 Zhenxing Cheng1,2
Zhenxing Cheng1,2 Simon T. Abrams1
Simon T. Abrams1 Julien Toh3Susan Siyu Wang4Zhi Wang2Qian Yu2Weiping Yu2*Cheng-Hock Toh1,5*
Julien Toh3Susan Siyu Wang4Zhi Wang2Qian Yu2Weiping Yu2*Cheng-Hock Toh1,5* Guozheng Wang1,2*
Guozheng Wang1,2*- 1Department of Clinical Infection, Microbiology and Immunology, Institute of Infection and Global Health, University of Liverpool, Liverpool, United Kingdom
- 2Medical School, Southeast University, Nanjing, China
- 3Wirral University Teaching Hospitals NHS Foundation Trust, Wirral, United Kingdom
- 4Royal London Hospital, London, United Kingdom
- 5Liverpool University Hospitals NHS Foundation Trust, Liverpool, United Kingdom
Sepsis was first described by the ancient Greek physicians over 2000 years ago. The pathophysiology of the disease, however, is still not fully understood and hence the mortality rate is still unacceptably high due to lack of specific therapies. In the last decade, great progress has been made by shifting the focus of research from systemic inflammatory response syndrome (SIRS) to multiple organ dysfunction syndrome (MODS). Sepsis has been re-defined as infection-induced MODS in 2016. How infection leads to MODS is not clear, but what mediates MODS becomes the major topic in understanding the molecular mechanisms and developing specific therapies. Recently, the mechanism of infection-induced extensive immune cell death which releases a large quantity of damage-associated molecular patterns (DAMPs) and their roles in the development of MODS as well as immunosuppression during sepsis have attracted much attention. Growing evidence supports the hypothesis that DAMPs, including high-mobility group box 1 protein (HMGB1), cell-free DNA (cfDNA) and histones as well as neutrophil extracellular traps (NETs), may directly or indirectly contribute significantly to the development of MODS. Here, we provide an overview of the mechanisms and consequences of infection-induced extensive immune cell death during the development of sepsis. We also propose a pivotal pathway from a local infection to eventual sepsis and a potential combined therapeutic strategy for targeting sepsis.
Sepsis
Sepsis is still the leading cause of death in most intensive care units (ICU) with an unacceptably high mortality rate (10–20%), although there has been a significant decrease in mortality rates in recent decades (from 1994 to 2014) (1, 2). Center for Disease Control in the United States estimated that over half a million people developed sepsis there per year with about a 1.5% increase per annum (3). A recent investigation of a cohort of 568 patients who died in six hospitals in the United States showed that sepsis presented in 300 patients (52.8%) and was the most common immediate cause of death in 198 patients (34.9%), indicating that sepsis is still the major cause of death in hospitals (4). For years, it was believed that high morbidity and mortality were due to systemic inflammatory response syndrome (SIRS), but many clinical trials to inhibit inflammation failed to improve survival (5–7). In 2016, sepsis has been redefined as multiple organ dysfunction syndrome (MODS) caused by a dysregulated host response to infection (8) and is now termed Sepsis-3. This has changed the focus from SIRS (9, 10) to MODS. Thus, finding what mediates MODS is now the major challenge in understanding the pathophysiology of sepsis (11).
Discovery of Immune Cell Death in Sepsis
Cheadle et al. (12) reported that a significant lymphopenia occurred in a group of trauma patients with sepsis. Years later, lymphopenia in sepsis began to attract increased attention (13–16). In human, depletion of both B cells and CD4+ T lymphocytes caused by sepsis-induced apoptosis were reported (16). In baboon and murine sepsis models, extensive apoptosis of lymphoid tissue was also found (17–19). Rapidly progressing lymphocyte exhaustion after severe sepsis has been widely recognized (20) and early circulating lymphocyte apoptosis was associated with poor outcome in patients with sepsis (21, 22). Thus, a number of research groups have focused on the role of altered cell death in contributing to MODS in sepsis and clinical trials for a new type of therapy has emerged (23–26).
Types of Immune Cell Death and Clinical Relevance
Lymphocyte death occurs in the spleen, thymus, and lymphoid tissues (27). The peripheral lymphocyte count is also dramatically reduced in both sepsis models and patients (16, 22, 24). Changes in the subsets of lymphocyte involved varies depending on the bacterial origin of sepsis (28), but there is no doubt that both T and B lymphocyte subsets are significantly changed by sepsis. CD3+, CD3+CD4+, and CD3+CD8+ lymphocyte counts drop significantly in septic patients, while CD3+CD4+ lymphocytes return to normal after 14 days in most patient survivors, but this is not true of the CD3+CD8+ counts (29). The ratio of Th1/Th2 helper cells has been found to be significantly lower in sepsis (30). Circulatory Th1, Th2, Th17, and Treg as well as Th1/CD4 + ratios are significantly lower in non-survivors compared to survivors (31). The αβ and γδ T cell subsets are all reduced in sepsis, but the CD3+ CD56+ γδ T cells show the largest decrease, and their loss is strongly associated with septic severity and mortality (32, 33). Sepsis causes progressive and profound depletion of B lymphocytes in patients (16). Thus, the percentage of CD19+CD23+ was significantly lower in patients who died of septic shock than in survivors (34). In a mouse poly-microbial sepsis model, substantial apoptosis of lamina propria B cells mediated by FasL has been reported (35).
Not only are B and T lymphocytes susceptible to programmed cell death, many other types of immune cells including neutrophils, macrophages and dendritic cells are also vulnerable to cell death in sepsis (22, 36, 37). Neutrophils are the first line of defense against invading bacteria. Neutrophils phagocytose bacteria or form neutrophil extracellular traps (NETs), and both these mechanisms are critical for clearance of invading bacteria (38). After taking up bacteria, neutrophils undergo a respiratory burst and die (39). NETs formation is also a novel program for cell death (40–42). Therefore, large numbers of neutrophils die during sepsis. In mouse models, apoptosis of mouse peritoneal macrophages may be due to the release of HMGB1 in sepsis (43). Dendritic cells have unique capabilities to regulate the activity and survival of T and B cells. Thus splenic interdigitating dendritic cells (IDCs) and follicular dendritic cells (FDCs) initially expand in sepsis. The FDCs expand to fill the entire lymphoid zone of spleen, which is otherwise occupied by B cells (44). Twelve hours after the onset, these dendritic cells undergo apoptosis (44). In contrast, natural killer (NK) cell counts increase in early sepsis and higher levels predict mortality in severe sepsis (45). Thus, the ratio of NK cells to CD4+ lymphocytes was used to predict the mortality of patients with sepsis (46). NK cells also contribute to the lethality of a murine model of sepsis, and NK cell-depleted and NK cell-deficient mice showed much high survival rates than wild type controls (47).
Mechanisms of Immune Cell Death
Apoptosis is the major mechanism of lymphocyte death in sepsis (35, 48). Both the death receptor and mitochondrial pathways activated by multiple triggers are involved in apoptosis of a broad range of subsets of lymphocyte (49). Apoptosis could occur via p53-dependent and -independent pathways (50). The increase in apoptosis in the thymus, spleen, lungs, and gut during poly-microbial sepsis of mice is mediated by FasL via death receptors, but not by endotoxins nor TNF-α (14, 35). Monocytes can induce Fas-mediated apoptosis of T lymphocytes (51). Caspase-1 is involved in apoptosis of splenic B lymphocytes (52). Activation of caspase-3 and externalization of phosphatidylserine in CD4+, CD8+, and CD19+ lymphocytes were reported in patients with sepsis (53). Activation of programmed cell death ligand 1 (PD-L1) pathway is involved in T cell exhaustion in patients with sepsis (54). In addition, endoplasmic reticulum (ER) stress can mediate lymphocyte apoptosis in sepsis (55). Bcl-2 is an anti-apoptosis protein and is found to be reduced in sepsis (56). Overexpression of Bcl-2 in septic mice provides protection by decreasing lymphocyte apoptosis (57, 58). In CD4+ T and B lymphocytes isolated from septic patients, the Bcl-2 protein was decreased but the expression of pro-apoptotic proteins Bim, Bid, and Bak were massively upregulated (23, 53). It has also been reported that overexpression of histamine H4 receptors counteracts the effect of NF-kB in contributing to splenic cell apoptosis in sepsis (59).
