 Derry K. Mercer*
Derry K. Mercer* Deborah A. O'Neil
Deborah A. O'Neil- NovaBiotics Ltd., Aberdeen, United Kingdom
The purpose of this review is to describe antifungal therapeutic candidates in preclinical and clinical development derived from, or directly influenced by, the immune system, with a specific focus on antimicrobial peptides (AMP). Although the focus of this review is AMP with direct antimicrobial effects on fungi, we will also discuss compounds with direct antifungal activity, including monoclonal antibodies (mAb), as well as immunomodulatory molecules that can enhance the immune response to fungal infection, including immunomodulatory AMP, vaccines, checkpoint inhibitors, interferon and colony stimulating factors as well as immune cell therapies. The focus of this manuscript will be a non-exhaustive review of antifungal compounds in preclinical and clinical development that are based on the principles of immunology and the authors acknowledge the incredible amount of in vitro and in vivo work that has been conducted to develop such therapeutic candidates.
Introduction
Medical and technological advances, improvements in hygiene and availability of vaccines to important life-threatening diseases means that since 1900 global average life expectancy has more than doubled and is now more than 70 years (1). Despite this, the prevalence of both life-threatening and superficial fungal infections has increased and has largely coincided with progress in the treatment of other diseases (2). Systemic fungal infections are significant causes of morbidity and mortality, responsible for the deaths of more than 1.6 million people per annum (3); comparable to tuberculosis and more than 3-fold higher than malaria. All fungal infections have risen in prevalence over recent decades, including allergic bronchopulmonary aspergillosis (ABPA) and superficial fungal infections, with the increased use of immunosuppressive medications for cancer and transplantation and patients with HIV/AIDS and other immunodeficiences (including genetic disorders), as well as indiscriminate antibiotic use, parenteral nutrition and permanent indwelling catheters. Climate change, pollution and environmental disruption are also considered likely to contribute to the increased incidence of fungal infection and fungal antigenicity (4–7). Defects in innate immune responses, including neutropenia, alveolar macrophage dysfunction, and mutations in STAT3 (resulting in autosomal dominant hyper IgE syndrome) and impaired NAPDH oxidase activity facilitate the development of pulmonary, and in some cases invasive, aspergillosis (8), whereas mutations in the gene for CARD9 (signaling adaptor protein for the C-type lectin receptor) results in increased susceptibility to many types of fungal infection, including dermatophytosis (9, 10). The reasons for the increased incidence of fungal infections over recent decades are beyond the scope of this manuscript and readers are directed to several excellent reviews on the subject (3, 4, 7, 11–15). Fungi are ubiquitous throughout nature and we are constantly exposed to these microbes from the environment via inhalation, ingestion or on epithelial surfaces including the skin and mucosae (16–21). Most fungi are not pathogenic to humans, and most of those that are do not cause life-threatening infections in immunocompetent individuals and such infections are relatively rare. Of the fungi that are able to colonise the human body, many co-exist (commensalism) without causing infection under normal circumstances, e. g. Candida spp. (22, 23). Candida spp., (~750,000 cases of invasive candidiasis/year) Cryptococcus spp. (~225,000 cases per annum in AIDS patients/year) and Aspergillus spp. (~3.75 million cases of chronic pulmonary or invasive aspergillosis/year) are responsible for a significant number of life-threatening fungal infections, whereas other fungi are responsible for substantial levels of systemic infection, including Pneumocystis spp. (~500,000 cases/year), Histoplasma spp. (~500,000 cases/year), Coccidioides spp. (~25,000 cases/year) and mucorales (>10,000 cases/year) (4, 11). Fungi cause superficial infections of the skin, hair, nails and mucosal membranes, including dermatophytes, Candida spp. and Malassezia spp. that are normally readily treatable. There are at least 1 billion cases of superficial fungal infection each year and this is both under-reported and increasing in incidence (3, 24). Dermatophytes are the main cause of superficial fungal infections and each year 20–25% of humans and animals suffer dermatophyte infections (25). Fungal exposure is also thought to contribute to allergies and worsening of asthma symptoms (e.g., ABPA), affecting millions of individuals worldwide (8, 26, 27). Difficulties in diagnosis, the limited antifungal armamentarium, the lack of any fungal vaccines and our limited understanding of the immune response to fungal infection all contribute to this disappointingly high level of morbidity and mortality (Table 1).
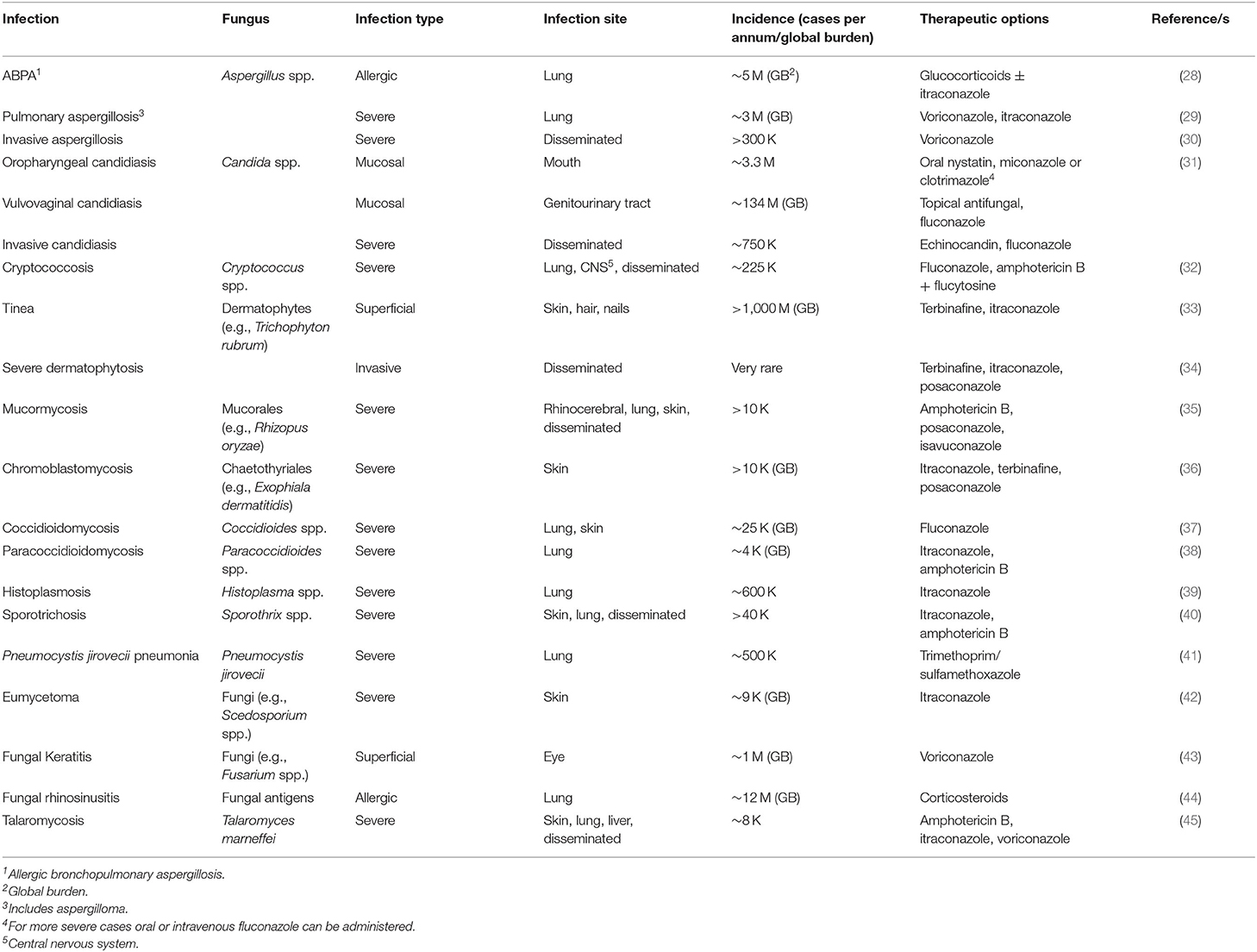
Table 1. Human fungal infections, incidence and treatment options [adapted from (3)].
There is a limited armamentarium of antifungal drugs for the treatment of fungal infection and significantly, drug resistant fungal infections are emerging as important clinical challenges (46–51). Currently available antifungals fall into a limited number of classes; polyenes (e.g., amphotericin B and nystatin), azoles (e.g., fluconazole, itraconazole, voriconazole, isavuconazole, efinaconazole and posaconazole), echinocandins (e.g., caspofungin, anidulafungin and micafungin), allylamines (e.g., terbinafine and naftifine) and other lesser used or topical therapies including flucytosine, ciclopirox olamine, tavaborole, amorolfine, butenafine, griseofulvin, tolnaftate and natamycin. Most serious fungal infections are treated with drugs from only 3 classes; azoles, echinocandins and the polyene, amphotericin B (46, 52) and therefore, resistance to one class of antifungal limits treatment options to a significant degree (51). Whilst resistance rates are low compared to those, for example, of the bacterial ESKAPE pathogens, ~3% of A. fumigatus are resistant to more than one azole, whereas 1.0–1.5% of Candida spp. are resistant to echinocandins and rates of resistance are increasing (47, 48, 51). Analogous to antibiotic resistance, antifungal resistance may be caused by acquired resistance mechanisms as well as primary resistance (also referred to as inherent resistance). For example, azole antifungals inhibit the ergosterol biosynthesis pathway (an essential component of the fungal cell membrane) by targeting lanosterol 14-α-demethylase, encoded by Erg11 in yeasts and Cyp51A/Cyp51B in filamentous fungi. Resistance to azole antifungals can be as a result of over-expression of the target gene (ERG11), loss of function of other enzymes involved in ergosterol biosynthesis (e.g., Δ-5,6-desaturase enzyme Erg3), up-regulation of multidrug transporters (e.g., Cdr1, Cdr2 and Mdr1 in Candida spp.), genome plasticity causing chromosomal duplications (aneuploidy) and the inherent resistance of C. auris to fluconazole (48). The recent emergence of C. auris, a predominantly nosocomial pathogen first isolated from a patient in 2009, is associated with high rates of mortality and antifungal resistance. In the US ~90% of C. auris isolates are fluconazole resistant, 30% are amphotericin B resistant, although <5% of isolates are resistant to echinocandins. Additionally, multi-drug resistance of C. auris has commonly been reported, as has its ability to persist following disinfection of surfaces (49, 50, 53).
Clearly, new therapeutic options for the treatment of fungal infections are urgently needed (54). The global antifungal drug market was valued at US $11.92 Bn in 2018 and is expected to grow to US $13.87 Bn by 2026 (fiormarkets.com, 2020)1. Understanding the immune responses to fungal infection is essential for the rational design of more effective therapies and therefore improved patient outcomes in the future. Depending on the site and type of infection, the immune response can mount fungus-specific and/or site-specific antifungal responses. The development of antifungal drug candidates that replace or correct defective elements or dysregulation in appropriate immune responses to fungal infection and/or enhance the host immune response appear to be logical starting points for the development of new antifungal therapies. Despite the prevalence of fungal infection, its significant morbidity and mortality and the increasing problem of antifungal resistance, antifungal drug development has been under-represented in the development of antimicrobials. The design and development of antifungal therapeutics is, arguably, more complex than the design of antibacterial drugs, as both humans and fungi are eukaryotes and therefore share many common cellular features (55). One of the most obvious differences between fungal and mammalian cells is the cell surface (cell membrane and wall in the case of fungi) and it is perhaps no surprise that the most successful antifungal drugs available today target fungal cell walls (echinocandins) or membranes (azoles, amphotericin B). If we are to design future generations of antifungal drugs, we should look to the immune system as this can readily distinguish between fungi and self and to target fungi for eradication. AMP are one such example of this and are ripe for exploitation as antifungal therapeutic candidates as we discuss in this review (56–59).
Innate Immunity and Human Fungal Infections
In immunocompetent individuals, innate immunity is the first-line of defence against invasive fungal infection. Host defence peptides (HDP), also termed antimicrobial peptides (AMP), form a key part of the innate immune response to infection and inflammation (60–62). HDP have been found at most sites in the human body, including the oral cavity, skin (including sweat and wound fluid), lungs, blood, tears, gastrointestinal tract, urinary tract & reproductive organs, breast milk and cerebrospinal fluid (63). A number of HDP are produced constitutively by epithelia and this basal level of HDP production can provide a first line of protection against fungal infection. Continuous interactions between fungal pathogen-associated molecular patterns (PAMP) and damage-associated molecular patterns (DAMP) and host pattern recognition receptors (PRR) initiate low levels of NF-κB activation that drives amplified expression of HDP-encoding genes (64–66). Upon greater levels of colonisation, inflammation and/or epithelial damage, expression of HDP genes, and concomitant HDP production, increases significantly (67, 68). For example, human β-defensins, cathelicidin and other HDP are considered integral to the innate immune response to fungal infection in the skin (69), whereas histatins are considered key effectors in the oral cavity (70). In addition to direct antimicrobial activity, HDP can act as immune modulators, for example, by promoting migration of neutrophils and monocytes to the site of infection, by upregulating tumour necrosis factor alpha (TNF-α) production and by chemoattraction of immature dendritic and T cells to modify the adaptive immune response (61, 62, 71). Perhaps unsurprisingly, most studies on the antimicrobial activities of AMP have focused on their antibacterial properties. Most research on antifungal AMP has been directed against Candida spp, especially C. albicans, with a smaller number of studies assessing activity against A. fumigatus, Cryptococcus neoformans and the questionably relevant S. cerevisiae. Thus, the direct antifungal activity of HDP, and most other AMP, may be significantly under-realised. In this review we will focus on the direct antifungal activity of AMP and anti-biofilm properties where relevant, but the immunomodulatory properties of HDP/AMP are largely beyond the scope of this manuscript and readers are directed to several excellent reviews on this subject (61, 62, 72–74).
Histatins
Histatins (Hst) are small histidine-rich HDP with an α-helical conformation in membranes. Histatins, and derivatives, have been investigated for their potential to treat localised infections, including vulvovaginal candidiasis, skin infections, cystic fibrosis lung infections, mucositis and gingivitis/periodontitis (75, 76). First isolated from human parotid saliva, Hst are also found in the saliva of other higher primates. Histatins are secreted by the parotid and submandibular salivary glands. Histatins comprise 12 structurally related members of which Hst-1 and Hst-3 are full-length proteins encoded by two genes, HTN1 (encoding Hst-1) and HTN3 (encoding Hst-3). The smaller proteins, Hst-2 (derived from Hst-1) and Hst-4 to−12 (derived from Hst-3), are generated by proteolytic cleavage of the parent Hst by salivary proteases during secretion (59, 77, 78).
Histatins comprise 3 main HDP (Hst -1, -3 and -5), of which Hst-5 (Figure 1A) has the most potent antifungal activity and can be found at concentrations of 15–30 μM in whole saliva (80). Fungicidal activity of Hst has been demonstrated against Candida spp. (albeit with little or no activity against C. glabrata), Cryptococcus neoformans and A. fumigatus (70, 81–83). In a study on the efficacy of Hst-5 on Candida spp. biofilms, Hst-5 was not effective against planktonic C. glabrata (2 isolates; IC50 > 100 μM). However, Hst-5 was effective against preformed biofilms of C. albicans and C. glabrata on poly(methyl methacrylate) discs, resulting in a 50% reduction in biofilm metabolic activity at concentrations of 1.7–62.5 μM (83, 84), albeit less effective than 0.12% chlorhexidine gluconate (84). Hst-1, -3 and -5 can also inhibit germination of C. albicans spores, leading to reduced virulence and ability to cause infection (85, 86).
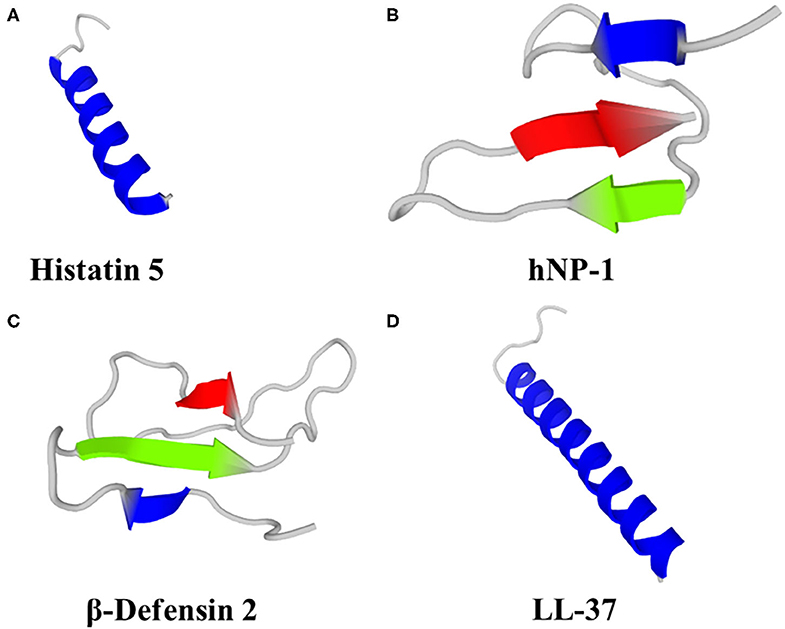
Figure 1. Predicted 3D structures of human HDP; (A) Histatin 5, (B) Neutrophil Peptide 1 (α-defensin), (C) β-Defensin 2 and (D) LL-37. Models were generated using PEP-FOLD 3 (79).
Unlike the membrane-active defensins and cathelicidin, Hst act at multiple levels by mechanisms of action conserved across the Hst family of AMPs. Histatins bind metal ions, including copper, and the presence of Cu improved the antifungal activity of Hst-5 against C. albicans (87). In C. albicans, Hst-5 binds to fungal cell wall glycans, predominantly β-1,3-glucan (88), and cell wall proteins Ssa1 & Ssa2. Hst-5 is transported into the cell via the fungal polyamine transporters Dur3 and Dur31 in an energy-dependent process (76), and it is the lack of these transporters that forms the basis of the lack of sensitivity of C. glabrata to Hst-5 (89). Hst-5 can also be internalized by endocytosis (76) and by direct uptake via interaction with the plasma membrane (90). Hst-5 causes release of K+, via the ion transporter Trk1, which causes osmotic imbalance and a consequent loss of cell volume and viability (76, 91). Hst also induce formation of reactive oxygen species (ROS), ATP efflux, inhibition of oxidative phosphorylation and metal ion chelation and these properties could contribute to the fungicidal activity of Hst-5 (76, 92–94). Human saliva also contains other non-immune proteins with antifungal properties, including lactoperoxidase, lactoferrin and lysozyme (95, 96). Interestingly, the antifungal caspofungin (inhibitor β-1,3-glucan biosynthesis) causes a loss of β-1,3-glucans in the Candida spp. cell wall, resulting in reduced susceptibility to Hst-5 (88).
Additionally, antibacterial properties of Hst-5 against ESKAPE pathogens have been demonstrated, including anti-biofilm properties (97). Hst may also exert their antimicrobial activities by inhibiting host and microbial proteases and may attenuate tissue damage and microbial propagation during the onset of disease (63). Hst have other functions in the oral cavity, including acceleration of wound healing, tooth enamel mineral homeostasis and pellicle formation (78, 98). Hst-1, -2 and -3, but not Hst-5, can promote re-epithelialization and angiogenesis during wound healing (78) and can prevent the translocation of bacteria across cell layers (99).
Defensins
There are three distinct families of defensins, α-, β- and θ-defensins which are cationic AMP characterised as three-stranded β-sheet folds stabilised by three conserved and regiospecific disulphide bridges. Humans produce only α- and β-defensins. In addition to their antimicrobial activity, roles in immunomodulation, fertility, development and wound healing have also been indicated (67, 100–102). The immunomodulatory activities of β-defensins include pro-inflammatory responses via recruitment (chemoattraction) of monocytes, macrophages, immature dendritic cells (DC) and T cells to sites of infection/inflammation, thereby providing a link between the innate and adaptive immune system (101, 103–106).
Humans produce six α-defensins; 4 are produced by neutrophils and some other myeloid cells [Human Neutrophil Peptides (HNP) 1–4] and a further two α-defensins (HD-5 and HD-6) are produced by the Paneth cells of the small intestine and some epithelial cells in the reproductive tract (HD-5 only) (101, 107). HNP-1 (Figure 1B) can kill C. albicans by depleting intracellular ATP (108) and was fungicidal against A. fumigatus (109). HNP-3 demonstrated limited activity against Cryptococcus neoformans planktonic cells and biofilms; 72 and 80% survival, respectively, after exposure to 8 μM HNP-3 for 30 min (110). HD-6 prevented adhesion of C. albicans to human intestinal epithelial cells, thereby preventing biofilm formation and cell invasion, but not hyphal transition. HD-6 functionality against C. albicans is dependent on the self-assembly properties of HD-6 and is non-lethal. HD-6 self-assembles into oligomers, termed nanonets, that entrap pathogens, including C. albicans, and prevent them from entering host cells (111).
Humans produce 4 β-defensins (hBD-1 - 4), primarily from epithelial cells that form biological barriers to pathogens at internal-external interfaces of the skin, gastrointestinal tract, respiratory tract and urogenital tract. Computational and bioinformatic approaches suggest at least 28 human β-defensin genes (112). Human β-defensins have direct antimicrobial activity, including via membrane permeabilization, against bacteria, fungi, viruses and unicellular parasites, as well as roles in immunomodulation, reproduction and pigmentation. Human β-defensin 1 is constitutively expressed, whereas hBD-2 (Figure 1C), 3 and -4 are induced in response to various stimuli, including inflammation and infection (101). hBD-1, hBD-2 and hBD-3 killed C. albicans by membrane permeabilization (113), hBD-2 was fungicidal against A. fumigatus (109) and hBD-3 was fungicidal against C. glabrata (114). hBD-1 in reduced form (i.e., lacking disulphide bridges) demonstrated activity against C. albicans, unlike the oxidised form, and is found in human colonic mucosa, small intestine crypts and skin epidermis (115). hBD-2 and hBD-3 reduced C. albicans adhesion by mediating elevation of Xog1 activity (116). hBD-2 and hBD-9 gene expression was induced by A. fumigatus and hBD-2 peptide co-localised with A. fumigatus conidia that had been phagocytosed by A549 cells (human alveolar basal epithelial adenocarcinoma cells), but not hyphae (117). Antifungal properties of hBD-1, hBD-2 and hBD-3 have been demonstrated against C. albicans (113), including antibiofilm properties of a 15 amino acid fragment from the C-terminus of hBD-3 (118). hBD-1 and hBD-3 were active against Cryptococcus neoformans planktonic cells and biofilms, albeit less effective against biofilms (110).
Cathelicidin
Cathelicidins are cationic HDP containing 12–80 aa (predominantly 23–37 aa) and adopt either α-helix or β-sheet secondary structures in amphipathic helices and include the single human cathelicidin, LL-37 (Figure 1D). The classification of cathelicidins as one family is due to the large evolutionary conserved N-terminal cathelin sequence. However, the highly variable C-terminal region is responsible for most of the broad-spectrum antimicrobial and immunomodulatory activities. Cathelicidin knockout mice were more susceptible to bacterial and viral infection, resulting in a higher morbidity and mortality (119–121). The myriad other properties of cathelicidin have been the subject of several recent reviews (68, 74, 122–124) and are beyond the scope of this manuscript.
The candidacidal activity of LL-37 has been demonstrated in a number of in vitro studies (125–129), but activity against other fungi has been demonstrated in a limited number of reports. Antifungal activity of LL-37 was demonstrated against T. rubrum (n = 2) and T. mentagrophytes (n = 2) with an MIC/MFC = 12.5–25 μM and was fungicidal against Malassezia furfur (25 μM) (130). LL-37 demonstrated antifungal activity (MIC <64 μM) against selected vaginal Candida spp. isolates (C. albicans, C. glabrata, C. krusei and C. parapsilosis), albeit the majority of isolates tested had MIC >64 μM, and was ineffective against preformed C. albicans biofilms at ≤32 μM. LL-37 (64 μM) was able to inhibit adhesion of C. albicans SC5314 to polystyrene and silicone surfaces, thereby preventing biofilm formation (128). LL-37 associated with the cell wall and/or membrane of C. albicans and caused membrane lysis, generation of ROS and release of ATP and other molecules (≤40 kDa) (131). Murine cathelicidin was fungicidal against Pneumocystis murina in a dose-dependent manner (10–50 mg/L) (132). The C. albicans cell wall β-1,3-exoglucanase, Xog1, interacts directly with LL-37 leading to elevated enzyme activity and subsequent cell wall remodelling and reduced adhesion of C. albicans to plastic surfaces (116), oral epidermoid OECM-1 cells and murine urinary bladder at concentrations that were not fungicidal (133). C. albicans that did not adhere were aggregated when LL-37 was bound to the cell surface, mediated by preferential binding to cell wall mannans and to a lesser extent chitin and cell wall glucans (133). Secreted aspartyl proteases (SAP1 – 4, 8 & 9) of C. albicans were able to hydrolyse LL-37 into smaller peptides in vitro and this correlated with a reduction in antifungal and immunomodulatory activity and may facilitate survival of C. albicans at sites where LL-37 is produced (134). Interestingly, the in vitro growth of A. fumigatus and A. flavus was stimulated by physiological concentrations of LL-37 (0.97–31.25 mg/L) found in the lung, whereas a scrambled analogue of LL-37 had no such effect (135).
