 Juliano Vilela Alves1
Juliano Vilela Alves1 Rafael Menezes da Costa1,2
Rafael Menezes da Costa1,2 Camila André Pereira1
Camila André Pereira1 Aline Garcia Fedoce1
Aline Garcia Fedoce1 Carlos Alberto Aguiar Silva3
Carlos Alberto Aguiar Silva3 Fernando Silva Carneiro1
Fernando Silva Carneiro1 Núbia Souza Lobato2
Núbia Souza Lobato2 Rita C. Tostes1*
Rita C. Tostes1*- 1Department of Pharmacology, Ribeirão Preto Medical School, University of São Paulo, São Paulo, Brazil
- 2Special Academic Unit of Health Sciences, Federal University of Jataí, Jataí, Brazil
- 3Department of Physiology, Ribeirão Preto Medical School, University of São Paulo, São Paulo, Brazil
Background: Both supraphysiological and subphysiological testosterone levels are associated with increased cardiovascular risk. Testosterone consumption at supraphysiological doses has been linked to increased blood pressure, left ventricular hypertrophy, vascular dysfunction, and increased levels of inflammatory markers. Activation of the NLRP3 inflammasome contributes to the production of proinflammatory cytokines, leading to cardiovascular dysfunction. We hypothesized that supraphysiological levels of testosterone, via generation of mitochondrial reactive oxygen species (mROS), activates the NLRP3 inflammasome and promotes vascular dysfunction.
Methods: Male, 12 week-old C57Bl/6J (WT) and NLRP3 knockout (NLRP3−/−) mice were used. Mice were treated with testosterone propionate [TP (10 mg/kg) in vivo] or vehicle for 30 days. In addition, vessels were incubated with testosterone [Testo (10−6 M, 2 h) in vitro]. Testosterone levels, blood pressure, vascular function (thoracic aortic rings), pro-caspase-1/caspase-1 and interleukin-1β (IL-1β) expression, and generation of reactive oxygen species were determined.
Results: Testosterone increased contractile responses and reduced endothelium-dependent vasodilation, both in vivo and in vitro. These effects were not observed in arteries from NLRP3−/− mice. Aortas of TP-treated WT mice (in vivo), as well as aortas from WT mice incubated with testo (in vitro), exhibited increased mROS levels and increased caspase-1 and IL-1β expression. These effects were not observed in arteries from NLRP3−/− mice. Flutamide [Flu, 10−5 M, androgen receptor (AR) antagonist], carbonyl cyanide m-chlorophenyl hydrazone (CCCP, 10−6 M, mitochondrial uncoupler) and MCC950 (MCC950, 10−6 M, a NLRP3 receptor inhibitor) prevented testosterone-induced mROS generation.
Conclusion: Supraphysiological levels of testosterone induce vascular dysfunction via mROS generation and NLRP3 inflammasome activation. These events may contribute to increased cardiovascular risk.
Introduction
Epidemiological studies have shown that men are at higher risk of cardiovascular disease than women, and sex steroids seem to contribute, at least in part, to this increased risk (1, 2). Over the past five decades, preclinical studies have produced a large body of data on the molecular mechanisms involved in testosterone effects in the cardiovascular system and/or how abnormal testosterone levels modify the risk of cardiovascular disease (3, 4).
At physiological levels, testosterone induces relaxation of many vascular beds, through its influence on the production or effects of endothelium-derived relaxant factors, including nitic oxide (NO) (5, 6) and prostacyclin (7). Testosterone also releases endothelium-derived substances that cause smooth muscle cells hyperpolarization by a mechanism that involves potassium channels (8). Endothelium-independent vascular effects of testosterone, including opening of voltage-sensitive potassium channels (Kv), small- and large- conductance calcium-sensitive potassium channels (SKCa and BKCa, respectively), have also been described (9, 10). Testosterone also has an important role in the regulation of cardiac function (11, 12). It affects cardiac contractility and relaxation, and cardiomyocyte repolarization. The latter effect results in the shortening of the action potential duration. Likewise, testosterone modulates immune/inflammatory responses, displaying protective effects against atherosclerosis (13), but also stimulating in vivo leukocyte-endothelial cell interactions, and contributing to increased leukocyte rolling and adhesion in male spontaneously hypertensive rats (14).
In the clinical setting, patients with low plasma levels of total testosterone (<300 ng/dL) undergo hormonal treatment to improve muscle performance, bone mineral density, cognitive and sexual function as well as to prevent metabolic syndrome and cardiovascular diseases (15). The Endocrine Society Clinical Practice Guideline has recommended a dose of 75–100 mg/week of testosterone in men with hypogonadism (16). On the other hand, a survey of drug abuse in bodybuilding and weightlifting sports reported the use of anabolic androgenic steroids at doses 5– 29 times greater than the usual supplemented doses (17) to boost up muscle mass and to reduce body fat (18). Despite the beneficial effects on skeletal muscle mass and strength (19, 20), cardiac hypertrophy with sudden cardiac death is often reported among athletes and bodybuilders taking anabolic androgenic steroids (21, 22). Testosterone users show slight left ventricular hypertrophy, even after discontinuation of prolonged high testosterone administration (23). In pre-clinical studies, experimental animals treated with high doses of testosterone enanthate exhibited cardiac (left ventricle) hypertrophy, fibrosis and apoptosis (increased caspase-3, a marker of cell apoptosis) (24). In addition, testosterone in supraphysiological concentrations alters the inflammatory state with an increase in circulating levels of pro-inflammatory cytokines as tumor necrosis factor alpha (TNFα), interleukin-1β (IL-1β) and IL-12 (25, 26).
Chronic inflammation or overactivation of the immune system is a central component in the development and complications of cardiovascular diseases (27). Inflammatory responses comprise a sequence of complex interactions between immune cells, such as neutrophils, lymphocytes, monocytes/macrophages, tissue cells (including vascular and cardiac cells), and a range of inflammatory mediators such as interleukins and chemokines (28). These interactions result in increased tissue production of soluble mediators such as complement system proteins, chemokines, cytokines and eicosanoids, accompanied by increased expression of cell adhesion molecules in circulating leukocytes and endothelial cells (29).
Activation of pattern recognition receptors (PRR) in innate immune cells is a key mechanism in the genesis and progression of cardiovascular diseases (30). Members of the nucleotide-binding oligomerization domain-(NOD)-leucine-rich repeats (NLRs) receptors lead to the formation of molecular platforms called inflammasomes (31). Inflammasomes activate cysteine proteases, known as caspases, which are involved in inflammatory and apoptotic processes. The NLRP3 inflammasome, a member of the NLRP subfamily, is expressed in cardiovascular cells and its activation contributes to cardiovascular damage (32). Several endogenous components activate the NLRP3 inflammasome (33–38). Among these components, reactive oxygen species (ROS), which are produced by NLRP3 activators and are essential secondary messengers in inflammatory pathways, are involved in NLRP3 inflammasome activation (39). Based on these observations and considering that testosterone stimulates ROS generation (40, 41), we hypothesized that high levels of testosterone induce vascular dysfunction via NLRP3 inflammasome activation.
Materials and Methods
Animals
All experimental protocols were performed in accordance with the National Council for Animal Experimentation Control and were approved by the Ethics Committee on Animal Use of the University of São Paulo, Ribeirao Preto, Brazil (Protocol n° 032/2018). Male C57BL/6J wild-type (WT) and NLRP3 knockout (NLRP3−/−) mice (12-week-old) were obtained from the Isogenic Breeding Unit at Ribeirao Preto Medical School, University of São Paulo, Ribeirao Preto, Brazil. Mice were maintained in a temperature (22 ± 1°C) and humidity (50–60%) controlled room on a 12-h light/dark cycle with ad libitum access to food and water.
WT and NLRP3−/− mice were treated with testosterone propionate (TP) 10 mg/kg or vehicle (peanut oil), subcutaneous injections, for 30 days. Animals were divided into four experimental groups: (1) WT_Vehicle; (2) WT_TP; (3) NLRP3−/−_Vehicle; (4) NLRP3−/−_TP.
Total Testosterone Levels
Plasma total testosterone levels were measured by IMMULITE 1000 Immunoassay System (Enzo Life Sciences). The samples and reagent containing the testosterone-conjugated alkaline phosphatase enzyme were distributed in 96-well plates. After 60 minutes (min) of incubation, the plates were washed to remove any remaining testosterone unbound fraction. The bound fraction was then quantified using the chemiluminescent dioxetane substrate.
