:25–39. doi: 10.1189/jlb.1212621
101. Jørgensen N, Persson G, Hviid TVF. The tolerogenic function of regulatory T cells in pregnancy and cancer. Front Immunol. (2019) 10:1–21. doi: 10.3389/fimmu.2019.00911
102. Liakou CI, Kamat A, Tang DN, Chen H, Sun J, Troncoso P, et al. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci USA. (2008) 105:14987–92. doi: 10.1073/pnas.0806075105
103. Lee HT, Lee SH, Heo YS. Molecular interactions of antibody drugs targeting PD-1, PD-L1, and CTLA-4 in immuno-oncology. Molecules. (2019) 4:1–16. doi: 10.3390/molecules24061190
104. Pernot S, Terme M, Voron T, Colussi O, Marcheteau E, Tartour E, et al. Colorectal cancer and immunity: what we know and perspectives. World J Gastroenterol. (2014) 20:3738–50. doi: 10.3748/wjg.v20.i14.3738
105. Ishida T, Ishii T, Inagaki A, Yano H, Komatsu H, Iida S, et al. Specific recruitment of CC chemokine receptor 4-positive regulatory T cells in Hodgkin lymphoma fosters immune privilege. Cancer Res. (2006) 66:5716–22. doi: 10.1158/0008-5472.CAN-06-0261
106. Shinohara T, Taniwaki M, Ishida Y, Kawaichi M, Honjo T. Structure and chromosomal localization of the human PD-1 gene (PDCD1). Genomics. (1994) 23:704–6. doi: 10.1006/geno.1994.1562
107. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. (1996) 8:765–72. doi: 10.1093/intimm/8.5.765
108. Kitano A, Ono M, Yoshida M, Noguchi E, Shimomura A, Shimoi T, et al. Tumour-infiltrating lymphocytes are correlated with higher expression levels of PD-1 and PD-L1 in early breast cancer. ESMO Open. (2017) 2:e000150. doi: 10.1136/esmoopen-2016-000150
109. Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, et al. Expression of programmed death 1 ligands by murine T Cells and APC. J Immunol. (2002) 169:5538–45. doi: 10.4049/jimmunol.169.10.5538
110. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
111. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. (2001) 2:261–8. doi: 10.1038/85330
112. Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, et al. IFN-α directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. (2011) 186:2772–9. doi: 10.4049/jimmunol.1003208
113. Shindo Y, Yoshimura K, Kuramasu A, Watanabe Y, Ito H, Kondo T, et al. Combination immunotherapy with 4-1BB activation and PD-1 blockade enhances antitumor efficacy in a mouse model of subcutaneous tumor. Anticancer Res. (2015) 35:129–36.
114. Dong Y, Sun Q, Zhang X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget. (2017) 8:2171–86. doi: 10.18632/oncotarget.13895
115. Henick BS, Herbst RS, Goldberg SB. The PD-1 pathway as a therapeutic target to overcome immune escape mechanisms in cancer. Expert Opin Ther Targets. (2014) 18:1407–20. doi: 10.1517/14728222.2014.955794
116. Gong J, Chehrazi-Raffle A, Reddi S, Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. (2018) 6:1–18. doi: 10.1186/s40425-018-0316-z
117. Patnaik A, Kang SP, Rasco D, Papadopoulos KP, Elassaiss-Schaap J, Beeram M, et al. Phase I study of pembrolizumab (MK-3475; Anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res. (2015) 21:4286–93. doi: 10.1158/1078-0432.CCR-14-2607
118. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csöszi T, Fülöp A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. (2016) 375:1823–33. doi: 10.1056/NEJMoa1606774
119. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
120. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. (2017) 18:1182–91. doi: 10.1016/S1470-2045(17)30422-9
121. Tran KQ, Zhou J, Durflinger KH, Langhan MM, Shelton TE, Wunderlich JR, et al. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J Immunother. (2008) 31:742–51. doi: 10.1097/CJI.0b013e31818403d5
122. Benmebarek MR, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci. (2019) 20:1283. doi: 10.3390/ijms20061283
123. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N Engl J Med. (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
124. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. (2006) 314:126–9. doi: 10.1126/science.1129003
125. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. (2006) 12(20 Pt 1):6106–15. doi: 10.1158/1078-0432.CCR-06-1183
126. Lamers CHJ, Willemsen R, Van Elzakker P, Van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, et al. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood. (2011) 117:72–82. doi: 10.1182/blood-2010-07-294520
127. Ramos CA, Dotti G. Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opin Biol Ther. (2011) 11:855–73. doi: 10.1517/14712598.2011.573476
128. Wang X, Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. (2016) 3:1–7. doi: 10.1038/mto.2016.15
129. Bishop MR, Maziarz RT, Waller EK, Jäger U, Westin JR, McGuirk JP, et al. Tisagenlecleucel in relapsed/refractory diffuse large B-cell lymphoma patients without measurable disease at infusion. Blood Adv. (2019) 3:2230–6. doi: 10.1182/bloodadvances.2019000151
130. Pituch KC, Miska J, Krenciute G, Panek WK, Li G, Rodriguez-Cruz T, et al. Adoptive transfer of IL13Rα2-specific chimeric antigen receptor T cells creates a pro-inflammatory environment in glioblastoma. Mol Ther. (2018) 26:986–95. doi: 10.1016/j.ymthe.2018.02.001
131. Miao H, Choi BD, Suryadevara CM, Sanchez-Perez L, Yang S, De Leon G, et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS One. (2014) 9:e94281. doi: 10.1371/journal.pone.0094281
132. Lázaro-Gorines R, Ruiz-de-la-Herrán J, Navarro R, Sanz L, Álvarez-Vallina L, Martínez-del-Pozo A, et al. A novel carcinoembryonic antigen (CEA)-targeted trimeric immunotoxin shows significantly enhanced antitumor activity in human colorectal cancer xenografts. Sci Rep. (2019) 9:11680. doi: 10.1038/s41598-019-48285-z
133. Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature. (2017) 545:423–31. doi: 10.1038/nature22395
134. Tang XY, Sun Y, Zhang A, Hu GL, Cao WW, Dan H, et al. Third-generation CD28/4-1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: a non-randomised, open-label phase I trial protocol. BMJ Open. (2016) 6:e013904. doi: 10.1136/bmjopen-2016-013904
135. Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. (2016) 128:1688–700. doi: 10.1182/blood-2016-04-711903
136. Perica K, Varela JC, Oelke M, Schneck J. Adoptive T cell immunotherapy for cancer. Rambam Maimonides Med J. (2015) 6:e0004. doi: 10.5041/RMMJ.10179
137. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. (2013) 36:133–51. doi: 10.1097/CJI.0b013e3182829903
138. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. (2014) 124:188–95. doi: 10.1182/blood-2014-05-552729
139. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
140. Pehlivan KC, Duncan BB, Lee DW. CAR-T cell therapy for acute lymphoblastic leukemia: Transforming the treatment of relapsed and refractory disease. Curr Hematol Malig Rep. (2018) 13:396–406. doi: 10.1007/s11899-018-0470-x
141. Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 Results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. (2017) 25:285–95. doi: 10.1016/j.ymthe.2016.10.020
142. Magee MS, Abraham TS, Baybutt TR, FlickingerJr JC, Ridge NA, Marszalowicz GP, et al. Human GUCY2C-targeted chimeric antigen receptor (CAR)-expressing T cells eliminate colorectal cancer metastases. Cancer Immunol Res. (2018) 6:509–16. doi: 10.1158/2326-6066.CIR-16-0362
143. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DAN, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. (2011) 19:620–6. doi: 10.1038/mt.2010.272
144. Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. (2017) 5:1–14. doi: 10.1186/s40425-017-0222-9
145. Zhang BL, Li D, Gong YL, Huang Y, Qin DY, Jiang L, et al. Preclinical evaluation of chimeric antigen receptor–modified T cells specific to epithelial cell adhesion molecule for treating colorectal cancer. Hum Gene Ther. (2019) 30:402–12. doi: 10.1089/hum.2018.229
146. Deng X, Gao F, Li N, Li Q, Zhou Y, Yang T, et al. Antitumor activity of NKG2D CAR-T cells against human colorectal cancer cells in vitro and in vivo. Am J Cancer Res. (2019) 9:945–58.
147. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. (2016) 3:1–7. doi: 10.1038/mto.2016.11
148. Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. (2019) 10:128. doi: 10.3389/fimmu.2019.00128
149. Abbasi AM, Chester KA, MacPherson AJ, Boxer GM, Begent RH, Malcolm AD. Localization of CEA messenger RNA by in situ hybridization in normal colonic mucosa and colorectal adenocarcinomas. J Pathol. (1992) 168:405–11. doi: 10.1002/path.1711680411
150. Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, et al. Phase I escalating-dose trial of CAR-T therapy targeting CEA + metastatic colorectal cancers. Mol Ther. (2017) 25:1248–58. doi: 10.1016/j.ymthe.2017.03.010
151. Chi X, Yang P, Zhang E, Jieyi G, Hui X, Li M, et al. Significantly increased anti−tumor activity of carcinoembryonic antigen−specific chimeric antigen receptor T cells in combination with recombinant human IL−12. Cancer Med. (2019) 8:4753–65. doi: 10.1002/cam4.2361
152. Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. (2016) 6:80–95. doi: 10.1158/2159-8290.CD-15-0224
153. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
154. Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM, et al. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther. (2002) 13:1971–80. doi: 10.1089/10430340260355374
155. Crespo J, Sun H, Welling TH, Tian Z, Zou WT. cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. (2013) 25:214–21. doi: 10.1016/j.coi.2012.12.003
156. Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, et al. Current progress in CAR-T cell therapy for solid tumors. Int J Biol Sci. (2019) 15:2548–60. doi: 10.7150/ijbs.34213
157. Xu Y, Yang Z, Horan LH, Zhang P, Liu L, Zimdahl B, et al. A novel antibody-TCR (AbTCR) platform combines Fab-based antigen recognition with gamma/delta-TCR signaling to facilitate T-cell cytotoxicity with low cytokine release. Cell Discov. (2018) 4:1–13. doi: 10.1038/s41421-018-0066-6
158. Barrett DM, Grupp SA, June CH. Chimeric antigen receptor– and TCR-Modified T cells enter main street and wall street. J Immunol. (2015) 195:755–61. doi: 10.4049/jimmunol.1500751
159. Garber K. Driving T-cell immunotherapy to solid tumors. Nat Biotechnol. (2018) 36:215–9. doi: 10.1038/nbt.4090
160. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. (2009) 114:535–46. doi: 10.1182/blood-2009-03-211714
161. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. (2011) 29:917–24. doi: 10.1200/JCO.2010.32.2537
162. Stadtmauer EA, Faitg TH, Lowther DE, Badros AZ, Chagin K, Dengel K, et al. Long−term safety and activity of NY-ESO−1 SPEAR T cells after autologous stem cell transplant for myeloma. Blood Adv. (2019) 3:2022–34. doi: 10.1182/bloodadvances.2019000194
163. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. (2013) 122:863–71. doi: 10.1182/blood-2013-03-490565
164. Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, et al. Adoptive transfer of MAGE-A4 T-cell receptor gene-transduced lymphocytes in patients with recurrent esophageal cancer. Clin Cancer Res. (2015) 21:2268–77. doi: 10.1158/1078-0432.CCR-14-1559
165. Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. (2015) 21:914–21. doi: 10.1038/nm.3910
166. ClinicalTrials.gov. (2020). Available online at: https://clinicaltrials.gov/ct2/home (accessed May 31, 2020).
167. Zhao L, Cao YJ. Engineered T cell therapy for cancer in the clinic. Front Immunol. (2019) 10:2250. doi: 10.3389/fimmu.2019.02250
168. Thomas S, Prendergast GC. Cancer vaccines: a brief overview. In: S Thomas editor. Methods in Molecular Biology. (Vol. 1403), New York, NY: Humana Press Inc (2016). p. 755–61. doi: 10.1007/978-1-4939-3387-7_43
169. Lollini PL, Cavallo F, Nanni P, Quaglino E. The promise of preventive cancer vaccines. Vaccines. (2015) 3:467–89. doi: 10.3390/vaccines3020467
170. Schlom J, Hodge JW, Palena C, Tsang KY, Jochems C, Greiner JW. Therapeutic cancer vaccines. Adv Cancer Res. (2014) 121:67–124. doi: 10.1016/B978-0-12-800249-0.00002-0
171. DeMaria PJ, Bilusic M. Cancer vaccines. Hematol Oncol Clin North Am. (2019) 33:199–214. doi: 10.1016/j.hoc.2018.12.001
172. Barreto L, Csizer Z, Sparkes JD. Evaluation of clinical data in bladder cancer immunotherapy with Connaught BCG (ImmuCyst). Dev Biol Stand. (1992) 77:229–31.
