 Isabelle Bekeredjian-Ding
Isabelle Bekeredjian-Ding- 1Division of Microbiology, Langen, Germany
- 2Institute for Medical Microbiology, Immunology and Parasitology, University Hospital Bonn, Bonn, Germany
Increasing antibiotic resistance in bacteria causing endogenous infections has entailed a need for innovative approaches to therapy and prophylaxis of these infections and raised a new interest in vaccines for prevention of colonization and infection by typically antibiotic resistant pathogens. Nevertheless, there has been a long history of failures in late stage clinical development of this type of vaccines, which remains not fully understood. This article provides an overview on present and past vaccine developments targeting nosocomial bacterial pathogens; it further highlights the specific challenges associated with demonstrating clinical efficacy of these vaccines and the facts to be considered in future study designs. Notably, these vaccines are mainly applied to subjects with preexistent immunity to the target pathogen, transient or chronic immunosuppression and ill-defined microbiome status. Unpredictable attack rates and changing epidemiology as well as highly variable genetic and immunological strain characteristics complicate the development. In views of the clinical need, re-thinking of the study designs and expectations seems warranted: first of all, vaccine development needs to be footed on a clear rationale for choosing the immunological mechanism of action and the optimal time point for vaccination, e.g., (1) prevention (or reduction) of colonization vs. prevention of infection and (2) boosting of a preexistent immune response vs. altering the quality of the immune response. Furthermore, there are different, probably redundant, immunological and microbiological defense mechanisms that provide protection from infection. Their interplay is not well-understood but as a consequence their effect might superimpose vaccine-mediated resolution of infection and lead to failure to demonstrate efficacy. This implies that improved characterization of patient subpopulations within the trial population should be obtained by pro- and retrospective analyses of trial data on subject level. Statistical and systems biology approaches could help to define immune and microbiological biomarkers that discern populations that benefit from vaccination from those where vaccines might not be effective.
Introduction
The increasing inefficacy of antibiotics due to antibiotic resistance of bacterial pathogens has triggered the desperate search for alternative therapies. While the discovery of new antibiotics is frequently halted by concerns about toxicity or metabolism or insufficient bioavailability and tissue penetration (1, 2), the development of phage therapies has been limited by concerns about the narrow host spectrum, which requires sophisticated susceptibility testing, and the induction of neutralizing antibodies upon repeated use (3). Obviously, this situation has raised a new interest to explore the potential of vaccines for prevention of colonization and infection by typically antibiotic resistant pathogens that typically acquire antibiotic resistance. However, despite a multitude of early developments and publications there has been a long history of failures in clinical development of this type of vaccines (4–6). This review will set its emphasis on providing insight into the reasons that led to discontinuation of vaccine development programs and the consequences for clinical trial design.
Target Pathogens for Vaccine-Based Approaches Against Nosocomial Bacterial Infections
Classical vaccine development has mainly focused on bacterial infections caused by toxin production (Tetanus, Diphtheria, Pertussis) or bacteria such as meningococci, pneumococci and Mycobacterium tuberculosis that cause severe, sometimes lethal infections and easily spread among the population. In the former category, disease is limited to the presence of toxins, e.g., it occurs upon infestation of Tetanus toxin in wounds or secretion of Diphtheria and Pertussis toxins by Corynebacteria and Bordetella species in the respiratory tract. The presence of toxin-neutralizing antibodies (induced by vaccination) mediates protection and can, thus, be quantified in international units (absolute correlate of protection) (7). It is further important to note that both these upper respiratory tract infections as well as the infections caused by meningococci, pneumococci, and M. tuberculosis are transmitted from human-to-human via droplets from nasal and respiratory secretions. Notably, the human is the main reservoir for transmission of these pathogens and, thus, vaccination has proven to be an efficient measure for protection on a population basis and containment of spread of these diseases.
In the context of antibiotic resistance, clinicians highlighted the importance of the ESKAPE pathogens, e.g., Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species (8, 9). This acronym summarized the most frequently encountered pathogens in hospital-acquired bacterial infections ranging from wound infections and ventilator-associated pneumonia to sepsis. Nevertheless, other infections such as those caused by Clostridioidales difficile have increased in frequency and antibiotic resistance rates (10) and have, thus, been added to the list of nosocomial pathogens and potential bacterial targets for vaccine development. They are now frequently referred to as ESCAPE pathogens (Enterococcus faecium, Staphylococcus aureus, C. difficile, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacteriaceae).
Characteristic Features of Nosocomial Bacterial Infections
Nosocomial infections are caused by bacterial pathogens either transmitted in the hospital environment or from commensals that were already present prior to hospitalization (endogenous infection). On an individual patient level, it is often cumbersome to follow up, which of these sources was causative, unless there is evidence for spread of a specific strain among patients. Moreover, the species causing hospital-acquired infections behave as facultative pathogens, indicating that they cause infections only in a subgroup of patients, under specific circumstances that are also hard to assess in the patient: a coincidence of transient (or chronic) immune suppression associated with age, co-morbidities and medical treatment, selection of resistant strains and dysbiosis caused by antibacterial therapies, transient (or chronic) disturbance of cutaneous and epithelial barriers, and possible displacement to other body areas (urinary tract infection or pneumonia caused by enteric bacteria). Lastly, the specific immune defense mechanisms and the ambiguous role of preexisting immune memory to microbiota are poorly understood.
Expectations and Concerns With Vaccination Against Nosocomial Bacterial Pathogens
One of the major drivers for development of vaccines against nosocomial bacterial pathogens is that antibiotics are no longer effective in all patients. Notably, for most of the ESCAPE pathogens the reservoirs include zoonotic and environmental habitats such as animal husbandry and wastewater, where they are subject to continuous antibiotic selection pressure. It is, therefore, nearly impossible to eradicate these pathogens or revert their resistance by immunization programs in humans. More comprehensive One Health strategies are needed to reduce the antimicrobial resistance burden arising from these sources (11). Nevertheless, vaccine-mediated prevention of nosocomial infections in patients could reduce antibiotic usage and resistance development in hospitals.
Thus, the expectation is to prevent transmission and infection, avoid antibiotic therapy and reduce development of or revert antibiotic resistance [reviewed in (12)]. Two examples may highlight that along with other antibiotic stewardship measures vaccine-based prevention of infections could have the potential to reduce antibiotic usage and—at least transiently—positively influence resistance trends:
1. An indirect effect is postulated in relation to the seasonal influenza burden: reduced antibiotic usage due to vaccine-induced protection against influenza (13) could result in reduced rates of C. difficile infection, which shows seasonal co-incidence with influenza (14, 15). However, this intriguing hypothesis remains to be confirmed.
2. The introduction of pneumococcal conjugate vaccines re-shaped the epidemiological representation of pneumococcal serotypes. Initially, the reduction in infections with antibiotic resistant serotypes suggested that vaccines could reduce antimicrobial resistance. However, long-term analyses revealed that the beneficial effects on antibiotic susceptibility profiles could not be maintained after serotype replacement (16–19).
One repeated concern has been that eradication of specific commensals might negatively affect the resident microbiota composition and the local immune response. On the one hand, absence of a previously colonizing pathogen and concomitant loss of continuous exposure of the immune system to this pathogen could weaken immune defense and increase susceptibility for infection with this pathogen (20). On the other hand, manipulation of the microbiome creates niches for replacement by foreign strains or other species as exemplified for S. aureus (21, 22), which can potentially affect susceptibility to infection. It can only be speculated whether these effects might have contributed to the clinical failure of V710 (Merck), the only vaccine formulation, so far, that was specifically targeting nasal colonization with S. aureus (23). Dedicated research is needed to provide a better understanding of the complex interrelationships of microbiota and the immune system as well as to ensure safety of future developments.
New Paradigms in Vaccine Development Targeting Nosocomial Bacterial Pathogens
One of the most obvious challenges for vaccine development is the high genetic diversity of strains typical of the commensal pathogens. The genetic heterogeneity translates into differences in chemical structure of variable proteins and polysaccharides and alters their immunogenicity. Immunological strain variability limits cross-reactivity of the immune response to variable surface proteins and polysaccharides and undermines vaccine-mediated cross-protection against strains not included in the vaccine design.
The lack of cross-protectivity has hindered the development of vaccines against several ESCAPE pathogens:
1. P. aeruginosa is an ubiquitously encountered environmental pathogen that favors humid environments. It can colonize the human mucosa in predisposed patients with altered or damaged epithelial barriers due to cystic fibrosis, ventilation (burn), wounds, and chemotherapy. Vaccines against P. aeruginosa were developed for three different target populations, e.g., cystic fibrosis, burn wounds and ventilator-associated pneumonia [reviewed in (5, 24)]. So far, the majority of vaccines that reached the clinical development stage targeted the lipopolysaccharide (LPS) and flagellar compounds with and without conjugation to protein carriers. These are highly immunogenic structures and antibodies against these compounds form part of the natural immune response in humans. Induction of LPS- and flagella-specific antibodies conferred protection in preclinical infection models. However, in the clinical trials, failure to prove efficacy was attributed to strain-dependent variability of LPS, Flagella and whole cell vaccines and insufficient coverage of the arsenal of different strains encountered in the patient population.
2. K. pneumoniae is a high-risk pathogen because it accumulates genetic resistance elements and easily spreads among patients, two features that favor the global spread of some highly virulent carbapenem-resistant strains. The species is characterized by the formation of a polysaccharide capsule, which acts as an important virulence factor. Early vaccines developments were, therefore, based on combinations of unconjugated and, later, conjugated capsular polysaccharide (CPS) antigens (5, 25). However, there are more than 70 serotypes with varying distribution worldwide. Although a multivalent vaccine covering 24 CPS was tested in a clinical trial (26). Although this vaccine was later tested in combination with 8-valent LPS vaccine against P. aeruginosa (27), development was later abandoned because it did not seem feasible to achieve protection against the multitude of different serotypes on a global level.
