 Jackline Joy Martín Lasola
Jackline Joy Martín Lasola Henry Kamdem
Henry Kamdem Michael W. McDaniel
Michael W. McDaniel Ryan M. Pearson
Ryan M. Pearson- 1Department of Microbiology and Immunology, University of Maryland School of Medicine, Baltimore, MD, United States
- 2Department of Pharmaceutical Sciences, University of Maryland School of Pharmacy, Baltimore, MD, United States
- 3Marlene and Stewart Greenebaum Comprehensive Cancer Center, University of Maryland School of Medicine, Baltimore, MD, United States
Inflammation is an essential component of a wide variety of disease processes and oftentimes can increase the deleterious effects of a disease. Finding ways to modulate this essential immune process is the basis for many therapeutics under development and is a burgeoning area of research for both basic and translational immunology. In addition to developing therapeutics for cellular and molecular targets, the use of biomaterials to modify innate and adaptive immune responses is an area that has recently sparked significant interest. In particular, immunomodulatory activity can be engineered into biomaterials to elicit heightened or dampened immune responses for use in vaccines, immune tolerance, or anti-inflammatory applications. Importantly, the inherent physicochemical properties of the biomaterials play a significant role in determining the observed effects. Properties including composition, molecular weight, size, surface charge, and others affect interactions with immune cells (i.e., nano-bio interactions) and allow for differential biological responses such as activation or inhibition of inflammatory signaling pathways, surface molecule expression, and antigen presentation to be encoded. Numerous opportunities to open new avenues of research to understand the ways in which immune cells interact with and integrate information from their environment may provide critical solutions needed to treat a variety of disorders and diseases where immune dysregulation is a key inciting event. However, to elicit predictable immune responses there is a great need for a thorough understanding of how the biomaterial properties can be tuned to harness a designed immunological outcome. This review aims to systematically describe the biological effects of nanoparticle properties—separate from additional small molecule or biologic delivery—on modulating innate immune cell responses in the context of severe inflammation and sepsis. We propose that nanoparticles represent a potential polypharmacological strategy to simultaneously modify multiple aspects of dysregulated immune responses where single target therapies have fallen short for these applications. This review intends to serve as a resource for immunology labs and other associated fields that would like to apply the growing field of rationally designed biomaterials into their work.
Introduction
Inflammation is a complex and essential homeostatic response to extrinsic and intrinsic damage. This process is responsible for everything from recognition of microbial breeches into sterile sites and tissue damage to clearance of the insulting microbe and resolution of the immune response. Host defense mechanisms act to mobilize immune cells and molecules into vascularized tissues with the objective to eliminate the source of cell injury. Acute inflammation has been noted since antiquity by the cardinal signs of rubor (redness), tumor (swelling), calor (heat), dolor (pain), and functio laesa (loss of function) (1). These cardinal signs may also be accompanied by systemic features triggered by cytokine release, such as fever, changes in the peripheral white blood cell count, and increases in clinically detectable acute phase reactants in the blood stream (2). Given the vast coordination of tissue sites and organ systems, inflammation requires a finely tuned, highly regulated physiologic process where a concerted regulatory network of cellular and chemical mediators exists to limit the extent, severity, and spread of inflammation. Failure to effectively and efficiently resolve this process leads to a state of chronic inflammation that can exacerbate disease and pathologic processes. As the role of inflammation becomes further appreciated as a major driver of pathogenesis in many diseases, the need for technologies capable of modulating vascular and immune responses during uncontrolled inflammation will become increasingly necessary.
In this review, we will facilitate our discussion of dysregulated inflammatory responses within the context of severe inflammation and sepsis. These serve as fitting models for understanding the inflammatory response and what occurs when it fails to resolve as expected (3, 4). Unfortunately, there are currently very few nanotechnology platforms that specifically investigate their utility for this indication. As such, we will address what is known about the physiologic pathways and mechanisms at play during inflammation through discussions of emerging technological developments to modulate inflammation in a variety of diseases. This will serve as a starting point to consider future nanotechnology prospects to improve patient health for those suffering from sepsis.
Here, we will specifically focus on technologies where the immune responses can be attributed to the inherent physicochemical properties of the engineered biomaterial itself (i.e., in the absence of any immunomodulatory small molecule or biologic). Although the immune response has previously been treated as something that must be overcome in the development of nanoparticles and microparticles (henceforth, referred to collectively as nanoparticles) for clinical usage, the rise of immunotherapy for vaccine and immune tolerance applications has shed new light on the ways nanoparticle physicochemical properties can be used for similar purposes to modify existing immune responses (5–8), especially for indications where dysfunctional or exaggerated inflammation and immunological processes are contributing factors. Lastly, these nanotechnology-based approaches will be discussed in the context of what is known about the biological processes during inflammation as appreciation of nano-bio interactions (9, 10) allows for development of design strategies for future biomaterial approaches. Our objective is to highlight emerging patterns in biomaterial designs for a variety of nanoparticle platforms and how they have been shown to regulate multifactorial immune responses by acting at various points in the inflammatory cascade. Given that inflammation is exceptionally complex, we propose nanoparticles as a polypharmacological approach to shift how we think about therapeutics for severe inflammation and sepsis.
The Persistence of Systemic Inflammation and Limitations of Sepsis Therapeutic Development
Inflammation is a powerful, multifactorial host defense mechanism intended to protect the host from microbial insult and tissue damage (11). As such, it is not only essential to the maintenance of homeostasis and protection but also can be deleterious on its own when regulatory mechanisms go awry. Examples of conditions characterized by dysregulated inflammatory responses include chronic inflammation, inappropriate fibrosis and scarring, and sepsis syndrome. These conditions demonstrate the delicate balance between damage and repair by which inflammation drives much of the pathology of common diseases including atherosclerosis, diabetes, neurodegenerative disorders, and others. Although situations of chronic inflammation and inappropriate wound healing are outside the scope of this review, sepsis syndrome provides a highly informative case study of what happens when acute inflammation continues unabated. When left unchecked, continuous inflammation leads to its own set of deleterious, systemic acute phase responses, and ineffectual regulatory responses.
Sepsis is a life-threatening condition of complex pathophysiological origin that develops due to an uncontrolled immune response during infection (12–14). Hallmark features of sepsis include profound acute pro-inflammatory cytokine responses, vascular endothelial leakage, and multi-organ failure (15). Concomitantly, a compensatory anti-inflammatory response develops in an attempt to resolve inflammation and promote tissue repair. This is accompanied by immune paralysis whereby antigen presenting cells (APCs) such as macrophages and dendritic cells lose their responsiveness to subsequent inflammatory challenge and significant alterations in immune cell apoptotic programming results in immunosuppression occurring with immune-mediated organ dysfunction (16–19). Beyond the dramatic physiologic changes, the human toll of sepsis and septic shock remains quite dramatic and accounted for over 2.5 million cases and $52 billion in aggregate costs between January 2010 and September 2016 alone (20). The global burden of sepsis is even greater with conservative estimates indicating that it is the leading cause of mortality and critical illness worldwide (13, 21). In 2017, it was estimated that 19.7% of all global deaths could be attributed to sepsis or sepsis-related causes (12). Because of this acute burden and the long-term physical, psychological, and cognitive disabilities for those who survive sepsis (22) efforts to improve treatment strategies and therapeutic approaches for those with and at risk of sepsis have been ongoing. While mainstays of treatment remain early administration of broad-spectrum antibiotics and intravenous fluids along with resuscitation, additional successful attempts to improve patient management for those with sepsis remain limited (23, 24).
Since the 1980's, over 100 therapeutic clinical trials have been conducted for sepsis and septic shock with little to no improved prospects for those affected. The general strategy for research and drug development within this space has been the generation of highly targeted agents that can be classified into categories based on known mechanisms at play in inflammation. These include as disparate and broad categories as anti-cytokines, anti-virulence factors, coagulopathy agents, anti-inflammatory agents, and even immune stimulators [reviewed nicely in (23–26)] with some of these examples being described in what follows. Of all the clinical trials, only activated protein C (drotrecogin alfa-activated), whose mechanism of action is an antithrombotic effect due to inhibition of factors Va and VIIIa in the coagulation cascade, was successfully licensed following a highly publicized clinical trial (27, 28). Yet, it was removed from the market a decade later due to further work showing not only a lack of a survival benefit for sepsis patients but also increased bleeding risks (29–32). With the inconclusive clinical trial data and controversial marketing strategies for drotrecogin alfa-activated, the example of activated protein C therapies and other failed clinical trials for targeted therapeutics raises the question, what are we missing in our strategies to treat sepsis? Various researchers have attempted to answer this question and have identified a few major themes (24–26): (1) preclinical models poorly recapitulate the complex physiological and molecular changes of sepsis syndrome; (2) patients with sepsis are plagued by a variety of initiating microbial infections and modes of entry; and, (3) patients are themselves very demographically complex based on age, sex, comorbidities, genetics, and infection site. However, one factor that remains underappreciated is the complex and redundant mechanisms at play to initiate the underlying bout of severe inflammation and the resultant sepsis. As such, any attempts to resolve the underlying dysregulated inflammation that triggers sepsis requires an approach that can address the redundancies of this highly coordinated defense mechanism. Elucidation of this multifactorial process requires ongoing work in preclinical models despite current recommendations to move away from such investigations (33).
Collectively, the efficacy of these single-agent, single-target therapeutics has not been as successful as preclinical models has suggested and when shown to be of some benefit, responses are highly dependent on their administration during a narrow treatment window with the associated immunosuppression rarely being addressed along with the vast pro-inflammatory response (24, 34). As such, safe and effective multi-targeted therapeutics for sepsis are critically needed to overcome the considerable heterogeneity of deficiencies at the cellular and molecular level that accumulate to result in deleterious tissue and multiorgan damage in sepsis. Modification of these vascular and immune cell responses using engineered nanoparticles is the basis for new therapies aimed to suppress inflammatory responses and functionally reprogram dysregulated cells and molecular pathways (7, 35, 36). In the following sections, we will describe approaches to treating severe inflammation and sepsis using nanoparticle strategies informed by the known cellular and molecular pathophysiology of inflammatory processes.
Nanoparticle Modulators of Integrated Vascular and Immune Inflammatory Responses
As a highly regulated process, deficient and/or overexaggerated responses at any step in the inflammatory cascade can result in serious deterioration in the health status of an individual. The inflammatory response can be characterized by the following processes: (1) recognition of the injurious agent, (2) regulation of the response (control), (3) recruitment of leukocytes, (4) removal of the agent, and (5) resolution (repair) (11). Throughout this process, microvascular tissue, innate immune cells, and circulating soluble mediators act to respond. Further, deficits in the adaptive immune system can contribute to the body's inability to control the infection and repair. Within each step of this response, points for intervention exist for therapeutics to alter the progression of inflammation and modulate the systemic responses (Figure 1). For decades, researchers have focused on developing single molecule or single pathway targeted therapeutics to modify highly specific regulatory nodes of the inflammatory response. As our discussion progresses, a number of these approaches will be discussed to compare and contrast with newer nanotechnologies driven by current biological understanding of inflammation. By evaluating the numerous approaches, the intention is to suggest future paths of therapeutic research and development to alter outcomes for those with severe inflammation and sepsis.
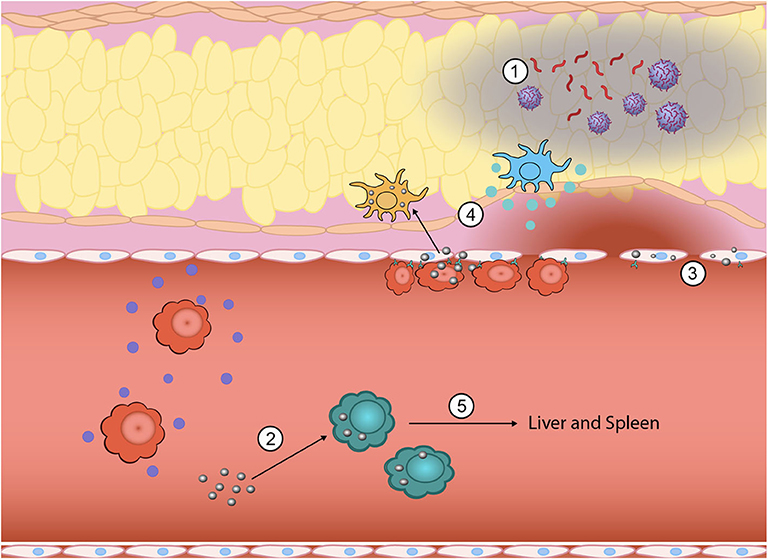
Figure 1. Inflammation is a highly complex, multistep process where nanoparticles can be engineered to intervene to tune the response at multiple points. During the initial generation of PAMPs and DAMPs, biomimetic nanoparticles have been used to sequester PAMPs and DAMPs from immune cell recognition (1). Innate immune cells that have taken up nanoparticles can be functionally reprogrammed from a pro-inflammatory phenotype (i.e., TNF-α, IL-1β, and IL-6 secreting) to an anti-inflammatory phenotype (2). The vascular endothelium also plays a key role in promoting inflammation and nanoparticles can be used to downregulate attachment of circulating immune cells and subsequent exudation (3). Nano-bio interactions can also alter direct homing to inflamed tissue sites by either eliminating chemokine production at the site (4) or redirecting inflammatory cells away from the inflamed site to the liver and spleen for elimination (5).
