 Ernest Y. Lee1,2
Ernest Y. Lee1,2 Yashes Srinivasan1
Yashes Srinivasan1 Jaime de Anda1
Jaime de Anda1 Lauren K. Nicastro3
Lauren K. Nicastro3 Çagla Tükel3
Çagla Tükel3 Gerard C. L. Wong1,4,5*
Gerard C. L. Wong1,4,5*- 1Department of Bioengineering, University of California, Los Angeles, Los Angeles, CA, United States
- 2UCLA-Caltech Medical Scientist Training Program, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, United States
- 3Department of Microbiology and Immunology, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, United States
- 4Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, CA, United States
- 5California Nano Systems Institute, University of California, Los Angeles, Los Angeles, CA, United States
Pathological self-assembly is a concept that is classically associated with amyloids, such as amyloid-β (Aβ) in Alzheimer's disease and α-synuclein in Parkinson's disease. In prokaryotic organisms, amyloids are assembled extracellularly in a similar fashion to human amyloids. Pathogenicity of amyloids is attributed to their ability to transform into several distinct structural states that reflect their downstream biological consequences. While the oligomeric forms of amyloids are thought to be responsible for their cytotoxicity via membrane permeation, their fibrillar conformations are known to interact with the innate immune system to induce inflammation. Furthermore, both eukaryotic and prokaryotic amyloids can self-assemble into molecular chaperones to bind nucleic acids, enabling amplification of Toll-like receptor (TLR) signaling. Recent work has shown that antimicrobial peptides (AMPs) follow a strikingly similar paradigm. Previously, AMPs were thought of as peptides with the primary function of permeating microbial membranes. Consistent with this, many AMPs are facially amphiphilic and can facilitate membrane remodeling processes such as pore formation and fusion. We show that various AMPs and chemokines can also chaperone and organize immune ligands into amyloid-like ordered supramolecular structures that are geometrically optimized for binding to TLRs, thereby amplifying immune signaling. The ability of amphiphilic AMPs to self-assemble cooperatively into superhelical protofibrils that form structural scaffolds for the ordered presentation of immune ligands like DNA and dsRNA is central to inflammation. It is interesting to explore the notion that the assembly of AMP protofibrils may be analogous to that of amyloid aggregates. Coming full circle, recent work has suggested that Aβ and other amyloids also have AMP-like antimicrobial functions. The emerging perspective is one in which assembly affords a more finely calibrated system of recognition and response: the detection of single immune ligands, immune ligands bound to AMPs, and immune ligands spatially organized to varying degrees by AMPs, result in different immunologic outcomes. In this framework, not all ordered structures generated during multi-stepped AMP (or amyloid) assembly are pathological in origin. Supramolecular structures formed during this process serve as signatures to the innate immune system to orchestrate immune amplification in a proportional, situation-dependent manner.
Introduction
Amyloids and antimicrobial peptides (AMPs) are two classes of proteins that have fascinating biophysical and structural properties. Until recently, they were thought to be distinct entities with vastly different functions. Amyloids were strictly pathologic and accumulation in tissues invariably led to diseases (1). In comparison, AMPs are considered essential components of the innate immune system, defending against invasive microbial infections and sounding the alarm to activate cellular-mediated immune responses (2, 3). Within the last 5–10 years, emerging work from collaborations between bioengineers, amyloid biologists, and immunologists has dramatically blurred the lines between amyloids and AMPs. AMPs and amyloids have strikingly similar structural and biophysical properties that enable them to self-assemble with immune ligands like DNA to amplify immune responses (4–6). Surprisingly, many amyloids possess hidden antimicrobial activity in addition to their known cytotoxic properties, suggesting a potential endogenous role in host defense (7, 8). AMPs and bacterial amyloids have also been implicated in the pathogenesis of autoimmune diseases like lupus and psoriasis (5, 9–13), parallel to the proinflammatory role of amyloids in neurodegeneration (14). Disentangling the molecular basis for the homeostatic and pathologic functions of both amyloids and AMPs has proven challenging (15).
The goal of this review is to highlight fundamental studies that showcase the unexpected similarities between amyloid and AMP self-assembly and discuss how these findings can transform our understanding of their functional roles in host defense, inflammation, and disease. While some effort has been made in the literature to compare and contrast amyloids and AMPs, it has been difficult to identify common themes due to the sheer diversity of sequences and structures in both classes of molecules (Figure 1). Here, we begin by first providing a short overview of AMPs and their known antimicrobial and immunomodulatory functions. We focus on recent work from our group that outlines a novel emerging paradigm for understanding how AMPs talk to the innate immune system. We find that AMPs self-assemble into amyloid-like protofibrils that act as molecular templates to scaffold canonical immune ligands into spatially periodic nanocomplexes, which amplify immune responses via pattern-recognition receptors (PRRs) such as the Toll-like receptors (TLRs) (Figure 2). We demonstrate how this paradigm is general to other immune proteins beyond AMPs such as chemokines as well as other TLRs. We then discuss implications for the synergistic role of AMPs in normal host defense as well as in autoimmunity. In the second part of the review, we compare AMP self-assembly to amyloid self-assembly in the contexts of antimicrobial and membrane-remodeling activity (Figure 3). Lastly, we summarize how the functional similarities between AMPs and amyloids extends to bacterial amyloids as well in the realm of immunomodulation. By borrowing lessons and tools from the AMP literature, we find that amyloids potentially have endogenous functions beyond their pathologic consequences. We conclude by suggesting future research directions that can integrate our knowledge of AMP and amyloid biology to uncover mechanisms of disease and develop new targeted therapies.
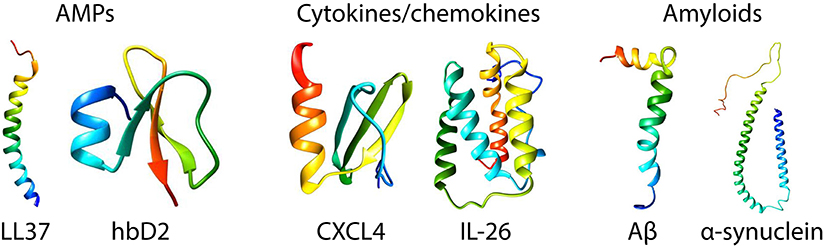
Figure 1. Structures of prototypical antimicrobial peptides, cytokines/chemokines, and amyloids. LL37 (22) and human β-defensin 2 (23) are canonical α-helical and β-sheet AMPs, respectively. CXCL4 (24) and IL-26 [homology model shown based on IL-19 (25)] are representative immune signaling molecules that also have known direct antimicrobial properties. Amyloid β (26) and α-synuclein (27) are the amyloids implicated in Alzheimer's disease and Parkinson's disease. The monomeric structures were taken from the Protein Data Bank (PDB) and visualized in Chimera (UCSF).
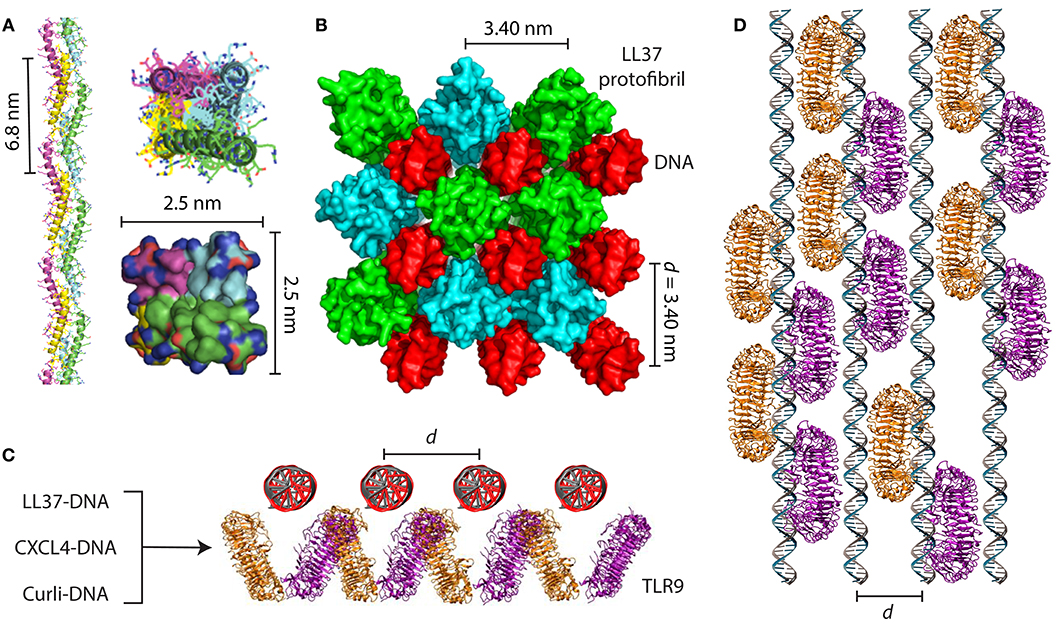
Figure 2. AMPs and amyloids organize immune ligands into spatially periodic nanocomplexes to amplify TLR activation. (A) LL37 self-assembles into a 4-fold amyloid-like superhelical protofibril in the presence of DNA. Hydrophobic residues are buried in the interior of the protofibril while cationic residues are exposed at the perimeter. (B) Structure of the LL37-DNA complex showing cross linking of spatially periodic DNA strands by LL37 protofibrils at an inter-DNA spacing of 3.40 nm, which is optimal for TLR9 binding and amplification of cytokine production. (C) End-on view and (D) top-down view of geometrically organized DNA immune complexes binding to clustered TLR9 in the endosomal membrane. In addition to the LL37-DNA complex, CXCL4-DNA complexes formed in scleroderma and curli-DNA complexes from Salmonella biofilms also demonstrate similar structural properties that enable amplification of TLR9 in immune cells and type I interferon production. (A,B) are adapted with permission from (4). (C,D) are adapted with permission from (44) and (10).
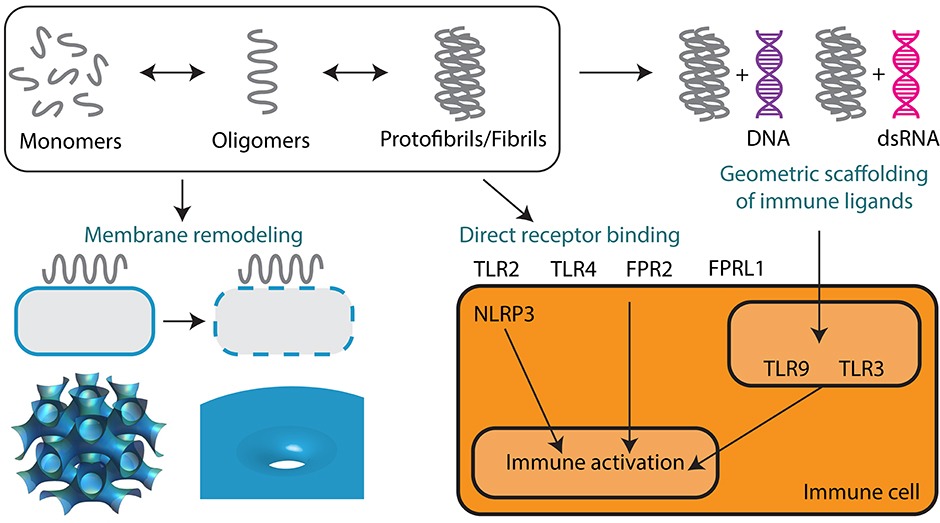
Figure 3. Supramolecular self-assembly of AMPs and amyloids enables membrane remodeling activity and immunomodulation. Monomers of AMPs and amyloids sequentially self-assemble into oligomers and protofibrils or fibrils. Oligomeric forms are predominantly responsible for mediating membrane permeation, including pore formation and membrane fusion leading to direct antimicrobial activity and cytotoxicity. Protofibrils and fibrils can signal to the innate immune system either by direct receptor binding or by the geometric scaffolding of immune ligands such as DNA and dsRNA. Both AMPs and amyloids can engage a broad range of immune receptors including TLR2, TLR3, TLR4, TLR9, FPR2, FPRL1, and NLRP3.