Other Human Antifungal AMP/HDP
A number of other human AMP/HDP possess documented antifungal activity, including RNases, psoriasin, dermcidin, lactoferricin, antileukprotease/secretory leukocyte protease inhibitor (SLPI), calprotectin, trappin-2/pre-elafin, granulysin, thrombocidins, hepcidins, α-melanocyte stimulating hormone, the chemokine CCL20, substance P, calcitonin gene-related peptide, neuropeptide Y, amyloid β-peptide and vasostatin-1 (136–152).
RNase 3 and RNase 7 demonstrated activity against C. albicans (MFC 2.5–5.0 μmol/L) (151), whereas dermcidin demonstrated pH-dependent activity against C. albicans with optimal activity at pH 5.5–6.5 (143). SLPI was active against A. fumigatus, including spores (137) and C. albicans (153). Hepcidins, Hepc20 and Hepc25, inhibited sporulation of A. fumigatus and A. niger and Hepc20 was fungicidal at 40 μM, whereas both Hepc20 and Hepc25 were only moderately antifungal against C. albicans at 30 μM (~1 log kill) (142). Hepc20 was fungicidal against a panel of C. glabrata (MIC 60–100 μM), which was enhanced in acidic conditions, whereas Hepc25 was not fungicidal (150, 154). The neuropeptides Substance P, Calcitonin gene-related peptide and Neuropeptide Y demonstrated activity against C. albicans (MIC 8.1, 63.1, and 46.5 mg/L, respectively) (146). Lactoferrin and peptides derived from it demonstrated broad-spectrum antifungal activity, including against important pathogenic moulds (e.g., Aspergillus spp., Alternaria spp., Fusarium spp., Absidia spp. and dermatophytes) and yeasts (e.g., Candida spp., Cryptococcus spp. and Exophiala spp.) (152). RNase 7, hBD-2 and psoriasin demonstrated activity against dermatophytes, including T. rubrum, T. mentagrophytes and Epidermophyton floccosum, albeit only psoriasin demonstrated significant activity against Microsporum canis (148). Psoriasin demonstrated broad-spectrum antifungal activity with a 90% MIC of ~2 μM against A. fumigatus, Malassezia furfur, M. canis, Rhizopus oryzae, Saccharomyces cerevisiae, T. rubrum and T. mentagrophytes, but was not active against C. albicans at concentrations up to 20 μM (155).
Novel Antifungal Peptides in Clinical and Preclinical Development
A number of synthetic AMP have been investigated as antifungal therapies (156, 157). AMP with antifungal activity show the same structural diversity as other AMP and include linear and cyclic peptides, lipopeptides and depsipeptides. Over 1100 putative endogenous AMP with antifungal activity have been described (The Antimicrobial Peptide Database; http://aps.unmc.edu/AP/main.php). Antifungal peptides may form α-helices, β-sheets or mixtures thereof and may be cysteine-stabilised. Some are rich in specific amino acids, contain non-natural amino acids or contain non-protein modifications including lipid and carbohydrate moieties. Therapeutic candidate antifungal peptides mostly have a membrane-lytic mechanism of action, but peptides with alternative and even multiple mechanisms of action have been investigated (57–59, 158–161). The structure/composition of fungal cellular membranes vary between species and between yeast and hyphal forms, but in general are more negatively charged than mammalian cell membranes and this may account for the specificity of membrane-active antifungal peptides (58). There have been a number of mechanisms of action both proposed and proven for the interactions of AMP with membranes including the formation of toroidal pores, barrel-stave pores (162), disordered toroidal pores (163), aggregate pores (164), the carpet model (peptide interaction with phospholipid head groups leading to membrane solubilisation) (165). Other less documented mechanisms of action include peptide-induced membrane curvature, induction of cubic lipid phases (166), membrane-thinning/thickening (167), membrane domain formation (168), membrane flip-flop (169), lipid clustering (170) and disruption of membrane potential (171).
NP213 (Novexatin®)
NP213 is a novel, first-in-class, synthetic AMP therapeutic candidate derived from HDP that was designed specifically as a topical therapy for the treatment of onychomycosis (fungal nail infection) by NovaBiotics Ltd. NP213 is a backbone-cyclised homopolymer of 7 L-arginine residues with a net charge of +7. NP213 is rapidly fungicidal against dermatophytes and other fungi causative of onychomycosis and is more active in the presence of human nail and keratin than in conventional antifungal susceptibility testing (RPMI-1640 liquid medium). NP213 was equally effective against dermatophyte spores and hyphae, unlike terbinafine, which demonstrated limited activity against spores, and demonstrated a 3 log kill within 3–4 h, compared to >24 h for terbinafine. NP213 is membranolytic and dependent on its positive charge for activity. NP213 was efficacious in ex vivo models of fungal nail infection, eradicating different Trichophyton rubrum isolates after only 28 d application, unlike ciclopirox and amorolfine (172). Preclinical and clinical safety and toxicity testing revealed no systemic exposure following topical application to the skin of mini-pigs or humans (including a maximum exposure study) with no NP213 detectable in plasma. In clinical trials, NP213 was safe and well tolerated. In two randomized, double-blind, placebo-controlled Phase IIa efficacy studies, daily application of NP213 for 28 d demonstrated clearance of infection in 43.3% (after 180 d; trial 1) and 56.5% (after 360 d; trial 2) of patients with mild-to-moderate onychomycosis (determined by culture) (173). NP213 has also been the subject of a Phase IIb study and further clinical studies are planned.
HXP124
HXP124 is an investigational novel AMP drug candidate in clinical development for the topical treatment of onychomycosis by Hexima Ltd. HXP124 is a novel plant defensin with a cysteine-stabilised αβ-motif structure. HXP124 demonstrated broad-spectrum fungicidal activity against clinically important human pathogens, including Candida spp., Cryptococcus spp., dermatophytes and other moulds. HXP penetrated human nails and was active in an ex vivo model of nail infection. Additionally, HXP124 demonstrated a favourable safety profile in preclinical testing (174). HXP124 has been the subject of a first in human Phase I/IIa trial to evaluate the safety, tolerability and efficacy of daily topical application for 6 weeks in otherwise healthy patients with mild-to-moderate toenail onychomycosis (Australian Clinical Trials ID: ACTRN12618000131257). HXP124 was safe and well-tolerated and substantially reduced the area of infection (>40%) in 15 of 41 patients (37%) analysed after 12 weeks, compared to only 3 of 17 patients (18%) in the vehicle-only group (6 weeks post-treatment) (https://hexima.com.au/).
CZEN-002
CZEN-002 is a synthetic octapeptide, (CKPV)2, derived from α-melanocyte stimulating hormone (α-MSH). α-MSH had previously demonstrated antifungal activity against C. albicans (175). CZEN-220 contains the C-terminal tripeptide (KPV) of α-MSH with a Cys-Cys linker to create an octapeptide. CZEN-002 was candidacidal against C. albicans, C. krusei and C. glabrata at sub-mM concentrations. CZEN-002 is not membranolytic (176). In a rat vaginitis model of C. albicans infection, CZEN-002 dose-dependently reduced the number of surviving C. albicans over 18 d. CZEN-002 inhibited C. albicans phagocytosis by macrophages and inhibited the production of pro-inflammatory cytokines including TNF-α, IL-1β and IL-6, while increasing arginase activity and the secretion of the anti-inflammatory cytokine IL-10, indicating anti-inflammatory properties (177). (CKPV)2 exhibited anti-inflammatory effects against human neutrophils (178) and inhibited TNF-α release form endotoxin-stimulated peripheral blood mononuclear cells in vitro and in vivo (179).
Zengen Inc., developed CZEN-002 for the topical treatment of vulvovaginal candidiasis as an intravaginal gel (56, 180). A phase I/IIa clinical trial reported 88.2% and 87.5% cure (KOH test and culture, respectively) in a total of 18 female VVC patients with VVC that completed the trial in 2004 (https://www.eurekalert.org/pub_releases/2004-05/z-zrp052404.php). A larger dose-ranging Phase IIb clinical trial was planned for 2005 in Canada & EU. The development status of CZEN-002 is not currently known.
P113
P113 (also known as PAC-113, PAC113 and P-113) is a synthetic amphipathic, α-helical 12 amino acid histatin 5 derivative (AKRHHGYKRKFH) with membrane-permeabilising activity against Candida spp. (181) and bacteria (182–185). P113 progressed through clinical development as a topical treatment for oral candidiasis. Complexation with zinc confers greater mechanical stability to the peptide (186). P113 represents the smallest fragment of histatin 5 that retains activity against Candida spp. that was comparable to the parent compound. An analogue of P113 containing D-amino acids, P113D, was equally active against C. albicans. Substitution of the 3 His residues with Phe or Tyr had little effect on activity against C. albicans (MIC 2.2–2.5 mg/L, but substitution of the 2 Arg and 2 Lys residues with Gln abrogated activity (MIC >80 mg/L) (181). P113 was candidacidal against Candida spp. (C. albicans, C. tropicalis, C. famata) in a time- and dose-dependent manner. A series of P113 derivatives have been designed, including a dimer and trimer. P113, P113 dimer and P113 trimer demonstrated limited cytotoxicity against human gingival epithelial cells (LD50 > 400 mg/L). The P113 dimer and trimer were more efficacious than P113 against C. albicans and C. krusei and similarly active against C. tropicalis, C. dubliniensis and C. parapsilosis, whereas C. glabrata was insensitive to all 3 peptides. The P113 trimer retained activity at high sodium acetate concentrations (31.25–93.75 mM), unlike P113 (187). P113, the dimer and the trimer, increased ROS generation and inhibited cellular respiration in C. albicans by targeting mitochondrial complex I. This activity was predominantly caused by inhibition of the NADH dehydrogenase in mitochondrial complex I. The P113 dimer and trimer were also able to target an alternative NADH dehydrogenase not present in mitochondrial complex I. The rapid killing by P113, dimer and trimer mostly occurs via ROS generation, rather than depletion of energy (188). In another study, Candida glabrata was not sensitive to P113 or other histatins and derivatives (189). As well as evidence for P113 causing membranolysis, similar to the histatins from which it is derived, P113 is rapidly taken up into the cytosol of Candida spp. after initial binding to the cell wall, and this process is facilitated by Ssa2p (Heat shock protein 70 (HSP70) chaperone) that can transfer cell wall-bound peptides to membrane permeases to specifically transport peptides into the cytosol. Thus, the antimicrobial activity of P-113 acts through binding to and destabilization of the microbial membrane and through a specific protein receptor on the microbial cell surface (190).
When the His residues at positions 4, 5, and 12 were replaced with the bulky, non-natural amino acids β-naphthylalanine (Nal-P113), salt sensitivity was less pronounced and activity against Candida spp. was retained. Such amino acid substitutions may improve activity under physiological salt concentrations (191). P113 was subject to proteolysis by C. albicans intracellular enzymes at Ala4 and Lys11, whereas P113D was not (192). Based on studies with Hst-5, the Lys residue at position 8 would be subject to cleavage by Candida spp. secreted aspartyl proteases, Sap2 and Sap9 (193). Additionally, histatins (and potentially P113) can form complexes with salivary proteins, e.g., salivary amylase, that can inhibit antifungal activity (194). A possible solution to improve the antifungal efficacy of P113 and other AMP in saliva is to formulate the peptides in delivery systems such as liposomes that facilitate gradual release and limit proteolysis (195). Interestingly, in a rat oral mucosal ulcer model, Nal-P113 increased expression of epidermal growth factor (EGF) and fibroblast growth factor-2 (FGF-2) and decreased the expression of transforming growth factor-β1 (TGF-β1), whereas in an in vitro wound healing assay, Nal-P113 promoted migration of human immortalized oral epithelial cells, indicating that application of Nal-P113 might be an effective therapeutic approach for recurrent aphthous stomatitis (196).
General Biologicals Corporation (GBC) currently market P113-containing compounds as part of their over-the-counter antibacterial “oh-care” range Whilst apparently continuing development of P113 for the treatment of oral candidiasis. A Phase I/IIa clinical trial demonstrated that P113 as an oral mouthrinse was generally safe and well-tolerated and similarly efficacious in curing oral candidiasis as the gold standard, as 37% of PAC-113 patients were assessed as clinically cured at day 14 compared to 36% of Nystatin patients (56, 180). A randomized, examiner-blinded, positive-controlled, parallel design Phase IIb clinical trial of PAC113 oral mouth rinse was carried out in 2008 in 223 HIV seropositive individuals with oral candidiasis and included 3 different concentrations of PAC113 (0.15, 0.075, and 0.0375%) compared to Nystatin oral suspension to determine whether there was elimination or a reduction in clinical signs and symptoms of oral candidiasis. Unfortunately, no results were posted for this trial (ClinicalTrials.gov Identifier: NCT00659971). In a double-blinded, randomized, controlled clinical trial to evaluate the safety and toxicity of three histatin (P-113) concentrations in gel formulations, and to assess potential clinical benefit on the development of gingivitis, 106 healthy subjects without gingivitis were enrolled. All formulations were safe and well-tolerated and efficacy data revealed that P113 gels applied twice daily may reduce experimental gingivitis in humans (197). In another phase 2 multi-centre clinical study, a P113 mouth rinse was safe and well-tolerated and reduced the development of gingivitis in 294 healthy subjects using the formulation twice daily in place normal oral hygiene procedures (198). In a double-blinded, randomized clinical study, 37 patients with moderate or severe chronic periodontitis were treated on one tooth with 20 mg/L Nal-P113 or placebo on days 0 and 3 and on day 7 teeth were sampled. Treatment with Nal-P113 improved periodontal clinical status, reduced plaque/biofilm formation compared to controls (199).
Omiganan
Omiganan (MX-226 or MBI-226) is a synthetic AMP (ILRWPWWPWRRK-amide) derived from indolicidin, originally isolated from bovine neutrophils, with antifungal (200, 201), antibacterial (200, 202), anti-biofilm (203, 204), antiviral (205) and immunomodulatory properties (206). Omiganan was active against Candida spp.; C. albicans (MIC 32–>512 mg/L; n = 104), C. glabrata (MIC 128–>512 mg/L; n = 27), C. krusei (MIC 16–256 mg/L; n = 26), C. parapsilosis (MIC 32–256 mg/L; n = 30) and C. tropicalis (MIC 8–64 mg/L; n = 27) (200) and moulds, including Aspergillus spp. (MIC 16–1,024 mg/L; n = 10), Curvularia spp., Fusarium spp., Paecilomyces variotii and Penicillium spp. (MIC 1–256 mg/L; n = 10) (201). 100 mg/L omiganan caused a 1–2 log kill against C. albicans (n = 3) within 1 h exposure (129). Interestingly, an omiganan analogue with the sequence reversed (KRRWPWWPWRLI-NH2) was more active against C. albicans (Forward MIC 128 mg/L; Reverse MIC 32–64 mg/L) and both were equally effective against A. niger ATCC16404 (MIC 64 mg/L) (207). An all D-enantiomer analogue of omiganan demonstrated the same antimicrobial activity as L-omiganan, but was less susceptible to skin proteases (t1/2 >120 min and t1/2 = 10 min, respectively) (208). In an ex vivo pig skin infection model, ≥0.1% (w/w) omiganan (in an aqueous gel) was active against C. albicans ATCC14053, causing a 2–3 log kill after 24 h, whereas in an in vivo guinea pig skin infection model 1% (w/w) omiganan cased a 2 log kill after 24 h (209).
Omiganan has been the subject of 16 clinical trials in the US and 10 in Europe (ClinicalTrials.gov clinicaltrialsregister.eu), probably making it the most studied AMP in humans, albeit all trials were for topical application, including acne vulgaris, rosacea and sebhorrhoeic dermatitis. Unfortunately, none of these clinical trials investigated the antifungal properties of omiganan, although one trial into the use of omiganan for the prevention of central venous catheter-related bloodstream infections described that they would test for fungaemia, bacteraemia and sepsis (ClinicalTrials.gov Identifier: NCT00027248). Unfortunately, no results for this study, sponsored by BioWest Therapeutics Inc, have been posted. In a later Phase III study of 1,859 hospitalised patients, omiganan 1% gel was compared to 10% povidone-iodine for the prevention of catheter infection/colonisation in patients with central venous catheters, but results were disappointing and the trial failed to achieve its primary efficacy end-point of reducing local catheter site infections (ClinicalTrials.gov Identifier: NCT00231153) (56, 180, 209). In two recent Phase II clinical trials sponsored by Cutanea Life Sciences (EudraCT Number: 2015-002724-16 & 2015-005553-13), the safety and efficacy of omiganan in the treatment of human papillomavirus-induced genital lesions (n = 12) or external ano-genital warts (n = 24) was assessed. Omiganan was safe and well tolerated by all patients. Human papillomavirus load significantly reduced after 12 weeks of treatment with omiganan compared to placebo, but only in the external ano-genital warts patients (205). Whilst clinical development of omiganan appears to be ongoing, omiganan has been proven to be safe and generally well-tolerated as a topical antimicrobial. Its efficacy has yet to be proven in the clinic. Carefully designed trials with appropriate efficacy/outcome measures and application of the peptide in appropriate formulations will be critical to ensure success and potential translation of this compound's promising in vitro antifungal data.
hLF1-11
The AMP hLF1-11 (GRRRRSVQWCA) comprises the first 11 amino acids of human lactoferrin and is a multi-functional peptide with antibacterial activity (185, 210, 211), antifungal activity (212) and immunomodulatory properties (213, 214). hLF1-11 demonstrated antifungal activity in vitro against C. albicans (MIC 22–44 mg/L; n = 11), including oral and vaginal isolates (212, 215). Pre-treatment of fluconazole-resistant C. albicans with non-candidacidal concentrations of hLF1-11 (4–8 μM) was synergistic with fluconazole, rendering this strain fluconazole sensitive. The combination of hLF1-11 and fluconazole was also effective against C. glabrata, C. krusei, C. tropicalis and C. parapsilosis (216). hLF1-11 caused mitochondrial calcium uptake which stimulated an increase in mitochondrial membrane potential and permeability, resulting in the synthesis and secretion of ATP and ROS production, leading to C. albicans cell death (217). hLF1-11 was also active against A. fumigatus hyphae (EC50 29 ± 5 μM) and spores (MIC 5 ± 4 μM) (218).
hLF1-11 (88–176 mg/L) prevented C. albicans biofilm formation with almost complete inhibition of metabolic activity, a 2 log reduction in cell viability (176 mg/L) and decreased expression of selected biofilm-associated genes. However, hLF1-11 demonstrated poor activity against pre-formed biofilms (215). hLF1-11 (88–176 mg/L) prevented C. parapsilosis (n = 3) biofilm formation and 55 mg/L hLF1-11 significantly reduced the amount of biofilm formed. When C. parapsilosis CP7 was allowed to adhere to the surface of 96-well plates or peripheral Teflon catheter pieces for 1.5 or 3 h, hLF1-11 (≥44 mg/L) significantly reduced the amount of biofilm formed and metabolic activity, whereas after being allowed to adhere for 6 h, 44 mg/L hLF1-11 was ineffective at preventing adhered cells developing into biofilms and both 44 and 88 mg/L hLF1-11 were ineffective when C. parapsilosis had been allowed to adhere for 24 h. Incubation of C. parapsilosis CP7 with 44 mg/L hLF1-11 led to reduced expression of the adhesin gene CpALS7, the biofilm formation-associated gene CpACE2 and the β-glucan synthase catalytic sub-unit 1 gene CpFSK1 (219). Coating of hLF1-11 onto titanium surfaces by atom transfer radical polymerization reduced adhesion of Streptococcus sanguinis, Lactobacillus salivarius and a mixed microflora derived from human dental plaque (220), whereas attachment of hLF1-11 to chitosan films via the cysteine residue increased the adhesion of Staphylococcus aureus ATCC33591 to the film, albeit with some reduction in viability (221). Thus, hLF1-11 may have application in prevention of infection of implanted medical devices provided careful consideration is given to the manner of surface attachment. hLF1-11 was not haemolytic at concentrations up to 200 mg/L and caused no significant loss of viability of murine osteoblast MC3T3-e1 cells at a concentration of 400 mg/L (222).
hLF1-11 demonstrated synergistic inhibition of C. albicans SC5314 in combination with caspofungin in vitro. When tested in the Galleria mellonella (wax moth) larva model of infection hLF1-11 was not toxic (≤100 mg/kg), but these concentrations were not effective at improving survival in larvae infected with C. albicans (2.8–3.0 × 105 cfu inoculum) and in this model, the combination of 25 mg/kg hLF-11 and 0.5 mg/kg caspofungin also resulted in no enhanced survival (223). In a neutropenic murine model of systemic candidiasis (established for 24 h) with a fluconazole-resistant C. albicans isolate 0.4 μg/kg hLF1-11 caused a ~1.5 log reduction in C. albicans kidney burden after 18 h and mice treated with up to 40 μg/kg hLF1-11 had smaller and fewer infectious foci in their kidneys and grew predominantly as yeast, unlike the hyphal growth observed in the kidneys of untreated mice. hLF1-11 was also able to inhibit the yeast-hyphal transition in vitro (217).
Exposure of monocytes to hLF1-11 during GM-CSF-driven differentiation is sufficient to direct differentiation of monocytes toward a macrophage subset characterized by both pro- and anti-inflammatory cytokine production (IL-10 and TNF-α) when subsequently exposed to heat-killed C. albicans and these macrophages also demonstrated increased responsiveness to bacterial lipopolysaccharide (LPS), lipoteichoic acid (LTA) and heat-killed C. albicans (213). Following intracellular uptake by monocytes, hLF1-11 bound to myeloperoxidase (MPO) and inhibited the chlorination and peroxidation activity of MPO (224). hLF1-11 also facilitated differentiation of human monocytes to dendritic cells (DC) with increased expression of HLA class II antigens and dectin-1 (a C. albicans PRR) and increased phagocytosis of C. albicans, but not Staphylococcus aureus. Upon stimulation with C. albicans, hLF1-11-differentiated DC produced increased amounts of ROS and the cytokines IL-6 and IL-10, but not IL-12p40 or TNF-α. Supernatants from hLF1–11-differentiated DCs caused CD4+ T cells to produce increased concentrations of IL-17, but reduced IFN-γ, following stimulation with C. albicans (214).
hLF1-11 has been the subject of 4 proposed human trials, sponsored by AM-Pharma, and registered on ClinicalTrials.gov although only one was completed. The completed trial was to determine the safety of a single intravenous dose of hLF1-11 (5 mg, single dose IV) in 8 autologous haematopoietic stem cell transplant recipients (HSCT) (ClinicalTrials.gov Identifier: NCT00509938). The safety and tolerability of hLF1-11 had to be established in HSCT recipients as they are at risk of developing, but have not yet developed, infectious complications due to invasive fungal disease. An earlier study in 48 healthy volunteers (36 hLF1-11 and 12 placebo) had established that single ascending intravenous doses (0.005–5 mg, single dose IV) and multiple intravenous doses (0.5 & 5 mg, single dose IV) were safe and well tolerated. HSCT patients differ from healthy volunteers as they have received myeloablative treatment which arrests haematopoiesis, resulting in neutropenia, but also causes mucosal barrier injury. Both of these predispose HSCT patients to fungal infections which typically occur during the week after transplant. It was therefore essential to know that hLF 1-11 is safe when given during neutropenia and mucosal barrier injury before infections ensue. A single 5 mg (single dose IV) dose was well-tolerated in patients with a side effect of elevated liver enzymes (alanine aminotransferase and aspartate aminotransferase) that was reversible and may have been related to treatment (225). A further study to determine the effect of multiple doses of hLF1-11 in HSCT patients (ClinicalTrials.gov Identifier: NCT00430469) was withdrawn by the sponsor prior to patient recruitment. Another of the withdrawn studies, one was a phase IIa, double-blind, randomized study to determine the tolerability and efficacy of hLF1-11 in patients with proven candidemia with concomitant fluconazole treatment (ClinicalTrials.gov Identifier: NCT00509834), but unfortunately the target patient population was not available. It is clear that hLF1-11 is generally safe and well-tolerated in healthy subjects and HSCT patients at the dose ranges tested thus far and that this peptide has in vitro and preclinical efficacy in models of fungal infection. It remains to be seen how effective this peptide can be in clinical use.
