 María Consuelo Bachmann1†
María Consuelo Bachmann1† Sofía Bellalta1†
Sofía Bellalta1† Roque Basoalto1†Fernán Gómez-Valenzuela1†Yorschua Jalil1†Macarena Lépez1†
Roque Basoalto1†Fernán Gómez-Valenzuela1†Yorschua Jalil1†Macarena Lépez1† Anibal Matamoros1,2†
Anibal Matamoros1,2† Rommy von Bernhardi1*
Rommy von Bernhardi1*- 1School of Medicine, Pontificia Universidad Católica de Chile, Santiago, Chile
- 2Institute of Biological Sciences (ICB), Federal University of Pará, Belem, Brazil
The aging process is driven by multiple mechanisms that lead to changes in energy production, oxidative stress, homeostatic dysregulation and eventually to loss of functionality and increased disease susceptibility. Most aged individuals develop chronic low-grade inflammation, which is an important risk factor for morbidity, physical and cognitive impairment, frailty, and death. At any age, chronic inflammatory diseases are major causes of morbimortality, affecting up to 5–8% of the population of industrialized countries. Several environmental factors can play an important role for modifying the inflammatory state. Genetics accounts for only a small fraction of chronic-inflammatory diseases, whereas environmental factors appear to participate, either with a causative or a promotional role in 50% to 75% of patients. Several of those changes depend on epigenetic changes that will further modify the individual response to additional stimuli. The interaction between inflammation and the environment offers important insights on aging and health. These conditions, often depending on the individual’s sex, appear to lead to decreased longevity and physical and cognitive decline. In addition to biological factors, the environment is also involved in the generation of psychological and social context leading to stress. Poor psychological environments and other sources of stress also result in increased inflammation. However, the mechanisms underlying the role of environmental and psychosocial factors and nutrition on the regulation of inflammation, and how the response elicited for those factors interact among them, are poorly understood. Whereas certain deleterious environmental factors result in the generation of oxidative stress driven by an increased production of reactive oxygen and nitrogen species, endoplasmic reticulum stress, and inflammation, other factors, including nutrition (polyunsaturated fatty acids) and behavioral factors (exercise) confer protection against inflammation, oxidative and endoplasmic reticulum stress, and thus ameliorate their deleterious effect. Here, we discuss processes and mechanisms of inflammation associated with environmental factors and behavior, their links to sex and gender, and their overall impact on aging.
General View
The systemic chronic low-grade inflammation observed in aged individuals has been coined as “inflammaging” (1), and leads to metabolic dysfunction, physical limitations, and frailty in older adults [reviewed in (2)]. Inflammaging leads to an increased secretion of interleukin 1beta (IL1β), interferons (IFNs), and tumor necrosis factor α (TNFα) (3). This inflammatory response appears to depend on biological factors like sex, being higher in older women, and is influenced by many environmental factors. The environment affects multiple biological mechanisms, epigenetics, mitochondrial function, cellular senescence, proteostasis, intercellular communication, metabolism, and inflammation (4). Several environmental links to chronic inflammation and age-related diseases have been shown including cardiovascular disease, type-2 diabetes, hypertension, and neurodegenerative disease (5). Their influence on chronic inflammation impacts aging across the individual’s lifespan [reviewed in (6)] and can have both beneficial and deleterious effects. For example, nutritional factors like western diet, associate with DNA damage and the impairment of its repair [discussed in (7)]. By the contrary, the Mediterranean diet shows beneficial effects, such as reduced inflammation, cardiovascular disease, and mortality (8). Several cytokines linked to inflammation, some of which target the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) pathway [reviewed in (9)] are also associated with metabolic changes [discussed in (10)]. Metabolic interventions like caloric restriction extends health- and lifespan in C. elegans, Drosophila, and mice.
Aging has also been linked to epigenetic changes affecting the regulation of expression of many genes. Several of the epigenetic mechanisms, including DNA methylation, histone modifications and miRNAs, are sensitive to the environment, change with age (11) and depend on the sex of the individual. Gene-environment interactions have been found in genes coding for proinflammatory cytokines such as IL1β and air pollution, being correlated to inflammation and increased risk of Parkinson’s disease (12).
Human aging and age-related chronic diseases have been linked to mitochondrial impairment (13), with decreased energy production and increased generation of radical oxygen species (ROS), and inflammation (14). Because inflammation appears to be involved in the molecular, phenotypic, and functional consequences of aging, a potential strategy to tackle pathological aging could be to intervene the inflammatory state of aging. Reducing chronic inflammation could prevent pathological aging phenotypes and their functional consequences. One readily accessible place for intervention is the modification of deleterious lifestyle factors. Obesity, for example, has a strong correlation with systemic inflammation, and together with insulin resistance are frequently observed in aging. Unhealthy diet, stress, use of drugs, exposure to pollution and sedentarism can lead to obesity, defective immunoregulation, and inflammatory cytokines production (15–17). Pollutants, especially those that are stored in adipose tissue, affect both inflammatory and metabolic pathway genes (18). In addition to health behavior (sleep, diet, physical activity), and exposure to environmental toxins [reviewed in (19)], an important, although lesser studied, part of the environmental context are social interactions. Exposure to psychosocial stress (20) and poor sleep (21) can contribute to elevated inflammation. Many sources of stress, not just traumatic events, may have pathophysiological implications, contributing to dysregulate the immune response, and have long lasting effects on aging.
The impact of environmental factors will depend on the combination of specific conditions, their timing along the lifespan, and their interaction with biological factors such as sex and the individual genetic background. Thus, aging and the risk for chronic diseases is the result of the combination of multiple factors. Understanding the interplay of these factors may offer the opportunity to design specific interventions.
Sex-Dependent Changes on Immune-Inflammatory Response
The inflammatory response is different in men and women. Adult females develop stronger innate and adaptive immune responses than males. These sex-related differences can determine the ability of immune cells to generate an effective inflammatory response, which translates into epidemiological differences on the prevalence of various pathologies, including allergies (22), asthma (23, 24), autoimmune diseases (25), anaphylaxis (26), neonatal sepsis (27), and cancer (28), among several pathologies. The immune response of women is polarized towards an increased production of Th2 cells, T regulatory cells (Treg), M2 macrophages, IL4, IL10, and GATA-3 cytokines, and decreased Th1, Th17, TBet, and RORγt lymphocytes (29–31). On the contrary, men show an immune response that depends on Th1 lymphocytes (32, 33), high IL33 production (34) and low levels of reactive mast cells (35). Men have also an increased response of microglia in the central nervous system (CNS) and an increased presence of TNFα and prostaglandins in response to inflammatory stimuli (36).
Mechanisms Involved in the Sex-Dependent Differences in Inflammatory Response
Differences in inflammatory response between men and women vary among specific tissues. In the CNS inflammation, women show greater levels of B-cell (CD19+, CD5+, CD1dhi B10) migration from the spleen to the site of injury than men, followed by an increase of macrophages/microglia (CD11b+, CD206), which appears to generate a lower neuroinflammatory response in female compared with male mice (37). In addition, women develop an increased immunoreactivity due to high numbers of IFN-producing dendritic cells (38, 39). Female mice tend to have M2 phenotype and activated eosinophils and mast cells show a higher reactivity than in male mice (35, 40). However, in response to an acute inflammatory stimulus, males produce higher amounts of inflammatory cytokines, CD8a+ neutrophil and T cells infiltration of the injury site (41). Conversely, the inflammatory microenvironment in female mice is characterized by an increased production of antibodies (42, 43) and a differential pattern migration of antibody-secreting cells (42).
The immune system responds differently in men and women not only because of the influence of sex hormones, but also differences in the patterns of autosomal methylation and X chromosome methylation, which determine distinctive profiles of gene expression (43, 44). Sex hormones exert antagonist effects on the immune system: Both estradiol and testosterone have a suppressive effect on the immune response (45).
Estrogen is the sex hormone with the greatest impact on the immune response, being described as one of the non-modifiable regulators of the immune system, due to its immunoregulatory and protective effects in many inflammatory models (46). However, this is contradictory with the fact that women have a higher prevalence of autoimmune diseases than men, although estrogens should be a protective condition (47).
