 Juan Garcia-Revilla
Juan Garcia-Revilla Tomas Deierborg
Tomas Deierborg Jose Luis Venero
Jose Luis Venero Antonio Boza-Serrano
Antonio Boza-Serrano- 1Departamento de Bioquímica y Biología Molecular, Facultad de Farmacia and Instituto de Biomedicina de Sevilla (IBiS), Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville, Spain
- 2Department of Experimental Medical Science, Experimental Neuroinflammation Laboratory, BMC, Lund University, Lund, Sweden
- 3Department of Experimental Medical Sciences, Experimental Dementia Research Laboratory, BMC, Lund University, Lund, Sweden
COVID-19 disease have become so far the most important sanitary crisis in the XXI century. In light of the events, any clinical resource should be considered to alleviate this crisis. Severe COVID-19 cases present a so-called cytokine storm as the most life-threatening symptom accompanied by lung fibrosis. Galectin-3 has been widely described as regulator of both processes. Hereby, we present compelling evidences on the potential role of galectin-3 in COVID-19 in the regulation of the inflammatory response, fibrosis and infection progression. Moreover, we provide a strong rationale of the utility of measuring plasma galectin-3 as a prognosis biomarker for COVID-19 patients and propose that inhibition of galectin-3 represents a feasible and promising new therapeutical approach.
Introduction
COVID-19 is a disease caused by the infection of a novel coronavirus known as SARS-CoV-2. COVID-19 has spread rapidly around the world causing a devastating pandemic with millions of people affected and thousands of lives gone (370,000 dead as per May 30, 2020) in what has become the main health and economic threat in the modern era. Most severe COVID-19 patients develop pneumonia and hyperinflammation likely related to a macrophage activation syndrome (1) commonly named “cytokine storm”. Linked to the inflammatory response, lung fibrosis emerges as a secondary event related to the progression of the pathology (2). Understanding the link between the hyperinflammation phase and fibrosis will give rise to new therapeutic targets especially beneficial in the most severe cases of COVID-19 (3). Given the pleiotropic roles of galectin-3 (gal3), especially those driving inflammatory-associated immune responses, fibrosis and hypoxia we propose the urgent need to decipher a potential pathological role of this lectin in severe cases of COVID-19 patients. Gal3 is a carbohydrate-binding protein expressed by macrophages, epithelial and alveolar cells in lungs (4). We would like to highlight three potential roles of gal3 in COVID-19 progression: (i) the macrophage-related hyperinflammation phase that drives the cytokine storm in most severe cases; (ii) the virus infection mechanism via the viral spike protein, given that its N-terminal domain has been suggested to evolve from a galectin origin (5); and (iii) the COVID-19-related lung fibrosis linked to the acute phase of diffuse alveolar damage, oedema, hypoxia, and inflammatory response. Therefore, gal3 encompasses unique COVID-19-associated pathophysiological features that deserve therapeutical attention associated with inflammatory response, infection mechanism, lung fibrosis and hypoxia. Importantly, the existence of clinically tolerable inhibitors for gal3 makes clinical trials feasible. Indeed, one of the gal3 inhibitors already available has proven its efficacy in Idiopathic lung fibrosis. Last, we wish to highlight the potential role of gal3 as a clinical biomarker tool. Measure of plasma gal3 levels has been used as a biomarker for several diseases with fibrotic or inflammatory features like heart failure. Here, we provide strong rationale to consider gal3 as a potential prognostic biomarker for severe COVID-19 cases.
Galectin-3 May Play a Key Role in Pulmonary Associated Inflammatory Response and Lung Fibrosis