173. Lamm DL, Blumenstein BA, Crawford, Montie JE, Scardino P, Grossman HB, et al. A randomized trial of intravesical doxorubicin and immunotherapy with Bacille Calmette–Guérin for transitional-cell carcinoma of the bladder. N Engl J Med. (1991) 325:1205–9. doi: 10.1056/NEJM199110243251703
174. Rentsch CA, Birkhäuser FD, Biot C, Gsponer JR, Bisiaux A, Wetterauer C, et al. Bacillus Calmette-Guérin strain differences have an impact on clinical outcome in bladder cancer immunotherapy. Eur Urol. (2014) 66:677–88. doi: 10.1016/j.eururo.2014.02.061
175. Witjes JA, Dalbagni G, Karnes RJ, Shariat S, Joniau S, Palou J, et al. The efficacy of BCG TICE and BCG Connaught in a cohort of 2,099 patients with T1G3 non–muscle-invasive bladder cancer. Urol Oncol Semin Orig Investig. (2016) 34:484.e19–e25. doi: 10.1016/j.urolonc.2016.05.033
176. Graddis TJ, McMahan CJ, Tamman J, Page KJ, Trager JB. Prostatic acid phosphatase expression in human tissues. Int J Clin Exp Pathol. (2011) 4:295–306.
177. Hammerstrom AE, Cauley DH, Atkinson BJ, Sharma P. Cancer immunotherapy: sipuleucel-T and beyond. Pharmacotherapy. (2011) 31:813–28. doi: 10.1592/phco.31.8.813
178. Dores GM, Bryant-Genevier M, Perez-Vilar S. Adverse events associated with the use of sipuleucel-T reported to the US Food and Drug Administration’s adverse event reporting system, 2010-2017. JAMA Netw Open. (2019) 2:e199249. doi: 10.1001/jamanetworkopen.2019.9249
179. Raman SS, Hecht JR, Chan E. Talimogene laherparepvec: review of its mechanism of action and clinical efficacy and safety. Immunotherapy. (2019) 11:705–23. doi: 10.2217/imt-2019-0033
180. Conry RM, Westbrook B, McKee S, Norwood TG. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum Vaccines Immunother. (2018) 14:839–46. doi: 10.1080/21645515.2017.1412896
181. Chesney J, Puzanov I, Collichio F, Singh P, Milhem MM, Glaspy J, et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. (2018) 36:1658–67. doi: 10.1200/JCO.2017.73.7379
182. Chesney J, Puzanov I, Collichio F, Milhem MM, Hauschild A, Chen L, et al. Patterns of response with talimogene laherparepvec in combination with ipilimumab or ipilimumab alone in metastatic unresectable melanoma. Br J Cancer. (2019) 121:417–20. doi: 10.1038/s41416-019-0530-6
183. Bilusic M, Heery CR, Arlen PM, Rauckhorst M, Apelian D, Tsang KY, et al. Phase I trial of a recombinant yeast-CEA vaccine (GI-6207) in adults with metastatic CEA-expressing carcinoma. Cancer Immunol Immunother. (2014) 63:225–34. doi: 10.1007/s00262-013-1505-8
184. Gulley JL, Arlen PM, Tsang KY, Yokokawa J, Palena C, Poole DJ, et al. Pilot study of vaccination with recombinant CEA-MUC-1-tricom pox viral-based vaccines in patients with metastatic carcinoma. Clin Cancer Res. (2008) 14:3060–9. doi: 10.1158/1078-0432.CCR-08-0126
185. Kaufman HL, Lenz HJ, Marshall J, Singh D, Garett C, Cripps C, et al. Combination chemotherapy and ALVAC-CEA/B7.1 vaccine in patients with metastatic colorectal cancer. Clin Cancer Res. (2008) 14:4843–9. doi: 10.1158/1078-0432.CCR-08-0276
186. Zhan W, Zhang Z, Zhang Y, Ma J, Wu T, Gu Y, et al. Prognostic value of MAGE-A9 expression in patients with colorectal cancer. Clin Res Hepatol Gastroenterol. (2016) 40:239–45. doi: 10.1016/J.CLINRE.2015.08.005
187. Zhang QM, He SJ, Shen N, Luo B, Fan R, Fu J, et al. Overexpression of MAGE-D4 in colorectal cancer is a potentially prognostic biomarker and immunotherapy target. Int J Clin Exp Pathol. (2014) 7:3918–27.