AMPs Organize Immune Ligands into Spatially Periodic Nanocomplexes to Amplify TLR Activation
AMPs are part of an ancient arm of the innate immune system that represents the first line of defense against microbial infections (2). AMPs are found in almost all living organisms including vertebrates, invertebrates, and plants (16–18), and can be broadly categorized by their secondary structures: the α-helical AMPs, β-sheet AMPs, AMPs with cross α-β structures, and extended linear peptides with specific enriched amino acids (19–21) (Figure 1). The prototypical human AMP is cathelicidin (LL37), which is an α-helical AMP with essential anti-infective and immunomodulatory functions (28, 29). Prototypical human β-sheet AMPs are the defensins. The mechanisms underpinning the antimicrobial activity of AMPs are thoroughly reviewed elsewhere but we briefly discuss it here (3, 30–32). In general, AMPs are cationic (+2 to +9) and amphiphilic with segregated groups of hydrophobic and polar/charged residues (2). These properties enable AMPs to electrostatically bind to negatively charged bacterial membranes and embed themselves into the membrane via hydrophobic interactions. Several models have been proposed for membrane permeation, including the “barrel-stave” model, “carpet” model, and “toroidal-pore” model (32). In the “barrel-stave” and “toroidal-pore” models, AMPs self-assemble into bundles that cylindrically insert into bacterial membranes to form aqueous pores, whereas in the “carpet” model, AMPs disintegrate the membrane via micellization (33). We have shown that AMP antimicrobial function correlates with its ability to induce negative Gaussian curvature (NGC) in bacterial membranes, a topological criterion for pore formation and membrane permeation (34). However, antimicrobial peptides are not only limited to membrane permeation. AMPs can also kill bacteria and fungi by disrupting metabolic gradients, inhibiting ribosomes, and binding to intracellular nucleic acids (35). However, the most underappreciated aspect of AMP function is their ability to amplify immune responses by autocrine signaling via PRRs such as TLRs. AMPs can signal through PRRs via direct binding. LL37 has been shown to be a chemoattractant for leukocytes by binding to the formyl peptide receptor-like 1 (FPRL1) (36). Furthermore, β-defensins are known to be chemotactic for monocytes and macrophages by binding to the CCR6 receptor (37), and β-defensin 2 is a known ligand for Toll-like receptor 4 (TLR4) (38). Despite this work, it was not known until recently whether AMPs could signal to PRRs without being direct ligands, or whether they could serve as chaperones by binding to immune ligands such as nucleic acids.
In a series of groundbreaking studies, Lande et al. showed that LL37 can break immune tolerance to self-DNA in diseases like lupus and psoriasis by forming insoluble complexes that are phagocytosed by immune cells. In these diseases, LL37 is overexpressed in the skin and blood and are predominantly produced by neutrophils and keratinocytes (39–41). LL37-DNA complexes are formed extracellularly and are internalized into the endosomes of plasmacytoid dendritic cells (pDCs), amplifying type I interferon (IFN-α) production by binding to Toll-like receptor 9 (TLR9). They also showed that other cationic AMPs in the skin possess a similar property, including the β-defensins and lysozyme (42). To understand the molecular basis for how LL37 and other AMPs signal through TLR9, we characterized the structures of numerous AMP-DNA complexes using X-ray scattering and correlated them with their ability to activate pDCs via TLR9 (43). We found that LL37 and β-defensins electrostatically self-assemble with DNA into spatially periodic grill-like nanostructures with well-defined inter-DNA spacings, and that the inter-DNA spacing within these complexes correlated directly with the quantitative degree of cytokine production (Figures 2A–D). The biophysics of the hierarchical electrostatic self-assembly of rigid polyelectrolytes like DNA has been well-described in the literature and is thoroughly discussed elsewhere (45–47). AMP-DNA complexes with spacings well-matched with the steric size of TLR9 enabled multivalent binding to clustered TLR9 on the endosomal membrane and IFN-α production orders of magnitude higher than expected from individual ligands (45). Surprisingly, this phenomenon was independent of the degree of endosomal uptake, suggesting that this differential response was solely due to differences in the nanostructures of the complexes. This conceptual transformation suggested that a much broader range of molecules could be predicted to activate TLR9 if they had the right physicochemical properties to organize and present DNA at optimal periodic positions that promote multivalent interactions with an ensemble of TLR9.
Inspired by this, we set out to discover general rules for how α-helical AMPs like LL37 can self-assemble into molecular templates for DNA binding and amplify immune responses. Previous work has shown that artificial patchy amphiphiles can be designed to self-assemble into various unique structures (48, 49). By combining computer simulations with X-ray structural characterization, we found that LL37 oligomerizes into a superhelical amyloid-like protofibril in the presence of DNA, with hydrophobic residues buried in the interior and outward-facing cationic residues (4) (Figures 2A,B). The LL37 protofibril cross-links DNA into a 4-fold coordinated lattice with inter-DNA spacings commensurate with the size of TLR9. We conducted experiments with other α-helical AMPs with different charge densities and hydrophobicities such as melittin (50) and buforin (51). We discovered that formation of this amyloid-like protofibril requires sufficient hydrophobicity to enable polymerization into a superhelix and cationic charge density well-matched to the high anionic charge density of DNA. Remarkably, we discovered that although melittin was able to form optimized complexes with DNA for TLR9 activation, its cytotoxicity to immune cells prevented cytokine production. By attenuating its cytolytic activity while retaining its ability to self-assemble into 4-fold coordinated nanocomplexes with DNA at the optimal inter-DNA spacing, we rescued its ability to activate TLR9 (4). This highlighted that there exist natural tradeoffs in antimicrobial and immunomodulatory functions of AMPs, and that we can deterministically modulate them by altering the AMP's physicochemical properties.
The next natural question to ask is whether this phenomenon is general to other immune ligands and innate immune receptors. Gallo and colleagues have previously shown that LL37 can break immune tolerance to double-stranded RNA (dsRNA) released from keratinocytes in psoriasis and other cutaneous diseases (52–55). Given the structural homology of TLR9 to Toll-like receptor 3 (TLR3) and DNA to dsRNA, respectively, we decided to map out the structural rules for immune activation of TLR3 by dsRNA complexes (56). We characterized the structures of numerous AMP-dsRNA complexes (LL37 and various truncated variants) and tested their ability to induce IL-6 production from psoriatic keratinocytes via TLR3. Cognate to LL37-DNA complexes, we found that LL37-dsRNA complexes formed nanocrystalline structures with well-defined inter-dsRNA spacings, and that complexes that maximally activated TLR3 had spacings perfectly matched with the steric size of TLR3. A mathematical model and computer simulation of TLR3 binding to spatially periodic AMP-dsRNA complexes recapitulated the experimental data and showed that both the inter-dsRNA spacing and the number of repeat units within the complexes were primary determinants of immune activation (56). This validated the idea that innate immune receptors like TLR9 and TLR3 can recognize both single ligands, as well as the crystallinity of spatially periodic, geometrically patterned ligands templated by molecular chaperones like AMPs.
As it turns out, this phenomenon is not limited to AMPs, but is rather general to other immune signaling proteins. Chemokines are a well-studied class of immune signaling molecules that are known to exert their biological activities by binding to G-protein coupled receptors (GPCRs) on the surface of immune cells. We discovered an unexpected signaling pathway for chemokine (C-X-C motif) ligand 4 (CXCL4)/platelet factor 4 (PF4) and its role in the pathogenesis of scleroderma. Interestingly, CXCL4 naturally self-assembles into an oligomeric homotetramer and has a cationic, amphipathic cross α-β structure that is homologous to that of defensin antimicrobial peptides (57) (Figure 1). It has also previously been shown to exert antimicrobial activity (58–62). CXCL4 is typically highly expressed in platelets and plays a key role in hemostasis and wound healing (63). CXCL4 is known to bind to anionic heparin, particularly in the context of heparin-induced thrombocytopenia (64–66), but its causal role in inflammatory diseases was unclear. We discovered that like LL37 and other AMPs, CXCL4 can self-assemble with microbial and self-DNA to form nanocomplexes to amplify IFN-α production via TLR9 within skin pDCs (Figures 2C,D). We identified CXCL4-DNA complexes in the blood and skin of scleroderma patients, and levels of these complexes correlated directly with the type I interferon signature (44). Surprisingly, this activity was independent of the canonical CXCL4 receptor, CXCR3. We predict that many other chemokines likely possess similar properties, since they share a structural backbone and have close physicochemical similarity, including the ability to self-assemble into oligomers. Taken together, our findings are consistent with a robust emerging conceptual framework where diverse classes of molecules can signal to the innate immune system by scaffolding endogenous immune ligands into spatially periodic nanocomplexes, rather than being direct agonists.
Synergy Between the Antimicrobial and Immunomodulatory Properties of AMPs and Chemokines
Thus far, we have demonstrated that AMPs and chemokines are multifunctional, and can exert direct antimicrobial activity and modulate immune responses via PRRs. Due to their cationicity and amphipathicity, AMPs are capable of directly killing microbes through membrane permeation, inhibition of metabolic machinery, and disruption of electrostatic gradients. However, the same physicochemical features allow them to also self-assemble into ordered nanocrystalline complexes with immune ligands such as DNA and dsRNA by functioning as structural scaffolds. These complexes can potently induce inflammation by amplifying Toll-like receptor activation via receptor clustering, and the crystallinity of these complexes can determine the degree of immune amplification (43). What are the consequences of this multifunctionality for host defense?
Synergy between the dual antimicrobial and immunomodulatory functions of many AMPs and chemokines enables them to play an important role in protection against infections and in mediating autoimmune disease and inflammation. Certain AMPs and chemokines are capable of lysing and killing bacteria and presenting fragments of bacteria such as DNA to innate immune receptors. For instance, Meller et al. demonstrated that interleukin 26 (IL-26), a cytokine secreted by human interleukin-17 producing helper T cells (TH17), both kills bacteria and promotes immune sensing of bacterial and host cell death, driving the potent antimicrobial and proinflammatory function of TH17 cells (67) (Figure 1). IL-26 is a highly cationic and amphipathic protein that possess broad-spectrum antimicrobial activity against several gram-negative bacterial strains including P. aeruginosa, E. coli, and K. pneumoniae, and gram-positive bacteria S. aureus (67, 68). IL-26, like AMPs, can oligomerize into multimers and lyse bacteria by forming pores in their membranes. The antimicrobial properties of TH17 cell-derived IL-26 helps explain why patients defective in TH17 cells are highly susceptible to S. aureus infections (69), and why depletion of TH17 cells during infection by simian immunodeficiency virus results in the dispersal of gut bacteria (70).