Vascular Function
After isoflurane anesthesia and mice euthanasia, the thoracic aortas were removed and transferred to a modified Krebs-Henseleit solution (4°C), with the following composition [(in mM): NaCl, 130; KCl, 4.7; NaHCO3, 14.9; KH2PO4, 1.18; MgSO4.7H2O, 1.17; Glucose 5.5; CaCl2.2H2O; 1.56; EDTA, 0.026]. Thoracic aortic rings (2 mm) were mounted on a myograph (model 620 M; Danish Myo Technology – DMT, Copenhagen, Denmark) containing Krebs-Henseleit solution gassed with 5% CO2/95% O2 to maintain a pH of 7.4 for isometric tension recording. After the stabilization period, arteries were stimulated with potassium chloride (120 mM KCl) to verify functional integrity. Endothelial integrity was confirmed by over 80% relaxation to acetylcholine [(ACh), endothelium-dependent vasodilator, 10−6 M] on vessels pre-contracted with phenylephrine [(PE), alpha-adrenergic agonist, 10−6 M]. All the aortic rings used in this study presented intact endothelium.
Cumulative concentration-effect curves for PE (10−10-10−4 M), ACh (10−10-10−4 M) and sodium nitroprusside [SNP (10−10-10−4 M)] were performed in all experimental groups. The in vitro effects of testosterone [incubation of vessels with testosterone 10−6 M, for 2 hours (h)] on NLRP3 inflammasome activation, mitochondrial ROS generation and androgen receptor (AR) activation were evaluated using a NLRP3 receptor inhibitor (MCC950, 10−6 M for 30 min), a mitochondrial oxidative phosphorylation uncoupler [carbonyl cyanide 3-chlorophenyl hydrazone (CCCP), 10−6 M for 30 min] and an androgen receptor antagonist [Flutamide (Flu), 10−5 M, for 30 min], respectively. To verify the in vivo effects of supraphysiological levels of testosterone on vascular function, we used thoracic aortic segments from mice treated with testosterone propionate [TP (10 mg/kg for 30 days)].
Determination of Cytokine Levels
IL-1β was quantified in the serum and thoracic aortas by Enzyme-Linked ImmunonoSorbent Assay [(ELISA) R&D Systems, MLB00C], which is based on antigen-antibody reactions detectable by enzymatic reactions.
Measurement of Reactive Oxygen Species
Dihydroethidine
ROS generation was determined using a qualitative method involving dihydroethidine (DHE), a non-fluorescent precursor to ethidium bromide, as previously described by Suzuki et al. (42). In the presence of ROS, dihydroethidine is oxidized inside the cell, producing the fluorescent compounds ethid (E) and 2-hydroxy ethid (EHO), which have an affinity for nuclear DNA. Aortas of WT and NLRP3−/− mice treated with TP or vehicle were isolated and quickly immersed in freezing medium. Using a cryostat (Leica, Germany), cross sections of the aorta (5 μm) were obtained and placed on silanized slides. The sections were incubated with DHE (5 × 10−6 M) for 30 min at 37°C in a humid chamber protected from light. After this period, the slides were observed in an optical microscope (ZEISS) equipped with a rhodamine filter and a photographic camera, using a fluorescence microscopy. ROS generation was quantified through the light density corrected by the area using the program ImageJ (National Institutes of Health).
Lucigenin
Superoxide anion () generation in thoracic aortas was measured by chemiluminescence assay. The adenine nicotinamide dinucleotide (NADH) which is expected to potentiate production by the respiratory chain via complex I, was used as the substrate (43). Aortas were incubated with testosterone [Testo (10−6 M) for 2 h] in the absence or presence of Flu (10−5 M) for 30 min, CCCP (10−6 M) for 30 min and MCC950 (10−6 M) for 30 min, which were added before the incubation with Testo. Aortas were then transferred into glass tubes containing 990 μL assay buffer (50 mM KH2PO4, 1 mM EGTA and 150 mM sucrose, pH 7.4) and 5 μL of lucigenin (5 × 10−6 M) for basal reading. After the baseline reading, 10 μL of NADH (10−6 M) was added. The Line TL Tube Luminometer (Titertek-Berthold®, Pforzheim, Germany) was used for quantification of superoxide anion generation and data were expressed in relative luminescence units (RLU)/protein concentration (mg).
Western Blotting
Protein expression of NLRP3 and caspase-1 was determined using protein analysis of thoracic aortas of in vivo and in vitro all groups. Samples were homogenized in lysis buffer and proteins were collected. Proteins (30 μg) were separated by electrophoresis on 10 or 12% polyacrylamide gels, transferred to 0.22 μm nitrocellulose membranes and blocked using 5% bovine serum albumin (BSA) in Tris buffered saline (TBS) and 0.1% Tween 20 for 1 h. Primary antibodies were incubated overnight at 4°C as follows: anti-NLRP3 (1:500 dilution; R&D Systems), anti-caspase-1 (1:1,000 dilution; Novus Biologicals), anti-β-actin-peroxidase (1:5,000 dilution; Sigma-Aldrich).
Blood Pressure
Mice were anesthetized with a mixture of isoflurane 2% and O2 for carotid artery cannulation. Subcutaneous administration of tramadol (12.5 mg/kg) in a single dose was performed to promote postoperative analgesia. Twenty-four hours later the catheter was coupled to a pressure transducer connected to an amplification system and a computer with an analog-to-digital interface (PowerLab/4SP, ADInstruments, Colorado Springs, CO). After stabilization, blood pressure was determined.
Drugs
Testosterone propionate, phenylephrine, acetylcholine, sodium nitroprusside, testosterone, flutamide, were purchased from Sigma Chemical Co (St. Louis, MO, USA), CCCP (Tocris®, Bristol, United Kingdom) and MCC950 (Avistron®, Bude, Cornwall, United Kingdom).
Statistical Analysis
For analysis of vascular reactivity, individual concentration-effect curves were plotted on a sigmoidal curve, by non-linear regression analysis. These curves, in turn, provide the maximum response value (Emax) and pEC50 (negative logarithm of the EC50 values - concentration that produces 50% of the maximum response). The values of Emax and pEC50 were compared using Student's t-test and two-way analysis of variance test (Two-way ANOVA), followed by the Tukey post-test. The results of the molecular experiments were analyzed by Student's t-test and Two-way ANOVA, followed by the Tukey post-test. Data were assessed for normality with Shapiro-Wilk test. Potential outliers were analyzed using the GraphPad Prism Outlier Test. In the present study, no outliers or data were excluded. The program GraphPad Prism, version 6.0 (GraphPad Software Inc., San. Diego, CA, USA) was used to analyze these parameters. The results were expressed as mean ± standard error of the mean (SEM). The acceptable level of significance was p < 0.05.
Results
Deletion of NLRP3 Prevents in vivo Effects of Supraphysiological Levels of Testosterone on Vascular Function
To address whether supraphysiological levels of testosterone promote vascular dysfunction via NLRP3 inflammasome, WT and NLRP3−/− mice were treated with TP (10 mg/kg for 30 days) or vehicle. TP treatment increased total testosterone plasma levels in both experimental groups. Supraphysiological levels of testosterone in the TP groups were confirmed by comparison with vehicle-treated mice. Testosterone also increased body mass and decreased epididymal fat in WT and NLRP3−/− mice in comparison to vehicle-treated mice. On the other hand, only TP-treated WT mice exhibited increased blood pressure (Table 1).

Table 1. Characteristics of WT and NLRP3−/− mice treated with TP or Vehicle.
Aortic rings from TP-treated WT mice exhibited increased PE-induced contractile responses compared to aortas from vehicle-treated WT mice (Figure 1A and Table 2). In addition, ACh-mediated endothelium-dependent vasodilation was decreased in aortic rings of TP-treated WT mice compared to those from vehicle-treated WT mice (Figure 1B and Table 2). Deletion of NLRP3 prevented testosterone-induced increased contractile responses to PE (Figure 1A and Table 2) as well as impaired ACh vasodilation (Figure 1B and Table 2). Aortas from TP-treated WT mice did not exhibit altered responses to SNP (Table 2).