There is no doubt that multiple factors are involved in lymphocyte apoptosis, but the detailed molecular mechanisms are still not fully understood. In addition, apoptosis has been the major focus of cell death in last two decades, but recently other processes have emerged, e.g., pyroptosis, necroptosis, ferroptosis, parthanatos, entotic cell death, NETotic cell death, immunogenic cell death, and mitotic catastrophe, to explain the complexity of cell death (60). Pyroptosis is caused by rapid plasma-membrane rupture by non-selective gasdermin-D pore and releases of DAMPs (61). Neutrophil and endothelial cell pyroptosis has been considered as a major pathological factor in sepsis (62, 63). Increased membrane permeabilization in necroptosis releases specific DAMPs, and lipid peroxidation in ferroptosis may be involved in renal failure (64–66). These regulated cell deaths may turn to necrosis if their resolution is delayed (67). The roles and mechanisms of different types of cell death in sepsis is far from clear and more work need to be done to understand how the immune cells die so extensively in sepsis.
Neutrophil respiratory burst and NETosis all involve generation of reactive oxygen species (ROS) and NADPH oxidase pays a critical role (40). Endotoxin reduced CD95-mediated neutrophil apoptosis occurs via cIAP-2 activation and the degradation of caspase-3 (68). The detailed molecular mechanisms of neutrophil respiratory burst, NETosis, and homeostasis will not be discussed in this review.
In summary, the types of cell death and underlying molecular mechanisms are still not fully understood, although the subpopulations of immune cells that die during sepsis is almost clarified.
Roles and Consequence of Immune Cell Death
It is known that the extent of immune cell death is strongly associated with severity and mortality of sepsis. However, the biological roles are still not clear. The direct cause-effect relationship of extensive immune cell death with sepsis has not yet been proven. When splenectomy to remove the largest lymph organ in mice prior to septic modeling was undertaken, it is found that this procedure protects mice against secondary sepsis (69, 70). This observation suggests that extensive splenocyte death is potentially pathogenic in sepsis. Neutrophil death, particularly NETosis, has been reported to be involved in the development of multiple organ failure in sepsis (71–73). Abrams et al. (74) recently showed that strong NETs formation mainly occurs in severe sepsis and is associated with disseminated intravascular coagulation (DIC) and ultimately poor outcomes. Patel et al. (75) recently showed that a reduction in ex vivo PMA-induced NETosis of neutrophils isolated from patients with severe sepsis is associated with poorer outcomes. This observation demonstrates the pathological role of in vivo NETs formation, a mechanism that eliminates the majority of pro-NETosis neutrophils. This result is also consistent with the current general consensus (72, 74). However, the pathological role of immune cell death in sepsis is still not fully understood, but the following mechanisms are widely considered to be very important.
DAMPs and Histone Release
The “danger” theory was proposed by Matzinger in 1994 (76) that damaged cells initiate immune responses by releasing substances were termed damage-associated molecular patterns (DAMPs) by Walter Land in 2003 (77). DAMPs represent danger-associated or damage-associated molecular patterns, which are released from the cell through activation of inflammasome or passively following cell death (78–80) and recognized by pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) NOD-like receptors, DNA sensors, C-type lectin receptor, and non-PRR DAMP receptors, including RAGE receptor (81). Many DAMPs that origin from extracellular matrix and different components or organelles of cells have been identified, such as histones, DNAs, HMGB1, heat shock protein, and ATP. More information can be found in a recent review (82). In sepsis, a large number of immune cell death releases a large quantity of DAMPs (83, 84). Similarly, NETs are released from neutrophils during inflammation (41). These NETs are brokendown into free DNA and histones and become a source of DAMPs (72, 85). DAMPs trigger the host's immune response, activate coagulation and mediate MODS (86–88). Therefore, they play a central role in development of sepsis and its progression (84, 89). DAMPs include a large group of molecules and are involved in different pathological processes during sepsis.
Release of chromatin protein HMGB1 triggers inflammation and mediates endotoxin lethality in mice (90, 91). HMGB1 facilitates LPS entering cells to trigger pyroptosis, which plays an important role in sepsis (63, 92, 93). In 2009, extracellular histones were shown to be major mediators of death in sepsis (94) and have attracted more and more attention. Extracellular histones bind to the cell membrane and form pores to allow calcium influx which leads to calcium overload, which directly damages cells that contacted (87, 88). Histones also induce rapid thrombocytopenia, increase thrombin generation and contribute to DIC (95–99). Anti-histone antibodies and non-anticoagulant heparin neutralize extracellular histones and improves survival in sepsis (87, 88, 99–101). Recently, the role of extracellular histones in the development of MODS in critical illnesses and animal models, including sepsis, pancreatitis, and trauma, has been demonstrated (86). Mitochondrial DNA released into the cytosol or outside cells also serves as DAMPs and play important roles in sepsis (11, 102). In addition, circulating cell-free DNA is associated with poor outcomes in patients with severe sepsis (103–106). The pathological roles of these cell-free DNAs are not clear but strengthening blood clots resistant to fibrin lysis may facilitate DIC development (107). A recent report shows increased S100 proteins, including A8/A9 and A12, which are types of DAMPs, are associated with a higher risk of death in patients with sepsis (108).
NETs Formation
Although NETs are an important source of DAMPs, NETs formation has specific roles in thrombosis, DIC and microcirculatory impairment. NETs formation induces organ injury and exacerbates the severity of sepsis (42, 73, 74, 109–112). Suppression of NETosis using PAD4 inhibitors or cleavage of NETs using DNase 1 improves survival in a murine sepsis model (113), but other reports showed the opposite effect (114, 115). Recently it has been reported that delayed, not early treatment with DNase 1 reduces organ injury and improves outcome in sepsis model (116). These observations strongly indicate the complex roles of NETs formation during sepsis.
Coagulopathy and DIC
Sepsis-induced coagulopathy and DIC play a major role in microcirculatory impairment and MODS development (117). DAMPs play important roles in septic coagulopathy (118). Extracellular histones are the most important DAMPs that promote coagulation activation by inducing rapid thrombocytopenia, enhancing thrombin generation, impairing thrombomodulin-dependent protein C activation, damaging endothelial cells and increasing tissue factor activity (95–99). cfDNA exert both pro- and anti-fibrinolytic effects and NETs serve as scaffolds for immunothrombosis and promote intracellular coagulation together with platelets (107, 119, 120). The overall consequence is the development of coagulopathy and DIC, which significantly enhance disease severity and worsen the outcomes (74, 86, 99, 104, 105).