Iseganan (IB-367)
Iseganan (IB-367) is a synthetic AMP containing 17 amino acid residues derived from protegrin I, part of the cathelicidin family of AMP, that has been in clinical development for the treatment of oral mucositis (226–228) and ventilator-associated pneumonia (229). Iseganan was selected as the most promising candidate for the prevention of oral mucositis based on a study of structure–activity relationships of synthetic protegrin analogues (230). Iseganan demonstrated antibacterial activity (231, 232), antifungal activity (233, 234), anti-parasitic (235), anti-biofilm activity (236) and both antibacterial and anti-endotoxin activity in rat models of septic shock (237). Iseganan was fungicidal against dermatophytes (MIC 8–16 mg/L (n = 20; MFC 16–32 mg/L (n = 20) (234). and C. albicans (MIC 4–8 mg/L; MFC 4–32 mg/L (n = 5), C. glabrata (MIC 2–16 mg/L; MFC 2–16 mg/L (n = 5), C. parapsilosis (MIC 8–32 mg/L; MFC 16–>128 mg/L (n = 5), C. krusei (MIC 4–16 mg/L; MFC 4–64 mg/L (n = 5) and C. tropicalis (MIC 2–4 mg/L; MFC 2–4 mg/L (n = 5) (233), although activity against A. fumigatus ATCC16404 was poor (MIC/MFC = 256 mg/L) (238). Local application of Iseganan (IB-367) reduced mucositis severity in a hamster model of oral mucositis which correlated with a >100-fold reduction in oral microflora densities in a dose-dependent manner (239).
A multi-centre double-blind, placebo-controlled Phase III trial to determine the efficacy of Iseganan HCl rinse in reducing the severity of oral mucositis in 323 patients (163 iseganan and 160 placebo) receiving stomatotoxic chemotherapy (PROMPT-CT trial). Iseganan (9 mg in 3 ml) was administered as a swish and swallow solution, six times daily for 21–28 d and was safe and well-tolerated. In this study, 43 and 33% of Iseganan and placebo patients, respectively, did not develop ulcerative oral mucositis. Iseganan patients experienced less mouth pain, throat pain and difficulty swallowing compared to placebo patients and experienced lower stomatitis scores (226). However, other studies failed to demonstrate a benefit of Iseganan in causing reduction in oral mucositis (227, 228). Stomatotoxic chemotherapy can induce changes in the oral microflora that may cause oral and systemic infections in myelosuppressed cancer patients and studies suggest that reduction of the microbial load in the oral cavity has some clinical benefit. A sub-analysis of the first trial was conducted to assess the antimicrobial activity of Iseganan in this patient population. Microbial cultures were generated before and after the daily Iseganan mouth rinse. Iseganan significantly reduced total microbial load in the oral cavity, mainly due to decreased numbers of streptococci and yeasts. This antifungal activity is of interest as oropharyngeal candidiasis is common in immunocompromised patients and some elderly populations (240). A multinational, double-blind, randomized, placebo-controlled trial of Iseganan (371 patients) applied topically to the oral cavity vs. placebo (354 patients) in intubated patients receiving mechanical ventilation for up to 14 d was conducted to determine the occurrence of microbiologically confirmed ventilator-associated bacterial pneumonia (VAP) measured among survivors up through Day 14 (ClinicalTrials.gov Identifier: NCT00118781). The peptide was deemed to be safe and well tolerated but the study's primary efficacy end-points were not met [no significant differences in the rate of VAP among survivors between patients treated with Iseganan (16%) and those treated with placebo (20%; p = 0.145) (229)]. The design of the study was potentially flawed due to the short exposure time of Iseganan to potential pathogens (241). Thus, as a proven safe and well tolerated candidate when applied topically in very sick patients with preclinical antifungal activity, Iseganan has the potential to be developed as an AMP for the treatment of oropharyngeal candidiasis and for topical application for the treatment of other fungal infections.
LTX-109
LTX-109 (LTX109, Lytixar, LTX 109) is an AMP peptidomimetic (Arg-Tbt-Arg-NH-EtPh) that was in clinical development by Lytix Biopharma AS. LTX-109 contains 2 arginine residues, a central modified tryptophan residue (2,5,7-tri(tert-butyl)tryptophan) and an ethylphenyl group at the C-terminus (242) with antibacterial (243, 244) and antifungal activity (245). LTX-109 was fungicidal against S. cerevisiae (MIC 8 mg/L), causing a 3 log kill within 60 min, and was also active against pre-formed S. cerevisiae biofilms. LTX-109 disrupted S. cerevisiae membrane integrity by a sphingolipid-dependent mechanism (245).
Topical LTX-109 has been the subject of 3 clinical trials in Gram-positive bacterial infections; nasal decolonisation of Staphylococcus aureus (Clinical trials identifier: NCT01158235), a role in non-bullous impetigo (Clinical trials identifier: NCT01803035) and Gram positive skin infections including patients with mild eczema/dermatoses such as atopic dermatitis (Clinical trials identifier: NCT01223222). The study for nasal decolonisation of Staphylococcus aureus was a randomized, double-blind, dose escalation phase I/IIa study conducted at a single centre to compare the efficacy, safety, tolerability, bioavailability and efficacy of 3 days nasal treatment with LTX-109 (TID) applied directly to the anterior nares vs. vehicle in persistent nasal carriers of Staphylococcus aureus. LTX-109 was safe and well-tolerated and treatment with LTX-109 resulted in a reduction in Staphylococcus aureus counts after only 1 day of application. A significant reduction of the number of CFU below the detection limit compared to the vehicle group was demonstrated in subjects treated with 2 and 5% LTX-109 after 2 days of treatment. The most frequently reported AEs related to the application site were itching, burning, pain, and redness (n = 26) and the subjects in the 2 and 5% LTX-109 treatment groups reported more of these symptoms than did the 1% or vehicle groups (246). Unfortunately, no results are available for the other 2 LTX-109 clinical trials. Given the positive clinical safety and tolerability data following topical application over multiple days in bacterial infection, together with promising antifungal activity in vitro, LTX-109 could be a promising candidate for the treatment of fungal infection.
Antibiofilm Peptides
The ability of fungi to form biofilms have been associated with high rates of morbidity and mortality, yet compared to bacterial biofilms and bacterial anti-biofilm compounds, the field of fungal biofilm research remains in its infancy. Fungal biofilms consist of adherent cells (on biotic or abiotic surfaces) surrounded by an extracellular matrix which can reduce antifungal efficacy and impair immune responses (247, 248). In addition to direct antifungal activity some AMP/HDP, in vitro, can prevent biofilm formation and/or eradicate preformed biofilms via mechanisms associated with fungal adhesion, cell wall perturbation, generation of ROS and gene regulation (59, 249). Although not yet in clinical use, the search for AMP with “druggable” antibiofilm properties remains ongoing (56, 250). For example, in the case of Cryptococcus neoformans biofilms, formation is dependent on the production of the polysaccharide capsule (251). hBD-1 and hBD-3 were active against Cryptococcus neoformans planktonic cells and biofilms, albeit less effective against biofilms (110), whereas lactoferrin was not effective against Cryptococcus neoformans biofilms (251). Hst-5 was effective against planktonic C. albicans (IC50 2.6–4.8 μM; n = 3), but not C. glabrata (IC50 > 100 μM; n = 2). However, Hst-5 was active against preformed biofilms of C. albicans and C. glabrata on poly(methyl methacrylate) discs, resulting in a 50% reduction in biofilm metabolic activity at concentrations of 1.7–6.9 μM (C. albicans; n = 3) and 31.2–62.5 μM (C. glabrata; n = 2) (83). LL-37 was able to prevent C. albicans biofilm formation on silicone elastomer discs (used in the manufacture of medical devices) at sub-MIC concentrations without a concomitant reduction in C. albicans viability, whereas LL-37 had no effect on pre-formed C. albicans biofilms (128). Thus, AMP have promise as anti-biofilm agents against fungi as well as bacteria.
Future Directions for Antifungal Peptide Design and Development
Antifungal Peptides in Preclinical Development
A limited number of AMP are in preclinical development for the treatment of fungal infections and have been extensively reviewed (156, 157, 252–254). In this section we will provide a non-exhaustive review of some of the later stage preclinical antifungal AMP candidates likely to be closer to clinical testing.
NP339 is a preclinical drug candidate being developed as an intravenous therapy for life threatening invasive fungal disease (bloodstream and deep tissue fungal infections) including those caused by yeasts and moulds that are resistant to existing antifungal therapies. An inhaled form of NP339 is also under development for direct delivery into the airways in patients with, or at risk of respiratory fungal infections, including Allergic Bronchial Pulmonary Aspergillosis (ABPA) and pulmonary fungal infections in cystic fibrosis patients. NP339 is a synthetic 2 kDa cationic linear AMP that has been engineered from β-defensins.
NP339 targets the fungal membrane and kills fungi by membrane disruption and cell lysis. This mechanism of action is specific to fungal cells and NP339 is not cytotoxic at significantly higher concentrations than are required to achieve antifungal activity. NP339 kills more rapidly than conventional classes of antifungals, including against metabolically active and inactive fungi and is also sporicidal. NP339 is active against a broad range of clinically relevant fungal pathogens, including Aspergillus spp., Candida spp. and Cryptococcus spp., as well as emerging fungal pathogens including Mucorales, Scedosporium spp. and Exophiala spp. (255). Nebulised NP339 as a monotherapy, or in combination with amphotericin B, elicited a reduction in lung burden relative to vehicle in murine models of invasive pulmonary aspergillosis (256).
In addition to P113 (see Section P113), Demegen had a second AMP, D2A21, in pre-clinical development (257). D2A21 was a synthetic peptide derived from cecropin (258) being investigated for a number of antimicrobial indications and was formulated as a topical gel (Demegel). D2A21 demonstrated in vitro antifungal activity against C. albicans, A. niger, Mucor spp. and T. mentagrophytes, as well as antibacterial, antiparasitic and potential anti-tumorigenic activity (257). Potential antimicrobial indications include fungal infections, sexually-transmitted infections caused by Chlamydia trachomatis (259), and Trichomonas vaginalis (260) (for which in vitro activity was demonstrated) and burn wound infections (261, 262), In an in vivo infected burn wound model in Wistar rats, D2A21 demonstrated significant antibacterial activity against P. aeruginosa infection, sterilized burn eschar and decreased the bacterial load in subeschar, leading to significantly improved survival (261, 262).
ETD151 is a preclinical AMP drug candidate derived from ARD1 (a heliomycin peptide), a naturally occurring AMP from the lepidopteran Heliothis virescens (tobacco budworm). ETD151, developed by EntoMed SA, is a 44 aa AMP intended for the treatment of serious invasive fungal infections affecting immunocompromised patients (263). ETD151 was derived from ARD1 by site-directed mutagenesis, following recombinant expression in Saccharomyces cerevisiae to create a peptide with increased cationicity (264). ETD151 demonstrated promising antifungal activity in vitro (MIC50 0.1–6.25 mg/L against C. albicans, Cryptococcus neoformans, A. fumigatus, F. solani and Scedosporium prolificans) (264). In murine models of systemic C. albicans or A. fumigatus infection, EDT-151 was effective when compared to amphotericin B and azoles and was non-toxic following intravenous administration (263). ETD151 has yet to enter clinical trials to the knowledge of the authors, however, most recently, the antifungal activity of ETD151 has been assessed against Botrytis cinerea, a necrotrophic plant pathogen responsible for gray mold disease, for use as a fungicide in crop protection (265).
Preclinical Activity Testing
Antimicrobial peptides, whether antifungal, antibacterial, antiparasitic or antiviral, cannot be developed through the same preclinical and clinical pathways as small molecule drugs. We cannot assume or expect that methods for determining antimicrobial activity that are employed in the development of antibiotics and other “small” molecule antimicrobials will be appropriate for the development of AMP as drug candidates. We say “small” as the authors acknowledge that many clinically used antimicrobials do not obey the traditional definition of small, i.e., <500 Da, from Lipinski's rule of five (266), but are nevertheless generally smaller than most AMP. Evaluation of AMP as antimicrobial drug candidates begins with in vitro antimicrobial susceptibility testing in which a number of key parameters need to be taken into consideration, including media composition, growth phase, oxygen, temperature and other biological matrices (Table 2) (267, 268). This also applies to in vitro cytotoxicity testing (269, 270), formulation and delivery considerations (see Section Formulation and Delivery) and the choice of models for in vivo testing (271). It is probable that with adequate consideration given to the factors outlined above and also appropriately designed clinical trials there would be significantly more AMP in preclinical and clinical development and the importance of this is described in detail in a new review of the subject (268).

Table 2. Factors influencing preclinical antimicrobial activity testing of AMP.
Rational Drug Design
As stated above, most manuscripts describing AMP &/or peptidomimetics focus on antibacterial properties, but when considering the AMP themselves, and not their target, most reports focus on the isolation of AMP from increasingly unusual organisms (272–274), library screening (275–277) and attempting to identify or modify AMP to have the highest possible level of antimicrobial activity (i.e., lowest MIC). This is perhaps reflected by the fact that The Antimicrobial Peptide Database (http://aps.unmc.edu/AP/main.php) now contains over 3100 entries. Despite this, no AMP has achieved approval by the regulatory authorities as an antimicrobial therapeutic in clinical practice.
Whilst our understanding of the biology and function of AMP remains incomplete, especially how peptides behave in complex biological systems, we are gaining sufficient insight that researchers are increasingly making use of this biological knowledge and even computational approaches to design novel, synthetic AMP (278–282). Novel, informed drug-design approaches to identify AMP is aided by the vast sequence space available (78, 283). Other approaches have taken known host defence peptides and attempted to optimise them using a variety of approaches (281, 284–286).
At a less complex level, rational drug design principles can be applied to designing AMP to target specific pathogens at specific anatomical sites. As described above, NP213 has completed Phase II clinical trials for the treatment of onychomycosis (173). NP213 was designed at the outset as an antifungal peptide, but one that also needed to have specific physicochemical properties that would facilitate penetration into human nail (172). Human nail is a highly effective biological barrier and delivery of therapeutics to the nail and nail bed is challenging (287, 288). Additionally, keratin, the major constituent of the nail, binds to and inactivates many of the existing small molecule antifungal classes, thus compromising therapy (289). AMP/HDP are expressed and produced in the nail (290–293) and several HDP/AMP are antifungal against dermatophytes, including LL-37 (130), hBD-2, RNase7 and Psoriasin (148). AMP therefore constituted a logical starting point for the design of a novel therapeutic for the treatment of onychomycosis. NP213 is highly hydrophilic and positively charged (net charge +7), properties that should facilitate nail penetration as the nail is a negatively-charged, concentrated hydrogel under physiological conditions (294). Additionally, NP213 is small compared to most AMP/HDP (7 aa vs. ~12–>50 aa) that are already known to penetrate nail (292, 293) and this low molecular weight should also facilitate nail penetration (295). One of the known drawbacks of peptide/protein therapeutic candidates is susceptibility to hydrolysis, especially proteolysis (296), which is of especial concern with respect to dermatophytes as they are known to produce multiple classes of proteases/peptidases that enable them to hydrolyse keratin (297, 298). NP213 is a cyclic peptide and therefore not prone to hydrolysis by exoproteases and the limited sequence diversity within NP213 limits the classes of endoproteases that could hydrolyse NP213 (https://www.ebi.ac.uk/merops). Therefore, even prior to peptide synthesis, NP213 had been designed to function as an antifungal at this unique site of infection.
Formulation and Delivery
In comparison to the considerable body of research focusing on the discovery of AMP and the optimisation of their activity, considerably less effort has been given to delivery systems, formulation or routes of administration for AMP. Formulation and delivery of AMP will play key roles in efficacy outcomes including reducing degradation of protease-susceptible AMP, limiting binding to plasma and other proteins and macromolecules, controlling dose-exposure parameters and even potentially targeting pathogens directly (e.g., intracellular pathogens or pathogens in biofilms). This topic merits a separate manuscript and several excellent reviews have already been written to that end (299–303).
As has been published widely, an issue for the development of certain peptide therapeutics is the potential for proteolysis, whether by proteases of host or microbial origin (304, 305). Infected tissue is often characterised by high levels of proteases, both microbially- and host-derived (306). Possible solutions to the problem of proteolysis include formulation of the peptide to afford protection from proteases, including liposomal formulations, as used for other drugs (307), use of non-natural or D-enantiomer amino acids (308, 309), design and development of peptidomimetics (310, 311) and multivalent peptides (312).
When considering formulation of AMP, the characteristics of both the AMP and the carrier require consideration. AMP charge (and its type and distribution), size, solubility, hydrophobicity and structure can affect loading and activity, as can the properties of the carrier including charge, pH, ionic strength, pore/mesh size, conjugation method (where appropriate). Formulation and delivery approaches that have been tested for AMP include the use of hydrogels (313–315), liposomal formulations (195, 316), carbon nanotubes (317), PEGylation (270, 299) and nanoparticles (318, 319). Appropriate formulation and delivery strategies may also allow us to resurrect and re-investigate some of the candidate AMP therapies that have previously been abandoned because in vivo and/or clinical efficacy was significantly diminished vs. in vitro data.
Other Antifungal Immunotherapeutics
The antifungal properties of endogenous HDP are such that these peptides are obvious templates for the design and development of synthetic therapeutic antifungal AMP. As described in preceding sections of this review, AMP have shown early promise as therapeutic candidates. The optimal clinical pathway (trial design, endpoints, formulation etc.) to demonstrate translation of their therapeutic potential into clinical use may not have been carved out as yet however, to the detriment of a number of molecules no longer in development as a result. There are, however, other potential immunotherapeutics that could be deployed alongside antifungal AMP and even existing classes of antifungal therapy; in each case to further enhance infection resolution and eradication. In particular, invasive (systemic) fungal infections predominantly affect immunocompromised patients and there are potential benefits in strengthening those aspects of the immune response that remain functional in these individuals in order to combat systemic fungal infection (30, 320, 321). In cases of invasive aspergillosis or systemic candidiasis, clinical practice guidelines recommend reduction or reversal of immune suppression (31, 322, 323), but in many cases this is simply not feasible due to the initial pathology in cases of stem cell malignancy. In some cases, the reversal of immune suppression can result in immune reconstitution inflammatory syndrome (IRIS), causing increased morbidity and mortality due to “cytokine storm” and an exaggerated host inflammatory response (324, 325). Identifying patients, therefore, for whom particular antifungal immunotherapies are appropriate is critical. It is essential to avoid overtly “boosting” any aspect of the host response in patients who are not entirely immunodeficient in order to mitigate potential immunotoxicity or hyperinflammation. Directly acting antifungal AMP with no host cell pharmacology are potentially the class of immunotherapy with broadest cross-patient applicability for fungal disease in this context. The development of biomarkers to predict responses to antifungal immunotherapy may be beneficial for broader, future adoption of fungal immunotherapy (326) and clinical trial design for these treatments will also require careful consideration as potential patient pools are likely to be limited compared to oncology trials where immunotherapeutics are more commonly used.
Adjunct immunotherapy strategies include the adoptive transfer of activated immune cells with antifungal activity, the administration of immune-activating cytokines in combination with antifungal therapy or the use of antibody therapy. Other approaches being studied include transfusion of leukocytes pre-loaded with antifungals, modulated T cells (e.g., stimulated ex vivo and re-infused) and investigation of potential vaccine strategies (321, 327–332). Some of these approaches will be described in subsequent sections.
Immunostimulatory Molecules
Interferon-γ
A number of clinical studies have demonstrated beneficial effects of recombinant interferon-γ (IFN-γ) administration in combination with antifungal therapy in immunocompromised patients with systemic fungal infections, including Candida spp. and Aspergillus spp. infection (n = 8 patients) (333), chronic granulomatous disease (CGD) (n = 130) (334–336), HIV infection (n = 173) (337–339), leukaemia (n = 5) (340, 341), and transplant patients (n = 7) (342), in a single patient with S. aureus liver abscess and invasive C. albicans infection (343), in a single patient with intracerebral aspergillosis (344), in two patients with progressive chronic pulmonary aspergillosis (345), and in two patients with idiopathic CD4 lymphopenia and cryptococcal meningitis (346). In the study of Delsing and co-workers, rIFN-γ administration partially restored immune function as evidenced by increased production of proinflammatory cytokines involved in antifungal defence by leukocytes (IL-1β, TNFα, IL-17, and IL-22) and increased human leukocyte antigen DR (HLA-DR) positive monocyte production in patients where levels were low (333). IFN-γ is FDA-approved for the treatment Chronic Granulomatous Disease patients at risk of invasive fungal and other infections in combination with antifungal therapy and Granulocyte-macrophage colony-stimulating factor (GM-CSF) (347).
Colony Stimulating Factors
In cancer patients with chemotherapy-associated neutropenia, the prophylactic use Granulocyte Colony-Stimulating Factor (G-CSF; e.g., filgrastim) is FDA-approved and results in a decrease in rates of infection and infection-related morbidity (all causes) in patients receiving cancer therapy or undergoing stem-cell transplantation, although the effect on infection-related mortality was moderate (348). In a clinical study of patients with haematological malignancy and suspected or proven systemic fungal infection, nearly twice as many responded to amphotericin B therapy with concomitant G-CSF compared to those receiving amphotericin B alone (349). Another small study (8 patients with leukaemia (n = 7) or breast cancer (n = 1) demonstrated that adjuvant therapy with G-CSF in addition to amphotericin B resulted in cure (n = 4), partial response (n = 2) or failure (n = 2), indicating potential utility of G-CSF in resolving fungal infection in patients with malignancy (350). In another study, G-CSF in combination with fluconazole resulted in faster infection resolution in non-neutropenic patients with invasive candidiasis/candidemia (324, 351). Treatment with G-CSF before chemotherapy resulted in a dose-dependent increase in the number of neutrophils and treatment after chemotherapy initiation reduced the number of days on which the neutrophil count was ≤1,000/μl, the number of days on which antibiotics were used to treat fever and the incidence and severity of mucositis was decreased (352). G-CSF also enhanced the respiratory burst response of human phagocytes in vitro to fungal conidia or yeast cells, but not hyphae (353).
Granulocyte-macrophage colony-stimulating factor (GM-CSF; e.g., sargramostim) promotes neutrophil, monocyte, macrophage and lymphocyte production, maturation, activation and migration (as well as progenitor cells), whereas G-CSF primarily affects myeloblasts and neutrophils and M-CSF primarily affects only monocytes. GM-CSF is licensed for the treatment of chemotherapy-associated neutropenia and stem cell transplantation (354, 355) and is likely to have advantages over G-CSF therapy due to its wider effects on fungi and the immune system (324). In a randomized trial of patients receiving allogenic haematopoietic stem cell transplantation HSCT, 100-day cumulative mortality and 100-day transplantation-related mortality were lower in patients receiving GM-CSF than receiving G-CSF and after 600 days of follow-up infection-related mortality and invasive fungal disease-related mortality was lower in the GM-CSF group compared to the G-CSF group (355). In other studies of acute myeloid leukaemia patients, administration of GM-CSF led to recovery of neutrophil counts and was associated with a more rapid clearance of infection when compared with a historical control group that did not receive GM-CSF (356), including fungal infections (357). In a small study of neutropenic patients with fungal infection, eight patients received amphotericin B and GM-CSF. Six patients responded to treatment, with four undergoing complete recovery, whereas the remaining two patients died of fungal infection. Although this study did not have controls, the survival rate is higher than would be infected from antifungal treatment alone (358). In a study of 11 AIDS patients with fluconazole-refractory oropharyngeal candidiasis that received GM-CSF and fluconazole, a mycological response was seen in seven patients and three patients were cured (359). Three patients with rhinocerebral zygomycosis were successfully treated with adjunctive GM-CSF when added to antifungal therapy (amphotericin B) and surgery (360). However, in a study of acute myelogenous leukaemia in elderly patients (55–75 years), GM-CSF therapy (114 patients) did not improve complete remission rates when compared to patients receiving placebo (126 patients), but did prolong disease-free survival and overall survival. The number of patients with infections, including serious fungal infections, was not different between the GM-CSF and placebo groups (361).
