 Geoffroy Méry1
Geoffroy Méry1 Anne-Laure Borel
Anne-Laure Borel Audrey Le Gouellec
Audrey Le Gouellec- 1Service Hospitalier Universitaire de Pneumologie Physiologie, CHU Grenoble-Alpes, La Tronche, France
- 2Service de Maladies Infectieuses et Tropicales, CHU Grenoble-Alpes, La Tronche, France
- 3Groupe de Recherche en Infectiologie Clinique, Université Grenoble Alpes, La Tronche, France
- 4UMR 5075—Institut de Biologie Structurale, Grenoble, France
- 5Service de Nutrition, Pole DIGIDUNE, CHU Grenoble-Alpes, La Tronche, France
- 6Hypoxia PathoPhysiology Laboratory, INSERM U1042, University Grenoble Alpes, La Tronche, France
- 7Laboratoire TIMC-TheREx UMR 5525 CNRS-Université Grenoble Alpes, La Tronche, France
- 8Service de Biochimie Biologie Moléculaire Toxicologie Environnementale, UM Biochimie des Enzymes et des Protéines, Institut de Biologie et Pathologie, C.H.U. Grenoble-Alpes, La Tronche, France
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the third coronavirus leading to a global health outbreak. Despite the high mortality rates from SARS-CoV-1 and Middle-East respiratory syndrome (MERS)-CoV infections, which both sparked the interest of the scientific community, the underlying physiopathology of the SARS-CoV-2 infection, remains partially unclear. SARS-CoV-2 shares similar features with SARS-CoV-1, notably the use of the angiotensin conversion enzyme 2 (ACE2) as a receptor to enter the host cells. However, some features of the SARS-CoV-2 pandemic are unique. In this work, we focus on the association between obesity, metabolic syndrome, and type 2 diabetes on the one hand, and the severity of COVID-19 infection on the other, as it seems greater in these patients. We discuss how adipocyte dysfunction leads to a specific immune environment that predisposes obese patients to respiratory failure during COVID-19. We also hypothesize that an ACE2-cleaved protein, angiotensin 1-7, has a beneficial action on immune deregulation and that its low expression during the SARS-CoV-2 infection could explain the severity of infection. This introduces angiotensin 1-7 as a potential candidate of interest in therapeutic research on CoV infections.
Introduction
Coronavirus (CoV) is a single-stranded RNA virus involved in human and animal diseases. The rare event of its transmission from avian and mammalian reservoirs (mostly bats) to the human host has led to widespread epidemics in recent years (1). Indeed, over the last two decades, three CoV outbreaks have forced human populations to change their perspectives regarding the control of emerging diseases and the importance of public health in general.
The first outbreak caused by severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1) occurred between November 2002 and July 2003, originating from China and then spreading worldwide (2). Although the symptoms of SARS-CoV-1 infection were in most cases non-specific, including lethargy, myalgia, and headache, the high mortality rate of 10% in case series was mostly related to respiratory failure due to acute respiratory distress syndrome (ARDS) (3, 4). The physiopathology underlying the severity of SARS-CoV-1 infection remained unclear after the epidemic due to insufficient sampling. A second CoV epidemic occurred in 2012 with Middle East respiratory syndrome (MERS)-CoV, which has mostly led to small-size outbreaks in the years ever since (5). Although it did not reach a pandemic status, MERS-CoV continues to infect humans, and the World Health Organization identified more than 850 patients who have died of related complications since its discovery (6). Indeed, MERS-CoV has a higher mortality rate in case series (case fatality rate of ~30%), mostly from respiratory failure, which has led to the identification of unique strategies of CoV infections to escape the immune response. Due to the ending of the SARS-CoV-1 epidemic and the somewhat limited number of cases of MERS-CoV in the recent years, understanding the mechanisms of CoV infections in humans has proven to be complex, and the conclusions drawn from in vitro experiments and animal models remain difficult to extrapolate.
In November 2019, cases of a pneumonia with atypical features were reported in Wuhan, China; in January 2020, SARS-CoV-2 was identified as the cause of this new CoV-induced disease (COVID-19), which became a worldwide pandemic in the following months (7). Although the mortality rates of this new COVID-19 are still being debated, ranging between 0.3 and 1.5%, it is still lower than those associated with SARS-CoV-1 and MERS-CoV infections. Patients suffering from severe SARS-CoV-2 infection could be healthy or only have mild comorbidities such as hypertension or diabetes (8). Most of all, severe cases due to respiratory failure occur 7–12 days after the first symptoms (9). Studies on COVID-19 have progressively stressed its similarities with previous CoV infections, mostly SARS-CoV-1, with the same unanswered questions regarding its physiopathology. One notable feature of this disease, already observed in previous CoV infections, is the high prevalence of obese patients among the most severe cases.
Here we seek to explore what underlies the link between immune response and respiratory failure in CoV infections on the one hand, and the current observation of obesity as a risk factor for severe outcome in COVID-19 on the other.
Physiopathology of Respiratory Failure in Covid-19
Most of the time, the need for intensive care during COVID-19 is secondary to the onset of ARDS (9), as defined by the Berlin criteria (bilateral shadowing on lung radiology, rapid deterioration of symptoms, and objective hypoxemia on blood samples). In the first published series, 30% of these ARDS cases were accompanied by septic shock or other organ dysfunction (8, 10).
The nature of COVID-19-induced ARDS is still under discussion. Interleukin (IL) dosages are usually very high, and hypoxemia is severe in COVID-19-induced ARDS, which matches the hyperinflammatory profile described by Calfee et al. (11). SARS-CoV-1-induced ARDS was associated with vascular leakage and neutrophilic alveolitis (12), both of which are compatible with a hyper-inflammatory profile. In COVID-19, some experts observed ventilatory abnormalities suggestive of microcirculatory involvement such as hypoxic pulmonary vasoconstriction or distal thrombosis (13, 14). This points to the contribution of several factors in respiratory failure, with experts also citing the possible involvement of genuine viral pneumonia as well as capillary thrombosis by neutrophil extracellular traps (NETs) (15). The reason for this respiratory outcome is most likely a complex interplay of multiple factors, which derive directly from CoV virulence.
Role of the Viral Gateway
The membrane protein angiotensin-converting enzyme 2 (ACE2) is used as an entry receptor by SARS-CoV-1 and SARS-CoV-2 (16, 17). It has been reported that SARS-CoV-2 has a greater affinity for ACE2 than SARS-CoV-1 due to the specific amino-acid composition in the receptor-binding domain of the spike protein (18). ACE2 is expressed at varying levels by most cells in the body but primarily in the small intestine, testis, kidney, heart, thyroid, and adipose tissue cells (19). The expression of ACE2 in adipocytes seems to be promoted by high fat diets (20). In the lungs, it is expressed by 2% of epithelial cells, increasing with cell differentiation, and it is mainly located on the apical (or luminal) pole, serving as an accessible anchor point to airborne contaminants (19).