Immune Suppression
As our understanding of the pathophysiology in sepsis has improved, we now know that the role of immunosuppression is more important than previously thought. IL-7, as an immune-adjuvant therapy that increased absolute lymphocyte counts and in circulating CD4+ and CD8+ T cells (3–4 fold), and T cell proliferation and activation (121), supports this contention. However, why IL-7 protected mice with sepsis but showed no effects on 28-days survival of patients with sepsis is not clear and further investigation is required (122). IL-15 is also reported to prevent apoptosis, reverse innate and adaptive immune dysfunction, and improve survival in murine models of sepsis (123). Changes associated with immunosuppression is more obvious in patients who died of sepsis than those who survived (31, 124). Immune cell death, particularly T and B lymphocytic apoptosis, is a major contributor to the development of immunosuppression (15, 32, 125), besides the usual anti-inflammatory cytokine release, such as that of IL-10 (126). Myeloid-derived suppressor cells (MDSCs) are closely related to neutrophils and monocytes. They are immature myeloid cells that have immunosuppressive functions and play important roles in the development of immunosuppression in sepsis (2, 127–129). DAMPs activate TLR-4 to enhance MDSCs accumulation (130). Many DAMPs possess both pro- and anti-inflammatory properties to induce both immune response and immunosuppression, which has been well-studied in trauma (131). Recently, the roles of PD-1 and PD-L1 in sepsis as key mediators of T-cell exhaustion in infections have been investigated (132, 133). Blocking PD-1 or PD-L1 inhibits lymphocytic apoptosis, reverses monocyte and immune dysfunction, and improves survival during sepsis (54, 134–136). Monneret et al. (137) demonstrated that after septic shock anti-inflammatory response became dominate with high IL-10 and low HLA-DR on monocytes, a surrogate marker of monocyte non-responsiveness (138). IL-7 and anti-PD-1 or blocking IL-10 reverse sepsis-induced immunosuppression, including increasing HLA-DR expression and IFN-γ production, and improve survival in mouse models (126, 139). Monitoring HLA-DR, PD-1, or PD-L1 may guide clinical immunotherapies (140). All available evidences showed no doubt that immunosuppression is the major pathological feature and immunotherapies will become a critical management in severe sepsis with poor outcomes.
In summary, the major consequence of immune cell death is the DAMPs release and NETs formation, both of which contribute to the development of coagulopathy and MODS. Another major consequence is immunosuppression. All these consequences are the major pathological changes during severe sepsis, strongly indicating that DAMPs and NETs are critical in the development of severe sepsis.
Inhibition of Immune Cell Death in Sepsis and Potential Downstream Therapy
Caspase inhibitors, which inhibit apoptosis and improve the survival of immune cells, have been demonstrated to improve survival in sepsis. Thus, caspase-3−/− mice have decreased levels of apoptosis (141, 142). Increasing anti-apoptotic proteins, such as Bcl-2, or decreasing pro-apoptotic proteins, such as Bim, reduces immune cell apoptosis and improves survival in septic animal models (57, 58, 142–145). The Protease Inhibitor (PI) class of antiretroviral agents also significantly improved survival of mouse septic models by reducing lymphocyte apoptosis (146). These anti-apoptosis therapies have been demonstrated in animal models (147), but there have been no successful clinical trials in humans as yet.
Therapies with immune modulators have attracted more attention in recent years. The success of the IL-7 clinical trial shed some light on the management of sepsis (121). Immunotherapy is potentially a major strategy (145, 148, 149), but the focus of research has shifted from simply suppressing the immune response to immune modulation and precision medicine based on immune status (148, 150–153). Targeting immune cell checkpoints during sepsis is also a potential therapeutic strategy (154).
Another promising strategy is to neutralize DAMPs, including histones, DNAs and HMGB1. Anti-histone therapy has been proposed by Xu et al. from 2009 (94). Anti-histone antibodies or heparin can neutralize extracellular histones and reduce their toxicity so as to increase survival rates in septic animal models, but no clinical trial has been reported yet (86–88, 94, 100). Normal heparin has anticoagulant activity which may cause side effects if it is used at a wrong time with high doses. Non-anticoagulant heparin has been developed and hold the promise for future clinical application (100, 155). DNase 1, used to digest free DNA or NETs, has also been shown to increase the survival rate of septic animal models (116, 156). Many reagents targeting HMGB1, its release or downstream pathways have been reported, but no drug has yet been fully developed for clinical management of sepsis (157, 158).
Correction of downstream events, such as coagulopathy, have been trialed. Activated protein C, an anti-coagulant enzyme, was used clinically for a few years, but was withdrawn from the market due to failure in randomized controlled trials (159). It is very difficult to justify the correct time to use anti-coagulants and fibrinolysis reagents, such as low-molecular-weight heparin, antithrombin, thrombomodulin, and tissue factor inhibitors (117). Therefore, anti-coagulant therapy for sepsis is difficult to use clinically. Developing therapies to target upstream events appears a better strategy.
Clinical Perspective
Sepsis was first described by the ancient Greek physicians. Despite millennia of experience with this illness, we are still investigating the nature of sepsis. In the last decade, great progress has been made by shifting the focus of research from SIRS to MODS. However, the pathophysiology of sepsis is still not fully understood, particularly the roles of extensive immune cell death and DAMPs. Many types of DAMPs could directly or indirectly mediate MODS by their cytotoxicity or by triggering inflammation and activating coagulation, respectively. Therefore, the axis of infection, immune response, immune cell death, DAMPs release and MODS could be the central pathological pathway in the transition of a local infection to sepsis (Figure 1).
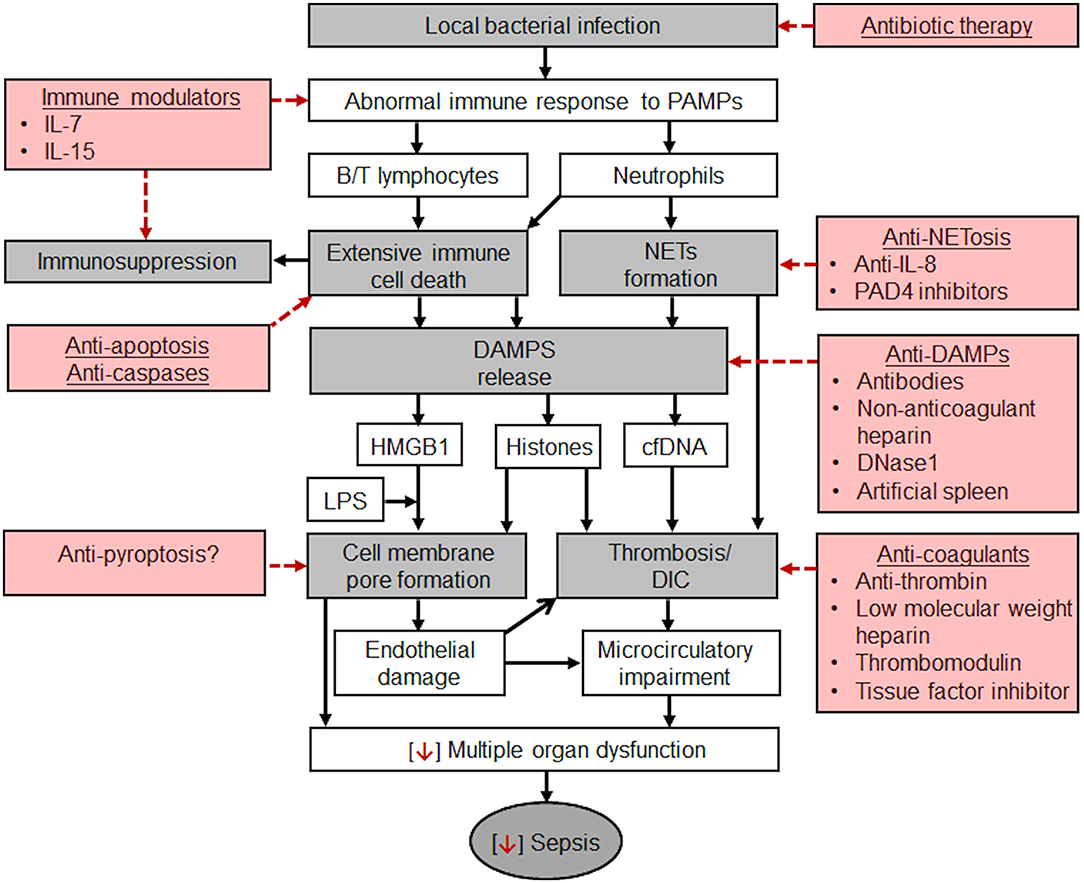
Figure 1. Potential pathological mechanisms of sepsis which develops from a local bacterial infection and potential therapeutic strategies. Gray boxes: pathways from a local infection to sepsis. Once a local bacterial infection causes host abnormal immune responses to pathogen-associated molecular pattern (PAMPs), extensive immune cell death, including B/T lymphocytes (spleen, thymus, lymphoid tissues, and peripheral blood), and neutrophils could occur and result in immunosuppression. Neutrophils could also form NETs. NETs and immune cell death could release a large quantity of DAMPs, mainly HMGB1, cfDNA, and histones. HMGB1 can delivers LPS into cells to trigger pyroptosis by forming pores in the cell membrane. Extracellular histones could also bind to cell membrane to form pores which may cause calcium overload and subsequently endothelial damage and organ injury. Indirectly, extracellular histones activate coagulation to form thrombi in the microvascular lumen to impair microcirculation. cfDNA could serve as scaffolds for thrombosis or stabilize thrombi by increasing their resistance to fibrinolysis. Microcirculatory impairment is the major feature of sepsis and a major contributor to MODS. Red boxes: Potential therapeutic strategies. Besides early diagnosis, prompt use of effective antibiotics, and supportive therapies, such as maintaining blood pressure and circulation, improving microcirculation, and protecting individual organs, the potential specific therapies include the combination of modulating immune status, preventing immune cell death and NETosis, neutralizing or clearing DAMPs. These new approaches could become the leading research directions in reducing the mortality rate of sepsis.