The sex-dependent difference in the immune response is time-, and estrogen dose-dependent (29). Variations on the estrogen concentration during the ovulatory cycle, puberty or menopause, can promote the development of immune-related diseases (48). Mice exposed to chronic estrogen-treatment generate hormone resistance, decreasing the clonal expansion of Treg lymphocytes in autoimmune diseases (49, 50).
Estrogen regulates immune response primarily through α- and β-estrogen receptors (ERα/β), mitogen-activated protein kinase (MAPK) pathways, estrogen-dependent 3′-5′-cyclic adenosine monophosphate (cAMP) response element-binding (CREB), and modifications in the production of cAMP in immune cells (51). In addition to estrogen receptors, the presence of IL receptors influences the type of immune response; female macrophages express greater amounts of IL4 receptors than males. IL4 receptors favor the M2 phenotype when stimulated by estrogen. In agreement with that, estrogen induces an increased expression of IL4 on naive CD4+ T cells (40, 52). For a better general view of estrogen´s mechanisms and effect on the innate immune system cells we recommend reviews that have extensively covered those topics (53–56).
Dependence on the Sex of the Immune Response Gene Expression
Sex regulates gene expression in multiple human tissues, in fact, one third of the autosomal genes that are expressed in a sex-biased manner exhibit androgen or estrogen hormonal response elements (57, 58). Sex hormones play a strong role in sexually dimorphic gene networks (59), inducing aberrant expression in immune response genes via differential methylation CCL18 CXCL5 IL5 (60). There are changes in the methylation pattern of sex-dependent immune response genes during embryonic development, which are reinforced in puberty by the estrogen-mediated induction of active forms of chromatins that are maintained during adulthood (61).
Immune response-related genes located in chromosomes 3 and X are differently expressed in B lymphocytes depending on the sex of the individual (62). Among the differentially expressed genes that are relevant for the immune/inflammatory response, can be mentioned the Toll-like signaling, cytokine receptors, Jak-STAT pathway and genes related to the activation of T-cell receptors (63). Phenotypically, the different pattern of gene expression may explain the greater female T-cell expandable capacity when exposed to an antigen (64).
Female T cells present higher activation and division capacities than their male counterparts. However, male T cells can develop greater infiltration potential and a lower self-reactive phenotype than female ones (65, 66). These differences could be due to the high expression of peroxisome proliferator-activated receptors (PPARs) (64), prostaglandins, and cyclooxygenase-2 (COX-2) in males (67).
The influence of sex on the immune response is observed throughout life and is accentuated with aging. In the neonatal stage, women have a lower concentration of regulatory T lymphocytes than men (68). During childhood, men develop a more intense immune response and are more likely to develop infections by various pathogens compared with women (69, 70). With increasing age, the dynamics and proportion of lymphocytes and myeloid cells differ depending on the sex due to the differential expression of 144 genes of the immune response in men and women (71). Also, in aged individuals, epigenomic changes generate a more robust innate and pro-inflammatory response in men and an increased activity in the adaptive immune response in women (72, 73). In recent times, during the COVID-19 pandemic, it has been observed that the infection by SARS-CoV-2 in older adults shows conspicuous differences; men have elevated plasma levels of IL8 and IL18 and a high amount of monocytes whereas women develop a robust activation of T lymphocytes (74). This differences in the immune response could explain the higher mortality of COVID19 in men than in women (75, 76).
To recapitulate, sex hormones and genetic expression patterns in men and women can generate distinct immune and inflammatory responses that determine singularities in the epidemiological distribution of immune diseases. Research protocols in immune response and inflammation must be redefined to avoid results biased by sex. Furthermore, research in women is urgently needed to define the efficacy for women of several therapies that were originally tested in men.
Nutrients and Inflammation: Role of the Diet and Polyunsaturated Fatty Acids
The increase in noncommunicable diseases (NCDs), such as obesity, hypertension and cancer as well as the low-grade chronic inflammation that characterizes most NCDs (77) can be affected by environmental factors that change the immune response. Lifestyle factors like nutrition can modulate the immune system. It has been reported in mice that western diet-induced systemic inflammation and reprogramming of myeloid cell precursors is mediated by the activation of the NLRP3 inflammasome, which is a key sensor of the innate immune system for metabolic danger signals, such as uric acid and cholesterol (78). Metabolic regulation appears to be very robust and long lasting, being reported that proper nutrition during pregnancy can reduce the risk for NCDs in the offspring even at adult age (79, 80).
The Impact of the Diet on the Immune Response and Inflammation
Some diet types can result in metabolic and epigenetic changes that affect immune function (81), as reported in populations that consume a high-fat and low-fiber western diet, who show a prevalence of NCDs higher than populations that consume a Mediterranean diet or a diet based on bioactive compounds, like the hydroxytyrosol in olive oil (82–84). There is evidence supporting the anti-inflammatory activity of phenolic extracts from olive oil, such as their ability to reduce lipopolysaccharide (LPS)-stimulated Nitric oxide (NO) production by the RAW-264.7 macrophage cell line. The hydroxytyrosol stearate and the hydroxytyrosol oleate decrease NO production in a concentration-dependent manner (85). In addition, olive oil extracts increase total plasma glutathione concentration (86), increasing the antioxidative response of the individual.
Nordic diet has many similarities with the Mediterranean diet, but its effects on low-grade chronic inflammation are less known. Both diets include abundant fruits, vegetables, whole grain products, fish and vegetable oil, but restrict saturated fat and red and processed meats (87, 88). Observational (89, 90) and interventional (91, 92) studies report an inverse association between the adherence to Nordic diet and the concentration of high sensitivity C-reactive protein (hsCRP). Single intervention studies reported beneficial effects, reducing IL1 receptor α (IL1Rα) (87) and Cathepsin S (93), and downregulation of inflammatory mediators in the adipose tissue (94) and peripheral blood mononuclear cells (PBMCs) (95). A key nutrient in fish are the n3 polyunsaturated fatty acids (PUFAs) (88). The Greenland Inuit population, which has a high dietary intake of n3-PUFAs, have a lower incidence of myocardial infarction than the Danish population (96). Numerous studies associate the cardioprotective effects of n-3 PUFAs to their effect on immunomodulation (97–99), and control of inflammation, including neuroinflammation during aging (100).
The Mechanism of the Anti-Inflammatory Effects of n3-PUFAs
n3-PUFAs can regulate the transcription and expression of inflammatory mediators such as cytokines, chemokines and adhesion molecules in cardiomyocytes, fibroblasts, endothelial cells, and monocyte-macrophages (101–104). Anti-inflammatory effect of eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA) and their biologically active metabolites (D and E Resolvins - mediators derived from omega-3 fatty acids, primarily EPA and DHA that block the production of proinflammatory mediators and regulate leukocyte trafficking to inflammatory sites) can be mediated through one of the mechanisms capable of reducing inflammation of RAW-264.7 cells and of primary intraperitoneal macrophages (105). One of the mechanisms is the activation of G-protein coupled receptors (GPR), ea. GPR120 inhibition of Toll-like receptor 4 (TLR4)-mediated inflammatory response, which blocks NFκB activation. The other is mediated by nuclear receptors, particularly PPARs-α/γ. DHA binds to PPARs with high affinity resulting in the activation of anti-inflammatory cascades (106), which appears to be responsible for the beneficial health effects (97). The inhibition of NFκB-mediated pro-inflammatory activity (107) is the common mechanism of immunomodulation by n3-PUFAs, being DHA more effective than EPA in reducing LPS-n3-PUFAs induced inflammatory cytokine production by macrophages (108).
n3-PUFAs are incorporated into phospholipid bilayers and in human atherosclerotic plaques. Their incorporation is associated with a reduction in the number of foam- and T cells, and a decrease in inflammation (109). The increased incorporation of n3-PUFAs in membranes affects both the innate and adaptive immune responses, impairing the maturation of dendritic cells and the function of macrophages, as well as the polarization and activation of T and B cells (110–112). It is well known that n3-PUFAs compete with n6-PUFAs for being incorporated into cell membranes and for the active sites of COX-2 and Lipoxygenase, resulting in the production of less potent pro-inflammatory or even anti-inflammatory mediators, such as the 3-series of prostaglandin and thromboxane (113). Resolvins reduce also neutrophil-derived ROS production, favoring neutrophil apoptosis and clearance by macrophages, and inhibit chemokine signaling (114). The partial agonist/antagonist activity of Resolvin E1 (RvE1) on the leukotriene B4 receptor on polymorphonuclear cells (PMNs), inhibits NFκB activation, reduces release of pro-inflammatory cytokines and reduces infiltration by PMN (115). Moreover, RvE1 reduces TNFα and IFNγ presence in the aortic wall, decreases the levels of the inflammatory marker CRP and reduces macrophage infiltration of the intima. Thus, RvE1 attenuates atherosclerosis and atherosclerotic plaque formation (116).