Halting Inflammation Before It Starts: Strategies to Sequester the Initiating Warning Signals of Inflammation
Initiation of inflammation requires recognition of the infectious microbe or products of cell and tissue damage. This work is accomplished by tissue macrophages, dendritic cells, and mast cells of the innate immune system, in addition to other sentinel cells resident in tissues that contain pattern recognition receptors (PRRs). These PRRs are unique in that they can recognize pathogen-associated and damage-associated molecular patterns (PAMPs and DAMPs, respectively) in a manner that triggers general molecular warning programs to initiate protective processes against the inciting insults (37, 38). These receptors include Toll-like receptors (TLRs) on the plasma and endosomal membranes, C-type lectin receptors (CLRs) on the plasma membrane, NOD-like receptors (NLRs) on the plasma and endosomal membranes and in the cytoplasm, RIG-I-like receptors (RLRs) in the cytoplasm, and AIM2-like receptors (ALRs) in the cytoplasm and nucleus (39). These receptors are unique in that they recognize cellular products exclusively produced by microbes, such as lipopolysaccharide (LPS) from Gram-negative bacteria or double-stranded RNA from specific viral classes, or cellular components that are only released by the host during times of tissue and cellular damage like high mobility group box 1 (HMGB1) (40, 41). These cellular responses are complemented with circulating proteins that serve as complementary humoral responses. Antibodies, complement proteins (42), and collectins (43–45) also recognize microbes, opsonize them, and target them for ingestion by phagocytes and activation of other immune cells via Fc receptors.
Given that inflammation starts with recognition of these PAMPs, DAMPs, and microbes, at the nascent site of inflammation, limiting the initiation of this process serves as a potential strategy by which to limit the severity of inflammation and halt progression to systemic inflammation. Traditional strategies to halt these initial stages of the inflammatory cascade have focused on neutralizing microbes, such as continuing antibiotic development or even utilization of human antiserum against microbes, such as against Escherichia coli (46). Newer approaches aim to bind and neutralize PAMPs, such as the development of monoclonal antibodies targeting the lipid A moiety of LPS (47–50) or direct antagonizing of the PRRs like trials conducted with eritoran, a TLR4 antagonist derived from lipid A (51, 52), or small molecule inhibitors of TLR signaling like TAK-242 (53).
More recent biomaterial strategies to prevent this initial recognition of microbial products and their ensuing damage are notable for biomimetic approaches (Figure 2 and Table 1) to sequester these initiators of inflammation and halt the cascade before it begins. Kunz et al. (55) developed cell-derived nanoparticles (CDNPs) to limit inflammation and showed that the CDNP platform was able to limit bacterial growth in vitro. CDNPs were generated via high-speed centrifugation of fibroblast cytoplasmic contents to isolate the desired exosomes. These exosomes were largely composed of proteins such as annexin A5, heat shock proteins, peroxiredoxines, with small traces of DNA and RNA, that showed preferential uptake by neutrophils, inflammatory monocytes, and macrophages, all key cells for the initiation of inflammation. This was correlated with decreased IL-6 levels in the peritoneum of mice with cecal ligation and puncture (CLP)-induced polymicrobial sepsis. Additional work showed that in an in vitro system, coincubation of these CDNPs with Pseudomonas aeruginosa resulted in direct decreases in bacterial colony-forming units, suggesting an additional bactericidal effect of the CDNPs. To contrast, a separate strategy by Thamphiwatana et al. (54) used a similar strategy of isolating immune cell components to drive protective responses against inflammation. The authors used poly(lactic-co-glycolic acid) (PLGA) as the core polymer for the particles and coated these particles with macrophage-derived cell membranes to prepare macrophage mimicking nanoparticles. As described in Figure 2, the authors show using both macrophages and endothelial cells (HUVECs), the ability of these macrophage mimicking nanoparticles to sequester LPS away from the PRRs of cells necessary to initiate the inflammatory cascade with an additional effect of also sequestering away inflammatory cytokines to prevent further inflammatory activation of macrophages and HUVECs. Using LPS-induced endotoxemia and E. coli-induced bacteremia murine models, these particles were shown to have a survival benefit specifically linked to the inclusion of the macrophage membranes in the particle formulation.
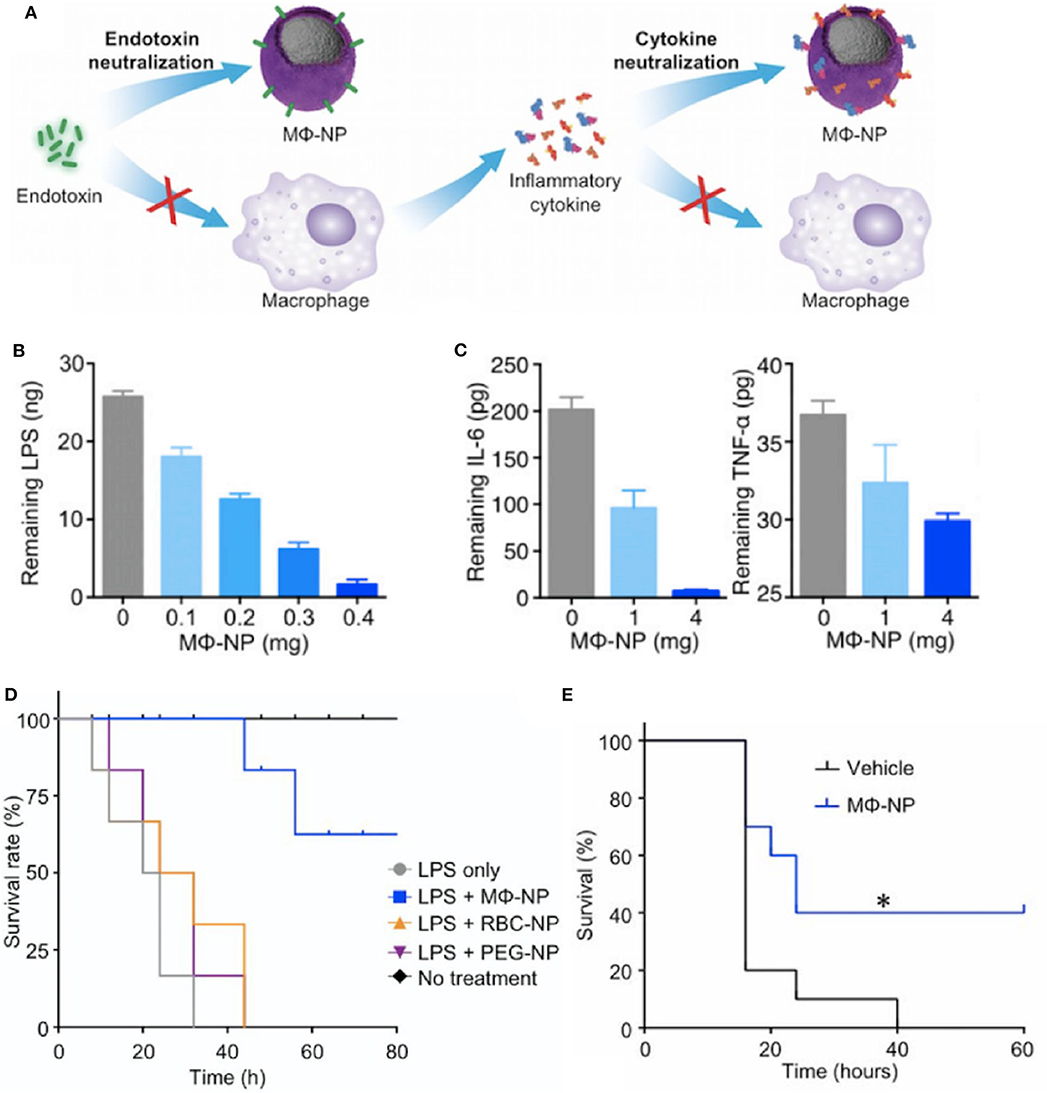
Figure 2. Macrophage mimicking nanoparticles (MΦ-NP) sequester bacteria derived endotoxin and subsequent inflammatory cytokines to limit inflammation associated damage (A). This results in a dose-dependent ability of the MΦ-NP to reduce free LPS (B) and pro-inflammatory cytokines such as IL-6 and TNF-α (C) in vitro. LPS-induced endotoxemia (D) and E. coli bacteremia (E) show a survival benefit specific to the biomimetic MΦ-NP, where *P < 0.05. Adapted from (54). Copyright (2017) National Academy of Science.
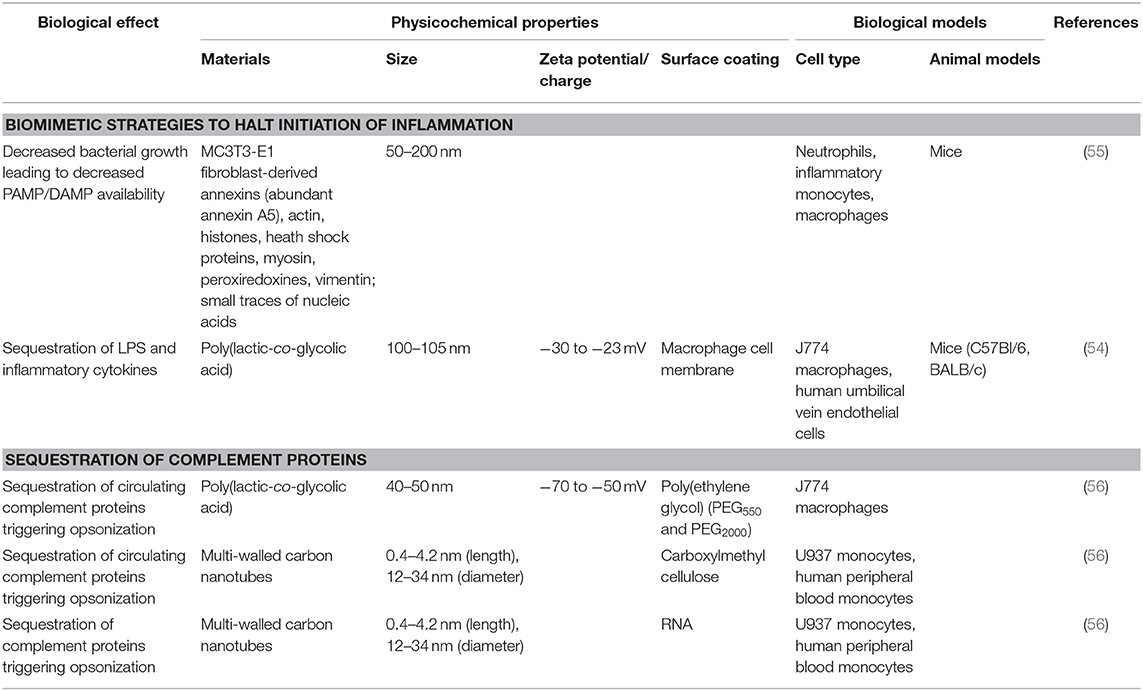
Table 1. Strategies to sequester the initiating warning signals of inflammation.
As noted above, in addition to direct cellular recognition of PAMPs and DAMPs, circulating proteins of the innate immune response can trigger activation of inflammatory pathways. Of particular note are those within the complement pathway where C3a, C4a, and C5a, serve as triggers of anaphylaxis and chemotaxis. Because of their key role as humoral mediators of inflammation, the interaction between biomaterials and the complement pathway are of great interest. In one study, PLGA particles were shown to trigger differential levels of complement activation based on the molecular weights of the poly(ethylene glycol) surface coating. By combining PEG550 with PEG2000 as the surface coating of PLGA particles, Pannuzzo et al. were able to limit generation of C5a and downstream complement components without altering particle uptake by macrophages (56). Another platform showed that multi-walled carbon nanotubes (CNTs) surface modified with carboxymethyl cellulose (CMC-CNT) or RNA (RNA-CNT) appear to serve as a type of sink for deposition of complement pathway proteins. This has the net effect of modifying the inherent pro-inflammatory responses of CNTs through analysis of dampened transcription of TNF-α and IL-1β in macrophages (57). In contrast, a study of CNTs showed that in combination with LPS activation, the pro-inflammatory effects of CNTs were mediated through inflammasome activation (58). This emphasizes the tunability of the immune response to CNTs in a manner dependent on their physicochemical properties. These varying responses to CNTs, with a particular emphasis on their effects on complement are described in a recent review (59).
Regulating the Regulators: Altering Production of Molecular Mediators of Inflammation
Due to its destructive potential, tight regulation of the initiation and progression of inflammation by its mediators is essential to limit deleterious effects beyond those necessary for eliminating the initial offending agent. As such, these mediators—including histamine, prostaglandins and leukotrienes, and cytokines and chemokines—are often targeted therapeutically to limit inflammation during disease processes (60, 61).
The vasoactive amine histamine is stored preformed in cells and is released upon mast cell degranulation (also blood basophils and platelets). This release allows for binding to the H1 receptor of microvascular endothelial cells to trigger arteriole dilation and increased venule vascular permeability. Due to histamine activity, it commonly serves as an anti-inflammatory target, particularly for allergy, and H1 receptor antagonists like diphenhydramine, loratadine, and cetirizine, are some of the most commonly used drugs for managing allergic reactions and acute inflammatory processes (62).
Arachidonic acid (AA) is found in membrane phospholipids and can be released from the membrane phospholipids (particularly from phospholipase A2, PLA2) upon activation to produce interesting classes of inflammation mediators, prostaglandins (PGs) and leukotrienes. Cyclooxygenases (COX-1 and COX-2) in mast cells, macrophages, and endothelial cells produce PGs to trigger vascular and systemic signs and symptoms of inflammation (63). PGE2 and PGD2 (mast cells) trigger vasodilation and increases permeability of postcapillary venules to allow for edema formation, whereas PGF2a stimulates uterine, bronchial, and small arteriole smooth muscle contraction. Prostacyclin (PGI2) is produced in vascular endothelium and serves as a vasodilator and potent inhibitor of platelet aggregator, in addition to serving as a potentiator of other mediators that increase vascular permeability and chemotaxis to sites of injury. Thromboxane (TxA2), produced in platelets, opposes the effects of prostacyclin in that it is a vasoconstrictor and a potent inducer of platelet aggregation. In addition to these local effects, prostaglandins are implicated in promoting the systemic symptoms of inflammation, namely pain and fever. In contrast, leukotrienes are produced in leukocytes and mast cells by lipoxygenases where LTB4 serves as a potent chemoattractant while LTC4, LTD4, and LTE4, serve to induce vasoconstriction, bronchospasm, and increased permeability of venules in a manner more potent than the initial histamine release from mast cell degranulation (64).