SARS-CoV-2 primarily induces a lung inflammation during acute infection. The severity of the disease has been associated to lung infiltrating immune cells causing two well-defined features that are connected: (i) a lung hyperinflammation phase that becomes systemic through the progression of the pathology and (ii) the consequent lung fibrosis (6). In addition, the production of pro-inflammatory cytokines (cytokine storm) is believed to be a key event in COVID-19 mortality and morbidity. Hence, immune suppression should be carefully considered (7) due to the clinical evidence of deterioration found in some patients under this condition associated to the disease. Several publications have already pointed out that high levels of pro-inflammatory cytokines are in relation with the severity of the pathology (8, 9). Recently, single-cell RNAseq analysis performed on different immune cells in lungs from COVID-19 patients has provided valuable information about the immune response of the disease. In this study by Liao and colleagues (10), gal3 appears to be elevated in proliferative T cells associated to severe condition of COVID-19 patients. Moreover, a subset of macrophages express several markers associated to fibrotic processes like TREM2 or SPP1 (10), two markers that have been consistently associated to gal3 (11, 12). Indeed, a relevant study aimed at identifying the molecular mechanism involved in fibrosis identified a subset of pro-fibrogenic macrophages where gal3 was one of the most upregulated genes in association with TREM2 and SPP1 (13). Moreover, we have demonstrated a critical role of gal3 in microglial proinflammatory response and importantly, the ability of gal3 to further bind to and activate TREM2 (14) and TLR4 (15) both reported to be involved in lung disease and fibrosis. TREM2 is suggested to prevent macrophage apoptosis and promote chronic inflammatory disease after lung viral infection (16). TLR4 is a leading actor in the resolution of the inflammatory response in pneumonia (17) and TLRs are essential in the antiviral response triggering a strong inflammation involving interferon related genes, interleukins, chemokines as well as gal3 expression (18). So that, the mutually non-exclusive roles of gal3, TREM2 and TLRs in the COVID-19-associated hyperinflammation phase and the aforementioned lung fibrosis are plausible (see Figure 1). Supporting this, inhibition or genetic manipulation of TLR4 reduces the proliferation capacity of Influenza-A Virus and the pro-inflammatory response linked to pneumonia (19) and acute lung injury (20). Moreover, TLR4 has been associated to fibroblast activation and the subsequent lung fibrosis while fibroblast-specific deletion of TLR4 in mice induced substantial reduction in lung fibrosis (21). The latest is one of the main outcomes of the hyperinflammation phase associated with severe COVID-19 cases supported by the concomitant upregulation of gal3, TNFα and IL-6 in lobar and bronchial pneumonia (22). For instance, high levels of IL-1ra and IL-6 cytokines in plasma samples of COVID-19 patients demonstrate a clear association between the severity of the immune response and fatal outcome (9). Indeed, blocking IL-6 can be effective (23) and it has been suggested to ameliorate the severity of the pathology in some of the most critical cases (24). Moreover, previous SARS-CoV infection also runs with a strong inflammatory response and fibrosis (25–27) highlighting the importance of this approach for SARS-CoV-2. Mechanistically, it has been proved that SARS-CoV can activate NLRP3 inflammasome (28) something that occurs in H5N1 influenza infection as well in a gal3-dependent manner driving IL-1β production (29). Additionally, NLRP3 activation controls serum levels of gal3 (30). Notably, there are ongoing therapies aimed at slowing down the cytokine storm by infusing IL−1R antagonist (Anakinra) (31) and IL-6R antagonist (Tocilizumab) (31, 32). In our microglia work, we have demonstrated gal3 to govern both IL-1, IL-6 and TNFα release (14, 33). Gal3 thus emerges as a feasible pharmacological target to minimize the threatening hyperinflammation phase and subsequent lung fibrosis in COVID-19 patients.
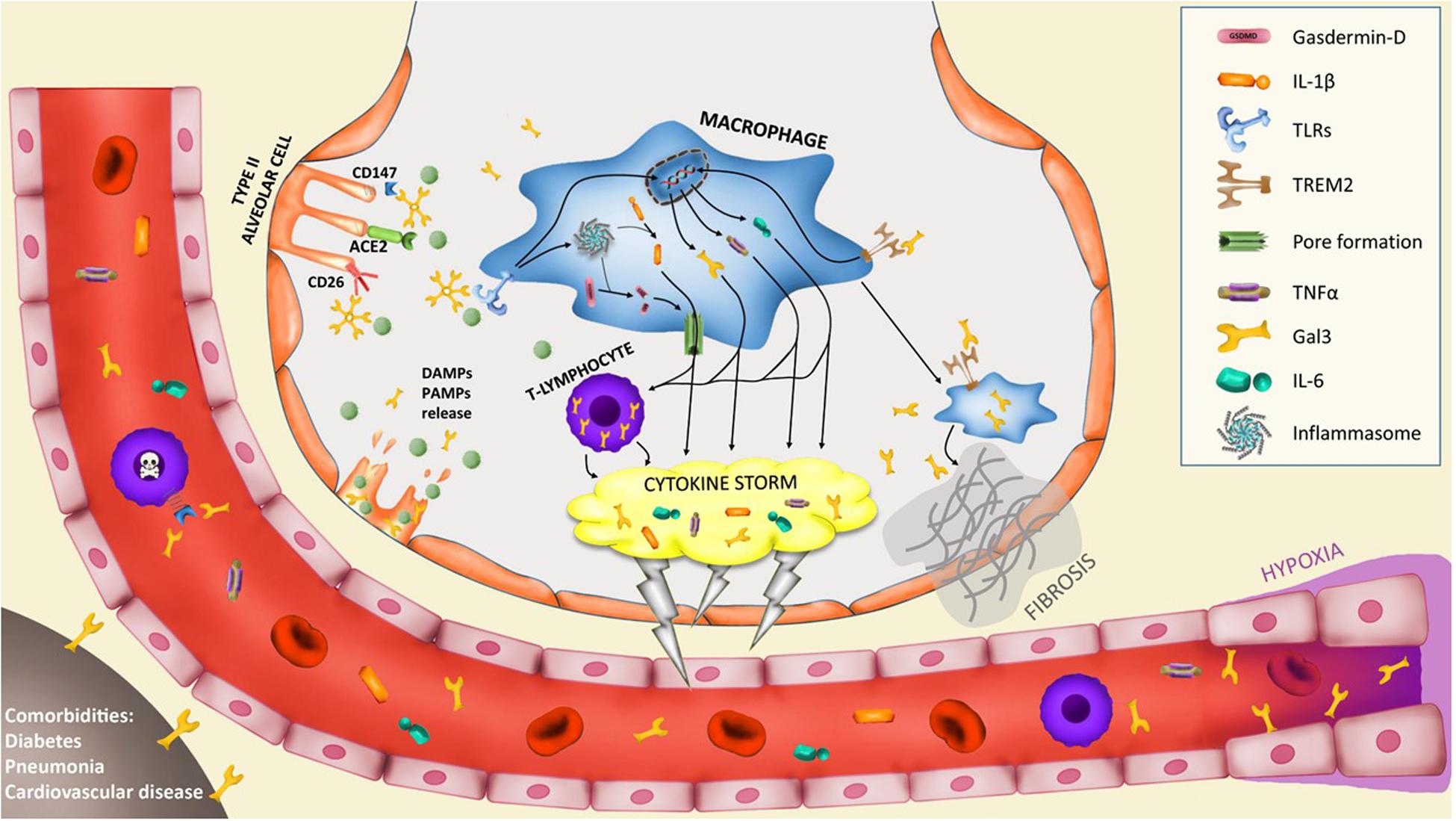
Figure 1. Potential implication of galectin-3 in the pathogenesis of severe COVID-19 cases. Infection of type II alveolar cells by SARS-CoV-2 is driven by ACE2-spike protein interaction and supported by CD147 and CD26 interaction. Gal3 can mediate ligand-receptor interaction by glycan recognition of spike protein and interaction with receptors. In particular, CD147-spike protein interaction promotes apoptosis of T-lymphocytes provoking systemic lymphocytopenia syndrome. Lysis of infected cells provoke the secretion of DAMPs and PAMPs recognized by alveolar macrophages receptors like triggering receptor expressed on myeloid cells 2 (TREM2) or toll-like receptors (TLRs) that trigger the upregulation of several cytokines including IL-6, TNFα and gal3. TLRs activation induces the assembly of the macromolecular platform known as inflammasome that promotes the cleavage of