Aging is associated with the activation of inflammatory signaling pathways (117, 118), which can be targeted by specific nutrients with anti-inflammatory effects, such as n3-PUFAs (119, 120). In the brain, the main n3-PUFA is DHA, representing 12–14% of total fatty acids (121). Aging and neurological disorders are associated with decreased levels and turn-over rate of brain n3-PUFAs (122–125). In aged mice, n3-PUFA supplementation and diets enriched in DHA have been reported to revert age-induced spatial memory deficits and impairment on learning and memory (126–128). In older adults, a low consumption of n3-PUFAs and decreased erythrocyte DHA levels are associated with cognitive impairment (129, 130). Dietary supplementation with DHA is positively correlated with an improvement in declarative memory test performance, improved cognitive function (131, 132) and a lower risk of developing neurological disorders (133). The probable mechanisms by which n3-PUFAs mediate their effects in the resolution of age-related neuroinflammation are the increased synthesis of n3-PUFA-derived RvD1 and decreased n6-PUFA-derived oxylipins, displaying an anti-inflammatory profile (134, 135).
To recapitulate, the evidence indicates that n3-PUFAs and their bioactive metabolites have immunomodulatory and anti-inflammatory properties. Potential cardioprotective lipid mediators, through multiple mechanisms, including changes in cell membranes composition, and modification of both cell signaling and gene expression, shift the pattern of lipid metabolites toward a more anti-inflammatory metabolite profile. Dietary habits may be essential regulators of the inflammatory profile and promote healthy aging, reinforcing the recommendation of a n3-PUFA rich diet.
The Impact of Psychological and Social Stress in the Inflammatory Response
The long term chronic psychological stress is increasing among the world’s population (136). Its circuit arises at high cortical centers through the limbic system to the hypothalamus, where corticotropin-releasing factor (CRF) is produced, which is responsible for inducing the pituitary gland to liberate adrenocorticotropic hormone (ACTH) that signals the adrenal cortex to synthesize and secrete glucocorticoids (GCs) (137). Stress also activates the sympathetic nervous system (SNS), particularly the adrenal medulla, activating chromaffin cells to produce epinephrine (EPI), a main stress hormone along with GCs. The latter plays a key regulation feature inhibiting the hypothalamic-pituitary-adrenal (HPA) axis through negative feedback at the pituitary gland, hypothalamus, and medial prefrontal cortex, reducing CRF secretion [rewieved in (138)].
Stress and Epigenome Changes
The interplay of social and environmental stressors induces inflammation through multiple biological mechanisms, including epigenetic factors (139). Studies in rats show that the methylation patterns of genes involved in the stress response, such as the glucocorticoid receptor (Nr3c1) and CRF, can be modified by psychosocial factors from early childhood (140). Similarly, early life adversity induces acute and long-lasting epigenetic modifications in Nr3c1 genes, regulating HPA axis and cytokine production, reinforcing the importance of the activation inputs during critical periods of development (137, 141).
Stress and Immune Response
Acute short-term emotional stress, such as speaking in public, leads to a transient increase in circulating inflammatory biomarkers and natural killer (NK) cells by the SNS catecholaminergic activity (142). On the contrary, chronic stress results in a reduction of cytotoxic NK activity, determining a poorer response to cytokines (143). Therefore, stress appears to have short term beneficial immune effects, whereas chronic stress in the absence of immune challenge has the opposite effect (138, 144), activating constantly the HPA axis with the consequent persistent elevation of systemic GCs and reduction of NK cell responsiveness to cytokines (143), affecting the balance of the T helper cell type 1/type 2 (Th1/Th2) cytokine networks, predisposing to a wide range of diseases (145). The stress magnitude has been associated with IL1β mRNA overexpression in peripheral PBMCs, providing a molecular mechanism by which psychological stress is translated into an immune system response (146, 147).
Chronic Stress and Chronic Inflammation
When stress becomes chronic, such as in depression, there is a maintained overproduction of inflammatory cytokines, which have been associated with GCs resistance. Immune cells become less sensitive to their anti-inflammatory effects because of their persistent secretion, leading to chronic low-grade inflammation (147, 148). Activation of the innate and adaptive immune system by chronic mild stressors increases inflammatory cytokines gene expression, maturation and trafficking of dendritic cells (DC), increased macrophage number and T cells recruitment and activation. Social stressors can induce an increase in inflammatory responses and a state of GCs resistance at different levels (144, 149).
Brain Inflammation
The acute repeated social defeat stress (RSDS) and chronic restraint stress (CRS) models induce an inflammatory response that results in neuroinflammation and depressive behavior (150). Stress activates the HPA axis and the sympatho-adreno-medullar (SAM) axis causing neuroinflammation by circulating cytokines that crossed the blood-brain barrier (BBB) at the circumventricular organs and by cytokine BBB transporters. An inflammatory response that promotes BBB permeability, allowing more inflammatory factors entering the brain, including CRF, metalloproteinase-9, IL6, and TNFα (150). Additionally, microglia produce chemokines that attract monocytes into the brain (150).
Endothelium Inflammation
Activation of SNS and HPA axis through continuous psychological stress dysregulate cytokine production, and together with the stress hormones corticosteroids and catecholamines, can affect endothelial adhesion molecules, causing endothelial damage (138). Corticosteroids could facilitate the infiltration of monocytes by increasing the expression of IL1 and IL6 receptors on endothelial cells. These monocytes and lymphocytes, after attaching to such sites, would commence the process of infiltration into the wall vessels, leading to foam cell formation and thrombotic events (138, 151).
Pancreas and Liver Inflammation
Chronic unpredictable mild stress (CUMS) decreases body mass and impairs the metabolism of carbohydrates and lipids. A model for CUMS showed an increased liver and pancreas protein-lipid peroxidation and protein oxidation (152). High ROS production in both organs could be a result of a response mechanism to stress at the cellular level. In the liver, protein oxidation can be due to the regulation of metabolic impairments by GCs and EPI (152). The antioxidant system of the liver is in general more efficient than the pancreas. However, it is insufficient to clear the reactive species increased as consequence of chronic stress, which could be due to alterations in the antioxidant enzymatic activity (138).
Chronic Stress and Aging
Altogether, stress appears to have short term beneficial effects on the immune function, whereas chronic stress (138, 144) activates persistently the HPA, elevating systemic GCs, and impairing the cytokine balance. The overproduction of inflammatory cytokines lead to GCs resistance driven by immune cells that lose their sensitivity to GCs, leading to a state of chronic low-grade inflammation (138, 145). This GCs imbalance, shares common features with aging, mediating an enhanced neuroinflammatory priming (153). The presence of psychological stress potentiates the defective immune response observed in aging, which at the same time conditionate an exaggerated sickness response to immune challenges (such as chronic stress). Thus, chronic stress contributes to the phenomenon of inflammaging, which promotes the development of several age-related pathologies, including atherosclerosis and diabetes among others [reviewed in (154)]. Additionally, there is an impairment of the antioxidant defense system to manage ROS production after chronic stress, resulting in the damage of various tissues (138). In addition, people exposed to chronic stress age rapidly, showing a faster telomere shortening in their cells (155–157). On the other hand, epigenetic changes acquired during critical developmental stages could shape chronic stress-response along the lifespan, either promoting or reducing pathological aging (139, 140).
Inflammatory Response Induced by Drug Abuse
Substance abuse, such as alcohol and drugs, are important triggers of chronic inflammatory processes (158, 159). The effects of alcohol on human health are complex and depend on multiple factors. However, many of those factors are associated with the generation of immunosuppression and increased morbimortality in heavy users. Those effects, which have been previously reviewed by Goral et al. (160) will not be discussed in this review. Here, we will describe the effect of cocaine and methamphetamine abuse. Both drugs are potent psychostimulants that, when repeatedly consumed, significantly disrupt the functioning of the CNS, and modify the regulation of the immune response, leading to a chronic neuroinflammatory state (161). In general, it is known that drug abuse, among other factors, increases NFκB transcription of multiple proinflammatory genes that spread across brain cell types further amplifying of NFκB transcription, as has been reviewed by Crews et al. (162).