Macrophage Colony-Stimulating Factor (M-CSF) can rapidly increase myeloid differentiation of haematopoietic stem cells. In a study of bone marrow transplant patients that developed invasive fungal infection and that received recombinant human M-CSF (rhM-CSF), survival was greater than historical patients not receiving rhM-CSF with Candida spp. infection, but not in patients with Aspergillus spp. infection or in any patients with Karnofsky scores of <20% (362, 363). Exogenous M-CSF was protective in murine models of Aspergillus spp. and Pseudomonas aeruginosa infection following haematopoietic stem cell or progenitor cell transplantation and was more efficacious than G-CSF (364). Synergy of M-CSF with fluconazole was observed in human monocyte-derived macrophages infected with Cryptococcus neoformans. M-CSF alone also reduced counts of Cryptococcus neoformans in this model (365) and in a murine model of Cryptococcus neoformans infection (366). In a rat model of acute candidiasis, administration of ≥0.1 mg/kg M-CSF with 0.3 mg/kg fluconazole enhanced survival (>30 d) compared with fluconazole alone (5 d) and similarly reduced C. albicans kidney burden in a chronic model of candidiasis (367). Conversely, another study of mice infected intravenously with C. albicans demonstrated that treatment with M-CSF exacerbated disease and led to significantly earlier death (368). Clearly, M-CSF has potential in the treatment of invasive fungal infection, either alone, or in combination with antifungal therapy, but more research is clearly required and it is possible that the effect may be dependent on the infecting pathogen. Thus, whilst showing clear promise the use of colony-stimulating factor therapy should be the subject of appropriately controlled clinical studies in patients with accurately diagnosed fungal infections and comparable antifungal therapeutic regimens.
Immune Checkpoint Inhibitors
Immune checkpoints are important regulators of immune homeostasis. Immune checkpoints consist of both stimulatory and inhibitory pathways that are important for maintaining self-tolerance and regulating the type, magnitude, and duration of the immune response (369, 370). Immune checkpoint therapies in oncology target regulatory pathways in T cells to enhance anti-tumour responses (370–373) and are used for the treatment of squamous-cell carcinoma and advanced melanoma. The checkpoint programmed cell death 1 (PD1) (a member of the B7-CD28 superfamily) is expressed on monocytes, natural killer cells, T- and B-lymphocytes. Binding of PD1 to the ligand PD1-L1 on myeloid cells impairs T-cell functions including cytokine production and cytotoxic activity, whereas blocking binding of PD1 to its ligand with an anti-PD1 antibody can restore immune function. Cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) is another immune checkpoint that can impair T-cell function and Ipilimumab (an anti-CTLA-4 antibody) was the first immune checkpoint inhibitor approved for the treatment of cancer (374). The PD1 and CTLA-4 pathways have roles to play in antifungal defences (374), as demonstrated in vitro in a murine model of Histoplasma capsulatum infection (PD1) (375) and in blood from patients with paracoccidioidomycosis (CTLA-4) (376). In a murine model of C. albicans sepsis, antibodies to PD1 and PD-L1 were effective at improving survival, as was an antibody to CTLA-4 in this model (377) and in a murine model of Cryptococcus neoformans infection (378). Nivolumab, an antibody drug that blocks PD1, was used successfully in combination with IFN-γ and antifungal therapy (liposomal amphotericin B and posaconazole) in a case of invasive mucormycosis following unsuccessful antifungal therapy for 28 days (379). The use of Nivolumab for immune checkpoint inhibition in sepsis (documented or suspected infection) has been the subject of a recent Phase 1b clinical trial (NCT02960854) (380).
Vaccines
It is estimated that vaccination prevented at least 10 million deaths globally between 2010 and 2015 (381). No fungal vaccine has yet been approved for use in humans although clinical trials of fungal vaccines have been reported and a number are in preclinical and clinical development (382, 383). Our ever-improving knowledge of the immune system ought to increase the likelihood of developing fungal vaccines, but a number of challenges exist and for a number of infectious diseases, treatment rather than vaccination remains the optimal strategy. Eliciting a protective response to immunisation in immunocompromised individuals who have developed/are at risk of invasive fungal infection is unlikely, particularly without risk of aggravating underlying disease and/or development of the fungal infection due to attenuated vaccine administration (382–384). Additionally, developing a vaccine against commensal microorganisms, e.g., Candida spp. could represent an additional problem (385). The high costs associated with vaccine development are a challenge considering that revenue will only be obtained from vaccinating only populations at risk of developing fungal infection, or in the case of endemic mycosis, only a limited patient population cannot attract sufficient investment (386).
A vaccine (NDV-3A) containing the N-terminal portion of the agglutinin-like sequence 3 (Als3) protein of C. albicans, is in development by NovaDigm Therapeutics for the prevention of recurrent vulvovaginal candidiasis (VVC). Als3 is a hyphal-specific virulence factor that mediates adherence to and invasion of human epithelial and vascular endothelial cells. In a Phase II randomized, double-blind, placebo-controlled clinical trial, NDV-3A demonstrated a statistically significant increase in the percentage of symptom-free patients at 12 months after vaccination and a doubling in the median time to first symptomatic episode for a subset of patients aged <40 years (ClinicalTrials.gov Identifiers: NCT01926028 and NCT02996448) (382, 387). Another vaccine, PEV7, has been the subject of a successful Phase I clinical trial (ClinicalTrials.gov Identifier: NCT01067131) for the prevention of recurrent VVC. PEV7 was developed by Pevion Biotech (rights subsequently acquired by NovaDigm Therapeutics) and contains recombinant secreted aspartyl protease 2 (rSAP-2) incorporated into influenza virisomes. Trial results demonstrated the generation of specific and functional B cell memory in 100% of the vaccinated women and a favourable safety profile (388). Earlier reports of an oral vaccine, D.651, for the prevention of VVC recurrence was prepared using ribosomes of C. albicans serotypes a and b. A Phase II clinical trial reported a good safety profile and efficacy, in which 13 of 20 patients taking the vaccine did not experience recurrence of VVC during the 6 months taking the vaccine (389). The current status of this vaccine is not known. In another study, a vaccine consisting of formaldehyde-killed spherules of Coccidioides immitis was tested in humans, but a statistically significant reduction of the incidence of infection was not observed in those vaccinated (390). A number of other fungal vaccines have been tested in animal models and are beyond the scope of this manuscript, but have been the subject of several recent reviews (328, 383, 388, 391–394). The vaccines described above represent the only ones to reach clinical trials to the best of the authors knowledge.
On a cautionary note, in some cases, live, attenuated fungi (Blastomyces dermatitidis and Histoplasma capsulatum) have demonstrated the induction of protective immunity in mice (395). Naturally, caution would be required before testing live attenuated fungi in immunocompromised individuals although live, attenuated vaccines are arguably much more appropriate candidates for vaccination against endemic fungal infections, such as histoplasmosis and sporotrichosis, in otherwise immunocompetent, healthy subjects.
Interestingly, heat-killed Saccharomyces cerevisiae administered as a vaccine was protective against systemic aspergillosis, candidiasis, cryptococcosis and coccidioidomycosis in mouse models (396), but to the best of our knowledge has not yet been tested in humans.
Antifungal Monoclonal Antibodies
Monoclonal antibodies (mAb) represent some of the world's best-selling therapeutics, of which more than 80 have received marketing approval and more than 100 are in development. In 2018 alone, twelve new mAb were approved by the FDA, representing 20% of the total number of approved drugs and sales of mAb were forecast to reach US $125 Bn by 2020. Most therapeutic monoclonal antibodies are used for the treatment of cancer or immunological disorders (397, 398). The development of monoclonal antibodies for the prevention and treatment of infectious diseases lags somewhat behind their development for other therapeutic areas, e.g., cancer and autoimmune diseases (399), and only three monoclonal antibodies have received approval for infectious disease prophylaxis or treatment; palivizumab for prevention of respiratory syncytial virus in high-risk infants (400); and obiltoxaximab (401) and raxibacumab (402) for prophylaxis and treatment of anthrax. The lack of development of mAb for infectious diseases may be because consensus on clinical end-points and definitions on conditions of use are lacking, as well as high costs associated with their development and lack of a cle arly defined market for these products (399). Fungal-specific mAb can mediate protection from fungal infection by direct action on fungal cells or via promotion of phagocytosis and complement activation. However, some mAb to fungi can be disease-enhancing or have no effect (403). Protective mAb against human fungal pathogens are currently in preclinical development (382), including examples with narrow spectrum reactivity [e.g., mAb 3D9.3 (anti-Als3) that specifically recognises C. albicans (404)] and broad-spectrum reactivity with a number of fungal pathogens (e.g., mAb C7 (anti-Als3) which inactivates germ tubes and spores of Candida spp., Cryptococcus neoformans, A. fumigatus and Scedosporium prolificans (405).
A murine mAb, 18B7, was raised against Cryptococcus neoformans and bound to capsular glucuronoxylomannan in infected mouse tissues (406). 18B7 was protective in a murine intraperitoneal model of Cryptococcus neoformans infection (407). In a human Phase I dose escalation study of human immunodeficiency virus (HIV)-infected patients who had been successfully treated for cryptococcal meningitis, the maximum tolerated dose was established as 1.0 mg/kg and serum cryptococcal antigen titres declined by a median of 3-fold at 2 weeks post-infusion. However, titres subsequently returned toward the baseline values by week 12, 3 of 4 subjects in the 1.0-mg/kg dosing cohort had a 0.5 log10 increase in HIV load and 18B7 was not detected in cerebrospinal fluid (408).
Interestingly, Rudkin et al. generated the first set of fully human anti-Candida spp. mAb isolated from B cells of patients suffering from candidiasis and that demonstrated morphology-specific, high avidity binding to the cell wall, including mAb specific for the C. albicans hyphal cell wall protein Hyr1. Cell wall mAb demonstrated cross-reactivity with other Candida spp., whereas anti-Hyr1 mAb were cross-reactive with only C. albicans. Importantly, tested mAb promoted phagocytosis of C. albicans by macrophages and reduced fungal burden in therapeutic or prophylactic murine models of disseminated candidiasis (409), but these have yet to be tested in humans. Efungumab (Mycograb) is a recombinant human mAb against fungal HSP90 with activity against C. albicans, C. krusei, C. tropicalis, C. glabrata, and C. parapsilosis (410, 411). In vitro studies revealed synergy with fluconazole, amphotericin B (AmB) and caspofungin, and in a murine model of systemic candidiasis, efungumab improved the killing of Candida spp. (C. albicans, C. krusei, and C. glabrata) in combination with AmB (412). However, the combination effect of efungumab and AmB was later revealed to be a nonspecific protein effect, as addition of efungumab or other unrelated proteins, including human serum, resulted in similar decreases in the MIC of AmB (413). Although clinical trials of this product were conducted, they were unsuccessful and development of this drug candidate has been abandoned.
Therefore, the potential for the use of mAb for treatment or prophylaxis against fungal infection remains a possibility, but large-scale clinical trials will be required to bring this promise to fruition.
Conclusions
Antimicrobial peptides are promising candidates as therapeutics for the treatment of fungal infection and are much needed in clinical practice due to the limited array of treatment options and increasing resistance to existing antifungals. Unfortunately, we are not seeing enough drug candidates making it through the drug development pipeline, as in vitro and in vivo testing approaches are not always appropriate and/or optimised for AMP (268). The same is true in part for clinical efficacy trials which must be appropriate for AMP (end points in particular). These factors are undoubtedly part of the reason behind there not being more AMP progressing through the drug development cycle and/or AMP candidates are confined to topical therapy status as delivery systems, formulation, routes of administration and duration of therapy for AMP have not been adequately optimised. The time is now coming for greater exploitation of AMP and other immunotherapeutics as antifungal drug candidates as we gain a greater understanding of how best to test these drug candidates in vitro and how regulatory pathways and clinical studies can be more accommodating for peptides (Table 3). As the global AMR crisis worsens and emerging fungal diseases increase, the potential of these drug candidates must be fulfilled sooner rather than later.
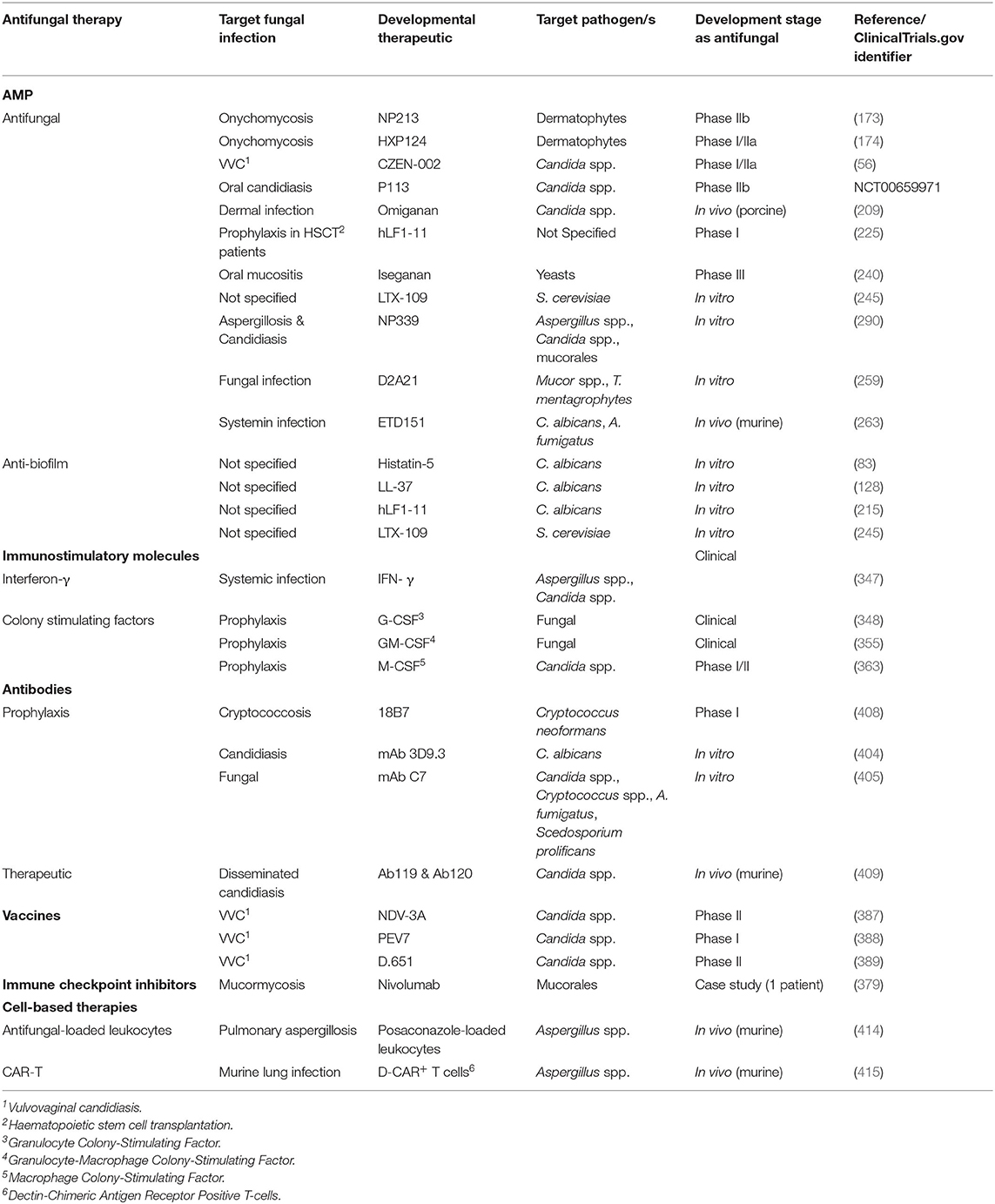
Table 3. Selected immunology-based approaches for the treatment of fungal infection.
Author Contributions
DM and DO'N contributed to the writing and editing of this manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
DM is an employee of NovaBiotics Ltd., and holds stock options. DO'N is a Director, shareholder, and employee of NovaBiotics.
Footnotes
1. ^Antifungal Drugs Market by Drug Class (Azoles, Echinocandins, Polyenes, Allyamines, Others), Indication, Dosage Form, Regions, Global Industry Analysis, Market Size, Share, Growth, Trends, and Forecast 2019 to 2026. Available online at: https://www.fiormarkets.com/report/antifungal-drugs-market-by-drug-class-azoles-echinocandins-407129.html
References
1. Roser M, Ortiz-Ospina E, Ritchie H. Life Expectancy. OurWorldInData.org (2020). Available online at: https://ourworldindata.org/life-expectancy (accessed January 1, 2020).
2. Brown GD, Wilson D. Mammalian innate immunity to fungal infection. Semin Cell Dev Biol. (2019) 89:1–2. doi: 10.1016/j.semcdb.2018.06.004
3. Bongomin F, Gago S, Oladele RO, Denning DW. Global and multi-national prevalence of fungal diseases-estimate precision. J Fungi. (2017) 3:57. doi: 10.3390/jof3040057
4. Benedict K, Richardson M, Vallabhaneni S, Jackson BR, Chiller T. Emerging issues, challenges, and changing epidemiology of fungal disease outbreaks. Lancet Infect Dis. (2017) 17:e403–11. doi: 10.1016/S1473-3099(17)30443-7
5. Bartemes KR, Kita H. Innate and adaptive immune responses to fungi in the airway. J Allergy Clin Immunol. (2018) 142:353–63. doi: 10.1016/j.jaci.2018.06.015
6. Casadevall A. Global catastrophic threats from the fungal kingdom : fungal catastrophic threats. Curr Top Microbiol Immunol. (2019) 424:21–32. doi: 10.1007/82_2019_161
7. Eguiluz-Gracia I, Mathioudakis AG, Bartel S, Vijverberg SJH, Fuertes E, Comberiati P, et al. The need for clean air: the way air pollution and climate change affect allergic rhinitis and asthma. Allergy. (2020). doi: 10.1111/all.14177
8. Mackel JJ, Steele C. Host defense mechanisms against Aspergillus fumigatus lung colonization and invasion. Curr Opin Microbiol. (2019) 52:14–9. doi: 10.1016/j.mib.2019.04.003
9. Lanternier F, Pathan S, Vincent QB, Liu L, Cypowyj S, Prando C, et al. Deep dermatophytosis and inherited CARD9 deficiency. N Engl J Med. (2013) 369:1704–14. doi: 10.1056/NEJMoa1208487
10. Drummond RA, Franco LM, Lionakis MS. Human CARD9: a critical molecule of fungal immune surveillance. Front Immunol. (2018) 9:1836. doi: 10.3389/fimmu.2018.01836
11. Enoch DA, Yang H, Aliyu SH, Micallef C. The changing epidemiology of invasive fungal infections. Methods Mol Biol. (2017) 1508:17–65. doi: 10.1007/978-1-4939-6515-1_2
12. Zhan P, Liu W. The changing face of dermatophytic infections worldwide. Mycopathologia. (2017) 182:77–86. doi: 10.1007/s11046-016-0082-8
13. Cilloniz C, Dominedo C, Alvarez-Martinez MJ, Moreno A, Garcia F, Torres A, et al. Pneumocystis pneumonia in the twenty-first century: HIV-infected versus HIV-uninfected patients. Expert Rev Anti Infect Ther. (2019) 17:787–801. doi: 10.1080/14787210.2019.1671823
14. Friedman DZP, Schwartz IS. Emerging fungal infections: new patients, new patterns, and new pathogens. J Fungi. (2019) 5:67. doi: 10.3390/jof5030067
15. Batista BG, Chaves MA, Reginatto P, Saraiva OJ, Fuentefria AM. Human fusariosis: an emerging infection that is difficult to treat. Rev Soc Bras Med Trop. (2020) 53:e20200013. doi: 10.1590/0037-8682-0013-2020
16. Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. Hidden killers: human fungal infections. Sci Transl Med. (2012) 4:165rv113. doi: 10.1126/scitranslmed.3004404
17. Brown GD, Denning DW, Levitz SM. Tackling human fungal infections. Science. (2012) 336:647. doi: 10.1126/science.1222236
18. Brown GD, May RC. Editorial overview: host-microbe interactions: fungi. Curr Opin Microbiol. (2017) 40:v–vii. doi: 10.1016/j.mib.2017.11.026
19. Li J, Vinh DC, Casanova JL, Puel A. Inborn errors of immunity underlying fungal diseases in otherwise healthy individuals. Curr Opin Microbiol. (2017) 40:46–57. doi: 10.1016/j.mib.2017.10.016
20. Lionakis MS, Levitz SM. Host control of fungal infections: lessons from basic studies and human cohorts. Annu Rev Immunol. (2018) 36:157–91. doi: 10.1146/annurev-immunol-042617-053318
21. Clark C, Drummond RA. The hidden cost of modern medical interventions: how medical advances have shaped the prevalence of human fungal disease. Pathogens. (2019) 8:45. doi: 10.3390/pathogens8020045
22. Limon JJ, Skalski JH, Underhill DM. Commensal fungi in health and disease. Cell Host Microbe. (2017) 22:156–65. doi: 10.1016/j.chom.2017.07.002
23. Paterson MJ, Oh S, Underhill DM. Host-microbe interactions: commensal fungi in the gut. Curr Opin Microbiol. (2017) 40:131–7. doi: 10.1016/j.mib.2017.11.012
24. Ameen M. Epidemiology of superficial fungal infections. Clin Dermatol. (2010) 28:197–201. doi: 10.1016/j.clindermatol.2009.12.005
25. Gnat S, Nowakiewicz A, Lagowski D, Zieba P. Host- and pathogen-dependent susceptibility and predisposition to dermatophytosis. J Med Microbiol. (2019) 68:823–36. doi: 10.1099/jmm.0.000982
26. Rudert A, Portnoy J. Mold allergy: is it real and what do we do about it? Expert Rev Clin Immunol. (2017) 13:823–35. doi: 10.1080/1744666X.2017.1324298
27. Wiesner DL, Klein BS. Lung epithelium: barrier immunity to inhaled fungi and driver of fungal-associated allergic asthma. Curr Opin Microbiol. (2017) 40:8–13. doi: 10.1016/j.mib.2017.10.007
28. Agarwal R, Sehgal IS, Dhooria S, Aggarwal AN. Developments in the diagnosis and treatment of allergic bronchopulmonary aspergillosis. Expert Rev Respir Med. (2016) 10:1317–34. doi: 10.1080/17476348.2016.1249853
29. Denning DW, Cadranel J, Beigelman-Aubry C, Ader F, Chakrabarti A, Blot S, et al. Chronic pulmonary aspergillosis: rationale and clinical guidelines for diagnosis and management. Eur Respir J. (2016) 47:45–68. doi: 10.1183/13993003.00583-2015
30. Cadena J, Thompson GR III, Patterson TF. Invasive aspergillosis: current strategies for diagnosis and management. Infect Dis Clin North Am. (2016) 30:125–42. doi: 10.1016/j.idc.2015.10.015
31. Pappas PG, Kauffman CA, Andes DR, Clancy CJ, Marr KA, Ostrosky-Zeichner L, et al. Clinical practice guideline for the management of candidiasis: 2016 update by the infectious diseases society of America. Clin Infect Dis. (2016) 62:e1–50. doi: 10.1093/cid/civ1194
32. Maziarz EK, Perfect JR. Cryptococcosis. Infect Dis Clin North Am. (2016) 30:179–206. doi: 10.1016/j.idc.2015.10.006
33. Moriarty B, Hay R, Morris-Jones R. The diagnosis and management of tinea. BMJ. (2012) 345:e4380. doi: 10.1136/bmj.e4380
34. Rouzaud C, Hay R, Chosidow O, Dupin N, Puel A, Lortholary O, et al. Severe dermatophytosis and acquired or innate immunodeficiency: a review. J Fungi. (2015) 2:4. doi: 10.3390/jof2010004
35. Lewis RE, Kontoyiannis DP. Epidemiology and treatment of mucormycosis. Fut Microbiol. (2013) 8:1163–75. doi: 10.2217/fmb.13.78
36. Queiroz-Telles F, de Hoog S, Santos DW, Salgado CG, Vicente VA, Bonifaz A, et al. Chromoblastomycosis. Clin Microbiol Rev. (2017) 30:233–76. doi: 10.1128/CMR.00032-16
37. Gabe LM, Malo J, Knox KS. Diagnosis and management of coccidioidomycosis. Clin Chest Med. (2017) 38:417–33. doi: 10.1016/j.ccm.2017.04.005
38. Martinez R. New trends in paracoccidioidomycosis epidemiology. J Fungi. (2017) 3:1. doi: 10.3390/jof3010001
39. Azar MM, Hage CA. Clinical perspectives in the diagnosis and management of histoplasmosis. Clin Chest Med. (2017) 38:403–15. doi: 10.1016/j.ccm.2017.04.004
40. Lopes-Bezerra LM, Mora-Montes HM, Zhang Y, Nino-Vega G, Rodrigues AM, de Camargo ZP, et al. Sporotrichosis between 1898 and 2017: the evolution of knowledge on a changeable disease and on emerging etiological agents. Med Mycol. (2018) 56(suppl_1):126–43. doi: 10.1093/mmy/myx103
41. Sokulska M, Kicia M, Wesolowska M, Hendrich AB. Pneumocystis jirovecii–from a commensal to pathogen: clinical and diagnostic review. Parasitol Res. (2015) 114:3577–85. doi: 10.1007/s00436-015-4678-6
42. Nenoff P, van de Sande WW, Fahal AH, Reinel D, Schofer H. Eumycetoma and actinomycetoma–an update on causative agents, epidemiology, pathogenesis, diagnostics and therapy. J Eur Acad Dermatol Venereol. (2015) 29:1873–83. doi: 10.1111/jdv.13008
43. Austin A, Lietman T, Rose-Nussbaumer J. Update on the management of infectious keratitis. Ophthalmology. (2017) 124:1678–89. doi: 10.1016/j.ophtha.2017.05.012
44. Tyler MA, Luong AU. Current understanding of allergic fungal rhinosinusitis. World J Otorhinolaryngol Head Neck Surg. (2018) 4:179–85. doi: 10.1016/j.wjorl.2018.08.003
45. Cao C, Xi L, Chaturvedi V. Talaromycosis (penicilliosis) due to Talaromyces (penicillium) marneffei: insights into the clinical trends of a major fungal disease 60 years after the discovery of the pathogen. Mycopathologia. (2019) 184:709–20. doi: 10.1007/s11046-019-00410-2
46. Robbins N, Wright GD, Cowen LE. Antifungal drugs: the current armamentarium and development of new agents. Microbiol Spectr. (2016) 4:FUNK-0002-2016. doi: 10.1128/microbiolspec.FUNK-0002-2016
47. Perlin DS, Rautemaa-Richardson R, Alastruey-Izquierdo A. The global problem of antifungal resistance: prevalence, mechanisms, and management. Lancet Infect Dis. (2017) 17:e383–92. doi: 10.1016/S1473-3099(17)30316-X
48. Revie NM, Iyer KR, Robbins N, Cowen LE. Antifungal drug resistance: evolution, mechanisms and impact. Curr Opin Microbiol. (2018) 45:70–6. doi: 10.1016/j.mib.2018.02.005
49. Chaabane F, Graf A, Jequier L, Coste AT. Review on antifungal resistance mechanisms in the emerging pathogen Candida auris. Front Microbiol. (2019) 10:2788. doi: 10.3389/fmicb.2019.02788
50. Hendrickson JA, Hu C, Aitken SL, Beyda N. Antifungal resistance: a concerning trend for the present and future. Curr Infect Dis Rep. (2019) 21:47. doi: 10.1007/s11908-019-0702-9
51. Berman J, Krysan DJ. Drug resistance and tolerance in fungi. Nat Rev Microbiol. (2020) 18:319–31. doi: 10.1038/s41579-019-0322-2
52. McKeny PT, Zito PM. Antifungal Antibiotics. Treasure Island, FL: StatPearls Publishing (2020). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK538168/ (accessed January, 2020).