pro-IL-1β into active IL-1β and the cleavage of gasdermin-D (GSDMD), thus promoting pores formation in the cell membrane and induce the secretion of IL-1β. This macrophage activation triggers the hyperinflammation phase called “cytokine storm” and is supported by lymphocyte infiltration and proliferation with high expression of gal3 that contributes to the cytokine storm. The macrophage activation is composed of several phenotypes. A subset of macrophages develop a pro-fibrotic phenotype controlled by TREM2 and gal3 expression that triggers lung fibrosis. Moreover, hypoxia triggers gal3 expression by endothelial and smooth muscle cells. Lastly, comorbidities like diabetes, pneumonia or cardiovascular diseases increase basal levels of plasma gal3 what would contribute to COVID-19 severe prognosis.
Galectin-3 as a Mediator for Viral Adhesion
Data obtained from SARS-CoV-2 genome revealed a spike glycoprotein as a key mediator of virus-host engagement and initiation of the infection (34). Indeed, novel glycation sites of this SARS-CoV-2 spike protein may explain the pandemic differences with previous SARS-CoV (35). Importantly, studies from N-terminal domain of spike protein determined that this domain likely evolved from a galectin precursor sharing most structural similarities with gal3 [see Ref. (36)] suggesting that gal3 could play a major role in SARS-CoV-2-host engagement. At present, only a possible relation has been suggested for coronavirus family and gal3. In an in vitro model of SARS-CoV-2 infection of human Caco-2 cells, gal3 protein levels increased 24 h after viral infection (37) what can be related to the increased transcriptional machinery due to its role in spliceosome, but also makes gal3 available for infections of nearby cells. The role of gal3 and glycation in virus infection has been observed in other viral infections like the influenza virus (38). Interestingly, gal3-virus interaction has been previously described in several viruses like herpesvirus-1 (HSV-1) infection, where gal3 binds to, at least, UL-46 viral protein promoting HSV-1 infection to host cells (39). Moreover, gal3-knockdown in keratinocytes decreased the infection of HSV-1, suggesting gal3 as a mediator of the viral infection process (39). Additionally, HSV-1 promotes the secretion of gal3 by the infected cells (40) exacerbating the initial infection. This mechanism also appears in diverse RNA-virus families. For instance, human immunodeficiency virus-1 (HIV-1) induces gal3 upregulation to later facilitate exosomes-mediated infection of nearby cells by its interaction with membrane fibronectin (41). Similarly, another retrovirus like HTLV-1 also takes advantage of gal3 to create a biofilm that facilitates the adhesion to new host cells (42). Moreover, influenza A virus and Streptococcus pneumonia bind to gal3 increasing their airway epithelial adhesion (43), suggesting a role of gal3 in primary and secondary airway infections in COVID-19 patients. In SARS-CoV-2 infection, angiotensin converting enzyme 2 (ACE2) has been proposed as the receptor for spike protein interaction (44). Despite no interaction has been described between gal3 and ACE2, gal3 has shown ability to bind to ACE (45) that shares the same extracellular domain with ACE2 (46), suggesting a potential interaction. Structural analysis of ACE2-Spike protein interaction has pointed out the relevance of ACE2 glycosylation (47). In addition, glycosylation of ACE2 has shown to be critical for viral invasion in previous coronavirus infections (48). Glycosylation sites also seem to be critical for the severity of SARS-CoV-2 infection in humans compared with other animal hosts (49). Moreover, a recent study identified at least two mutations in humans that incorporate new glycosylation sites in ACE2 receptor linked with an increased risk of COVID-19 (50). Nevertheless, other receptors have been proposed to interact with viral-spike protein like CD26 (34), a membrane glycoprotein previously involved in MERS epidemic (51), and CD147 (52), both known to bind gal3 (53, 54) (see Figure 1). Indeed, SARS-CoV-2 interaction with CD147 has been linked with apoptosis in T-lymphocytes what contribute to the leukocytopenia observed in severe COVID-19 patients (55) as part of the mechanisms of evasion from the immune system. In view of this data suggesting an involvement of gal3 in SARS-CoV-2 pathogenesis, clinical gal3 studies are urgently warranted to determine the clinical benefits of gal3 inhibition.