As an answer to the previous failures and the eminent clinical need, we have recently witnessed a change in paradigm: new vaccine developments no longer focus on broad coverage of strains but narrow the spectrum to individual, epidemiologically relevant strains known to harbor resistance to carbapenems. These developments include
1. Targeting of the capsule of K. pneumoniae based on a recently described semi-synthetic hexasaccharide-glycoconjugate (with CRM197 as protein carrier). The glyoconjugate was shown to induce antibodies with opsonophagocytic activity against carbapenem-resistant K. pneumoniae strains in vitro (28) and monoclonal antibodies derived thereof promoted protection against the K. pneumoniae ST258 strain in vivo (29).
2. Strain-specific LPS (O-glycan)-based developments of monoclonal antibodies targeting K. pneumoniae strains such as ST258 and E. coli strains ST131 (30– 32) have been developed to protect from epidemic strains with high transmission potential, antibiotic resistance and severe disease manifestation. Notably, serotype-specific vaccines have also been proposed for targeting O111 E. coli due to this serotype's high representation in toxin producing E. coli (EHEC, STEC, EPEC, and EAEC) (33, 34). To avoid toxicity in these vaccines O111 LPS was conjugated to carrier proteins.
3. Recent data further highlighted that small glycan motifs detected in a broad range of strains (and species) are natural antibody targets and could have the potential to serve as vaccine antigens (35–38).
Altogether, this development suggests that in light of global spread of antibiotic resistant strains exploring strain-specific vaccines may be worthwhile and abandoning the concept of broad strain coverage by vaccines may facilitate targeted vaccine development in the AMR field.
The Immunological Mechanism of Action
The immunological mechanism of action of a vaccine usually refers to a known immune correlate of protection. The identification of this parameter requires investigation of the natural immune responses leading to the resolution of infection and vaccine-related immune protection. The currently licensed vaccines for prevention of bacterial infections either generate toxin-neutralizing antibodies, or enable clearance of bacteria via formation of bacterial immune complexes and subsequent phagocytosis. The studies sometimes use “immunological surrogates of protection” to correlate immunogenicity with protection and to provide an absolute quantifiable value for protection by measuring specific serum antibody titers and defining threshold levels (7). However, for bacterial pathogens causing nosocomial infections the correlates of protection remain unknown. Additionally, for most pathogens it is unclear whether the preexisting, natural immune response is protective.
For vaccine development it is of primary importance to understand, which type of immune response is needed to achieve the intended effect, e.g., prevention (or reduction) of colonization vs. prevention of infection and disease. However, there is a current lack of understanding of the immune response to most of the nosocomial bacterial pathogens.
Immunization with endogenous S. aureus and E. coli strains in parallel to the initiation of an antibiotic therapy prevented recolonization with these strains in mice, highlighting the efficacy of a systemic immune response in targeting these pathogens (39). However, at present it is unclear to which degree preformed natural immune responses are protective and whether Th2-dominated responses should considered as tolerogenic due to their failure to clear the commensal pathogens from the mucosal surfaces. It is, thus, very relevant to establish whether vaccine-mediated boosting of preexisting immune responses is sufficient for protective action of a vaccine or whether vaccines need to re-shape the immune response. While a Th2 response, which is linked to antibody production, may be effective in promoting protection against toxin-mediated diseases such as tetanus and diphtheria, this may be insufficient for infections where inflammatory T cell-mediated immunity (Th1/Th17) is required for immune defense. This could imply that de novo formation of immune memory or re-education of an established immune response by a vaccine could be required to induce protection.
There are three fundamental mechanisms responsible for vaccine-mediated protection:
1. Neutralization of toxins as drivers of disease: In the attempt to develop vaccines against nosocomial bacterial infections, many different strategies have been evaluated. Prominent examples for toxin-based vaccination strategies are vaccines targeting C. difficile toxins tcdA and tcdB to prevent manifestation of C. difficile infection (CDI). Additionally, some vaccine prototypes (and monoclonal antibodies) are targeting S. aureus toxins such as alpha toxin (hemolysin A), staphylococcal eneterotoxin B (SEB) and other secreted toxins such as leukocidins [reviewed in (4, 5, 40–42)]. The general assumption is that raising the level of neutralizing antibodies against toxins prevents invasive disease and lowers disease severity. It can further be speculated that immunization with C. difficile or S. aureus inactivated toxin antigens relies on a preexisting humoral immune response to the natural toxins and acts as a booster vaccination. Similarly, anti-toxin antibodies can be generated by immunization against toxins from toxigenic E. coli (43) while this approach is not feasible for protection against non-toxigenic E. coli such as extraintestinal E. coli harboring extended spectrum betalactamases (ESBL), which are often referred to as ExPEC.
2. Opsonophagocytosis of infecting pathogens: vaccines targeting bacterial surface molecules usually act by promoting opsonophagocytosis through antibodies and subsequent intracellular lysis of the bacterial pathogens. This mechanism is exploited by nearly all vaccine developments ranging from whole cells to formulations consisting of single or combined antigens. Multiple targets have been described for most ESCAPE pathogens [reviewed in (4, 5, 24, 41, 44)]. These include polysaccharides such as LPS, CPS, conserved glycan motifs and highly conserved immunogenic surface proteins such as outer membrane proteins (OMP). Nevertheless, only few vaccines targeting bacterial surface antigens from nosocomial bacterial pathogens have reached the stage of clinical development, among these mainly S. aureus vaccines (5, 40, 41, 45).
3. Shaping T cell immunity: the role of T cell-mediated immunity in defense against extracellular bacteria has been neglected although some of these pathogens, e.g., A. baumanii and S. aureus, reside intracellularly (46–50). Furthermore, different types of T cell responses might be required depending on the body compartment (51, 52). For example, Th17 responses are relevant in the skin and mucosal surfaces in defense against S. aureus (53, 54), and have also been found to be important for clearance of ESCAPE pathogens such as Klebsiella spp., P. aeruginosa and A. baumanii (55–64). Thus, vaccine formulations targeting colonizing pathogens might need to be optimized to induce Th17 responses (24, 58, 65). However, due to the paucity of data, further investigation is needed to identify the specific T cell responses required for protection, and understand, whether vaccine-induced long-term immunity is preferable to acute induction of immune memory in short interval to infection.
Although initially, the LPS content in outer membrane vesicles (OMV) was regarded a safety issue, OMV have gained acceptance as vaccine components after licensing of the MenB vaccine Bexsero (66). These vesicles are physiologically released by Gram negative bacteria and embed several surface antigens in a lipophilic vesicular structure. They resemble bacterial cells because they combine protein antigens, polysaccharides and molecules with innate immune stimulatory properties such as LPS, lipopeptides, lipoteichoic acid and peptidoglycan. Next to the induction of opsonizing antibodies, OMV trigger Th1/Th17 cell responses. For ESCAPE pathogens several OMV-based approaches to vaccination have been evaluated at the preclinical stage (66). The diversity of these approaches to OMV-based or related vaccines can be exemplified by summarizing those evaluated for A. baumannii (summarized in Figure 1) [reviewed in (67–69)].
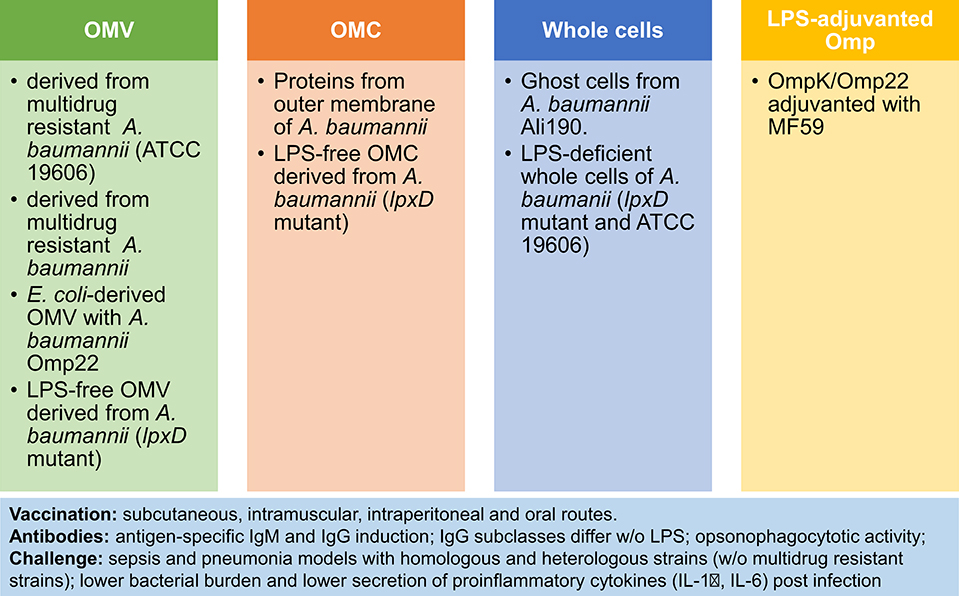
Figure 1. OMV-based and related approaches to vaccination against A. baumannii. Four immunization strategies have been tested in preclinical models of sepsis and pneumonia: (1) Outer Membrane Vesicles (OMV) (70–73); (2) Outer Membrane Complex (OMC) (74, 75); (3) Whole cells (76, 77); (4) LPS-adjuvanted Omp (78). Comparison of vaccines reveals higher potency of LPS-containing vaccines. Notably, the vaccine response is characterized by antibody induction and reduced pro-inflammatory responses after challenge, next to improved survival and lower bacterial burden post-infection.