Targeting this central pathological pathway is already underway. However, fully understand the pathophysiology of sepsis is still the first task toward the success in clinical management.
Discussion
There is no doubt that extensive immune cell death is a major driver of sepsis. This mainly involves T and B lymphocyte apoptosis in the spleen, thymus, lymphoid tissues, and circulation. Neutrophil apoptosis, respiratory burst, and NETosis are also involved in this event. Macrophages and dendritic cells may also be involved, but their contributions may be negligible. However, the mechanism of how bacterial infection leads to extensive immune cell death is still not fully understood. Moreover, significant gaps still exist in our understanding of how extensive immune cell death proceeds to the development of sepsis. The obvious consequence of immune cell death would be immunosuppression but no direct link has been demonstrated. It is clear that the release of large quantities of DAMPs can enhance inflammation, directly damage endothelial cells, impair microcirculation and cause multiple organ injury, but to what extent these DAMPs contribute to the development of sepsis is still unclear. Some DAMPs, such as histones and NETs, strongly activate coagulation and eventually lead to DIC. Therefore, the importance of DAMPs in sepsis development and progression cannot be underestimated. In the future, targeting the axis of immune cell death-DAMPs release-and microcirculatory impairment, will become themost comprehensive strategy to reduce the unacceptably high mortality rate of sepsis.
Author Contributions
ZC, ZW, and QY wrote the first draft. JT and SW edited the reference. SA drew the diagram. WY, C-HT, and GW supervised the work and edited final version of paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by British Heart Foundation (PG/14/19/30751 and PG/16/65/32313), and Scholarships from China Scholarship Council (CSC:20160609156) and Southeast University (YBJJ1740) to ZC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, et al. International forum of acute care. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med. (2016) 193:259–72. doi: 10.1164/rccm.201504-0781OC
2. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
3. Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. (2001) 29:1303–10. doi: 10.1097/00003246-200107000-00002
4. Rhee C, Jones TM, Hamad Y, Pande A, Varon J, O'Brien C, et al. Prevalence, underlying causes, and preventability of sepsis-associated mortality in US Acute Care Hospitals. JAMA Netw Open. (2019) 2:e187571. doi: 10.1001/jamanetworkopen.2018.7571
5. Hunt A. Sepsis: an overview of the signs, symptoms, diagnosis, treatment and pathophysiology. Emerg Nurse. (2019) 27:32–41. doi: 10.7748/en.2019.e1926
6. Balk RA. Systemic inflammatory response syndrome (SIRS): where did it come from and is it still relevant today? Virulence. (2014) 5:20–6. doi: 10.4161/viru.27135
7. Marik PE, Taeb AM. SIRS, qSOFA and new sepsis definition. J Thorac Dis. (2017) 9:943–45. doi: 10.21037/jtd.2017.03.125
8. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
9. Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. (1992) 101:1644–55. doi: 10.1378/chest.101.6.1644
10. American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. (1992) 20:864–74. doi: 10.1097/00003246-199206000-00025
11. Harrington JS, Choi AMK, Nakahira K. Mitochondrial DNA in Sepsis. Curr Opin Crit Care. (2017) 23:284–90. doi: 10.1097/MCC.0000000000000427
12. Cheadle WG, Pemberton RM, Robinson D, Livingston DH, Rodriguez JL, Polk HC Jr. Lymphocyte subset responses to trauma and sepsis. J Trauma. (1993) 35:844–9. doi: 10.1097/00005373-199312000-00007
13. Rajan G, Sleigh JW. Lymphocyte counts and the development of nosocomial sepsis. Intensive Care Med. (1997) 23:1187. doi: 10.1007/s001340050482
14. Hiramatsu M, Hotchkiss RS, Karl IE, Buchman TG. Cecal ligation and puncture (CLP) induces apoptosis in thymus, spleen, lung, and gut by an endotoxin and TNF-independent pathway. Shock. (1997) 7:247–53. doi: 10.1097/00024382-199704000-00002
15. Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. (1999) 27:1230–51. doi: 10.1097/00003246-199907000-00002
16. Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE Jr, Hui JJ, Chang KC, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol. (2001) 166:6952–63. doi: 10.4049/jimmunol.166.11.6952
17. Efron PA, Tinsley K, Minnich DJ, Monterroso V, Wagner J, Lainee P, et al. Increased lymphoid tissue apoptosis in baboons with bacteremic shock. Shock. (2004) 21:566–71. doi: 10.1097/01.shk.0000126648.58732.8c
18. Boomer JS, Shuherk-Shaffer J, Hotchkiss RS, Green JM. A prospective analysis of lymphocyte phenotype and function over the course of acute sepsis. Crit Care. (2012) 16:R112. doi: 10.1186/cc11404
19. Lang JD, Matute-Bello G. Lymphocytes, apoptosis and sepsis: making the jump from mice to humans. Crit Care. (2009) 13:109. doi: 10.1186/cc7144
20. Monneret G, Venet F. A rapidly progressing lymphocyte exhaustion after severe sepsis. Crit Care. (2012) 16:140. doi: 10.1186/cc11416
21. Le Tulzo Y., Pangault C, Gacouin A, Guilloux V, Tribut O, Amiot L, et al. Early circulating lymphocyte apoptosis in human septic shock is associated with poor outcome. Shock. (2002) 18:487–94. doi: 10.1097/00024382-200212000-00001
22. Roth G, Moser B, Krenn C, Brunner M, Haisjackl M, Almer G, et al. Susceptibility to programmed cell death in T-lymphocytes from septic patients: a mechanism for lymphopenia and Th2 predominance. Biochem Biophys Res Commun. (2003) 308:840–6. doi: 10.1016/S0006-291X(03)01482-7
23. Wesche DE, Lomas-Neira JL, Perl M, Chung CS, Ayala A. Leukocyte apoptosis and its significance in sepsis and shock. J Leukoc Biol. (2005) 78:325–37. doi: 10.1189/jlb.0105017
24. de Pablo R, Monserrat J, Prieto A, Alvarez-Mon M. Role of circulating lymphocytes in patients with sepsis. Biomed Res Int. (2014) 2014:671087. doi: 10.1155/2014/671087
25. Ammer-Herrmenau C, Kulkarni U, Andreas N, Ungelenk M, Ravens S, Hubner C, et al. Sepsis induces long-lasting impairments in CD4+ T-cell responses despite rapid numerical recovery of T-lymphocyte populations. PLoS One. (2019) 14:e0211716. doi: 10.1371/journal.pone.0211716