Upon bacterial killing, TH17 cell-derived IL-26 triggers potent immune activation. IL-26 forms nanocrystalline complexes with bacterial DNA released during the antimicrobial response. These complexes are internalized into the endosomal compartments of pDCs and induce an amplified production of IFN-α via recruitment and super-selective binding of TLR9 receptors. Type I interferons are responsible for driving many proinflammatory responses, including CD8+ T cell activation (71, 72), TH1 cell differentiation (72), NK cell activation, dendritic cell maturation (73, 74), and promotion of antibody-secreting plasma cells (75). Consequently, their production has been shown to be beneficial in the context of extracellular bacterial infections, including the resolution and control of infections caused by P. aeruginosa, S. pneumoniae, and E. coli (76, 77), and reducing inflammation in mouse models of bacterial sepsis (78). In addition to serving as a direct antimicrobial, IL-26 has evolved the ability to amplify and regulate innate and adaptive responses to extracellular bacteria. Its dual functionality allows our immune system to more effectively clear bacterial infections. Modulating the endogenous activity of IL-26 may offer promising strategies to enhance our natural host defense against microbes.
IL-26 and CXCL4 are likely several of many examples of multifunctional molecules that play a synergistic role in host defense against microbes via direct killing and immunomodulation, in addition to their other homeostatic functions. Recently, other interferons like IFN-β (79) and IFN-γ (80) were shown to exhibit direct antimicrobial properties in addition to their known immunomodulatory functions. These findings suggest that the nature has evolved a way to bioconjugate multiple distinct functions into the same amino acid sequence (81), and that understanding how the immune system works requires us to examine these hidden functions.
Comparison of AMP and Amyloid Self-Assembly
Here, we draw comparisons between the self-assembly of AMPs and the classical self-assembly of amyloids. Amyloids constitute a broad class of proteins that have the unique ability to aggregate into fibrils with characteristic secondary structures. The structural, physicochemical, and biological properties of AMPs are similar to those of many amyloid proteins. The majority of amyloids have a β-sheet secondary structure, but recently a subset of α-helical amyloids was identified (82, 83). Amyloids can be broadly categorized into those of eukaryotic and prokaryotic origins. Human endogenous amyloids are associated with over 50 distinct disease processes, the most famous of which is amyloid β-peptide (Aβ) in Alzheimer's disease (AD) (Figure 1). More and more proteins are being discovered to have amyloidogenic properties. Whether amyloids play a causal role in disease or are merely a consequence of disease is hotly debated. However, amyloids have unequivocally been shown to exhibit direct cytotoxic activity against human cells. The best data is available for Aβ, but many other amyloids have been shown to self-assemble into structures that can disrupt membranes (84) and signal to the immune system (Figure 3).
Aβ is the main component of amyloid plaques found within neurons in AD brains and is thought to induce cytotoxicity leading to neuronal cell death (85) via multiple mechanisms (86–89). Traditionally, Aβ has been characterized as a functional catabolic byproduct of amyloid precursor protein (APP) without much evidence for a possible endogenous homeostatic function (90). However, recent in vitro studies have shown that Aβ can exhibit AMP-like direct antimicrobial activity by disrupting membranes (91) and may play a role as an effector molecule of innate immunity, exhibiting broad-spectrum activity against several common and clinically relevant organisms (92) (Figure 3). In a directly related study, Kumar et al. highlighted the potent antimicrobial activity of Aβ and demonstrated its biological relevance in host defense through in vivo models of infection. Aβ expression is associated with increased host survival in both nematode and mouse models of bacterial (93) and viral infection (94). Low Aβ expression resulted in greater death of APP-KO mice after infection. The protective role of Aβ can be attributed to classic AMP mechanisms characterized by reduced microbial adhesion, bacterial membrane disruption, and entrapment of microbes by Aβ fibrils (93). Alternatively, low levels of fibrillar Aβ may signal to the immune system and elicit inflammation to keep the immune system or the infection in check. Low levels of Aβ can get cleared without amyloid deposition. Nonetheless, these data imply that Aβ possesses a normally protective role in host defense that, when dysregulated, can lead to neurodegenerative disease. Aβ may normally function as an endogenous inducible AMP that is cleared upon resolution of inflammation. However, when dysregulated in the right of genetic or environmental context, Aβ instead forms toxic amyloid oligomers leading to neuronal cell death and eventually deposits leading to chronic inflammation (95).
It is important to note that genetic factors may also be involved in the dysregulation of Aβ production in addition to environmental factors like bacterial and viral infections (96, 97). Overexpression of APP on chromosome 21 is associated with AD, and individuals with Down syndrome (Trisomy 21) are at a higher risk of AD relative to the population (96). In addition, Aβ from individuals with the “Arctic” mutation (E693G 669 on APP) tends to self-assemble into protofibrils at a much higher rate than the wild type protein (98). A larger number of additional genetic polymorphisms have been identified which affect Aβ cytotoxicity (99), but their consequences on Aβ in host defense is currently unknown. It is also possible that genetic polymorphisms in other immune and inflammatory genes can alter Aβ production and contribute to AD. For example, the apolipoprotein gene ApoE4 is another major genetic risk factor for AD (100, 101), and deficient clearance of Aβ is associated with disease (102). Further work will be required to elucidate how these genetic changes affect the function of Aβ in host defense and inflammation.
Similarly, while AMPs are typically protective, dysregulation of AMP expression can lead to host cell toxicity, degenerative pathologies, and chronic inflammation and autoimmunity as described above (103–105) (Figure 4). For example, LL37 is a human cathelicidin AMP essential for normal immune function and protection against lethal infections (106). However, at elevated physiological concentrations, it is cytotoxic to host smooth muscle cells (107) and implicated in the pathogenesis of late-stage diseases including atherosclerosis, rheumatoid arthritis, and systemic lupus erythematosus (29). Interestingly, certain AMPs are deposited as amyloids in common human amyloidopathies including isolated atrial and senile seminal vesicle amyloidosis (7, 92, 108). In fact, a large number of naturally occurring AMPs including LL37 (4, 109), lysozyme (110), protegrin-1 (111), plant defensins (112), temporins (113, 114), histatin 5 (115), HAL-2 (116), uperin 3.5 (117), dermaseptin S9 (118), Cn-AMP2 (119), and longipin (120) and apolipoprotein A-I (121) from invertebrates form amyloids or amyloid-like fibrils in vitro and in vivo. A number of synthetic amyloid-inspired peptides have been designed primarily as novel broad-spectrum antibiotics (83, 122), and many AMPs are known to oligomerize before or upon membrane binding and pore formation (123, 124).
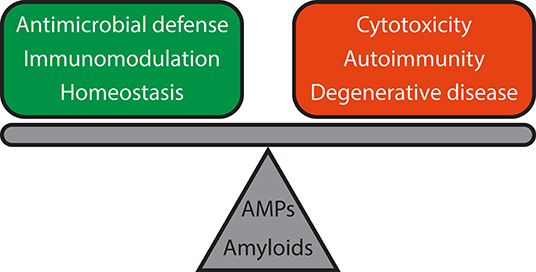
Figure 4. Functional reciprocity of AMPs and amyloids and consequences for host defense and immune signaling. Both AMPs and amyloids are involved in antimicrobial defense, immunomodulation, and homeostasis, but dysregulation can lead to adverse outcomes such as cytotoxicity, chronic inflammation, and autoimmune diseases like lupus, and degenerative diseases like Alzheimer's disease and Parkinson's disease. Further work will be required to elucidate the mechanisms of how such a delicate balance is attained.
The potential protective effects of host-generated amyloids have only recently emerged (7, 8, 125) despite recognition of the association between chronic bacterial infections and amyloidosis for nearly a century (1). Findings related to the role Aβ plays in neuronal innate immune defense may extend to proteins associated with amyloidopathies other than AD, several of which have been shown to exhibit antimicrobial activity (18, 108, 126–129). Pathways that regulate innate immunity in AD and other amyloidopathies may serve as novel targets for therapeutic intervention. Parkinson's disease (PD)-associated α-synuclein has been long-studied as a model system for amyloid-mediated cytotoxicity (130–134) due to its propensity for membrane interactions (135, 136) via its N-terminal helix (137, 138) (Figure 1). Recently, it was shown to be antimicrobial against a variety of bacteria and fungi (139). Unexpectedly, it was found to be also involved in the chemoattraction of immune cells, suggesting a potential endogenous role in host defense (140). In human patients with chronic gut inflammation, α-synuclein was found to be upregulated in enteric neurons (141), a fascinating finding given that PD often begins in the gut as constipation before neurologic symptoms appear (142, 143). Disruption of the ability of α-synuclein to self-assemble into oligomers on neuronal membranes appears to be a potential therapeutic strategy in a nematode model of PD (144). Beyond Aβ and α-synuclein, several other amyloids or their fragments have been shown to have antimicrobial or membrane-lytic properties, including tau (145), islet amyloid polypeptide (IAPP) (146–148), human prion protein (128), superoxide dismutase (127), and endostatin (149). The functional bacterial amyloid curli, which is a key stromal component of Salmonella biofilms (150), was also shown to form cytotoxic oligomeric intermediates (151).
Interestingly, a recent machine learning tool originally trained to identify antimicrobial activity in α-helical AMPs identified a subset of naturally occurring amyloid peptides that possess predicted membrane-permeating activity (33, 152–154), among numerous other classes of molecules (155, 156). This demonstrates that data-driven approaches may be helpful in further identifying amyloids that are involved in host defense, but it is clear that much more work needs to be done to validate the extent and relevance of that function.
Immunomodulatory Aspects of Amyloids and Similarity to AMPs
The functional similarities between AMPs and Aβ amyloids extend to bacterial amyloids as well. In bacterial biofilms, bacterial amyloids form the building blocks of the biofilm extracellular matrix alongside extracellular DNA (eDNA) (157). In a series of landmark papers, Tükel and colleagues showed that the biofilm amyloid curli from Salmonella and E. coli activated TLR2 (158–160) (Figure 3). Subsequent studies have shown that TLR2/TLR1 heterocomplex recognized the fibrillar structure of amyloids from both prokaryotic and eukaryotic origin including curli, Aβ and serum amyloid A (SAA) (158, 160–162). In the case of curli, the adaptor molecule CD14 further enhanced the recognition of curli via the TLR2/TLR1 heterocomplex (163). These data instigated further studies investigating whether the conserved fibrillar structure of amyloids serve as a pathological molecular signature for the innate immune system. Consistent with this idea, fibrillar curli (164), Aβ (14), serum amyloid A (165), and IAPP (166) elicited IL-1β cytokine production by directly activating the NLRP3 inflammasome in macrophages. This process impacts the innate immune system in multiple ways: (1) TLR2 activation initiated the pre-IL-1β production and amyloid internalization, (2) NLRP3 inflammasome activation by cytosolic fibrils activated caspase1 and cleaved the pre-IL-1β into mature IL-1β (164). In addition to TLR2, possible activation of TLR4 and TLR6 by Aβ was also reported (167, 168). However, it is not known whether the observed activation of TLR4 and TLR6 was due to the generation of additional Aβ structural conformations during in vitro fibrillization or any other contaminating factors. In invertebrates, amyloid formation is key to activation of the innate immune system and host defense. SAA from marine bivalves resembling SAA from vertebrates is a potent acute phase protein and are induced upon bacteria infection (169). In insects such as Heliothis virescens, the functional amyloid P102 is synthesized and released to protect against pathogens such as bacteria and parasites. This can occur in response to lipopolysaccharide stimulation (170). The secreted amyloid layer acts as a molecular scaffold to promote localized melanin synthesis and immune cell adhesion to foreign invaders (171). However, it is unknown whether they play a role in direct receptor binding.