Consequences for Design of Efficacy Studies
One major factor complicating vaccine trials for nosocomial pathogens is that the patients affected are elderly individuals. They suffer of co-morbidities and are at risk for immune suppression through both medical intervention and immunosenescence. Their immune status is difficult to assess with current diagnostic methods but it predisposes for infections and antibiotic therapy. A study on A. baumannii pneumonia in aged mice illustrates that mortality increases with age, while efficacies of treatment, both antibiotics and vaccination, decrease because both rely on functional immune responses (79). Similarly, the human vaccine response is compromised by the immunodeficiencies caused by aging of the immune system [reviewed in (80, 81)].
The clinical development of vaccines for prevention of hospital-acquired infections is linked to a history of failures. Today, the major challenges are well-understood and variables such as immune deficiency due to aging of the immune system are being taken into account and reflected by new vaccine formulations (82, 83). Nevertheless, it remains difficult for trial design to predict some known factors that seem out of control and hit rates are often lower than expected (84). As summarized in Table 1 the main enemy in trial design for this type of vaccines is time.
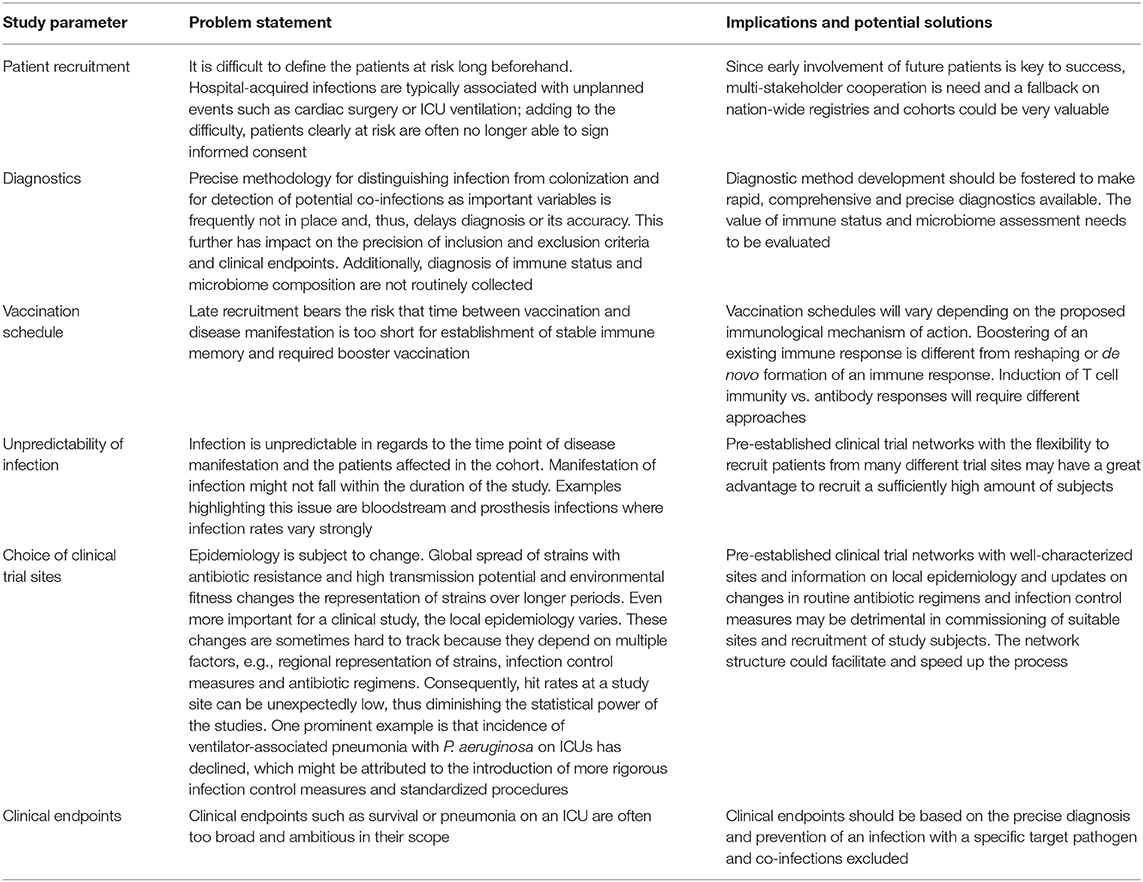
Table 1. Challenges in clinical trial design for vaccine development to prevent hospital-acquired infections.
Controlled Human Infection Models
One alternative to obtain efficacy data is to develop controlled human infection models (CHIM) and use these models for evaluation of vaccines (Human Challenge Trials, HCT) (85). Lately, this type of studies has gained more importance for proof-of-concept studies, licensing and prequalification of vaccines. In particular, indications where epidemiology of disease does not allow the timely execution of the clinical efficacy trials these studies have become relevant for decision making of regulators and developers. However, there are significant limitations to this type of trials that need to be considered:
• The safety of the study participants is the most important requirement: infection has to be controlled, e.g., appropriate treatment has to be available and clearance of the pathogen guaranteed.
• Clinical endpoints must be clinically relevant and reflect the natural course of disease
• The choice of infectious dose, the virulence of the challenge pathogen and severity of disease manifestation should resemble natural course of infection without compromising the safety of the study participant.
• The number of study participants is typically small.
• 100% colonization rates or 100% infection rates may be required to obtain significant results but may be difficult to achieve in practice.
• The studies have limited value if the challenge agent does not reflect the epidemiologically relevant spectrum of strains encountered in real life.
• Standardization of procedures (administration and manufacturing of the challenge agent, the challenge agent itself, disease severity scores, primary and secondary endpoints) is essential for comparability of the studies and vaccines but often not in place.
In vaccine development HCT studies are well-established for many indications. For bacteria, CHIM have mainly been used in the development of vaccines against enteric pathogens among them enterotoxigenic E. coli (ETEC), Shigella spp. and V. cholera. The learnings included that HCT results do not predict vaccine effectiveness in field studies (86). Substantial efforts for standardization have been made to achieve high quality and reliability of results in ETEC and Shigella studies (87–89).
To date, no human challenge models have been published for evaluation of vaccines for hospital-acquired bacterial infections. A few studies addressed the colonization potential of S. aureus (ST398) (90) and non-toxigenic C. difficile (91, 92). Although proof-of-concept studies could be supportive, accounting for the heterogeneity of strains, patients (immune status, dysbiosis, barrier function of epithelia) and disease course would not be feasible in this setting. This questions the relevance of data collected in HCT and, in this context, limits utility to CHIM studies related to understanding of the role of the immune response and microbiota in prevention and resolution of infection and, potentially, very specific research questions such as decolonization of patients colonized with multidrug resistant pathogens.
Synergies and Redundancies of Microbiological and Immunological Defense Mechanisms
Another aspect that influences clinical trials to a so far unknown extent is that host defense is prepared to fight infections with two different strategies: next to the host immune system reconstitution and resilience of the host microbiome limits spread and promotes clearance of the infecting pathogen (93). Although many studies have addressed the interplay of both of these systems in the healthy setting the synergies and redundancies are is not well-understood in the context of an infection and is even less controllable in the clinical trial setting. These functionally distinct, but redundant, immunological and microbiological defense mechanisms have evolved to secure protection from infection. Their presence, absence or resurgence (resilience of the microbiome, recovery of the immune response) could superimpose vaccine-mediated resolution of infection resulting in failure to demonstrate efficacy in the clinical trial.
Recent reports indicate that the microbiota influence the vaccine response. From studies in low income countries we learned that many factors including the microbiota composition can influence the response to oral vaccination (94). It is further well-known that the microbiota are essential for the development of the intestinal immune system. Among them, segmented filamentous bacteria have been associated with recruitment of Th17 cells to the gut (95). Furthermore, the abundance of specific microbiota correlated with higher immunogenicity, e.g., Actinobacteria, in particular the Bifidobacteria genus, were found to increase immunogenicity of OPV in Bangladesh and low representation of Bacteroidetes and high abundance of Bacilli (Streptococcus bovis) correlated with better vaccine response and seroconversion in rural Ghana (96, 97). Additionally, treatment of mice with antibiotics reduced the polio replication and infectivity in mice, a phenomenon well in line with previous findings that the microbiota enhance uptake and replication of enteric viruses (98, 99). However, this was not observed in a trial using azithromycin in children (100). Similarly to Polio vaccine virus, shedding of the Rota virus was increased if subjects were treated with antibiotics before vaccination but no differences in IgA levels were observed (101, 102). By contrast, pretreatment with antibiotics reduced the neutralizing antibody response to influenza vaccination, indicating that systemic immune responses are also affected by the lack of microbiota (103).
The pathogens causing nosocomial infections usually reside on the mucosal surfaces. On the one hand, microbiota colonizing the mucosa constantly stimulate epithelia and innate immune cells, trigger the production of antimicrobial peptides and IgA and shape the T and B cell repertoires [reviewed in (104)]. On the other hand, these factors play an important role in regulating the quantity and composition of the local microbiome as well as the defense against invading pathogens (105, 106). Notably, the presence of IgA protects mice from DSS colitis and polymicrobial sepsis (107, 108).
Recent evidence suggests that administration of antibiotics leads to loss of IgA secretion. This was demonstrated in the respiratory tract where IgA-deficiency was associated with increased susceptibility to infection with P. aeruginosa (109). Similarly, in a humanized mouse model of IgA nephropathy treatment with antibiotics prevented renal deposition of IgA complexes, which was proposed to be due to a reduction of the intestinal microbiota and the concomitant loss of the microbiota-specific IgA (110).