ACE2 is a key enzyme of the renin-angiotensin system, converting angiotensin 2 (Ang2) into Ang1-7. Ang2 binds to a receptor, the angiotensin type 1 receptor (AT1R), a transmembrane G protein-coupled receptor, which is found in a large variety of cells, ranging from smooth muscle cells, endometrium, and myocardium to blood cells, renal interstitial, and glomeruli. The activation of AT1R has several effects: for example, vasoconstriction, vascular permeability, macrophage maturation, and pro-inflammatory cytokine release. During the resolution phase of the inflammation, Ang2 promotes tumor growth factor beta production and fibroblast proliferation, leading to fibrosis and inadequate healing of the wounded tissue (21).
An antagonistic pathway of the Ang2-derived effects results from the binding of Ang1-7 to the mitochondrial assembly (MAS) receptor. MAS receptor is a ubiquitous G-protein-coupled receptor, implicated, among others, in retina development (22), muscle wasting (23), and benign prostate hyperplasia (24). Activation of the MAS receptor by Ang1-7 induces vasodilatation by a nitric-oxide-dependent mechanism (25, 26) and reduces oxidative stress induced by Ang2 in vascular injuries (27). In macrophages, it promotes an anti-inflammatory profile (28), for example, by lowering pro-inflammatory cytokine production, notably IL-6 and tumor necrosis factor alpha (TNFα). Ang1-7 has also shown beneficial effects in inflammation resolution and fibrosis, notably in kidney and myocardial disease (21, 29). The binding of ACE2 by SARS-CoV-2 prevents it from exerting its enzymatic activities, resulting in decreased anti-inflammatory Ang1-7 production and the accumulation of pro-inflammatory Ang2 (16, 17). This results in high cytokine titers, neutrophil infiltration, and endothelial dysfunction in the lungs, potentially predisposing for ARDS.
As early as 2004, ACE2 tampering was suggested to be an important mechanism in SARS-CoV-1 infection (30, 31). It was only later discovered that CoV possesses very specific mechanisms to escape the host's immunity (32). These mechanisms, in addition to the pro-inflammatory response secondary to ACE2 binding, might act as a trigger for a sustained and uncontrolled inflammatory response, leading to ARDS.
Immune Polarization and Its Consequences During CoV Infections
In general, an efficient antiviral response is driven by T-helper lymphocytes (LTh) with a specific polarization such as LTh1 and LTh2. LTh1 refers to a polarization in which LTh primarily promotes cytotoxic lymphocytes (CTL) and natural killers (NK) for the control and destruction of infected cells as well as the release of specific cytokines, such as type 1 interferon (INF-1) by innate immune cells.
INF-1 is produced by infected cells and innate immune cells after recognizing the viral pathogen-associated molecular patterns (PAMPS), such as single-strained or uncapped RNA, using cytoplasmic pattern-recognition receptors (PRR). In particular, toll-like receptor 3 (TLR3) induces Toll/interleukin-1 receptor domain-containing adapter-inducing interferon-β (TRIF). Hosts deficient in either TLR3 or TRIF are more susceptible to viral injuries and thus more at risk of developing ARDS during CoV infections (33).
INF-1 activates the Janus kinase-signal transducers and activators of transcription (JAK-STAT) pathway, resulting in the modulation of hundreds of interferon-sensitive genes and notably in the synthesis of specific cytokines, preferably oriented toward viral control and clearance (34).
Most of these steps, involved in INF-1 signaling, are blocked by CoV infections. This evolution trait is probably due to the presence of a constitutive INF-1 production in bats (principal reservoir of CoV). CoV infections are expert evaders of this antiviral response (35). Their escape plan revolves around three main mechanisms:
- First, hiding viral RNA from cytoplasmic PRR. After entering the cell, SARS-CoV-1 shields its RNA by forming, inside the host's endoplasmic reticulum, a large network of double-membrane vesicles isolated from the cytosol (36, 37). The modified capping of the viral RNA 2′-O-methylation also prevents the binding to an important cytosolic PRR (38).
- Next, direct tampering of the PRR-related enzymes. For example, the papain-like protein in CoV can modify the ubiquitinylation profile of TLR7 (39) or other antiviral-related PRR (40). Moreover, S protein triggers IL-1R-associated kinase and peroxisome proliferator-associated receptor gamma, subsequently downregulating interferon regulatory factor 7 activity (41). In addition, the jamming of TLR3 phosphorylation reduces the PRR activity, while blocking most of the INF-1 production pathways.
- Lastly, the non-structural protein 1 in both MERS and SARS-CoV-1 can selectively degrade host RNA via endonucleolytic activity against which the viral RNA is protected (42, 43).
The many mechanisms used by CoV probably leave the infected cells in a defensive cul-de-sac where they are incapable of developing an efficient antiviral response. On the one hand, viral PAMPS do not result in INF-1 production. On the other hand, non-viral PAMPS such as debris from cell lysis still stimulate the immune response. This could lead to inappropriate cytokine environments that lack INF-1 and are thus less effective against viruses, as seen in COVID-19 (44).
Indeed, during COVID-19 infection, most patients exhibit a specific cytokine profile, associating innate immunity chemokines (such as monocyte chemoattractant protein 3 and interferon gamma-induced protein 10 (IP-10), which are suggestive of macrophage activation and epithelial suffering), and pro-inflammatory macrophage-produced cytokines such as IL-6 (45). Moreover, CoV infections can directly induce the activation of nuclear factor kappa B (NFkB), notably by tampering with the TNF receptor-associated factor 3 pathway (TRAF3) via its open reading frame 3a. Activation by ubiquitination of TRAF3 also promotes the de novo development of the NOD-like receptor pyrin domain containing protein 3 (NLRP3) inflammasome and the production of IL-1β and IL-18 (46). This cytokine production promotes macrophage activation and INF-3, although it does not salvage a deficient polarization of the adaptive immunity toward LTh1 and its subsequent efficient antiviral response. High plasma levels of IL-6 and the absence of INF-1 have been noted in severe patients (47), illustrating a sustained innate response that fails to achieve viral clearance and triggers ARDS.
However, this sustained inflammation without LTh polarization might not be the only profile to bypass the antiviral cul-de-sac. Some patients infected by MERS-CoV demonstrated a polarization of the immune profile toward a LTh17-mediated response. Faure et al. compared two cases of MERS with different outcomes (48); the patient with a fatal outcome had an early increase in IL-17 and IL-23 titers (hallmarks of LTh17 polarization), whereas the surviving patient had a spike in INF-1 but no indication of LTh17 polarization. LTh17 are effective actors in the clearance of extracellular microorganisms such as fungi and bacteria, but poorly effective against viral pathogens (49). In general, viral PAMPS do not usually polarize the immune response to LTh17.