188. Morse MA, Niedzwiecki D, Marshall JL, Garrett C, Chang DZ, Aklilu M, et al. A randomized phase II study of immunization with dendritic cells modified with poxvectors encoding CEA and MUC1 compared with the same poxvectors plus GM-CSF for resected metastatic colorectal cancer. Ann Surg. (2013) 258:879–86. doi: 10.1097/SLA.0b013e318292919e
189. Tang CK, Katsara M, Apostolopoulos V. Strategies used for MUC1 immunotherapy: human clinical studies. Expert Rev Vaccines. (2008) 7:963–75. doi: 10.1586/14760584.7.7.963
190. DiPaola RS, Plante M, Kaufman H, Petrylak DP, Israeli R, Lattime E, et al. A phase I trial of pox PSA vaccines (PROSTVAC®-VF) with B7-1, ICAM-1, and LFA-3 co-stimulatory molecules (TRICOMTM) in patients with prostate cancer. J Transl Med. (2006) 4:1–5. doi: 10.1186/1479-5876-4-1
191. Hodge JW, McLaughlin JP, Kantor JA, Schlom J. Diversified prime and boost protocols using recombinant vaccinia virus and recombinant non-replicating avian pox virus to enhance T-cell immunity and antitumor responses. Vaccine. (1997) 15:759–68. doi: 10.1016/s0264-410x(96)00238-1
192. Saha A, Chatterjee SK, Foon KA, Celis E, Bhattacharya-Chatterjee M. Therapy of established tumors in a novel murine model transgenic for human carcinoembryonic antigen and HLA-A2 with a combination of anti-idiotype vaccine and CTL peptides of carcinoembryonic antigen. Cancer Res. (2007) 67:2881–92. doi: 10.1158/0008-5472.CAN-06-3045
193. Hensel JA, Khattar V, Ashton R, Ponnazhagan S. Recombinant AAV-CEA tumor vaccine in combination with an immune adjuvant breaks tolerance and provides protective immunity. Mol Ther Oncolytics. (2019) 12:41–8. doi: 10.1016/j.omto.2018.12.004
194. Lamm DL. BCG immunotherapy for transitional-cell carcinoma in situ of the bladder. Oncology (Williston Park). (1995) 9:947–55.
195. Burch PA, Breen JK, Buckner JC, Gastineau DA, Kaur JA, Laus RL, et al. Priming tissue-specific cellular immunity in a phase I trial of autologous dendritic cells for prostate cancer. Clin Cancer Res. (2000) 6:2175–82.
196. Burch PA, Croghan GA, Gastineau DA, Jones LA, Kaur JS, Kylstra JW, et al. Immunotherapy (APC8015, provenge®) targeting prostatic acid phosphatase can induce durable remission of metastatic androgen-independent prostate cancer: a phase 2 trial. Prostate. (2004) 60:197–204. doi: 10.1002/pros.20040
197. Small EJ, Schellhammer PF, Higano CS, Redfern CH, Nemunaitis JJ, Valone FH, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. (2006) 24:3089–94. doi: 10.1200/JCO.2005.04.5252
198. Beer TM, Bernstein GT, Corman JM, Glode LM, Hall SJ, Poll WL, et al. Randomized trial of autologous cellular immunotherapy with sipuleucel-T in androgen-dependent prostate cancer. Clin Cancer Res. (2011) 17:4558–67. doi: 10.1158/1078-0432.CCR-10-3223
199. Hu JCC, Coffin RS, Davis CJ, Graham NJ, Groves N, Guest PJ, et al. A phase I study of OncoVEX GM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. (2006) 12:6737–47. doi: 10.1158/1078-0432.CCR-06-0759
200. Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol. (2009) 27:5763–71. doi: 10.1200/JCO.2009.24.3675
201. Kantoff PW, Schuetz TJ, Blumenstein BA, Michael GL, Bilhartz DL, Wyand M, et al. Overall survival analysis of a phase II randomized controlled trial of a poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. (2010) 28:1099–105. doi: 10.1200/JCO.2009.25.0597
202. Kantoff PW, Gulley JL, Pico-Navarro C. Revised overall survival analysis of a phase II, randomized, double-blind, controlled study of PROSTVAC in men with metastatic castration-resistant prostate cancer. J Clin Oncol. (2017) 35:124–5. doi: 10.1200/JCO.2016.69.7748