26. Hotchkiss RS, Colston E, Yende S, Angus DC, Moldawer LL, Crouser ED, et al. Immune checkpoint inhibition in sepsis: a phase 1b randomized, placebo-controlled, single ascending dose study of antiprogrammed cell death-ligand 1 antibody (BMS-936559). Crit Care Med. (2019) 47:632–42. doi: 10.1097/CCM.0000000000003685
27. Ayala A, Herdon CD, Lehman DL, DeMaso CM, Ayala CA, Chaudry IH. The induction of accelerated thymic programmed cell death during polymicrobial sepsis: control by corticosteroids but not tumor necrosis factor. Shock. (1995) 3:259–67. doi: 10.1097/00024382-199504000-00003
28. Holub M, Kluckova Z, Helcl M, Prihodov J, Rokyta R, Beran O. Lymphocyte subset numbers depend on the bacterial origin of sepsis. Clin Microbiol Infect. (2003) 9:202–11. doi: 10.1046/j.1469-0691.2003.00518.x
29. Monserrat J, de Pablo R, Reyes E, Diaz D, Barcenilla H, Zapata MR, et al. Clinical relevance of the severe abnormalities of the T cell compartment in septic shock patients. Crit Care. (2009) 13:R26. doi: 10.1186/cc7731
30. Ferguson NR, Galley HF, Webster NR. T helper cell subset ratios in patients with severe sepsis. Intensive Care Med. (1999) 25:106–9. doi: 10.1007/s001340050795
31. Wu HP, Chung K, Lin CY, Jiang BY, Chuang DY, Liu YC. Associations of T helper 1, 2, 17 and regulatory T lymphocytes with mortality in severe sepsis. Inflamm Res. (2013) 62:751–63. doi: 10.1007/s00011-013-0630-3
32. Venet F, Bohe J, Debard AL, Bienvenu J, Lepape A, Monneret G. Both percentage of gammadelta T lymphocytes and CD3 expression are reduced during septic shock. Crit Care Med. (2005) 33:2836–40. doi: 10.1097/01.CCM.0000189745.66585.AE
33. Andreu-Ballester JC, Tormo-Calandin C, Garcia-Ballesteros C, Perez-Griera J, Amigo V, Almela-Quilis A, et al. Association of γδ T cells with disease severity and mortality in septic patients. Clin Vaccine Immunol. (2013) 20:738–46. doi: 10.1128/CVI.00752-12
34. Monserrat J, de Pablo R, Diaz-Martin D, Rodriguez-Zapata M, de la Hera A, Prieto A, et al. Early alterations of B cells in patients with septic shock. Crit Care. (2013) 17:R105. doi: 10.1186/cc12750
35. Chung CS, Wang W, Chaudry IH, Ayala A. Increased apoptosis in lamina propria B cells during polymicrobial sepsis is FasL but not endotoxin mediated. Am J Physiol Gastrointest Liver Physiol. (2001) 280:G812–8. doi: 10.1152/ajpgi.2001.280.5.G812
36. Luan YY, Yao YM, Xiao XZ, Sheng ZY. Insights into the apoptotic death of immune cells in sepsis. J Interferon Cytokine Res. (2015) 35:17–22. doi: 10.1089/jir.2014.0069
37. Swan R, Chung CS, Albina J, Cioffi W, Perl M, Ayala A. Polymicrobial sepsis enhances clearance of apoptotic immune cells by splenic macrophages. Surgery. (2007) 142:253–61. doi: 10.1016/j.surg.2007.04.005
38. Sonego F, Castanheira FV, Ferreira RG, Kanashiro A, Leite CA, Nascimento DC, et al. Paradoxical roles of the neutrophil in sepsis: protective and deleterious. Front Immunol. (2016) 7:155. doi: 10.3389/fimmu.2016.00155
39. Dahlgren C, Karlsson A. Respiratory burst in human neutrophils. J Immunol Methods. (1999) 232:3–14. doi: 10.1016/S0022-1759(99)00146-5
40. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. (2007) 176:231–41. doi: 10.1083/jcb.200606027
41. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
42. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
43. Zhu XM, Yao YM, Liang HP, Liu F, Dong N, Yu Y, et al. Effect of high mobility group box-1 protein on apoptosis of peritoneal macrophages. Arch Biochem Biophys. (2009) 492:54–61. doi: 10.1016/j.abb.2009.09.016
44. Tinsley KW, Grayson MH, Swanson PE, Drewry AM, Chang KC, Karl IE, et al. Sepsis induces apoptosis and profound depletion of splenic interdigitating and follicular dendritic cells. J Immunol. (2003) 171:909–14. doi: 10.4049/jimmunol.171.2.909
45. Andaluz-Ojeda D, Iglesias V, Bobillo F, Almansa R, Rico L, Gandia F, et al. Early natural killer cell counts in blood predict mortality in severe sepsis. Crit Care. (2011) 15:R243. doi: 10.1186/cc10501
46. Giamarellos-Bourboulis EJ, Tsaganos T, Spyridaki E, Mouktaroudi M, Plachouras D, Vaki I, et al. Early changes of CD4-positive lymphocytes and NK cells in patients with severe Gram-negative sepsis. Crit Care. (2006) 10:R166. doi: 10.1186/cc5111
47. Badgwell B, Parihar R, Magro C, Dierksheide J, Russo T, Carson WE 3rd. Natural killer cells contribute to the lethality of a murine model of Escherichia coli infection. Surgery. (2002) 132:205–12. doi: 10.1067/msy.2002.125311
48. Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. (2006) 6:813–22. doi: 10.1038/nri1943
49. Hotchkiss RS, Osmon SB, Chang KC, Wagner TH, Coopersmith CM, Karl IE. Accelerated lymphocyte death in sepsis occurs by both the death receptor and mitochondrial pathways. J Immunol. (2005) 174:5110–8. doi: 10.4049/jimmunol.174.8.5110
50. Hotchkiss RS, Tinsley KW, Hui JJ, Chang KC, Swanson PE, Drewry AM, et al. p53-dependent and -independent pathways of apoptotic cell death in sepsis. J Immunol. (2000) 164:3675–80. doi: 10.4049/jimmunol.164.7.3675
51. Daigneault M, De Silva TI, Bewley MA, Preston JA, Marriott HM, Mitchell AM, et al. Monocytes regulate the mechanism of T-cell death by inducing Fas-mediated apoptosis during bacterial infection. PLoS Pathog. (2012) 8:e1002814. doi: 10.1371/journal.ppat.1002814
52. Sarkar A, Hall MW, Exline M, Hart J, Knatz N, Gatson NT, et al. Caspase-1 regulates Escherichia coli sepsis and splenic B cell apoptosis independently of interleukin-1β and interleukin-18. Am J Respir Crit Care Med. (2006) 174:1003–10. doi: 10.1164/rccm.200604-546OC
53. Weber SU, Schewe JC, Lehmann LE, Muller S, Book M, Klaschik S, et al. Induction of Bim and Bid gene expression during accelerated apoptosis in severe sepsis. Crit Care. (2008) 12:R128. doi: 10.1186/cc7088
54. Chang K, Svabek C, Vazquez-Guillamet C, Sato B, Rasche D, Wilson S, et al. Targeting the programmed cell death 1: programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit Care. (2014) 18:R3. doi: 10.1186/cc13176