Given the central role of AA metabolites in inflammation, pharmacologic inhibitors of AA metabolism are widespread in the pharmacopeia. Corticosteroids are an essential class of drugs that can prevent the initial release of AA by phospholipase activity in addition to a series of other proposed mechanisms of action. Non-steroidal anti-inflammatory drugs (NSAIDs) like naproxen and ibuprofen are common over the counter and prescription medication that serve as COX inhibitors to limit inflammation, while lipoxygenase inhibitors and leukotriene receptor antagonists serve as therapeutic strategies in asthma management due to their specific induction of bronchial smooth muscle contraction (63).
In contrast to the non-specific, broad activities of corticosteroids or the highly specific COX inhibitors utilized for asthma management, the applicability of these strategies for severe inflammation and sepsis have been of limited utility. A variety of clinical trials using corticosteroids have shown inconclusive results ranging from benefit with hydrocortisone and fludrocortisone (65) to no overall effect with hydrocortisone (66–70), methylprednisolone, or dexamethasone (71) with some clinical studies concluding corticosteroid strategies with methylprednisolone to actually be a detriment to survival (72). Similarly, a trial for ibuprofen, a common NSAID that serves as an unselective COX inhibitor, showed no effect on mortality in severe sepsis (73).
The limited successes in utilizing these anti-inflammatory strategies in sepsis, has left open the opportunity for biomaterials to serve a role in modifying these immune mediators. Often, due to the inherent capability of materials to be easily altered to better bind a broad variety of metabolites in the bloodstream. An interesting approach is one taken by O'Brien et al. (74, 75) where poly(N-isopropylacrylamide) (NIPAm) particles generated in combination with other acrylamide moieties were synthesized to alter the affinity of the protein corona for a variety of plasma components (Figure 3). This desire to “tune” the corona for high affinity and selectivity to a variety of biomacromolecules showed that although NIPAm-based particles showed little affinity for plasma proteins, the hydrophobicity of NIPAm-based particles allowed for them to interact favorably with lipophilic molecules. This later was used to show that they could be used for lipid-bound toxin sequestration and neutralization, such as whole honey-bee venom containing a significant amount of venomous PLA2. It would be interesting to see this work expanded to see if this sequestration and neutralization strategy via protein corona tuning could be applied to neutralizing endogenous lipid species released during inflammation such as the AA metabolites described above that are produced upon vascular endothelial activation.
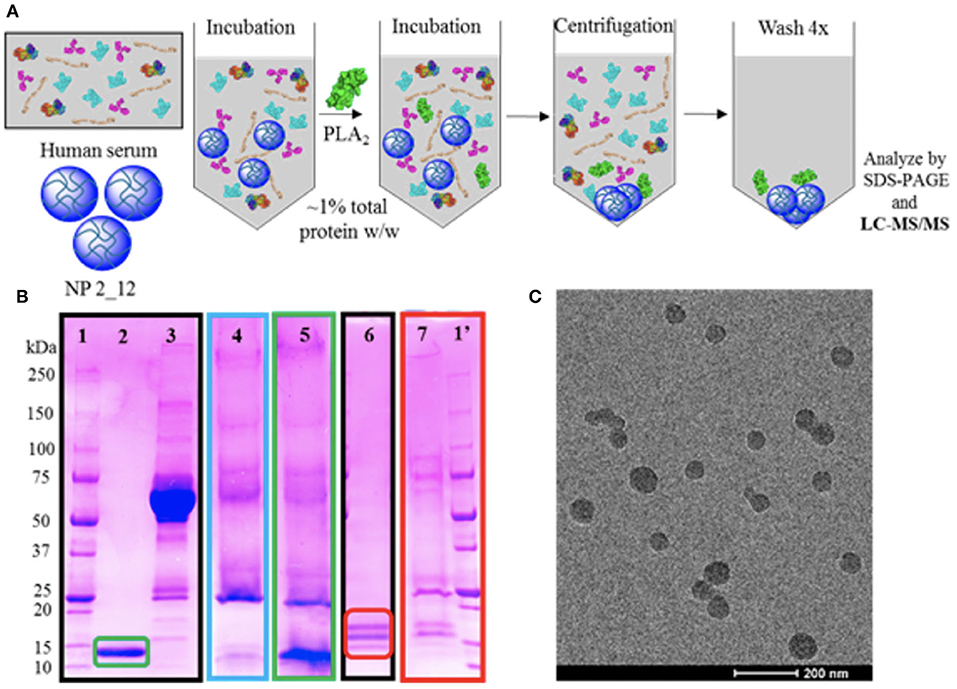
Figure 3. Selectivity experiments and TEM characterization of nanoparticles for targeted sequestration of venom proteins. Polymer composition was optimized to enable specificity toward venom yet avoid serum protein binding. Strategy for assessing selectivity of nanoparticles to venom (A). Selectivity assessment via SDS-PAGE visualization (B) of (1/1′) ladder; (2) purified PLA2 from Naja mossambica venom; (3) serum control; (4) nanoparticle in serum only; (5) nanoparticle incubated in serum and PLA2 from N. mossambica venom; (6) purified PLA2 from honey-bee venom; (7) nanoparticle incubated in ovine plasma and PLA2 from honey-bee venom. Unstained TEM image of nanoparticle for sequestration of venom (C). Reprinted with permission from (74). Copyright (2016) American Chemical Society.
Similar to the role played by AA metabolites in regulating vascular activity, cytokines and chemokines are proteins produced and secreted by a variety of cell types (activated lymphocytes, macrophages, dendritic cells; also, endothelial, epithelial, and connective tissue cells) to regulate immune and inflammatory activity. In acute inflammation, tumor necrosis factor alpha (TNF-α) and interleukin-1 beta (IL-1β) are essential for leukocyte recruitment by their promotion of endothelial adhesion and diapedesis. Given this activity, research on cytokine receptor blockade has produced therapeutic drug classes that have broad effects at modifying autoimmune disease outcomes. Beyond activating the endothelium and leukocytes, TNF-α, IL-1β, and IL-6 combine to induce the systemic acute phase response that is implicated in the development of sepsis (76–78). In comparison, chemokines serve to attract specific cells to the inflammatory site with individual chemokines of note being IL-8 from macrophages and endothelial cells that drive neutrophil recruitment (79), while monocyte chemoattractant protein (MCP-1) (80), macrophage inflammatory protein-1α (MIP-1α) (81), and regulated and normal T cell expressed and secreted (RANTES) (82), attract monocytes, eosinophils, basophils, and lymphocytes.
The elucidation of molecular mechanisms involved in leukocyte recruitment and migration during inflammation have led to major developments in the generation of therapeutic targets for a variety of inflammation-mediated diseases. Although first discovered to play a role in the pathogenesis of sepsis (83, 84), therapies to directly block TNF-α and IL-1β signaling have done more to change the progression and day-to-day symptomatology for patients with a variety of autoimmune (60, 85) and dermatological conditions (86). In contrast, a variety of clinical trials utilizing anti-TNF monoclonal antibodies (26) or even fusion proteins to neutralize TNF-α, like lenercept (87, 88), or etanercept (89), which is in common clinical usage today, have shown no benefit and even harm for patients with septic shock in the case of etanercept (89). This has also been shown with anakinra, an IL-1β decoy receptor, which failed to conclusively show a survival benefit for patients with sepsis or septic shock (90–92).
Interestingly, biomaterials have also been shown to have inherent capabilities to alter immune cells to downregulate key chemotactic molecules (Figure 1 and Table 2) at play in recruiting leukocytes to inflammatory sites. As described above, key players of this process include the cytokines TNF-α and IL-1β. Inhibition of the innate immune cell's capability to secrete TNF-α and IL-1β serves to achieve a similar end as halting initiation of inflammatory signaling and shows the ability to act following activation of inflamed immune cells. Multiple groups have shown a capability to utilize biomaterials to affect this alteration in a variety of inflammation models and suggest a diversity of strategies to impart a similar net effect to modify the molecular regulators of immune activity during inflammation. Using poly(lactic acid) (PLA) and PLGA as nanoparticle cores with poly(ethylene-alt-maleic acid) (PEMA) and poly(vinyl alcohol) (PVA) as surfactants, Casey et al. (93), showed that polymer-based biomaterials, lacking any small molecule or biologic for therapeutic effect, have the inherent capability of suppressing cytokine secretion from bone marrow-derived macrophages and dendritic cells following LPS or CpG-DNA stimulation (Figure 4). This effect occurred in a surface charge-dependent manner and used polymeric particles in the 350–500 nm diameter range. Furthermore, these materials imparted a survival benefit in a murine LPS-induced endotoxemia model for sepsis. Remarkably, similar results were observed using completely different material composition. In another study, 2 nm gold core nanoparticles with a surface coating of hydroxylated tetraethylene glycol (TEGOH) (98)—again without any delivery of small molecules, peptides, or nucleic acid products—showed a survival benefit in a sepsis model. These nanoparticles were characterized as having an overall net neutral charge and the in vitro suppression of TNF-α production in monocytes appeared regardless of choice of surfactant (the TEGOH described above or the hydrophobic tetraethylene glycol coating, ZDiPen). Interestingly, using a murine LPS-induced endotoxemia model showed that only the TEGOH-coated gold nanoparticles showed the similar suppression of TNF-α production, whereas the hydrophobic ZDiPen failed to recapitulate the cytokine response. This further demonstrates the importance of designing nanoparticles with appropriate physicochemical properties followed by relevant in vitro and in vivo testing to obtain a comprehensive understanding of their effects on desired immunological outcomes.
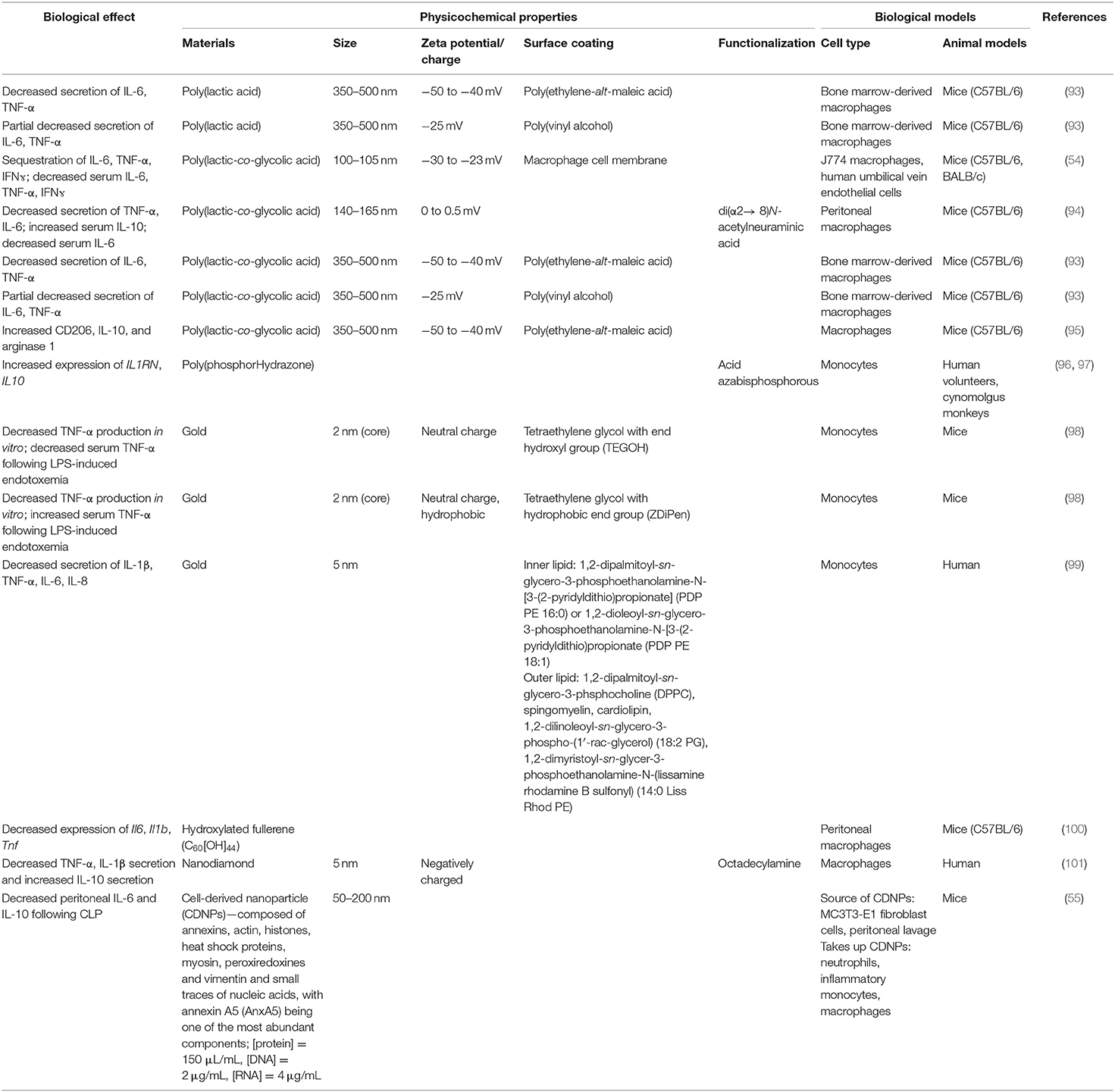
Table 2. Methods to alter cytokine availability.