Galectin-3 as a Biomarker and Therapeutic Target in COVID-19 Infection
As previously stated, lung fibrosis and hypoxia are a key feature related to the most severe cases of COVID-19 patients. Indeed, lung fibrosis has been found in autopsies in fatal COVID-19 cases (56). Lung fibrosis can be a secondary event consequence of the hyperinflammation phase in COVID-19. Both processes are connected mainly by the immune system and the activity of pro-inflammatory macrophage (1). Single-cell analysis has revealed a pro-fibrotic macrophage phenotype in the bronchoalveolar lavage fluid from patients with severe COVID-19 where lung fibrosis is present (10). Use of antifibrotic drugs has already been recently proposed [see Ref. (3)] as a treatment for severe COVID-19 patients. Among them, gal3 appears to be the most relevant due to its role in the inflammation and viral infection process. Gal3 antagonists have been developed against macrophage activation and inflammation associated to lung fibrosis showing positive results (57). Importantly, gal3 is already a therapeutic target in Idiopathic Pulmonary Fibrosis (IPF) with very promising results. Furthermore, a gal3 inhibitor (TD139, Galecto Biotech) has already passed phase I/IIa and proven its efficacy and safety (NCT03832946). We have successfully demonstrated the high efficiency of this drug in microglia cultures in response to well-defined pro-inflammatory agents (14, 15, 33). The availability of specific gal3 inhibitor in advanced clinical phases urge for clinical studies of the role of gal3 in SARS-CoV-2 pathogenesis. Hence, trying out interventions with gal3 inhibition could potentially be life saving for critically ill COVID-19 patients, and additionally support the lung tissue in the chronic phase for surviving patients.
Among the broad symptoms spectrum of COVID-19 patients, hypoxemia has shown the better correlation with disease severity. However, patients often maintain a “normal” lung function at the first stages of the disease while hypoxia appears as consequence of pulmonary vascular impairments that leads to a symptomatology that resembles the pulmonary arterial hypertension (PAH) (58, 59). Both COVID-19 and PAH develop hypoxia as one of their most relevant features and gal3 expression has been shown to be tightly regulated in hypoxic processes, making gal3 an attractive therapeutic target. For instance, Hao et al. (60) demonstrated the role of gal3 in hypoxia-induced PAH. Using small transferring RNA to inhibit gal3, they reduced the increased ventricular pressure, hypoxia-induced inflammatory response and alleviated ventricle hypertrophy of mice with hypoxia-induced PAH (60). Furthermore, Luo et al. corroborated that hypoxic condition in PAH triggers gal3 levels, promoting cell proliferation, inflammatory response and fibrotic driven tissue remodeling (61). In the same study, inhibition of gal3 reduced PAH (61). Similarly, Fulton and colleagues demonstrated that genetic or pharmacological inhibition of gal3 resulted in reduced pulmonary hypertension in vivo in a model of hypoxia-induced PAH (62). Recently, Fulton’s lab demonstrated that vascular smooth muscle cells are one of the main sources of gal3, which corroborates its implication in proliferation, apoptosis and fibrosis associated to PAH (63). Altogether, these studies highlight the importance of hypoxia-induced gal3 in PAH and lung function, being a potential target to counteract severe COVID-19 events.