55. Ma T, Han L, Gao Y, Li L, Shang X, Hu W, et al. The endoplasmic reticulum stress-mediated apoptosis signal pathway is involved in sepsis-induced abnormal lymphocyte apoptosis. Eur Surg Res. (2008) 41:219–25. doi: 10.1159/000135631
56. Haendeler J, Messmer UK, Brune B, Neugebauer E, Dimmeler S. Endotoxic shock leads to apoptosis in vivo and reduces Bcl-2. Shock. (1996) 6:405–9. doi: 10.1097/00024382-199612000-00004
57. Iwata A, Stevenson VM, Minard A, Tasch M, Tupper J, Lagasse E, et al. Over-expression of Bcl-2 provides protection in septic mice by a trans effect. J Immunol. (2003) 171:3136–41. doi: 10.4049/jimmunol.171.6.3136
58. Hotchkiss RS, Swanson PE, Knudson CM, Chang KC, Cobb JP, Osborne DF, et al. Overexpression of Bcl-2 in transgenic mice decreases apoptosis and improves survival in sepsis. J Immunol. (1999) 162:4148–56. doi: 10.1097/00024382-199806001-00219
59. Matsuda N, Teramae H, Futatsugi M, Takano K, Yamamoto S, Tomita K, et al. Up-regulation of histamine H4 receptors contributes to splenic apoptosis in septic mice: counteraction of the antiapoptotic action of nuclear factor-κB. J Pharmacol Exp Ther. (2010) 332:730–7. doi: 10.1124/jpet.109.163543
60. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-018-0102-y
61. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. (2009) 7:99–109. doi: 10.1038/nrmicro2070
62. Singla S, Machado RF. Death of the endothelium in sepsis: understanding the crime scene. Am J Respir Cell Mol Biol. (2018) 59:3–4. doi: 10.1165/rcmb.2018-0051ED
63. Liu L, Sun B. Neutrophil pyroptosis: new perspectives on sepsis. Cell Mol Life Sci. (2019) 76:2031–42. doi: 10.1007/s00018-019-03060-1
64. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. (2013) 38:209–23. doi: 10.1016/j.immuni.2013.02.003
65. Zhu H, Santo A, Jia Z, Robert Li Y. GPx4 in bacterial infection and polymicrobial sepsis: involvement of ferroptosis and pyroptosis. React Oxyg Species (Apex). (2019) 7:154–60. doi: 10.20455/ros.2019.835
66. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. (2014) 16:1180–91. doi: 10.1038/ncb3064
67. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defense against infection. Nat Rev Immunol. (2017) 17:151–64. doi: 10.1038/nri.2016.147
68. Mica L, Harter L, Trentz O, Keel M. Endotoxin reduces CD95-induced neutrophil apoptosis by cIAP-2-mediated caspase-3 degradation. J Am Coll Surg. (2004) 199:595–602. doi: 10.1016/j.jamcollsurg.2004.05.272
69. Drechsler S, Zipperle J, Rademann P, Jafarmadar M, Klotz A, Bahrami S, et al. Splenectomy modulates early immuno-inflammatory responses to trauma-hemorrhage and protects mice against secondary sepsis. Sci Rep. (2018) 8:14890. doi: 10.1038/s41598-018-33232-1
70. Huston JM, Wang H, Ochani M, Ochani K, Rosas-Ballina M, Gallowitsch-Puerta M, et al. Splenectomy protects against sepsis lethality and reduces serum HMGB1 levels. J Immunol. (2008) 181:3535–9. doi: 10.4049/jimmunol.181.5.3535
71. Brown KA, Brain SD, Pearson JD, Edgeworth JD, Lewis SM, Treacher DF. Neutrophils in development of multiple organ failure in sepsis. Lancet. (2006) 368:157–69. doi: 10.1016/S0140-6736(06)69005-3
72. Denning NL, Aziz M, Gurien SD, Wang P. DAMPs and NETs in Sepsis. Front Immunol. (2019) 10:2536. doi: 10.3389/fimmu.2019.02536
73. Colon DF, Wanderley CW, Franchin M, Silva CM, Hiroki CH, Castanheira FVS, et al. Neutrophil extracellular traps (NETs) exacerbate severity of infant sepsis. Crit Care. (2019) 23:113. doi: 10.1186/s13054-019-2407-8
74. Abrams ST, Morton B, Alhamdi Y, Alsabani M, Lane S, Welters ID, et al. A Novel assay for neutrophil extracellular trap formation independently predicts disseminated intravascular coagulation and mortality in critically ill patients. Am J Respir Crit Care Med. (2019) 200:869–80. doi: 10.1164/rccm.201811-2111OC
75. Patel JM, Sapey E, Parekh D, Scott A, Dosanjh D, Gao F, et al. Sepsis induces a dysregulated neutrophil phenotype that is associated with increased mortality. Mediators Inflamm. (2018) 2018:4065362. doi: 10.1155/2018/4065362
76. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
77. Land W. Allograft injury mediated by reactive oxygen species: from conserved proteins of Drosophila to acute and chronic rejection of human transplants. Part II: Role of reactive oxygen species in the induction of the heat shock response as a regulator of innate. Transpl Rev. (2003) 17:31–44. doi: 10.1053/trre.2003.2
78. Relja B, Land WG. Damage-associated molecular patterns in trauma. Eur J Trauma Emerg Surg. (2019). doi: 10.1007/s00068-019-01235-w. [Epub ahead of print].
79. Venereau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol. (2015) 6:422. doi: 10.3389/fimmu.2015.00422
80. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) 464:104–7. doi: 10.1038/nature08780
81. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
82. Roh J, Sohn D. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. (2018) 18:e27. doi: 10.4110/in.2018.18.e27
83. Rajaee A, Barnett R, Cheadle WG. Pathogen- and danger-associated molecular patterns and the cytokine response in sepsis. Surg Infect (Larchmt). (2018) 19:107–116. doi: 10.1089/sur.2017.264
84. Matzinger P. The danger model: a renewed sense of self. Science. (2002) 296:301–5. doi: 10.1126/science.1071059
85. Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol. (2012) 189:2689–95. doi: 10.4049/jimmunol.1201719
86. Cheng Z, Abrams ST, Alhamdi Y, Toh J, Yu W, Wang G, et al. Circulating histones are major mediators of multiple organ dysfunction syndrome in acute critical illnesses. Crit Care Med. (2019) 47:e677–e84. doi: 10.1097/CCM.0000000000003839
87. Alhamdi Y, Abrams ST, Cheng Z, Jing S, Su D, Liu Z, et al. Circulating histones are major mediators of cardiac injury in patients with sepsis. Crit Care Med. (2015) 43:2094–103. doi: 10.1097/CCM.0000000000001162
88. Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med. (2013) 187:160–9. doi: 10.1164/rccm.201206-1037OC
89. Gentile LF, Moldawer LL. DAMPs, PAMPs, and the origins of SIRS in bacterial sepsis. Shock. (2013) 39:113–4. doi: 10.1097/SHK.0b013e318277109c
90. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. (2002) 418:191–5. doi: 10.1038/nature00858
91. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. (1999) 285:248–51.