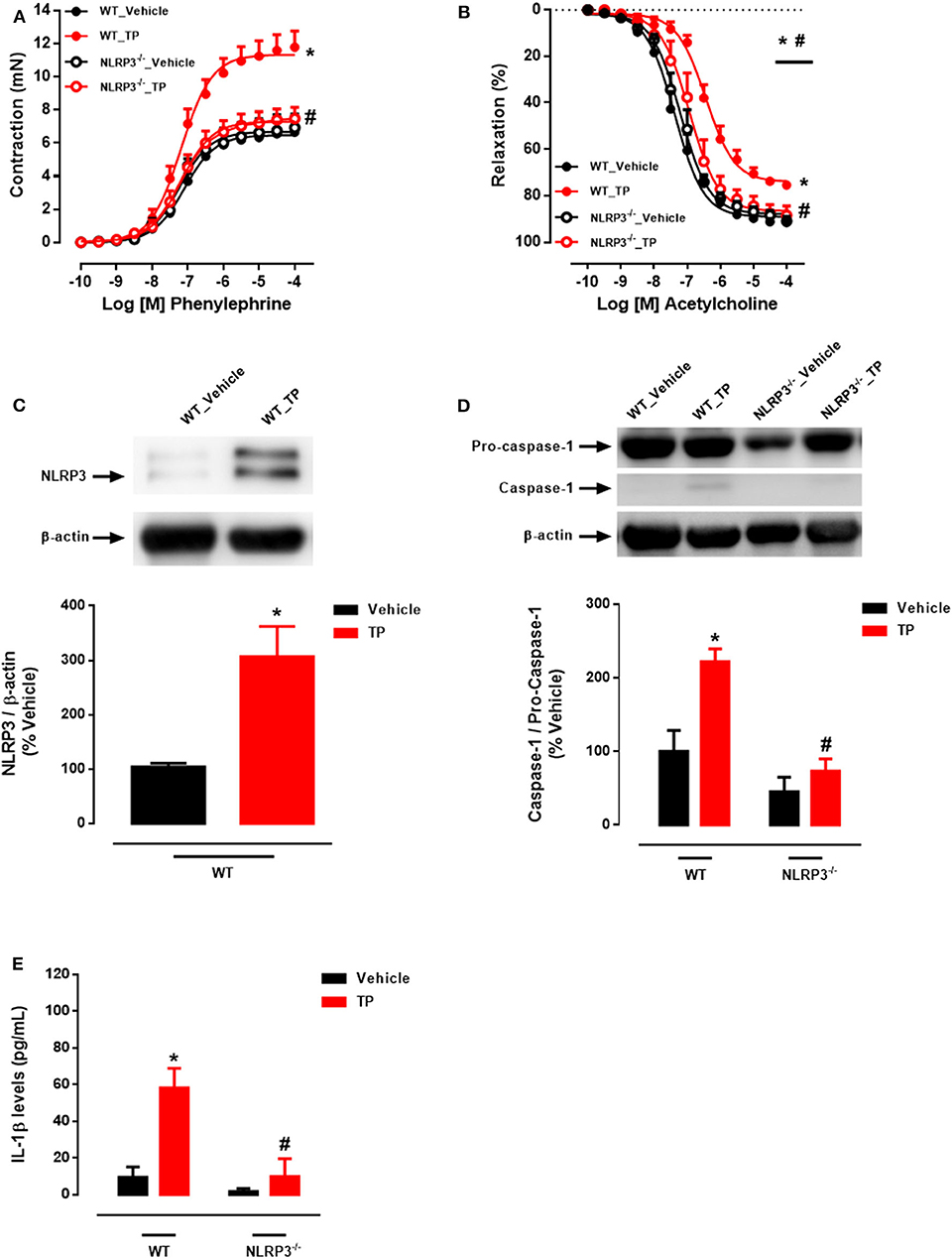
Figure 1. Testosterone propionate treatment induces vascular dysfunction via NLRP3 inflammasome (in vivo experiments). Concentration-response curves to phenylephrine - PE (A) and acetylcholine - ACh (B) were performed in aortic rings; NLRP3 (C) and caspase-1 (D) expression was determined in thoracic aortas; IL-1β levels (E) in the serum of WT and NLRP3−/− mice treated with testosterone propionate (10 mg/Kg for 30 days). Data are expressed as mean ± SEM (n = 3–10). *p < 0.05 vs. WT_Vehicle; #p < 0.05 vs. WT_TP.
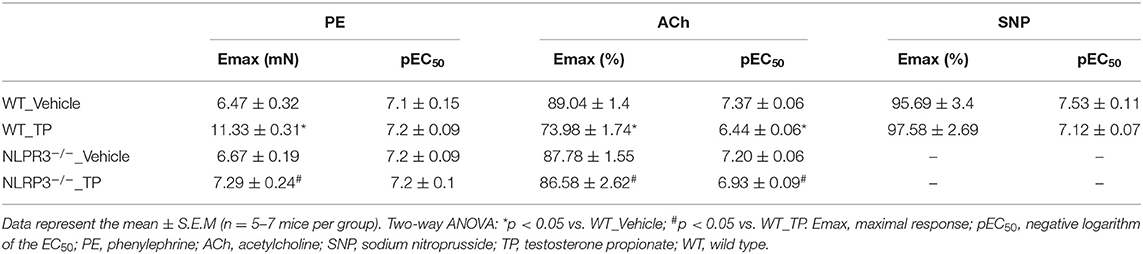
Table 2. Maximal response and pEC50 values for PE-induced contraction and ACh- and SNP-induced relaxation in aortas of WT and NLRP3−/− mice treated with TP or Vehicle.
Activation of the NLRP3 inflammasome by testosterone was then evaluated. In vivo, treatment of WT mice with TP increased NLRP3 expression (Figure 1C) and caspase-1 activation (Figure 1D) in thoracic aortas. Serum IL-1β levels were increased in TP-treated WT mice (Figure 1E). Lack of NLRP3 prevented increased caspase-1 activation and IL-1β release in TP-treated mice (Figures 1D,E).
Vascular Dysfunction Induced by in vitro Treatment of Aortic Rings With Testosterone Involves NLRP3 Inflammasome Activation
Mechanisms by which NLRP3 inflammasome contributes to testosterone-induced vascular dysfunction were investigated in aortic rings, from WT and NLRP3−/− mice, incubated with testosterone (10−6 M for 2 h). PE induced concentration-dependent contractions that were increased in testosterone-treated aortic rings compared to vehicle-treated rings (Figure 2A and Table 3). In addition, ACh-mediated vasodilation was decreased in aortic rings incubated with testosterone compared to vehicle-treated aortas (Figure 2B and Table 3). Lack of NLRP3 receptor completely prevented testosterone-induced increased contractile responses to PE (Figure 2A and Table 3) and impaired ACh-induced vasodilation (Figure 2B and Table 3). Testosterone did not alter vasodilator responses to SNP (Table 3). Activation of the NLRP3 inflammasome was also evaluated in these vessels. In vitro, testosterone increased NLRP3 expression (Figure 2C), caspase-1 activation (Figure 2D) and IL-1β levels (Figure 2E) expression in thoracic aortas. Aortas from NLRP3−/− mice did not exhibit increased caspase-1 activation (Figure 2D) or IL-1β expression (Figure 2E) in response to testosterone.
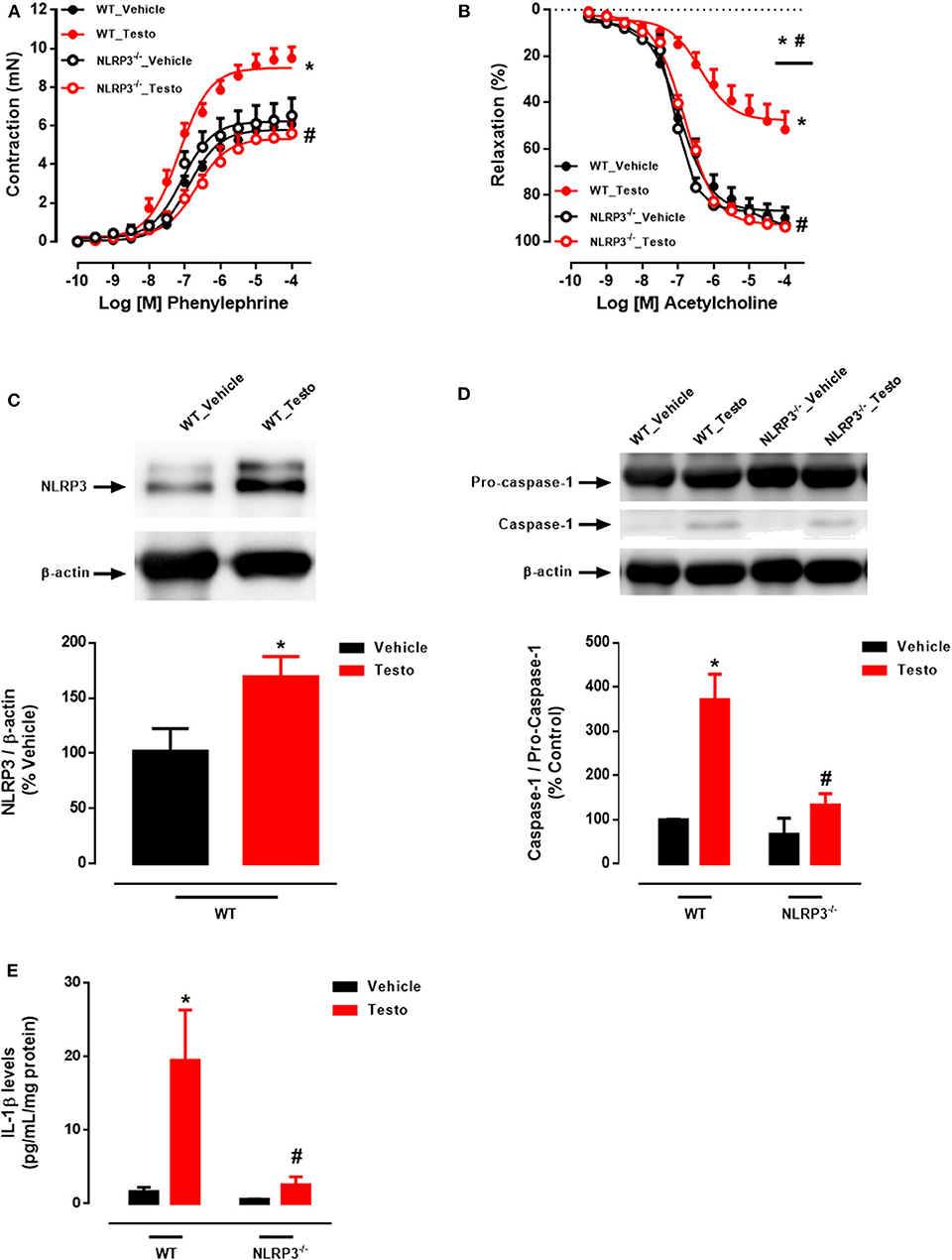
Figure 2. Testosterone induces vascular dysfunction via NLRP3 inflammasome (in vitro experiments). Concentration-response curves to phenylephrine - PE (A) and acetylcholine - ACh (B); NLRP3 (C), caspase-1 (D) and IL-1β levels (E) expression, all determined in thoracic aortas incubated with testosterone (10−6 M for 2 h) from WT and NLRP3−/− mice. Data are expressed as mean ± SEM (n = 3–10). *p < 0.05 vs. WT_Vehicle; #p < 0.05 vs. WT_Testo.