Cocaine
Cocaine (benzoylmethylecgonine according to the International Common Denomination) is a strong stimulant tropane alkaloid that acts by modulating the catecholaminergic neurotransmitter dopamine. Studies of the striatum of mice after the administration of various drugs showed that 1 h after administration of 25 mg/kg cocaine, there is a significant increase in gene arrays for Hypoxia-inducible factor 1 (HIF-1), transcription factors, and cytokine receptors (IL6r, TNFα). Two hours after cocaine administration, there is an increased gene expression for various TNF receptors, inducible NO synthase (iNOS) and adhesion molecules (163). In the nucleus accumbens of mice stimulated with cocaine, there is a significant increase in matrix metalloproteinase 28 (MMP28), Macrophage Colony Stimulating Factor (MCSF) and Major Histocompatibility Complex II (MHC-II) (164). The brain of human subjects consuming cocaine shows an increased density of macrophages and activated microglia (165). Cocaine induces the activation of microglia through the endoplasmic reticulum stress and autophagy pathways (166). Studies of human and rodent immune cell populations after cocaine administration show decreased numbers of T lymphocytes, modulation of NK activity and cytokine production (167).
Among brain glial cells, astrocytes are the most abundant, and perform critical functions, being involved in neurogenesis, promotion of neuronal survival, elimination of free radicals, and the production of NO to maintain neuronal homeostasis (166). Nevertheless, astrocytes can also be activated by toxic stimuli, leading to a new phenotype called “reactive astrocytes”, similar with the changes observed after inflammatory activation. This phenomenon has been described in various neuropsychiatric disorders, such as Alzheimer’s and Parkinson’s disease, amyotrophic lateral sclerosis and multiple sclerosis (166). The reactivity of astrocytes to toxic stimuli, such as cocaine, infection or disease, potentiates the neuroinflammatory process (168).
Methamphetamine
Methamphetamine (desoxyephedrine; METH) is a synthetic adrenergic agonist with psychostimulatory effects, structurally related to the ephedrine alkaloid and adrenaline. Studies on the effect of METH are limited. However, it has been determined that its abuse affects the immune response. Animals exposed to both acute and chronic METH use show alkalization of normally acidic organelles in immune cells, inhibition of antigen presentation, and impairment of phagocytosis (169). METH also generates mitochondrial oxidative damage, dysfunction of T lymphocytes and decreased production of antibodies and cytokines (159).
METH has effects in various tissues (170). In the lungs, the number of T lymphocytes decreases compared with that of untreated animals, indicating a reduction in circulating CD3+ cells, and levels of IL6 and IL10 increases. In the spleen, recruitment of PMN and the number of Ly-6G+ and F4/80+ are increased, whereas CD3+ cells are significantly reduced. In addition, levels of TNFα, IFNγ, IL6, and IL12 are higher than those of control mice. In the liver, there is an increase of T lymphocytes and macrophages, hepatocellular atrophy, and increased levels of IFNγ, TNFα, IL1β, -4, -6, -10, and -12 in the group exposed to METH compared with control animals (170).
In the CNS, METH can induce the activation of calpains and caspases; the production of ROS with the subsequent induction of oxidative stress, and the release of high amounts of glutamate, causing excitotoxicity (171). Recently, Raineri et al. reported that METH induces activation of astrocytes and microglia, increasing the levels of IL6 and TNFα mRNA and its receptor (TNFR1) in the mouse striatum and hippocampus (172, 173).
Drug Use and Aging
Medical advances have resulted in the increment of the average life expectancy in developed countries. The aging of the population is associated with an increase in the number of older people using drugs of abuse. From 2000 to 2012, the number of cocaine users aged 55 or older that required treatment for drug addiction in the US increased by 63% (174, 175). Aging is associated with low-grade basal inflammation that can be compounded by substance use. As cocaine exposure is associated with elevated inflammation and altered immune functioning, the presence of cocaine use disorder might exacerbate inflammatory processes in aging adults (176). A recent report by Soder et al, compared the levels of inflammation (through the neutrophil to lymphocyte ratio) in older adults with cocaine use disorder (CUD) and in healthy older adults, finding that the group with CUD had a significantly higher baseline level of inflammation (176). The use of illegal drugs such as cocaine or methamphetamine has not been shown to affect cognitive function in older adults at the clinical level. However, the evaluation of the cognitive function of young drug users reveals a decreased performance compared with healthy young people. In fact, the cognitive function of young drug users is similar to that of adults older than 60 years of age (174, 177, 178).
In summary, both cocaine and METH can directly impair the immune response, induce the activation of glial cells and stimulate the release of pro-inflammatory mediators in the CNS. All those effects cause relevant changes in glial cell regulation and inflammatory activation, triggering chronic neuroinflammation and potentiating pathological aging.
Induction of an Uncontrolled Inflammatory Process by Air Pollution
Air pollution has become an important threat to public health. Air pollutants consider a mixture of gases such as nitrogen oxides (NOx), sulphur oxides (SOx), tropospheric ozone (O3), volatile organic compounds (VOCs), and particulate matter (PM) (179). PM can enter the respiratory tract leading to severe in situ damage as well as inducing additional systemic effects (180). The World Health Organization (WHO) suggests a maximum annual exposure of 10 µg/m³ of PM2.5, however, the exposure of 90% of the world’s population exceeds the proposed limit (181). Exposure to air pollutants is associated with increased morbimortality associated with respiratory, cardiovascular, metabolic, neurological, carcinogenic and autoimmune diseases (17, 182–184). Inflammation is the main pathophysiological mechanism induced by air pollutants.
Oxidative Stress
In terms of the molecular and cellular mechanism induced by pollutants, PM and SOx can generate ROS, inducing oxidative stress, together with mitochondrial dysfunction and the consequent energy deprivation (185–187). As a direct consequence, NFκB and MAPK inflammatory pathways are activated, triggering an innate immune activation (188, 189). Despite the attempts to resolve the inflammatory event, the outcome appears to be an imbalance in lymphocyte homeostasis and immune system dysregulation, with inhibition of Th1 and Treg lymphocytes (190). There is also an increase of Th2 lymphocytes and recruitment of eosinophils, resulting in respiratory disorders such as asthma (186, 191, 192). In parallel, PM deactivates the nuclear factor erythroid 2 pathway (Nrf2), involved in antioxidant regulation and prevention of oxidative stress, a necessary process for the resolution of inflammation. Therefore, to maintain oxidation-reduction reactions becomes impossible, becoming a breaking point towards increased ROS production and the non-resolution of the inflammatory event (193).
Activation of the Aryl Hydrocarbon Receptor
Another mechanism of action of pollutants is the activation of the aryl hydrocarbon receptor (AhR) by toxic agents. The binding of PM to AhR increases circulating Th17 and decreases Treg lymphocytes. Increase in Th17 associates to the release of IL17, promoting an abrupt increase of Th2 lymphocyte response. These changes promote the dysregulation of the immune response associated with the development of autoimmune processes (193). Aberrant increases in Th17 may result in increased inflammation, with consequences such as asthma and acute respiratory failure syndrome (ARDS), due to neutrophil infiltration and tissue damage (194). Studies suggest the existence of a decline in Treg levels and, therefore, an inability to suppress Th1, Th2 and phagocyte responses (195, 196). In addition, exposure to PM has been associated with fibrotic events, where IL17 increases synthesis and secretion of collagen in the lung parenchyma (197, 198). In addition, it has been described that PM also induces the expression of TGFβ, directly promoting fibroblast differentiation, which could also induce collagen deposition followed by a lower antifibrotic process in the liver (199).
Epigenetic Regulation
Pollutants may promote direct DNA damage through oxidation of nitrogenous bases. Hu and Yu described in a 2019 paper different mechanisms and changes in miRNA expression that comprise specific targets of DNA methyltransferases, which can impair the methylation of tumor suppressor genes (200). Furthermore, urban populations show increased levels of mitochondrial methylation genes due to PM exposure (201). There is evidence of the existence of methylation, acetylation and phosphorylation of histones H3 and H4, markers found in genes involved in the activation of immune cells and cardiovascular diseases (200, 202–205). Altogether, air pollutants can generate DNA adducts promoting carcinogenesis and deteriorate telomerase activity, as reviewed by Martens and Nawrot (2016), and contributing to continuous DNA damage and premature aging (206, 207).