The association of severe outcome and inappropriate cytokine environment in CoV infection suggests a link with immune polarization, as a result of the “cul-de-sac” of antiviral response induced by the CoV escaping strategies. The resulting inefficient immune profile leads to a sustained viral exposure and persistent inflammatory state. In addition to the pro-inflammatory signals mediated by ACE2 inhibition, this sustained and inappropriate immune activation might be strongly involved in the development of ARDS.
Implications of Covid-19 in Obese Patients
Obesity is a common condition, affecting up to 30% of adults in Western countries. It is defined by a body mass index (BMI) >30 kg/m2, irrespective of the location of the adipose tissue. However, all profiles of obesity are not equivalent in terms of their consequences. Indeed, abdominal (or visceral) obesity (estimated by the waist circumference or waist-to-hip ratio), in which visceral fat predominates, is more associated with metabolic disorders such as type 2 diabetes or hypertension, compared to “metabolically healthy” obesity, in which subcutaneous fat predominates.
Early observations in the SARS-CoV-2 epidemics suggested obesity to be a risk factor to COVD-19, or at least to severe forms of the disease (50). In our retrospective cohort, we observed more than 60% of patients with overweight or obesity (n = 155) (Figure 1). In a retrospective cohort, Simonnet et al. showed an increasing risk of intensive care unit (ICU) admission in COVID-19 patients as BMI increased, independently of other metabolic disorders (51), which was subsequently confirmed by other teams (52, 53).
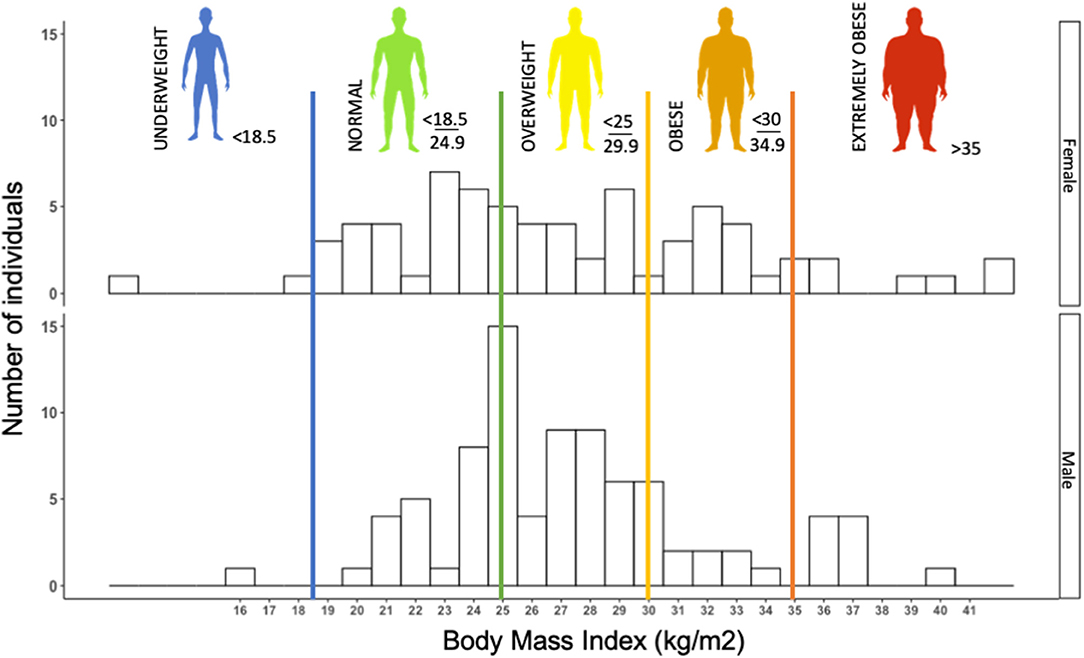
Figure 1. Histogram of the distribution of body mass index (BMI) (kg/m2) in 155 consecutive patients (female and male) admitted to Grenoble University Hospital for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (retrospective cohort study). Histogram illustrating that a majority of COVID patients, more precisely, 64% of COVID-19 patients were overweight or obese (BMI > 25 kg/m2). The median BMI of females (F) and males (M) was 26.30 and 27.08 kg/m2, respectively.
Thus, obesity appears to be a risk factor for presenting a severe form of COVID-19. It should be mentioned that once in ICU, obesity is known to confer a survival advantage, termed the “obesity paradox” (54). Patients with a BMI > 25 kg/m2 seem to survive mechanical ventilation and severe septic states significantly better than patients with a normal or low BMI (55, 56), presumably due to their elevated muscle mass, which represents a metabolic reserve in the hypermetabolic state of ICU patients (54, 57). It is not yet known whether once admitted to ICU, obese COVID-19 patients also present a better prognosis than patients of normal body weight.
Adipokines
The scientific observations of the last two decades have placed obesity in a complex pathological framework centered around the deregulation of adipocyte, which is far from the naive idea of a simple diet-induced condition (58).
White adipose tissue (WAT) is now recognized as an independent endocrine organ, whose main role is to regulate and store the energy provided by food. However, the hormones released by WAT, specific to the adipocyte and known as adipokines, reach a large variety of organs and modulate an extensive range of functions, from appetite control to inflammatory response (29). Leptin is the leading adipokine, whose anorexigen properties regulate satiety and food intake. Leptin levels in blood are proportionate to the amount of WAT and increase with BMI. Interestingly, the leptin receptor (LEPR) on immune cells mostly activates JAK-STAT and NFkB dependent pathways, except in neutrophils, macrophages, and antigen presenting cells, which all express a particular form of LEPR. Leptin promotes migration in the WAT of resident macrophages and induces their polarization toward a pro-inflammatory profile or a classical activated macrophage (M1) profile, and unbalances the LTh profiles, by reducing regulatory T-cells and promoting LTh17 polarization (59). Adiponectin is another adipokine, whose levels increase in proportion to subcutaneous fat but decrease with visceral fat accumulation. It favors whole-body insulin sensitivity, fatty-acid oxidation and diminishes the hepatic neo-glucogenesis pathways (60). Adiponectin promotes primarily LTh1 polarization, hence antiviral inflammation. Other adipokines, such as lipocalin-2, down-regulate inflammatory LTh altogether by promoting regulatory lymphocytes. Adipokines form a large family regularly counting new members over the last few years, all of which reveal complex and multiple implications in the regulation of energy storage and release, adipose tissue regulation and rather ubiquitous cellular metabolism (61).