At present, no clear prognosis biomarker have been identified while age and comorbidities are the unique clues in the prognosis of the disease. Considering that development of a hyperinflammatory phase is the main feature of the severe COVID-19 cases and given that immune cells can release gal3 during the inflammatory process (14, 15),we propose that it can be used as a biomarker of the inflammatory status in COVID-19 patients. Indeed, gal3 is elevated in plasma under strong inflammatory conditions (64). For instance, plasma levels of gal3 have shown strong correlation with disease progression in lung fibrosis (65) where post-mortem levels of lung gal3 can also correlate with the inflammatory status (66). Notably, cardiovascular diseases, diabetes or pneumonia are among the main risk factors for severe COVID-19 patients, all having in common elevated levels of gal3 (67, 68). Importantly, Mattioli and colleagues have shown that plasma from COVID-19 patients present elevated levels of gal3 (69) what supports that gal3 could be a good prognostic marker for severe COVID-19 and that elevated plasma levels of gal3 can participate in triggering the cytokine storm observed in severe COVID-19 patients. Consequently, plasma gal3 levels should be considered a clinical tool for inflammatory prognosis in COVID-19.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Author Contributions
JG-R and AB-S have contributed equally in the writing, design, and preparation of the manuscript. JV and TD have contributed in reviewing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Swedish Research Council (2019-06333, AB-S), and the Strong Research Environment MultiPark (Multidisciplinary Research in Parkinson’s and Alzheimer’s Disease at Lund University), Bagadilico (Linné consortium sponsored by the Swedish Research Council), the Swedish Alzheimer’s Foundation, Swedish Brain Foundation, A.E. Berger Foundation, Gyllenstiern-ska Krapperup Foundation, the Royal Physiographic Society, Crafoord Foundation, Olle Engkvist Byggmästare Foundation, Wiberg Foundation, G&J Kock Foundation, Stohnes Foundation, and Swedish Dementia Association and the Medical Faculty at Lund University. This perspective was also funded by the Spanish Ministerio de Ciencia, Innovación y Universidades/FEDER/UE RTI2018-098645-B-100, FEDER I+D+i-USE US-1265062, and US-1264806. JG-R has been funded by a grant from the Spanish Ministerio de Economia y Competitividad SAF2015-64171-R (MINECO/FEDER, EU).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. McGonagle, D, Sharif, K, O’Regan, A, Bridgewood, C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev. (2020) 19:102537. doi: 10.1016/j.autrev.2020.102537
2. Spagnolo, P, Balestro, E, Aliberti, S, Cocconcelli, E, Biondini, D, Casa, GD, et al. Pulmonary fibrosis secondary to COVID-19: a call to arms? Lancet Respir Med. (2020) 8:750–52. doi: 10.1016/s2213-2600(20)30222-8
3. George, PM, Wells, AU, Jenkins, RG. Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. Lancet Respir Med. (2020) 8:807–15. doi: 10.1016/s2213-2600(20)30225-3
4. Reyfman, PA, Walter, JM, Joshi, N, Anekalla, KR, McQuattie-Pimentel, AC, Chiu, S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med. (2019) 199:1517–36. doi: 10.1164/rccm.201712-2410OC
5. Peng, G, Sun, D, Rajashankar, KR, Qian, Z, Holmes, KV, Li, F. Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc Natl Acad Sci USA. (2011) 108:10696–701. doi: 10.1073/pnas.1104306108
6. Merad, M, Martin, JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. (2020) 20:355–62. doi: 10.1038/s41577-020-0331-4
7. Ritchie, AI, Singanayagam, A. Immunosuppression for hyperinflammation in COVID-19: a double-edged sword? Lancet. (2020) 395:1111. doi: 10.1016/s0140-6736(20)30691-7
8. Huang, C, Wang, Y, Li, X, Ren, L, Zhao, J, Hu, Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/s0140-6736(20)30183-5
9. Yang, Y, Shen, C, Li, J, Yuan, J, Yang, M, Wang, F, et al. Exuberant elevation of IP-10, MCP-3 and IL-1ra during SARS-CoV-2 infection is associated with disease severity and fatal outcome. MedRxiv. (2020). [Preprint]. doi: 10.1101/2020.03.02.20029975
10. Liao, M, Liu, Y, Yuan, J, Wen, Y, Xu, G, Zhao, J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
11. Krasemann, S, Madore, C, Cialic, R, Baufeld, C, Calcagno, N, El Fatimy, R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. (2017) 47:566–81.e9. doi: 10.1016/j.immuni.2017.08.008