Previously, we showed that AMPs like LL37 can self-assemble into an amyloid-like superhelical protofibril to present spatially ordered DNA to TLR9, and that AMP self-assembly with immune ligands can enable signaling through a broad range of PRRs without being direct agonists. Interestingly, nucleic acids have previously been shown to accelerate amyloid fibrillation and serve as molecular templates for self-assembly (172, 173). AD amyloids like Aβ in particular have a propensity to bind to DNA (174) and co-localize within nuclei of affected cells (175, 176). Autoimmune responses to Aβ-containing amyloid structures have been described in AD patients (177). PD-associated α-synuclein fibrils have the ability to self-assemble with DNA (178). Surprisingly, another endogenous amyloid serum amyloid P component (SAP) was shown to be protective against lupus by binding to DNA to prevent formation of anti-DNA antibodies (179, 180), suggesting that perhaps different amyloids are involved in regulating inflammation and recognition of immune ligands. Previously, we showed how structural scaffolding of immune ligands like DNA by AMPs and amyloids dramatically affects immune outcomes (10, 43, 56). AMP-DNA complexes with inter-DNA spacings well-matched with the size of TLR9 amplifies cytokine production, but those with spacings that are much smaller or larger can actually inhibit TLR9 activation and inflammation (4, 43, 45). SAP may potentially regulate inflammation by out-competing binding of proinflammatory amyloids to DNA. This challenges the notion that amyloid assembly is strictly proinflammatory or pathologic.
The ability of amyloids to act as a carrier for nucleic acids to promote endosomal TLR signaling was only recently discovered. Di Domizio et al. showed that artificially formed amyloid fibrils bound to DNA to form amyloid-DNA complexes (181). When administered systemically, these amyloid-DNA promoted systemic autoimmunity, autoantibody production, and lupus-like syndromes in mice by amplifying TLR9 activation in pDCs (6) (Figure 3). A similar observation was made with curli proteins and eDNA found at close proximity in the extracellular matrix of the biofilm. Curli and eDNA formed irreversible complexes together. Similar to what was observed with human amyloids, DNA accelerated the self-assembly process of bacterial amyloid curli (182). Incorporation of DNA into curli rendered DNA resistant to enzymatic degradation. Systemic administration of curli-DNA complexes induced autoantibody production and type I interferon production (12) suggesting that complexes of curli-like bacterial amyloids with DNA may promote inflammatory disorders (183). These findings are fascinating in the setting of our previous work showing that LL37 self-assembles into amyloid-like protofibrils to amplify TLR9 activation. We set out to examine the structures of curli-DNA complexes and found that, similar to LL37 and other AMPs and chemokines, curli was able to organize DNA into geometrically optimal nanostructures to amplify TLR9 activation (Figures 2C,D). Immune activation occurred via a two-step process—curli-DNA complexes were first internalized into immune cells via binding to TLR2 (158–160) and then activated TLR9 once inside the endosome leading to the generation of type I interferons (5). Engagement of TLR2 and TLR9 also contributed to the autoantibody production through unknown mechanisms.
For the longest time, it has been known that infections initiate and/or exacerbate autoimmune diseases. However, the mechanisms of how infections trigger autoimmunity remained a mystery. Besides curli producing enteric bacteria, many important human pathogens such as Borrelia burgdorferi (184), Mycobacterium tuberculosis (185), Pseudomonas aeruginosa (186, 187), and Staphylococcus aureus (188) also produce amyloids. Individuals infected with these pathogens develop some form of autoimmune sequelae such as inflammatory arthritis (13). Phenol soluble modulins (PSMs) from Staphylococcus biofilms (189–191) and Fap amyloids from Pseudomonas biofilms (186) have been studied concisely, but at present, the mechanisms of DNA binding by other functional amyloids remain unclear, and it remains to be seen whether this has consequences for immune signaling and inflammation. Nevertheless, extracellular DNA is known to facilitate the formation of functional amyloids in Staphylococcus biofilms (192), and PSMs are known to bind directly to human formyl peptide receptor 2 (FPR2) (193). Together, these studies strongly suggests a link between chronic bacterial infections, biofilms, and autoimmune diseases (13, 194) (Figure 4). By therapeutically targeting curli amyloid fibers (195), disruption could potentially eradicate bacterial biofilms and secondary autoimmunity.
Formation of amyloid deposits by subunits of different amyloid fibrils is termed as cross-seeding. The co-existence of combinations of α-synuclein, tau, prion protein, and Aβ have all been observed in amyloid deposits in humans (144). In the past several years, few studies also investigated cross-seeding events and a possible link between neurodegenerative diseases and bacterial amyloids. Cross-seeding between SAA and curli was reported in a mouse model of secondary amyloidosis (147). Recent studies have shown that curli can also seed the self-assembly of human α-synuclein (6, 196–198). Colonization of α-synuclein-overexpressing mice with curli-producing E. coli exacerbates motor impairment and GI dysfunction, and promotes α-synuclein deposition in the brain (199). However, the spatial interactions between bacterial and host amyloids that would allow for cross-seeding and how these interactions could be influenced by binding to nucleic acids to induce inflammation still remains unknown. We feel that this is an area that should attract and reward attention.
Conclusions and Outlook
In this review, we discussed the unique functional reciprocity of amyloids and antimicrobial peptides, and how supramolecular self-assembly changes our understanding of their respective roles in host defense and immune activation. We outlined recent work highlighting novel molecular mechanisms for AMP-mediated immunomodulation via TLRs, and implications for antimicrobial responses and inflammatory diseases. We then compared AMP and amyloid self-assembly in the contexts of antimicrobial and membrane-remodeling activity, cytotoxicity, and immune signaling using LL37, Aβ, and curli as fundamental examples.
By critically examining the AMP and amyloid literature together, we discover several convergent themes. First, the amphiphilic properties unique to AMPs and amyloids enable them to cooperatively self-assemble into supramolecular nanostructures to modulate the innate immune system and defend against microbial infections. AMPs, which were thought of as only having antimicrobial function, are now known to modulate innate immune receptors by forming amyloid-like protofibrils and scaffolding canonical immune ligands like DNA and RNA into geometrically organized patterns (Figure 2). Recognition of these complexes by the immune system drives autoimmunity in diseases like lupus, psoriasis, and scleroderma. In a parallel direction, functional bacterial amyloids such as curli from Salmonella has shown how these stromal biofilm proteins organize eDNA into cognate spatially ordered complexes to induce autoimmunity in diseases like lupus. Further studies will be required to map out the immune activation landscape of both eukaryotic and prokaryotic amyloids and their distinct mechanisms (Figure 3). For example, exploring how amyloids bind to other immune ligands and identifying the structural rules for immune activation would be incredibly fascinating, analogous to our work with AMP self-assembly. Can we adapt this paradigm to explain autoimmune sequelae of other bacterial infections? We imagine that lessons learned from work on the α-helical AMPs can inform new research directions for α-helical amyloids such as the Staphylococcus PSMs, and vice versa. Similarly, our strong understanding of the self-assembly of β-sheet amyloids may inform a better understanding about how β-sheet rich AMPs and AMP-like molecules such as chemokines oligomerize.
Second, the revolutionary work demonstrating that Aβ, which has no known primary function, is an AMP that protects the nervous system against bacterial and fungal infections fundamentally challenges our view of endogenous human amyloids as solely pathologic. This model of Aβ activity suggests that excessive β amyloid deposition in AD and pathogenesis may not necessarily arise from an intrinsic abnormal propensity for Aβ to aggregate, but rather as a consequence of dysregulation of the brain's normal host defense system against invasive infections, similar to how dysregulation of AMP expression and production in tissues can adversely lead to autoimmune diseases (Figure 4). The discovery that α-synuclein, which also has no previous known primary function, is a chemoattractant and is induced to alert the immune system during gut infections opens up incredible opportunities for discovery. Are there other amyloids with hidden antimicrobial activity with potential roles in host defense? What are the primary roles of other endogenous amyloids?
We are just beginning to elucidate the role of supramolecular assembly in immune recognition and modulation. Recent studies have shown that innate immune receptor adaptor proteins like melanoma differentiation-associated protein 5 (MDA5), which senses cytosolic dsRNA, can self-assemble into amyloid-like helical filaments in the presence of dsRNA (200, 201). Helical filament assemblies can also be observed in the signaling pathways of the RIG-I-like receptors (RLRs), AIM2-like receptors (ALRs), and mitochondrial antiviral-signaling protein (MAVS) (202–204). Given that we know how AMPs and amyloids self-assemble with nucleic acids to talk to TLRs, further work will be required to illuminate how they interact with filamentous assemblies of cytoplasmic immune receptors. In summary, we hope that this review will serve to highlight the advances, opportunities, and outlook for the AMP and amyloid communities, and stimulate collaborations between AMP and amyloid biologists, immunologists, as well as bioengineers.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
EL was supported by the Systems and Integrative Biology Training Program (NIH T32GM008185), the Medical Scientist Training Program (NIH T32GM008042), and the Dermatology Scientist Training Program (NIH T32AR071307) at UCLA. EL was also supported by an Early Career Research Grant from the National Psoriasis Foundation. JA was supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1650604. ÇT was supported by NIH AI137541, AI132996, AI148770, and AI151893. GW was supported by NIH R01AI143730, NIH R01AI052453, NSF DMR1808459, and the National Psoriasis Foundation 20194384. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med. (1997) 337:898–909. doi: 10.1056/NEJM199709253371306
2. Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. (2002) 415:389–95. doi: 10.1038/415389a
3. Yeaman MR, Yount NY. Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev. (2003) 55:27–55. doi: 10.1124/pr.55.1.2
4. Lee EY, Zhang C, Di Domizio J, Jin F, Connell W, Hung M, et al. Helical antimicrobial peptides assemble into protofibril scaffolds that present ordered dsDNA to TLR9. Nat Commun. (2019) 10:1012. doi: 10.1038/s41467-019-08868-w
5. Tursi SA, Lee EY, Medeiros NJ, Lee MH, Nicastro LK, Buttaro B, et al. Bacterial amyloid curli acts as a carrier for DNA to elicit an autoimmune response via TLR2 and TLR9. PLoS Pathog. (2017) 13:e1006315. doi: 10.1371/journal.ppat.1006315
6. Di Domizio J, Dorta-Estremera S, Gagea M, Ganguly D, Meller S, Li P, et al. Nucleic acid-containing amyloid fibrils potently induce type I interferon and stimulate systemic autoimmunity. Proc Natl Acad Sci USA. (2012) 109:14550–5. doi: 10.1073/pnas.1206923109
8. Kagan BL, Jang H, Capone R, Arce FT, Ramachandran S, Lal R, et al. Antimicrobial properties of amyloid peptides. Mol Pharmaceutics. (2011) 9:708–17. doi: 10.1021/mp200419b
9. Gilliet M, Lande R. Antimicrobial peptides and self-DNA in autoimmune skin inflammation. Curr Opin Immunol. (2008) 20:401–7. doi: 10.1016/j.coi.2008.06.008