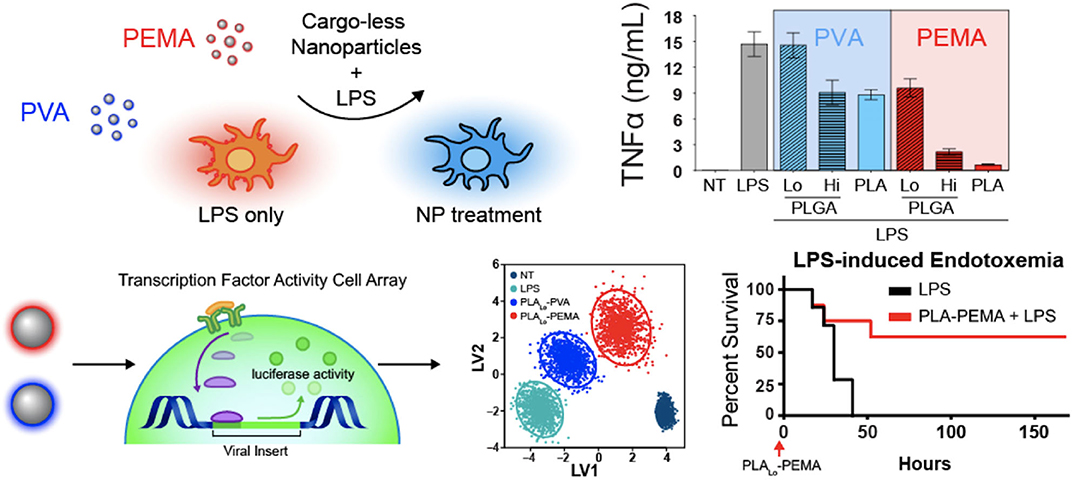
Figure 4. Immunomodulatory effects of nanoparticles. Nanoparticle-dependent inflammatory cytokine suppression of innate immune cells when stimulated with LPS (top). Dynamic transcription factor activity of bone marrow-derived macrophages treated with particles followed by LPS stimulation and improved survival in lethal LPS-induced endotoxemia model. PVA, neutral charge. PEMA, negative charge. Adapted from (93). Copyright (2019) Elsevier.
Recruiting Leukocytes to Inflammatory Sites: Biomaterial-Driven Modification of Cellular Trafficking Patterns
A major role of lipid and peptide mediators for inflammation is to facilitate the recruitment of leukocytes to the sites of inflammation, which is necessary to eliminate the source of the infection and/or tissue damage. However, infiltration of these leukocytes can also further injure the inflamed tissue or nearby healthy tissue through the inherent activity of the neutrophils, inflammatory monocytes, macrophages, and other leukocytes. In order to limit their damage at the inflamed site beyond what is needed to control the infection or initial tissue damage, understanding how these sites are accessed from the vasculature and modification of the accessibility of these tissues can be of benefit in developing therapeutic strategies (Figure 1 and Table 3).
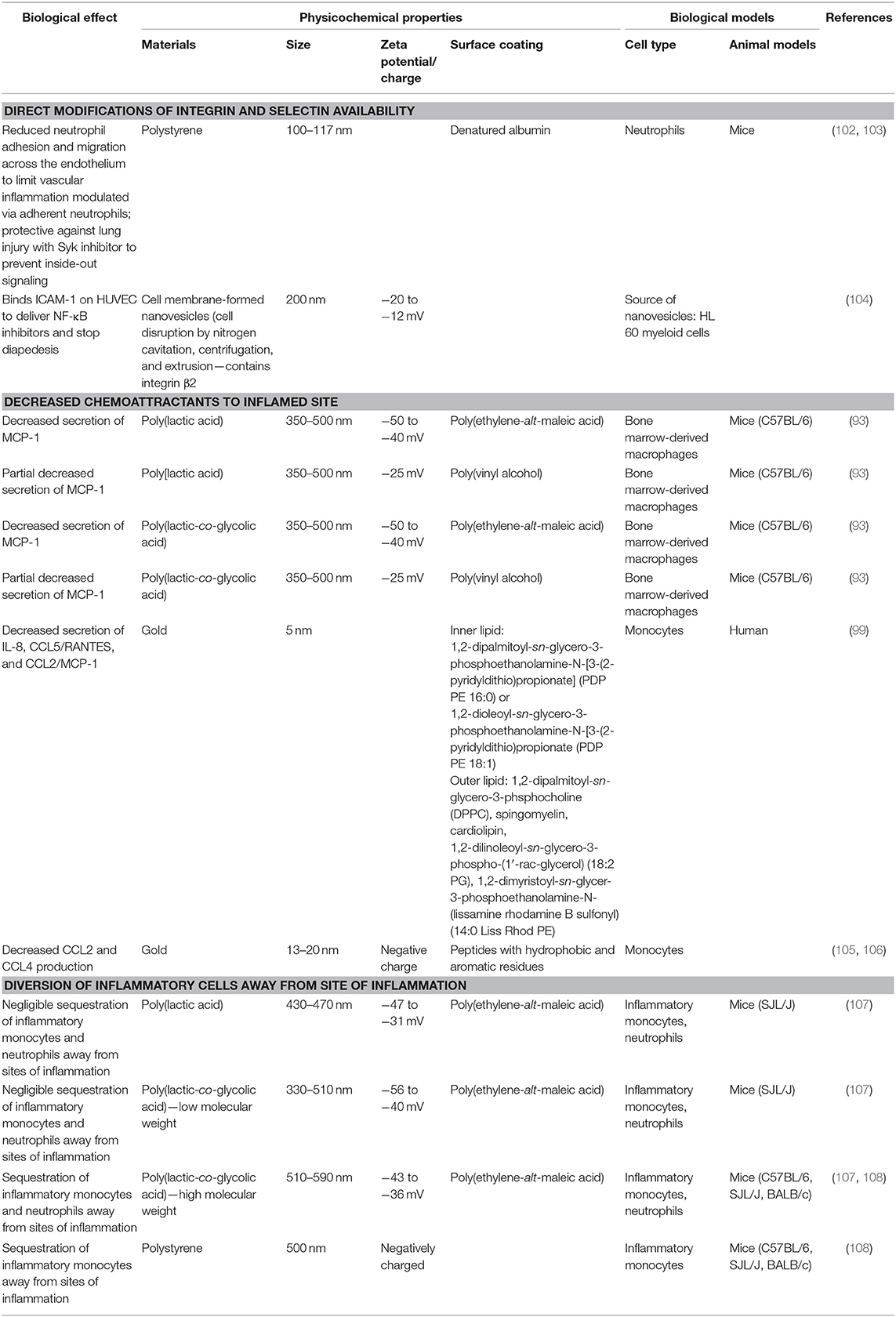
Table 3. Modifiers of cellular trafficking patterns.
Initiation of removal of the injury source requires dilation of the small blood vessels to allow for increased blood flow, increased permeability of the microvasculature, and emigration of the leukocytes from the microcirculation to accumulate in the inflamed tissue (109). Recruitment of leukocytes involves interactions between the vasculature and the immune response. Interjection at any of these steps through altered signaling and inhibition of chemical mediators or redirection and sequestration of leukocytes can alter the course of inflammation to limit the damage it inflicts.
Following sentinel cell recognition of breaches of normal tissue, the vascular mediators described above are mobilized to induce vasodilation while other chemical mediators trigger increased permeability of the postcapillary venules (110). This increased vessel diameter and loss of fluid slows blood flow and allows for concentration of blood cells at the site of tissue injury. As the stasis matures, it is accompanied by increasing amounts of immune mediators at the site to aid in exudation of leukocytes. Additionally, the vascular endothelium expresses increased levels of adhesion molecules that allow for leukocytes, particularly neutrophils, to accumulate along the endothelium and allow for emigration from the vasculature into the injured tissue.
Activation of the vascular endothelium results in selectin upregulation mediated by release of cytokines and chemokines by tissue macrophages, mast cells, and endothelial cells in response to injury. In particular, TNF-α and IL-1β act on endothelial cells of the postcapillary venules near the infection to trigger expression of E-selectin and ligands for L-selectin. Histamine and thrombin also play a role and stimulate P-selectin redistribution from endothelial cell granules (Weibel-Palade bodies) to the cell surface. In contrast, firm adhesion is mediated by integrins whose expression are also under the influence of TNF-α and IL-1β. In the case of endothelium, these cytokines induce expression of vascular cell adhesion molecule 1 (VCAM-1, the ligand for β1 integrin VLA-4) and intercellular adhesion molecule 1 (ICAM-1, the ligand for the β2 integrins LFA-1 and Mac-1). Under normal conditions, the binding of the integrins to VCAM-1 and ICAM-1 are relatively low affinity, but under the influence of cytokines binding to the rolling leukocytes, VLA-4 and LFA-1 are converted to a high affinity state that allows for firm binding of the leukocytes to the endothelial surface (111).
Within the field of biomaterials, numerous groups have attempted to alter these vascular interactions to reduce inflammation [reviewed nicely in (112, 113)] but most have focused on delivery of therapeutics rather than investigation of physicochemical interactions that may modify endothelial behavior. In a few notable examples (102, 103), polystyrene in combination with denatured albumin modulated neutrophil adherence to the vasculature. This interaction aided in delivery of Syk inhibitors to prevent the inside-out signaling that increases leukocyte adhesion to the endothelium. By altering cellular trafficking through making the endothelium less sticky, these studies showed a protective effect against lung injury mediated through alterations in neutrophil activity. Similarly, direct delivery of NF-κB inhibitors to could be achieved through a biomimetic approach. Gao et al. (104) used myeloid cell-derived nanovesicles containing β2 integrins to bind directly to ICAM-1 on HUVECs. This showed a two-fold effect by firstly physically blocking further binding of other leukocytes while also delivering NF-κB inhibitors at the site to stop additional leukocyte diapedesis across the endothelium. Thus, developing a method to use inflammatory cell derivatives to block recruitment of leukocytes is a strategy similar in concept to that of cell membrane-coated nanoparticles described above by Thamphiwatana et al. (54).
Prevention of leukocyte binding is key to stopping cellular infiltration of the inflamed site and multiple mechanisms are simultaneously at play to encourage this process. In addition to the molecular regulators described above, chemokines are also simultaneously stimulating diapedesis through the interendothelial spaces along a concentration gradient toward the site of injury or infection where chemokines are actively being produced. Exogenous chemoattractants include bacterial products such as peptides with N-formylmethionine as the terminal amino acid and some bacteria-specific lipids while endogenous chemoattractants include a variety of chemokines (such as IL-8), proteins of the complement system (particularly C5a), and arachidonic acid (AA) metabolites (namely LTB4). Again, biomaterial approaches have shown an ability to modify these chemokine responses without utilization of drug delivery. In the same Casey article from 2019 (93), in addition to modification of cytokine responses induced my nanoparticle uptake, similarly MCP-1 secretion was shown to be decreased suggesting a global reprogramming of functional responses upon uptake of PLA- and PLGA-based negatively charged particles (Figure 4). In parallel to Moyano et al. (98), modification of chemokines from monocytes can also be achieved with gold-based particles where affected chemokines are dependent on choice of surface coating with lipid-based substrates (99) to decrease chemokine release of IL-8, CCL5/RANTES, and CCL2/MCP-1 vs. decoration of gold particles with peptides containing aromatic and hydrophobic residues to impart a decrease in production of CCL2 and CCL4 (105, 106).
Uniquely, another approach in the literature by Getts et al. (108) bypasses the process of leukocyte migration. Rather than alter the cellular function of inflammatory cells, they showed that PLGA and polystyrene (PS) particles with negatively charged PEMA coating were actively taken up by MARCO+ inflammatory monocytes to induce trafficking of these cells away from sites of tissue injury in multiple disease models (including West Nile virus-induced encephalitis, experimental autoimmune encephalomyelitis (EAE), and cardiac infarction). In each of these disease processes, excessive inflammation is implicated as a major source of disease pathogenesis. Whereby uptake of these particles targeted the offending inflammatory monocytes to be actively removed from the circulation and sequestered in the spleen for degradation. As a result of this redirection, this strategy aided in sparing of the end-organs in these disease models most at risk for damage and failure. Another study demonstrated that the composition of nanoparticles, PLGA (high or low molecular weight) vs. PLA, affected their interactions with neutrophils and monocytes in vitro and in vivo. Using the EAE mouse model, it was demonstrated that high molecular weight PLGA particles significantly improved disease scores compared to controls (107).
The same nanoparticle approach was taken by Park et al. (95) to abrogate paralysis-induced secondary to traumatic spinal cord injury (SCI). Using the same 500 nm diameter PLGA particles that had been shown to trigger sequestration of inflammatory macrophages and neutrophils away from the injury site (108), a non-invasive strategy was devised to alter the functional capacity of the immune cells at the SCI site and drive a predominantly regenerative phenotype at the SCI (Figure 5). Indeed, as seen with the preceding work, the nanoparticle-containing cells were predominantly sequestered and targeted for destruction at the spleen, but, in combination with spinal cord injury a protective population of M2-like macrophages expressing CD206 selectively homed at the site of injury in a way absent for sham injured mice. With this wound repair phenotype predominating at the SCI site, nanoparticle-mediated promotion of axonal regrowth and remyelination was shown, further emphasizing a therapeutic value to the presence of the materials themselves to engineer the dominant immune response at the site of injury.