53. Kenters N, Kiernan M, Chowdhary A, Denning DW, Peman J, Saris K, et al. Control of Candida auris in healthcare institutions: Outcome of an International Society for Antimicrobial Chemotherapy expert meeting. Int J Antimicrob Agents. (2019) 54:400–6. doi: 10.1016/j.ijantimicag.2019.08.013
54. Su H, Han L, Huang X. Potential targets for the development of new antifungal drugs. J Antibiot. (2018) 71:978–91. doi: 10.1038/s41429-018-0100-9
55. Roemer T, Krysan DJ. Antifungal drug development: challenges, unmet clinical needs, and new approaches. Cold Spring Harb Perspect Med. (2014) 4:a019703. doi: 10.1101/cshperspect.a019703
56. Duncan VMS, O'Neil DA. Commercialization of antifungal peptides. Fung Biol Rev. (2013) 26:156–65. doi: 10.1016/j.fbr.2012.11.001
57. Mercer DK, O'Neil DA. Peptides as the next generation of anti-infectives. Future Med Chem. (2013) 5:315–37. doi: 10.4155/fmc.12.213
58. Rautenbach M, Troskie AM, Vosloo JA. Antifungal peptides: To be or not to be membrane active. Biochimie. (2016) 130:132–45. doi: 10.1016/j.biochi.2016.05.013
59. Oshiro KGN, Rodrigues G, Monges BED, Cardoso MH, Franco OL. Bioactive peptides against fungal biofilms. Front Microbiol. (2019) 10:2169. doi: 10.3389/fmicb.2019.02169
60. Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. (2005) 3:238–50. doi: 10.1038/nrmicro1098
61. Haney EF, Straus SK, Hancock REW. Reassessing the host defense peptide landscape. Front Chem. (2019) 7:43. doi: 10.3389/fchem.2019.00043
62. van der Does AM, Hiemstra PS, Mookherjee N. Antimicrobial host defence peptides: immunomodulatory functions and translational prospects. Adv Exp Med Biol. (2019) 1117:149–71. doi: 10.1007/978-981-13-3588-4_10
63. Bastos P, Trindade F, da Costa J, Ferreira R, Vitorino R. Human antimicrobial peptides in bodily fluids: current knowledge and therapeutic perspectives in the postantibiotic era. Med Res Rev. (2018) 38:101–46. doi: 10.1002/med.21435
64. Swidergall M, Ernst JF. Interplay between Candida albicans and the antimicrobial peptide armory. Eukaryot Cell. (2014) 13:950–7. doi: 10.1128/EC.00093-14
65. Gow NAR, Latge JP, Munro CA. The fungal cell wall: structure, biosynthesis, and function. Microbiol Spectr. (2017) 5:FUNK-0035-2016. doi: 10.1128/9781555819583.ch12
66. Patin EC, Thompson A, Orr SJ. Pattern recognition receptors in fungal immunity. Semin Cell Dev Biol. (2019) 89:24–33. doi: 10.1016/j.semcdb.2018.03.003
67. Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol. (2005) 6:551–7. doi: 10.1038/ni1206
68. Vandamme D, Landuyt B, Luyten W, Schoofs L. A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell Immunol. (2012) 280:22–35. doi: 10.1016/j.cellimm.2012.11.009
69. Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol. (2012) 12:503–16. doi: 10.1038/nri3228
70. Tsai H, Bobek LA. Human salivary histatins: promising anti-fungal therapeutic agents. Crit Rev Oral Biol Med. (1998) 9:480–97. doi: 10.1177/10454411980090040601
71. Steinstraesser L, Kraneburg U, Jacobsen F, Al-Benna S. Host defense peptides and their antimicrobial-immunomodulatory duality. Immunobiology. (2011) 216:322–33. doi: 10.1016/j.imbio.2010.07.003
72. Mansour SC, Pena OM, Hancock RE. Host defense peptides: front-line immunomodulators. Trends Immunol. (2014) 35:443–50. doi: 10.1016/j.it.2014.07.004
73. Pfalzgraff A, Brandenburg K, Weindl G. Antimicrobial peptides and their therapeutic potential for bacterial skin infections and wounds. Front Pharmacol. (2018) 9:281. doi: 10.3389/fphar.2018.00281
74. van Harten RM, van Woudenbergh E, van Dijk A, Haagsman HP. Cathelicidins: immunomodulatory antimicrobials. Vaccines. (2018) 6:63. doi: 10.3390/vaccines6030063
75. Rothstein DM, Helmerhorst EJ, Spacciapoli P, Oppenheim FG, Friden P. Histatin-derived peptides: potential agents to treat localised infections. Expert Opin Emerg Drugs. (2002) 7:47–59. doi: 10.1517/14728214.7.1.47
76. Puri S, Edgerton M. How does it kill - Understanding the candidacidal mechanism of salivary Histatin 5. Eukaryot Cell. (2014). doi: 10.1128/EC.00095-14
77. Edgerton M, Koshlukova SE, Lo TE, Chrzan BG, Straubinger RM, Raj PA. Candidacidal activity of salivary histatins. Identification of a histatin 5-binding protein on Candida albicans. J Biol Chem. (1998) 273:20438–47. doi: 10.1074/jbc.273.32.20438
78. Torres P, Castro M, Reyes M, Torres VA. Histatins, wound healing, and cell migration. Oral Dis. (2018) 24:1150–60. doi: 10.1111/odi.12816
79. Lamiable A, Thevenet P, Rey J, Vavrusa M, Derreumaux P, Tuffery P. PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. (2016) 44:W449–54. doi: 10.1093/nar/gkw329
80. Campese M, Sun X, Bosch JA, Oppenheim FG, Helmerhorst EJ. Concentration and fate of histatins and acidic proline-rich proteins in the oral environment. Arch Oral Biol. (2009) 54:345–53. doi: 10.1016/j.archoralbio.2008.11.010
81. Troxler RF, Offner GD, Xu T, Vanderspek JC, Oppenheim FG. Structural relationship between human salivary histatins. J Dent Res. (1990) 69:2–6. doi: 10.1177/00220345900690010101
82. Helmerhorst EJ, Reijnders IM, van't Hof W, Simoons-Smit I, Veerman EC, Amerongen AV. Amphotericin B- and fluconazole-resistant Candida spp., Aspergillus fumigatus, and other newly emerging pathogenic fungi are susceptible to basic antifungal peptides. Antimicrob Agents Chemother. (1999) 43:702–4. doi: 10.1128/AAC.43.3.702
83. Konopka K, Dorocka-Bobkowska B, Gebremedhin S, Duzgunes N. Susceptibility of Candida biofilms to histatin 5 and fluconazole. Antonie Van Leeuwenhoek. (2010) 97:413–7. doi: 10.1007/s10482-010-9417-5
84. Pusateri CR, Monaco EA, Edgerton M. Sensitivity of Candida albicans biofilm cells grown on denture acrylic to antifungal proteins and chlorhexidine. Arch Oral Biol. (2009) 54:588–94. doi: 10.1016/j.archoralbio.2009.01.016
85. Xu T, Levitz SM, Diamond RD, Oppenheim FG. Anticandidal activity of major human salivary histatins. Infect Immun. (1991) 59:2549–54. doi: 10.1128/IAI.59.8.2549-2554.1991
86. Lin AL, Shi Q, Johnson DA, Patterson TF, Rinaldi MG, Yeh CK. Further characterization of human salivary anticandidal activities in a human immunodeficiency virus-positive cohort by use of microassays. Clin Diagn Lab Immunol. (1999) 6:851–5. doi: 10.1128/CDLI.6.6.851-855.1999
87. Conklin SE, Bridgman EC, Su Q, Riggs-Gelasco P, Haas KL, Franz KJ. Specific histidine residues confer histatin peptides with copper-dependent activity against Candida albicans. Biochemistry. (2017) 56:4244–55. doi: 10.1021/acs.biochem.7b00348
88. Jang WS, Bajwa JS, Sun JN, Edgerton M. Salivary histatin 5 internalization by translocation, but not endocytosis, is required for fungicidal activity in Candida albicans. Mol Microbiol. (2010) 77:354–70. doi: 10.1111/j.1365-2958.2010.07210.x
89. Tati S, Jang WS, Li R, Kumar R, Puri S, Edgerton M. Histatin 5 resistance of Candida glabrata can be reversed by insertion of Candida albicans polyamine transporter-encoding genes DUR3 and DUR31. PLoS ONE. (2013) 8:e61480. doi: 10.1371/journal.pone.0061480
90. Helmerhorst EJ, Breeuwer P, van't Hof W, Walgreen-Weterings E, Oomen LC, Veerman EC, et al. The cellular target of histatin 5 on Candida albicans is the energized mitochondrion. J Biol Chem. (1999) 274:7286–91. doi: 10.1074/jbc.274.11.7286
91. Baev D, Li XS, Dong J, Keng P, Edgerton M. Human salivary histatin 5 causes disordered volume regulation and cell cycle arrest in Candida albicans. Infect Immun. (2002) 70:4777–84. doi: 10.1128/IAI.70.9.4777-4784.2002
92. Koshlukova SE, Lloyd TL, Araujo MW, Edgerton M. Salivary histatin 5 induces non-lytic release of ATP from Candida albicans leading to cell death. J Biol Chem. (1999) 274:18872–9. doi: 10.1074/jbc.274.27.18872
93. Helmerhorst EJ, Troxler RF, Oppenheim FG. The human salivary peptide histatin 5 exerts its antifungal activity through the formation of reactive oxygen species. Proc Natl Acad Sci USA. (2001) 98:14637–42. doi: 10.1073/pnas.141366998
94. Baev D, Rivetta A, Vylkova S, Sun JN, Zeng GF, Slayman CL, et al. The TRK1 potassium transporter is the critical effector for killing of Candida albicans by the cationic protein, Histatin 5. J Biol Chem. (2004) 279:55060–72. doi: 10.1074/jbc.M411031200
95. Woods CM, Hooper DN, Ooi EH, Tan LW, Carney AS. Human lysozyme has fungicidal activity against nasal fungi. Am J Rhinol Allergy. (2011) 25:236–40. doi: 10.2500/ajra.2011.25.3631
96. Nakano M, Suzuki M, Wakabayashi H, Hayama K, Yamauchi K, Abe F, et al. Synergistic anti-candida activities of lactoferrin and the lactoperoxidase system. Drug Discov Ther. (2019) 13:28–33. doi: 10.5582/ddt.2019.01010
97. Du H, Puri S, McCall A, Norris HL, Russo T, Edgerton M. Human salivary protein histatin 5 has potent bactericidal activity against ESKAPE pathogens. Front Cell Infect Microbiol. (2017) 7:41. doi: 10.3389/fcimb.2017.00041
98. Siqueira WL, Margolis HC, Helmerhorst EJ, Mendes FM, Oppenheim FG. Evidence of intact histatins in the in vivo acquired enamel pellicle. J Dent Res. (2010) 89:626–30. doi: 10.1177/0022034510363384
99. van Dijk IA, Ferrando ML, van der Wijk AE, Hoebe RA, Nazmi K, de Jonge WJ, et al. Human salivary peptide histatin-1 stimulates epithelial and endothelial cell adhesion and barrier function. FASEB J. (2017) 31:3922–33. doi: 10.1096/fj.201700180R
100. Silva PM, Goncalves S, Santos NC. Defensins: antifungal lessons from eukaryotes. Front Microbiol. (2014) 5:97. doi: 10.3389/fmicb.2014.00097
101. Machado LR, Ottolini B. An evolutionary history of defensins: a role for copy number variation in maximizing host innate and adaptive immune responses. Front Immunol. (2015) 6:115. doi: 10.3389/fimmu.2015.00115
102. Polesello V, Segat L, Crovella S, Zupin L. Candida infections and human defensins. Protein Pept Lett. (2017) 24:747–56. doi: 10.2174/0929866524666170807125245
103. Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. (2002) 298:1025–9. doi: 10.1126/science.1075565
104. Funderburg N, Lederman MM, Feng Z, Drage MG, Jadlowsky J, Harding CV, et al. Human -defensin-3 activates professional antigen-presenting cells via Toll-like receptors 1 and 2. Proc Natl Acad Sci USA. (2007) 104:18631–5. doi: 10.1073/pnas.0702130104
105. Rohrl J, Yang D, Oppenheim JJ, Hehlgans T. Human beta-defensin 2 and 3 and their mouse orthologs induce chemotaxis through interaction with CCR2. J Immunol. (2010) 184:6688–94. doi: 10.4049/jimmunol.0903984
106. Barabas N, Rohrl J, Holler E, Hehlgans T. Beta-defensins activate macrophages and synergize in pro-inflammatory cytokine expression induced by TLR ligands. Immunobiology. (2013) 218:1005–11. doi: 10.1016/j.imbio.2012.11.007
107. Lehrer RI, Lu W. alpha-Defensins in human innate immunity. Immunol Rev. (2012) 245:84–112. doi: 10.1111/j.1600-065X.2011.01082.x
108. Edgerton M, Koshlukova SE, Araujo MW, Patel RC, Dong J, Bruenn JA. Salivary histatin 5 and human neutrophil defensin 1 kill Candida albicans via shared pathways. Antimicrob Agents Chemother. (2000) 44:3310–6. doi: 10.1128/AAC.44.12.3310-3316.2000
109. Okamoto T, Tanida T, Wei B, Ueta E, Yamamoto T, Osaki T. Regulation of fungal infection by a combination of amphotericin B and peptide 2, a lactoferrin peptide that activates neutrophils. Clin Diagn Lab Immunol. (2004) 11:1111–9. doi: 10.1128/CDLI.11.6.1111-1119.2004
110. Martinez LR, Casadevall A. Cryptococcus neoformans cells in biofilms are less susceptible than planktonic cells to antimicrobial molecules produced by the innate immune system. Infect Immun. (2006) 74:6118–23. doi: 10.1128/IAI.00995-06
111. Chairatana P, Nolan EM. Human alpha-defensin 6: a small peptide that self-assembles and protects the host by entangling microbes. Acc Chem Res. (2017) 50:960–7. doi: 10.1021/acs.accounts.6b00653
112. Weinberg A, Jin G, Sieg S, McCormick TS. The yin and yang of human beta-defensins in health and disease. Front Immunol. (2012) 3:294. doi: 10.3389/fimmu.2012.00294
113. Krishnakumari V, Rangaraj N, Nagaraj R. Antifungal activities of human beta-defensins HBD-1 to HBD-3 and their C-terminal analogs Phd1 to Phd3. Antimicrob Agents Chemother. (2009) 53:256–60. doi: 10.1128/AAC.00470-08
114. Inthanachai T, Thammahong A, Edwards SW, Virakul S, Kiatsurayanon C, Chiewchengchol D. The inhibitory effect of human beta-defensin-3 on Candida Glabrata isolated from patients with candidiasis. Immunol Invest. (2020). doi: 10.1080/08820139.2020.1755307. [Epub ahead of print].
115. Schroeder BO, Wu Z, Nuding S, Groscurth S, Marcinowski M, Beisner J, et al. Reduction of disulphide bonds unmasks potent antimicrobial activity of human beta-defensin 1. Nature. (2011) 469:419–23. doi: 10.1038/nature09674
116. Chang HT, Tsai PW, Huang HH, Liu YS, Chien TS, Lan CY. LL37 and hBD-3 elevate the beta-1,3-exoglucanase activity of Candida albicans Xog1p, resulting in reduced fungal adhesion to plastic. Biochem J. (2012) 441:963–70. doi: 10.1042/BJ20111454
117. Alekseeva L, Huet D, Femenia F, Mouyna I, Abdelouahab M, Cagna A, et al. Inducible expression of beta defensins by human respiratory epithelial cells exposed to Aspergillus fumigatus organisms. BMC Microbiol. (2009) 9:33. doi: 10.1186/1471-2180-9-33
118. Lim SM, Ahn KB, Kim C, Kum JW, Perinpanayagam H, Gu Y, et al. Antifungal effects of synthetic human beta-defensin 3-C15 peptide. Restor Dent Endod. (2016) 41:91–7. doi: 10.5395/rde.2016.41.2.91
119. Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, et al. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature. (2001) 414:454–7. doi: 10.1038/35106587
120. Iimura M, Gallo RL, Hase K, Miyamoto Y, Eckmann L, Kagnoff MF. Cathelicidin mediates innate intestinal defense against colonization with epithelial adherent bacterial pathogens. J Immunol. (2005) 174:4901–7. doi: 10.4049/jimmunol.174.8.4901
121. Chromek M, Slamova Z, Bergman P, Kovacs L, Podracka L, Ehren I, et al. The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat Med. (2006) 12:636–41. doi: 10.1038/nm1407
122. Fabisiak A, Murawska N, Fichna J. LL-37: Cathelicidin-related antimicrobial peptide with pleiotropic activity. Pharmacol Rep. (2016) 68:802–8. doi: 10.1016/j.pharep.2016.03.015
123. Sun L, Wang W, Xiao W, Yang H. The roles of cathelicidin LL-37 in inflammatory bowel disease. Inflamm Bowel Dis. (2016) 22:1986–91. doi: 10.1097/MIB.0000000000000804
124. Chen X, Zou X, Qi G, Tang Y, Guo Y, Si J, et al. Roles and mechanisms of human cathelicidin LL-37 in cancer. Cell Physiol Biochem. (2018) 47:1060–73. doi: 10.1159/000490183
125. den Hertog AL, van Marle J, van Veen HA, Van't Hof W, Bolscher JG, Veerman EC, et al. Candidacidal effects of two antimicrobial peptides: histatin 5 causes small membrane defects, but LL-37 causes massive disruption of the cell membrane. Biochem J. (2005) 388(Pt 2):689–95. doi: 10.1042/BJ20042099
126. Lopez-Garcia B, Lee PH, Yamasaki K, Gallo RL. Anti-fungal activity of cathelicidins and their potential role in Candida albicans skin infection. J Invest Dermatol. (2005) 125:108–15. doi: 10.1111/j.0022-202X.2005.23713.x
127. Ordonez SR, Amarullah IH, Wubbolts RW, Veldhuizen EJ, Haagsman HP. Fungicidal mechanisms of cathelicidins LL-37 and CATH-2 revealed by live-cell imaging. Antimicrob Agents Chemother. (2014) 58:2240–8. doi: 10.1128/AAC.01670-13
128. Scarsini M, Tomasinsig L, Arzese A, D'Este F, Oro D, Skerlavaj B. Antifungal activity of cathelicidin peptides against planktonic and biofilm cultures of Candida species isolated from vaginal infections. Peptides. (2015) 71:211–21. doi: 10.1016/j.peptides.2015.07.023
129. Durnas B, Wnorowska U, Pogoda K, Deptula P, Watek M, Piktel E, et al. Candidacidal activity of selected ceragenins and human cathelicidin LL-37 in experimental settings mimicking infection sites. PLoS ONE. (2016) 11:e0157242. doi: 10.1371/journal.pone.0157242
130. Lopez-Garcia B, Lee PH, Gallo RL. Expression and potential function of cathelicidin antimicrobial peptides in dermatophytosis and tinea versicolor. J Antimicrob Chemother. (2006) 57:877–82. doi: 10.1093/jac/dkl078
131. Wong JH, Ng TB, Legowska A, Rolka K, Hui M, Cho CH. Antifungal action of human cathelicidin fragment (LL13-37) on Candida albicans. Peptides. (2011) 32:1996–2002. doi: 10.1016/j.peptides.2011.08.018
132. Elsegeiny W, Zheng M, Eddens T, Gallo RL, Dai G, Trevejo-Nunez G, et al. Murine models of Pneumocystis infection recapitulate human primary immune disorders. JCI Insight. (2018) 3:e91894. doi: 10.1172/jci.insight.91894
133. Tsai PW, Yang CY, Chang HT, Lan CY. Human antimicrobial peptide LL-37 inhibits adhesion of Candida albicans by interacting with yeast cell-wall carbohydrates. PLoS ONE. (2011) 6:e17755. doi: 10.1371/journal.pone.0017755
134. Rapala-Kozik M, Bochenska O, Zawrotniak M, Wolak N, Trebacz G, Gogol M, et al. Inactivation of the antifungal and immunomodulatory properties of human cathelicidin LL-37 by aspartic proteases produced by the pathogenic yeast Candida albicans. Infect Immun. (2015) 83:2518–30. doi: 10.1128/IAI.00023-15
135. Sheehan G, Bergsson G, McElvaney NG, Reeves EP, Kavanagh K. The human cathelicidin antimicrobial peptide LL-37 promotes the growth of the pulmonary pathogen Aspergillus fumigatus. Infect Immun. (2018). doi: 10.1128/IAI.00097-18
136. Murthy AR, Lehrer RI, Harwig SS, Miyasaki KT. In vitro candidastatic properties of the human neutrophil calprotectin complex. J Immunol. (1993) 151:6291–301.