203. Gulley JL, Borre M, Vogelzang NJ, Ng S, Agarwal N, Parker CC, et al. Phase III trial of PROSTVAC in asymptomatic or minimally symptomatic metastatic castration-resistant prostate cancer. J Clin Oncol. (2019) 37:1051–61. doi: 10.1200/JCO.18.02031
204. Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Pérez-Gracia JL, et al. Cytokines in clinical cancer immunotherapy. Br J Cancer. (2019) 120:6–15. doi: 10.1038/s41416-018-0328-y
205. Rusch T, Bayry J, Werner J, Shevchenko I, Bazhin AV. Immunotherapy as an option for cancer treatment. Arch Immunol Ther Exp (Warsz). (2018) 66:89–96. doi: 10.1007/s00005-017-0491-5
206. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. (2018) 9:7204–18. doi: 10.18632/oncotarget.23208
207. Ferreira VL, Borba HHL, Bonetti AF, Leonart LP, Pontarolo R. Cytokines and Interferons: Types and Functions. Autoantibodies and Cytokines. London: IntechOpen (2019). doi: 10.5772/intechopen.74550
208. García-Martínez E, Smith M, Buqué A, Aranda F, Peña FA, Ivars A, et al. Trial watch: immunostimulation with recombinant cytokines for cancer therapy. Oncoimmunology. (2018) 7:e1433982. doi: 10.1080/2162402X.2018.1433982
209. McDermott DF, Cheng SC, Signoretti S, Margolin KA, Clark JI, Sosman JA, et al. The high-dose aldesleukin “select” trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res. (2015) 21:561–8. doi: 10.1158/1078-0432.CCR-14-1520
210. Eigentler TK, Gutzmer R, Hauschild A, Heinzerling L, Schadendorf D, Nashan D, et al. Adjuvant treatment with pegylated interferon α-2a versus low-dose interferon α-2a in patients with high-risk melanoma: a randomized phase III DeCOG trial. Ann Oncol. (2016) 27:1625–32. doi: 10.1093/annonc/mdw225
211. Habermann TM, Andersen JW, Cassileth PA, Bennett JM, Oken MM. Sequential administration of recombinant interferon alpha and deoxycoformycin in the treatment of hairy cell leukaemia. Br J Haematol. (1992) 80:466–71. doi: 10.1111/j.1365-2141.1992.tb04559.x
212. Lipton JH, Khoroshko N, Golenkov A, Abdulkadyrov K, Nair K, Raghunadharao D, et al. Phase II, randomized, multicenter, comparative study of peginterferon–α–2a (40 kD) (Pegasys®) versus interferon α-2a (Roferon®-A) in patients with treatment-naïve, chronic-phase chronic myelogenous leukemia. Leuk Lymphoma. (2007) 48:497–505. doi: 10.1080/10428190601175393
213. Biesma B, Willemse PHB, Mulder NH, Verschueren RCJ, Kema IP, de Bruijn HWA, et al. Recombinant interferon alpha-2b in patients with metastatic apudomas: effect on tumours and tumour markers. Br J Cancer. (1992) 66:850–5. doi: 10.1038/bjc.1992.372
214. Flanigan RC, Salmon SE, Blumenstein BA, Bearman SI, Roy V, McGrath PC, et al. Nephrectomy followed by interferon alfa-2b compared with interferon alfa-2b alone for metastatic renal-cell cancer. N Engl J Med. (2001) 345:1655–9. doi: 10.1056/NEJMoa003013
215. Michallet M, Maloisel F, Delain M, Hellmann A, Rosas A, Silver RT, et al. Pegylated recombinant interferon alpha-2b vs recombinant interferon alpha-2b for the initial treatment of chronic-phase chronic myelogenous leukemia: A phase III study. Leukemia. (2004) 18:309–15. doi: 10.1038/sj.leu.2403217
Keywords: colorectal carcinoma, immunotherapy, FDA, T-cells, antibodies, cytokines, treatments
Citation: Johdi NA and Sukor NF (2020) Colorectal Cancer Immunotherapy: Options and Strategies. Front. Immunol. 11:1624. doi: 10.3389/fimmu.2020.01624
Received: 10 January 2020; Accepted: 17 June 2020;
Published: 18 September 2020.
Edited by:
Alexandr Bazhin, LMU Munich University Hospital, GermanyReviewed by:
Cristina Maccalli, Sidra Medicine, QatarDouglas Clayton Palmer, National Cancer Institute (NCI), United States
Copyright © 2020 Johdi and Sukor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nor Adzimah Johdi, YWR6aW1haEBwcHVrbS51a20uZWR1Lm15