Temporal and Concentration Effects Over Inflammatory Mediators
In vivo studies suggest that the inflammatory activation is dose- and time-dependent. Mice exposed to PM show that both variables are determinant for the outcome. However, inflammatory effects and major genetic changes appear to be especially dependent on the exposure to high concentrations of PM. One possible explanation is that a prolonged exposure could induce an adaptive response of the inflammatory activation (208), which may be mediated by the inactivation of the Nrf2 pathway, generating a loss of antioxidant capacity and deregulation of the immune system (193). The resolution appears to depend on the exposure context. Acute exposure would result in high levels of ROS and damage, whereas prolonged stimulation, even a low-grade one, generates a constant production of ROS and chronic low-grade inflammation (187), consequent with the potentiation of disease risk and an epigenetic age acceleration (206), promoting pathological aging.
Direct causes of the deregulation of the inflammatory resolution process resulting from inhaled contaminants are still unknown, however, the burden of associated chronic diseases is expected to increase. It is mandatory to intensify environmental policies specifically in lower-middle-income countries in prevention of the development of inflammatory conditions and the subsequent chronic diseases.
Aging, Epigenetic and Immuno-Inflammatory Imbalance
Aging, characterized by a progressive loss of cellular functions, is an inevitable physiologic process inherent to all living beings (209). The number of older adults is increasing. During the next 30 years, up to 22% of the world population will be older than 60 years (210). This demographic change is accompanied by a higher incidence of NCDs accumulated in the aging population (211). Therefore, various strategies have been proposed to improve the health and quality of life of older adults (212), along with recommendations for the development of Public Policies that support the fiscal expenditure resulting from NCDs (213).
One of the most studied events of aging is the impairment of the immune system, characterized by an aberrant-increased activation of the innate immunity (214, 215), and high levels of circulatory inflammatory mediators that establish an inflammatory environment, and a decrease of the adaptive immune response (216, 217) and a decrease of the adaptive immune response (214) due to this low-grade chronic inflammation (214, 218), which together would promote the inflammaging phenomenon (219). Interestingly, it is proposed that age would not be the cause per se of these diseases associated with aging (214). Thus, there is a deterioration of the immune system’s response to external stimuli, which depends on the individual’s history (218). Also, several epigenetic mechanisms can modulate the immune response in aging, enhancing changes in intercellular communication that could perpetuate inflammatory events (220). On the other hand, it is described that epigenetic clocks would be useful to analyze mechanisms associated with this environmental influence (221). Finally, they would be capable of modulating the immune response in aging, enhancing changes in intercellular communication that could perpetuate inflammatory events (220).
Aging and Systemic Inflammation
Multiple age-dependent changes play important roles in the promotion of NCDs, with increased oxidative stress standing out as one of the main mechanisms. Over the last two decades, evidence has revealed that increased oxidative stress and inflammation are involved in various NCDs such as Alzheimer‘s disease (219), rheumatoid arthritis (222), cardiovascular diseases (223, 224), and cancer (225), among others. Also, recent studies propose that the activation of NFκB signaling pathways could be the main driver of these associations (226–229). Interestingly, De Almeida et al. showed different sources of low-grade chronic inflammation that promote cardiovascular disease (226). In the CNS, high levels of ROS lead to the activation of astrocytes and microglia, further increasing the overproduction of ROS and proinflammatory cytokines that promote the development of neurodegenerative changes (217, 230, 231). In fact, several systemic biomarkers appear to be associated with neuroinflammation and the development of CNS diseases associated with aging (230). These modifications trigger the phenotype of senescent or aged cells characterized as SASP (216, 232) extensively studied in the context of the deleterious effects of aging. However, SASP is also essential for remodeling and promoting wound healing, which requires a strict control of the inflammatory response, thus avoiding the induction of cell aging phenotypes that contribute to the development of chronic inflammatory diseases (233).
Mechanisms Associated With the Immune Imbalance
The immune imbalance in aging occurs due to various alterations in cellular behavior and phenotype, which cause functional deficiencies in immune cells (3). For example, this context induces polarization of macrophages towards an inflammatory phenotype characterized by strong activation of the inflammasome (234). Thus, these events could induce IL1β and TNFα release, changes in the chemoattraction of neutrophils mediated by the reduction of the intercellular adhesion molecule 1 (ICAM-1) expression, and the aberrant activation of the phosphoinositide lipid kinase-3 (PI3K) (235). Also, there is a decrease in the expression of pattern recognition receptors (PRR), which leads to the activation of proinflammatory signaling promoting tissue damage (215, 216). Finally, the reduced level of certain hormones due to the impaired hypothalamic function causes the loss of muscle mass and an increase in adipose tissue, further contributing to the release of inflammatory cytokines and changes in metabolism (236). Despite the remarkable effort being made to understand the basis of the processes underlying the inflammatory imbalance during aging, it is not fully understood.
Role of Epigenetics in the Immune Imbalance
In aging, there are cumulative epigenetic changes that promote low-grade inflammation (220, 237), including a decrease in the global genome methylation, with increased methylation in specific regions, as those with repressive histone marks of CD8+ and CD4+ T cells (238) and bivalent chromatin domains (239) and histone acetylation and methylation. However, the influence of genomic methylation during aging remains undetermined (237). Several studies correlate the methylation of multiple sites on CpG islands with the increase of the low-grade inflammation marker, CRP (220, 232, 240). Nonetheless, Stevenson et al. propose that the DNA methylation could be better associated with the low-grade chronic inflammation than CRP (237). In addition, the age-related mitochondrial dysfunction, with the resulting oxidative stress and decreased ATP production (241), affect the expression and activity of DNA methyltransferases, which are responsible for maintaining the methylation pattern of DNA (242). The reduced methylation results in the demethylation of the TNFα promoter in leukocytes and macrophages (243) and the adhesion of immune cells to the endothelium (244). Also, many epigenetic events contribute to the differentiation of proinflammatory T cells, Th17 (220), which can compromise immunocompetence, associated with repression of differentiation of immune cells, loss of Treg function (240) and the alteration of the hematopoietic stem cells differentiation (245).
Thus, epigenetic mechanisms appear to have a major role in the inflammatory imbalance, which are associated with the accumulation of damage in time that ultimately leads to the perpetuation of a constant inflammatory response.
Modulation of the Inflammatory Activation Through Physical Exercise
According to the WHO, 60% of the world population is sedentary, lacking the benefits of physical exercise (246). Conditions such as sedentarism, unhealthy diet, overweight, obesity and aging induce chronic low-grade inflammation. Physical exercise increases the anti-inflammatory potential and reduces the pro-inflammatory effect (247). This equilibrium is partly modulated through TLRs (248), which are fundamental for the recognition of PRRs, including the damage-associated molecular patterns (DAMPs) and the induction of an inflammatory response in the absence of pathogens.
Anti-Inflammatory Exercise
There is evidence that in young people, physical exercise decreases TLRs expression, co-stimulatory molecules CD80/CD86, and MHCII (248, 249) in CD14+ monocytes. Physical exercise also affects the adipose tissue. Exercising reduces TLR4 mRNA expression and TNFα production in adipocytes (250, 251) in obese mice. Chronic physical exercise decreases TNFα and TLR4 gene expression in the skeletal muscle (252). The evidence suggests that obesity- or cerebral ischemia-induced neuroinflammation, which are associated with the overexpression of TLR2 and TLR4, may be reduced by physical exercise through the reduction of TLRs expression as well as their downstream signaling molecules (TNFα, IL1β, MyD88, TRAF6, 552 TAK1, and NFκB), together with the reduced microglial activation (253, 254). There is evidence that cigarette smoking induces inflammatory status [reviewed in (255)]. However, exercise training reduces smoke-induced inflammation. In that sense, training for 30 min with endurance exercise for 5 days in smoke-exposed mice demonstrated that therapeutic exercise training significantly reduces the expression of IL1β and TNFα mRNA in rectus femoris (256).