137. Tomee JF, Hiemstra PS, Heinzel-Wieland R, Kauffman HF. Antileukoprotease: an endogenous protein in the innate mucosal defense against fungi. J Infect Dis. (1997) 176:740–7. doi: 10.1086/514098
138. Stenger S, Hanson DA, Teitelbaum R, Dewan P, Niazi KR, Froelich CJ, et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science. (1998) 282: 1–125. doi: 10.1126/science.282.5386.121
139. Cutuli M, Cristiani S, Lipton JM, Catania A. Antimicrobial effects of alpha-MSH peptides. J Leukoc Biol. (2000) 67:233–9. doi: 10.1002/jlb.67.2.233
140. Krijgsveld J, Zaat SA, Meeldijk J, van Veelen PA, Fang G, Poolman B, et al. Thrombocidins, microbicidal proteins from human blood platelets, are C-terminal deletion products of CXC chemokines. J Biol Chem. (2000) 275:20374–81. doi: 10.1074/jbc.275.27.20374
141. Lugardon K, Raffner R, Goumon Y, Corti A, Delmas A, Bulet P, et al. Antibacterial and antifungal activities of vasostatin-1, the N-terminal fragment of chromogranin A. J Biol Chem. (2000) 275:10745–53. doi: 10.1074/jbc.275.15.10745
142. Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. (2001) 276:7806–10. doi: 10.1074/jbc.M008922200
143. Schittek B, Hipfel R, Sauer B, Bauer J, Kalbacher H, Stevanovic S, et al. Dermcidin: a novel human antibiotic peptide secreted by sweat glands. Nat Immunol. (2001) 2:1133–7. doi: 10.1038/ni732
144. Yang D, Chen Q, Hoover DM, Staley P, Tucker KD, Lubkowski J, et al. Many chemokines including CCL20/MIP-3alpha display antimicrobial activity. J Leukoc Biol. (2003) 74:448–55. doi: 10.1189/jlb.0103024
145. Baranger K, Zani ML, Chandenier J, Dallet-Choisy S, Moreau T. The antibacterial and antifungal properties of trappin-2. (pre-elafin) do not depend on its protease inhibitory function. FEBS J. (2008) 275:2008–20. doi: 10.1111/j.1742-4658.2008.06355.x
146. El Karim IA, Linden GJ, Orr DF, Lundy FT. Antimicrobial activity of neuropeptides against a range of micro-organisms from skin, oral, respiratory and gastrointestinal tract sites. J Neuroimmunol. (2008) 200:11–6. doi: 10.1016/j.jneuroim.2008.05.014
147. Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS ONE. (2010) 5:e9505. doi: 10.1371/journal.pone.0009505
148. Fritz P, Beck-Jendroschek V, Brasch J. Inhibition of dermatophytes by the antimicrobial peptides human beta-defensin-2, ribonuclease 7 and psoriasin. Med Mycol. (2012) 50:579–84. doi: 10.3109/13693786.2012.660203
149. Mehra T, Koberle M, Braunsdorf C, Mailander-Sanchez D, Borelli C, Schaller M. Alternative approaches to antifungal therapies. Exp Dermatol. (2012) 21:778–82. doi: 10.1111/exd.12004
150. Lombardi L, Maisetta G, Batoni G, Tavanti A. Insights into the antimicrobial properties of hepcidins: advantages and drawbacks as potential therapeutic agents. Molecules. (2015) 20:6319–41. doi: 10.3390/molecules20046319
151. Salazar VA, Arranz-Trullen J, Navarro S, Blanco JA, Sanchez D, Moussaoui M, et al. Exploring the mechanisms of action of human secretory RNase 3 and RNase 7 against Candida albicans. Microbiologyopen. (2016) 5:830–45. doi: 10.1002/mbo3.373
152. Fernandes KE, Carter DA. The antifungal activity of lactoferrin and its derived peptides: mechanisms of action and synergy with drugs against fungal pathogens. Front Microbiol. (2017) 8:2. doi: 10.3389/fmicb.2017.00002
153. Curvelo JA, Barreto AL, Portela MB, Alviano DS, Holandino C, Souto-Padron T, et al. Effect of the secretory leucocyte proteinase inhibitor (SLPI) on Candida albicans biological processes: a therapeutic alternative? Arch Oral Biol. (2014) 59:928–37. doi: 10.1016/j.archoralbio.2014.05.007
154. Tavanti A, Maisetta G, Del Gaudio G, Petruzzelli R, Sanguinetti M, Batoni G, et al. Fungicidal activity of the human peptide hepcidin 20 alone or in combination with other antifungals against Candida glabrata isolates. Peptides. (2011) 32:2484–7. doi: 10.1016/j.peptides.2011.10.012
155. Hein KZ, Takahashi H, Tsumori T, Yasui Y, Nanjoh Y, Toga T, et al. Disulphide-reduced psoriasin is a human apoptosis-inducing broad-spectrum fungicide. Proc Natl Acad Sci USA. (2015) 112:13039–44. doi: 10.1073/pnas.1511197112
156. Greber KE, Dawgul M. Antimicrobial peptides under clinical trials. Curr Top Med Chem. (2017) 17:620–8. doi: 10.2174/1568026616666160713143331
157. Koo HB, Seo J. Antimicrobial peptides under clinical investigation. Peptide Sci. (2019) 111:e24122. doi: 10.1002/pep2.24122
158. van der Weerden NL, Bleackley MR, Anderson MA. Properties and mechanisms of action of naturally occurring antifungal peptides. Cell Mol Life Sci. (2013) 70:3545–70. doi: 10.1007/s00018-013-1260-1
159. Nawrot R, Barylski J, Nowicki G, Broniarczyk J, Buchwald W, Gozdzicka-Jozefiak A. Plant antimicrobial peptides. Folia Microbiol. (2014) 59:181–96. doi: 10.1007/s12223-013-0280-4
160. Ciociola T, Giovati L, Conti S, Magliani W, Santinoli C, Polonelli L. Natural and synthetic peptides with antifungal activity. Future Med Chem. (2016) 8:1413–33. doi: 10.4155/fmc-2016-0035
161. Faruck MO, Yusof F, Chowdhury S. An overview of antifungal peptides derived from insect. Peptides. (2016) 80:80–8. doi: 10.1016/j.peptides.2015.06.001
162. Mihajlovic M, Lazaridis T. Antimicrobial peptides in toroidal and cylindrical pores. Biochim Biophys Acta. (2010) 1798:1485–93. doi: 10.1016/j.bbamem.2010.04.004
163. Sengupta D, Leontiadou H, Mark AE, Marrink SJ. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim Biophys Acta. (2008) 1778:2308–17. doi: 10.1016/j.bbamem.2008.06.007
164. Bechinger B, Lohner K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim Biophys Acta. (2006) 1758:1529–39. doi: 10.1016/j.bbamem.2006.07.001
165. Pouny Y, Rapaport D, Mor A, Nicolas P, Shai Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry. (1992) 31:12416–23. doi: 10.1021/bi00164a017
166. Haney EF, Nathoo S, Vogel HJ, Prenner EJ. Induction of non-lamellar lipid phases by antimicrobial peptides: a potential link to mode of action. Chem Phys Lipids. (2010) 163:82–93. doi: 10.1016/j.chemphyslip.2009.09.002
167. Grage SL, Afonin S, Kara S, Buth G, Ulrich AS. Membrane thinning and thickening induced by membrane-active amphipathic peptides. Front Cell Dev Biol. (2016) 4:65. doi: 10.3389/fcell.2016.00065
168. Epand RM, Epand RF. Bacterial membrane lipids in the action of antimicrobial agents. J Pept Sci. (2011) 17:298–305. doi: 10.1002/psc.1319
169. Matsuzaki K, Murase O, Fujii N, Miyajima K. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry. (1996) 35:11361–8. doi: 10.1021/bi960016v
170. Finger S, Kerth A, Dathe M, Blume A. The efficacy of trivalent cyclic hexapeptides to induce lipid clustering in PG/PE membranes correlates with their antimicrobial activity. Biochim Biophys Acta. (2015) 1848:2998–3006. doi: 10.1016/j.bbamem.2015.09.012
171. Yasir M, Dutta D, Willcox MDP. Mode of action of the antimicrobial peptide Mel4 is independent of Staphylococcus aureus cell membrane permeability. PLoS ONE. (2019) 14:e0215703. doi: 10.1371/journal.pone.0215703
172. Mercer DK, Stewart CS, Miller L, Robertson J, Duncan VMS, O'Neil DA. Improved methods for assessing therapeutic potential of antifungal agents against dermatophytes and their application in the development of NP213, a novel onychomycosis therapy candidate. Antimicrob Agents Chemother. (2019) 63:e02117-18. doi: 10.1128/AAC.02117-18
173. Mercer DK, Robertson J, Miller L, Stewart CS, O'Neil DA. NP213 (Novexatin): a unique therapy candidate with a differentiated safety and efficacy profile. Med Mycol. (2020). doi: 10.1093/mmy/myaa015. [Epub ahead of print].
174. van der Weerden N, Hayes B, McKenna J, Bleackley M, McCorkelle O, Weaver S, et al. The plant defensin HXP124 has the potential to be a safe and effective topical treatment for onychomycosis. In: 20th ISHAM Congress. Amsterdam, Netherlands: The International Society for Human and Animal Mycology (2018).
175. Csato M, Kenderessy AS, Dobozy A. Enhancement of Candida albicans killing activity of separated human epidermal cells by alpha-melanocyte stimulating hormone. Br J Dermatol. (1989) 121:145–7. doi: 10.1111/j.1365-2133.1989.tb01415.x
176. Catania A, Grieco P, Randazzo A, Novellino E, Gatti S, Rossi C, et al. Three-dimensional structure of the alpha-MSH-derived candidacidal peptide [Ac-CKPV]2. J Pept Res. (2005) 66:19–26. doi: 10.1111/j.1399-3011.2005.00265.x
177. Ji HX, Zou YL, Duan JJ, Jia ZR, Li XJ, Wang Z, et al. The synthetic melanocortin (CKPV)2 exerts anti-fungal and anti-inflammatory effects against Candida albicans vaginitis via inducing macrophage M2 polarization. PLoS ONE. (2013) 8:e56004. doi: 10.1371/journal.pone.0056004
178. Capsoni F, Ongari A, Colombo G, Turcatti F, Catania A. The synthetic melanocortin (CKPV)2 exerts broad anti-inflammatory effects in human neutrophils. Peptides. (2007) 28:2016–22. doi: 10.1016/j.peptides.2007.08.001
179. Gatti S, Carlin A, Sordi A, Leonardi P, Colombo G, Fassati LR, et al. Inhibitory effects of the peptide (CKPV)2 on endotoxin-induced host reactions. J Surg Res. (2006) 131:209–14. doi: 10.1016/j.jss.2005.08.009
180. Fjell CD, Hiss JA, Hancock RE, Schneider G. Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov. (2011) 11:37–51. doi: 10.1038/nrd3591
181. Rothstein DM, Spacciapoli P, Tran LT, Xu T, Roberts FD, Dalla Serra M, et al. Anticandida activity is retained in P-113, a 12-amino-acid fragment of histatin 5. Antimicrob Agents Chemother. (2001) 45:1367–73. doi: 10.1128/AAC.45.5.1367-1373.2001
182. Sajjan US, Tran LT, Sole N, Rovaldi C, Akiyama A, Friden PM, et al. P-113D, an antimicrobial peptide active against Pseudomonas aeruginosa, retains activity in the presence of sputum from cystic fibrosis patients. Antimicrob Agents Chemother. (2001) 45:3437–44. doi: 10.1128/AAC.45.12.3437-3444.2001
183. Giacometti A, Cirioni O, Kamysz W, D'Amato G, Silvestri C, Del Prete MS, et al. In vitro activity of the histatin derivative P-113 against multidrug-resistant pathogens responsible for pneumonia in immunocompromised patients. Antimicrob Agents Chemother. (2005) 49:1249–52. doi: 10.1128/AAC.49.3.1249-1252.2005
184. Welling MM, Brouwer CP, van 't Hof W, Veerman EC, Amerongen AV. Histatin-derived monomeric and dimeric synthetic peptides show strong bactericidal activity towards multidrug-resistant Staphylococcus aureus in vivo. Antimicrob Agents Chemother. (2007) 51:3416–9. doi: 10.1128/AAC.00196-07
185. Huo L, Zhang K, Ling J, Peng Z, Huang X, Liu H, et al. Antimicrobial and DNA-binding activities of the peptide fragments of human lactoferrin and histatin 5 against Streptococcus mutans. Arch Oral Biol. (2011) 56:869–76. doi: 10.1016/j.archoralbio.2011.02.004
186. Di Giampaolo A, Luzi C, Casciaro B, Bozzi A, Mangoni ML, Aschi M. P-113 peptide: New experimental evidences on its biological activity and conformational insights from molecular dynamics simulations. Biopolymers. (2014) 102:159–67. doi: 10.1002/bip.22452
187. Lin GY, Chen HF, Xue YP, Yeh YC, Chen CL, Liu MS, et al. The Antimicrobial Peptides P-113Du and P-113Tri Function against Candida albicans. Antimicrob Agents Chemother. (2016) 60:6369–73. doi: 10.1128/AAC.00699-16
188. Xue YP, Kao MC, Lan CY. Novel mitochondrial complex I-inhibiting peptides restrain NADH dehydrogenase activity. Sci Rep. (2019) 9:13694. doi: 10.1038/s41598-019-50114-2
189. Helmerhorst EJ, Venuleo C, Beri A, Oppenheim FG. Candida glabrata is unusual with respect to its resistance to cationic antifungal proteins. Yeast. (2005) 22:705–14. doi: 10.1002/yea.1241
190. Jang WS, Li XS, Sun JN, Edgerton M. The P-113 fragment of histatin 5 requires a specific peptide sequence for intracellular translocation in Candida albicans, which is independent of cell wall binding. Antimicrob Agents Chemother. (2008) 52:497–504. doi: 10.1128/AAC.01199-07
191. Yu HY, Tu CH, Yip BS, Chen HL, Cheng HT, Huang KC, et al. Easy strategy to increase salt resistance of antimicrobial peptides. Antimicrob Agents Chemother. (2011) 55:4918–21. doi: 10.1128/AAC.00202-11
192. Ruissen AL, Groenink J, Krijtenberg P, Walgreen-Weterings E, van 't Hof W, Veerman EC, et al. Internalisation and degradation of histatin 5 by Candida albicans. Biol Chem. (2003) 384:183–90. doi: 10.1515/BC.2003.020
193. Ikonomova SP, Moghaddam-Taaheri P, Jabra-Rizk MA, Wang Y, Karlsson AJ. Engineering improved variants of the antifungal peptide histatin 5 with reduced susceptibility to Candida albicans secreted aspartic proteases and enhanced antimicrobial potency. FEBS J. (2018) 285:146–59. doi: 10.1111/febs.14327
194. Moffa EB, Mussi MC, Xiao Y, Garrido SS, Machado MA, Giampaolo ET, et al. Histatin 5 inhibits adhesion of C. albicans to reconstructed human oral epithelium. Front Microbiol. (2015) 6:885. doi: 10.3389/fmicb.2015.00885
195. Zambom CR, da Fonseca FH, Crusca EJr, da Silva PB, Pavan FR, Chorilli M, et al. A novel antifungal system with potential for prolonged delivery of histatin 5 to limit growth of Candida albicans. Front Microbiol. (2019) 10:1667. doi: 10.3389/fmicb.2019.01667
196. Liu N, Guan S, Wang H, Li C, Cheng J, Yu H, et al. The antimicrobial peptide Nal-P-113 exerts a reparative effect by promoting cell proliferation, migration, and cell cycle progression. Biomed Res Int. (2018) 2018:7349351. doi: 10.1155/2018/7349351
197. Paquette DW, Simpson DM, Friden P, Braman V, Williams RC. Safety and clinical effects of topical histatin gels in humans with experimental gingivitis. J Clin Periodontol. (2002) 29:1051–8. doi: 10.1034/j.1600-051X.2002.291201.x
198. Van Dyke T, Paquette D, Grossi S, Braman V, Massaro J, D'Agostino R, et al. Clinical and microbial evaluation of a histatin-containing mouthrinse in humans with experimental gingivitis: a phase-2 multi-center study. J Clin Periodontol. (2002) 29:168–76. doi: 10.1034/j.1600-051x.2002.290212.x
199. Wang H, Ai L, Zhang Y, Cheng J, Yu H, Li C, et al. The Effects of antimicrobial peptide Nal-P-113 on inhibiting periodontal pathogens and improving periodontal status. Biomed Res Int. (2018) 2018:1805793. doi: 10.1155/2018/1805793
200. Sader HS, Fedler KA, Rennie RP, Stevens S, Jones RN. Omiganan pentahydrochloride (MBI 226), a topical 12-amino-acid cationic peptide: spectrum of antimicrobial activity and measurements of bactericidal activity. Antimicrob Agents Chemother. (2004) 48:3112–8. doi: 10.1128/AAC.48.8.3112-3118.2004
201. Fritsche TR, Rhomberg PR, Sader HS, Jones RN. Antimicrobial activity of omiganan pentahydrochloride against contemporary fungal pathogens responsible for catheter-associated infections. Antimicrob Agents Chemother. (2008) 52:1181–9. doi: 10.1128/AAC.01475-07
202. Faccone D, Veliz O, Corso A, Noguera M, Martinez M, Payes C, et al. Antimicrobial activity of de novo designed cationic peptides against multi-resistant clinical isolates. Eur J Med Chem. (2014) 71:31–5. doi: 10.1016/j.ejmech.2013.10.065
203. Zapotoczna M, Forde E, Hogan S, Humphreys H, O'Gara JP, Fitzgerald-Hughes D, et al. Eradication of Staphylococcus aureus biofilm infections using synthetic antimicrobial peptides. J Infect Dis. (2017) 215:975–83. doi: 10.1093/infdis/jix062
204. Jaskiewicz M, Neubauer D, Kazor K, Bartoszewska S, Kamysz W. Antimicrobial activity of selected antimicrobial peptides against planktonic culture and biofilm of Acinetobacter baumannii. Probiotics Antimicrob Proteins. (2019) 11:317–24. doi: 10.1007/s12602-018-9444-5
205. Rijsbergen M, Rijneveld R, Todd M, Feiss GL, Kouwenhoven STP, Quint KD, et al. Results of phase 2 trials exploring the safety and efficacy of omiganan in patients with human papillomavirus-induced genital lesions. Br J Clin Pharmacol. (2019). doi: 10.1111/bcp.14181. [Epub ahead of print].
206. Lorenzi T, Trombettoni MMC, Ghiselli R, Paolinelli F, Gesuita R, Cirioni O, et al. Effect of omiganan on colonic anastomosis healing in a rat model of peritonitis. Am J Transl Res. (2017) 9:3374–86.
207. Neubauer D, Jaskiewicz M, Migon D, Bauer M, Sikora K, Sikorska E, et al. Retro analog concept: comparative study on physico-chemical and biological properties of selected antimicrobial peptides. Amino Acids. (2017) 49:1755–71. doi: 10.1007/s00726-017-2473-7
208. Ng SMS, Teo SW, Yong YE, Ng FM, Lau QY, Jureen R, et al. Preliminary investigations into developing all-D Omiganan for treating Mupirocin-resistant MRSA skin infections. Chem Biol Drug Des. (2017) 90:1155–60. doi: 10.1111/cbdd.13035
209. Rubinchik E, Dugourd D, Algara T, Pasetka C, Friedland HD. Antimicrobial and antifungal activities of a novel cationic antimicrobial peptide, omiganan, in experimental skin colonisation models. Int J Antimicrob Agents. (2009) 34:457–61. doi: 10.1016/j.ijantimicag.2009.05.003
210. Nibbering PH, Ravensbergen E, Welling MM, van Berkel LA, van Berkel PH, Pauwels EK, et al. Human lactoferrin and peptides derived from its N terminus are highly effective against infections with antibiotic-resistant bacteria. Infect Immun. (2001) 69:1469–76. doi: 10.1128/IAI.69.3.1469-1476.2001
211. Morici P, Florio W, Rizzato C, Ghelardi E, Tavanti A, Rossolini GM, et al. Synergistic activity of synthetic N-terminal peptide of human lactoferrin in combination with various antibiotics against carbapenem-resistant Klebsiella pneumoniae strains. Eur J Clin Microbiol Infect Dis. (2017) 36:1739–48. doi: 10.1007/s10096-017-2987-7
212. Lupetti A, Paulusma-Annema A, Welling MM, Senesi S, van Dissel JT, Nibbering PH. Candidacidal activities of human lactoferrin peptides derived from the N terminus. Antimicrob Agents Chemother. (2000) 44:3257–63. doi: 10.1128/AAC.44.12.3257-3263.2000
213. van der Does AM, Bogaards SJ, Jonk L, Wulferink M, Velders MP, Nibbering PH. The human lactoferrin-derived peptide hLF1-11 primes monocytes for an enhanced TLR-mediated immune response. Biometals. (2010) 23:493–505. doi: 10.1007/s10534-010-9322-4
214. van der Does AM, Hensbergen PJ, Bogaards SJ, Cansoy M, Deelder AM, van Leeuwen HC, et al. The human lactoferrin-derived peptide hLF1-11 exerts immunomodulatory effects by specific inhibition of myeloperoxidase activity. J Immunol. (2012) 188:5012–9. doi: 10.4049/jimmunol.1102777
215. Morici P, Fais R, Rizzato C, Tavanti A, Lupetti A. Inhibition of Candida albicans biofilm formation by the synthetic lactoferricin derived peptide hLF1-11. PLoS ONE. (2016) 11:e0167470. doi: 10.1371/journal.pone.0167470
216. Lupetti A, Paulusma-Annema A, Welling MM, Dogterom-Ballering H, Brouwer CP, Senesi S, et al. Synergistic activity of the N-terminal peptide of human lactoferrin and fluconazole against Candida species. Antimicrob Agents Chemother. (2003) 47:262–7. doi: 10.1128/AAC.47.1.262-267.2003
217. Lupetti A, Brouwer CP, Bogaards SJ, Welling MM, de Heer E, Campa M, et al. Human lactoferrin-derived peptide's antifungal activities against disseminated Candida albicans infection. J Infect Dis. (2007) 196:1416–24. doi: 10.1086/522427
218. Lupetti A, van Dissel JT, Brouwer CP, Nibbering PH. Human antimicrobial peptides' antifungal activity against Aspergillus fumigatus. Eur J Clin Microbiol Infect Dis. (2008) 27:1125–9. doi: 10.1007/s10096-008-0553-z
219. Fais R, Di Luca M, Rizzato C, Morici P, Bottai D, Tavanti A, et al. The N-terminus of human lactoferrin displays anti-biofilm activity on Candida parapsilosis in lumen catheters. Front Microbiol. (2017) 8:2218. doi: 10.3389/fmicb.2017.02218
220. Godoy-Gallardo M, Wang Z, Shen Y, Manero JM, Gil FJ, Rodriguez D, et al. Antibacterial coatings on titanium surfaces: a comparison study between in vitro single-species and multispecies biofilm. ACS Appl Mater Interfaces. (2015) 7:5992–6001. doi: 10.1021/acsami.5b00402
221. Costa F, Maia S, Gomes J, Gomes P, Martins MC. Characterization of hLF1-11 immobilization onto chitosan ultrathin films, and its effects on antimicrobial activity. Acta Biomater. (2014) 10:3513–21. doi: 10.1016/j.actbio.2014.02.028
222. Stallmann HP, Faber C, Bronckers AL, de Blieck-Hogervorst JM, Brouwer CP, Amerongen AV, et al. Histatin and lactoferrin derived peptides: antimicrobial properties and effects on mammalian cells. Peptides. (2005) 26:2355–9. doi: 10.1016/j.peptides.2005.05.014
223. MacCallum DM, Desbois AP, Coote PJ. Enhanced efficacy of synergistic combinations of antimicrobial peptides with caspofungin versus Candida albicans in insect and murine models of systemic infection. Eur J Clin Microbiol Infect Dis. (2013) 32:1055–62. doi: 10.1007/s10096-013-1850-8
224. van der Does AM, Joosten SA, Vroomans E, Bogaards SJ, van Meijgaarden KE, Ottenhoff TH, et al. The antimicrobial peptide hLF1-11 drives monocyte-dendritic cell differentiation toward dendritic cells that promote antifungal responses and enhance Th17 polarization. J Innate Immun. (2012) 4:284–92. doi: 10.1159/000332941
225. van der Velden WJ, van Iersel TM, Blijlevens NM, Donnelly JP. Safety and tolerability of the antimicrobial peptide human lactoferrin 1-11 (hLF1-11). BMC Med. (2009) 7:44. doi: 10.1186/1741-7015-7-44
226. Giles FJ, Miller CB, Hurd DD, Wingard JR, Fleming TR, Sonis ST, et al. A phase III, randomized, double-blind, placebo-controlled, multinational trial of iseganan for the prevention of oral mucositis in patients receiving stomatotoxic chemotherapy (PROMPT-CT trial). Leuk Lymphoma. (2003) 44:1165–72. doi: 10.1080/1042819031000079159
227. Giles FJ, Rodriguez R, Weisdorf D, Wingard JR, Martin PJ, Fleming TR, et al. A phase III, randomized, double-blind, placebo-controlled, study of iseganan for the reduction of stomatitis in patients receiving stomatotoxic chemotherapy. Leuk Res. (2004) 28:559–65. doi: 10.1016/j.leukres.2003.10.021
228. Trotti A, Garden A, Warde P, Symonds P, Langer C, Redman R, et al. A multinational, randomized phase III trial of iseganan HCl oral solution for reducing the severity of oral mucositis in patients receiving radiotherapy for head-and-neck malignancy. Int J Radiat Oncol Biol Phys. (2004) 58:674–81. doi: 10.1016/S0360-3016(03)01627-4
229. Kollef M, Pittet D, Sanchez Garcia M, Chastre J, Fagon JY, Bonten M, et al. A randomized double-blind trial of iseganan in prevention of ventilator-associated pneumonia. Am J Respir Crit Care Med. (2006) 173:91–7. doi: 10.1164/rccm.200504-656OC