10. Lee EY, Lee MW, Wong GCL. Modulation of toll-like receptor signaling by antimicrobial peptides. Semin Cell Dev Biol. (2019) 88:173–84. doi: 10.1016/j.semcdb.2018.02.002
11. Zhang L-J, Gallo RL. Antimicrobial peptides. Curr Biol. (2016) 26:R14–9. doi: 10.1016/j.cub.2015.11.017
12. Gallo PM, Rapsinski GJ, Wilson RP, Oppong GO, Sriram U, Goulian M, et al. Amyloid-DNA composites of bacterial biofilms stimulate autoimmunity. Immunity. (2015) 42:1171–84. doi: 10.1016/j.immuni.2015.06.002
13. Nicastro L, Tükel Ç. Bacterial amyloids: the link between bacterial infections and autoimmunity. Trends Microbiol. (2019) 27:954–63. doi: 10.1016/j.tim.2019.07.002
14. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. (2008) 9:857–65. doi: 10.1038/ni.1636
15. Landreh M, Johansson J, Jörnvall H. Separate molecular determinants in amyloidogenic and antimicrobial peptides. J Mol Biol. (2014) 426:2159–66. doi: 10.1016/j.jmb.2014.03.005
16. Bulet P, Stöcklin R, Menin L. Anti-microbial peptides: from invertebrates to vertebrates. Immunol Rev. (2004) 198:169–84. doi: 10.1111/j.0105-2896.2004.0124.x
17. Tincu JA, Taylor SW. Antimicrobial peptides from marine invertebrates. Antimicrob Agents Chemother. (2004) 48:3645–54. doi: 10.1128/AAC.48.10.3645-3654.2004
18. Wiesner J, Vilcinskas A. Antimicrobial peptides: the ancient arm of the human immune system. Virulence. (2010) 1:440–64. doi: 10.4161/viru.1.5.12983
19. Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. (2003) 3:710–20. doi: 10.1038/nri1180
20. Brown KL, Hancock REW. Cationic host defense (antimicrobial) peptides. Curr Opin Immunol. (2006) 18:24–30. doi: 10.1016/j.coi.2005.11.004
21. Lehrer RI, Ganz T. Antimicrobial peptides in mammalian and insect host defence. Curr Opin Immunol. (1999) 11:23–7. doi: 10.1016/S0952-7915(99)80005-3
22. Wang G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J Biol Chem. (2008) 283:32637–43. doi: 10.1074/jbc.M805533200
23. Hoover DM, Rajashankar KR, Blumenthal R, Puri A, Oppenheim JJ, Chertov O, et al. The structure of human beta-defensin-2 shows evidence of higher order oligomerization. J Biol Chem. (2000) 275:32911–8. doi: 10.1074/jbc.M006098200
24. Zhang X, Chen L, Bancroft DP, Lai CK, Maione TE. Crystal structure of recombinant human platelet factor 4. Biochemistry. (1994) 33:8361–6. doi: 10.1021/bi00193a025
25. Chang C, Magracheva E, Kozlov S, Fong S, Tobin G, Kotenko S, et al. Crystal structure of interleukin-19 defines a new subfamily of helical cytokines. J Biol Chem. (2003) 278:3308–13. doi: 10.1074/jbc.M208602200
26. Crescenzi O, Tomaselli S, Guerrini R, Salvadori S, D'Ursi AM, Temussi PA, et al. Solution structure of the Alzheimer amyloid beta-peptide (1-42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur J Biochem. (2002) 269:5642–8. doi: 10.1046/j.1432-1033.2002.03271.x
27. Rao JN, Jao CC, Hegde BG, Langen R, Ulmer TS. A combinatorial NMR and EPR approach for evaluating the structural ensemble of partially folded proteins. J Am Chem Soc. (2010) 132:8657–68. doi: 10.1021/ja100646t
28. Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, et al. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature. (2001) 414:454–7. doi: 10.1038/35106587
29. Kahlenberg JM, Kaplan MJ. Little peptide, big effects: the role of LL-37 in inflammation and autoimmune disease. J Immunol. (2013) 191:4895–901. doi: 10.4049/jimmunol.1302005
30. Shai Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta. (1999) 1462:55–70. doi: 10.1016/S0005-2736(99)00200-X
31. Oren Z, Shai Y. Mode of action of linear amphipathic α-helical antimicrobial peptides. Biopolymers. (1998) 47:451–63. doi: 10.1002/(SICI)1097-0282(1998)47:6<451::AID-BIP4>3.0.CO;2-F
32. Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. (2005) 3:238–50. doi: 10.1038/nrmicro1098
33. Lee EY, Lee MW, Fulan BM, Ferguson AL, Wong GCL. What can machine learning do for antimicrobial peptides, and what can antimicrobial peptides do for machine learning? Interface Focus. (2017) 7:20160153. doi: 10.1098/rsfs.2016.0153
34. Schmidt NW, Mishra A, Lai GH, Davis M, Sanders LK, Tran D, et al. Criterion for amino acid composition of defensins and antimicrobial peptides based on geometry of membrane destabilization. J Am Chem Soc. (2011) 133:6720–7. doi: 10.1021/ja200079a
35. Park CB, Kim HS, Kim SC. Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem Biophys Res Commun. (1998) 244:253–7. doi: 10.1006/bbrc.1998.8159
36. Kurosaka K, Chen Q, Yarovinsky F, Oppenheim JJ, Yang D. Mouse cathelin-related antimicrobial peptide chemoattracts leukocytes using formyl peptide receptor-like 1/mouse formyl peptide receptor-like 2 as the receptor and acts as an immune adjuvant. J Immunol. (2005) 174:6257–65. doi: 10.4049/jimmunol.174.10.6257
37. Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, et al. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science. (1999) 286:525–8. doi: 10.1126/science.286.5439.525
38. Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. (2002) 298:1025–9. doi: 10.1126/science.1075565
39. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang Y-H, Homey B, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. (2007) 449:564–9. doi: 10.1038/nature06116
40. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra19. doi: 10.1126/scitranslmed.3001180
41. Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, Conrad C, et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun. (2014) 5:5621. doi: 10.1038/ncomms6621
42. Lande R, Chamilos G, Ganguly D, Demaria O, Frasca L, Durr S, et al. Cationic antimicrobial peptides in psoriatic skin cooperate to break innate tolerance to self-DNA. Eur J Immunol. (2015) 45:203–13. doi: 10.1002/eji.201344277
43. Schmidt NW, Jin F, Lande R, Curk T, Xian W, Lee C, et al. Liquid-crystalline ordering of antimicrobial peptide-DNA complexes controls TLR9 activation. Nat Mater. (2015) 14:696–700. doi: 10.1038/nmat4298
44. Lande R, Lee EY, Palazzo R, Marinari B, Pietraforte I, Santos GS, et al. CXCL4 assembles DNA into liquid crystalline complexes to amplify TLR9-mediated interferon-α production in systemic sclerosis. Nat Commun. (2019) 10:1731. doi: 10.1038/s41467-019-09683-z
45. Lee EY, Lee CK, Schmidt NW, Jin F, Lande R, Curk T, et al. A review of immune amplification via ligand clustering by self-assembled liquid-crystalline DNA complexes. Adv Colloid Interface Sci. (2016) 232:17–24. doi: 10.1016/j.cis.2016.02.003
46. Wong GCL, Pollack L. Electrostatics of strongly charged biological polymers: ion-mediated interactions and self-organization in nucleic acids and proteins. Annu Rev Phys Chem. (2010) 61:171–89. doi: 10.1146/annurev.physchem.58.032806.104436
47. Wong GCL. Electrostatics of rigid polyelectrolytes. Curr Opin Colloid Interface Sci. (2006) 11:310–5. doi: 10.1016/j.cocis.2006.12.003
48. Chen Q, Bae SC, Granick S. Directed self-assembly of a colloidal kagome lattice. Nature. (2011) 469:381–4. doi: 10.1038/nature09713
49. Jiang S, Chen Q, Tripathy M, Luijten E, Schweizer KS, Granick S. Janus particle synthesis and assembly. Adv Mater Weinheim. (2010) 22:1060–71. doi: 10.1002/adma.200904094
50. Terwilliger TC, Eisenberg D. The structure of melittin. II. Interpretation of the structure. J Biol Chem. (1982) 257:6016–22.
51. Park CB, Yi KS, Matsuzaki K, Kim MS, Kim SC. Structure-activity analysis of buforin II, a histone H2A-derived antimicrobial peptide: the proline hinge is responsible for the cell-penetrating ability of buforin II. Proc Natl Acad Sci USA. (2000) 97:8245–50. doi: 10.1073/pnas.150518097
52. Bernard JJ, Cowing-Zitron C, Nakatsuji T, Muehleisen B, Muto J, Borkowski AW, et al. Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nat Med. (2012) 18:1286–90. doi: 10.1038/nm.2861
53. Adase CA, Borkowski AW, Zhang L-J, Williams MR, Sato E, Sanford JA, et al. Non-coding double-stranded RNA and antimicrobial peptide LL-37 induce growth factor expression from keratinocytes and endothelial cells. J Biol Chem. (2016) 291:11635–46. doi: 10.1074/jbc.M116.725317
54. Zhang L-J, Sen GL, Ward NL, Johnston A, Chun K, Chen Y, et al. Antimicrobial peptide LL37 and MAVS signaling drive interferon-β production by epidermal keratinocytes during skin injury. Immunity. (2016) 45:119–30. doi: 10.1016/j.immuni.2016.06.021
55. Takahashi T, Kulkarni NN, Lee EY, Zhang L-J, Wong GCL, Gallo RL. Cathelicidin promotes inflammation by enabling binding of self-RNA to cell surface scavenger receptors. Sci Rep. (2018) 8:4032. doi: 10.1038/s41598-018-22409-3
56. Lee EY, Takahashi T, Curk T, Dobnikar J, Gallo RL, Wong GCL. Crystallinity of double-stranded RNA-antimicrobial peptide complexes modulates toll-like receptor 3-mediated inflammation. ACS Nano. (2017) 11:12145–55. doi: 10.1021/acsnano.7b05234
57. Chen Y-P, Wu H-L, Boyé K, Pan C-Y, Chen Y-C, Pujol N, et al. Oligomerization state of CXCL4 chemokines regulates G protein-coupled receptor activation. ACS Chem Biol. (2017) 12:2767–78. doi: 10.1021/acschembio.7b00704
58. Yount NY, Cohen SE, Kupferwasser D, Waring AJ, Ruchala P, Sharma S, et al. Context mediates antimicrobial efficacy of kinocidin congener peptide RP-1. PLoS ONE. (2011) 6:e26727. doi: 10.1371/journal.pone.0026727
59. Yeaman MR, Yount NY, Waring AJ, Gank KD, Kupferwasser D, Wiese R, et al. Modular determinants of antimicrobial activity in platelet factor-4 family kinocidins. Biochim Biophys Acta. (2007) 1768:609–19. doi: 10.1016/j.bbamem.2006.11.010
60. Yang D, Chen Q, Hoover DM, Staley P, Tucker KD, Lubkowski J, et al. Many chemokines including CCL20/MIP-3α display antimicrobial activity. J Leukocyte Biol. (2003) 74:448–55. doi: 10.1189/jlb.0103024
61. Xiong YQ, Bayer AS, Elazegui L, Yeaman MR. A synthetic congener modeled on a microbicidal domain of thrombin- induced platelet microbicidal protein 1 recapitulates staphylocidal mechanisms of the native molecule. Antimicrob Agents Chemother. (2006) 50:3786–92. doi: 10.1128/AAC.00038-06
62. Yeaman MR. Platelets: at the nexus of antimicrobial defence. Nat Rev Microbiol. (2014) 12:426–37. doi: 10.1038/nrmicro3269
63. Struyf S, Salogni L, Burdick MD, Vandercappellen J, Gouwy M, Noppen S, et al. Angiostatic and chemotactic activities of the CXC chemokine CXCL4L1 (platelet factor-4 variant) are mediated by CXCR3. Blood. (2011) 117:480–8. doi: 10.1182/blood-2009-11-253591
64. Arepally GM. Heparin-induced thrombocytopenia. Blood. (2017) 129:2864–72. doi: 10.1182/blood-2016-11-709873
65. Warkentin TE. Heparin-induced thrombocytopenia. Curr Opin Crit Care. (2015) 21:576–85. doi: 10.1097/MCC.0000000000000259
66. Bloom MB, Johnson J, Volod O, Lee EY, White T, Margulies DR. Improved prediction of HIT in the SICU using an improved model of the Warkentin 4-T system: 3-T. Am J Surg. (2020) 219:54–7. doi: 10.1016/j.amjsurg.2019.07.039
67. Meller S, Di Domizio J, Voo KS, Friedrich HC, Chamilos G, Ganguly D, et al. TH17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol. (2015) 16:970–9. doi: 10.1038/ni.3211
68. Hör S, Pirzer H, Dumoutier L, Bauer F, Wittmann S, Sticht H, et al. The T-cell lymphokine interleukin-26 targets epithelial cells through the interleukin-20 receptor 1 and interleukin-10 receptor 2 chains. J Biol Chem. (2004) 279:33343–51. doi: 10.1074/jbc.M405000200
69. Ma CS, Chew GYJ, Simpson N, Priyadarshi A, Wong M, Grimbacher B, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. (2008) 205:1551–7. doi: 10.1084/jem.20080218
70. Raffatellu M, Santos RL, Verhoeven DE, George MD, Wilson RP, Winter SE, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med. (2008) 14:421–8. doi: 10.1038/nm1743
71. Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, et al. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol. (2003) 4:1009–15. doi: 10.1038/ni978
72. Hibbert L, Pflanz S, de Waal Malefyt R, Kastelein RA. IL-27 and IFN-α signal via Stat1 and Stat3 and induce T-Bet and IL-12Rβ 2 in naive T cells. J Interferon Cytokine Res. (2003) 23:513–22. doi: 10.1089/10799900360708632
73. Santini SM, Lapenta C, Logozzi M, Parlato S, Spada M, Di Pucchio T, et al. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J ExpMed. (2000) 191:1777–88. doi: 10.1084/jem.191.10.1777
74. Luft T, Pang KC, Thomas E, Hertzog P, Hart D, Trapani J, et al. Type I IFNs enhance the terminal differentiation of dendritic cells. J Immunol. (1998) 161:1947–53.