92. Kim HM, Kim YM. HMGB1: LPS Delivery Vehicle for Caspase-11-Mediated Pyroptosis. Immunity. (2018) 49:582–4. doi: 10.1016/j.immuni.2018.09.021
93. Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, et al. The endotoxin delivery protein HMGB1 mediates caspase-11-dependent lethality in sepsis. Immunity. (2018) 49:740–53.e7. doi: 10.1016/j.immuni.2018.08.016
94. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. (2009) 15:1318–21. doi: 10.1038/nm.2053
95. Fuchs TA, Bhandari AA, Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood. (2011) 118:3708–14. doi: 10.1182/blood-2011-01-332676
96. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. (2011) 118:1952–61. doi: 10.1182/blood-2011-03-343061
97. Ammollo CT, Semeraro F, Xu J, Esmon NL, Esmon CT. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J Thromb Haemost. (2011) 9:1795–803. doi: 10.1111/j.1538-7836.2011.04422.x
98. Gould TJ, Lysov Z, Swystun LL, Dwivedi DJ, Zarychanski R, Fox-Robichaud AE, et al. Canadian critical care translational biology, extracellular histones increase tissue factor activity and enhance thrombin generation by human blood monocytes. Shock. (2016) 46:655–62. doi: 10.1097/SHK.0000000000000680
99. Alhamdi Y, Abrams ST, Lane S, Wang G, Toh CH. Histone-associated thrombocytopenia in patients who are critically ill. JAMA. (2016) 315:817–9. doi: 10.1001/jama.2016.0136
100. Wildhagen KC, Garcia de Frutos P, Reutelingsperger CP, Schrijver R, Areste C, Ortega-Gomez A, et al. Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood. (2014) 123:1098–101. doi: 10.1182/blood-2013-07-514984
101. Abrams ST, Zhang N, Dart C, Wang SS, Thachil J, Guan Y, et al. Human CRP defends against the toxicity of circulating histones. J Immunol. (2013) 191:2495–502. doi: 10.4049/jimmunol.1203181
102. Baudouin SV, Saunders D, Tiangyou W, Elson JL, Poynter J, Pyle A, et al. Mitochondrial DNA and survival after sepsis: a prospective study. Lancet. (2005) 366:2118–21. doi: 10.1016/S0140-6736(05)67890-7
103. Dwivedi DJ, Toltl LJ, Swystun LL, Pogue J, Liaw KL, Weitz JI, et al. Canadian critical care translational biology, prognostic utility and characterization of cell-free DNA in patients with severe sepsis. Crit Care. (2012) 16:R151. doi: 10.1186/cc11466
104. Jackson Chornenki NL, Coke R, Kwong AC, Dwivedi DJ, Xu MK, McDonald E, et al. Comparison of the source and prognostic utility of cfDNA in trauma and sepsis. Intensive Care Med Exp. (2019) 7:29. doi: 10.1186/s40635-019-0251-4
105. Saukkonen K, Lakkisto P, Pettila V, Varpula M, Karlsson S, Ruokonen E, et al. Finnsepsis study, cell-free plasma DNA as a predictor of outcome in severe sepsis and septic shock. Clin Chem. (2008) 54:1000–7. doi: 10.1373/clinchem.2007.101030
106. Long Y, Zhang Y, Gong Y, Sun R, Su L, Lin X, et al. Diagnosis of sepsis with cell-free DNA by next-generation sequencing technology in ICU patients. Arch Med Res. (2016) 47:365–71. doi: 10.1016/j.arcmed.2016.08.004
107. Gould TJ, Vu TT, Stafford AR, Dwivedi DJ, Kim PY, Fox-Robichaud AE, et al. Cell-free DNA modulates clot structure and impairs fibrinolysis in sepsis. Arterioscler Thromb Vasc Biol. (2015) 35:2544–53. doi: 10.1161/ATVBAHA.115.306035
108. Dubois C, Marce D, Faivre V, Lukaszewicz AC, Junot C, Fenaille F, et al. High plasma level of S100A8/S100A9 and S100A12 at admission indicates a higher risk of death in septic shock patients. Sci Rep. (2019) 9:15660. doi: 10.1038/s41598-019-52184-8
109. Czaikoski PG, Mota JM, Nascimento DC, Sonego F, Castanheira FV, Melo PH, et al. Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS One. (2016) 11:e0148142. doi: 10.1371/journal.pone.0148142
110. Luo L, Zhang S, Wang Y, Rahman M, Syk I, Zhang E, et al. Proinflammatory role of neutrophil extracellular traps in abdominal sepsis. Am J Physiol Lung Cell Mol Physiol. (2014) 307:L586–96. doi: 10.1152/ajplung.00365.2013
111. O'Brien XM, Biron BM, Reichner JS. Consequences of extracellular trap formation in sepsis. Curr Opin Hematol. (2017) 24:66–71. doi: 10.1097/MOH.0000000000000303
112. Kumar S, Gupta E, Kaushik S, Srivastava VK, Saxena J, Mehta S, et al. Quantification of NETs formation in neutrophil and its correlation with the severity of sepsis and organ dysfunction. Clin Chim Acta. (2019) 495:606–10. doi: 10.1016/j.cca.2019.06.008
113. Biron BM, Chung CS, O'Brien XM, Chen Y, Reichner JS, Ayala A. Cl-amidine prevents histone 3 citrullination and neutrophil extracellular trap formation, and improves survival in a murine sepsis model. J Innate Immun. (2017) 9:22–32. doi: 10.1159/000448808
114. Lee SK, Kim SD, Kook M, Lee HY, Ghim J, Choi Y, et al. Phospholipase D2 drives mortality in sepsis by inhibiting neutrophil extracellular trap formation and down-regulating CXCR2. J Exp Med. (2015) 212:1381–90. doi: 10.1084/jem.20141813
115. Meng W, Paunel-Gorgulu A, Flohe S, Hoffmann A, Witte I, MacKenzie C, et al. Depletion of neutrophil extracellular traps in vivo results in hypersusceptibility to polymicrobial sepsis in mice. Crit Care. (2012) 16:R137. doi: 10.1186/cc11442
116. Mai SH, Khan M, Dwivedi DJ, Ross CA, Zhou J, Gould TJ, et al. Delayed but not early treatment with DNase reduces organ damage and improves outcome in a murine model of sepsis. Shock. (2015) 44:166–72. doi: 10.1097/SHK.0000000000000396
117. Iba T, Levy JH, Raj A, Warkentin TE. Advance in the management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J Clin Med. (2019) 8:728. doi: 10.3390/jcm8050728
118. Liaw PC, Ito T, Iba T, Thachil J, Zeerleder S. DAMP and DIC: the role of extracellular DNA and DNA-binding proteins in the pathogenesis of DIC. Blood Rev. (2016) 30:257–61. doi: 10.1016/j.blre.2015.12.004
119. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
120. McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. (2017) 129:1357–67. doi: 10.1182/blood-2016-09-741298
121. Francois B, Jeannet R, Daix T, Walton AH, Shotwell MS, Unsinger J, et al. Interleukin-7 restores lymphocytes in septic shock: the IRIS-7 randomized clinical trial. JCI Insight. (2018) 3:e98960. doi: 10.1172/jci.insight.98960
122. Shindo Y, Fuchs AG, Davis CG, Eitas T, Unsinger J, Burnham CD, et al. Interleukin 7 immunotherapy improves host immunity and survival in a two-hit model of Pseudomonas aeruginosa pneumonia. J Leukoc Biol. (2017) 101:543–54. doi: 10.1189/jlb.4A1215-581R
123. Inoue S, Unsinger J, Davis CG, Muenzer JT, Ferguson TA, Chang K, et al. IL-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J Immunol. (2010) 184:1401–9. doi: 10.4049/jimmunol.0902307
124. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. (2011) 306:2594–605. doi: 10.1001/jama.2011.1829
125. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. (1997) 390:350–1. doi: 10.1038/37022
126. Hiraki S, Ono S, Tsujimoto H, Kinoshita M, Takahata R, Miyazaki H, et al. Neutralization of interleukin-10 or transforming growth factor-beta decreases the percentages of CD4+ CD25+ Foxp3+ regulatory T cells in septic mice, thereby leading to an improved survival. Surgery. (2012) 151:313–22. doi: 10.1016/j.surg.2011.07.019