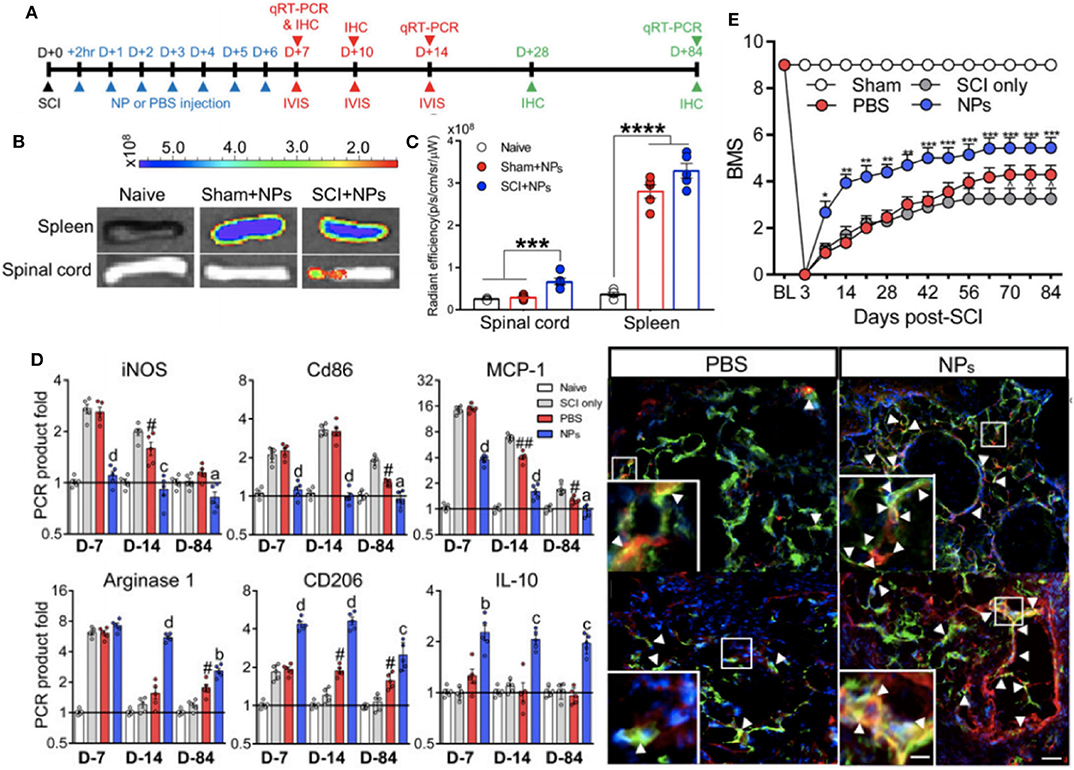
Figure 5. Non-invasive strategy to alter immune cell responses to enhance spinal cord injury (SCI) recovery with in vivo biodistribution and analysis of nanoparticles. Experimental timeline for the study (A). In vivo images from spinal cord and spleen at 1 day post-injection (B). Fluorescence quantification of imaging in (B), where ***P < 0.001 and ****P < 0.0001 (C). Immunomodulation of macrophages as assessed with RT-qPCR data for pro-inflammatory and anti-inflammatory genes at multiple timepoints post-SCI and immunodetection of M2 macrophages (yellow color) within bridge following SCI (aP < 0.05, bP < 0.01, cP < 0.001, and dP < 0.0001 compared to the PBS group, and #P < 0.05 and ##P < 0.01 relative to the SCI only group) (D). Functional recovery of locomotor activity from SCI, where *P < 0.05, **P < 0.01, and ***P < 0.001 compared the the PBS group, and ∧P < 0.05 relative to the SCI only group (E). Adapted from (95). Copyright (2019) National Academy of Sciences.
Removing the Offending Microbes and Damaged Tissue: Developing Ways to Limit Collateral Damage
Once neutrophils and monocytes arrive at the injured tissue, recognition of microbes, or dead cells by TLRs and other PRRs drives leukocyte activation with phagocytosis and intracellular killing resulting in clearing of microbes and dead cells (114). As stated in the previous section, however, many of the mechanisms by which these cells clear microbes and dead cells are non-specific and can cause harm to healthy surrounding tissue. Because of this, strategies that can limit this collateral damage in combination with the methods described above can help to alleviate the most destructive organ damage seen with severe inflammation and sepsis.
Phagocytosis requires recognition and attachment by the leukocyte of the agent to be ingested, engulfment of the agent with a phagocytic vacuole, and killing or degradation of the extracellular products taken up by the cell. Recognition is often performed by mannose receptors, scavenger receptors, and a variety of opsonin receptors that can bind and ingest microbes. Of these the macrophage mannose receptor (MMR or CD206) from the lectin family binds terminal mannose and fucose residues of glycoproteins and glycolipids uniquely found on microbial cell walls to drive their phagocytosis (115). This parallels PRR-specific recognition of microbial PAMPs amid ignorance of molecular characteristics of mammalian cells. In a more generalized manner, scavenger receptors constantly sample the environment and can bind and mediate endocytosis of a variety of microbes in addition to oxidized or acetylated low-density lipoprotein (LDL) that fail to interact with the primary LDL receptor. Macrophage integrins, like Mac-1 introduced above (CD11b/CD18) can also bind microbes for phagocytosis. Coating of microbes by opsonins (particularly IgG antibodies, C3b from the complement system, and mannose-binding lectin) greatly increase the efficiency phagocytosis due to high-affinity receptors for opsonins on the cell surface of neutrophils and monocytes (116).
Alteration in CD206 is essential to the phagocytic capability of macrophages and is easily altered by nanoparticle formulations. As described above, Park et al. (95) showed increased levels of CD206, as well as other markers of M2-like macrophages such as IL-10 and arginase-1 at the site of spinal cord injury. In contrast, using peripheral blood monocytes from human volunteers and cynomolgus monkeys, Fruchon et al. (96, 97) show that another nanoparticle formulation using poly(phosphorHydrazone) functionalized with acid azabisphosphorous increased expression of MRC1 complemented with increased cell surface expression of the protein product, CD206.
Once microbes and necrotic debris have been engulfed, final killing and clearance by neutrophils and macrophages requires highly regulated microbicidal activity within phagocytic compartments driven by generation of reactive oxygen and nitrogen species (ROS and nitric oxide, NO, respectively) and lysosomal enzymes (117, 118). ROS production is dependent upon the rapid assembly and activation of NADPH oxidase on the phagosomal membrane. In neutrophils, evolution of superoxide (O2·) hydrogen peroxide (H2O2) is acted upon by myeloperoxidase (MPO) under the influence of halides like Cl− to convert H2O2 to hypochlorite (). These reactions in combination drive halogenation of microbial components or oxidation of microbial proteins and lipids. In addition to this efficient H2O2-MPO-halide system, H2O2 can also be converted to hydroxyl radicals (−OH·) to also drive modification of cellular lipids, proteins, and nucleic acids, thus destroying microbes. Similarly, NO is produced from arginine by inducible nitric oxide synthase (iNOS) in macrophages and neutrophils following activation by cytokines (e.g., IFNɤ) or microbial products. NO can then react with O2 to form the highly reactive free radical peroxynitrite (ONOO−) to damage the lipids, proteins, and nucleic acids of microbes in a manner similar to ROS. Additional intracellular microbicidal activity is driven by lysosomal enzymes contained in lysosomal granules that contribute to microbial killing and vast amounts of tissue damage.
Although multiple groups have shown an ability to decrease ROS production in vitro (98, 100), Soh et al. (119) introduce an interesting twist in monocytes by using ceria-zirconia nanoparticles to actively scavenge ROS given the faster conversion of ceria-zirconia nanoparticles to convert between the Ce3+ and Ce4+ oxidation states of ceria nanoparticles alone. In LPS-induced endotoxemia rat models and CLP-induced bacteremia mouse models, this increase in ROS and NO scavenging had a net effect of increasing animal survival. At a tissue level, this increased survival is correlated to sparing of the liver and lungs from LPS-associated ROS and NO immune damage with intravenous LPS administration and the gastrointestinal tract of damage associated with similar bactericidal immune mechanisms in widespread polymicrobial bacteremia.
Additional mechanisms at play in microbial killing include neutrophil extracellular traps (NETs) that are composed of extracellular fibrillar networks with a high concentration of antimicrobial substances at the site of infection. These have the ability to trap microbes within the fibrils in response to bacteria and fungi and inflammatory mediators such as cytokines and chemokines, complement proteins, and ROS. NETs are viscous in nature due to neutrophil nuclei loss during NET formation leading to extracellular chromatin binding and concentrating granule proteins and these NETs have been shown in the literature to be particularly destructive during sepsis as they are broken down (120). Recent murine work has shown that antibody-mediated stabilization of NETs prevents release of their captured bacteria and additional toxic NET contents has shown to be protective during sepsis (121), suggesting further opportunities to design biomaterials to aid in minimizing in a controlled fashion the deleterious effects of this necessary microbicidal mechanism.
Resolution of Inflammation
Given the powerful host defense mechanisms at play during inflammation, resolution of the response needs to be tightly controlled to prevent deleterious consequences. Although complete resolution of inflammation is ideal, other consequences of inflammation include connective tissue replacement for healed tissues (scarring or fibrosis) and chronic inflammation.
Among endogenous modulators of inflammation, many are closely related to those driving the inflammatory response. Another AA metabolite class, lipoxins, serve to aid in resolution of inflammation by preventing leukocyte recruitment. LXA4 and LXB4 serve to prevent neutrophil chemotaxis and adhesion during the presence of both neutrophils and platelets at the site of inflammation. Among cytokines, transforming growth factor-beta (TGF-β) and IL-10 are generally regarded as having anti-inflammatory activity. With some nanoparticle strategies, direct induction of IL-10 production (55, 94, 96, 97, 101) has been possible with a variety of biomaterial composition approaches (Table 4). Additionally, the complement system contains a number of regulatory components with even more soluble protein mediators of resolving inflammation include resolvins, protectins, and maresins (129).
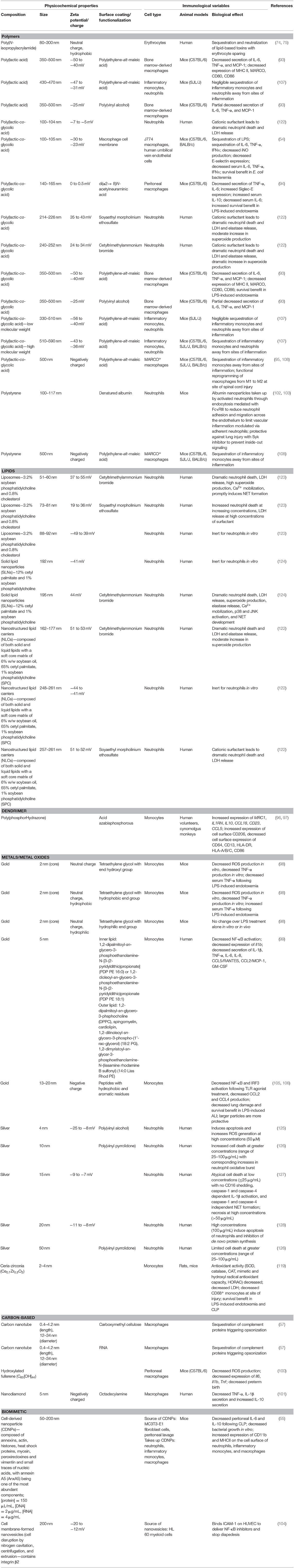
Table 4. Empirical relationships determined between biomaterial physicochemical properties and immune cell activity.
Because of the destructive nature of lysosomal enzymes, antiproteases are also present in the serum and tissue fluids to limit inflammation-associated lysosomal damage. Of these, α1-antitrypsin is a major inhibitor of neutrophil elastase and α2-macroglobulin is another found in serum and various secretions. Additionally, neutrophils themselves have very short lives and turnover of inflammatory cells and the produced mediators of inflammation following removal of the provoked injury are key to resolution. Of note, however, is that in cases of sepsis neutrophil apoptosis is delayed but their function is impaired. Under normal conditions circulating neutrophil have a short half-life (7–12 h in vivo) but this is increased downstream of LPS- and C5a-mediated neutrophil activation. This is attributed to a combination of pro-survival cell signaling, including decreased activation of caspase-8 (130) leading to an accumulation of nuclear factor myeloid nuclear differentiation antigen (MNDA) in parallel with accumulation of Mcl-1 (131), increased anti-apoptotic Bcl-xL (132), decreased pro-apoptotic Bim (133), and increased phosphorylation of Bad downstream of Akt activation (134–136). The net result of these combined molecular mediators is decreased neutrophil apoptosis. This long-lived neutrophil population in sepsis is also characteristic for its impaired transmigration to the site of inflammation. Rather than limiting the damaging effects of neutrophils solely to the site of tissue microbes and injury, neutrophils in sepsis are marked by aberrant neutrophil localization into remote organs where they can inflict damage and further augment the damage of inflammation (137–139).
Given the dramatic destruction inflicted by dysregulated trafficking of long-lived neutrophils during sepsis, it is of benefit to generate therapeutic strategies that can eliminate neutrophils while minimizing the collateral damage inflicted by these cells (behaving as they are expected to do) in aberrant tissue sites. As such, strategies in the literature that were originally intended as studies of the toxicity of nanoparticles provide hints of ways to normalize neutrophil behavior and limit organ dysfunction. With this in mind, Table 4 reiterates the studies discussed above and summarized in Tables 1–3 with an emphasis on cataloging features in biomaterial design. This allows for emphasizing the relationship between the physicochemical characteristics of the chosen materials and the resultant biological effects from the perspective of immune responses at the cellular and, when available, animal model level. As an example of how fine-tuning of physicochemical properties can be harnessed for desired biological effects, the Girard Lab provides an elegant series of studies that stresses this point. This group has shown (127, 128) with human neutrophils that silver nanoparticles in the range of 15–20 nm induced apoptosis and atypical cell death of neutrophils with the ability to inhibit de novo protein synthesis. In related studies, silver nanoparticles were further coated with either PVA (125) or poly(vinyl pyrrolidone) (PVP) (126) to show a size-dependence to cell death induction. Indeed, smaller nanoparticles (4–10 nm) showed the most dramatic cell death in a manner dependent on neutrophil oxidative burst, while even small variations in nanoparticle size (50 nm) abrogated the neutrophil cell death. As this series of studies tell us, each element of material design, from the core material to size to even the choice of surfactant, can impart a dramatic change in the functional responses of innate immune cells further highlighting the importance of cataloging physicochemical characteristics to enable rational design strategies for immunomodulation.
Polypharmacological Strategies for Severe Inflammation and Sepsis
The dysregulation that develops due to sepsis affects cellular phenotypes and gene expression profiles in both transient and long-term manners. In humans, LPS administration resulted in 3,714 genes being differentially expressed in blood leukocytes as early as 2 h post exposure with a near complete resolution of clinical perturbations within 24 h post challenge (140). Similar genomic studies in mice corroborate the vast genetic alterations and have identified over 1,900 differentially expressed genes following LPS challenge (141). Sepsis survivors generally suffer from additional morbidities including higher risk of readmission, cardiovascular disease, cognitive impairment, and death for years following sepsis. Epigenetic mechanisms such as DNA methylation, histone modifications, and non-coding RNAs are also perturbed in sepsis and are associated with increased mortality due to their contributions to long-term immunosuppression (142). Given that thousands of genes are differentially expressed during sepsis, the number of tractable therapeutic options that aim to augment or abrogate single molecular targets is out of the scope of practical and experimental possibilities.