75. Jego G, Palucka AK, Blanck J-P, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. (2003) 19:225–34. doi: 10.1016/S1074-7613(03)00208-5
76. Parker D, Cohen TS, Alhede M, Harfenist BS, Martin FJ, Prince A. Induction of type I interferon signaling by Pseudomonas aeruginosa is diminished in cystic fibrosis epithelial cells. Am J Respir Cell Mol Biol. (2012) 46:6–13. doi: 10.1165/rcmb.2011-0080OC
77. Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol. (2007) 178:3126–33. doi: 10.4049/jimmunol.178.5.3126
78. Venet F, Huang X, Chung C-S, Chen Y, Ayala A. Plasmacytoid dendritic cells control lung inflammation and monocyte recruitment in indirect acute lung injury in mice. Am J Pathol. (2010) 176:764–73. doi: 10.2353/ajpath.2010.090765
79. Kaplan A, Lee MW, Wolf AJ, Limon JJ, Becker CA, Ding M, et al. Direct antimicrobial activity of IFN-β. J Immunol. (2017) 198:4036–45. doi: 10.4049/jimmunol.1601226
80. Yount NY, Weaver DC, Lee EY, Lee MW, Wang H, Chan LC, et al. Unifying structural signature of eukaryotic α-helical host defense peptides. Proc Natl Acad Sci USA. (2019) 116:6944–53. doi: 10.1073/pnas.1819250116
81. Lee MW, Lee EY, Wong GCL. What can pleiotropic proteins in innate immunity teach us about bioconjugation and molecular design? Bioconjug Chem. (2018) 29:2127–39. doi: 10.1021/acs.bioconjchem.8b00176
82. Tayeb-Fligelman E, Tabachnikov O, Moshe A, Goldshmidt-Tran O, Sawaya MR, Coquelle N, et al. The cytotoxic Staphylococcus aureus PSMα3 reveals a cross-α amyloid-like fibril. Science. (2017) 355:831–3. doi: 10.1126/science.aaf4901
83. Zhang S-Q, Huang H, Yang J, Kratochvil HT, Lolicato M, Liu Y, et al. Designed peptides that assemble into cross-α amyloid-like structures. Nat Chem Biol. (2018) 14:870–5. doi: 10.1038/s41589-018-0105-5
84. Friedman R, Pellarin R, Caflisch A. Amyloid aggregation on lipid bilayers and its impact on membrane permeability. J Mol Biol. (2009) 387:407–15. doi: 10.1016/j.jmb.2008.12.036
85. Hardy JA, Higgins GA. Alzheimers-disease - the amyloid cascade hypothesis. Science. (1992) 256:184–5. doi: 10.1126/science.1566067
86. Chen JX, Yan SD. Amyloid-beta-induced mitochondrial dysfunction. J Alzheimers Dis. (2007) 12:177–84. doi: 10.3233/JAD-2007-12208
87. Chen X, Petranovic D. Amyloid-β peptide-induced cytotoxicity and mitochondrial dysfunction in yeast. FEMS Yeast Res. (2015) 15:fov061. doi: 10.1093/femsyr/fov061
88. Cha M-Y, Han S-H, Son SM, Hong H-S, Choi Y-J, Byun J, et al. Mitochondria-specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS ONE. (2012) 7:e34929. doi: 10.1371/journal.pone.0034929
89. Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol Med. (2008) 14:45–53. doi: 10.1016/j.molmed.2007.12.002
90. Storey E, Cappai R. The amyloid precursor protein of Alzheimer's disease and the Abeta peptide. Neuropathol Appl Neurobiol. (1999) 25:81–97. doi: 10.1046/j.1365-2990.1999.00164.x
91. Ambroggio EE, Kim DH, Separovic F, Barrow CJ, Barnham KJ, Bagatolli LA, et al. Surface behavior and lipid interaction of Alzheimer beta-amyloid peptide 1-42: a membrane-disrupting peptide. Biophys J. (2005) 88:2706–13. doi: 10.1529/biophysj.104.055582
92. Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS ONE. (2010) 5:e9505. doi: 10.1371/journal.pone.0009505
93. Kumar DKV, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci Transl Med. (2016) 8:340ra72. doi: 10.1126/scitranslmed.aaf1059
94. Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer's disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron. (2018) 99:56–63.e3. doi: 10.1016/j.neuron.2018.06.030
95. Welling MM, Nabuurs RJA, van der Weerd L. Potential role of antimicrobial peptides in the early onset of Alzheimer's disease. Alzheimers Dement. (2015) 11:51–7. doi: 10.1016/j.jalz.2013.12.020
96. Tanzi RE, Bertram L. New frontiers in Alzheimer's disease genetics. Neuron. (2001) 32:181–4. doi: 10.1016/S0896-6273(01)00476-7
97. Ashford JW, Mortimer JA. Non-familial Alzheimer's disease is mainly due to genetic factors. J Alzheimers Dis. (2002) 4:169–77. doi: 10.3233/JAD-2002-4307
98. Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, et al. The 'Arctic‘ APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nat Neurosci. (2001) 4:887–93. doi: 10.1038/nn0901-887
99. van Nostrand WE. The influence of the amyloid ß-protein and its precursor in modulating cerebral hemostasis. Biochim Biophys Acta. (2016) 1862:1018–26. doi: 10.1016/j.bbadis.2015.10.020
100. Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. (1993) 90:9649–53. doi: 10.1073/pnas.90.20.9649
101. Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA. (1993) 90:8098–102. doi: 10.1073/pnas.90.17.8098
102. Wildsmith KR, Holley M, Savage JC, Skerrett R, Landreth GE. Evidence for impaired amyloid β clearance in Alzheimer's disease. Alzheimers Res Ther. (2013) 5:33. doi: 10.1186/alzrt187
103. Yamaguchi Y, Nagase T, Tomita T, Nakamura K, Fukuhara S, Amano T, et al. Beta-defensin overexpression induces progressive muscle degeneration in mice. Am J Physiol Cell Physiol. (2007) 292:C2141–9. doi: 10.1152/ajpcell.00295.2006
104. Reinholz M, Ruzicka T, Schauber J. Cathelicidin LL-37: an antimicrobial peptide with a role in inflammatory skin disease. Ann Dermatol. (2012) 24:126–35. doi: 10.5021/ad.2012.24.2.126
105. Cao Y, Chtarbanova S, Petersen AJ, Ganetzky B. Dnr1 mutations cause neurodegeneration in Drosophila by activating the innate immune response in the brain. Proc Natl Acad Sci USA. (2013) 110:E1752–60. doi: 10.1073/pnas.1306220110
106. Pütsep K, Carlsson G, Boman HG, Andersson M. Deficiency of antibacterial peptides in patients with morbus Kostmann: an observation study. Lancet. (2002) 360:1144–49. doi: 10.1016/S0140-6736(02)11201-3
107. Ciornei CD, Tapper H, Bjartell A, Sternby NH, Bodelsson M. Human antimicrobial peptide LL-37 is present in atherosclerotic plaques and induces death of vascular smooth muscle cells: a laboratory study. BMC Cardiovasc Disord. (2006) 6:49–12. doi: 10.1186/1471-2261-6-49
108. Krause A, Liepke C, Meyer M, Adermann K, Forssmann WG, Maronde E. Human natriuretic peptides exhibit antimicrobial activity. Eur J Med Res. (2001) 6:215–8. Available online at: https://pubmed.ncbi.nlm.nih.gov/11410403/
109. Engelberg Y, Landau M. The human LL-37(17-29) antimicrobial peptide reveals a functional supramolecular nanostructure. bioRxiv. 18:2020.02.04.933432 (2020). doi: 10.1101/2020.02.04.933432
110. Swaminathan R, Ravi VK, Kumar S, Kumar MVS, Chandra N. Lysozyme: a model protein for amyloid research. Adv Protein Chem Struct Biol. (2011) 84:63–111. doi: 10.1016/B978-0-12-386483-3.00003-3
111. Jang H, Arce FT, Mustata M, Ramachandran S, Capone R, Nussinov R, et al. Antimicrobial protegrin-1 forms amyloid-like fibrils with rapid kinetics suggesting a functional link. Biophys J. (2011) 100:1775–83. doi: 10.1016/j.bpj.2011.01.072
112. Garvey M, Meehan S, Gras SL, Schirra HJ, Craik DJ, van der Weerden NL, et al. A radish seed antifungal peptide with a high amyloid fibril-forming propensity. Biochim Biophys Acta. (2013) 1834:1615–23. doi: 10.1016/j.bbapap.2013.04.030
113. Mahalka AK, Kinnunen PKJ. Binding of amphipathic alpha-helical antimicrobial peptides to lipid membranes: lessons from temporins B and L. Biochim Biophys Acta. (2009) 1788:1600–9. doi: 10.1016/j.bbamem.2009.04.012
114. Code C, Domanov YA, Killian JA, Kinnunen PKJ. Activation of phospholipase A2 by temporin B: formation of antimicrobial peptide-enzyme amyloid-type cofibrils. Biochim Biophys Acta. (2009) 1788:1064–72. doi: 10.1016/j.bbamem.2009.03.002
115. Schnaider L, Rosenberg A, Kreiser T, Kolusheva S, Gazit E, Berman J. Peptide self-assembly is linked to antibacterial, but not antifungal, activity of histatin 5 derivatives. mSphere. (2020) 5:943. doi: 10.1128/mSphere.00021-20
116. Wang J, Li Y, Wang X, Chen W, Sun H, Wang J. Lipopolysaccharide induces amyloid formation of antimicrobial peptide HAL-2. Biochim Biophys Acta. (2014) 1838:2910–18. doi: 10.1016/j.bbamem.2014.07.028
117. Calabrese AN, Liu Y, Wang T, Musgrave IF, Pukala TL, Tabor RF, et al. The amyloid fibril-forming properties of the amphibian antimicrobial peptide Uperin 3.5. Chembiochem. (2016) 17:239–46. doi: 10.1002/cbic.201500518
118. Caillon L, Killian JA, Lequin O, Khemtémourian L. Biophysical investigation of the membrane-disrupting mechanism of the antimicrobial and amyloid-like peptide dermaseptin S9. PLoS ONE. (2013) 8:e75528. doi: 10.1371/journal.pone.0075528
119. Gour S, Kaushik V, Kumar V, Bhat P, Yadav SC, Yadav JK. Antimicrobial peptide (Cn-AMP2) from liquid endosperm of Cocos nucifera forms amyloid-like fibrillar structure. J Pept Sci. (2016) 22:201–7. doi: 10.1002/psc.2860
120. Sayegh RSR, Batista I de FC, Melo RL de, Riske KA, Daffre S, Montich G, et al. Longipin: an amyloid antimicrobial peptide from the harvestman Acutisoma longipes (Arachnida: Opiliones) with preferential affinity for anionic vesicles. PLoS ONE. (2016) 11:e0167953. doi: 10.1371/journal.pone.0167953