Physical exercise has been used as a therapeutic tool in chronic pathological conditions. In that sense, obese older adults (body mass index 38 ± 2 kg/m2; 69 ± 1 years) undergoing an exercise program consisting in physical therapy, endurance, and resistance for 90 min, 3 days per week, show a reduced expression of TLR4, IL6, and TNFα mRNA in skeletal muscle (257). In older adults, 8-week physical exercise reduces the expression of TLR4 and TLR2, as well as TLRs downstream mediators, such as MyD88, p65, pp38, TRIF, IKKi/IKKϵ, IRF3, and pIRF7 in PBMCs (258). Similarly, dendritic cells from multiple sclerosis patients undergoing an exercise (endurance and resistance) program for 12 weeks reduce TNFα and MMP9 secretion when stimulated with a TLR4 ligand (LPS in combination with IFNγ, or a TLR7 ligand) (259), suggesting that long-term physical exercise decrease TLR responsiveness.
Pro-Inflammatory Exercise
On the other hand, high-intensity physical exercise in untrained individuals induces inflammation, resulting in the increased expression of TLR4, AP1, NFκB, and p65 in mice myocardium and in adipose tissue (260–262). Physical exercise associated with eccentric contractions causes expression of TLR and NFκB in skeletal muscle and liver in rats (263, 264). Furthermore, this phenomenon induces muscle damage, which can increase chemotaxis, attracting NK, CD8+ 559 T cells, macrophages and neutrophils to the site of injury, promoting the production of COX560 2, iNOS, monocyte chemotactic protein-1 (MCP-1), TNFα, IL6, and IL1β, in addition to the production of ROS and the activation of NFκB (265, 266). In healthy young males, one session of intense endurance exercise (1 h intense cycling immediately followed by 1 h intense running), increases plasmatic concentrations of IL6 and IL10, in addition to increased gene expression of proinflammatory IL1 receptor (IL1R) and TLR signaling pathways. Moreover, plasma myoglobin changes in correlation with neutrophil TLR4 gene expression (r= 0.74), suggesting that their transcriptional activity was particularly induced by DAMPs (267). Therefore, inflammation and muscle damage are mainly associated with the type and intensity of the exercise, with loads that exceed individual physical abilities.
Exercise, Epigenetic Regulation, and Inflammation
Chronic physical exercise generates epigenetic modifications. The physical exercise associated with an energy expenditure >500 kilocalories per week, results in hypomethylation of the IL10 gene and hypermethylation of the TNFα gene (268), with an inverse correlation between TNFα methylation and TNFα mRNA expression (269). The methylation of the caspase recruitment domain (ASC) of the apoptosis-associated speck-like protein gene, the main regulator of inflammasome and promoter of the activation of IL1β and IL18, decreases with aging. However, older adults who maintain physical exercises regularly express higher levels of ASC methylation than subjects not exercising, which would imply a decreased release of inflammatory cytokines (270–273). Similarly, in review a 6-month walk training can induce hypermethylation of the NFκB-2 gene, suppressing inflammation through the inhibition of the NFκB pathway (274).
Discussion
As life expectancy increases, age-related diseases thrive. Aging is a complex multifactorial process of molecular and cellular decline that renders individuals susceptible to disease and death. Maintenance of cell integrity, cell metabolism and host-defense mechanisms are tightly regulated by the surrounding microenvironment. A growing body of evidence in different biological models has contributed towards identifying biological mechanisms that ward off structural and functional deterioration. These data offer us insights into healthy aging. Molecular integrity of the genome, telomere length, epigenetic stability, and protein homeostasis are all features linked to more youthful stages (regardless of the age), associated with mitochondrial fitness, metabolic regulation, efficient intercellular communication, stem cell renewal, and regenerative capacity in tissues. A good understanding of the environmental and endogenous mechanisms that underlie age-related normal and deleterious changes, and how these pathways interconnect, remains a major challenge for slowing pathological aging while extending older adults’ healthy lifespan.
The study of the environmental influence on the development of complex-chronic diseases shows that in addition to genetic predisposition, the pathogenesis is promoted by changes in metabolism and behavior, cellular environment, and epigenetic regulation patterns. The type of nutrient, or environmental cytokine milieu dramatically affects not only the homoeostasis of tissues but also of complete organs and even of the whole individual. Thus, tissue stress, malfunction, and damage may induce inflammation alarm responses, which result either in resolution of tissue damage, restoration of normal cell function or development of chronic disease (Figure 1). Older adults often present inflammaging, characterized by increased levels of pro-inflammatory cytokines IL1, IL6, IL8, TNFα/CRP (275). However, the cellular sources of these cytokines are partially unknown. The increased inflammatory cytokines have been proposed to be a driver of unsuccessful aging (increased morbidity, degenerative processes, or frailty) and shortened health-span. The inflammatory scenario is complex and occurs in response to various internal and environmental stimuli (Figure 1) mediated mainly by a high level of pro-inflammatory cytokines. Indeed, in healthy aging, increased production of the anti-inflammatory cytokines TGFβ and IL10, can regulate the pro-inflammatory state (276, 277).
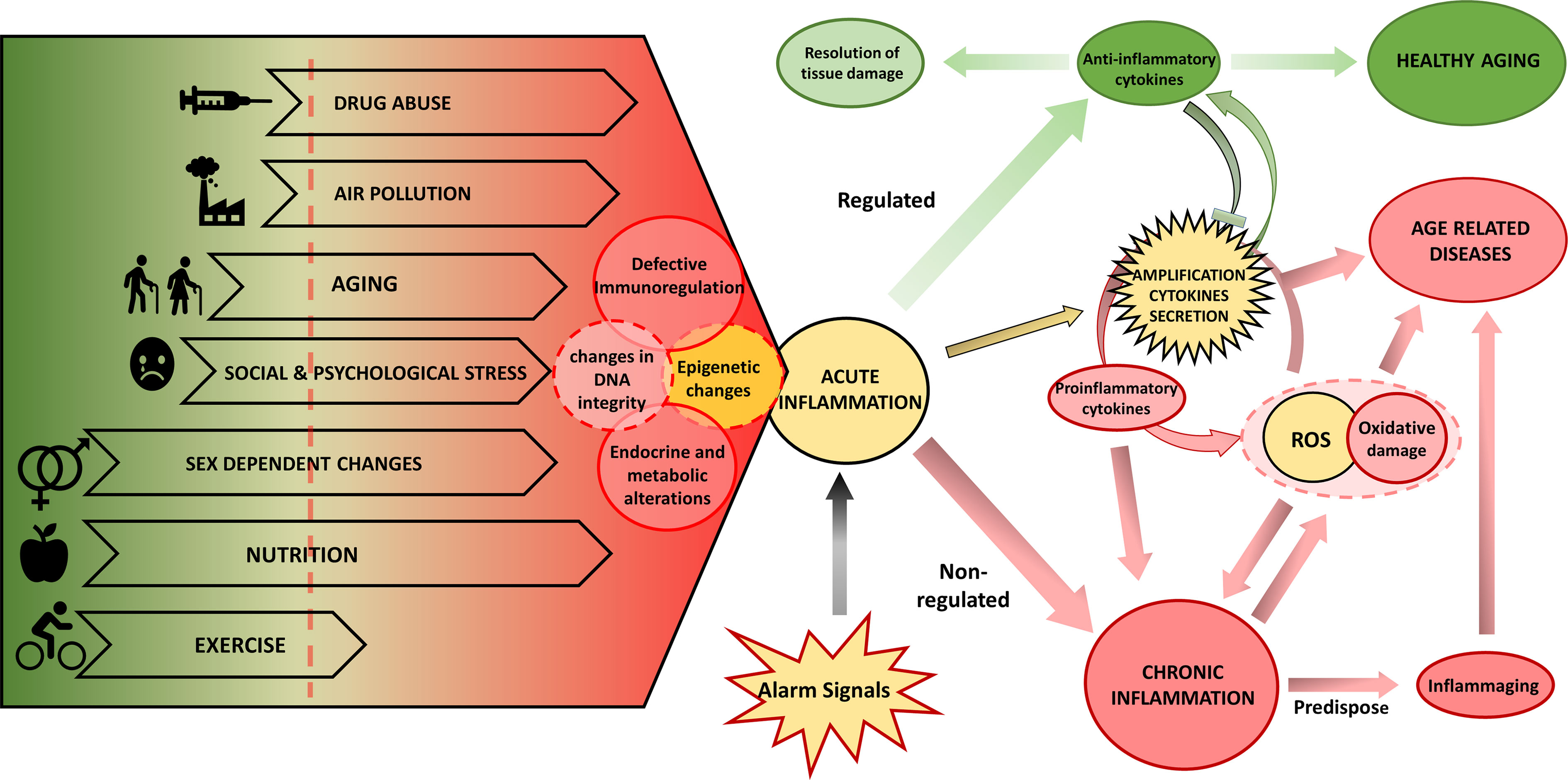
Figure 1 Biological and environmental factors determining the inflammatory response and the aging phenotype. Endogenous and environmental factors can be mostly beneficial (in green) and deleterious (in red) or can have both beneficial and deleterious effects depending on the specific context. The interplay of lifespan endogenous and environmental factors regulates the aging phenotype depending on DNA damage, epigenetic changes, and inflammation. These drivers can induce functional aging hallmarks: changes in endocrine and metabolic regulation, and defective immune regulation that will further determine the response of the individual. In yellow we show processes that can participate in both protection and damage. Exposure to various alarm signals induce an acute inflammation that, when associated with deleterious environmental and biological factors, potentiates chronic inflammation, which can be further promoted by excess ROS production and oxidative stress that results from mitochondrial dysfunction or NOX2 activity, leading to inflammaging and eventually to age-related disease. On the contrary, in the presence of protective environmental and biological factors, the initial inflammatory activation will be resolved and lead to a healthy aging process. ROS, reactive oxygen species.