Unlike subcutaneous fat, visceral fat accumulation, also described as “abdominal obesity,” is characterized by a dysfunctional profile of adipokines associated with a rise in pro-inflammatory signals. The triggers of this dysfunction is believed to be a metabolic stress in the presence of nutrient excess and a hypoxic stress caused by hypertrophic visceral adipocytes, due to an increase in cells' size and low neovascularization, via a mobilization of Hypoxia Inducible Factor 1 (62). Unlike visceral fat, subcutaneous fat expansion is hyperplasic and is not correlated with low-grade inflammation (63). In severe abdominal obesity, the adipokine profile is unbalanced in favor of leptin production and low-grade inflammation at the expense of adiponectin, or lipocalin-2. This deregulation of the adipokine profile links various disorders associated with metabolic diseases, such as insulin-resistance, to inflammatory manifestations, as described in rheumatoid arthritis (64).
Ang1-7 takes an active role in regulating the effects of adipokines. Its involvement was reviewed by Lelis et al., with an exhaustive approach and emphasis on other adipokines that will not be described here, such as sirtuin and resistin (29). A strong interest in Ang1-7 has already arisen from these observations, particularly in the field of atherosclerosis and non-alcoholic fatty liver disease, in which Ang1-7 seems beneficial. In a concise article, Mori et al. hypothesized that the disruption of the renin-angiontensin system by the virus could impair the energetic functions of these pathways during SARS-CoV infections (65). We suggest that the tampering with such pathways could also lead to abnormalities in the inflammatory response observed in severe CoV infections through their influence on immune regulation and cytokine production.
Meta-Inflammation in Obese Patients and Viral Response
Adipocyte dysfunction in visceral fat is correlated to low-grade persistent inflammation, known as meta-inflammation, which is suspected to be the starting point or an early factor in metabolic disorders associated with severe obesity (63). This meta-inflammation is mostly driven by the leptin-activated M1 macrophages in WAT. WAT-resident macrophages exhibit pro-inflammatory behavior, producing IL-1β, IL-6, and TNFα. The precursor of IL-1β is cleaved into bioactive IL-1β by the NLRP3 inflammasome, as a result of the NFkB pathway activation, which is induced by both pro-inflammatory and hypoxic signals originating from the adipocytes (58). Adiponectin can inhibit NFkB activation, but as mentioned above, depending on the obesity severity and profile, the effects of adiponectin can easily be overwhelmed by those of leptin (66).
Leptin also polarizes hematopoiesis directly in the bone marrow, promoting granulocyte, and erythroblast lines (the latter probably acts as a protective mechanism against hypoxia) at the expense of lymphocytes (67). When neutrophils are mature and circulating, leptin also promotes their survival on a dose-dependent scale (68). Higher levels of neutrophils have thus been observed in obese patients, possibly making the neutrophil recruitment during an inflammatory process more potent than in patients with a normal BMI (69).
Besides suffering from a pro-inflammatory environment, which favors macrophage activation and neutrophil production, obese patients exhibit abnormal responses to viral infection. As summarized by Honce et al., during influenza infections, obese patients tend to have greater neutrophil activation and NET development, contributing to capillary damage and thrombosis. Such phenomena have been extensively found in COVID-19 patients (70). Their inflammatory response is also characterized by a lack of INF-1 production as well as a strong cytokine production, notably IL-6, IP-10, and type 3 INF, which are elevated in severe COVID-19. Interestingly, patients with visceral fat accumulation also tend to have a lower TLR3 expression in adipocytes, muscle cells, and adipose tissue-resident macrophages, as well as a concomitant lower production of cytokines following exposure to viral PAMPS (71–73). This suggests that their baseline profile resembles that found in severe CoV infections, in which the antiviral response is less efficient, but the overall inflammation is higher than in other viral infections.
Finally, both obesity and metabolic disorders are associated with vascular dysfunction. At the acute phase of lung infection, this could result in microcirculatory abnormalities, as suggested by intensive care physicians, and increased lung edema.
Discussion
Patients with visceral fat accumulation, type 2 diabetes (74), and hypertension are not the only subjects at a higher risk of severe SARS-CoV-2 infection. When considering metabolic disorders separately, diabetes, non-alcoholic liver disease, and obstructive sleep disorders have been recently reported as risk factors for a severe outcome (74–76). This suggests that the metabolic dysfunction associated with these disorders more than obesity alone might be involved in the severity of the disease in these patients.
When comparing the effects of Ang1-7 and the inflammatory environment of patients with adipocyte dysregulation and metabolic disorders, an interesting pattern emerges. All the immunological features arising from the adipocyte dysfunction—(i.e., M1 macrophage polarization with IL-6 and TNFα production), and neutrophil promotion—may contribute to the development of ARDS and thus be countered by the activation of the Ang1-7/MAS receptor axis. Ang1-7 also favors a strong capillary barrier and a beneficial oxidative profile, which are altered in patients with visceral fat activation and could help to prevent ARDS. This leads us to two hypotheses: either patients with metabolic disorders, primarily visceral fat accumulation, have a constitutional lower titer of Ang1-7, as suggested by some observations (77), and a resulting higher inflammation; or the Ang1-7 levels in these patients are preserved and restrain the baseline inflammation. In the first case, the inappropriate inflammatory response, added to the diminished activation of TLR3 in obese patients, leads to unrestrained inflammation. However, if Ang1-7 is present in these patients and limits the meta-inflammation, acting as a guardrail, the antagonization of ACE2 by SARS-CoV-1 and 2 in addition to the lack of de novo Ang1-7 production could exacerbate the meta-inflammation and contribute to the severe septic states of obese patients with COVID-19, as illustrated in Figure 2. In both cases, the supplementation of Ang1-7 in these patients might improve fitness upon SARS-CoV infection.