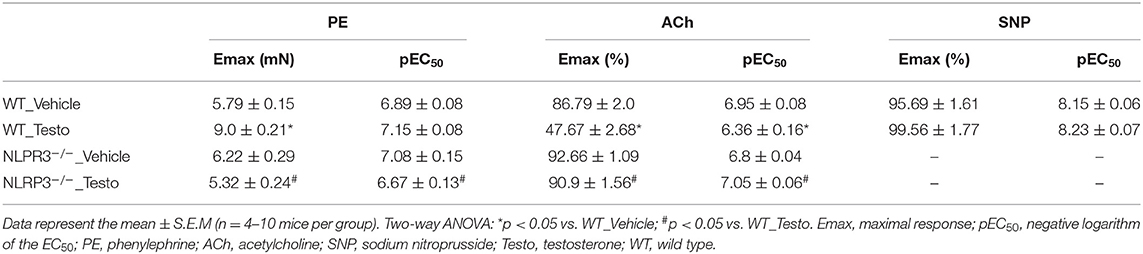
Table 3. Maximal response and pEC50 values for PE-induced contraction and ACh- and SNP-induced relaxation in aortas from WT and NLRP3−/− mice stimulated in vitro with Testo or Vehicle.
Pharmacological Inhibition of NLRP3 Inflammasome, Androgen Receptors and ROS Prevents Testosterone-Induced Vascular Dysfunction
The contribution of the NLRP3 inflammasome to testosterone-induced vascular dysfunction was further evaluated using a selective NLRP3 inhibitor, MCC950. MCC950 prevented the increased contractile responses to PE (Figure 3A and Table 4) and partially prevented the impairment of ACh-induced vasodilation (Figure 3B and Table 4). No differences were observed between reactivity of MCC950- and vehicle-treated aortas of WT mice.
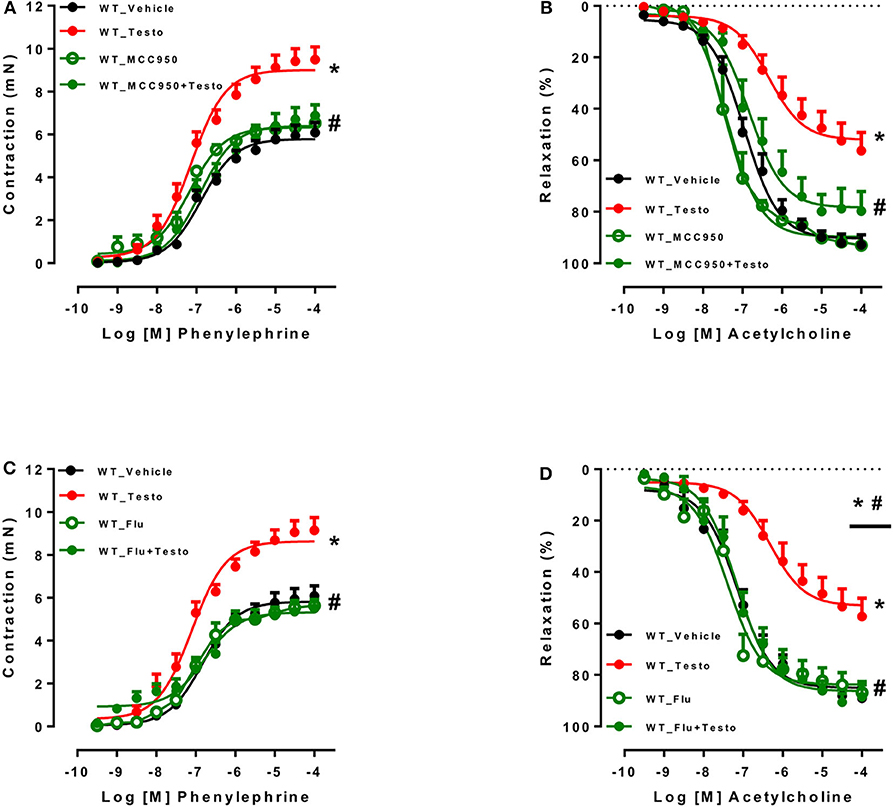
Figure 3. Pharmacological inhibition of NLRP3 inflammasome and androgen receptor prevents testosterone-induced vascular dysfunction. Concentration-response curves to phenylephrine - PE (A,C) and acetylcholine - ACh (B,D) were performed in aortic rings incubated with vehicle (WT_Vehicle) or testosterone (10−6 M for 2 h) (WT_Testo). The effects of MCC950 (10−6 M for 30 min) and Flutamide [Flu (10−5 M for 30 min)] on testosterone-induced vascular changes are shown in (A,B) - WT_MCC950 and WT_MCC950+Testo - and (C,D) - WT_Flu and WT_Flu+Testo - respectively. Data are expressed as mean ± SEM (n = 3–10). *p < 0.05 vs. WT_Vehicle; #p < 0.05 vs. WT_Testo.
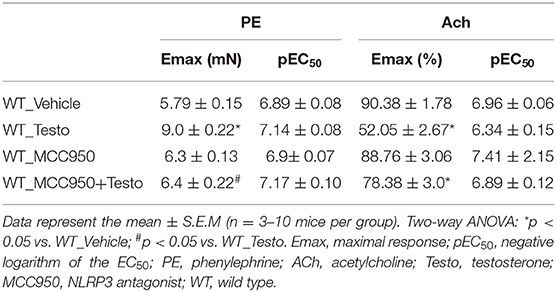
Table 4. Maximal response and pEC50 values for PE-induced contraction and ACh-induced relaxation in aortas incubated with Testo, MCC950, MCC950+Testo or Vehicle from WT mice.
To determine whether AR mediate testosterone-induced vascular dysfunction, Flu, an AR antagonist, was used. Flu prevented testosterone-induced increased contractile responses to PE (Figure 3C and Table 5) and the impaired ACh vasodilation (Figure 3D and Table 5). No differences were observed between vascular reactivity of thoracic aortic rings of WT mice incubated with Flu and vehicle.
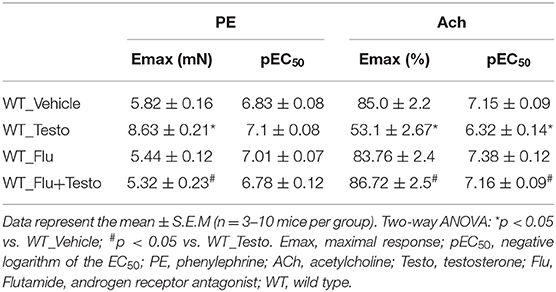
Table 5. Maximal response and pEC50 values for PE-induced contraction and ACh-induced relaxation in aortas incubated with Testo, Flu, Flu+Testo or Vehicle from WT mice.
Supraphysiological Testosterone Levels Induce Vascular Generation of Mitochondria-Derived Reactive Oxygen Species
Initially, ROS generation in thoracic aortas of WT and NLRP3−/− mice treated with TP or vehicle was determined. TP treatment increased ROS vascular generation in WT mice, but not in NLRP3−/− mice (Figure 4A).