230. Chen J, Falla TJ, Liu H, Hurst MA, Fujii CA, Mosca DA, et al. Development of protegrins for the treatment and prevention of oral mucositis: structure-activity relationships of synthetic protegrin analogues. Pept Sci. (2000) 55:88–98. doi: 10.1002/1097-0282(2000)55:1<88::AID-BIP80>3.0.CO;2-K
231. Mosca DA, Hurst MA, So W, Viajar BS, Fujii CA, Falla TJ. IB-367, a protegrin peptide with in vitro and in vivo activities against the microflora associated with oral mucositis. Antimicrob Agents Chemother. (2000) 44:1803–8. doi: 10.1128/AAC.44.7.1803-1808.2000
232. Simonetti O, Cirioni O, Ghiselli R, Orlando F, Silvestri C, Mazzocato S, et al. In vitro activity and in vivo animal model efficacy of IB-367 alone and in combination with imipenem and colistin against Gram-negative bacteria. Peptides. (2014) 55C:17–22. doi: 10.1016/j.peptides.2014.01.029
233. Barchiesi F, Giacometti A, Cirioni O, Arzeni D, Kamysz W, Silvestri C, et al. In-vitro activity of the synthetic protegrin IB-367 alone and in combination with antifungal agents against clinical isolates of Candida spp. J Chemother. (2007) 19:514–8. doi: 10.1179/joc.2007.19.5.514
234. Simonetti O, Silvestri C, Arzeni D, Cirioni O, Kamysz W, Conte I, et al. In vitro activity of the protegrin IB-367 alone and in combination compared with conventional antifungal agents against dermatophytes. Mycoses. (2014) 57:233–9. doi: 10.1111/myc.12148
235. Landa A, Jimenez L, Willms K, Jimenez-Garcia LF, Lara-Martinez R, Robert L, et al. Antimicrobial peptides (Temporin A and Iseganan IB-367): effect on the cysticerci of Taenia crassiceps. Mol Biochem Parasitol. (2009) 164:126–30. doi: 10.1016/j.molbiopara.2008.12.006
236. Ghiselli R, Giacometti A, Cirioni O, Mocchegiani F, Silvestri C, Orlando F, et al. Pretreatment with the protegrin IB-367 affects Gram-positive biofilm and enhances the therapeutic efficacy of linezolid in animal models of central venous catheter infection. JPEN J Parenter Enteral Nutr. (2007) 31:463–8. doi: 10.1177/0148607107031006463
237. Giacometti A, Cirioni O, Ghiselli R, Mocchegiani F, D'Amato G, Del Prete MS, et al. Administration of protegrin peptide IB-367 to prevent endotoxin induced mortality in bile duct ligated rats. Gut. (2003) 52:874–8. doi: 10.1136/gut.52.6.874
238. Rodziewicz-Motowidlo S, Mickiewicz B, Greber K, Sikorska E, Szultka L, Kamysz E, et al. Antimicrobial and conformational studies of the active and inactive analogues of the protegrin-1 peptide. FEBS J. (2010) 277:1010–22. doi: 10.1111/j.1742-4658.2009.07544.x
239. Loury D, Embree JR, Steinberg DA, Sonis ST, Fiddes JC. Effect of local application of the antimicrobial peptide IB-367 on the incidence and severity of oral mucositis in hamsters. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. (1999) 87:544–51. doi: 10.1016/S1079-2104(99)70131-9
240. Elad S, Epstein JB, Raber-Durlacher J, Donnelly P, Strahilevitz J. The antimicrobial effect of Iseganan HCl oral solution in patients receiving stomatotoxic chemotherapy: analysis from a multicenter, double-blind, placebo-controlled, randomized, phase III clinical trial. J Oral Pathol Med. (2012) 41:229–34. doi: 10.1111/j.1600-0714.2011.01094.x
241. van Saene H, van Saene J, Silvestri L, de la Cal M, Sarginson R, Zandstra D. Iseganan failure due to the wrong pharmaceutical technology. Chest. (2007) 132:1412. doi: 10.1378/chest.07-0172
242. Isaksson J, Brandsdal BO, Engqvist M, Flaten GE, Svendsen JS, Stensen W. A synthetic antimicrobial peptidomimetic (LTX 109): stereochemical impact on membrane disruption. J Med Chem. (2011) 54:5786–95. doi: 10.1021/jm200450h
243. Saravolatz LD, Pawlak J, Johnson L, Bonilla H, Saravolatz LD II, Fakih MG, et al. In vitro activities of LTX-109, a synthetic antimicrobial peptide, against methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, daptomycin-nonsusceptible, and linezolid-nonsusceptible Staphylococcus aureus. Antimicrob Agents Chemother. (2012) 56:4478–82. doi: 10.1128/AAC.00194-12
244. Saravolatz LD, Pawlak J, Martin H, Saravolatz S, Johnson L, Wold H, et al. Postantibiotic effect and postantibiotic sub-MIC effect of LTX-109 and mupirocin on Staphylococcus aureus blood isolates. Lett Appl Microbiol. (2017) 65:410–3. doi: 10.1111/lam.12792
245. Bojsen R, Torbensen R, Larsen CE, Folkesson A, Regenberg B. The synthetic amphipathic peptidomimetic LTX109 is a potent fungicide that disturbs plasma membrane integrity in a sphingolipid dependent manner. PLoS ONE. (2013) 8:e69483. doi: 10.1371/journal.pone.0069483
246. Nilsson AC, Janson H, Wold H, Fugelli A, Andersson K, Hakangard C, et al. LTX-109 is a novel agent for nasal decolonization of methicillin-resistant and -sensitive Staphylococcus aureus. Antimicrob Agents Chemother. (2015) 59:145–51. doi: 10.1128/AAC.03513-14
247. Reichhardt C, Stevens DA, Cegelski L. Fungal biofilm composition and opportunities in drug discovery. Future Med Chem. (2016) 8:1455–68. doi: 10.4155/fmc-2016-0049
248. Wu S, Wang Y, Liu N, Dong G, Sheng C. Tackling fungal resistance by biofilm inhibitors. J Med Chem. (2017) 60:2193–211. doi: 10.1021/acs.jmedchem.6b01203
249. Nett JE, Andes DR. Fungal biofilms: in vivo models for discovery of anti-biofilm drugs. Microbiol Spectr. (2015) 3:E30. doi: 10.1128/microbiolspec.MB-0008-2014
250. Delattin N, Brucker K, Cremer K, Cammue BP, Thevissen K. Antimicrobial peptides as a strategy to combat fungal biofilms. Curr Top Med Chem. (2017) 17:604–12. doi: 10.2174/1568026616666160713142228
251. Martinez LR, Casadevall A. Specific antibody can prevent fungal biofilm formation and this effect correlates with protective efficacy. Infect Immun. (2005) 73:6350–62. doi: 10.1128/IAI.73.10.6350-6362.2005
252. Nicola AM, Albuquerque P, Paes HC, Fernandes L, Costa FF, Kioshima ES, et al. Antifungal drugs: new insights in research & development. Pharmacol Ther. (2019) 195:21–38. doi: 10.1016/j.pharmthera.2018.10.008
253. Fernandez de Ullivarri M, Arbulu S, Garcia-Gutierrez E, Cotter PD. Antifungal peptides as therapeutic agents. Front Cell Infect Microbiol. (2020) 10:105. doi: 10.3389/fcimb.2020.00105
254. Mookherjee N, Anderson MA, Haagsman HP, Davidson DJ. Antimicrobial host defence peptides: functions and clinical potential. Nat Rev Drug Discov. (2020) 19:311–32. doi: 10.1038/s41573-019-0058-8
255. Katvars LK, Smith DW, Duncan VMS, Simpson L, Fraser-Pitt D, Enderby B, et al. Novamycin. (NP339) as a novel approach against respiratory fungal infections. In: 27th European Congress of Clinical Microbiology and Infectious Diseases. Vienna (2017).
256. Mercer DK, Duncan VMS, Katvars LK, Smith DW, Shaw T, Holden K, et al. Antifungal activity of Novamycin (NP339) in vivo in respiratyory models of fungal infection. In: 27th European Congress of Clinical Microbiology and Infectious Diseases. Vienna: ECCMID (2017).
257. Inc. D. Demegen. (2013). Available online at: http://www.demegen.com/index.htm (accessed April 24, 2020).
258. Schwab U, Gilligan P, Jaynes J, Henke D. In vitro activities of designed antimicrobial peptides against multidrug-resistant cystic fibrosis pathogens. Antimicrob Agents Chemother. (1999) 43:1435–40. doi: 10.1128/AAC.43.6.1435
259. Ballweber LM, Jaynes JE, Stamm WE, Lampe MF. In vitro microbicidal activities of cecropin peptides D2A21 and D4E1 and gel formulations containing 0.1 to 2% D2A21 against Chlamydia trachomatis. Antimicrob Agents Chemother. (2002) 46:34–41. doi: 10.1128/AAC.46.1.34-41.2002
260. Lushbaugh WB, Blossom AC, Shah PH, Banga AK, Jaynes JM, Cleary JD, et al. Use of intravaginal microbicides to prevent acquisition of Trichomonas vaginalis infection in Lactobacillus-pretreated, estrogenized young mice. Am J Trop Med Hyg. (2000) 63:284–9. doi: 10.4269/ajtmh.2000.63.284
261. Chalekson CP, Neumeister MW, Jaynes J. Improvement in burn wound infection and survival with antimicrobial peptide D2A21 (Demegel). Plast Reconstr Surg. (2002) 109:1338–43. doi: 10.1097/00006534-200204010-00020
262. Chalekson CP, Neumeister MW, Jaynes J. Treatment of infected wounds with the antimicrobial peptide D2A21. J Trauma. (2003) 54:770–4. doi: 10.1097/01.TA.0000047047.79701.6D
263. Andres E. Cationic antimicrobial peptides in clinical development, with special focus on thanatin and heliomicin. Eur J Clin Microbiol Infect Dis. (2012) 31:881–8. doi: 10.1007/s10096-011-1430-8
264. Landon C, Barbault F, Legrain M, Menin L, Guenneugues M, Schott V, et al. Lead optimization of antifungal peptides with 3D NMR structures analysis. Protein Sci. (2004) 13:703–13. doi: 10.1110/ps.03404404
265. Aumer T, Voisin SN, Knobloch T, Landon C, Bulet P. Impact of an antifungal insect defensin on the proteome of the phytopathogenic fungus botrytis cinerea. J Proteome Res. (2020) 19:1131–46. doi: 10.1021/acs.jproteome.9b00638
266. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. (2001) 46:3–26. doi: 10.1016/S0169-409X(00)00129-0
267. Ersoy SC, Heithoff DM, Barnes L, Tripp GK, House JK, et al. Correcting a fundamental flaw in the paradigm for antimicrobial susceptibility testing. EBioMedicine. (2017) 20:173–81. doi: 10.1016/j.ebiom.2017.05.026
268. Mercer DK, Torres MDT, Duay SS, Lovie E, Simpson L, Von Kockritz-Blickwede M, et al. Antimicrobial susceptibility testing of antimicrobial peptides to better predict efficacy. Front Cell Infect Microbiol. (2020) 10:326. doi: 10.3389/fcimb.2020.00326
269. Gautam A, Chaudhary K, Singh S, Joshi A, Anand P, Tuknait A, et al. Hemolytik: a database of experimentally determined hemolytic and non-hemolytic peptides. Nucleic Acids Res. (2014) 42(Database issue):D444–9. doi: 10.1093/nar/gkt1008
270. Singh S, Papareddy P, Morgelin M, Schmidtchen A, Malmsten M. Effects of PEGylation on membrane and lipopolysaccharide interactions of host defense peptides. Biomacromolecules. (2014) 15:1337–45. doi: 10.1021/bm401884e
271. Brunetti J, Falciani C, Bracci L, Pini A. Models of in-vivo bacterial infections for the development of antimicrobial peptide-based drugs. Curr Top Med Chem. (2017) 17:613–9. doi: 10.2174/1568026616666160713143017
272. Chaparro E, da Silva PIJ. Lacrain: the first antimicrobial peptide from the body extract of the Brazilian centipede Scolopendra viridicornis. Int J Antimicrob Agents. (2016) 48:277–85. doi: 10.1016/j.ijantimicag.2016.05.015
273. Diniz LCL, Miranda A, da Silva PIJr. Human antimicrobial peptide isolated from triatoma infestans haemolymph, Trypanosoma cruzi-transmitting vector. Front Cell Infect Microbiol. (2018) 8:354. doi: 10.3389/fcimb.2018.00354
274. Roscetto E, Contursi P, Vollaro A, Fusco S, Notomista E, Catania MR. Antifungal and anti-biofilm activity of the first cryptic antimicrobial peptide from an archaeal protein against Candida spp. clinical isolates. Sci Rep. (2018) 8:17570. doi: 10.1038/s41598-018-35530-0
275. Blondelle SE, Lohner K. Optimization and high-throughput screening of antimicrobial peptides. Curr Pharm Des. (2010) 16:3204–11. doi: 10.2174/138161210793292438
276. Ashby M, Petkova A, Gani J, Mikut R, Hilpert K. Use of peptide libraries for identification and optimization of novel antimicrobial peptides. Curr Top Med Chem. (2017) 17:537–53. doi: 10.2174/1568026616666160713125555
277. Bosso M, Standker L, Kirchhoff F, Munch J. Exploiting the human peptidome for novel antimicrobial and anticancer agents. Bioorg Med Chem. (2018) 26:2719–26. doi: 10.1016/j.bmc.2017.10.038
278. Lipkin R, Lazaridis T. Computational studies of peptide-induced membrane pore formation. Philos Trans R Soc Lond B Biol Sci. (2017) 372:20160219. doi: 10.1098/rstb.2016.0219
279. Cipcigan F, Carrieri AP, Pyzer-Knapp EO, Krishna R, Hsiao YW, Winn M, et al. Accelerating molecular discovery through data and physical sciences: applications to peptide-membrane interactions. J Chem Phys. (2018) 148:241744. doi: 10.1063/1.5027261
280. Pfeil MP, Pyne ALB, Losasso V, Ravi J, Lamarre B, Faruqui N, et al. Tuneable poration: host defense peptides as sequence probes for antimicrobial mechanisms. Sci Rep. (2018) 8:14926. doi: 10.1038/s41598-018-33289-y
281. Porto WF, Irazazabal L, Alves ESF, Ribeiro SM, Matos CO, Pires AS, et al. In silico optimization of a guava antimicrobial peptide enables combinatorial exploration for peptide design. Nat Commun. (2018) 9:1490. doi: 10.1038/s41467-018-03746-3
282. Yount NY, Weaver DC, Lee EY, Lee MW, Wang H, Chan LC, et al. Unifying structural signature of eukaryotic alpha-helical host defense peptides. Proc Natl Acad Sci USA. (2019) 116:6944–53. doi: 10.1073/pnas.1819250116
283. Tucker AT, Leonard SP, DuBois CD, Knauf GA, Cunningham AL, Wilke CO, et al. Discovery of next-generation antimicrobials through bacterial self-screening of surface-displayed peptide libraries. Cell. (2018) 172:618–28 e613. doi: 10.1016/j.cell.2017.12.009
284. Wang C, Shen M, Gohain N, Tolbert WD, Chen F, Zhang N, et al. Design of a potent antibiotic peptide based on the active region of human defensin 5. J Med Chem. (2015) 58:3083–93. doi: 10.1021/jm501824a
285. Wang G, Narayana JL, Mishra B, Zhang Y, Wang F, Wang C, et al. Design of antimicrobial peptides: progress made with human cathelicidin LL-37. Adv Exp Med Biol. (2019) 1117:215–40. doi: 10.1007/978-981-13-3588-4_12
286. Zhou J, Liu Y, Shen T, Chen L, Zhang C, Cai K, et al. Antimicrobial activity of the antibacterial peptide PMAP-36 and its analogues. Microb Pathog. (2019) 136:103712. doi: 10.1016/j.micpath.2019.103712
287. Neubert RH, Gensbugel C, Jackel A, Wartewig S. Different physicochemical properties of antimycotic agents are relevant for penetration into and through human nails. Pharmazie. (2006) 61:604–7.
288. Davies-Strickleton H, Cook J, Hannam S, Bennett R, Gibbs A, Edwards D, et al. Assessment of the nail penetration of antifungal agents, with different physico-chemical properties. PLoS ONE. (2020) 15:e0229414. doi: 10.1371/journal.pone.0229414
289. Matsuda Y, Sugiura K, Hashimoto T, Ueda A, Konno Y, Tatsumi Y. Efficacy coefficients determined using nail permeability and antifungal activity in keratin-containing media are useful for predicting clinical efficacies of topical drugs for onychomycosis. PLoS ONE. (2016) 11:e0159661. doi: 10.1371/journal.pone.0159661
290. Mercer DK, Sairi T, Sroka E, Lamont H, Lawrie Y, O'Neil DA. Expression of innate immune defence genes in healthy and onychomycotic nail and stratum corneum. Br J Dermatol. (2017) 177:279–81. doi: 10.1111/bjd.15063
291. Dorschner RA, Lopez-Garcia B, Massie J, Kim C, Gallo RL. Innate immune defense of the nail unit by antimicrobial peptides. J Am Acad Dermatol. (2004) 50:343–8. doi: 10.1016/j.jaad.2003.09.010
292. Brasch J, Morig A, Neumann B, Proksch E. Expression of antimicrobial peptides and toll-like receptors is increased in tinea and pityriasis versicolor. Mycoses. (2014) 57:147–52. doi: 10.1111/myc.12118
293. Zaikovska O, Pilmane M, Kisis J. Morphopathological aspects of healthy nails and nails affected by onychomycosis. Mycoses. (2014) 57:531–6. doi: 10.1111/myc.12191
294. Hao J, Li SK. Permeability of the nail plate. In: Murthy SN, Maibach HI, editors. Topical Nail Products and Ungual Drug Delivery. Boca Raton, FL: CRC Press (2013). p. 37–60.
295. Kobayashi Y, Komatsu T, Sumi M, Numajiri S, Miyamoto M, Kobayashi D, et al. In vitro permeation of several drugs through the human nail plate: relationship between physicochemical properties and nail permeability of drugs. Eur J Pharm Sci. (2004) 21:471–7. doi: 10.1016/j.ejps.2003.11.008
296. Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. (2015) 20:122–8. doi: 10.1016/j.drudis.2014.10.003
297. Monod M. Secreted proteases from dermatophytes. Mycopathologia. (2008) 166:285–94. doi: 10.1007/s11046-008-9105-4
298. Mercer DK, Stewart CS. Keratin hydrolysis by dermatophytes. Med Mycol. (2019) 57:13–22. doi: 10.1093/mmy/myx160
299. Nordstrom R, Malmsten M. Delivery systems for antimicrobial peptides. Adv Colloid Interface Sci. (2017) 242:17–34. doi: 10.1016/j.cis.2017.01.005
300. Piotrowska U, Sobczak M, Oledzka E. Current state of a dual behaviour of antimicrobial peptides-Therapeutic agents and promising delivery vectors. Chem Biol Drug Des. (2017) 90:1079–93. doi: 10.1111/cbdd.13031
301. Javia A, Amrutiya J, Lalani R, Patel V, Bhatt P, Misra A. Antimicrobial peptide delivery: an emerging therapeutic for the treatment of burn and wounds. Ther Deliv. (2018) 9:375–86. doi: 10.4155/tde-2017-0061
302. Makowski M, Silva IC, Pais do Amaral C, Goncalves S, Santos NC. Advances in lipid and metal nanoparticles for antimicrobial peptide delivery. Pharmaceutics. (2019) 11:588. doi: 10.3390/pharmaceutics11110588
303. Thapa RK, Diep DB, Tonnesen HH. Topical antimicrobial peptide formulations for wound healing: current developments and future prospects. Acta Biomater. (2020) 103:52–67. doi: 10.1016/j.actbio.2019.12.025
304. Werle M, Bernkop-Schnurch A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids. (2006) 30:351–67. doi: 10.1007/s00726-005-0289-3
305. Lee AC, Harris JL, Khanna KK, Hong JH. A comprehensive review on current advances in peptide drug development and design. Int J Mol Sci. (2019) 20:2383. doi: 10.3390/ijms20102383
306. Marshall NC, Finlay BB, Overall CM. Sharpening host defenses during infection: proteases cut to the chase. Mol Cell Proteomics. (2017) 16(4 Suppl. 1):S161–71. doi: 10.1074/mcp.O116.066456
307. Steimbach LM, Tonin FS, Virtuoso S, Borba HH, Sanches AC, Wiens A, et al. Efficacy and safety of amphotericin B lipid-based formulations-A systematic review and meta-analysis. Mycoses. (2017) 60:146–54. doi: 10.1111/myc.12585
308. Hamamoto K, Kida Y, Zhang Y, Shimizu T, Kuwano K. Antimicrobial activity and stability to proteolysis of small linear cationic peptides with D-amino acid substitutions. Microbiol Immunol. (2002) 46:741–9. doi: 10.1111/j.1348-0421.2002.tb02759.x
309. Oliva R, Chino M, Pane K, Pistorio V, De Santis A, Pizzo E, et al. Exploring the role of unnatural amino acids in antimicrobial peptides. Sci Rep. (2018) 8:8888. doi: 10.1038/s41598-018-27231-5
310. Bolt HL, Eggimann GA, Jahoda CAB, Zuckermann RN, Sharples GJ, Cobb SL. Exploring the links between peptoid antibacterial activity and toxicity. Medchemcomm. (2017) 8:886–96. doi: 10.1039/C6MD00648E
311. Kuppusamy R, Willcox M, Black DS, Kumar N. Short cationic peptidomimetic antimicrobials. Antibiotics. (2019) 8:44. doi: 10.3390/antibiotics8020044
312. Lakshminarayanan R, Liu S, Li J, Nandhakumar M, Aung TT, Goh E, et al. Synthetic multivalent antifungal peptides effective against fungi. PLoS ONE. (2014) 9:e87730. doi: 10.1371/journal.pone.0087730
313. Zhou C, Li P, Qi X, Sharif AR, Poon YF, Cao Y, et al. A photopolymerized antimicrobial hydrogel coating derived from epsilon-poly-L-lysine. Biomaterials. (2011) 32:2704–12. doi: 10.1016/j.biomaterials.2010.12.040
314. Hakansson J, Bjorn C, Lindgren K, Sjostrom E, Sjostrand V, Mahlapuu M. Efficacy of the novel topical antimicrobial agent PXL150 in a mouse model of surgical site infections. Antimicrob Agents Chemother. (2014) 58:2982–4. doi: 10.1128/AAC.00143-14
315. Kong EF, Tsui C, Boyce H, Ibrahim A, Hoag SW, Karlsson AJ, et al. Development and in vivo evaluation of a novel histatin-5 bioadhesive hydrogel formulation against oral candidiasis. Antimicrob Agents Chemother. (2016) 60:881–9. doi: 10.1128/AAC.02624-15
316. Ahmad I, Perkins WR, Lupan DM, Selsted ME, Janoff AS. Liposomal entrapment of the neutrophil-derived peptide indolicidin endows it with in vivo antifungal activity. Biochim Biophys Acta. (1995) 1237:109–14. doi: 10.1016/0005-2736(95)00087-J
317. Nellore BP, Kanchanapally R, Pedraza F, Sinha SS, Pramanik A, Hamme AT, et al. Bio-conjugated CNT-bridged 3D porous graphene oxide membrane for highly efficient disinfection of pathogenic bacteria and removal of toxic metals from water. ACS Appl Mater Interfaces. (2015) 7:19210–8. doi: 10.1021/acsami.5b05012
318. Liu L, Xu K, Wang H, Tan PK, Fan W, Venkatraman SS, et al. Self-assembled cationic peptide nanoparticles as an efficient antimicrobial agent. Nat Nanotechnol. (2009) 4:457–63. doi: 10.1038/nnano.2009.153
319. Bajaj M, Pandey SK, Nain T, Brar SK, Singh P, Singh S, et al. Stabilized cationic dipeptide capped gold/silver nanohybrids: Towards enhanced antibacterial and antifungal efficacy. Colloids Surf B Biointerfaces. (2017) 158:397–407. doi: 10.1016/j.colsurfb.2017.07.009
320. Kohler JR, Hube B, Puccia R, Casadevall A, Perfect JR. Fungi that infect humans. Microbiol Spectr. (2017) 5:a019273. doi: 10.1128/9781555819583.ch39
321. Eades CP, Armstrong-James DPH. Invasive fungal infections in the immunocompromised host: mechanistic insights in an era of changing immunotherapeutics. Med Mycol. (2019) 57(Supplement_3):S307–17. doi: 10.1093/mmy/myy136
322. Patterson TF, Thompson GR III, Denning DW, Fishman JA, Hadley S, Herbrecht R, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the infectious diseases society of America. Clin Infect Dis. (2016) 63:e1–60. doi: 10.1093/cid/ciw444
323. Sam QH, Yew WS, Seneviratne CJ, Chang MW, Chai LYA. Immunomodulation as therapy for fungal infection: are we closer? Front Microbiol. (2018) 9:1612. doi: 10.3389/fmicb.2018.01612
324. Scriven JE, Tenforde MW, Levitz SM, Jarvis JN. Modulating host immune responses to fight invasive fungal infections. Curr Opin Microbiol. (2017) 40:95–103. doi: 10.1016/j.mib.2017.10.018
325. Delliere S, Guery R, Candon S, Rammaert B, Aguilar C, Lanternier F, et al. Understanding pathogenesis and care challenges of immune reconstitution inflammatory syndrome in fungal infections. J Fungi. (2018) 4:139. doi: 10.3390/jof4040139
326. Schmidt C. The benefits of immunotherapy combinations. Nature. (2017) 552:S67–9. doi: 10.1038/d41586-017-08702-7
327. Armstrong-James D, Harrison TS. Immunotherapy for fungal infections. Curr Opin Microbiol. (2012) 15:434–9. doi: 10.1016/j.mib.2012.06.001
328. Datta K, Hamad M. Immunotherapy of fungal infections. Immunol Invest. (2015) 44:738–76. doi: 10.3109/08820139.2015.1093913
329. Armstrong-James D, Brown GD, Netea MG, Zelante T, Gresnigt MS, van de Veerdonk FL, et al. Immunotherapeutic approaches to treatment of fungal diseases. Lancet Infect Dis. (2017) 17:e393–402. doi: 10.1016/S1473-3099(17)30442-5
330. Loreto ES, Tondolo JSM, Alves SH, Santurio JM. Immunotherapy for fungal infections. In: Metodiev K, editor. Immunotherapy: Myths, Reality, Ideas, Future. Intech Open (2017). p. 291–322.