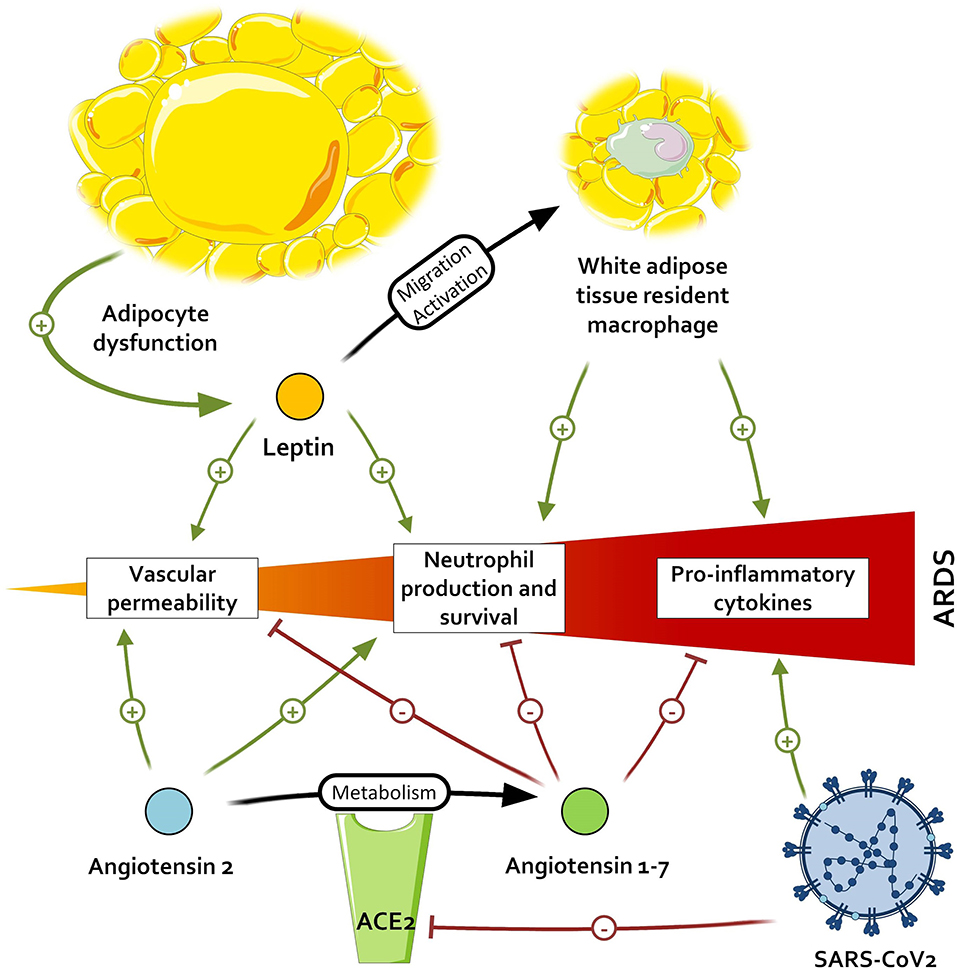
Figure 2. Impact of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) on pathways promoting acute respiratory distress syndrome (ARDS). By inactivating the angiotensin conversion enzyme 2 (ACE2), SARS-CoV-2 leads to an accumulation of angiotensin 2 and a lower dosage of angiotensin 1-7, respectively resulting in the higher promotion and lower inhibition of pro-inflammatory signals.
ACE2 deficiency has already been explored by some research teams to better understand the potential metabolic benefits of conversion enzyme inhibitors used in hypertension, among others. Their studies highlighted the association between ACE2 deficiency and higher titers of pro-inflammatory cytokines in obese mice, as well as in mice with glucose intolerance (78), which is closely correlated with meta-inflammation (79). Other studies correlate ACE2 deficiency with epicardial inflammation (80). This suggests that the Ang1-7/MAS axis allows a better control of inflammation in obese patients.
TLR4 is a receptor to LPS and leads to NFkB activation and (among others) hepatic inflammation. When administered orally to rats fed with a high-fat diet, Ang1-7 lowered hepatic inflammation, notably through a modulation of a metabolic pathway involving TLR4 (81). Moreover, promoting the effects of the Ang1-7/MAS receptor axis using medication also improves the aforementioned cytokines and oxidative stress in obese mice, with a protective effect against diabetic cardiomyopathy (82).
Ang1-7 is already in the spotlight of scientific research given its beneficial effects in preventing the development of metabolic disorders and obesity (83). We believe that our literature review highlights the beneficial effects of Ang1-7 on meta-inflammation in preexisting obesity and its potential involvement in inflammatory response and viral clearance, notably against SARS-CoV-2. Modulation of the renin-angiotensin system has been mentioned by others to explain the severity of COVID-19. A recent study found a lower mortality and intubation risk during COVID-19 among elderly patients treated with nifedipine or amlodipine (84), although the study sample was small and most of the accessible data do not suggest a strong connection (85, 86). However, these drugs interfere with AT1R and not with the genuine production of Ang1-7.
In obese patients with COVID-19, this hypothesis should be considered. Oral or parenteral Ang1-7 supplementation could be a therapeutic option to diminish the low-grade systemic inflammation due to adipocyte dysfunction and attenuate the severity of ACE2-mediated injuries consecutive to SARS infection. Parenteral Ang1-7 has already been used in human research on account of its property to enhance acetylcholine-mediated vasodilatation in endothelia, with safe outcomes (87).
Conclusion
COVID-19 is a viral disease with remarkable characteristics given its high severity in obese patients and its ability to tamper ACE2 metabolism. We believe that more than being just an incentive to accelerate research on viral infection, COVID-19 also presents an opportunity to respond to questions that were previously considered to be too intricate or complex, such as non-septic inflammation or the immune system communication underlying metabolic disorders. Understanding the multiple and interrelated factors linking SARS-CoV-2 infection, angiotensin metabolism, global inflammation, and metabolic disorders such as type 2 diabetes and obesity should provide us with a better insight into the way in which these conditions and physiological states interact outside of an acute aggression.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors within respect of General Data Protection Regulation, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Comité Ethique du Centre Investigation clinique (CECIC). Subjects were all informed and did not oppose, written consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
GM and AL conceptualized the idea, provided discussion, feedback, and organize the plan of the article. GM, A-LB, OE, and AL wrote the paper. GM, A-LB, OE, BT, and AL reviewed the different version of paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Foundation of Grenoble Alpes University, Grenoble, France.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors thank Mrs. Victoria Grace, from English Publications, for the careful editing of the publication. We kindly thank P. Audoin, C. Clape, A. Metz, and I El Amrani for their support regarding the reglementary procedures.
References
1. Cheng VCC, Lau SKP, Woo PCY, Yuen KY. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin Microbiol Rev. (2007) 20:660–94. doi: 10.1128/CMR.00023-07
2. WHO. SARS (Severe Acute Respiratory Syndrome). Available online at: https://www.who.int/ith/diseases/sars/en/
3. Peiris JSM, Lai ST, Poon LLM, Guan Y, Yam LYC, Lim W, et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. (2003) 361:1319–25. doi: 10.1016/S0140-6736(03)13077-2
4. Hui DS, Wong P, Wang C. SARS: clinical features and diagnosis. Respirology. (2003) 8:S20–4. doi: 10.1046/j.1440-1843.2003.00520.x
5. Mackay IM, Arden KE. MERS coronavirus: diagnostics, epidemiology and transmission. Virol J. (2015) 12:222. doi: 10.1186/s12985-015-0439-5
6. WHO. Middle East Respiratory Syndrome Coronavirus (MERS-CoV). (2020). Available online at: https://www.who.int/emergencies/mers-cov/en/ (accessed May 15, 2020).
7. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. (2020) 382:727–33. doi: 10.1056/NEJMoa2001017
8. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
9. Phua J, Weng L, Ling L, Egi M, Lim C-M, Divatia JV, et al. Intensive care management of coronavirus disease 2019 (COVID-19): challenges and recommendations. Lancet Respir Med. (2020) 8:506–17. doi: 10.1016/S2213-2600(20)30161-2
10. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. (2020) 395:507–13. doi: 10.1016/S0140-6736(20)30211-7
11. Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA, et al. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med. (2014) 2:611–20. doi: 10.1016/S2213-2600(14)70097-9
12. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. (2016) 19:181–93. doi: 10.1016/j.chom.2016.01.007
13. Gattinoni L, Chiumello D, Rossi S. COVID-19 pneumonia: ARDS or not? Crit Care. (2020) 24:154. doi: 10.1186/s13054-020-02880-z
14. Rello J, Storti E, Belliato M, Serrano R. Clinical phenotypes of SARS-CoV-2: implications for clinicians and researchers. Eur Respir J. (2020) 55:2001028. doi: 10.1183/13993003.01028-2020
15. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med. (2020) 217:652. doi: 10.1084/jem.20200652
16. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat Med. (2005) 11:875–9. doi: 10.1038/nm1267
17. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80.e8. doi: 10.1016/j.cell.2020.02.052
18. Tai W, He L, Zhang X, Pu J, Voronin D, Jiang S, et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol. (2020) 17:613–20. doi: 10.1038/s41423-020-0400-4
19. Li M-Y, Li L, Zhang Y, Wang X-S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty. (2020) 9:45. doi: 10.1186/s40249-020-00662-x
20. Gupte M, Boustany-Kari CM, Bharadwaj K, Police S, Thatcher S, Gong MC, et al. ACE2 is expressed in mouse adipocytes and regulated by a high-fat diet. Am J Physiol Regul Integr Comp Physiol. (2008) 295:R781–8. doi: 10.1152/ajpregu.00183.2008
21. Simões e Silva A, Silveira K, Ferreira A, Teixeira M. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol. (2013) 169:477–92. doi: 10.1111/bph.12159
22. Vaajanen A, Kalesnykas G, Vapaatalo H, Uusitalo H. The expression of Mas-receptor of the renin–angiotensin system in the human eye. Graefes Arch Clin Exp Ophthalmol. (2015) 253:1053–9. doi: 10.1007/s00417-015-2952-z
23. Morales MG, Abrigo J, Meneses C, Cisternas F, Simon F, Cabello-Verrugio C. Expression of the Mas receptor is upregulated in skeletal muscle wasting. Histochem Cell Biol. (2015) 143:131–41. doi: 10.1007/s00418-014-1275-1
24. Singh Y, Gupta G, Sharma R, Matta Y, Mishra A, Pinto T, de JA, et al. Embarking effect of ACE2-angiotensin 1-7/Mas receptor axis in benign prostate hyperplasia. Crit Rev Eukaryot Gene Expr. (2018) 28:115–24. doi: 10.1615/CritRevEukaryotGeneExpr.2018021364
25. Zhang C, Jiang Z, Shao C. Clinical characteristics of allergic bronchopulmonary aspergillosis. Clin Respir J. (2020) 131:1108–9. doi: 10.1111/crj.13147
26. Sobrino A, Vallejo S, Novella S, Lázaro-Franco M, Mompeón A, Bueno-Betí C, et al. Mas receptor is involved in the estrogen-receptor induced nitric oxide-dependent vasorelaxation. Biochem Pharmacol. (2017) 129:67–72. doi: 10.1016/j.bcp.2017.01.012
27. Lin L, Liu X, Xu J, Weng L, Ren J, Ge J, et al. Mas receptor mediates cardioprotection of angiotensin-(1-7) against Angiotensin II-induced cardiomyocyte autophagy and cardiac remodelling through inhibition of oxidative stress. J Cell Mol Med. (2016) 20:48–57. doi: 10.1111/jcmm.12687
28. Hammer A, Yang G, Friedrich J, Kovacs A, Lee D-H, Grave K, et al. Role of the receptor Mas in macrophage-mediated inflammation in vivo. Proc Natl Acad Sci USA. (2016) 113:14109–14. doi: 10.1073/pnas.1612668113
29. Lelis D, de F, Freitas DF, de Machado AS, Crespo TS, Santos SHS. Angiotensin-(1-7), adipokines and inflammation. Metab Clin Exp. (2019) 95:36–45. doi: 10.1016/j.metabol.2019.03.006
30. Lau Y-L. SARS: future research and vaccine. Paediatr Respir Rev. (2004) 5:300–3. doi: 10.1016/j.prrv.2004.07.005
31. Lau YL, Peiris JSM. Pathogenesis of severe acute respiratory syndrome. Curr Opin Immunol. (2005) 17:404–10. doi: 10.1016/j.coi.2005.05.009
32. Law HKW, Cheung CY, Ng HY, Sia SF, Chan YO, Luk W, et al. Chemokine up-regulation in SARS-coronavirus–infected, monocyte-derived human dendritic cells. Blood. (2005) 106:2366–74. doi: 10.1182/blood-2004-10-4166
33. Totura AL, Whitmore A, Agnihothram S, Schäfer A, Katze MG, Heise MT, et al. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio. (2015) 6:15. doi: 10.1128/mBio.00638-15
34. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. (2014) 14:36–49. doi: 10.1038/nri3581
35. Kindler E, Thiel V, Weber F. Chapter seven - interaction of SARS and MERS coronaviruses with the antiviral interferon response. In J. Ziebuhr, editor. Advances in Virus Research Coronaviruses (Cambridge, MA: Academic Press). p. 219–43. doi: 10.1016/bs.aivir.2016.08.006
36. Hemert MJ, van Worm SHE, van den Knoops K, Mommaas AM, Gorbalenya AE, Snijder EJ. SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLOS Pathogens. (2008) 4:e1000054. doi: 10.1371/journal.ppat.1000054
37. Knoops K, Kikkert M, Worm SHE, van den Zevenhoven-Dobbe JC, Meer Y, van der Koster AJ, et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLOS Biol. (2008) 6:e226. doi: 10.1371/journal.pbio.0060226
38. Menachery VD, Yount BL, Josset L, Gralinski LE, Scobey T, Agnihothram S, et al. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-O-methyltransferase activity. J Virol. (2014) 88:4251–64. doi: 10.1128/JVI.03571-13