121. Villarroel F, Bastías A, Casado A, Amthauer R, Concha MI. Apolipoprotein A-I, an antimicrobial protein in Oncorhynchus mykiss: evaluation of its expression in primary defence barriers and plasma levels in sick and healthy fish. Fish Shellfish Immunol. (2007) 23:197–209. doi: 10.1016/j.fsi.2006.10.008
122. Schnaider L, Brahmachari S, Schmidt NW, Mensa B, Shaham-Niv S, Bychenko D, et al. Self-assembling dipeptide antibacterial nanostructures with membrane disrupting activity. Nat Commun. (2017) 8:1365. doi: 10.1038/s41467-017-01447-x
123. Arnusch CJ, Branderhorst H, de Kruijff B, Liskamp RMJ, Breukink E, Pieters RJ. Enhanced membrane pore formation by multimeric/oligomeric antimicrobial peptides. Biochemistry. (2007) 46:13437–42. doi: 10.1021/bi7015553
124. Nguyen LT, Vogel HJ. Structural perspectives on antimicrobial chemokines. Front Immunol. (2012) 3:384. doi: 10.3389/fimmu.2012.00384
125. Torrent M, Pulido D, Nogués MV, Boix E. Exploring new biological functions of amyloids: bacteria cell agglutination mediated by host protein aggregation. PLoS Pathog. (2012) 8:e1003005. doi: 10.1371/journal.ppat.1003005
126. Wang L, Liu Q, Chen J-C, Cui Y-X, Zhou B, Chen Y-X, et al. Antimicrobial activity of human islet amyloid polypeptides: an insight into amyloid peptides' connection with antimicrobial peptides. Biol Chem. (2012) 393:641–6. doi: 10.1515/hsz-2012-0107
127. Pasupuleti M, Davoudi M, Malmsten M, Schmidtchen A. Antimicrobial activity of a C-terminal peptide from human extracellular superoxide dismutase. BMC Res Notes. (2009) 2:136–6. doi: 10.1186/1756-0500-2-136
128. Pasupuleti M, Roupe M, Rydengård V, Surewicz K, Surewicz WK, Chalupka A, et al. Antimicrobial activity of human prion protein is mediated by its N-terminal region. PLoS One. (2009) 4:e7358. doi: 10.1371/journal.pone.0007358
129. Vernekar V, Velhal S, Bandivdekar A. Evaluation of cystatin C activities against HIV. Indian J Med Res. (2015) 141:423–30. doi: 10.4103/0971-5916.159282
130. Stefanovic AND, Stöckl MT, Claessens MMAE, Subramaniam V. α-Synuclein oligomers distinctively permeabilize complex model membranes. FEBS J. (2014) 281:2838–50. doi: 10.1111/febs.12824
131. Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. (2013) 14:38–48. doi: 10.1038/nrn3406
132. Tosatto L, Andrighetti AO, Plotegher N, Antonini V, Tessari I, Ricci L, et al. Alpha-synuclein pore forming activity upon membrane association. Biochim Biophys Acta. (2012) 1818:2876–83. doi: 10.1016/j.bbamem.2012.07.007
133. Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, et al. Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry. (2001) 40:7812–9. doi: 10.1021/bi0102398
134. Volles MJ, Lansbury PT. Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson's disease-linked mutations and occurs by a pore-like mechanism. Biochemistry. (2002) 41:4595–602. doi: 10.1021/bi0121353
135. Shi Z, Sachs JN, Rhoades E, Baumgart T. Biophysics of α-synuclein induced membrane remodelling. Phys Chem Chem Phys. (2015) 17:15561–8. doi: 10.1039/C4CP05883F
136. Pfefferkorn CM, Jiang Z, Lee JC. Biophysics of α-synuclein membrane interactions. Biochim Biophys Acta. (2012) 1818:162–71. doi: 10.1016/j.bbamem.2011.07.032
137. Bartels T, Ahlstrom LS, Leftin A, Kamp F, Haass C, Brown MF, et al. The N-Terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophysj. (2010) 99:2116–24. doi: 10.1016/j.bpj.2010.06.035
138. Vamvaca K, Volles MJ, Lansbury PT Jr. The first N-terminal amino acids of α-synuclein are essential for α-helical structure formation in vitro and membrane binding in yeast. J Mol Biol. (2009) 389:413–24. doi: 10.1016/j.jmb.2009.03.021
139. Park S-C, Moon JC, Shin SY, Son H, Jung YJ, Kim N-H, et al. Functional characterization of alpha-synuclein protein with antimicrobial activity. Biochem Biophys Res Commun. (2016) 478:924–8. doi: 10.1016/j.bbrc.2016.08.052
140. Wang S, Chu C-H, Stewart T, Ginghina C, Wang Y, Nie H, et al. α-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc Natl Acad Sci USA. (2015) 112:E1926–35. doi: 10.1073/pnas.1417883112
141. Stolzenberg E, Berry D, Yang D, Lee EY, Kroemer A, Kaufman S, et al. A role for neuronal α-synuclein in gastrointestinal immunity. J Innate Immun. (2017) 9:456–63. doi: 10.1159/000477990
142. Grathwohl SA, Steiner JA, Britschgi M, Brundin P. Mind the gut: secretion of α-synuclein by enteric neurons. J Neurochem. (2013) 125:487–90. doi: 10.1111/jnc.12191
143. Lebouvier T, Chaumette T, Damier P, Coron E, Touchefeu Y, Vrignaud S, et al. Pathological lesions in colonic biopsies during Parkinson's disease. Gut. (2008) 57:1741–3. doi: 10.1136/gut.2008.162503
144. Perni M, Galvagnion C, Maltsev A, Meisl G, Müller MBD, Challa PK, et al. A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc Natl Acad Sci USA. (2017) 114:E1009–17. doi: 10.1073/pnas.1610586114
145. Kobayashi N, masuda J, kudoh J, shimizu N, yoshida T. Binding sites on tau proteins as components for antimicrobial peptides. Biocontrol Sci. (2008) 13:49–56. doi: 10.4265/bio.13.49
146. Last NB, Rhoades E, Miranker AD. Islet amyloid polypeptide demonstrates a persistent capacity to disrupt membrane integrity. Proc Natl Acad Sci USA. (2011) 108:9460–5. doi: 10.1073/pnas.1102356108
147. Engel MFM, Khemtémourian L, Kleijer CC, Meeldijk HJD, Jacobs J, Verkleij AJ, et al. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc Natl Acad Sci USA. (2008) 105:6033–8. doi: 10.1073/pnas.0708354105
148. Last NB, Miranker AD. Common mechanism unites membrane poration by amyloid and antimicrobial peptides. Proc Natl Acad Sci USA. (2013) 110:6382–7. doi: 10.1073/pnas.1219059110
149. Zhao H, Jutila A, Nurminen T, Wickström SA, Keski-Oja J, Kinnunen PKJ. Binding of endostatin to phosphatidylserine-containing membranes and formation of amyloid-like fibers. Biochemistry. (2005) 44:2857–63. doi: 10.1021/bi048510j
150. Michelle M, Barnhart MRC. Curli biogenesis and function. Annu Rev Microbiol. (2006) 60:131–47. doi: 10.1146/annurev.micro.60.080805.142106
151. Nicastro LK, Tursi SA, Le LS, Miller AL, Efimov A, Buttaro B, et al. Cytotoxic Curli intermediates form during Salmonella biofilm development. J Bacteriol. (2019) 201:7. doi: 10.1128/JB.00095-19
152. Lee EY, Fulan BM, Wong GCL, Ferguson AL. Mapping membrane activity in undiscovered peptide sequence space using machine learning. Proc Natl Acad Sci USA. (2016) 113:13588–93. doi: 10.1073/pnas.1609893113
153. Lee EY, Wong GCL, Ferguson AL. Machine learning-enabled discovery and design of membrane-active peptides. Bioorg Med Chem. (2018) 26:2708–18. doi: 10.1016/j.bmc.2017.07.012
154. Lee MW, Lee EY, Ferguson AL, Wong GCL. Machine learning antimicrobial peptide sequences: Some surprising variations on the theme of amphiphilic assembly. Curr Opin Colloid Interface Sci. (2018) 38:204–13. doi: 10.1016/j.cocis.2018.11.003
155. Silvestre-Roig C, Braster Q, Wichapong K, Lee EY, Teulon JM, Berrebeh N, et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature. (2019) 569:236–40. doi: 10.1038/s41586-019-1167-6
156. Lee MW, Lee EY, Lai GH, Kennedy NW, Posey AE, Xian W, et al. Molecular motor dnm1 synergistically induces membrane curvature to facilitate mitochondrial fission. ACS Cent Sci. (2017) 3:1156–67. doi: 10.1021/acscentsci.7b00338
157. Taglialegna A, Lasa I, Valle J, O'Toole GA. Amyloid structures as biofilm matrix scaffolds. J Bacteriol. (2016) 198:2579–88. doi: 10.1128/JB.00122-16
158. Tükel Ç, Nishimori JH, Wilson RP, Winter MG, Keestra AM, Van Putten JPM, et al. Toll-like receptors 1 and 2 cooperatively mediate immune responses to curli, a common amyloid from enterobacterial biofilms. Cell Microbiol. (2010) 12:1495–505. doi: 10.1111/j.1462-5822.2010.01485.x
159. Tükel Ç, Raffatellu M, Humphries AD, Wilson RP, Andrews Polymenis HL, Gull T, et al. CsgA is a pathogen-associated molecular pattern of Salmonella enterica serotype Typhimurium that is recognized by Toll-like receptor 2. Mol Microbiol. (2005) 58:289–304. doi: 10.1111/j.1365-2958.2005.04825.x
160. Tükel Ç, Wilson RP, Nishimori JH, Pezeshki M, Chromy BA, Bäumler AJ. Responses to amyloids of microbial and host origin are mediated through toll-like receptor 2. Cell Host Microbe. (2009) 6:45–53. doi: 10.1016/j.chom.2009.05.020
161. Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, et al. TLR2 is a primary receptor for Alzheimer's amyloid β peptide to trigger neuroinflammatory activation. J Immunol. (2012) 188:1098–107. doi: 10.4049/jimmunol.1101121
162. Cheng N, He R, Tian J, Ye PP, Ye RD. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J Immunol. (2008) 181:22–6. doi: 10.4049/jimmunol.181.1.22
163. Rapsinski GJ, Newman TN, Oppong GO, van Putten JPM, Tükel Ç. CD14 protein acts as an adaptor molecule for the immune recognition of Salmonella curli fibers. J Biol Chem. (2013) 288:14178–88. doi: 10.1074/jbc.M112.447060