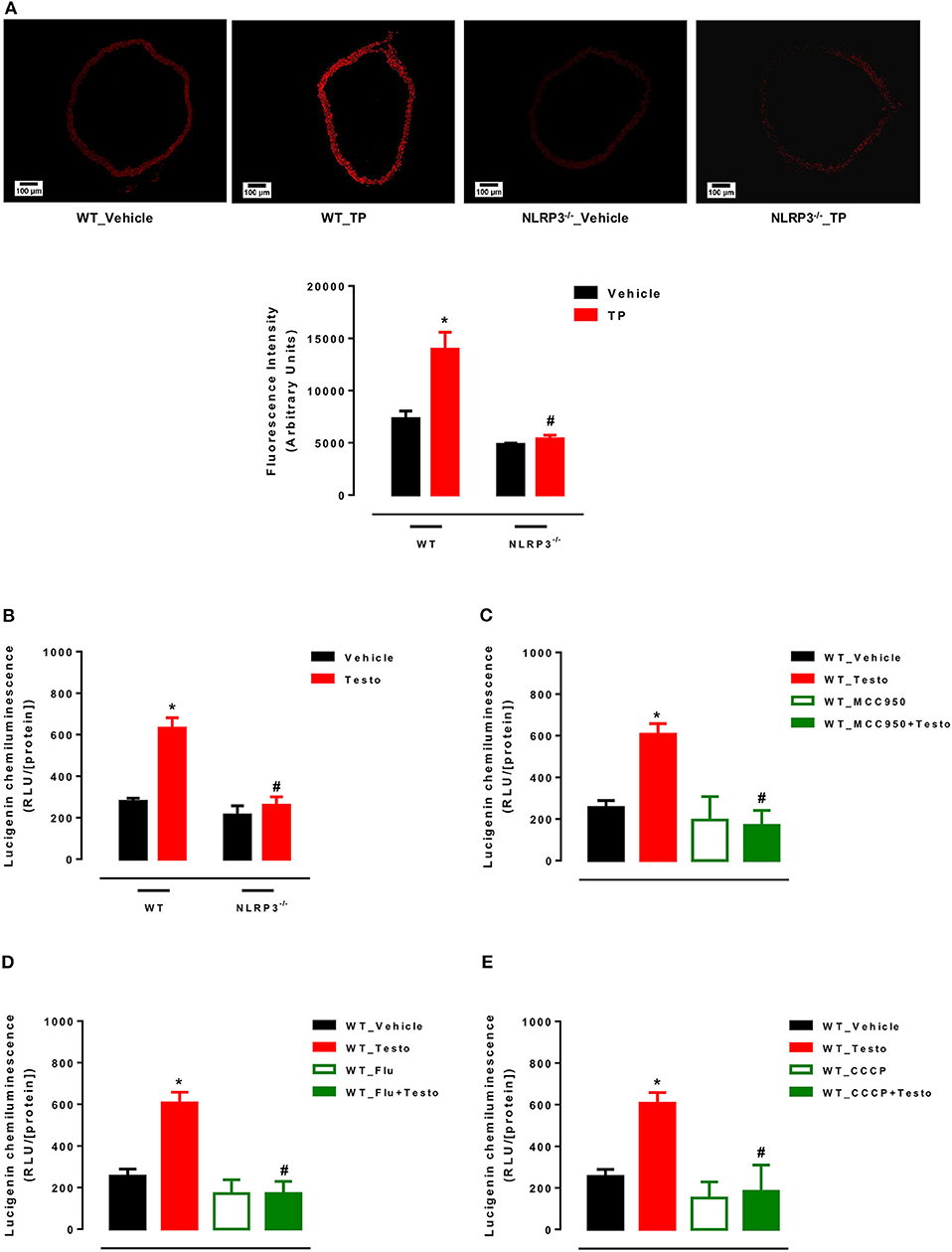
Figure 4. Supraphysiological testosterone levels induce vascular generation of reactive oxygen species. Vascular ROS generation was measured by dihydroethidine in aortas from WT_Vehicle, WT_TP, NLRP3−/−_Vehicle and NLRP3−/−_TP mice (A); superoxide anion generation – measured by lucigenin in vessels incubated with Testo or vehicle (WT_Vehicle, WT_Testo, NLRP3−/−_Vehicle and NLRP3−/−_Testo) (B); effects of MCC950 (C), Flu (D) and CCCP (E) on testosterone effects in aortas isolated from WT mice and incubated with vehicle or testosterone. Data are expressed as mean ± SEM (n = 5). *p < 0.05 vs. WT_Vehicle; #p < 0.05 vs. WT_TP; #p < 0.05 vs. WT_Testo.
Mitochondrial ROS generation was also determined in thoracic aortas of WT and NLRP3−/− mice incubated with testosterone or vehicle. Testosterone increased mROS in aortas from WT mice, but not in NLRP3−/− aortas (Figure 4B). The contribution of NLRP3 inflammasome (Figure 4C), AR (Figure 4D) and mitochondrial electron transport chain (Figure 4E) to ROS generation was also determined in vitro in aortas from WT mice. Mitochondrial uncoupling by CCCP, an inhibitor of oxidative phosphorylation, NLRP3 deletion and blockade of AR abrogated testosterone-induced vascular ROS generation.
To determine whether mitochondrial ROS contribute to the vascular effects of testosterone, experiments were performed in the presence of the mitochondrial uncoupler CCCP. CCCP prevented testosterone-induced increased in contractile responses to PE (Figure 5A and Table 6) and the impaired ACh vasodilation (Figure 5B and Table 6). No differences were observed between vascular reactivity of thoracic aortic rings of WT mice treated with CCCP and vehicle.
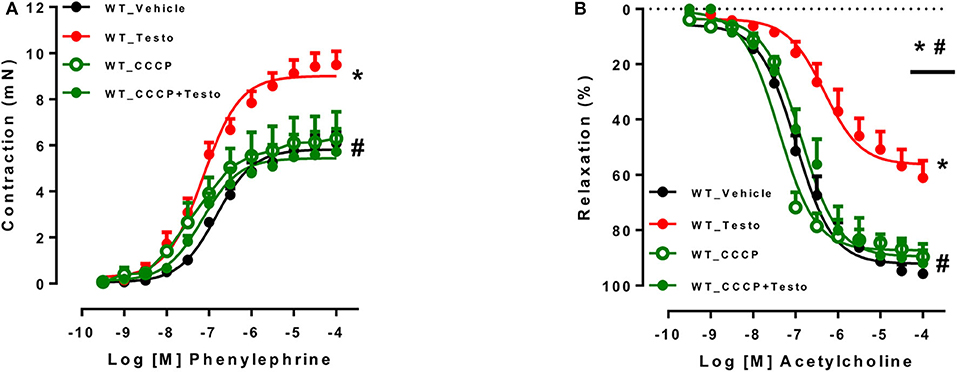
Figure 5. Testosterone induces vascular dysfunction via the generation of mitochondrial reactive oxygen species. Concentration-response curves to phenylephrine - PE (A) and acetylcholine - ACh (B) were performed in aortic rings incubated with testosterone (10−6 M for 2 h) or vehicle isolated from WT mice. The effects of CCCP (10−6 M for 30 min) on testosterone effects were determined. Data are expressed as mean ± SEM (n = 3–10). *p < 0.05 vs. WT_Vehicle; #p < 0.05 vs. WT_Testo.
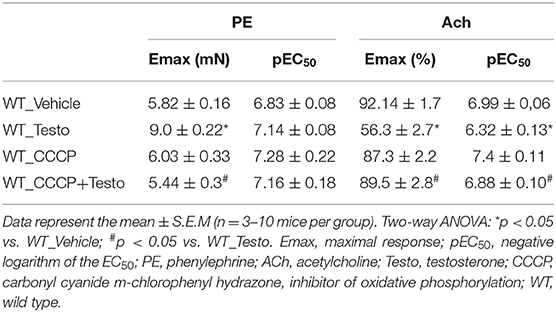
Table 6. Maximal response and pEC50 values of PE-induced contraction and ACh-induced relaxation in aortas incubated with Testo, CCCP, CCCP+Testo or Vehicle from WT mice.
Discussion
The primary goal of the present study was to uncover whether supraphysiological levels of testosterone induce NLRP3 inflammasome activation and consequent vascular dysfunction. In vitro studies using thoracic aortas and in vivo treatment of WT and NLRP3−/− mice demonstrated that supraphysiological levels of testosterone activate the NLRP3 inflammasome in vascular cells and cause vascular dysfunction. Our study provides in vivo evidence that NLRP3 inflammasome activation mediates proinflammatory effects of elevated levels of testosterone, thereby contributing to vascular dysfunction. Antagonism of testosterone receptors and inhibition of mitochondrial ROS generation prevent testosterone-induced vascular dysfunction in vitro. These findings, for the first time, demonstrate a key role for both increased ROS-mediated signaling and activation of NLRP3 inflammasome to vascular damage induced by testosterone.