Research into the impact of environmental factors on inflammaging is at an early stage and the involved mechanisms are not completely understood. Several hypotheses have been developed to explain the chronic inflammation: aging-related increase of stress (278) and oxidative stress (279), DNA damage in senescent cells [reviewed in (280)], and stem cell aging (281). The proposed mechanisms are likely interdependent, resulting in the generation of ROS causing oxidative damage and amplification of the cytokines secretion, thus perpetuating a vicious circle of systemic inflammation where tissue injury and healing mechanisms proceed in parallel while damage slowly accumulates over the lifespan of the individuals. Endocrine and metabolic alterations are linked to the shift towards a pro-inflammatory profile, which could explain some age-related pathologies, such as Alzheimer’s and Parkinson’s disease, osteoporosis, diabetes, cancer, and frailty (282, 283).
Regarding stress-induced immune modifications, new evidence suggests that cross talk signals between the CNS, endocrine and immune system are required for optimal response to stress [discussed in (284)]. Various stressors can affect the activity and regulation of immune cells via direct regulation by the autonomic and peptidergic system or through the release of neuroendocrine mediators. Moreover, neuronal catecholamines modulate immune cell functions. These interactions are bidirectional, cytokines produced by immune cells, such as IL1, can modulate the production of corticotropin-releasing hormone (CRH) by the hypothalamus. Chronic diseases are favored by some modern living conditions, such as the intake of high-caloric foods and the low level of physical activity, or endogenous signals produced by the chronic stress of modern life. There are many challenges in conducting research on biosocial processes, which will define novel disease-trigger factors.
Tailor-made approaches will depend on genetics, epigenetics and a constellation of factors depending on the historical as well as the present exposure to the environment. Although environmental factors also express themselves as epigenetic changes, the combinatorial effect of the multiple factors generates complex patterns of epigenetic regulation, and the concomitant exposure to environmental factors can further modify the individual response.
Author Contributions
All authors contributed equally on the conception of the work, the analysis of literature and preparing the content of the review. RBe drafted and organized the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was partially supported by Project FONDECYT 1171645 from CONICYT Program of Chile and the Santander Universia Research Award in aging research (RBe). SB, YJ, and FG acknowledge partial support from CONICYT-PFCHA/Doctorado Nacional 2019-Folio 21191070, 21191025 and 21190421, respectively. RBa and JM acknowledge partial support from CONICYT-PFCHA/Doctorado Nacional 2020-Folio 21201751 and 21201210, respectively.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ACTH, adrenocorticotropic hormone; Ahr, aryl hydrocarbon receptor; AMPK, 5’ AMP-activated protein kinase; ARDS, acute respiratory distress syndrome; ASC, caspase recruitment domain; BBB, blood-brain barrier; cAMP, cyclic adenosine monophosphate; CNS, central nervous system; COX-2: cyclooxygenase-2; CREB, cAMP response element-binding; CRF, corticotropin releasing factor; CRS, chronic restraint stress; CUMS, chronic unpredictable mild stress model; DAMPs, damage-associated molecular patterns; DC, dendritic cells; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; ERα/β, α- and β-estrogen receptors; GCs, glucocorticoids; GPR, G-protein coupled receptors; HPA, hypothalamic-hypophysis-adrenal; hsCRP, high sensitivity C-reactive protein; ICAM-1, intercellular adhesion molecule;,IFN, interferon; IL1Rα, IL1 receptor α; IL1β, interleukin 1β; iNOS, inducible NO synthase; LGCI, low-grade chronic inflammation; LPS, lipopolysaccharide; MAPK, mitogen activated protein kinase; MCP1, monocyte chemotactic protein-1; MCSF, macrophage Colony Stimulating Factor; METH, methamphetamine; MHC-II, major histocompatibility complex II; MMP28: matrix metalloproteinase 28; NCDs, noncommunicable diseases; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; NK, natural killer; NO, nitric oxide; NOx, nitrogen oxides; Nrf2, nuclear factor erythroid 2 pathway; O3, tropospheric ozone; PBMCs, peripheral blood mononuclear cells; PI3K, phosphoinositide lipid kinase-3K; PM, particulate matter; PMNs, polymorphonuclear cells; PPAR, peroxisome proliferator-activated receptor; PRR, pattern recognition receptors; PUFAs, polyunsaturated fatty acids; ROS: radical oxygen species; RSDS, repeated social defeat stress; RvE1, resolvin E1; SAM, sympatho-adreno-medullar axis; SASP: senescence-associated secretory phenotype; SDR, social disruption; SNS, sympathetic nervous system; SOx, sulphur oxides; TGFβ, transforming growth factor β; TLR4, toll-like receptor 4; TNFR1, TNFα receptor 1; TNFα, tumor necrosis factor-α; Treg, regulatory T cells; VOCs, volatile organic compounds; WHO, World Health Organization.