Microbiological and Immunological Susceptibility to C. difficile Infection
Colonization with C. difficile occurs at very early age and is detectable in 80% of newborns in their first month of life. It decreases to colonization rates of 3% (comparable with adults) by the end of the first year of life (111). Thus, trained innate immunity is probably established during this early phase of life and shapes the tolerogenic immune response to the colonizing pathogen. As for other hospital-acquired infections in the elderly the risk for CDI is increased, which might be attributed to both immunosenescence (112) and age-related changes in the gut microbiota (113). However, the relative contribution to susceptibility remains ill-defined for both factors and is complicated by the reciprocal regulation of intestinal microbiota and mucosal immunity. Figure 2 summarizes the major events contributing to CDI susceptibility.
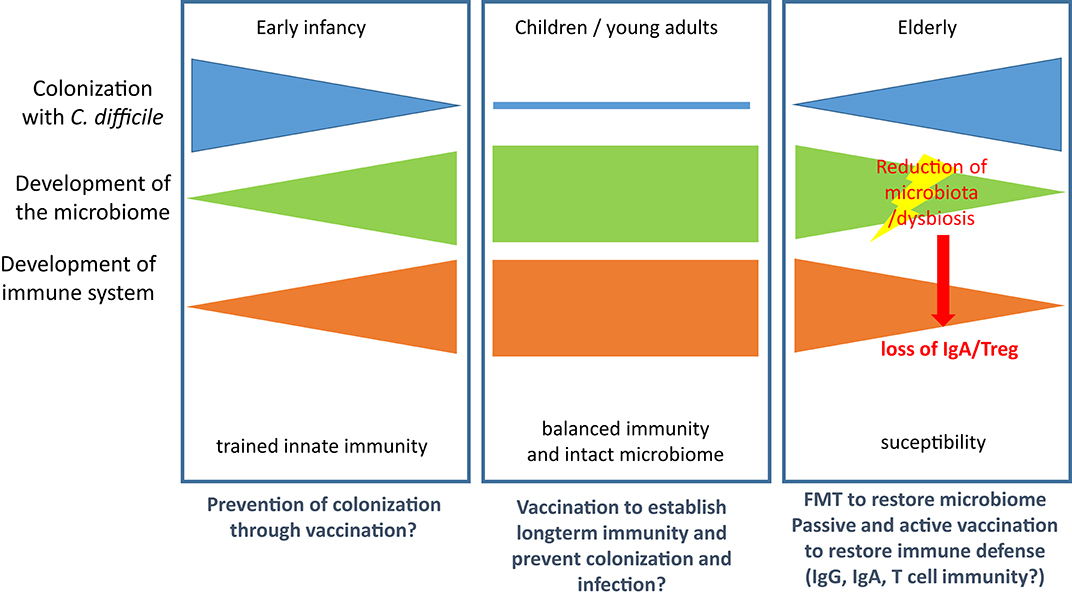
Figure 2. Microbiological and immunological defense against CDI. (Left): Colonization is established in early infancy but regresses in the first year of life with both the maturation of the immune system and the development of the full microbiome. (Middle): In children and young adults the immune system is balanced and the microbiome intact. Both factors control growth of C. diffcile and toxin production. (Right): In the elderly population the microbiome and the immune system are both subject to age-related changes, which leads to increased susceptibility for CDI. Treatment with antibiotics results in reduction of microbiota and dysbiosis and enables growth of C. difficile. Loss of microbiota-mediated stimulation of immune cells leads to loss of IgA secretion and Treg and thereby facilitates CDI and the associated inflammatory processes. Therapeutic options are provided below the panels.
Based on the findings described above it can be speculated that an antibiotic-induced IgA-deficiency could account for susceptibility to C. difficile infection (CDI) (114). High titers of toxin (TcdA and TcdB)-specific antibodies, in particular IgA in serum and feces, correlate with protection against CDI, while low titers or absence of toxin-specific IgG and IgA were found in patients with acute or recurrent CDI and in non-colonized individuals (115–118). These data indicated that patients with transient deficiency in IgA might be more susceptible for infection. Well in line with this observation Bezlotoxumab, a monoclonal antibodies directed against TcdB, prevented recurrence of CDI, highlighting the value of antibody-mediated toxin neutralization (119, 120).
The role of cellular immunity in CDI is less well-understood. Notably, HIV+ individuals with low CD4+ T cell counts and homozygotes with a Q223R mutation in the leptin receptor, which abrogates synthesis of IL-23, a cytokine that induces formation of Th17 cells, have an increased risk for CDI (121, 122). It is further known that high levels of T cell-derived cytokines (IFNγ and IL-5) in peripheral blood correlate with less severe disease manifestation (123). IL-23 is elevated in feces and intestinal biopsies of CDI patients (124, 125) and patients with recurrent CDI display increased numbers of Th1 and Th17 cells in peripheral blood (126). The role of these inflammatory T cell subsets is, however, controversial. Recent data highlight their contribution to immune pathology of CDI rather than a protective role (127). It has further been suggested that microbiota induce Treg and regulate the balance between Treg and Th17 cells (128). In analogy to IgA, the reduction of microbiota by antibiotics could, thus, increase susceptibility to CDI and, in particular, contribute to inflammation and immune pathology by relaxing Treg-mediated suppression and allowing increased formation of Th17 cells.
Although these data argue for an important contribution of both T and B lymphocytes to the immune response in CDI, Leslie et al. recently demonstrated in a mouse model that clearance of C. difficile due to resilience of the microbiome occurs in the absence of adaptive immune responses (129), arguing for a non-essential role of immune defense in this context. Furthermore, colonization with non-toxigenic strains can prevent colonization with toxigenic strains of C. difficile and, thus, prevent disease, which was demonstrated in the hamster and in recurrent human CDI (92, 130). Similarly, fecal microbiota transplantation (FMT) has become a success story in treatment of CDI (131). Nevertheless, recent studies indicate that non-immune, soluble factors such as such as butyrate and bile salts or bacteriophages might play an underestimated role in reconstitution of the microbiota after CDI (131–134). However, the model developed by Leslie et al. did not consider the effects of aging of the microbiome (113). It is, therefore, likely that age-related changes in the immune system and the microbiota facilitate colonization with C. diffcile and development of CDI and that regeneration of one or both systems drives resolution if infection (Figure 2).
Conclusions and Perspectives
These multiple findings highlight the complexity behind the endeavor to develop vaccines for nosocomial bacterial infections. The heterogeneity of patients, bacterial strains, disease course and hospital epidemiology have complicated vaccine development in this area and have led to discontinuation of vaccine development programs. However, the power of vaccine prevention has been demonstrated in many occasions. Thus, re-thinking the strategies may be warranted but failures should not be accepted without further refurbishing on available and new scientific data. For new developments, the definition of pathogen-specific clinical endpoints and the suitability of the immunological mechanism of action are key to success. Figure 3 summarizes the interconnection of the relevant parameters, which influence the trail design and final indication.
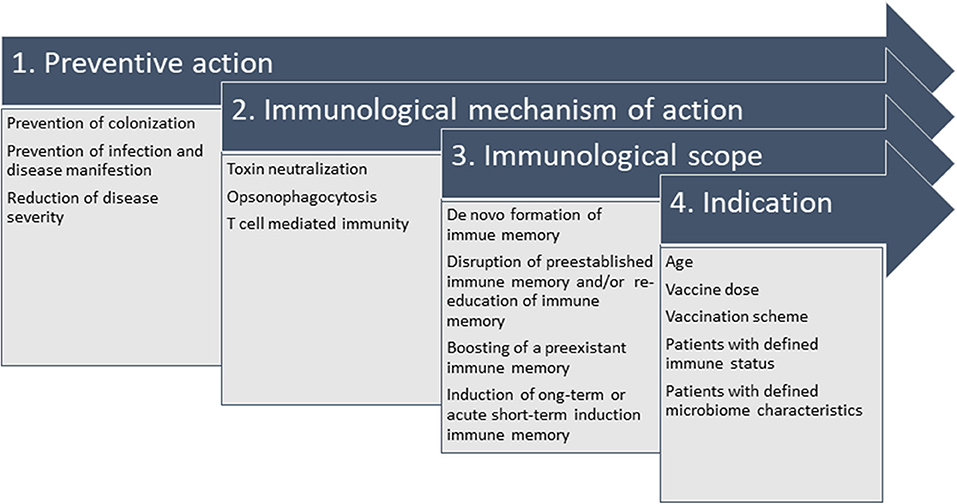
Figure 3. Interconnection of parameters relevant to design the vaccination strategy against nosocomial bacterial pathogens. It is crucial to define the desired preventive action (1), which defines the immunological mechanism of action (2) when set in context with the available knowledge of pathophysiology, e.g., intra- and extracellular survival, correlate of protection (or a surrogate, if unknown). In the specific case of hospital-acquired infections, colonization in youth or in elderly patients at risk precedes infection and vaccine design needs to take into account that natural immune responses to colonizing pathogens might not be protective. Thus, they might need refurbishing with the immunological scope (3) of strengthening preexistent immune responses (“booster”), establishing long-term protective immune memory or promoting immunity by acute intervention. All considerations generate the indication (4), e.g., all details on administration (e.g., vaccination scheme and dosage) and indication for defined patient populations (age indication, immune status, microbiome).