12. Zhou, Y, Song, WM, Andhey, PS, Swain, A, Levy, T, Miller, KR, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med. (2020) 26:131–42. doi: 10.1038/s41591-019-0695-9
13. Ramachandran, P, Dobie, R, Wilson-Kanamori, JR, Dora, EF, Henderson, BEP, Luu, NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. (2019) 575:512–8. doi: 10.1038/s41586-019-1631-3
14. Boza-Serrano, A, Ruiz, R, Sanchez-Varo, R, García-Revilla, J, Yang, Y, Jimenez-Ferrer, I, et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. (2019) 138:251–73. doi: 10.1007/s00401-019-02013-z
15. Burguillos, MA, Svensson, M, Schulte, T, Boza-Serrano, A, Garcia-Quintanilla, A, Kavanagh, E, et al. Microglia-secreted galectin-3 acts as a toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. (2015) 10:1626–38. doi: 10.1016/j.celrep.2015.02.012
16. Wu, K, Byers, DE, Jin, X, Agapov, E, Alexander-Brett, J, Patel, AC, et al. TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J Exp Med. (2015) 212:681–97. doi: 10.1084/jem.20141732
17. Yang, HZ, Wang, JP, Mi, S, Liu, HZ, Cui, B, Yan, HM, et al. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am J Pathol. (2012) 180:275–92. doi: 10.1016/j.ajpath.2011.09.019
18. Guo, YR, Cao, QD, Hong, ZS, Tan, YY, Chen, SD, Jin, HJ, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak - an update on the status. Mil Med Res. (2020) 7:11. doi: 10.1186/s40779-020-00240-0
19. Dai, JP, Wang, QW, Su, Y, Gu, LM, Deng, HX, Chen, XX, et al. Oxymatrine inhibits influenza a virus replication and inflammation via TLR4, p38 MAPK and NF-κB Pathways. Int J Mol Sci. (2018) 19:965. doi: 10.3390/ijms19040965
20. Shirey, KA, Lai, W, Scott, AJ, Lipsky, M, Mistry, P, Pletneva, LM, et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature. (2013) 497:498–502. doi: 10.1038/nature12118
21. Bhattacharyya, S, Wang, W, Qin, W, Cheng, K, Coulup, S, Chavez, S, et al. TLR4-dependent fibroblast activation drives persistent organ fibrosis in skin and lung. JCI Insight. (2018) 3:98850. doi: 10.1172/jci.insight.98850
22. Tian, F, Chen, LP, Yuan, G, Zhang, AM, Jiang, Y, Li, S. Differences of TNF-α, IL-6 and Gal-3 in lobar pneumonia and bronchial pneumonia caused by mycoplasma pneumoniae. Technol Health Care. (2020) 20:1–9. doi: 10.3233/thc-192011
23. Tanaka, T, Narazaki, M, Kishimoto, T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy. (2016) 8:959–70. doi: 10.2217/imt-2016-0020
24. Zhao, M. Cytokine storm and immunomodulatory therapy in COVID-19: role of chloroquine and anti-IL-6 monoclonal antibodies. Int J Antimicrob Agents. (2020) 20:105982. doi: 10.1016/j.ijantimicag.2020.105982
25. Zuo, W, Zhao, X, Chen, Y-G. SARS coronavirus and lung fibrosis. In: SK Lal editor. Molecular Biology of the SARS-Coronavirus. Berlin: Springer (2010). p. 247–58.
26. Wong, CK, Lam, CW, Wu, AK, Ip, WK, Lee, NL, Chan, IH, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. (2004) 136:95–103. doi: 10.1111/j.1365-2249.2004.02415.x
27. He, L, Ding, Y, Zhang, Q, Che, X, He, Y, Shen, H, et al. Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE2+ cells in SARS patients: relation to the acute lung injury and pathogenesis of SARS. J Pathol. (2006) 210:288–97. doi: 10.1002/path.2067
28. Shi, CS, Nabar, NR, Huang, NN, Kehrl, JH. SARS-coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. (2019) 5:101. doi: 10.1038/s41420-019-0181-7
29. Chen, YJ, Wang, SF, Weng, IC, Hong, MH, Lo, TH, Jan, JT, et al. Galectin-3 enhances avian H5N1 influenza a virus-induced pulmonary inflammation by promoting NLRP3 inflammasome activation. Am J Pathol. (2018) 188:1031–42. doi: 10.1016/j.ajpath.2017.12.014
30. Chen, Y, Wang, H, Shen, J, Deng, R, Yao, X, Guo, Q, et al. Gasdermin D drives the nonexosomal secretion of galectin-3, an insulin signal antagonist. J Immunol. (2019) 203:2712–23. doi: 10.4049/jimmunol.1900212
31. Adam, ML, Boothby, A, Gertner, E. Continuous intravenous anakinra infusion to calm the cytokine storm in macrophage activation syndrome. ACR Open Rheumatol. (2020) 2:276–82. doi: 10.1002/acr2.11135
32. Zhang, C, Wu, Z, Li, JW, Zhao, H, Wang, GQ. The cytokine release syndrome (CRS) of severe COVID-19 and Interleukin-6 receptor (IL-6R) antagonist Tocilizumab may be the key to reduce the mortality. Int J Antimicrob Agents. (2020) 20:105954. doi: 10.1016/j.ijantimicag.2020.105954