Multiple target-based approaches should be considered to improve patient outcomes in sepsis. A single timepoint nor single cytokine/receptor intervention is unlikely to be successful on a broad range of patients with diverse conditions that have led to the state of sepsis (143). The complexity of disease states offers a range of potential molecular targets, as well as numerous other factors including the time of treatment administration and the combination of drugs. Providing further evidence for multi-target approaches, Cockrell and An developed computational algorithms and predicted the necessity for a multi-target therapy for the treatment of sepsis (144). The specificity to which small molecules and biologics modulate immune responses at a single-target level or through non-specific mechanisms limits their utility in treating the underlying dysfunction encoded in immune cells during and following sepsis. Due to the lack of conceivable small molecules or biologics, nanoparticles are uniquely positioned to achieve this goal due to their highly controllable physicochemical properties, targetability, and immune-modulating properties (7).
A polypharmacological strategy has the potential to address the redundant molecular, cellular, and tissue functions during inflammation but anti-inflammatory and anti-coagulants are neither innocuous nor without potential adverse effects in combination or alone. Of particular note is that morbidity and mortality associated with sepsis and septic shock tends to be most severe within the geriatric and pediatric population, two groups where polypharmacy can be especially deleterious in combination with existing comorbidities or developmental concerns (145–149). Given these concerns with a multi-drug approach, other strategies that can work with multimodal mechanisms of action and minimize adverse effects are ideal and ongoing research with biomaterials serves as an exciting area to deliver on some of these strategies. As such, biomaterials and what is known about the cellular and tissue effects of their physicochemical properties will serve as the focus of the remainder of this review.
There are advantages and disadvantages to each strategy in the management or cure of disease. However, particularly noteworthy in polypharmacology is the reduction in treatment complexity, reduced side effects, and reduced or altogether eliminated drug–drug interactions, in addition to improved patient compliance. Also, given that a single agent can simultaneously affect multiple targets in the same tissue (by default, both pharmacophores must co-localize), partial modulation of targets that are synergistically linked suggests that reduced doses may be sufficient to elicit full therapeutic efficacy, widening the therapeutic windows.
Conclusion and Future Prospects for Biomaterial-Driven Immune Modulation
Developing strategies to control severe inflammation and sepsis remains a healthcare priority. Given the toll sepsis and septic shock plays in increasing healthcare costs and the continuing staggering rates of mortality and long-term morbidity for those affected, it is essential that strategies to improve patient outcomes are informed by the pathophysiology of dysregulated inflammation. As laid out in the review, although sepsis can be triggered by one of numerous types of bacteria breeching initial defenses at a variety of tissue sites, the course of inflammation itself, although complex, has stereotypic physiological processes that provide opportunities for intervention (Figure 1). From the survey of studies included in this review, diverse strategies have been implemented that attempt to address each stage: (1) limiting initial activation of innate immune cells (Figure 2 and Table 1), (2) regulating pro-inflammatory mediators (Figures 3, 4 and Table 2), (3) inhibiting further leukocyte recruitment (Figures 1, 4, 5 and Table 3), (4) removing the initiating microbe and signals for inflammation, and (5) regulating mediators of resolution (Figures 3, 4 and Table 2). Among these works, the strategies with the most promise are those that attempt to affect multiple stages of this process. Indeed, the complex and parallel physiologic responses that have been thus far accounted for during sepsis show that effective management of sepsis requires a multi-targeted approach.
As we have put forth, biomaterials and the generation of nanotechnology-based approaches has the potential to allow for finely tuned engineering of immune responses based on experimentally determined rational design principles. Through elucidation of the principles at play in development of these biomaterials and nanoparticle platforms, the potential exists to generate multi-targeted therapeutics that meet our specific needs based on physicochemical properties deemed significant (e.g., composition, size, charge, and others) as summarized in Table 5. With the maturation of nanotechnology-based immune engineering, several outstanding questions remain to be addressed by all stakeholders in the field including development of biologically relevant animal models, standardization of GMP manufacturing procedures, standardization of formulations with potential implications for pharmacokinetics and pharmacodynamics, and further guidance from regulatory agencies in regard to the nanocarriers themselves. It is our hope that in the upcoming years, these design principles are further developed and adopted in the field as these questions for scalability of nanotechnology are addressed. Future biomaterial designs will be informed by the immunology it intends to assist and, vice versa, the immunology continues to provide new avenues of exploration for the application of biomaterials to improve human health. This interface promises to expand the development of nano-based therapeutics as well as to further the basic understanding of nano-bio interactions and their implications for therapeutic strategies.
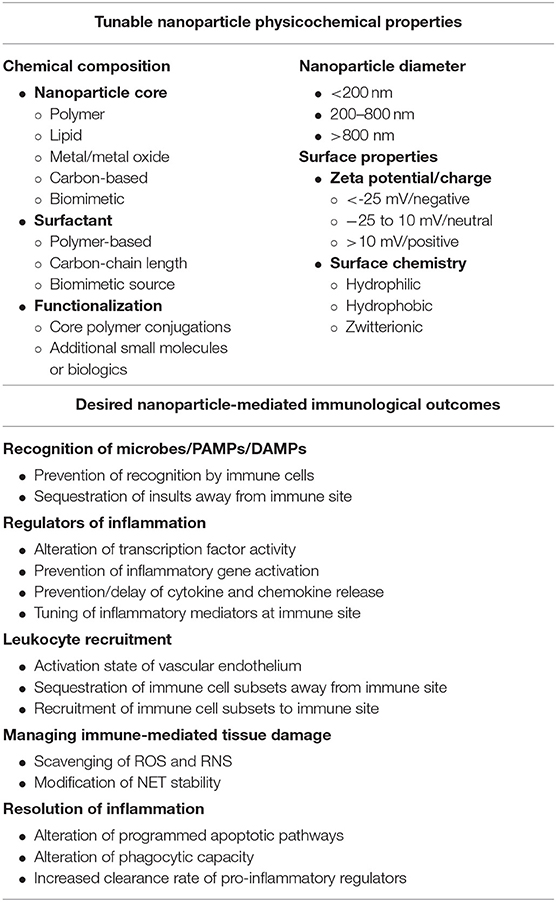
Table 5. Nanoparticle physicochemical properties and desired immune responses to consider when designing biomaterials to fine tune inflammatory responses.
Author Contributions
JL and RP conceptualized the manuscript, wrote the text, drew and adapted figures and tables, and approved the manuscript. HK and MM assisted with gathering, reviewing references, and approved the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by startup funds by the University of Maryland School of Pharmacy, the University of Maryland Baltimore Institute for Clinical and Translational Research (ICTR) Accelerated Translational Incubator Pilot Grant (NIH #1UL1TR003098), and a New Investigator Award from the American Association of Colleges of Pharmacy (AACP) to RP. Additional support was provided by the NIAID-NIH Signaling Pathways in Innate Immunity Training Program (NIH #T32AI095190) and NHLBI-NIH Interdisciplinary Training Program in Cardiovascular Disease (NIH #T32HL007698) to JL.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Heidland A, Klassen A, Rutkowski P, Bahner U. The contribution of rudolf virchow to the concept of inflammation: what is still of importance? J Nephrol. (2006) 19(Suppl.10):S102–9.
2. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. (1999) 340:448–54. doi: 10.1056/NEJM199902113400607
3. Venet F, Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol. (2018) 14:121–37. doi: 10.1038/nrneph.2017.165
4. Lelubre C, Vincent JL. Mechanisms and treatment of organ failure in sepsis. Nat Rev Nephrol. (2018) 14:417–27. doi: 10.1038/s41581-018-0005-7
5. Mitchell MJ, Jain RK, Langer R. Engineering and physical sciences in oncology: challenges and opportunities. Nat Rev Cancer. (2017) 17:659–75. doi: 10.1038/nrc.2017.83
6. Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov. (2019) 18:175–96. doi: 10.1038/s41573-018-0006-z
7. Pearson RM, Casey LM, Hughes KR, Miller SD, Shea LD. In vivo reprogramming of immune cells: Technologies for induction of antigen-specific tolerance. Adv Drug Deliv Rev. (2017) 114:240–55. doi: 10.1016/j.addr.2017.04.005
8. Pearson RM, Podojil JR, Shea LD, King NJC, Miller SD, Getts DR. Overcoming challenges in treating autoimmuntity: development of tolerogenic immune-modifying nanoparticles. Nanomedicine. (2019) 18:282–91. doi: 10.1016/j.nano.2018.10.001
9. Pearson RM, Hsu H-j, Bugno J, Hong S. Understanding nano-bio interactions to improve nanocarriers for drug delivery. MRS Bulletin. (2014) 39:227. doi: 10.1557/mrs.2014.9
10. Keshavan S, Calligari P, Stella L, Fusco L, Delogu LG, Fadeel B. Nano-bio interactions: a neutrophil-centric view. Cell Death Dis. (2019) 10:569. doi: 10.1038/s41419-019-1806-8
11. Kumar V, Abbas AK, Aster JC. Robbins and Cotran pathologic basis of disease. 9th ed. Philadelphia, PA: Elsevier/Saunders. (2015). p. 1391.
12. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the global burden of disease study. Lancet. (2020) 395:200–11. doi: 10.1016/S0140-6736(19)32989-7
13. Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, et al. Assessment of global incidence and mortality of hospital-treated sepsis. Current Estimates and Limitations. Am J Respir Crit Care Med. (2016) 193:259–72. doi: 10.1164/rccm.201504-0781OC
14. Jensen IJ, Sjaastad FV, Griffith TS, Badovinac VP. Sepsis-induced T cell immunoparalysis: the ins and outs of impaired T cell immunity. J Immunol. (2018) 200:1543–53. doi: 10.4049/jimmunol.1701618
15. Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers. (2016) 2:16045. doi: 10.1038/nrdp.2016.45
16. Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest. (2016) 126:23–31. doi: 10.1172/JCI82224
17. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. (2013) 13:260–8. doi: 10.1016/S1473-3099(13)70001-X
18. Cross D, Drury R, Hill J, Pollard AJ. Epigenetics in sepsis: understanding its role in endothelial dysfunction, immunosuppression, and potential therapeutics. Front Immunol. (2019) 10:1363. doi: 10.3389/fimmu.2019.01363
19. Sonego F, Castanheira FV, Ferreira RG, Kanashiro A, Leite CA, Nascimento DC, et al. Paradoxical roles of the neutrophil in sepsis: protective and deleterious. Front Immunol. (2016) 7:155. doi: 10.3389/fimmu.2016.00155
20. Paoli CJ, Reynolds MA, Sinha M, Gitlin M, Crouser E. Epidemiology and costs of sepsis in the united states-an analysis based on timing of diagnosis and severity level. Crit Care Med. (2018) 46:1889–97. doi: 10.1097/CCM.0000000000003342
21. Vincent JL, Marshall JC, Namendys-Silva SA, Francois B, Martin-Loeches I, Lipman J, et al. Assessment of the worldwide burden of critical illness: the intensive care over nations (ICON) audit. Lancet Respir Med. (2014) 2:380–6. doi: 10.1016/S2213-2600(14)70061-X
22. Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. (2010) 304:1787–94. doi: 10.1001/jama.2010.1553
23. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ. (2016) 353:i1585. doi: 10.1136/bmj.i1585
24. Fink MP, Warren HS. Strategies to improve drug development for sepsis. Nat Rev Drug Discov. (2014) 13:741–58. doi: 10.1038/nrd4368
25. Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med. (2014) 20:195–203. doi: 10.1016/j.molmed.2014.01.007
27. Matthay MA. Severe sepsis–a new treatment with both anticoagulant and antiinflammatory properties. N Engl J Med. (2001) 344:759–62. doi: 10.1056/NEJM200103083441009
28. Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. (2001) 344:699–709. doi: 10.1056/NEJM200103083441001
29. Eichacker PQ, Natanson C, Danner RL. Surviving sepsis–practice guidelines, marketing campaigns, and eli lilly. N Engl J Med. (2006) 355:1640–2. doi: 10.1056/NEJMp068197
30. Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. (2012) 366:2055–64. doi: 10.1056/NEJMoa1202290
31. Marti-Carvajal AJ, Sola I, Gluud C, Lathyris D, Cardona AF. Human recombinant protein C for severe sepsis and septic shock in adult and paediatric patients. Cochrane Database Syst Rev. (2012) 12:CD004388. doi: 10.1002/14651858.CD004388.pub5
32. Lai PS, Thompson BT. Why activated protein C was not successful in severe sepsis and septic shock: are we still tilting at windmills? Curr Infect Dis Rep. (2013) 15:407–12. doi: 10.1007/s11908-013-0358-9
33. NAGMSC Working Group on Sepsis. NAGMSC Working Group on Sepsis Final Report. Bethesda, MD: National Institute of General Medical Sciences; National Institutes of Health. (2019).