39. Li S-W, Wang C-Y, Jou Y-J, Huang S-H, Hsiao L-H, Wan L, et al. SARS coronavirus papain-like protease inhibits the TLR7 signaling pathway through removing Lys63-linked polyubiquitination of TRAF3 and TRAF6. Int J Mol Sci. (2016) 17:678. doi: 10.3390/ijms17050678
40. Hu Y, Li W, Gao T, Cui Y, Jin Y, Li P, et al. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J Virol. (2017) 91:16. doi: 10.1128/JVI.02143-16
41. Al-Qahtani AA, Lyroni K, Aznaourova M, Tseliou M, Al-Anazi MR, Al-Ahdal MN, et al. Middle east respiratory syndrome corona virus spike glycoprotein suppresses macrophage responses via DPP4-mediated induction of IRAK-M and PPARγ. Oncotarget. (2017) 8:9053–66. doi: 10.18632/oncotarget.14754
42. Huang C, Lokugamage KG, Rozovics JM, Narayanan K, Semler BL, Makino S. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathog. (2011) 7:2433. doi: 10.1371/journal.ppat.1002433
43. Lokugamage KG, Narayanan K, Nakagawa K, Terasaki K, Ramirez SI, Tseng C-TK, et al. Middle east respiratory syndrome coronavirus nsp1 inhibits host gene expression by selectively targeting mRNAs transcribed in the nucleus while sparing mRNAs of cytoplasmic origin. J Virol. (2015) 89:10970–81. doi: 10.1128/JVI.01352-15
44. Frieman M, Yount B, Heise M, Kopecky-Bromberg SA, Palese P, Baric RS. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J Virol. (2007) 81:9812–24. doi: 10.1128/JVI.01012-07
45. Yang Y, Shen C, Li J, Yuan J, Wei J, Huang F, et al. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J Allergy Clin Immunol. (2020) 146:119–27. doi: 10.1016/j.jaci.2020.04.027
46. Fung S-Y, Yuen K-S, Ye Z-W, Chan C-P, Jin D-Y. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: lessons from other pathogenic viruses. Emerg Microbes Infect. (2020) 9:558–70. doi: 10.1080/22221751.2020.1736644
47. McGonagle D, Sharif K, O'Regan A, Bridgewood C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev. (2020) 19:102537. doi: 10.1016/j.autrev.2020.102537
48. Faure E, Poissy J, Goffard A, Fournier C, Kipnis E, Titecat M, et al. Distinct immune response in two MERS-CoV-infected patients: can we go from bench to bedside? PLoS ONE. (2014) 9:e88716. doi: 10.1371/journal.pone.0088716
49. Raucci F, Mansour AA, Casillo GM, Saviano A, Caso F, Scarpa R, et al. Interleukin-17A (IL-17A), a key molecule of innate and adaptive immunity, and its potential involvement in COVID-19-related thrombotic and vascular mechanisms. Autoimmun Rev. (2020) 19:102572. doi: 10.1016/j.autrev.2020.102572
50. Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med. (2020) 382:1199–207. doi: 10.1056/NEJMoa2001316
51. Simonnet A, Chetboun M, Poissy J, Raverdy V, Noulette J, Duhamel A, et al. High prevalence of obesity in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) requiring invasive mechanical ventilation. Obesity. 28:1195–9. doi: 10.1002/oby.22831
52. Caussy C, Pattou F, Wallet F, Simon C, Chalopin S, Telliam C, et al. Prevalence of obesity among adult inpatients with COVID-19 in France. Lancet Diabetes Endocrinol. (2020) 8:562–4. doi: 10.1016/S2213-8587(20)30160-1
53. Dietz W, Santos-Burgoa C. Obesity and its implications for COVID-19 mortality. Obesity. (2020) 28:1005. doi: 10.1002/oby.22818
54. Borel A-L, Schwebel C, Planquette B, Vésin A, Garrouste-Orgeas M, Adrie C, et al. Initiation of nutritional support is delayed in critically ill obese patients: a multicenter cohort study. Am J Clin Nutr. (2014) 100:859–66. doi: 10.3945/ajcn.114.088187
55. Nie W, Zhang Y, Jee SH, Jung KJ, Li B, Xiu Q. Obesity survival paradox in pneumonia: a meta-analysis. BMC Med. (2014) 12:61. doi: 10.1186/1741-7015-12-61
56. Zhi G, Xin W, Ying W, Guohong X, Shuying L. “Obesity paradox” in acute respiratory distress syndrome: asystematic review and meta-analysis. PLoS ONE. (2016) 11:e0163677. doi: 10.1371/journal.pone.0163677
57. Wilson MR, Petrie JE, Shaw MW, Hu C, Oakley CM, Woods SJ, et al. High-fat feeding protects mice from ventilator-induced lung injury, via neutrophil-independent mechanisms. Crit Care Med. (2017) 45:e831–9. doi: 10.1097/CCM.0000000000002403
58. de Heredia FP, Gómez-Martínez S, Marcos A. Obesity, inflammation and the immune system. Proc Nutr Soc. (2012) 71:332–8. doi: 10.1017/S0029665112000092
59. Abella V, Scotece M, Conde J, Pino J, Gonzalez-Gay MA, Gómez-Reino JJ, et al. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat Rev Rheumatol. (2017) 13:100–9. doi: 10.1038/nrrheum.2016.209
60. Fang H, Judd RL. Adiponectin regulation and function. Compr Physiol. (2018) 8:1031–63. doi: 10.1002/cphy.c170046
61. Francisco V, Ruiz-Fernández C, Pino J, Mera A, González-Gay MA, Gómez R, et al. Adipokines: linking metabolic syndrome, the immune system, and arthritic diseases. Biochem Pharmacol. (2019) 165:196–206. doi: 10.1016/j.bcp.2019.03.030
62. Trayhurn P, Alomar SY. Oxygen deprivation and the cellular response to hypoxia in adipocytes—perspectives on white and brown adipose tissues in obesity. Front Endocrinol. (2015) 6:19. doi: 10.3389/fendo.2015.00019
63. Daryabor G, Kabelitz D, Kalantar K. An update on immune dysregulation in obesity-related insulin resistance. Scand J Immunol. (2019) 89:e12747. doi: 10.1111/sji.12747
64. Francisco V, Pino J, Gonzalez-Gay MA, Mera A, Lago F, Gómez R, Mobasheri A, et al. Adipokines and inflammation: is it a question of weight? Br J Pharmacol. (2018) 175:1569–79. doi: 10.1111/bph.14181