33. Boza-Serrano, A, Reyes, JF, Rey, NL, Leffler, H, Bousset, L, Nilsson, U, et al. The role of Galectin-3 in α-synuclein-induced microglial activation. Acta Neuropathol Commun. (2014) 2:156. doi: 10.1186/s40478-014-0156-0
34. Vankadari, N, Wilce, JA. Emerging WuHan (COVID-19) coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect. (2020) 9:601–4. doi: 10.1080/22221751.2020.1739565
35. Kumar, S, Maurya, VK, Prasad, AK, Bhatt, MLB, Saxena, SK. Structural, glycosylation and antigenic variation between 2019 novel coronavirus (2019-nCoV) and SARS coronavirus (SARS-CoV). Virusdisease. (2020) 31:13–21. doi: 10.1007/s13337-020-00571-5
36. Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. (2016) 3:237–61. doi: 10.1146/annurev-virology-110615-042301
37. Bojkova, D, Klann, K, Koch, B, Widera, M, Krause, D, Ciesek, S, et al. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature. (2020) 583:469–72. doi: 10.1038/s41586-020-2332-7
38. York, IA, Stevens, J, Alymova, IV. Influenza virus N-linked glycosylation and innate immunity. Biosci Rep. (2019) 39:BSR20171505. doi: 10.1042/bsr20171505
39. Woodward, AM, Mauris, J, Argüeso, P. Binding of transmembrane mucins to galectin-3 limits herpesvirus 1 infection of human corneal keratinocytes. J Virol. (2013) 87:5841–7. doi: 10.1128/jvi.00166-13
40. King, RD, Lubinski, JM, Friedman, HM. Herpes simplex virus type 1 infection increases the carbohydrate binding activity and the secretion of cellular galectin-3. Arch Virol. (2009) 154:609–18. doi: 10.1007/s00705-009-0351-7
41. Kulkarni, R, Prasad, A. Exosomes derived from HIV-1 infected dcs mediate viral trans-infection via fibronectin and galectin-3. Sci Rep. (2017) 7:14787. doi: 10.1038/s41598-017-14817-8
42. Pais-Correia, AM, Sachse, M, Guadagnini, S, Robbiati, V, Lasserre, R, Gessain, A, et al. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat Med. (2010) 16:83–9. doi: 10.1038/nm.2065
43. Nita-Lazar, M, Banerjee, A, Feng, C, Amin, MN, Frieman, MB, Chen, WH, et al. Desialylation of airway epithelial cells during influenza virus infection enhances pneumococcal adhesion via galectin binding. Mol Immunol. (2015) 65:1–16. doi: 10.1016/j.molimm.2014.12.010
44. Ou, X, Liu, Y, Lei, X, Li, P, Mi, D, Ren, L, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. (2020) 11:1620. doi: 10.1038/s41467-020-15562-9
45. Kovak, MR, Saraswati, S, Schoen, DJ, Diekman, AB. Investigation of galectin-3 function in the reproductive tract by identification of binding ligands in human seminal plasma. Am J Reprod Immunol. (2014) 72:403–12. doi: 10.1111/aji.12273
46. Clarke, NE, Turner, AJ. Angiotensin-converting enzyme 2: the first decade. Int J Hypertens. (2012) 2012:307315. doi: 10.1155/2012/307315
47. Yan, R, Zhang, Y, Li, Y, Xia, L, Guo, Y, Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. (2020) 367:1444–8. doi: 10.1126/science.abb2762
48. Zhao, X, Guo, F, Comunale, MA, Mehta, A, Sehgal, M, Jain, P, et al. Inhibition of endoplasmic reticulum-resident glucosidases impairs severe acute respiratory syndrome coronavirus and human coronavirus NL63 spike protein-mediated entry by altering the glycan processing of angiotensin I-converting enzyme 2. Antimicrob Agents Chemother. (2015) 59:206–16. doi: 10.1128/aac.03999-14
49. Zhai, X, Sun, J, Yan, Z, Zhang, J, Zhao, J, Zhao, Z, et al. Comparison of Severe acute respiratory syndrome coronavirus 2 spike protein binding to ACE2 receptors from human, pets, farm animals, and putative intermediate hosts. J Virol. (2020) 94:20. doi: 10.1128/jvi.00831-20
50. Gupta, R, Charron, J, Stenger, CL, Painter, J, Steward, H, Cook, TW, et al. SARS-CoV-2 (COVID-19) structural and evolutionary dynamicome: Insights into functional evolution and human genomics. J Biol Chem. (2020) 25:14873. doi: 10.1074/jbc.RA120.014873
51. Raj, VS, Mou, H, Smits, SL, Dekkers, DH, Müller, MA, Dijkman, R, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. (2013) 495:251–4. doi: 10.1038/nature12005
52. Wang, K, Chen, W, Zhou, Y-S, Lian, J-Q, Zhang, Z, Du, P, et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. BioRxiv. (2020). [Preprint]. doi: 10.1101/2020.03.14.988345
53. Kovak, MR, Saraswati, S, Goddard, SD, Diekman, AB. Proteomic identification of galectin-3 binding ligands and characterization of galectin-3 proteolytic cleavage in human prostasomes. Andrology. (2013) 1:682–91. doi: 10.1111/j.2047-2927.2013.00099.x