For vaccines targeting K. pneumoniae and E. coli there is a trend to focus on epidemiologically relevant strains. A next step could be the development vaccine formulations that trigger specific, protective T cell responses such as Th17 cells in the mucosa. However, future research will need to define the type of T cell responses required and the route of immunization needed to establish protection in different body compartments. Next to the quality of the T cell response it will be relevant to understand the optimal time point for vaccination: (1) whether it is preferable that vaccines build on establishment of long-term immunity, or, (2) whether immunization at short temporal distance to onset of infection is favorable because acute formation of T cell immunity is more effective in promoting protection against infection, although the vaccine response in the latter case may be short-lived.
Despite the higher cost, a more detailed characterization of individual patients and patient subpopulations within the trial population seems warranted to ensure that the results obtained in the clinical trials are meaningful. One further step could be to identify the patient population that benefits from vaccination. Pro- and retrospective analyses of trial data on subject level could help to define the characteristics of these patient collectives and improve stratification in future trials. This concept is not new but well in line with the current understanding of personalized medicine and individualized treatment concepts that have recently been introduced to the field of infectious diseases, antibiotics and vaccination (135–137). In light of the interdependency of immune status and microbiome resilience, the influence of these factors on clinical trial success needs to be investigated more thoroughly.
Author Contributions
IB-D developed the concept, performed the analyses, and wrote the paper.
Funding
This research was funded by the Paul-Ehrlich-Institut and the EU/EFPIA Innovative Medicines Initiative 2 Joint Undertaking project Collaboration for prevention and treatment of MDR bacterial infections (COMBINE) IMI2 JU (grant agreement # 853967).
Disclaimer
Views expressed in this review are the personal view of the author and do not reflect the position of the PEI in its regulatory capacity.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gajdács M. The concept of an ideal antibiotic: implications for drug design. Molecules. (2019) 24:892. doi: 10.3390/molecules24050892
2. Rolain JM, Baquero F. The refusal of the Society to accept antibiotic toxicity: missing opportunities for therapy of severe infections. Clin Microbiol Infect. (2016) 22:423–7. doi: 10.1016/j.cmi.2016.03.026
3. Krut O, Bekeredjian-Ding I. Contribution of the immune response to phage therapy. J Immunol. (2018) 200:3037–44. doi: 10.4049/jimmunol.1701745
4. Knisely JM, Liu B, Ranallo RT, Zou L. Vaccines for healthcare-associated infections: promise and challenge. Clin Infect Dis. (2016) 63:657–62. doi: 10.1093/cid/ciw333
5. Pletz MW, Uebele J, Götz K, Hagel S, Bekeredjian-Ding I. Vaccines against major ICU pathogens: where do we stand? Curr Opin Crit Care. (2016) 22:470–6. doi: 10.1097/MCC.0000000000000338
6. Gagneux-Brunon A, Lucht F, Launay O, Berthelot P, Botelho-Nevers E. Vaccines for healthcare-associated infections: present, future, and expectations. Expert Rev Vaccines. (2018) 17:421–33. doi: 10.1080/14760584.2018.1470507
7. Plotkin SA. Correlates of protection induced by vaccination. Clin Vaccine Immunol. (2010) 17:1055–65. doi: 10.1128/CVI.00131-10
8. Pendleton JN, Gorman SP, Gilmore BF. Clinical relevance of the ESKAPE pathogens. Expert Rev Anti Infect Ther. (2013) 11:297–308. doi: 10.1586/eri.13.12
9. Rice LB. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis. (2008) 197:1079–81. doi: 10.1086/533452
10. Peng Z, Jin D, Kim HB, Stratton CW, Wu B, Tang Y-W, et al. Update on antimicrobial resistance in Clostridium difficile: resistance mechanisms and antimicrobial susceptibility testing. J Clin Microbiol. (2017) 55:1998–2008. doi: 10.1128/JCM.02250-16
11. McEwen SA, Collignon PJ. Antimicrobial resistance: a one health perspective. Microbiol Spectr. (2018) 6:1–2. doi: 10.1128/9781555819804.ch25
12. Jansen KU, Anderson AS. The role of vaccines in fighting antimicrobial resistance (AMR). Hum Vaccin Immunother. (2018) 14:2142–9. doi: 10.1080/21645515.2018.1476814
13. Kwong JC, Maaten S, Upshur RE, Patrick DM, Marra F. The effect of universal influenza immunization on antibiotic prescriptions: an ecological study. Clin Infect Dis. (2009) 49:750–6. doi: 10.1086/605087
14. Polgreen PM, Yang M, Bohnett LC, Cavanaugh JE. A time-series analysis of Clostridium difficile and its seasonal association with influenza. Infect Control Hosp Epidemiol. (2010) 31:382–7. doi: 10.1086/651095
15. Brown KA, Daneman N, Arora P, Moineddin R, Fisman DN. The co-seasonality of pneumonia and influenza with Clostridium difficile infection in the United States, 1993-2008. Am J Epidemiol. (2013) 178:118–25. doi: 10.1093/aje/kws463
16. Imöhl M, Möller J, Reinert RR, Perniciaro S, van der Linden M, Aktas O. Pneumococcal meningitis and vaccine effects in the era of conjugate vaccination: results of 20 years of nationwide surveillance in Germany. BMC Infect Dis. (2015) 15:61. doi: 10.1186/s12879-015-0787-1
17. Picazo JJ. Management of antibiotic-resistant Streptococcus pneumoniae infections and the use of pneumococcal conjugate vaccines. Clin Microbiol Infect. (2009) 15(Suppl. 3):4–6. doi: 10.1111/j.1469-0691.2009.02723.x
18. Dagan R. Impact of pneumococcal conjugate vaccine on infections caused by antibiotic-resistant Streptococcus pneumoniae. Clin Microbiol Infect. (2009) 15(Suppl. 3):16–20. doi: 10.1111/j.1469-0691.2009.02726.x
19. Imöhl M, Reinert RR, van der Linden M. Antibiotic susceptibility rates of invasive pneumococci before and after the introduction of pneumococcal conjugate vaccination in Germany. Int J Med Microbiol. (2015) 305:776–83. doi: 10.1016/j.ijmm.2015.08.031
20. Wertheim HF, Vos MC, Ott A, van Belkum A, Voss A, Kluytmans JA, et al. Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. Lancet. (2004) 364:703–5. doi: 10.1016/S0140-6736(04)16897-9
21. Sakr A, Brégeon F, Mège J-L, Rolain J-M, Blin O. Staphylococcus aureus nasal colonization: an update on mechanisms, epidemiology, risk factors, and subsequent infections. Front Microbiol. (2018) 9:2419. doi: 10.3389/fmicb.2018.02419
22. Khamash DF, Voskertchian A, Milstone AM. Manipulating the microbiome: evolution of a strategy to prevent S. aureus disease in children. J Perinatol. (2018) 38:105–9. doi: 10.1038/jp.2017.155
23. McNeely TB, Shah NA, Fridman A, Joshi A, Hartzel JS, Keshari RS, et al. Mortality among recipients of the Merck V710 Staphylococcus aureus vaccine after postoperative S. aureus infections: an analysis of possible contributing host factors. Hum Vaccin Immunother. (2014) 10:3513–6. doi: 10.4161/hv.34407
24. Priebe GP, Goldberg JB. Vaccines for Pseudomonas aeruginosa: a long and winding road. Expert Rev Vaccines. (2014) 13:507–19. doi: 10.1586/14760584.2014.890053
25. Kobayashi SD, De Leo FR. Re-evaluating the potential of immunoprophylaxis and/or immunotherapy for infections caused by multidrug resistant Klebsiella pneumoniae. Future Microbiol. (2018) 13:1343–6. doi: 10.2217/fmb-2018-0189
26. Cryz SJ, Cross AS, Sadoff GC, Que JU. Human IgG and IgA subclass response following immunization with a polyvalent Klebsiella capsular polysaccharide vaccine. Eur J Immunol. (1988) 18:2073–5. doi: 10.1002/eji.1830181230
27. Campbell WN, Hendrix E, Cryz S, Cross AS. Immunogenicity of a 24-valent Klebsiella capsular polysaccharide vaccine and an eight-valent Pseudomonas O-polysaccharide conjugate vaccine administered to victims of acute trauma. Clin Infect Dis. (1996) 23:179–81. doi: 10.1093/clinids/23.1.179
28. Seeberger PH, Pereira CL, Khan N, Xiao G, Diago-Navarro E, Reppe K, et al. A semi-synthetic glycoconjugate vaccine candidate for carbapenem-resistant Klebsiella pneumoniae. Angew Chem Int Ed Engl. (2017) 56:13973–8. doi: 10.1002/anie.201700964
29. Diago-Navarro E, Motley MP, Ruiz-Peréz G, Yu W, Austin J, Seco BM, et al. Novel, broadly reactive anticapsular antibodies against carbapenem-resistant Klebsiella pneumoniae protect from infection. mBio. (2018) 9:e01005-18. doi: 10.1128/mBio.01005-18
30. Szijártó V, Guachalla LM, Hartl K, Varga C, Banerjee P, Stojkovic K, et al. Both clades of the epidemic KPC-producing Klebsiella pneumoniae clone ST258 share a modified galactan O-antigen type. Int J Med Microbiol. (2016) 306:89–98. doi: 10.1016/j.ijmm.2015.12.002
31. Szijártó V, Guachalla LM, Visram ZC, Hartl K, Varga C, Mirkina I, et al. Bactericidal monoclonal antibodies specific to the lipopolysaccharide O antigen from multidrug-resistant Escherichia coli clone ST131-O25b:H4 elicit protection in mice. Antimicrob Agents Chemother. (2015) 59:3109–16. doi: 10.1128/AAC.04494-14
32. Szijártó V, Guachalla LM, Hartl K, Varga C, Badarau A, Mirkina I, et al. Endotoxin neutralization by an O-antigen specific monoclonal antibody: a potential novel therapeutic approach against Klebsiella pneumoniae ST258. Virulence. (2017) 8:1203–15. doi: 10.1080/21505594.2017.1279778
33. Andrade GR, New RR, Sant'Anna OA, Williams NA, Alves RC, Pimenta DC, et al. A universal polysaccharide conjugated vaccine against O111 E. coli. Hum Vaccin Immunother. (2014) 10:2864–74. doi: 10.4161/21645515.2014.972145
34. Santos MF, New RR, Andrade GR, Ozaki CY, Sant'Anna OA, Mendonça-Previato L, et al. Lipopolysaccharide as an antigen target for the formulation of a universal vaccine against Escherichia coli O111 strains. Clin Vaccine Immunol. (2010) 17:1772–80. doi: 10.1128/CVI.00232-10
35. Pabst O, Slack E. IgA and the intestinal microbiota: the importance of being specific. Mucosal Immunol. (2020) 13:12–21. doi: 10.1038/s41385-019-0227-4
36. Rollenske T, Szijarto V, Lukasiewicz J, Guachalla LM, Stojkovic K, Hartl K, et al. Cross-specificity of protective human antibodies against Klebsiella pneumoniae LPS O-antigen. Nat Immunol. (2018) 19:617–24. doi: 10.1038/s41590-018-0106-2
37. Micoli F, Costantino P, Adamo R. Potential targets for next generation antimicrobial glycoconjugate vaccines. FEMS Microbiol Rev. (2018) 42:388–423. doi: 10.1093/femsre/fuy011
38. Frenck RW, Ervin J, Chu L, Abbanat D, Spiessens B, Go O, et al. Safety and immunogenicity of a vaccine for extra-intestinal pathogenic Escherichia coli (ESTELLA): a phase 2 randomised controlled trial. Lancet Infect Dis. (2019) 19:631–40. doi: 10.1016/S1473-3099(18)30803-X
39. Garfias-López JA, Castro-Escarpuli G, Cárdenas PE, Moreno-Altamirano MM, Padierna-Olivos J, Sánchez-García FJ. Immunization with intestinal microbiota-derived Staphylococcus aureus and Escherichia coli reduces bacteria-specific recolonization of the intestinal tract. Immunol Lett. (2018) 196:149–54. doi: 10.1016/j.imlet.2018.02.007
40. Giersing BK, Dastgheyb SS, Modjarrad K, Moorthy V. Status of vaccine research and development of vaccines for Staphylococcus aureus. Vaccine. (2016) 34:2962–6. doi: 10.1016/j.vaccine.2016.03.110
41. Redi D, Raffaelli CS, Rossetti B, Luca A de, Montagnani F. Staphylococcus aureus vaccine preclinical and clinical development: current state of the art. New Microbiol. (2018) 41:208–13.