331. Davies R, O'Dea K, Gordon A. Immune therapy in sepsis: are we ready to try again? J Intensive Care Soc. (2018) 19:326–44. doi: 10.1177/1751143718765407
332. Lauruschkat CD, Einsele H, Loeffler J. Immunomodulation as a therapy for aspergillus infection: current status and future perspectives. J Fungi. (2018) 4:137. doi: 10.3390/jof4040137
333. Delsing CE, Gresnigt MS, Leentjens J, Preijers F, Frager FA, Kox M, et al. Interferon-gamma as adjunctive immunotherapy for invasive fungal infections: a case series. BMC Infect Dis. (2014) 14:166. doi: 10.1186/1471-2334-14-166
334. TICGDCS G. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. The International Chronic Granulomatous Disease Cooperative Study Group. N Engl J Med. (1991) 324:509–16. doi: 10.1056/NEJM199102213240801
335. Pasic S, Abinun M, Pistignjat B, Vlajic B, Rakic J, Sarjanovic L, et al. Aspergillus osteomyelitis in chronic granulomatous disease: treatment with recombinant gamma-interferon and itraconazole. Pediatr Infect Dis J. (1996) 15:833–4. doi: 10.1097/00006454-199609000-00021
336. Saulsbury FT. Successful treatment of aspergillus brain abscess with itraconazole and interferon-gamma in a patient with chronic granulomatous disease. Clin Infect Dis. (2001) 32:E137–9. doi: 10.1086/320158
337. Riddell LA, Pinching AJ, Hill S, Ng TT, Arbe E, Lapham GP, et al. A phase III study of recombinant human interferon gamma to prevent opportunistic infections in advanced HIV disease. AIDS Res Hum Retroviruses. (2001) 17:789–97. doi: 10.1089/088922201750251981
338. Bodasing N, Seaton RA, Shankland GS, Pithie A. Gamma-interferon treatment for resistant oropharyngeal candidiasis in an HIV-positive patient. J Antimicrob Chemother. (2002) 50:765–6. doi: 10.1093/jac/dkf206
339. Jarvis JN, Meintjes G, Rebe K, Williams GN, Bicanic T, Williams A, et al. Adjunctive interferon-gamma immunotherapy for the treatment of HIV-associated cryptococcal meningitis: a randomized controlled trial. AIDS. (2012) 26:1105–13. doi: 10.1097/QAD.0b013e3283536a93
340. Poynton CH, Barnes RA, Rees J. Interferon gamma and granulocyte-macrophage colony-stimulating factor for the treatment of hepatosplenic candidosis in patients with acute leukemia. Clin Infect Dis. (1998) 26:239–40. doi: 10.1086/517077
341. Dignani MC, Rex JH, Chan KW, Dow G, de Magalhaes-Silverman M, Maddox A, et al. Immunomodulation with interferon-gamma and colony-stimulating factors for refractory fungal infections in patients with leukemia. Cancer. (2005) 104:199–204. doi: 10.1002/cncr.21142
342. Armstrong-James D, Teo IA, Shrivastava S, Petrou MA, Taube D, Dorling A, et al. Exogenous interferon-gamma immunotherapy for invasive fungal infections in kidney transplant patients. Am J Transplant. (2010) 10:1796–803. doi: 10.1111/j.1600-6143.2010.03094.x
343. Malmvall BE, Follin P. Successful interferon-gamma therapy in a chronic granulomatous disease (CGD) patient suffering from Staphylococcus aureus hepatic abscess and invasive Candida albicans infection. Scand J Infect Dis. (1993) 25:61–6. doi: 10.1080/00365549309169671
344. Ellis M, Watson R, McNabb A, Lukic ML, Nork M. Massive intracerebral aspergillosis responding to combination high dose liposomal amphotericin B and cytokine therapy without surgery. J Med Microbiol. (2002) 51:70–5. doi: 10.1099/0022-1317-51-1-70
345. Kelleher P, Goodsall A, Mulgirigama A, Kunst H, Henderson DC, Wilson R, et al. Interferon-gamma therapy in two patients with progressive chronic pulmonary aspergillosis. Eur Respir J. (2006) 27:1307–10. doi: 10.1183/09031936.06.00021705
346. Netea MG, Brouwer AE, Hoogendoorn EH, Van der Meer JW, Koolen M, Verweij PE, et al. Two patients with cryptococcal meningitis and idiopathic CD4 lymphopenia: defective cytokine production and reversal by recombinant interferon- gamma therapy. Clin Infect Dis. (2004) 39:e83–7. doi: 10.1086/425121
347. Miller CH, Maher SG, Young HA. Clinical use of interferon-gamma. Ann N Y Acad Sci. (2009) 1182:69–79. doi: 10.1111/j.1749-6632.2009.05069.x
348. Sung L, Nathan PC, Alibhai SM, Tomlinson GA, Beyene J. Meta-analysis: effect of prophylactic hematopoietic colony-stimulating factors on mortality and outcomes of infection. Ann Intern Med. (2007) 147:400–11. doi: 10.7326/0003-4819-147-6-200709180-00010
349. Hazel DL, Newland AC, Kelsey SM. Malignancy: granulocyte colony stimulating factor increases the efficacy of conventional amphotericin in the treatment of presumed deep-seated fungal infection in neutropenic patients following intensive chemotherapy or bone marrow transplantation for haematological malignancies. Hematology. (1999) 4:305–11. doi: 10.1080/10245332.1999.11746453
350. Bodey GP, Anaissie E, Gutterman J, Vadhan-Raj S. Role of granulocyte-macrophage colony-stimulating factor as adjuvant therapy for fungal infection in patients with cancer. Clin Infect Dis. (1993) 17:705–7. doi: 10.1093/clinids/17.4.705
351. Kullberg BJ, Vandewoude K, Herbrecht R, Jacobs F, Aoun M, Kujath P. A double-blind, randomized, placebo-controlled phase II study of filgrastim. (recombinant granulocyte colony-stimulating factor) in combination with fluconazole for the treatment of invasive candidiasis and candidemia in nonneutropenic patients. In: 38th Interscience Conference on Antimicrobial Agents and Chemotherapy. San Diego, CA: American Society for Microbiology (1998).
352. Gabrilove JL, Jakubowski A, Scher H, Sternberg C, Wong G, Grous J, et al. Effect of granulocyte colony-stimulating factor on neutropenia and associated morbidity due to chemotherapy for transitional-cell carcinoma of the urothelium. N Engl J Med. (1988) 318:1414–22. doi: 10.1056/NEJM198806023182202
353. Pursell K, Verral S, Daraiesh F, Shrestha N, Skariah A, Hasan E, et al. Impaired phagocyte respiratory burst responses to opportunistic fungal pathogens in transplant recipients: in vitro effect of r-metHuG-CSF (Filgrastim). Transpl Infect Dis. (2003) 5:29–37. doi: 10.1034/j.1399-3062.2003.00004.x
354. Mehta HM, Malandra M, Corey SJ. G-CSF and GM-CSF in neutropenia. J Immunol. (2015) 195:1341–9. doi: 10.4049/jimmunol.1500861
355. Wan L, Zhang Y, Lai Y, Jiang M, Song Y, Zhou J, et al. Effect of granulocyte-macrophage colony-stimulating factor on prevention and treatment of invasive fungal disease in recipients of allogeneic stem-cell transplantation: a prospective multicenter randomized phase IV trial. J Clin Oncol. (2015) 33:3999–4006. doi: 10.1200/JCO.2014.60.5121
356. Rowe JM, Andersen JW, Mazza JJ, Bennett JM, Paietta E, Hayes FA, et al. A randomized placebo-controlled phase III study of granulocyte-macrophage colony-stimulating factor in adult patients. (> 55 to 70 years of age) with acute myelogenous leukemia: a study of the Eastern Cooperative Oncology Group (E1490). Blood. (1995) 86:457–62. doi: 10.1182/blood.V86.2.457.bloodjournal862457
357. Buchner T, Hiddemann W, Koenigsmann M, Zuhlsdorf M, Wormann B, Boeckmann A, et al. Recombinant human granulocyte-macrophage colony-stimulating factor after chemotherapy in patients with acute myeloid leukemia at higher age or after relapse. Blood. (1991) 78:1190–7. doi: 10.1182/blood.V78.5.1190.1190
358. Bodey GP, Anaissie E, Gutterman J, Vadhan-Raj S. Role of granulocyte-macrophage colony-stimulating factor as adjuvant treatment in neutropenic patients with bacterial and fungal infection. Eur J Clin Microbiol Infect Dis. (1994) 13 (Suppl. 2):S18–22. doi: 10.1007/BF01971991
359. Vazquez JA, Hidalgo JA, De Bono S. Use of sargramostim (rh-GM-CSF) as adjunctive treatment of fluconazole-refractory oropharyngeal candidiasis in patients with AIDS: a pilot study. HIV Clin Trials. (2000) 1:23–9. doi: 10.1310/LF5T-WYY7-0U3E-G8BQ
360. Garcia-Diaz JB, Palau L, Pankey GA. Resolution of rhinocerebral zygomycosis associated with adjuvant administration of granulocyte-macrophage colony-stimulating factor. Clin Infect Dis. (2001) 32:e145–50. doi: 10.1086/320767
361. Witz F, Sadoun A, Perrin MC, Berthou C, Briere J, Cahn JY, et al. A placebo-controlled study of recombinant human granulocyte-macrophage colony-stimulating factor administered during and after induction treatment for de novo acute myelogenous leukemia in elderly patients. Groupe Ouest Est Leucemies Aigues Myeloblastiques (GOELAM). Blood. (1998) 91:2722–30.
362. Nemunaitis J, Meyers JD, Buckner CD, Shannon-Dorcy K, Mori M, Shulman H, et al. Phase I trial of recombinant human macrophage colony-stimulating factor in patients with invasive fungal infections. Blood. (1991) 78:907–13. doi: 10.1182/blood.V78.4.907.907
363. Nemunaitis J, Shannon-Dorcy K, Appelbaum FR, Meyers J, Owens A, Day R, et al. Long-term follow-up of patients with invasive fungal disease who received adjunctive therapy with recombinant human macrophage colony-stimulating factor. Blood. (1993) 82:1422–7. doi: 10.1182/blood.V82.5.1422.1422
364. Kandalla PK, Sarrazin S, Molawi K, Berruyer C, Redelberger D, Favel A, et al. M-CSF improves protection against bacterial and fungal infections after hematopoietic stem/progenitor cell transplantation. J Exp Med. (2016) 213:2269–79. doi: 10.1084/jem.20151975
365. Nassar F, Brummer E, Stevens DA. Macrophage colony-stimulating factor (M-CSF) induction of enhanced anticryptococcal activity in human monocyte-derived macrophages: synergy with fluconazole for killing. Cell Immunol. (1995) 164:113–8. doi: 10.1006/cimm.1995.1149
366. Nassar F, Brummer E, Stevens DA. Effect of in vivo macrophage colony-stimulating factor on fungistasis of bronchoalveolar and peritoneal macrophages against Cryptococcus neoformans. Antimicrob Agents Chemother. (1994) 38:2162–4. doi: 10.1128/AAC.38.9.2162
367. Vitt CR, Fidler JM, Ando D, Zimmerman RJ, Aukerman SL. Antifungal activity of recombinant human macrophage colony-stimulating factor in models of acute and chronic candidiasis in the rat. J Infect Dis. (1994) 169:369–74. doi: 10.1093/infdis/169.2.369
368. Hume DA, Denkins Y. The deleterious effect of macrophage colony-stimulating factor (CSF-1) on the pathology of experimental candidiasis in mice. Lymphokine Cytokine Res. (1992) 11:95–8.
369. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
370. Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. (2018) 11:39. doi: 10.1186/s13045-018-0582-8
371. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. (2015) 372:2521–32. doi: 10.1056/NEJMoa1503093
372. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. (2015) 348:56–61. doi: 10.1126/science.aaa8172
373. Ferris RL, Blumenschein GJr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. (2016) 375:1856–67. doi: 10.1056/NEJMoa1602252
374. Mellinghoff SC, von Bergwelt-Baildon M, Schosser HA, Cornely OA. A novel approach to candidemia? The potential role of checkpoint inhibition. Med Mycol. (2019) 57:151–4. doi: 10.1093/mmy/myy089
375. Lazar-Molnar E, Gacser A, Freeman GJ, Almo SC, Nathenson SG, Nosanchuk JD. The PD-1/PD-L costimulatory pathway critically affects host resistance to the pathogenic fungus Histoplasma capsulatum. Proc Natl Acad Sci USA. (2008) 105:2658–63. doi: 10.1073/pnas.0711918105
376. Campanelli AP, Martins GA, Souto JT, Pereira MS, Livonesi MC, Martinez R, et al. Fas-Fas ligand (CD95-CD95L) and cytotoxic T lymphocyte antigen-4 engagement mediate T cell unresponsiveness in patients with paracoccidioidomycosis. J Infect Dis. (2003) 187:1496–505. doi: 10.1086/374646
377. Chang KC, Burnham CA, Compton SM, Rasche DP, Mazuski RJ, McDonough JS, et al. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit Care. (2013) 17:R85. doi: 10.1186/cc12711
378. McGaha T, Murphy JW. CTLA-4 down-regulates the protective anticryptococcal cell-mediated immune response. Infect Immun. (2000) 68:4624–30. doi: 10.1128/IAI.68.8.4624-4630.2000
379. Grimaldi D, Pradier O, Hotchkiss RS, Vincent JL. Nivolumab plus interferon-gamma in the treatment of intractable mucormycosis. Lancet Infect Dis. (2017) 17:18. doi: 10.1016/S1473-3099(16)30541-2
380. Hotchkiss RS, Colston E, Yende S, Crouser ED, Martin GS, Albertson T, et al. Immune checkpoint inhibition in sepsis: a Phase 1b randomized study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of nivolumab. Intensive Care Med. (2019) 45:1360–71. doi: 10.1007/s00134-019-05704-z
382. Sui X, Yan L, Jiang YY. The vaccines and antibodies associated with Als3p for treatment of Candida albicans infections. Vaccine. (2017) 35:5786–93. doi: 10.1016/j.vaccine.2017.08.082
383. Nami S, Mohammadi R, Vakili M, Khezripour K, Mirzaei H, Morovati H. Fungal vaccines, mechanism of actions and immunology: a comprehensive review. Biomed Pharmacother. (2019) 109:333–44. doi: 10.1016/j.biopha.2018.10.075
384. Medici NP, Del Poeta M. New insights on the development of fungal vaccines: from immunity to recent challenges. Mem Inst Oswaldo Cruz. (2015) 110:966–73. doi: 10.1590/0074-02760150335
385. Fidel PL Jr, Cutler JE. Prospects for development of a vaccine to prevent and control vaginal candidiasis. Curr Infect Dis Rep. (2011) 13:102–7. doi: 10.1007/s11908-010-0143-y
386. Spellberg B. Vaccines for invasive fungal infections. F1000 Med Rep. (2011) 3:13. doi: 10.3410/M3-13
387. Edwards JE Jr, Schwartz MM, Schmidt CS, Sobel JD, Nyirjesy P, Schodel F, et al. A fungal immunotherapeutic vaccine (NDV-3A) for treatment of recurrent vulvovaginal candidiasis-a phase 2 randomized, double-blind, placebo-controlled trial. Clin Infect Dis. (2018) 66:1928–36. doi: 10.1093/cid/ciy185
388. De Bernardis F, Graziani S, Tirelli F, Antonopoulou S. Candida vaginitis: virulence, host response and vaccine prospects. Med Mycol. (2018) 56(suppl_1):26–31. doi: 10.1093/mmy/myx139
389. Levy DA, Bohbot JM, Catalan F, Normier G, Pinel AM, Dussourd d'Hinterland L. Phase II study of D.651, an oral vaccine designed to prevent recurrences of vulvovaginal candidiasis. Vaccine. (1989) 7:337–40. doi: 10.1016/0264-410X(89)90197-7
390. Pappagianis D. Evaluation of the protective efficacy of the killed Coccidioides immitis spherule vaccine in humans. The Valley Fever Vaccine Study Group. Am Rev Respir Dis. (1993) 148:656–60. doi: 10.1164/ajrccm/148.3.656
391. Santos E, Levitz SM. Fungal vaccines and immunotherapeutics. Cold Spring Harb Perspect Med. (2014) 4:a019711. doi: 10.1101/cshperspect.a019711
392. Kirkland TN. The quest for a vaccine against coccidioidomycosis: a neglected disease of the Americas. J Fungi. (2016) 2:34. doi: 10.3390/jof2040034
393. Levitz SM. Aspergillus vaccines: hardly worth studying or worthy of hard study? Med Mycol. (2017) 55:103–8. doi: 10.1093/mmy/myw081
394. Caballero Van Dyke MC, Wormley FL Jr. A call to arms: quest for a cryptococcal vaccine. Trends Microbiol. (2018) 26:436–46. doi: 10.1016/j.tim.2017.10.002
395. Wuthrich M, Filutowicz HI, Warner T, Deepe GSJr, Klein BS. Vaccine immunity to pathogenic fungi overcomes the requirement for CD4 help in exogenous antigen presentation to CD8+ T cells: implications for vaccine development in immune-deficient hosts. J Exp Med. (2003) 197:1405–16. doi: 10.1084/jem.20030109
396. Martinez M, Clemons KV, Stevens DA. Heat-killed yeast as a pan-fungal vaccine. Methods Mol Biol. (2017) 1625:23–30. doi: 10.1007/978-1-4939-7104-6_2
397. Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs. (2015) 7:9–14. doi: 10.4161/19420862.2015.989042
398. Castelli MS, McGonigle P, Hornby PJ. The pharmacology and therapeutic applications of monoclonal antibodies. Pharmacol Res Perspect. (2019) 7:e00535. doi: 10.1002/prp2.535
399. Sparrow E, Friede M, Sheikh M, Torvaldsen S. Therapeutic antibodies for infectious diseases. Bull World Health Organ. (2017) 95:235–7. doi: 10.2471/BLT.16.178061
400. Wong SK, Li A, Lanctot KL, Paes B. Adherence and outcomes: a systematic review of palivizumab utilization. Expert Rev Respir Med. (2018) 12:27–42. doi: 10.1080/17476348.2018.1401926
401. Nagy CF, Leach TS, King A, Guttendorf R. Safety, pharmacokinetics, and immunogenicity of obiltoxaximab after intramuscular administration to healthy humans. Clin Pharmacol Drug Dev. (2018) 7:652–60. doi: 10.1002/cpdd.410
402. Tsai CW, Morris S. Approval of raxibacumab for the treatment of inhalation anthrax under the US Food and Drug Administration “Animal Rule”. Front Microbiol. (2015) 6:1320. doi: 10.3389/fmicb.2015.01320
403. Casadevall A, Pirofski LA. Immunoglobulins in defense, pathogenesis, and therapy of fungal diseases. Cell Host Microbe. (2012) 11:447–56. doi: 10.1016/j.chom.2012.04.004
404. Beucher B, Marot-Leblond A, Billaud-Nail S, Oh SH, Hoyer LL, Robert R. Recognition of Candida albicans Als3 by the germ tube-specific monoclonal antibody 3D9.3. FEMS Immunol Med Microbiol. (2009) 55:314–23. doi: 10.1111/j.1574-695X.2008.00502.x
405. Moragues MD, Omaetxebarria MJ, Elguezabal N, Sevilla MJ, Conti S, Polonelli L, et al. A monoclonal antibody directed against a Candida albicans cell wall mannoprotein exerts three anti-C. albicans activities. Infect Immun. (2003) 71:5273–9. doi: 10.1128/IAI.71.9.5273-5279.2003
406. Casadevall A, Cleare W, Feldmesser M, Glatman-Freedman A, Goldman DL, Kozel TR, et al. Characterization of a murine monoclonal antibody to Cryptococcus neoformans polysaccharide that is a candidate for human therapeutic studies. Antimicrob Agents Chemother. (1998) 42:1437–46. doi: 10.1128/AAC.42.6.1437
407. Mukherjee J, Scharff MD, Casadevall A. Protective murine monoclonal antibodies to Cryptococcus neoformans. Infect Immun. (1992) 60:4534–41. doi: 10.1128/IAI.60.11.4534-4541.1992
408. Larsen RA, Pappas PG, Perfect J, Aberg JA, Casadevall A, Cloud GA, et al. Phase I evaluation of the safety and pharmacokinetics of murine-derived anticryptococcal antibody 18B7 in subjects with treated cryptococcal meningitis. Antimicrob Agents Chemother. (2005) 49:952–8. doi: 10.1128/AAC.49.3.952-958.2005
409. Rudkin FM, Raziunaite I, Workman H, Essono S, Belmonte R, MacCallum DM, et al. Single human B cell-derived monoclonal anti-Candida antibodies enhance phagocytosis and protect against disseminated candidiasis. Nat Commun. (2018) 9:5288. doi: 10.1038/s41467-018-07738-1
410. Matthews RC, Rigg G, Hodgetts S, Carter T, Chapman C, Gregory C, et al. Preclinical assessment of the efficacy of mycograb, a human recombinant antibody against fungal HSP90. Antimicrob Agents Chemother. (2003) 47:2208–16. doi: 10.1128/AAC.47.7.2208-2216.2003
411. Hodgetts S, Nooney L, Al-Akeel R, Curry A, Awad S, Matthews R, et al. Efungumab and caspofungin: pre-clinical data supporting synergy. J Antimicrob Chemother. (2008) 61:1132–9. doi: 10.1093/jac/dkn075
412. Bugli F, Cacaci M, Martini C, Torelli R, Posteraro B, Sanguinetti M, et al. Human monoclonal antibody-based therapy in the treatment of invasive candidiasis. Clin Dev Immunol. (2013) 2013:403121. doi: 10.1155/2013/403121
413. Richie DL, Ghannoum MA, Isham N, Thompson KV, Ryder NS. Nonspecific effect of Mycograb on amphotericin B MIC. Antimicrob Agents Chemother. (2012) 56:3963–4. doi: 10.1128/AAC.00435-12
414. Baistrocchi SR, Lee MJ, Lehoux M, Ralph B, Snarr BD, Robitaille R, et al. Posaconazole-loaded leukocytes as a novel treatment strategy targeting invasive pulmonary Aspergillosis. J Infect Dis. (2017) 215:1734–41. doi: 10.1093/infdis/jiw513
Keywords: antimicrobial peptide, host defence peptide, innate immunity, antifungal, immunotherapeutics
Citation: Mercer DK and O'Neil DA (2020) Innate Inspiration: Antifungal Peptides and Other Immunotherapeutics From the Host Immune Response. Front. Immunol. 11:2177. doi: 10.3389/fimmu.2020.02177
Received: 30 May 2020; Accepted: 10 August 2020;
Published: 17 September 2020.
Edited by:
Thanh Kha Phan, La Trobe University, AustraliaReviewed by:
Agostinho Carvalho, University of Minho, PortugalFengliang Jin, South China Agricultural University, China
Copyright © 2020 Mercer and O'Neil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Derry K. Mercer, ZGVycnlAbm92YWJpb3RpY3MuY28udWs=