54. Priglinger, CS, Szober, CM, Priglinger, SG, Merl, J, Euler, KN, Kernt, M, et al. Galectin-3 induces clustering of CD147 and integrin-β1 transmembrane glycoprotein receptors on the RPE cell surface. PLoS One. (2013) 8:e70011. doi: 10.1371/journal.pone.0070011
55. Wang, X, Xu, W, Hu, G, Xia, S, Sun, Z, Liu, Z, et al. SARS-CoV-2 infects T lymphocytes through its spike protein-mediated membrane fusion. Cell Mol Immunol. (2020) 7:1–3. doi: 10.1038/s41423-020-0424-9
56. Zhang, T, Sun, LX, Feng, RE. [Comparison of clinical and pathological features between severe acute respiratory syndrome and coronavirus disease 2019]. Zhonghua Jie He He Hu Xi Za Zhi. (2020) 43:E040. doi: 10.3760/cma.j.cn112147-20200311-00312
57. Delaine, T, Collins, P, MacKinnon, A, Sharma, G, Stegmayr, J, Rajput, VK, et al. Galectin-3-binding glycomimetics that strongly reduce bleomycin-induced lung fibrosis and modulate intracellular glycan recognition. Chembiochem. (2016) 17:1759–70. doi: 10.1002/cbic.201600285
58. Rello, J, Storti, E, Belliato, M, Serrano, R. Clinical phenotypes of SARS-CoV-2: implications for clinicians and researchers. Eur Respir J. (2020) 55:1028. doi: 10.1183/13993003.01028-2020
59. Pagnesi, M, Baldetti, L, Beneduce, A, Calvo, F, Gramegna, M, Pazzanese, V, et al. Pulmonary hypertension and right ventricular involvement in hospitalised patients with COVID-19. Heart. (2020) doi: 10.1136/heartjnl-2020-317355
60. Hao, M, Li, M, Li, W. Galectin-3 inhibition ameliorates hypoxia-induced pulmonary artery hypertension. Mol Med Rep. (2017) 15:160–8. doi: 10.3892/mmr.2016.6020
61. Luo, H, Liu, B, Zhao, L, He, J, Li, T, Zha, L, et al. Galectin-3 mediates pulmonary vascular remodeling in hypoxia-induced pulmonary arterial hypertension. J Am Soc Hypertens. (2017) 11:673.e–83.e. doi: 10.1016/j.jash.2017.07.009
62. Barman, SA, Chen, F, Li, X, Haigh, S, Stepp, DW, Kondrikov, D, et al. Galectin-3 promotes vascular remodeling and contributes to pulmonary hypertension. Am J Respir Crit Care Med. (2018) 197:1488–92. doi: 10.1164/rccm.201711-2308LE
63. Barman, SA, Li, X, Haigh, S, Kondrikov, D, Mahboubi, K, Bordan, Z, et al. Galectin-3 is expressed in vascular smooth muscle cells and promotes pulmonary hypertension through changes in proliferation, apoptosis, and fibrosis. Am J Physiol Lung Cell Mol Physiol. (2019) 316:L784–97. doi: 10.1152/ajplung.00186.2018
64. Ten Oever, J, Giamarellos-Bourboulis, EJ, van de Veerdonk, FL, Stelma, FF, Simon, A, Janssen, M, et al. Circulating galectin-3 in infections and non-infectious inflammatory diseases. Eur J Clin Microbiol Infect Dis. (2013) 32:1605–10. doi: 10.1007/s10096-013-1919-4
65. Ho, JE, Gao, W, Levy, D, Santhanakrishnan, R, Araki, T, Rosas, IO, et al. Galectin-3 is associated with restrictive lung disease and interstitial lung abnormalities. Am J Respir Crit Care Med. (2016) 194:77–83. doi: 10.1164/rccm.201509-1753OC
66. Yorulmaz, H, Ozkok, E, Kaptan, E, Ates, G, Tamer, S. Therapeutic effects of simvastatin on Galectin-3 and oxidative stress parameters in endotoxemic lung tissue. Biosci Rep. (2018) 38:3. doi: 10.1042/bsr20180308
67. Hara, A, Niwa, M, Noguchi, K, Kanayama, T, Niwa, A, Matsuo, M, et al. Galectin-3 as a next-generation biomarker for detecting early stage of various diseases. Biomolecules. (2020) 10:389. doi: 10.3390/biom10030389
68. Sato, S, Ouellet, N, Pelletier, I, Simard, M, Rancourt, A, Bergeron, MG. Role of galectin-3 as an adhesion molecule for neutrophil extravasation during streptococcal pneumonia. J Immunol. (2002) 168:1813–22. doi: 10.4049/jimmunol.168.4.1813
Keywords: COVID-19, cytokine storm, fibrosis, galectin-3, biomarker
Citation: Garcia-Revilla J, Deierborg T, Venero JL and Boza-Serrano A (2020) Hyperinflammation and Fibrosis in Severe COVID-19 Patients: Galectin-3, a Target Molecule to Consider. Front. Immunol. 11:2069. doi: 10.3389/fimmu.2020.02069
Received: 12 June 2020; Accepted: 29 July 2020;
Published: 18 August 2020.
Edited by:
Emira Ayroldi, University of Perugia, ItalyReviewed by:
Antonella Naldini, University of Siena, ItalyLu-Gang Yu, University of Liverpool, United Kingdom
Copyright © 2020 Garcia-Revilla, Deierborg, Venero and Boza-Serrano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Garcia-Revilla, amdhcmNpYTExNEB1cy5lcw==; Antonio Boza-Serrano, YW50b25pby5ib3phX3NlcnJhbm9AbWVkLmx1LnNl