34. Delano MJ, Ward PA. The immune system's role in sepsis progression, resolution, and long-term outcome. Immunol Rev. (2016) 274:330–53. doi: 10.1111/imr.12499
35. Fontana F, Figueiredo P, Bauleth-Ramos T, Correia A, Santos HA. Immunostimulation and immunosuppression: nanotechnology on the brink. Small Methods. (2018) 2:1700347. doi: 10.1002/smtd.201700347
36. Gammon JM, Jewell CM. Engineering immune tolerance with biomaterials. Adv Healthc Mater. (2019) 8:e1801419. doi: 10.1002/adhm.201801419
37. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
38. Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev. (2009) 227:221–33. doi: 10.1111/j.1600-065X.2008.00731.x
39. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. (2015) 33:257–90. doi: 10.1146/annurev-immunol-032414-112240
40. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. (2012) 249:158–75. doi: 10.1111/j.1600-065X.2012.01146.x
41. Yang H, Tracey KJ. Targeting HMGB1 in inflammation. Biochim Biophys Acta. (2010) 1799:149–56. doi: 10.1016/j.bbagrm.2009.11.019
42. Nesargikar PN, Spiller B, Chavez R. The complement system: history, pathways, cascade and inhibitors. Eur J Microbiol Immunol. (2012) 2:103–11. doi: 10.1556/EuJMI.2.2012.2.2
43. van de Wetering JK, van Golde LM, Batenburg JJ. Collectins: players of the innate immune system. Eur J Biochem. (2004) 271:1229–49. doi: 10.1111/j.1432-1033.2004.04040.x
44. Gupta G, Surolia A. Collectins: sentinels of innate immunity. Bioessays. (2007) 29:452–64. doi: 10.1002/bies.20573
45. Nayak A, Dodagatta-Marri E, Tsolaki AG, Kishore U. An insight into the diverse roles of surfactant proteins, SP-A and SP-D in innate and adaptive immunity. Front Immunol. (2012) 3:131. doi: 10.3389/fimmu.2012.00131
46. Ziegler EJ, McCutchan JA, Fierer J, Glauser MP, Sadoff JC, Douglas H, et al. Treatment of gram-negative bacteremia and shock with human antiserum to a mutant escherichia coli. N Engl J Med. (1982) 307:1225–30. doi: 10.1056/NEJM198211113072001
47. McCloskey RV, Straube RC, Sanders C, Smith SM, Smith CR. Treatment of septic shock with human monoclonal antibody HA-1A. A randomized, double-blind, placebo-controlled trial. CHESS trial study group. Ann Intern Med. (1994) 121:1–5. doi: 10.7326/0003-4819-121-1-199407010-00001
48. Greenman RL, Schein RM, Martin MA, Wenzel RP, MacIntyre NR, Emmanuel G, et al. A controlled clinical trial of E5 murine monoclonal IgM antibody to endotoxin in the treatment of gram-negative sepsis. The XOMA sepsis study group. JAMA. (1991) 266:1097–102. doi: 10.1001/jama.1991.03470080067031
49. Bone RC, Balk RA, Fein AM, Perl TM, Wenzel RP, Reines HD, et al. A second large controlled clinical study of E5, a monoclonal antibody to endotoxin: results of a prospective, multicenter, randomized, controlled trial. The E5 sepsis study group. Crit Care Med. (1995) 23:994–1006. doi: 10.1097/00003246-199506000-00003
50. Angus DC, Birmingham MC, Balk RA, Scannon PJ, Collins D, Kruse JA, et al. E5 murine monoclonal antiendotoxin antibody in gram-negative sepsis: a randomized controlled trial. E5 study investigators. JAMA. (2000) 283:1723–30. doi: 10.1001/jama.283.13.1723
51. Tidswell M, Tillis W, Larosa SP, Lynn M, Wittek AE, Kao R, et al. Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit Care Med. (2010) 38:72–83. doi: 10.1097/CCM.0b013e3181b07b78
52. Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. (2013) 309:1154–62. doi: 10.1001/jama.2013.2194
53. Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med. (2010) 38:1685–94. doi: 10.1097/CCM.0b013e3181e7c5c9
54. Thamphiwatana S, Angsantikul P, Escajadillo T, Zhang Q, Olson J, Luk BT, et al. Macrophage-like nanoparticles concurrently absorbing endotoxins and proinflammatory cytokines for sepsis management. Proc Natl Acad Sci USA. (2017) 114:11488–93. doi: 10.1073/pnas.1714267114
55. Kunz N, Xia BT, Kalies KU, Klinger M, Gemoll T, Habermann JK, et al. Cell-derived nanoparticles are endogenous modulators of sepsis with therapeutic potential. Shock. (2017) 48:346–54. doi: 10.1097/SHK.0000000000000855
56. Pannuzzo M, Esposito S, Wu LP, Key J, Aryal S, Celia C, et al. Overcoming nanoparticle-mediated complement activation by surface PEG pairing. Nano Lett. (2020) 20:4312–21. doi: 10.1021/acs.nanolett.0c01011
57. Pondman KM, Sobik M, Nayak A, Tsolaki AG, Jakel A, Flahaut E, et al. Complement activation by carbon nanotubes and its influence on the phagocytosis and cytokine response by macrophages. Nanomedicine. (2014) 10:1287–99. doi: 10.1016/j.nano.2014.02.010
58. Meunier E, Coste A, Olagnier D, Authier H, Lefevre L, Dardenne C, et al. Double-walled carbon nanotubes trigger IL-1beta release in human monocytes through Nlrp3 inflammasome activation. Nanomedicine. (2012) 8:987–95. doi: 10.1016/j.nano.2011.11.004
59. Pondman KM, Salvador-Morales C, Paudyal B, Sim RB, Kishore U. Interactions of the innate immune system with carbon nanotubes. Nanoscale Horiz. (2017) 2:174–86. doi: 10.1039/C6NH00227G
60. Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. (2012) 11:633–52. doi: 10.1038/nrd3800
61. Kaufmann SHE, Dorhoi A, Hotchkiss RS, Bartenschlager R. Host-directed therapies for bacterial and viral infections. Nat Rev Drug Discov. (2018) 17:35–56. doi: 10.1038/nrd.2017.162
62. Du Buske LM. Clinical comparison of histamine H1-receptor antagonist drugs. J Allergy Clin Immunol. (1996) 98:S307–18. doi: 10.1016/S0091-6749(96)80116-3
63. Khanapure SP, Garvey DS, Janero DR, Letts LG. Eicosanoids in inflammation: biosynthesis, pharmacology, and therapeutic frontiers. Curr Top Med Chem. (2007) 7:311–40. doi: 10.2174/156802607779941314
64. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. (2015) 15:511–23. doi: 10.1038/nri3859
65. Annane D, Sebille V, Charpentier C, Bollaert PE, Francois B, Korach JM, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. (2002) 288:862–71. doi: 10.1001/jama.288.7.862
66. Bollaert PE, Charpentier C, Levy B, Debouverie M, Audibert G, Larcan A. Reversal of late septic shock with supraphysiologic doses of hydrocortisone. Crit Care Med. (1998) 26:645–50. doi: 10.1097/00003246-199804000-00010
67. Oppert M, Schindler R, Husung C, Offermann K, Graf KJ, Boenisch O, et al. Low-dose hydrocortisone improves shock reversal and reduces cytokine levels in early hyperdynamic septic shock. Crit Care Med. (2005) 33:2457–64. doi: 10.1097/01.CCM.0000186370.78639.23
68. Sprung CL, Annane D, Keh D, Moreno R, Singer M, Freivogel K, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. (2008) 358:111–24. doi: 10.1056/NEJMoa071366
69. Arabi YM, Aljumah A, Dabbagh O, Tamim HM, Rishu AH, Al-Abdulkareem A, et al. Low-dose hydrocortisone in patients with cirrhosis and septic shock: a randomized controlled trial. CMAJ. (2010) 182:1971–7. doi: 10.1503/cmaj.090707
70. Briegel J, Forst H, Haller M, Schelling G, Kilger E, Kuprat G, et al. Stress doses of hydrocortisone reverse hyperdynamic septic shock: a prospective, randomized, double-blind, single-center study. Crit Care Med. (1999) 27:723–32. doi: 10.1097/00003246-199904000-00025
71. Sprung CL, Caralis PV, Marcial EH, Pierce M, Gelbard MA, Long WM, et al. The effects of high-dose corticosteroids in patients with septic shock. A prospective, controlled study. N Engl J Med. (1984) 311:1137–43. doi: 10.1056/NEJM198411013111801
72. Bone RC, Fisher CJ Jr, Clemmer TP, Slotman GJ, Metz CA, Balk RA. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med. (1987) 317:653–8. doi: 10.1056/NEJM198709103171101
73. Bernard GR, Wheeler AP, Russell JA, Schein R, Summer WR, Steinberg KP, et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. The ibuprofen in sepsis study group. N Engl J Med. (1997) 336:912–8. doi: 10.1056/NEJM199703273361303
74. O'Brien J, Lee SH, Onogi S, Shea KJ. Engineering the protein corona of a synthetic polymer nanoparticle for broad-spectrum sequestration and neutralization of venomous biomacromolecules. J Am Chem Soc. (2016) 138:16604–7. doi: 10.1021/jacs.6b10950
75. O'Brien J, Shea KJ. Tuning the protein corona of hydrogel nanoparticles: the synthesis of abiotic protein and peptide affinity reagents. Acc Chem Res. (2016) 49:1200–10. doi: 10.1021/acs.accounts.6b00125
77. Dinarello CA. Anti-cytokine therapies in response to systemic infection. J Investig Dermatol Symp Proc. (2001) 6:244–50. doi: 10.1046/j.0022-202x.2001.00046.x
78. Dinarello CA. The proinflammatory cytokines interleukin-1 and tumor necrosis factor and treatment of the septic shock syndrome. J Infect Dis. (1991) 163:1177–84. doi: 10.1093/infdis/163.6.1177
79. Harada A, Sekido N, Akahoshi T, Wada T, Mukaida N, Matsushima K. Essential involvement of interleukin-8 (IL-8) in acute inflammation. J Leukoc Biol. (1994) 56:559–64. doi: 10.1002/jlb.56.5.559
80. Leonard EJ, Skeel A, Yoshimura T. Biological aspects of monocyte chemoattractant protein-1 (MCP-1). Adv Exp Med Biol. (1991) 305:57–64. doi: 10.1007/978-1-4684-6009-4_7
81. Wolpe SD, Davatelis G, Sherry B, Beutler B, Hesse DG, Nguyen HT, et al. Macrophages secrete a novel heparin-binding protein with inflammatory and neutrophil chemokinetic properties. J Exp Med. (1988) 167:570–81. doi: 10.1084/jem.167.2.570
82. Lam HS, Ng PC. Biochemical markers of neonatal sepsis. Pathology. (2008) 40:141–8. doi: 10.1080/00313020701813735
83. Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. (1985) 229:869–71. doi: 10.1126/science.3895437
84. Vilcek J. First demonstration of the role of TNF in the pathogenesis of disease. J Immunol. (2008) 181:5–6. doi: 10.4049/jimmunol.181.1.5
85. Scallon B, Cai A, Solowski N, Rosenberg A, Song XY, Shealy D, et al. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. (2002) 301:418–26. doi: 10.1124/jpet.301.2.418
86. Scheinfeld N. A comprehensive review and evaluation of the side effects of the tumor necrosis factor alpha blockers etanercept, infliximab and adalimumab. J Dermatolog Treat. (2004) 15:280–94. doi: 10.1080/09546630410017275
87. Abraham E, Glauser MP, Butler T, Garbino J, Gelmont D, Laterre PF, et al. p55 tumor necrosis factor receptor fusion protein in the treatment of patients with severe sepsis and septic shock. A randomized controlled multicenter trial. Ro 45-2081 study group. JAMA. (1997) 277:1531–8. doi: 10.1001/jama.1997.03540430043031
88. Abraham E, Laterre PF, Garbino J, Pingleton S, Butler T, Dugernier T, et al. Lenercept (p55 tumor necrosis factor receptor fusion protein) in severe sepsis and early septic shock: a randomized, double-blind, placebo-controlled, multicenter phase III trial with 1,342 patients. Crit Care Med. (2001) 29:503–10. doi: 10.1097/00003246-200103000-00006
89. Fisher CJ Jr, Agosti JM, Opal SM, Lowry SF, Balk RA, Sadoff JC, et al. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The soluble TNF receptor sepsis study group. N Engl J Med. (1996) 334:1697–702. doi: 10.1056/NEJM199606273342603
90. Fisher CJ Jr, Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra sepsis syndrome study group. JAMA. (1994) 271:1836–43. doi: 10.1001/jama.1994.03510470040032
91. Fisher CJ Jr, Marra MN, Palardy JE, Marchbanks CR, Scott RW, Opal SM. Human neutrophil bactericidal/permeability-increasing protein reduces mortality rate from endotoxin challenge: a placebo-controlled study. Crit Care Med. (1994) 22:553–8. doi: 10.1097/00003246-199404000-00008
92. Opal SM, Fisher CJ Jr, Dhainaut JF, Vincent JL, Brase R, Lowry SF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The interleukin-1 receptor antagonist sepsis investigator group. Crit Care Med. (1997) 25:1115–24. doi: 10.1097/00003246-199707000-00010
93. Casey LM, Kakade S, Decker JT, Rose JA, Deans K, Shea LD, et al. Cargo-less nanoparticles program innate immune cell responses to toll-like receptor activation. Biomaterials. (2019) 218:119333. doi: 10.1016/j.biomaterials.2019.119333
94. Spence S, Greene MK, Fay F, Hams E, Saunders SP, Hamid U, et al. Targeting Siglecs with a sialic acid-decorated nanoparticle abrogates inflammation. Sci Transl Med. (2015) 7:303ra140. doi: 10.1126/scitranslmed.aab3459
95. Park J, Zhang Y, Saito E, Gurczynski SJ, Moore BB, Cummings BJ, et al. Intravascular innate immune cells reprogrammed via intravenous nanoparticles to promote functional recovery after spinal cord injury. Proc Natl Acad Sci USA. (2019) 116:14947–54. doi: 10.1073/pnas.1820276116