65. Mori J, Oudit GY, Lopaschuk GD. SARS-CoV-2 perturbs the Renin-Angiotensin System and energy metabolism. Am J Physiol Endocrinol Metab. (2020) 319:E43–7. doi: 10.1152/ajpendo.00219.2020
66. Dikmen K, Bostanci H, Gobut H, Yavuz A, Alper M, Kerem M. Recombinant adiponectin inhibits inflammation processes via NF-kB pathway in acute pancreatitis. Bratisl Lek Listy. (2018) 119:619–24. doi: 10.4149/BLL_2018_110
67. Claycombe K, King LE, Fraker PJ. A role for leptin in sustaining lymphopoiesis and myelopoiesis. Proc Natl Acad Sci USA. (2008) 105:2017–21. doi: 10.1073/pnas.0712053105
68. Bruno A, Conus S, Schmid I, Simon H-U. Apoptotic pathways are inhibited by leptin receptor activation in neutrophils. J Immunol. (2005) 174:8090–6. doi: 10.4049/jimmunol.174.12.8090
69. Shah TJ, Leik CE, Walsh SW. Neutrophil infiltration and systemic vascular inflammation in obese women. Reprod Sci. (2010) 17:116–24. doi: 10.1177/1933719109348252
70. Honce R, Schultz-Cherry S. Impact of obesity on influenza A virus pathogenesis, immune response, and evolution. Front Immunol. (2019) 10:71. doi: 10.3389/fimmu.2019.01071
71. Fabre O, Breuker C, Amouzou C, Salehzada T, Kitzmann M, Mercier J, et al. Defects in TLR3 expression and RNase L activation lead to decreased MnSOD expression and insulin resistance in muscle cells of obese people. Cell Death Dis. (2014) 5:e1136. doi: 10.1038/cddis.2014.104
72. Latorre J, Moreno-Navarrete JM, Sabater M, Buxo M, Rodriguez-Hermosa JI, Girones J, et al. Decreased TLR3 in hyperplastic adipose tissue, blood and inflamed adipocytes is related to metabolic inflammation. Cell Physiol Biochem. (2018) 51:1051–68. doi: 10.1159/000495487
73. Teran-Cabanillas E, Montalvo-Corral M, Caire-Juvera G, Moya-Camarena SY, Hernández J. Decreased interferon-α and interferon-β production in obesity and expression of suppressor of cytokine signaling. Nutrition. (2013) 29:207–12. doi: 10.1016/j.nut.2012.04.019
74. Guo W, Li M, Dong Y, Zhou H, Zhang Z, Tian C, et al. Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes Metab Res Rev. (2020) 29:207–12. doi: 10.1002/dmrr.3319
75. Gao F, Zheng KI, Wang X-B, Yan H-D, Sun Q-F, Pan K-H, et al. Metabolic associated fatty liver disease increases COVID-19 disease severity in non-diabetic patients. J Gastroenterol Hepatol. (2020). doi: 10.1111/jgh.15112. [Epub ahead of print].
76. Pazarli AC, Ekiz T, Ilik F. Coronavirus disease 2019 and obstructive sleep apnea syndrome. Sleep Breath. (2020) 1. doi: 10.1007/s11325-020-02087-0. [Epub ahead of print].
77. Oliveira Andrade JM, Paraíso AF, Garcia ZM, Ferreira AVM, Sinisterra RDM, Sousa FB, et al. Cross talk between angiotensin-(1-7)/Mas axis and sirtuins in adipose tissue and metabolism of high-fat feed mice. Peptides. (2014) 55:158–65. doi: 10.1016/j.peptides.2014.03.006
78. Thatcher SE, Gupte M, Hatch N, Cassis LA. Deficiency of ACE2 in bone-marrow-derived cells increases expression of TNF-α in adipose stromal cells and augments glucose intolerance in obese C57BL/6 mice. Int J Hypertens. (2012) 2012:762094. doi: 10.1155/2012/762094
79. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. (2006) 116:3015–25. doi: 10.1172/JCI28898
80. Patel VB, Mori J, McLean BA, Basu R, Das SK, Ramprasath T, et al. ACE2 deficiency worsens epicardial adipose tissue inflammation and cardiac dysfunction in response to diet-induced obesity. Diabetes. (2016) 65:85–95. doi: 10.2337/db15-0399
81. Santos SHS, Andrade JMO, Fernandes LR, Sinisterra RDM, Sousa FB, Feltenberger JD, et al. Oral Angiotensin-(1-7) prevented obesity and hepatic inflammation by inhibition of resistin/TLR4/MAPK/NF-κB in rats fed with high-fat diet. Peptides. (2013) 46:47–52. doi: 10.1016/j.peptides.2013.05.010
82. Sukumaran V, Tsuchimochi H, Tatsumi E, Shirai M, Pearson JT. Azilsartan ameliorates diabetic cardiomyopathy in young db/db mice through the modulation of ACE-2/ANG 1-7/Mas receptor cascade. Biochem Pharmacol. (2017) 144:90–9. doi: 10.1016/j.bcp.2017.07.022
83. Santos SHS, Andrade JMO. Angiotensin 1-7: a peptide for preventing and treating metabolic syndrome. Peptides. (2014) 59:34–41. doi: 10.1016/j.peptides.2014.07.002
84. Solaimanzadeh I. Nifedipine and Amlodipine Are Associated With Improved Mortality and Decreased Risk for Intubation and Mechanical Ventilation in Elderly Patients Hospitalized for COVID-19. Cureus. 12:e8069. doi: 10.7759/cureus.8069
85. Kai H, Kai M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors-lessons from available evidence and insights into COVID-19. Hypertens Res. (2020) 43:648–54. doi: 10.1038/s41440-020-0455-8
86. Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin–angiotensin–aldosterone system inhibitors in patients with Covid-19. N Engl J Med. (2020) 382:1653–9. doi: 10.1056/NEJMsr2005760
Keywords: coronavirus, SARS-CoV, ACE2, obesity, adipocyte, metabolic syndrome, inflammation
Citation: Méry G, Epaulard O, Borel A-L, Toussaint B and Le Gouellec A (2020) COVID-19: Underlying Adipokine Storm and Angiotensin 1-7 Umbrella. Front. Immunol. 11:1714. doi: 10.3389/fimmu.2020.01714
Received: 10 June 2020; Accepted: 29 June 2020;
Published: 21 July 2020.
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Oreste Gualillo, Servicio Gallego de Salud, SpainP. Trayhurn, University of Liverpool, United Kingdom
Copyright © 2020 Méry, Epaulard, Borel, Toussaint and Le Gouellec. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Audrey Le Gouellec, YWxlZ291ZWxsZWNAY2h1LWdyZW5vYmxlLmZy