Whereas, the consequences of testosterone deprivation in the cardiovascular system have been investigated (44), the effects of supraphysiological levels of the hormone have been surprisingly little studied. Evidence indicates that supraphysiological levels of testosterone affect the function and structure of the cardiovascular system. Sprague-Dawley rats treated with supraphysiological doses of testosterone show eccentric cardiac hypertrophy mediated by ERK 1/2 and mTOR phosphorylation (45). Increased systemic testosterone also decreases relaxation of human pulmonary arteries and veins (46).
In the present study, we addressed direct and long-term effects of testosterone on NLRP3 inflammasome-mediated vascular dysfunction. The innate and adaptive immune responses significantly influence both acute and chronic changes in cardiovascular phenotype that lead to clinical cardiovascular abnormalities (47–50).
Members of the NLR family have emerged as important sensors involved in the immune responses to pathogens and inflammatory diseases. NLRP3, a well-characterized member of the NLR family, regulates the assembly of the inflammasome, a multimeric complex protein that activates inflammatory caspase-1, which cleaves pro-interleukin-1β (pro-IL-1β) and pro-IL-18 into their mature and biologically active forms (51). Vascular cells can detect and respond to a variety of signals that are indicative of cell damage, including environmental irritants, endogenous danger signals, pathogens, and mitochondria-derived ROS, leading to the release of cytokines, chemokines and hormones (52, 53). The present study supports that the NLRP3 inflammasome is involved in the vascular dysfunction triggered by high testosterone levels. First, NLRP3 gene deletion prevented testosterone-induced hypercontractility of vascular smooth muscle (VSM) and endothelial dysfunction both in vitro and in vivo. Vascular alterations in response to testosterone in vitro were also partially prevented in the presence of MCC950, further indicating that NLRP3 inflammasome is key to vascular damage induced by testosterone. The observations that both chronic treatment of mice with testosterone propionate and incubation of aortas with testosterone increase vascular caspase-1 expression as well as IL-1β levels provide further support to the idea that testosterone induces vascular NLRP3 inflammasome activation.
Our group recently demonstrated that NLRP3 receptor inhibition restores the functional integrity of resistance mesenteric arteries of diabetic animals (38). Previous studies also showed that IL-1β, a key immunoregulatory and proinflammatory cytokine produced by the inflammasome, reduces endothelium-dependent relaxation, increases vascular contractility, as well as VSM cells migration and proliferation with consequent vascular remodeling (37, 54–56).
ROS production, especially from the mitochondria, triggers NLRP3 inflammasome activation (57). Testosterone induces long-term ROS production by genomic mechanisms in a time- and concentration-dependent manner. Also, it stimulates short-term ROS production by unique nongenomic mechanisms in VSM cells from hypertensive animals (40). In that the present study in vitro treatment of vascular segments with testosterone increased vascular ROS generation. Interestingly, chronic treatment with TP and incubation of arteries with testosterone increased IL-1β, the end product of NLRP3 inflammasome activation (58). Finally, the findings that vascular ROS generation in response to testosterone incubation is prevented by MCC950, Flu and CCCP, imply AR and mitochondria on testosterone-induced ROS generation in VSM cells (41).
To determine NLRP3 inflammasome activation in testosterone-induced ROS generation and vascular dysfunction in vivo, we used NLRP3−/− mice treated with TP. Our analysis revealed that inflammasome signaling is critical to testosterone-induced vascular dysfunction. Lack of NLRP3 inflammasome abolished testosterone-induced ROS generation and IL-1β production, indicating an interplay between ROS and NLRP3, as trigger and effector molecules.
The role of mitochondrial ROS in the vascular effects of high testosterone concentrations were further investigated in our study. CCCP prevented the deleterious effects of testosterone on vascular function. This is consistent with earlier data from our group, showing that mitochondrial ROS from the perivascular adipose tissue (PVAT) of obese mice induces vascular dysfunction (59).
AR modulate inflammatory processes and activation of components of the immune system. Accordingly, male mice lacking AR in monocytes/macrophages exhibit less eosinophil recruitment and lung inflammation due to impaired M2 polarization (60). On the other hand, animals with deletion of AR in macrophages/monocytes are protected from atherosclerosis (61) and depletion of AR has protective effects on abdominal aortic aneurysm development in WT mice (62). In this study Flu prevented testosterone effects on vascular function and ROS generation, supporting that AR contribute to testosterone-induced activation of inflammasome and oxidative stress. In addition to inhibition of mitochondrial ROS generation and NLRP3 inflammasome activation, blockade of AR prevented impaired vascular contractile and relaxant responses. The evidence that testosterone, via AR, promotes calcium (Ca2+) influx (63) and that intracellular Ca2+ is crucial for the generation of mitochondrial ROS (64) is consistent with our hypothesis that supraphysiological levels of testosterone induces vascular dysfunction via increased mitochondrial ROS-mediated activation of NLRP3 inflammasome. Since testosterone activates other receptors, including GPRC6A and GPER (65), further studies are warranted for a more detailed investigation on the relative contribution of AR to the vascular complications induced by supraphysiological concentrations of testosterone. Meanwhile, our study highlights classical AR in the pathogenesis of vascular dysfunction.
The current study has strengths and limitations. The experiments rigorously followed stringent quality criteria in experimental research such as randomization, manipulation and evaluations performed in a blinded fashion, controlled physiological parameters and use of control groups of health animals. A major limitation of the current study was the in vivo approach, which restricts mechanistic investigations of tissue-specific inflammatory pathways, since it does not allow identification of the specific cell types responsible for the observed systemic changes. Considering that the primary goal of this study was to directly investigate mechanisms of impaired vascular dysfunction induced by high systemic levels of testosterone, to overcome this limitation we also addressed the effects of testosterone on isolated vessels. The analysis of molecular mechanisms involved in testosterone-induced activation of NLRP3 was supported by the in vitro approaches. However, results observed on in vitro assays, although important, often fail to translate to similar results in vivo. Additionally, although DHE has been extensively used DHE to detect in cells or systems, the interference from other oxidative radicals prevents a fine tune quantification of (66, 67). Since every method has shortcomings, we used a combination of DHE, lucigenin and pharmacological inhibitors to better evaluate production. In the present study we have not used aromatase or 5-alpha reductase inhibitors to determine whether estradiol or dihydrotestosterone contribute to testosterone effects. However, in previous studies, testosterone effects were not blocked or modified by anastrozole (aromatase inhibitor). Finally, the time of treatment as well as the dose of testosterone were based on previous studies that investigated other metabolic and cardiovascular parameters in different experimental models.
Conclusion
Our findings provide evidence that supraphysiological levels of testosterone impairs vascular function via activation of the NLRP3 inflammasome in vascular cells. The generation of mitochondrial ROS is crucial for activation of the NLRP3 inflammasome. Pharmacologic inhibition or genetic deletion of the NLRP3 in mice protects from testosterone-induced vascular dysfunction. Our study highlights the importance of NLRP3 inflammasome in vascular dysfunction promoted by supraphysiological levels of testosterone.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by Ethics Committee on Animal Use (CEUA) of the University of São Paulo, Ribeirao Preto, Brazil (Protocol n° 032/2018).
Author Contributions
JA, RC, NL, and RT designed the study. JA, RC, CP, AF, and CS conducted the experiments. JA, RC, and RT provided supplies and analytical tools. JA, RC, CP, and AF performed the data analysis. JA, RC, FC, NL, and RT wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the São Paulo Research Foundation (FAPESP) under grant agreement no 2013/08216-2 (Center for Research in Inflammatory Diseases - CRID), CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Vishva Dixit (Genentech) for providing the NLRP3−/− mice used in this study [CITAR: (68)].
Abbreviations
ACh, acetylcholine; AR, androgen receptor; BKCa, large-conductance voltage- and calcium-activated potassium channels; BSA, bovine serum albumin; CCCP, carbonyl cyanide m-chlorophenyl hydrazone; CONCEA, National Council for Animal Experimentation Control; DHE, dihydroethidine; Emax, maximum response; Flu, flutamide; IL-12, interleukin-12; IL-1β, interleukin-1β; KCa, calcium-activated potassium channels; KCl, potassium chloride; Kv, voltage-sensitive potassium channels; MCC950, NLRP3 receptor inhibitor; mROS, mitochondrial reactive oxygen species; NADH, adenine nicotinamide dinucleotide; NLRP3−/−, NLRP3 knockout mouse; NLRs, NOD-like receptors; NO, nitric oxide; NOD, nucleotide-binding oligomerization domain-like receptors; , superoxide anion; PE, phenylephrine; pEC50, negative logarithm of the EC50; pro-IL-18, pro-interleukin-18; pro-IL-1β, pro-interlukin-1β; PRR, pattern recognition receptors; PVAT, perivascular adipose tissue; RLU, relative luminescence units; ROS, reactive oxygen species; SEM, standard error of the mean; SKCa, small-conductance voltage- and calcium-activated potassium channels; SNP, sodium nitroprusside; TBS, Tris-buffered saline; Testo, testosterone; TNFα, tumor necrosis factor alpha; TP, testosterone propionate; Vehicle, peanut oil; VSM, vascular smooth muscle; WT, wild type.