96. Fruchon S, Mouriot S, Thiollier T, Grandin C, Caminade AM, Turrin CO, et al. Repeated intravenous injections in non-human primates demonstrate preclinical safety of an anti-inflammatory phosphorus-based dendrimer. Nanotoxicology. (2015) 9:433–41. doi: 10.3109/17435390.2014.940406
97. Fruchon S, Poupot M, Martinet L, Turrin CO, Majoral JP, Fournie JJ, et al. Anti-inflammatory and immunosuppressive activation of human monocytes by a bioactive dendrimer. J Leukoc Biol. (2009) 85:553–62. doi: 10.1189/jlb.0608371
98. Moyano DF, Liu Y, Ayaz F, Hou S, Puangploy P, Duncan B, et al. Immunomodulatory effects of coated gold nanoparticles in LPS-stimulated in vitro and in vivo murine model systems. Chem. (2016) 1:320–7. doi: 10.1016/j.chempr.2016.07.007
99. Foit L, Thaxton CS. Synthetic high-density lipoprotein-like nanoparticles potently inhibit cell signaling and production of inflammatory mediators induced by lipopolysaccharide binding Toll-like receptor 4. Biomaterials. (2016) 100:67–75. doi: 10.1016/j.biomaterials.2016.05.021
100. Wakimoto T, Uchida K, Mimura K, Kanagawa T, Mehandjiev TR, Aoshima H, et al. Hydroxylated fullerene: a potential antiinflammatory and antioxidant agent for preventing mouse preterm birth. Am J Obstet Gynecol. (2015) 213:708 e1–9. doi: 10.1016/j.ajog.2015.07.017
101. Pentecost AE, Witherel CE, Gogotsi Y, Spiller KL. Anti-inflammatory effects of octadecylamine-functionalized nanodiamond on primary human macrophages. Biomater Sci. (2017) 5:2131–43. doi: 10.1039/C7BM00294G
102. Wang Z, Li J, Cho J, Malik AB. Prevention of vascular inflammation by nanoparticle targeting of adherent neutrophils. Nat Nanotechnol. (2014) 9:204–10. doi: 10.1038/nnano.2014.17
103. Chu D, Gao J, Wang Z. Neutrophil-mediated delivery of therapeutic nanoparticles across blood vessel barrier for treatment of inflammation and infection. ACS Nano. (2015) 9:11800–11. doi: 10.1021/acsnano.5b05583
104. Gao J, Chu D, Wang Z. Cell membrane-formed nanovesicles for disease-targeted delivery. J Control Release. (2016) 224:208–16. doi: 10.1016/j.jconrel.2016.01.024
105. Yang H, Fung SY, Xu S, Sutherland DP, Kollmann TR, Liu M, et al. Amino acid-dependent attenuation of toll-like receptor signaling by peptide-gold nanoparticle hybrids. ACS Nano. (2015) 9:6774–84. doi: 10.1021/nn505634h
106. Gao W, Wang Y, Xiong Y, Sun L, Wang L, Wang K, et al. Size-dependent anti-inflammatory activity of a peptide-gold nanoparticle hybrid in vitro and in a mouse model of acute lung injury. Acta Biomater. (2019) 85:203–17. doi: 10.1016/j.actbio.2018.12.046
107. Saito E, Kuo R, Pearson RM, Gohel N, Cheung B, King NJC, et al. Designing drug-free biodegradable nanoparticles to modulate inflammatory monocytes and neutrophils for ameliorating inflammation. J Control Release. (2019) 300:185–96. doi: 10.1016/j.jconrel.2019.02.025
108. Getts DR, Terry RL, Getts MT, Deffrasnes C, Muller M, van Vreden C, et al. Therapeutic inflammatory monocyte modulation using immune-modifying microparticles. Sci Transl Med. (2014) 6:219ra7. doi: 10.1126/scitranslmed.3007563
109. Leick M, Azcutia V, Newton G, Luscinskas FW. Leukocyte recruitment in inflammation: basic concepts and new mechanistic insights based on new models and microscopic imaging technologies. Cell Tissue Res. (2014) 355:647–56. doi: 10.1007/s00441-014-1809-9
110. Xiao L, Liu Y, Wang N. New paradigms in inflammatory signaling in vascular endothelial cells. Am J Physiol Heart Circ Physiol. (2014) 306:H317–25. doi: 10.1152/ajpheart.00182.2013
111. Liao JK. Linking endothelial dysfunction with endothelial cell activation. J Clin Invest. (2013) 123:540–1. doi: 10.1172/JCI66843
112. Kelley WJ, Safari H, Lopez-Cazares G, Eniola-Adefeso O. Vascular-targeted nanocarriers: design considerations and strategies for successful treatment of atherosclerosis and other vascular diseases. Wiley Interdiscip Rev Nanomed Nanobiotechnol. (2016) 8:909–26. doi: 10.1002/wnan.1414
113. Calin M, Manduteanu I. Emerging nanocarriers-based approaches to diagnose and red uce vascular inflammation in atherosclerosis. Curr Med Chem. (2017) 24:550–67. doi: 10.2174/0929867324666161123091627
114. Kieser KJ, Kagan JC. Multi-receptor detection of individual bacterial products by the innate immune system. Nat Rev Immunol. (2017) 17:376–90. doi: 10.1038/nri.2017.25
115. Azad AK, Rajaram MV, Schlesinger LS. Exploitation of the macrophage mannose receptor (CD206) in infectious disease diagnostics and therapeutics. J Cytol Mol Biol. (2014) 1:1000003. doi: 10.13188/2325-4653.1000003
116. Silva MT, Correia-Neves M. Neutrophils and macrophages: the main partners of phagocyte cell systems. Front Immunol. (2012) 3:174. doi: 10.3389/fimmu.2012.00174
117. Rosen H, Crowley JR, Heinecke JW. Human neutrophils use the myeloperoxidase-hydrogen peroxide-chloride system to chlorinate but not nitrate bacterial proteins during phagocytosis. J Biol Chem. (2002) 277:30463–8. doi: 10.1074/jbc.M202331200
118. Nauseef WM. Myeloperoxidase in human neutrophil host defence. Cell Microbiol. (2014) 16:1146–55. doi: 10.1111/cmi.12312
119. Soh M, Kang DW, Jeong HG, Kim D, Kim DY, Yang W, et al. Ceria-zirconia nanoparticles as an enhanced multi-antioxidant for sepsis treatment. Angew Chem Int Ed Engl. (2017) 56:11399–403. doi: 10.1002/anie.201704904
120. Yipp BG, Kubes P. NETosis: how vital is it? Blood. (2013) 122:2784–94. doi: 10.1182/blood-2013-04-457671
121. Gollomp K, Sarkar A, Harikumar S, Seeholzer SH, Arepally GM, Hudock K, et al. Fc-modified HIT-like monoclonal antibody as a novel treatment for sepsis. Blood. (2020) 135:743–54. doi: 10.1182/blood.2019002329
122. Hwang TL, Aljuffali IA, Lin CF, Chang YT, Fang JY. Cationic additives in nanosystems activate cytotoxicity and inflammatory response of human neutrophils: lipid nanoparticles versus polymeric nanoparticles. Int J Nanomedicine. (2015) 10:371–85. doi: 10.2147/IJN.S73017
123. Hwang TL, Hsu CY, Aljuffali IA, Chen CH, Chang YT, Fang JY. Cationic liposomes evoke proinflammatory mediator release and neutrophil extracellular traps (NETs) toward human neutrophils. Colloids Surf B Biointerfaces. (2015) 128:119–26. doi: 10.1016/j.colsurfb.2015.02.022
124. Hwang TL, Aljuffali IA, Hung CF, Chen CH, Fang JY. The impact of cationic solid lipid nanoparticles on human neutrophil activation and formation of neutrophil extracellular traps (NETs). Chem Biol Interact. (2015) 235:106–14. doi: 10.1016/j.cbi.2015.04.011
125. Paino IM, Zucolotto V. Poly(vinyl alcohol)-coated silver nanoparticles: activation of neutrophils and nanotoxicology effects in human hepatocarcinoma and mononuclear cells. Environ Toxicol Pharmacol. (2015) 39:614–21. doi: 10.1016/j.etap.2014.12.012
126. Soares T, Ribeiro D, Proenca C, Chiste RC, Fernandes E, Freitas M. Size-dependent cytotoxicity of silver nanoparticles in human neutrophils assessed by multiple analytical approaches. Life Sci. (2016) 145:247–54. doi: 10.1016/j.lfs.2015.12.046
127. Liz R, Simard JC, Leonardi LB, Girard D. Silver nanoparticles rapidly induce atypical human neutrophil cell death by a process involving inflammatory caspases and reactive oxygen species and induce neutrophil extracellular traps release upon cell adhesion. Int Immunopharmacol. (2015) 28:616–25. doi: 10.1016/j.intimp.2015.06.030
128. Poirier M, Simard JC, Antoine F, Girard D. Interaction between silver nanoparticles of 20 nm (AgNP20) and human neutrophils: induction of apoptosis and inhibition of de novo protein synthesis by AgNP20 aggregates. J Appl Toxicol. (2014) 34:404–12. doi: 10.1002/jat.2956
129. Serhan CN. Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J. (2017) 31:1273–88. doi: 10.1096/fj.201601222R
130. Jia SH, Parodo J, Kapus A, Rotstein OD, Marshall JC. Dynamic regulation of neutrophil survival through tyrosine phosphorylation or dephosphorylation of caspase-8. J Biol Chem. (2008) 283:5402–13. doi: 10.1074/jbc.M706462200
131. Milot E, Fotouhi-Ardakani N, Filep JG. Myeloid nuclear differentiation antigen, neutrophil apoptosis and sepsis. Front Immunol. (2012) 3:397. doi: 10.3389/fimmu.2012.00397
132. Guo RF, Sun L, Gao H, Shi KX, Rittirsch D, Sarma VJ, et al. In vivo regulation of neutrophil apoptosis by C5a during sepsis. J Leukoc Biol. (2006) 80:1575–83. doi: 10.1189/jlb.0106065
133. Guo RF, Riedemann NC, Sun L, Gao H, Shi KX, Reuben JS, et al. Divergent signaling pathways in phagocytic cells during sepsis. J Immunol. (2006) 177:1306–13. doi: 10.4049/jimmunol.177.2.1306
134. Perianayagam MC, Balakrishnan VS, Pereira BJ, Jaber BL. C5a delays apoptosis of human neutrophils via an extracellular signal-regulated kinase and bad-mediated signalling pathway. Eur J Clin Invest. (2004) 34:50–6. doi: 10.1111/j.1365-2362.2004.01273.x
135. Simon HU. Targeting apoptosis in the control of inflammation. Eur Respir J Suppl. (2003) 44:20s–1s. doi: 10.1183/09031936.03.00000603b
136. Simon HU. Neutrophil apoptosis pathways and their modifications in inflammation. Immunol Rev. (2003) 193:101–10. doi: 10.1034/j.1600-065X.2003.00038.x
137. Souto FO, Alves-Filho JC, Turato WM, Auxiliadora-Martins M, Basile-Filho A, Cunha FQ. Essential role of CCR2 in neutrophil tissue infiltration and multiple organ dysfunction in sepsis. Am J Respir Crit Care Med. (2011) 183:234–42. doi: 10.1164/rccm.201003-0416OC
138. Souto FO, Zarpelon AC, Staurengo-Ferrari L, Fattori V, Casagrande R, Fonseca MJ, et al. Quercetin reduces neutrophil recruitment induced by CXCL8, LTB4, and fMLP: inhibition of actin polymerization. J Nat Prod. (2011) 74:113–8. doi: 10.1021/np1003017
139. Speyer CL, Gao H, Rancilio NJ, Neff TA, Huffnagle GB, Sarma JV, et al. Novel chemokine responsiveness and mobilization of neutrophils during sepsis. Am J Pathol. (2004) 165:2187–96. doi: 10.1016/S0002-9440(10)63268-3
140. Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, et al. A network-based analysis of systemic inflammation in humans. Nature. (2005) 437:1032–7. doi: 10.1038/nature03985
141. Genster N, Ostrup O, Schjalm C, Eirik Mollnes T, Cowland JB, Garred P. Ficolins do not alter host immune responses to lipopolysaccharide-induced inflammation in vivo. Sci Rep. (2017) 7:3852. doi: 10.1038/s41598-017-04121-w
142. Carson WF, Cavassani KA, Dou Y, Kunkel SL. Epigenetic regulation of immune cell functions during post-septic immunosuppression. Epigenetics. (2011) 6:273–83. doi: 10.4161/epi.6.3.14017
143. Remick DG. Cytokine therapeutics for the treatment of sepsis: why has nothing worked? Curr Pharm Des. (2003) 9:75–82. doi: 10.2174/1381612033392567
144. Cockrell RC, An G. Examining the controllability of sepsis using genetic algorithms on an agent-based model of systemic inflammation. PLoS Comput Biol. (2018) 14:e1005876. doi: 10.1371/journal.pcbi.1005876
145. Golchin N, Frank SH, Vince A, Isham L, Meropol SB. Polypharmacy in the elderly. J Res Pharm Pract. (2015) 4:85–8. doi: 10.4103/2279-042X.155755
146. Maher RL, Hanlon J, Hajjar ER. Clinical consequences of polypharmacy in elderly. Expert Opin Drug Saf. (2014) 13:57–65. doi: 10.1517/14740338.2013.827660
147. Horace AE, Ahmed F. Polypharmacy in pediatric patients and opportunities for pharmacists' involvement. Integr Pharm Res Pract. (2015) 4:113–26. doi: 10.2147/IPRP.S64535
148. Morden NE, Goodman D. Pediatric polypharmacy: time to lock the medicine cabinet? Arch Pediatr Adolesc Med. (2012) 166:91–2. doi: 10.1001/archpediatrics.2011.162
Keywords: nanoparticles, microparticles, biomaterials, innate immunity, macrophage, neutrophil, sepsis, inflammation
Citation: Lasola JJM, Kamdem H, McDaniel MW and Pearson RM (2020) Biomaterial-Driven Immunomodulation: Cell Biology-Based Strategies to Mitigate Severe Inflammation and Sepsis. Front. Immunol. 11:1726. doi: 10.3389/fimmu.2020.01726
Received: 13 March 2020; Accepted: 29 June 2020;
Published: 04 August 2020.
Edited by:
Evan Alexander Scott, Northwestern University, United StatesReviewed by:
Anthony George Tsolaki, Brunel University London, United KingdomXiuling Lu, University of Connecticut, United States
Copyright © 2020 Lasola, Kamdem, McDaniel and Pearson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryan M. Pearson, cnBlYXJzb25AcngudW1hcnlsYW5kLmVkdQ==