42. Bagnoli F. Staphylococcus aureus toxin antibodies: good companions of antibiotics and vaccines. Virulence. (2017) 8:1037–42. doi: 10.1080/21505594.2017.1295205
43. Rojas-Lopez M, Monterio R, Pizza M, Desvaux M, Rosini R. Intestinal pathogenic Escherichia coli: insights for vaccine development. Front Microbiol. (2018) 9:440. doi: 10.3389/fmicb.2018.00440
44. Péchiné S, Bruxelle JF, Janoir C, Collignon A. Targeting Clostridium difficile surface components to develop immunotherapeutic strategies against Clostridium difficile infection. Front Microbiol. (2018) 9:1009. doi: 10.3389/fmicb.2018.01009
45. Proctor RA. Is there a future for a Staphylococcus aureus vaccine? Vaccine. (2012) 30:2921–7. doi: 10.1016/j.vaccine.2011.11.006
46. Sato Y, Unno Y, Miyazaki C, Ubagai T, Ono Y. Multidrug-resistant Acinetobacter baumannii resists reactive oxygen species and survives in macrophages. Sci Rep. (2019) 9:17462. doi: 10.1038/s41598-019-53846-3
47. Parra-Millán R, Guerrero-Gómez D, Ayerbe-Algaba R, Pachón-Ibáñez ME, Miranda-Vizuete A, Pachón J, et al. Intracellular trafficking and persistence of Acinetobacter baumannii requires transcription factor EB. mSphere. (2018) 3:e00106-18. doi: 10.1128/mSphere.00106-18
48. Horn J, Stelzner K, Rudel T, Fraunholz M. Inside job: Staphylococcus aureus host-pathogen interactions. Int J Med Microbiol. (2018) 308:607–24. doi: 10.1016/j.ijmm.2017.11.009
49. Löffler B, Tuchscherr L, Niemann S, Peters G. Staphylococcus aureus persistence in non-professional phagocytes. Int J Med Microbiol. (2014) 304:170–6. doi: 10.1016/j.ijmm.2013.11.011
50. Bist P, Dikshit N, Koh TH, Mortellaro A, Tan TT, Sukumaran B. The Nod1, Nod2, and Rip2 axis contributes to host immune defense against intracellular Acinetobacter baumannii infection. Infect Immun. (2014) 82:1112–22. doi: 10.1128/IAI.01459-13
51. O'Brien EC, McLoughlin RM. Considering the 'Alternatives' for next-generation anti-Staphylococcus aureus vaccine development. Trends Mol Med. (2019) 25:171–84. doi: 10.1016/j.molmed.2018.12.010
52. Bekeredjian-Ding I. Deciphering the significance of the T-cell response to Staphylococcus aureus. Fut Microbiol. (2017) 12:1023–6. doi: 10.2217/fmb-2017-0138
53. Montgomery CP, Daniels M, Zhao F, Alegre M-L, Chong AS, Daum RS. Protective immunity against recurrent Staphylococcus aureus skin infection requires antibody and interleukin-17A. Infect Immun. (2014) 82:2125–34. doi: 10.1128/IAI.01491-14
54. Archer NK, Harro JM, Shirtliff ME. Clearance of Staphylococcus aureus nasal carriage is T cell dependent and mediated through interleukin-17A expression and neutrophil influx. Infect Immun. (2013) 81:2070–5. doi: 10.1128/IAI.00084-13
55. Amezcua Vesely MC, Pallis P, Bielecki P, Low JS, Zhao J, Harman CC, et al. Effector TH17 cells give rise to long-lived TRM cells that are essential for an immediate response against bacterial infection. Cell. (2019) 178:1176–88.e15. doi: 10.1016/j.cell.2019.07.032
56. Clemente AM, Castronovo G, Antonelli A, D'Andrea MM, Tanturli M, Perissi E, et al. Differential Th17 response induced by the two clades of the pandemic ST258 Klebsiella pneumoniae clonal lineages producing KPC-type carbapenemase. PLoS ONE. (2017) 12:e0178847. doi: 10.1371/journal.pone.0178847
57. Kim G-L, Lee S, Kim S-J, Lee S-O, Pyo S, Rhee D-K. Pulmonary colonization resistance to pathogens via noncanonical Wnt and interleukin-17A by intranasal pep27 mutant immunization. J Infect Dis. (2018) 217:1977–86. doi: 10.1093/infdis/jiy158
58. Kumar P, Chen K, Kolls JK. Th17 cell based vaccines in mucosal immunity. Curr Opin Immunol. (2013) 25:373–80. doi: 10.1016/j.coi.2013.03.011
59. Baker SM, McLachlan JB, Morici LA. Immunological considerations in the development of Pseudomonas aeruginosa vaccines. Hum Vaccin Immunother. (2019) 16:412–8. doi: 10.1080/21645515.2019.1650999
60. Bayes HK, Ritchie ND, Evans TJ. Interleukin-17 is required for control of chronic lung infection caused by Pseudomonas aeruginosa. Infect Immun. (2016) 84:3507–16. doi: 10.1128/IAI.00717-16
61. Wu W, Huang J, Duan B, Traficante DC, Hong H, Risech M, et al. Th17-stimulating protein vaccines confer protection against Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med. (2012) 186:420–7. doi: 10.1164/rccm.201202-0182OC
62. Yan Z, Yang J, Hu R, Hu X, Chen K. Acinetobacter baumannii infection and IL-17 mediated immunity. Mediat Inflamm. (2016) 2016:9834020. doi: 10.1155/2016/9834020
63. Priebe GP, Walsh RL, Cederroth TA, Kamei A, Coutinho-Sledge YS, Goldberg JB, et al. IL-17 is a critical component of vaccine-induced protection against lung infection by lipopolysaccharide-heterologous strains of Pseudomonas aeruginosa. J Immunol. (2008) 181:4965–75. doi: 10.4049/jimmunol.181.7.4965
64. Chen K, McAleer JP, Lin Y, Paterson DL, Zheng M, Alcorn JF, et al. Th17 cells mediate clade-specific, serotype-independent mucosal immunity. Immunity. (2011) 35:997–1009. doi: 10.1016/j.immuni.2011.10.018
65. Daum RS, Spellberg B. Progress toward a Staphylococcus aureus vaccine. Clin Infect Dis. (2012) 54:560–7. doi: 10.1093/cid/cir828
66. van der Pol L, Stork M, van der Ley P. Outer membrane vesicles as platform vaccine technology. Biotechnol J. (2015) 10:1689–706. doi: 10.1002/biot.201400395
67. Chen W. Current advances and challenges in the development of Acinetobacter vaccines. Hum Vaccin Immunother. (2015) 11:2495–500. doi: 10.1080/21645515.2015.1052354
68. Perez F, Bonomo RA. Vaccines for Acinetobacter baumannii: thinking “out of the box”. Vaccine. (2014) 32:2537–9. doi: 10.1016/j.vaccine.2014.03.031
69. Pachón J, McConnell MJ. Considerations for the development of a prophylactic vaccine for Acinetobacter baumannii. Vaccine. (2014) 32:2534–6. doi: 10.1016/j.vaccine.2013.10.064
70. McConnell MJ, Rumbo C, Bou G, Pachón J. Outer membrane vesicles as an acellular vaccine against Acinetobacter baumannii. Vaccine. (2011) 29:5705–10. doi: 10.1016/j.vaccine.2011.06.001
71. Huang W, Yao Y, Long Q, Yang X, Sun W, Liu C, et al. Immunization against multidrug-resistant Acinetobacter baumannii effectively protects mice in both pneumonia and sepsis models. PLoS ONE. (2014) 9:e100727. doi: 10.1371/journal.pone.0100727
72. Huang W, Wang S, Yao Y, Xia Y, Yang X, Li K, et al. Employing Escherichia coli-derived outer membrane vesicles as an antigen delivery platform elicits protective immunity against Acinetobacter baumannii infection. Sci Rep. (2016) 6:37242. doi: 10.1038/srep37242
73. Pulido MR, García-Quintanilla M, Pachón J, McConnell MJ. Immunization with lipopolysaccharide-free outer membrane complexes protects against Acinetobacter baumannii infection. Vaccine. (2018) 36:4153–6. doi: 10.1016/j.vaccine.2018.05.113
74. Pulido MR, García-Quintanilla M, Pachón J, McConnell MJ. A lipopolysaccharide-free outer membrane vesicle vaccine protects against Acinetobacter baumannii infection. Vaccine. (2020) 38:719–24. doi: 10.1016/j.vaccine.2019.11.043
75. McConnell MJ, Domínguez-Herrera J, Smani Y, López-Rojas R, Docobo-Pérez F, Pachón J. Vaccination with outer membrane complexes elicits rapid protective immunity to multidrug-resistant Acinetobacter baumannii. Infect Immun. (2011) 79:518–26. doi: 10.1128/IAI.00741-10
76. Sheweita SA, Batah AM, Ghazy AA, Hussein A, Amara AA. A new strain of Acinetobacter baumannii and characterization of its ghost as a candidate vaccine. J Infect Public Health. (2019) 12:831–42. doi: 10.1016/j.jiph.2019.05.009
77. García-Quintanilla M, Pulido MR, Pachón J, McConnell MJ. Immunization with lipopolysaccharide-deficient whole cells provides protective immunity in an experimental mouse model of Acinetobacter baumannii infection. PLoS ONE. (2014) 9:e114410. doi: 10.1371/journal.pone.0114410
78. Yang A-Q, Yang H-Y, Guo S-J, Xie Y-E. MF59 adjuvant enhances the immunogenicity and protective immunity of the OmpK/Omp22 fusion protein from Acineterbacter baumannii through intratracheal inoculation in mice. Scand J Immunol. (2019) 90:e12769. doi: 10.1111/sji.12769