References
1. George J, Rapsomaniki E, Pujades-Rodriguez M, Shah AD, Denaxas S, Herrett E, et al. How does cardiovascular disease first present in women and men? Incidence of 12 cardiovascular diseases in a contemporary cohort of 1 937 360 people. Circulation. (2015) 132:1320–8. doi: 10.1161/CIRCULATIONAHA.114.013797
2. Groban L, Lindsey SH, Wang H, Alencar AK. Sex and gender differences in cardiovascular disease. Sex Diff Physiol. (2016) 1:61–87. doi: 10.1016/B978-0-12-802388-4.00005-7
3. Kloner RA, Carson C, Dobs A, Kopecky S, Mohler ER. Testosterone and cardiovascular disease. J Am Coll Cardiol. (2016) 67:545–57. doi: 10.1016/j.jacc.2015.12.005
4. Gagliano-Jucá T, Basaria S. Testosterone replacement therapy and cardiovascular risk. Nat Rev Cardiol. (2019) 16:555–74. doi: 10.1038/s41569-019-0211-4
5. Skogastierna C, Hotzen M, Rane A, Ekström L. A supraphysiological dose of testosterone induces nitric oxide production and oxidative stress. Eur J Prev Cardiol. (2014) 21:1049–54. doi: 10.1177/2047487313481755
6. Hotta Y, Kataoka T, Kimura K. Testosterone deficiency and endothelial dysfunction: Nitric oxide, asymmetric dimethylarginine, and endothelial progenitor cells. Sex Med Rev. (2019) 7:661–8. doi: 10.1016/j.sxmr.2019.02.005
7. Marrachelli VG, Miranda FJ, Centeno JM, Salom JB, Torregrosa G, Jover-Mengual T, et al. Role of NO-synthases and cyclooxygenases in the hyperreactivity of male rabbit carotid artery to testosterone under experimental diabetes. Pharmacol Res. (2010) 61:62–70. doi: 10.1016/j.phrs.2009.06.008
8. Honda H, Unemoto T, Kogo H. Different mechanisms for testosterone-induced relaxation of aorta between normotensive and spontaneously hypertensive rats. Hypertension. (1999) 34:1232–6. doi: 10.1161/01.HYP.34.6.1232
9. Yu J, Akishita M, Eto M, Ogawa S, Son BK, Kato S, et al. Androgen receptor-dependent activation of endothelial nitric oxide synthase in vascular endothelial cells: role of phosphatidylinositol 3-kinase/akt pathway. Endocrinology. (2010) 151:1822–8. doi: 10.1210/en.2009-1048
10. Campelo AE, Cutini PH, Massheimer VL. Testosterone modulates platelet aggregation and endothelial cell growth through nitric oxide pathway. J Endocrinol. (2012) 213:77–87. doi: 10.1530/JOE-11-0441
11. Drici MD, Burklow TR, Haridasse V, Glazer RI, Woosley RL. Sex hormones prolong the QT interval and downregulate potassium channel expression in the rabbit heart. Circulation. (1996) 94:1471–4. doi: 10.1161/01.CIR.94.6.1471
12. Song M, Helguera G, Eghbali M, Zhu N, Zarei MM, Olcese R, et al. Remodeling of Kv4.3 potassium channel gene expression under the control of sex hormones. J Biol Chem. (2001) 276:31883–90. doi: 10.1074/jbc.M101058200
13. Wilhelmson AS, Lantero Rodriguez M, Svedlund Eriksson E, Johansson I, Fogelstrand P, Stubelius A, et al. Testosterone protects against atherosclerosis in male mice by targeting thymic epithelial cells—brief report. Arterioscler Thrombos Vasc Biol. (2018) 38:1519–27. doi: 10.1161/ATVBAHA.118.311252
14. Filgueira FP, Lobato NDS, DosSantos RA, Oliveira MA, Akamine EH, Tostes RC, et al. Endogenous testosterone increases leukocyte–endothelial cell interaction in spontaneously hypertensive rats. Life Sci. (2012) 90:689–94. doi: 10.1016/j.lfs.2012.03.009
15. Bassil N, Alkaade S, Morley JE. The benefits and risks of testosterone replacement therapy: a review. Ther Clin Risk Manag. (2009) 5:427. doi: 10.2147/TCRM.S3025
16. Bhasin S, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, et al. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. (2010) 95:2536–59. doi: 10.1210/jc.2009-2354
17. Perry PJ, Lund BC, Deninger MJ, Kutscher EC, Schneider J. Anabolic steroid use in weightlifters and bodybuilders: an internet survey of drug utilization. Clin J Sport Med. (2005) 15:326–30. doi: 10.1097/01.jsm.0000180872.22426.bb
18. Nordström A, Högström G, Eriksson A, Bonnerud P, Tegner Y, Malm C. Higher muscle mass but lower gynoid fat mass in athletes using anabolic androgenic steroids. J Strength Cond Res. (2012) 26:246–50. doi: 10.1519/JSC.0b013e318218daf0
19. Bhasin S, Storer TW, Berman N, Callegari C, Clevenger B, Phillips J. The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. New Engl J Med. (1996) 335:1–7. doi: 10.1056/NEJM199607043350101
20. Vermeulen A, Goemaere S, Kaufman JM. Testosterone, body composition and aging. J Endocrinol Investig. (1999) 22:110–6.
21. Sullivan ML, Martinez CM, Gennis P, Gallagher EJ. The cardiac toxicity of anabolic steroids. Progr Cardiovasc Dis. (1998) 41:1–15. doi: 10.1016/S0033-0620(98)80019-4
22. Frati P, Busardo FP, Cipolloni L, Dominicis E, Fineschi V. Anabolic androgenic steroid (AAS) related deaths: autoptic, histopathological and toxicological findings. Curr Neuropharmacol. (2015) 13:146–59. doi: 10.2174/1570159X13666141210225414
23. Urhausen A, Albers T, Kindermann W. Are the cardiac effects of anabolic steroid abuse in strength athletes reversible? Heart. (2004) 90:496–501. doi: 10.1136/hrt.2003.015719
24. Papamitsou T, Barlagiannis D, Papaliagkas V, Kotanidou E, Dermentzopoulou-Theodoridou M. Testosterone-induced hypertrophy, fibrosis and apoptosis of cardiac cells–an ultrastructural and immunohistochemical study. Med Sci Monit. (2011) 17:BR266. doi: 10.12659/MSM.881930
25. Posma E, Moes H, Heineman MJ, Faas MM. The effect of testosterone on cytokine production in the specific and non-specific immune response. Am J Reprod Immunol. (2004) 52:237–43. doi: 10.1111/j.1600-0897.2004.00216.x
26. Ren X, Fu X, Zhang X, Chen S, Huang S, Yao L, et al. Testosterone regulates 3T3-L1 pre-adipocyte differentiation and epididymal fat accumulation in mice through modulating macrophage polarization. Biochem Pharmacol. (2017) 140:73–88. doi: 10.1016/j.bcp.2017.05.022
27. Lopez-Candales A, Burgos PMH, Hernandez-Suarez DF, Harris D. Linking chronic inflammation with cardiovascular disease: from normal aging to the metabolic syndrome. J Nat Sci. (2017) 3:e341.
28. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. (2018) 9:7204. doi: 10.18632/oncotarget.23208
29. Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. (2015) 160:816–27. doi: 10.1016/j.cell.2015.02.010
30. Wang X, Yi F. Implication of pattern-recognition receptors in cardiovascular diseases. Antioxid Redox Signal. (2015) 22:1130–45. doi: 10.1089/ars.2014.6184
31. Patel MN, Carroll RG, Galván-Peña S, Mills EL, Olden R, Triantafilou M. Inflammasome priming in sterile inflammatory disease. Trends Mol Med. (2017) 23:165–80. doi: 10.1016/j.molmed.2016.12.007
32. Zhou W, Chen C, Chen Z, Liu L, Jiang J, Wu Z. NLRP3: a novel mediator in cardiovascular disease. J Immunol Res. (2018) 2018:5702103. doi: 10.1155/2018/5702103
33. Kahlenberg J, Dubyak GR. Mechanisms of caspase-1 activation by P2X 7 receptor-mediated K+ release. Am J Physiol Cell Physiol. (2004) 286:1100–8. doi: 10.1152/ajpcell.00494.2003
34. Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: first line of the immune response to cell stress. Cell. (2006) 126:659–62. doi: 10.1016/j.cell.2006.08.002
35. Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci USA. (2008) 105:9035–40. doi: 10.1073/pnas.0803933105
36. Jin C, Flavell RA. Molecular mechanism of NLRP3 inflammasome activation. J Clin Immunol. (2010) 30:628–31. doi: 10.1007/s10875-010-9440-3
37. Bruder-Nascimento T, Ferreira NS, Zanotto CZ, Ramalho F, Pequeno IO, Olivon VC, et al. NLRP3 inflammasome mediates aldosterone-induced vascular damage. Circulation. (2016) 134:1866–80. doi: 10.1161/CIRCULATIONAHA.116.024369
38. Ferreira NS, Bruder-Nascimento T, Pereira CA, Zanotto CZ, Prado DS, Silva JF, et al. NLRP3 inflammasome and mineralocorticoid receptors are associated with vascular dysfunction in type 2 diabetes mellitus. Cells. (2019) 8:1595. doi: 10.3390/cells8121595
39. Abais JM, Xia M, Zhang Y, Boini KM, Li PL. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal. (2015) 22:1111–29. doi: 10.1089/ars.2014.5994
40. Chignalia AZ, Schuldt EZ, Camargo LL, Montezano AC, Callera GE, Laurindo FR, et al. Testosterone induces vascular smooth muscle cell migration by NADPH oxidase and c-Src–dependent pathways. Hypertension. (2012) 59:1263–71. doi: 10.1161/HYPERTENSIONAHA.111.180620
41. Lopes RAM, Neves KB, Pestana CR, Queiroz AL, Zanotto CZ, Chignalia AZ, et al. Testosterone induces apoptosis in vascular smooth muscle cells via extrinsic apoptotic pathway with mitochondria-generated reactive oxygen species involvement. Am J Physiol Heart Circ Physiol. (2014) 306:1485–94. doi: 10.1152/ajpheart.00809.2013
42. Suzuki H, Swei A, Zweifach BW, Schmid-Schönbein GW. In vivo evidence for microvascular oxidative stress in spontaneously hypertensive rats: hydroethidine microfluorography. Hypertension. (1995) 25:1083–9. doi: 10.1161/01.HYP.25.5.1083
43. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. (2012) 48:158–67. doi: 10.1016/j.molcel.2012.09.025
44. Maggio M, Basaria S. Welcoming low testosterone as a cardiovascular risk factor. Int J Impotence Resarch. (2009) 21:261–4. doi: 10.1038/ijir.2009.25
45. Pirompol P, Teekabut V, Weerachatyanukul W, Bupha-Intr T, Wattanapermpool J. Supra-physiological dose of testosterone induces pathological cardiac hypertrophy. J Endocrinol. (2016) 229:13–23. doi: 10.1530/JOE-15-0506
46. Rowell KO, Hall J, Pugh PJ, Jones TH, Channer KS, Jones RD, et al. Testosterone acts as an efficacious vasodilator in isolated human pulmonary arteries and veins: evidence for a biphasic effect at physiological and supra-physiological concentrations. J Endocrinol Investig. (2009) 32:718–23. doi: 10.1007/BF03346526
47. Mann DL. The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ Res. (2011) 108:1133–45. doi: 10.1161/CIRCRESAHA.110.226936
48. Fernández-Ruiz I. Immune system and cardiovascular disease. Nat Rev Cardiol. (2016) 13:503. doi: 10.1038/nrcardio.2016.127
49. Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. (2018) 18:733–44. doi: 10.1038/s41577-018-0065-8
50. Dal Lin C, Tona F, Osto E. The crosstalk between the cardiovascular and the immune system. Vasc Biol. (2019) 1:83–8. doi: 10.1530/VB-19-0023
51. Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
52. Schultz K, Murthy V, Tatro JB, Beasley D. Endogenous interleukin-1α promotes a proliferative and proinflammatory phenotype in human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. (2007) 292:2927–34. doi: 10.1152/ajpheart.00700.2006
53. Földes G, Liu A, Badiger R, Paul-Clark M, Moreno L, Lendvai Z, et al. Innate immunity in human embryonic stem cells: comparison with adult human endothelial cells. PLoS ONE. (2010) 5:e10501. doi: 10.1371/journal.pone.0010501
54. Dorrance AM. Interleukin 1-beta (IL-1β) enhances contractile responses in endothelium-denuded aorta from hypertensive, but not normotensive, rats. Vasc Pharmacol. (2007) 47:160–5. doi: 10.1016/j.vph.2007.05.007
55. Jimenez-Altayo F, Briones AM, Giraldo J, Planas AM, Salaices M, Vila E, et al. Increased superoxide anion production by interleukin-1β impairs nitric oxide-mediated relaxation in resistance arteries. J Pharmacol Exp Ther. (2006) 316:42–52. doi: 10.1124/jpet.105.088435
56. Aguado A, Rodríguez C, Martínez-Revelles S, Avendaño MS, Zhenyukh O, Orriols M, et al. HuR mediates the synergistic effects of angiotensin II and IL-1β on vascular COX-2 expression and cell migration. Br J Pharmacol. (2015) 172:3028–42. doi: 10.1111/bph.13103
57. An N, Gao Y, Zhang H, Wang L, Tian C, Yuan M, et al. Regulatory mechanisms of the NLRP3 inflammasome, a novel immune-inflammatory marker in cardiovascular diseases. Front Immunol. (2019) 10:1592. doi: 10.3389/fimmu.2019.01592
58. Ginnan R, Jourd'heuil FL, Guikema B, Simons M, Singer HA, Jourd'heuil D. NADPH oxidase 4 is required for interleukin-1β-mediated activation of protein kinase Cδ and downstream activation of c-jun N-terminal kinase signaling in smooth muscle. Free Radic Biol Med. (2013) 54:125–34. doi: 10.1016/j.freeradbiomed.2012.09.026
59. da Costa RM, Fais RS, Dechandt CR, Louzada-Junior P, Alberici LC, Lobato NS, et al. Increased mitochondrial ROS generation mediates the loss of the anti-contractile effects of perivascular adipose tissue in high-fat diet obese mice Br J Pharmacol. (2017) 174:3527–41. doi: 10.1111/bph.13687
60. Becerra-Díaz M, Strickland AB, Keselman A, Heller NM. Androgen and androgen receptor as enhancers of M2 macrophage polarization in allergic lung inflammation. J Immunol. (2018) 201:2923–33. doi: 10.4049/jimmunol.1800352
61. Huang CK, Pang H, Wang L, Niu Y, Luo J, Chang E, et al. New therapy via targeting androgen receptor in monocytes/macrophages to battle atherosclerosis. Hypertension. (2014) 63:1345–53. doi: 10.1161/HYPERTENSIONAHA.113.02804
62. Huang CK, Luo J, Lai KP, Wang R, Pang H, Chang E, et al. Androgen receptor promotes abdominal aortic aneurysm development via modulating inflammatory interleukin-1α and transforming growth factor-β1 expression. Hypertension. (2015) 66:881–91. doi: 10.1161/HYPERTENSIONAHA.115.05654
63. Steinsapir J, Socci R, Reinach P. Effects of androgen on intracellular calcium of LNCaP cells. Biochem Biophys Res Commun. (1991) 179:90–6. doi: 10.1016/0006-291X(91)91338-D
64. Bertero E, Maack C. Calcium signaling and reactive oxygen species in mitochondria. Circ Res. (2018) 122:1460–78. doi: 10.1161/CIRCRESAHA.118.310082
65. Cruz-Topete D, Dominic P, Stokes KY. Uncovering sex-specific mechanisms of action of testosterone and redox balance. Redox Biol. (2020) 31:101490. doi: 10.1016/j.redox.2020.101490
66. Daiber A, Oelze M, Steven S, Kröller-Schön S, Münzel T. Taking up the cudgels for the traditional reactive oxygen and nitrogen species detection assays and their use in the cardiovascular system. Redox Biol. (2017) 12:35–49. doi: 10.1016/j.redox.2017.02.001
67. Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med. (2012) 52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030
Keywords: testosterone, androgen receptor, NLRP3 inflammasome, reactive oxygen species, vascular dysfunction
Citation: Alves JV, da Costa RM, Pereira CA, Fedoce AG, Silva CAA, Carneiro FS, Lobato NS and Tostes RC (2020) Supraphysiological Levels of Testosterone Induce Vascular Dysfunction via Activation of the NLRP3 Inflammasome. Front. Immunol. 11:1647. doi: 10.3389/fimmu.2020.01647
Received: 01 March 2020; Accepted: 19 June 2020;
Published: 31 July 2020.
Edited by:
Trine N. Jorgensen, Case Western Reserve University, United StatesReviewed by:
Roger Lyrio Santos, Federal University of Espirito Santo, BrazilHong Liu, Central South University, China
Copyright © 2020 Alves, da Costa, Pereira, Fedoce, Silva, Carneiro, Lobato and Tostes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rita C. Tostes, cnRvc3Rlc0B1c3AuYnI=