164. Rapsinski GJ, Wynosky-Dolfi MA, Oppong GO, Tursi SA, Wilson RP, Brodsky IE, et al. Toll-like receptor 2 and NLRP3 cooperate to recognize a functional bacterial amyloid, curli. Infect Immun. (2015) 83:693–701. doi: 10.1128/IAI.02370-14
165. Niemi K, Teirilä L, Lappalainen J, Rajamäki K, Baumann MH, Öörni K, Wolff H, et al. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J Immunol. (2011) 186:6119–28. doi: 10.4049/jimmunol.1002843
166. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. (2010) 11:897–904. doi: 10.1038/ni.1935
167. Tang S-C, Lathia JD, Selvaraj PK, Jo D-G, Mughal MR, Cheng A, et al. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp Neurol. (2008) 213:114–21. doi: 10.1016/j.expneurol.2008.05.014
168. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. (2010) 11:155–61. doi: 10.1038/ni.1836
169. Rosani U, Domeneghetti S, Gerdol M, Franzoi M, Pallavicini A, Venier P. Serum amyloid A in marine bivalves: an acute phase and innate immunity protein. Dev Comp Immunol. (2016) 59:136–44. doi: 10.1016/j.dci.2016.01.019
170. Grimaldi A, Tettamanti G, Girardello R, Pulze L, Valvassori R, Malagoli D, et al. Functional amyloid formation in LPS activated cells from invertebrates to vertebrates. Isj Invertebrate Survival J. (2014) 11:286–97. Available online at: http://www.isj.unimore.it/index.php/ISJ/article/view/329
171. Falabella P, Riviello L, Pascale M, Lelio ID, Tettamanti G, Grimaldi A, et al. Functional amyloids in insect immune response. Insect Biochem Mol Biol. (2012) 42:203–11. doi: 10.1016/j.ibmb.2011.11.011
172. Liu C, Zhang Y. Nucleic acid-mediated protein aggregation and assembly. Adv Protein Chem Struct Biol. (2011) 84:1–40. doi: 10.1016/B978-0-12-386483-3.00005-7
173. Braun S, Humphreys C, Fraser E, Brancale A, Bochtler M, Dale TC. Amyloid-associated nucleic acid hybridisation. PLoS ONE. (2011) 6:e19125. doi: 10.1371/journal.pone.0019125
174. Yu H, Ren J, Qu X. Time-dependent DNA condensation induced by amyloid beta-peptide. Biophys J. (2007) 92:185–91. doi: 10.1529/biophysj.106.093559
175. Camero S, Ayuso JM, Barrantes A, Benítez MJ, Jiménez JS. Specific binding of DNA to aggregated forms of Alzheimer's disease amyloid peptides. Int J Biol Macromol. (2013) 55:201–6. doi: 10.1016/j.ijbiomac.2013.01.007
176. Goers J, Manning-Bog AB, McCormack AL, Millett IS, Doniach S, Di Monte DA. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry. (2003) 42:8465–71. doi: 10.1021/bi0341152
177. Gruden MA, Davudova TB, Mališauskas M, Zamotin VV, Sewell RDE, Voskresenskaya NI, et al. Autoimmune responses to amyloid structures of Aβ (25-35) peptide and human lysozyme in the serum of patients with progressive Alzheimer's disease. DEM. (2004) 18:165–71. doi: 10.1159/000079197
178. Cherny D, Hoyer W, Subramaniam V, Jovin TM. Double-stranded DNA stimulates the fibrillation of α-synuclein in vitro and is associated with the mature fibrils: an electron microscopy study. J Mol Biol. (2004) 344:929–38. doi: 10.1016/j.jmb.2004.09.096
179. Sørensen IJ, Holm Nielsen E, Schrøder L, Voss A, Horváth L, Svehag SE. Complexes of serum amyloid P component and DNA in serum from healthy individuals and systemic lupus erythematosus patients. J Clin Immunol. (2000) 20:408–15. doi: 10.1023/a:1026478914129
180. Voss A, Nielsen EH, Svehag SE, Junker P. Serum amyloid P component-DNA complexes are decreased in systemic lupus erythematosus. Inverse association with anti-dsDNA antibodies. J Rheumatol. (2008) 35:625–30. Available online at: https://pubmed.ncbi.nlm.nih.gov/18278838/
181. Di Domizio J, Zhang R, Stagg LJ, Gagea M, Zhuo M, Ladbury JE, et al. Binding with nucleic acids or glycosaminoglycans converts soluble protein oligomers to amyloid. J Biol Chem. (2012) 287:736–47. doi: 10.1074/jbc.M111.238477
182. Mao X, Li K, Liu M, Wang X, Zhao T, An B, et al. Directing curli polymerization with DNA origami nucleators. Nat Commun. (2019) 10:1395. doi: 10.1038/s41467-019-09369-6
183. Spaulding CN, Dodson KW, Chapman MR, Hultgren SJ. Fueling the fire with fibers: bacterial amyloids promote inflammatory disorders. Cell Host and Microbe. (2015) 18:1–2. doi: 10.1016/j.chom.2015.06.013
184. Ohnishi S, Koide A, Koide S. Solution conformation and amyloid-like fibril formation of a polar peptide derived from a beta-hairpin in the OspA single-layer beta-sheet. J Mol Biol. (2000) 301:477–89. doi: 10.1006/jmbi.2000.3980
185. Alteri CJ, Xicohténcatl-Cortes J, Hess S, Caballero-Olín G, Girón JA, Friedman RL. Mycobacterium tuberculosis produces pili during human infection. Proc Natl Acad Sci USA. (2007) 104:5145–50. doi: 10.1073/pnas.0602304104
186. Dueholm MS, Petersen SV, Sønderkær M, Larsen P, Christiansen G, Hein KL, et al. Functional amyloid in Pseudomonas. Mol Microbiol. (2010) 77:1009–20. doi: 10.1111/j.1365-2958.2010.07269.x
187. Bleem A, Christiansen G, Madsen DJ, Maric H, Strømgaard K, Bryers JD, et al. Protein engineering reveals mechanisms of functional amyloid formation in Pseudomonas aeruginosa biofilms. J Mol Biol. (2018) 430:3751–63. doi: 10.1016/j.jmb.2018.06.043
188. Schwartz K, Syed AK, Stephenson RE, Rickard AH, Boles BR. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. (2012) 8:e1002744. doi: 10.1371/journal.ppat.1002744
189. Peschel A, Otto M. Phenol-soluble modulins and staphylococcal infection. Nat Rev Microbiol. (2013) 11:667–73. doi: 10.1038/nrmicro3110
190. Zheng Y, Joo H-S, Nair V, Le KY, Otto M. Do amyloid structures formed by Staphylococcus aureus phenol-soluble modulins have a biological function? Int J Med Microbiol. (2017) 308:675–82. doi: 10.1016/j.ijmm.2017.08.010
191. Björnsdottir H, Dahlstrand Rudin A, Klose FP, Elmwall J, Welin A, Stylianou M, et al. Phenol-Soluble modulin α peptide toxins from aggressive Staphylococcus aureus induce rapid formation of neutrophil extracellular traps through a reactive oxygen species-independent pathway. Front Immunol. (2017) 8:257. doi: 10.3389/fimmu.2017.00257
192. Schwartz K, Ganesan M, Payne DE, Solomon MJ, Boles BR. Extracellular DNA facilitates the formation of functional amyloids in Staphylococcus aureus biofilms. Mol Microbiol. (2016) 99:123–34. doi: 10.1111/mmi.13219
193. Kretschmer D, Gleske A-K, Rautenberg M, Wang R, Köberle M, Bohn E, et al. Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell Host Microbe. (2010) 7:463–73. doi: 10.1016/j.chom.2010.05.012
194. Tursi SA, Tükel Ç. Curli-containing enteric biofilms inside and out: matrix composition, immune recognition, and disease implications. Microbiol Mol Biol Rev. (2018) 82:217. doi: 10.1128/MMBR.00028-18
195. Tursi SA, Puligedda RD, Szabo P, Nicastro LK, Miller AL, Qiu C, et al. Salmonella Typhimurium biofilm disruption by a human antibody that binds a pan-amyloid epitope on curli. Nat Commun. (2020) 11:1007–13. doi: 10.1038/s41467-020-14685-3
196. Chorell E, Andersson E, Evans ML, Jain N, Götheson A, Åden J, Chapman MR, et al. Bacterial chaperones CsgE and CsgC differentially modulate human α-synuclein amyloid formation via transient contacts. PLoS ONE. (2015) 10:e0140194. doi: 10.1371/journal.pone.0140194
197. Friedland RP, Chapman MR. The role of microbial amyloid in neurodegeneration. PLoS Pathog. (2017) 13:e1006654. doi: 10.1371/journal.ppat.1006654
198. Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, Hamilton AM, et al. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci. (2020) 23:327–36. doi: 10.1038/s41593-020-0589-7
199. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell. (2016) 167:1469–80.e12. doi: 10.1016/j.cell.2016.11.018
200. Berke IC, Yu X, Modis Y, Egelman EH. MDA5 assembles into a polar helical filament on dsRNA. Proc Natl Acad Sci USA. (2012) 109:18437–41. doi: 10.1073/pnas.1212186109
201. Peisley A, Lin C, Wu B, Orme-Johnson M, Liu M, Walz T, et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci USA. (2011) 108:21010–21015. doi: 10.1073/pnas.1113651108
202. Sohn J, Hur S. Filament assemblies in foreign nucleic acid sensors. Curr Opin Struct Biol. (2016) 37:134–44. doi: 10.1016/j.sbi.2016.01.011
203. Cadena C, Hur S. Filament-like assemblies of intracellular nucleic acid sensors: commonalities and differences. Mol Cell. (2019) 76:243–54. doi: 10.1016/j.molcel.2019.09.023
Keywords: antimicrobial peptides, amyloids, self-assembly, Toll-like receptors, innate immunity, autoimmune diseases, neurodegenerative diseases
Citation: Lee EY, Srinivasan Y, de Anda J, Nicastro LK, Tükel Ç and Wong GCL (2020) Functional Reciprocity of Amyloids and Antimicrobial Peptides: Rethinking the Role of Supramolecular Assembly in Host Defense, Immune Activation, and Inflammation. Front. Immunol. 11:1629. doi: 10.3389/fimmu.2020.01629
Received: 01 May 2020; Accepted: 17 June 2020;
Published: 31 July 2020.
Edited by:
Mark Hulett, La Trobe University, AustraliaReviewed by:
Fengliang Jin, South China Agricultural University, ChinaFelix Ngosa Toka, Warsaw University of Life Sciences, Poland
Copyright © 2020 Lee, Srinivasan, de Anda, Nicastro, Tükel and Wong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerard C. L. Wong, Z2Nsd29uZ0BzZWFzLnVjbGEuZWR1