79. Gu H, Liu D, Zeng X, Peng L-S, Yuan Y, Chen Z-F, et al. Aging exacerbates mortality of Acinetobacter baumannii pneumonia and reduces the efficacies of antibiotics and vaccine. Aging. (2018) 10:1597–608. doi: 10.18632/aging.101495
80. Crooke SN, Ovsyannikova IG, Poland GA, Kennedy RB. Immunosenescence and human vaccine immune responses. Immun Ageing. (2019) 16:25. doi: 10.1186/s12979-019-0164-9
81. McElhaney JE, Kuchel GA, Zhou X, Swain SL, Haynes L. T-cell immunity to influenza in older adults: a pathophysiological framework for development of more effective vaccines. Front Immunol. (2016) 7:41. doi: 10.3389/fimmu.2016.00041
82. Ciabattini A, Nardini C, Santoro F, Garagnani P, Franceschi C, Medaglini D. Vaccination in the elderly: the challenge of immune changes with aging. Semin Immunol. (2018) 40:83–94. doi: 10.1016/j.smim.2018.10.010
83. Baay M, Bollaerts K, Verstraeten T. A systematic review and meta-analysis on the safety of newly adjuvanted vaccines among older adults. Vaccine. (2018) 36:4207–14. doi: 10.1016/j.vaccine.2018.06.004
84. Maragakis LL, Tucker MG, Miller RG, Carroll KC, Perl TM. Incidence and prevalence of multidrug-resistant acinetobacter using targeted active surveillance cultures. JAMA. (2008) 299:2513–4. doi: 10.1001/jama.299.21.2513
85. Roestenberg M, Hoogerwerf M-A, Ferreira DM, Mordmüller B, Yazdanbakhsh M. Experimental infection of human volunteers. Lancet Infect Dis. (2018) 18:e312–22. doi: 10.1016/S1473-3099(18)30177-4
86. Shirley D-AT, McArthur MA. The utility of human challenge studies in vaccine development: lessons learned from cholera. Vaccine. (2011) 2011:3–13. doi: 10.2147/VDT.S23634
87. Porter CK, Lynen A, Riddle MS, Talaat K, Sack D, Gutiérrez RL, et al. Clinical endpoints in the controlled human challenge model for Shigella: a call for standardization and the development of a disease severity score. PLoS ONE. (2018) 13:e0194325. doi: 10.1371/journal.pone.0194325
88. Hanevik K, Chen WH, Talaat KR, Porter C, Bourgeois L. The way forward for ETEC controlled human infection models (CHIMs). Vaccine. (2019) 37:4794–9. doi: 10.1016/j.vaccine.2019.01.003
89. Porter CK, Gutierrez RL, Kotloff KL. Clinical endpoints for efficacy studies. Vaccine. (2019) 37:4814–22. doi: 10.1016/j.vaccine.2019.03.051
90. Slingerland BC, Tavakol M, McCarthy AJ, Lindsay JA, Snijders SV, Wagenaar JA, et al. Survival of Staphylococcus aureus ST398 in the human nose after artificial inoculation. PLoS ONE. (2012) 7:e48896. doi: 10.1371/journal.pone.0048896
91. Villano SA, Seiberling M, Tatarowicz W, Monnot-Chase E, Gerding DN. Evaluation of an oral suspension of VP20621, spores of nontoxigenic Clostridium difficile strain M3, in healthy subjects. Antimicrob Agents Chemother. (2012) 56:5224–9. doi: 10.1128/AAC.00913-12
92. Gerding DN, Meyer T, Lee C, Cohen SH, Murthy UK, Poirier A, et al. Administration of spores of nontoxigenic Clostridium difficile strain M3 for prevention of recurrent C. difficile infection: a randomized clinical trial. JAMA. (2015) 313:1719–27. doi: 10.1001/jama.2015.3725
93. Sommer F, Anderson JM, Bharti R, Raes J, Rosenstiel P. The resilience of the intestinal microbiota influences health and disease. Nat Rev Microbiol. (2017) 15:630–8. doi: 10.1038/nrmicro.2017.58
94. Parker EP, Ramani S, Lopman BA, Church JA, Iturriza-Gómara M, Prendergast AJ, et al. Causes of impaired oral vaccine efficacy in developing countries. Fut Microbiol. (2018) 13:97–118. doi: 10.2217/fmb-2017-0128
95. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
96. Huda MN, Lewis Z, Kalanetra KM, Rashid M, Ahmad SM, Raqib R, et al. Stool microbiota and vaccine responses of infants. Pediatrics. (2014) 134:e362–72. doi: 10.1542/peds.2013-3937
97. Harris VC, Armah G, Fuentes S, Korpela KE, Parashar U, Victor JC, et al. Significant correlation between the infant gut microbiome and rotavirus vaccine response in rural ghana. J Infect Dis. (2017) 215:34–41. doi: 10.1093/infdis/jiw518
98. Pfeiffer JK, Virgin HW. Viral immunity. transkingdom control of viral infection and immunity in the mammalian intestine. Science. (2016) 351:eaad5872. doi: 10.1126/science.aad5872
99. Kuss SK, Best GT, Etheredge CA, Pruijssers AJ, Frierson JM, Hooper LV, et al. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science. (2011) 334:249–52. doi: 10.1126/science.1211057
100. Grassly NC, Praharaj I, Babji S, Kaliappan SP, Giri S, Venugopal S, et al. The effect of azithromycin on the immunogenicity of oral poliovirus vaccine: a double-blind randomised placebo-controlled trial in seronegative Indian infants. Lancet Infect Dis. (2016) 16:905–14. doi: 10.1016/S1473-3099(16)30023-8
101. Vlasova AN, Takanashi S, Miyazaki A, Rajashekara G, Saif LJ. How the gut microbiome regulates host immune responses to viral vaccines. Curr Opin Virol. (2019) 37:16–25. doi: 10.1016/j.coviro.2019.05.001
102. Harris VC, Haak BW, Handley SA, Jiang B, Velasquez DE, Hykes BL, et al. Effect of antibiotic-mediated microbiome modulation on rotavirus vaccine immunogenicity: a human, randomized-control proof-of-concept trial. Cell Host Microbe. (2018) 24:197–207.e4. doi: 10.1016/j.chom.2018.07.005
103. Hagan T, Cortese M, Rouphael N, Boudreau C, Linde C, Maddur MS, et al. Antibiotics-driven gut microbiome perturbation alters immunity to vaccines in humans. Cell. (2019) 178:1313–28.e13. doi: 10.1016/j.cell.2019.08.010
104. Zhao Q, Elson CO. Adaptive immune education by gut microbiota antigens. Immunology. (2018) 154:28–37. doi: 10.1111/imm.12896
105. Donaldson GP, Ladinsky MS, Yu KB, Sanders JG, Yoo BB, Chou W-C, et al. Gut microbiota utilize immunoglobulin A for mucosal colonization. Science. (2018) 360:795–800. doi: 10.1126/science.aaq0926
106. Schofield WB, Palm NW. Gut microbiota: IgA protects the pioneers. Curr Biol. (2018) 28:R1117–9. doi: 10.1016/j.cub.2018.08.019
107. Wilmore JR, Gaudette BT, Gomez Atria D, Hashemi T, Jones DD, Gardner CA, et al. Commensal microbes induce serum IgA responses that protect against polymicrobial sepsis. Cell Host Microbe. (2018) 23:302–11.e3. doi: 10.1016/j.chom.2018.01.005
108. Gupta S, Basu S, Bal V, Rath S, George A. Gut IgA abundance in adult life is a major determinant of resistance to dextran sodium sulfate-colitis and can compensate for the effects of inadequate maternal IgA received by neonates. Immunology. (2019) 158:19–34. doi: 10.1111/imm.13091
109. Robak OH, Heimesaat MM, Kruglov AA, Prepens S, Ninnemann J, Gutbier B, et al. Antibiotic treatment-induced secondary IgA deficiency enhances susceptibility to Pseudomonas aeruginosa pneumonia. J Clin Invest. (2018) 128:3535–45. doi: 10.1172/JCI97065
110. Chemouny JM, Gleeson PJ, Abbad L, Lauriero G, Boedec E, Le Roux K, et al. Modulation of the microbiota by oral antibiotics treats immunoglobulin a nephropathy in humanized mice. Nephrol Dial Transplant. (2019) 34:1135–44. doi: 10.1093/ndt/gfy323
111. Nicholson MR, Thomsen IP, Edwards KM. Controversies surrounding Clostridium difficile infection in infants and young children. Children. (2014) 1:40–7. doi: 10.3390/children1010040
112. Shin JH, High KP, Warren CA. Older is not wiser, immunologically speaking: effect of aging on host response to Clostridium difficile infections. J Gerontol A Biol Sci Med Sci. (2016) 71:916–22. doi: 10.1093/gerona/glv229
113. Biagi E, Candela M, Turroni S, Garagnani P, Franceschi C, Brigidi P. Ageing and gut microbes: perspectives for health maintenance and longevity. Pharmacol Res. (2013) 69:11–20. doi: 10.1016/j.phrs.2012.10.005
114. Kempf VA, Roeb E editors. Leitsymptom Diarrhö: Chapter 20: Immunprophylaxe zur Prävention von Durchfallerkrankungen. English: Immune Prophylaxis for Prevention of Diarrhea. Berlin; Boston, MA: De Gruyter (2019).