127. Schrijver IT, Theroude C, Roger T. Myeloid-derived suppressor cells in sepsis. Front Immunol. (2019) 10:327. doi: 10.3389/fimmu.2019.00327
128. Kulkarni U, Herrmenau C, Win SJ, Bauer M, Kamradt T. IL-7 treatment augments and prolongs sepsis-induced expansion of IL-10-producing B lymphocytes and myeloid-derived suppressor cells. PLoS One. (2018) 13:e0192304. doi: 10.1371/journal.pone.0192304
129. Uhel F, Azzaoui I, Gregoire M, Pangault C, Dulong J, Tadie JM, et al. Early Expansion of circulating granulocytic myeloid-derived suppressor cells predicts development of nosocomial infections in patients with sepsis. Am J Respir Crit Care Med. (2017) 196:315–27. doi: 10.1164/rccm.201606-1143OC
130. Tsukamoto H, Kozakai S, Kobayashi Y, Takanashi R, Aoyagi T, Numasaki M, et al. Impaired antigen-specific lymphocyte priming in mice after Toll-like receptor 4 activation via induction of monocytic myeloid-derived suppressor cells. Eur J Immunol. (2019) 49:546–63. doi: 10.1002/eji.201847805
131. Vourc'h M, Roquilly A, Asehnoune K. Trauma-induced damage-associated molecular patterns-mediated remote organ injury and immunosuppression in the acutely ill patient. Front Immunol. (2018) 9:1330. doi: 10.3389/fimmu.2018.01330
132. Zhang Q, Qi Z, Bo L, Li CS. Programmed cell death-1/programmed death-ligand 1 blockade improves survival of animals with sepsis: a systematic review and meta-analysis. Biomed Res Int. (2018) 2018:1969474. doi: 10.1155/2018/1969474
133. Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U S A. (2009) 106:6303–8. doi: 10.1073/pnas.0809422106
134. Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, Hotchkiss RS. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol. (2010) 88:233–40. doi: 10.1189/jlb.0110037
135. Zhang Y, Zhou Y, Lou J, Li J, Bo L, Zhu K, et al. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care. (2010) 14:R220. doi: 10.1186/cc9354
136. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. (2013) 13:862–74. doi: 10.1038/nri3552
137. Monneret G, Finck ME, Venet F, Debard AL, Bohe J, Bienvenu J, et al. The anti-inflammatory response dominates after septic shock: association of low monocyte HLA-DR expression and high interleukin-10 concentration. Immunol Lett. (2004) 95:193–8. doi: 10.1016/j.imlet.2004.07.009
138. Henry BM, Vikse J, Benoit S, Favaloro EJ, Lippi G. Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: A novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin Chim Acta. (2020) 507:167–73. doi: 10.1016/j.cca.2020.04.027
139. Shindo Y, Unsinger J, Burnham CA, Green JM, Hotchkiss RS. Interleukin-7 and anti-programmed cell death 1 antibody have differing effects to reverse sepsis-induced immunosuppression. Shock. (2015) 43:334–43. doi: 10.1097/SHK.0000000000000317
140. Ono S, Tsujimoto H, Hiraki S, Aosasa S. Mechanisms of sepsis-induced immunosuppression and immunological modification therapies for sepsis. Ann Gastroenterol Surg. (2018) 2:351–8. doi: 10.1002/ags3.12194
141. Hotchkiss RS, Chang KC, Swanson PE, Tinsley KW, Hui JJ, Klender P, et al. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat Immunol. (2000) 1:496–501. doi: 10.1038/82741
142. Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, et al. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci U S A. (1999) 96:14541–6. doi: 10.1073/pnas.96.25.14541
143. Schwulst SJ, Muenzer JT, Peck-Palmer OM, Chang KC, Davis CG, McDonough JS, et al. Bim siRNA decreases lymphocyte apoptosis and improves survival in sepsis. Shock. (2008) 30:127–34. doi: 10.1097/shk.0b013e318162cf17
144. Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. (2003) 348:138–50. doi: 10.1056/NEJMra021333
145. Hotchkiss RS, Opal S. Immunotherapy for sepsis–a new approach against an ancient foe. N Engl J Med. (2010) 363:87–9. doi: 10.1056/NEJMcibr1004371
146. Weaver JG, Rouse MS, Steckelberg JM, Badley AD. Improved survival in experimental sepsis with an orally administered inhibitor of apoptosis. FASEB J. (2004) 18:1185–91. doi: 10.1096/fj.03-1230com
147. Wesche-Soldato DE, Swan RZ, Chung CS, Ayala A. The apoptotic pathway as a therapeutic target in sepsis. Curr Drug Targets. (2007) 8:493–500. doi: 10.2174/138945007780362764
148. van der Poll T. Immunotherapy of sepsis. Lancet Infect Dis. (2001) 1:165–74. doi: 10.1016/S1473-3099(01)00093-7
149. Nasraway SA. The problems and challenges of immunotherapy in sepsis. Chest. (2003) 123:451S−9S. doi: 10.1378/chest.123.5_suppl.451S
150. Peters van Ton AM, Kox M, Abdo WF, Pickkers P. Precision immunotherapy for sepsis. Front Immunol. (2018) 9:1926. doi: 10.3389/fimmu.2018.01926
151. Lee WL. Immunotherapy for sepsis: a good idea or another dead end? Anesthesiology. (2018) 129:5–7. doi: 10.1097/ALN.0000000000002237
152. Prucha M, Zazula R, Russwurm S. Immunotherapy of sepsis: blind alley or call for personalized assessment? Arch Immunol Ther Exp (Warsz). (2017) 65:37–49. doi: 10.1007/s00005-016-0415-9
153. Leentjens J, Kox M, van der Hoeven JG, Netea MG, Pickkers P. Immunotherapy for the adjunctive treatment of sepsis: from immunosuppression to immunostimulation. Time for a paradigm change? Am J Respir Crit Care Med. (2013) 187:1287–93. doi: 10.1164/rccm.201301-0036CP
154. Patil NK, Guo Y, Luan L, Sherwood ER. Targeting immune cell checkpoints during sepsis. Int J Mol Sci. (2017) 18:2413. doi: 10.3390/ijms18112413
155. Hogwood J, Pitchford S, Mulloy B, Page C, Gray E. Heparin and non-anticoagulant heparin attenuate histone-induced inflammatory responses in whole blood. PLoS One. (2020) 15:e0233644. doi: 10.1371/journal.pone.0233644
156. Laukova L, Konecna B, Babickova J, Wagnerova A, Meliskova V, Vlkova B, et al. Exogenous deoxyribonuclease has a protective effect in a mouse model of sepsis. Biomed Pharmacother. (2017) 93:8–16. doi: 10.1016/j.biopha.2017.06.009
157. Wang H, Ward MF, Sama AE. Targeting HMGB1 in the treatment of sepsis. Expert Opin Ther Targets. (2014) 18:257–68. doi: 10.1517/14728222.2014.863876
158. Fink MP. Bench-to-bedside review: High-mobility group box 1 and critical illness. Crit Care. (2007) 11:229. doi: 10.1186/cc6088
Keywords: sepsis, extensive immune cell death, damage-associated molecular patterns (DAMPs), multiple organ dysfunction syndrome (MODS), extracellular histones, immunosuppression
Citation: Cheng Z, Abrams ST, Toh J, Wang SS, Wang Z, Yu Q, Yu W, Toh C-H and Wang G (2020) The Critical Roles and Mechanisms of Immune Cell Death in Sepsis. Front. Immunol. 11:1918. doi: 10.3389/fimmu.2020.01918
Received: 14 February 2020; Accepted: 16 July 2020;
Published: 25 August 2020.
Edited by:
Walter Gottlieb Land, Université de Strasbourg, FranceReviewed by:
Jaewoo Lee, Duke University, United StatesJulien Pottecher, Hôpitaux Universitaires de Strasbourg, France
Copyright © 2020 Cheng, Abrams, Toh, Wang, Wang, Yu, Yu, Toh and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiping Yu, eXV3ZWlwaW5nQHNldS5lZHUuY24=; Cheng-Hock Toh, dG9oQGxpdmVycG9vbC5hYy51aw==; Guozheng Wang, d2FuZ2dAbGl2ZXJwb29sLmFjLnVr