115. Rees WD, Steiner TS. Adaptive immune response to Clostridium difficile infection: a perspective for prevention and therapy. Eur J Immunol. (2018) 48:398–406. doi: 10.1002/eji.201747295
116. Kyne L, Warny M, Qamar A, Kelly CP. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N Engl J Med. (2000) 342:390–7. doi: 10.1056/NEJM200002103420604
117. Kelly CP. Immune response to Clostridium difficile infection. Eur J Gastroenterol Hepatol. (1996) 8:1048–53. doi: 10.1097/00042737-199611000-00004
118. Warny M, Vaerman JP, Avesani V, Delmée M. Human antibody response to Clostridium difficile toxin A in relation to clinical course of infection. Infect Immun. (1994) 62:384–9. doi: 10.1128/IAI.62.2.384-389.1994
119. Schmoldt A, Benthe HF, Haberland G. Digitoxin metabolism by rat liver microsomes. Biochem Pharmacol. (1975) 24:1639–41. doi: 10.1016/0006-2952(75)90094-5
120. Oksi J, Aalto A, Säilä P, Partanen T, Anttila V-J, Mattila E. Real-world efficacy of bezlotoxumab for prevention of recurrent Clostridium difficile infection: a retrospective study of 46 patients in five university hospitals in Finland. Eur J Clin Microbiol Infect Dis. (2019) 38:1947–52. doi: 10.1007/s10096-019-03630-y
121. Collini PJ, Bauer M, Kuijper E, Dockrell DH. Clostridium difficile infection in HIV-seropositive individuals and transplant recipients. J Infect. (2012) 64:131–47. doi: 10.1016/j.jinf.2011.12.003
122. Madan R, Guo X, Naylor C, Buonomo EL, Mackay D, Noor Z, et al. Role of leptin-mediated colonic inflammation in defense against Clostridium difficile colitis. Infect Immun. (2014) 82:341–9. doi: 10.1128/IAI.00972-13
123. Yu H, Chen K, Sun Y, Carter M, Garey KW, Savidge TC, et al. Cytokines are markers of the Clostridium difficile-induced inflammatory response and predict disease severity. Clin Vaccine Immunol. (2017) 24:e00037-17. doi: 10.1128/CVI.00037-17
124. Buonomo EL, Madan R, Pramoonjago P, Li L, Okusa MD, Petri WA. Role of interleukin 23 signaling in Clostridium difficile colitis. J Infect Dis. (2013) 208:917–20. doi: 10.1093/infdis/jit277
125. Darkoh C, Turnwald BP, Koo HL, Garey KW, Jiang Z-D, Aitken SL, et al. Colonic immunopathogenesis of Clostridium difficile infections. Clin Vaccine Immunol. (2014) 21:509–17. doi: 10.1128/CVI.00770-13
126. Yacyshyn MB, Reddy TN, Plageman LR, Wu J, Hollar AR, Yacyshyn BR. Clostridium difficile recurrence is characterized by pro-inflammatory peripheral blood mononuclear cell (PBMC) phenotype. J Med Microbiol. (2014) 63:1260–73. doi: 10.1099/jmm.0.075382-0
127. Buonomo EL, Petri WA. The microbiota and immune response during Clostridium difficile infection. Anaerobe. (2016) 41:79–84. doi: 10.1016/j.anaerobe.2016.05.009
128. Pandiyan P, Bhaskaran N, Zou M, Schneider E, Jayaraman S, Huehn J. Microbiome dependent regulation of tregs and Th17 cells in mucosa. Front Immunol. (2019) 10:426. doi: 10.3389/fimmu.2019.00426
129. Leslie JL, Vendrov KC, Jenior ML, Young VB. The gut microbiota is associated with clearance of Clostridium difficile infection independent of adaptive immunity. mSphere. (2019) 4:e00698-18. doi: 10.1128/mSphereDirect.00698-18
130. Merrigan MM, Sambol SP, Johnson S, Gerding DN. New approach to the management of Clostridium difficile infection: colonisation with non-toxigenic C. difficile during daily ampicillin or ceftriaxone administration. Int J Antimicrob Agents. (2009) 33:S46–50. doi: 10.1016/S0924-8579(09)70017-2
131. Khoruts A, Sadowsky MJ. Understanding the mechanisms of faecal microbiota transplantation. Nat Rev Gastroenterol Hepatol. (2016) 13:508–16. doi: 10.1038/nrgastro.2016.98
132. Mullish BH, McDonald JA, Pechlivanis A, Allegretti JR, Kao D, Barker GF, et al. Microbial bile salt hydrolases mediate the efficacy of faecal microbiota transplant in the treatment of recurrent Clostridioides difficile infection. Gut. (2019) 68:1791–800. doi: 10.1136/gutjnl-2018-317842
133. Brown JR-M, Flemer B, Joyce SA, Zulquernain A, Sheehan D, Shanahan F, et al. Changes in microbiota composition, bile and fatty acid metabolism, in successful faecal microbiota transplantation for Clostridioides difficile infection. BMC Gastroenterol. (2018) 18:131. doi: 10.1186/s12876-018-0860-5
134. Ott SJ, Waetzig GH, Rehman A, Moltzau-Anderson J, Bharti R, Grasis JA, et al. Efficacy of sterile fecal filtrate transfer for treating patients with Clostridium difficile infection. Gastroenterology. (2017) 152:799–811.e7. doi: 10.1053/j.gastro.2016.11.010
135. Salzer HJ, Wassilew N, Köhler N, Olaru ID, Günther G, Herzmann C, et al. Personalized medicine for chronic respiratory infectious diseases: tuberculosis, nontuberculous mycobacterial pulmonary diseases, and chronic pulmonary aspergillosis. Respiration. (2016) 92:199–214. doi: 10.1159/000449037
136. Moser C, Lerche CJ, Thomsen K, Hartvig T, Schierbeck J, Jensen PØ, et al. Antibiotic therapy as personalized medicine - general considerations and complicating factors. APMIS. (2019) 127:361–71. doi: 10.1111/apm.12951
Keywords: nosocomial infection, vaccine, bacterial, colonization, immune, antibiotic resistance
Citation: Bekeredjian-Ding I (2020) Challenges for Clinical Development of Vaccines for Prevention of Hospital-Acquired Bacterial Infections. Front. Immunol. 11:1755. doi: 10.3389/fimmu.2020.01755
Received: 09 February 2020; Accepted: 30 June 2020;
Published: 05 August 2020.
Edited by:
Winfried F. Pickl, Medical University of Vienna, AustriaReviewed by:
Eliana Marina Coccia, Istituto Superiore di Sanità (ISS), ItalyArun Kumar, Coalition for Epidemic Preparedness Innovations (CEPI), Norway
Copyright © 2020 Bekeredjian-Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabelle Bekeredjian-Ding, aXNhYmVsbGUuYmVrZXJlZGppYW4tZGluZ0BwZWkuZGU=
†IB-D is a member of the German Center for Infectious Disease Research (DZIF) and the IMI2 JU COMBINE (The Collaboration for Prevention and Treatment of Multi-Drug Resistant Bacterial Infection) consortium