References
1. Schrager MA, Metter EJ, Simonsick E, Ble A, Bandinelli S, Lauretani F, et al. Sarcopenic obesity and inflammation in the InCHIANTI study. J Appl Physiol (2007) 102:919–25. doi: 10.1152/japplphysiol.00627.2006
2. Stout MB, Justice JN, Nicklas BJ, Kirkland JL. Physiological aging: Links among adipose tissue dysfunction, diabetes, and frailty. Physiology (2017) 32:9–19. doi: 10.1152/physiol.00012.2016
3. Salminen A, Kaarniranta K, Kauppinen A. Immunosenescence: the potential role of myeloid − derived suppressor cells ( MDSC ) in age − related immune deficiency. Cell Mol Life Sci (2019) 76:1901–18. doi: 10.1007/s00018-019-03048-x
4. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153:1194. doi: 10.1016/j.cell.2013.05.039
5. Bai B, Ban B, Liu Z, Zhang MM, Tan BK, Chen J. Circulating C1q complement/TNF-related protein (CTRP) 1, CTRP9, CTRP12 and CTRP13 concentrations in Type 2 diabetes mellitus: In vivo regulation by glucose. PloS One (2017) 12:1–12. doi: 10.1371/journal.pone.0172271
6. Bektas A, Schurman SH, Sen R, Ferrucci L. Aging, inflammation and the environment. Exp Gerontol (2018) 105:10–8. doi: 10.1016/j.exger.2017.12.015
7. Slyskova J, Lorenzo Y, Karlsen A, Carlsen MH, Novosadova V, Blomhoff R, et al. Both genetic and dietary factors underlie individual differences in DNA damage levels and DNA repair capacity. DNA Repair (Amst) (2014) 16:66–73. doi: 10.1016/j.dnarep.2014.01.016
8. García-Calzón S, Martínez-González MA, Razquin C, Corella D, Salas-Salvadó J, Alfredo Martínez J, et al. Pro12Ala polymorphism of the PPARγ2 gene interacts with a Mediterranean diet to prevent telomere shortening in the PREDIMED-NAVARRA randomized trial. Circ Cardiovasc Genet (2015) 8:91–9. doi: 10.1161/CIRCGENETICS.114.000635
9. Olivieri F, Rippo MR, Monsurrò V, Salvioli S, Capri M, Procopio AD, et al. MicroRNAs linking inflamm-aging, cellular senescence and cancer. Ageing Res Rev (2013) 12:1056–68. doi: 10.1016/j.arr.2013.05.001
10. López-Otín C, Galluzzi L, Freije JMP, Madeo F, Kroemer G. Metabolic Control of Longevity. Cell (2016) 166:802–21. doi: 10.1016/j.cell.2016.07.031
11. Franceschi C, Salvioli S, Garagnani P, de Eguileor M, Monti D, Capri M. Immunobiography and the heterogeneity of immune responses in the elderly: A focus on inflammaging and trained immunity. Front Immunol (2017) 8:982. doi: 10.3389/fimmu.2017.00982
12. Lee A, Gilbert RM. Epidemiology of Parkinson Disease. Neurol Clin (2016) 34:955–65. doi: 10.1016/j.ncl.2016.06.012
13. Wang Y, Hekimi S. Mitochondrial dysfunction and longevity in animals: Untangling the knot. Sci (80- ) (2015) 350:1204–7. doi: 10.1126/science.aac4357
14. Faria A, Persaud SJ. Cardiac oxidative stress in diabetes: Mechanisms and therapeutic potential. Pharmacol Ther (2017) 172:50–62. doi: 10.1016/j.pharmthera.2016.11.013
15. Cáceres L, Paz ML, Garcés M, Calabró V, Magnani ND, Martinefski M, et al. NADPH oxidase and mitochondria are relevant sources of superoxide anion in the oxinflammatory response of macrophages exposed to airborne particulate matter. Ecotoxicol Environ Saf (2020) 205:111186. doi: 10.1016/j.ecoenv.2020.111186
16. Cervellati F, Woodby B, Benedusi M, Ferrara F, Guiotto A, Valacchi G. Evaluation of oxidative damage and Nrf2 activation by combined pollution exposure in lung epithelial cells. Environ Sci Pollut Res (2020) 27:31841–53. doi: 10.1007/s11356-020-09412-w
17. Zhao CN, Xu Z, Wu GC, Mao YM, Liu LN, Qian --W, et al. Emerging role of air pollution in autoimmune diseases. Autoimmun Rev (2019) 18:607–14. doi: 10.1016/j.autrev.2018.12.010
18. Kim J, Copley SD. Inhibitory cross-talk upon introduction of a new metabolic pathway into an existing metabolic network. Proc Natl Acad Sci USA (2012) 109:E2856–64. doi: 10.1073/pnas.1208509109
19. Evans GW. Child Development and the Physical Environment. Annu Rev Psychol (2006) 57:423–51. doi: 10.1146/annurev.psych.57.102904.190057
20. Miller GE, Chen E, Parker KJ. Psychological Stress in Childhood and Susceptibility to the Chronic Diseases of Aging: Moving Towards a Model ofBehavioral and Biological Mechanisms. Psychol Bull (2012) 137:959–97. doi: 10.1037/a0024768.Psychological
21. Quist JS, Sjödin A, Chaput JP, Hjorth MF. Sleep and cardiometabolic risk in children and adolescents. Sleep Med Rev (2016) 29:76–100. doi: 10.1016/j.smrv.2015.09.001
22. Loh W, Tang MLK. The epidemiology of food allergy in the global context. Int J Environ Res Public Health (2018) 15:2043. doi: 10.3390/ijerph15092043
23. Baibergenova A, Thabane L, Akhtar-Danesh N, Levine M, Gafni A, Leeb K. Sex differences in hospital admissions from emergency departments in asthmatic adults: A population-based study. Ann Allergy Asthma Immunol (2006) 96:666–72. doi: 10.1016/S1081-1206(10)61063-0
24. Fagan JK, Scheff PA, Hryhorczuk D, Ramakrishnan V, Ross M, Persky V. Prevalence of asthma and other allergic diseases in an adolescent population: association with gender and race. Ann Allergy Asthma Immunol (2001) 86:177–84. doi: 10.1016/S1081-1206(10)62688-9
25. Ji J, Sundquist J, Sundquist K. Gender-specific incidence of autoimmune diseases from national registers. J Autoimmun (2016) 69:102–6. doi: 10.1016/j.jaut.2016.03.003
26. Turner PJ, Jerschow E, Umasunthar T, Lin R, Campbell DE, Boyle RJ. Fatal Anaphylaxis: Mortality Rate and Risk Factors. J Allergy Clin. Immunol Pract (2017) 5:1169–78. doi: 10.1016/j.jaip.2017.06.031
27. Lukacs SL, Schrag SJ. Clinical sepsis in neonates and young infants, United States, 1988-2006. J Pediatr (2012) 160:960–965.e1. doi: 10.1016/j.jpeds.2011.12.023
28. Gwak JM, Jang MH, Kim D II, Seo AN, Park SY. Prognostic value of tumor-associated macrophages according to histologic locations and hormone receptor status in breast cancer. PloS One (2015) 10:1–14. doi: 10.1371/journal.pone.0125728
29. Chen P, Wang D-B, Liang Y-M. Evaluation of estrogen in endometriosis patients: Regulation of GATA-3 in endometrial cells and effects on Th2 cytokines. J Obstet Gynaecol Res (2016) 42:669–77. doi: 10.1111/jog.12957
30. Haghmorad D, Salehipour Z, Nosratabadi R, Rastin M, Kokhaei P, Mahmoudi MB, et al. Medium-dose estrogen ameliorates experimental autoimmune encephalomyelitis in ovariectomized mice. J Immunotoxicol (2016) 13:885–96. doi: 10.1080/1547691X.2016.1223768
31. Wei C, Mei J, Tang L, Liu Y, Li D, Li M, et al. 1-Methyl-tryptophan attenuates regulatory T cells differentiation due to the inhibition of estrogen-IDO1-MRC2 axis in endometriosis. Cell Death Dis (2016) 7:e2489–9. doi: 10.1038/cddis.2016.375
32. Bouman A, Schipper M, Heineman MJ, Faas MM. Gender difference in the non-specific and specific immune response in humans. Am J Reprod Immunol (2004) 52:19–26. doi: 10.1111/j.1600-0897.2004.00177.x
33. Roved J, Westerdahl H, Hasselquist D. Sex differences in immune responses: Hormonal effects, antagonistic selection, and evolutionary consequences. Horm Behav (2017) 88:95–105. doi: 10.1016/j.yhbeh.2016.11.017
34. Russi AE, Ebel ME, Yang Y, Brown MA. Male-specific IL-33 expression regulates sex-dimorphic EAE susceptibility. Proc Natl Acad Sci USA (2018) 115:E1520–9. doi: 10.1073/pnas.1710401115
35. Mackey E, Ayyadurai S, Pohl CS, D’Costa S, Li Y, Moeser AJ. Sexual dimorphism in the mast cell transcriptome and the pathophysiological responses to immunological and psychological stress. Biol Sex Differ (2016) 7:1–19. doi: 10.1186/s13293-016-0113-7
36. Villapol S, Faivre V, Joshi P, Moretti R, Besson VC, Charriaut-Marlangue C. Early sex differences in the immune-inflammatory responses to neonatal ischemic stroke. Int J Mol Sci (2019) 20:3809–24. doi: 10.3390/ijms20153809
37. Seifert HA, Benedek G, Liang J, Nguyen H, Kent G, Vandenbark AA, et al. Sex differences in regulatory cells in experimental stroke. Cell Immunol (2017) 318:49–54. doi: 10.1016/j.cellimm.2017.06.003
38. Ziegler SM, Beisel C, Sutter K, Griesbeck M, Hildebrandt H, H. Hagen S, et al. Human pDCs display sex-specific differences in type I interferon subtypes and interferon α/β receptor expression. Eur J Immunol (2017) 47:251–6. doi: 10.1002/eji.201646725
39. Hannah MF, Bajic VB, Klein SL. Sex differences in the recognition of and innate antiviral responses to Seoul virus in Norway rats. Brain Behav Immun (2008) 22:503–16. doi: 10.1016/j.bbi.2007.10.005
40. Keselman A, Fang X, White PB, Heller NM. Estrogen Signaling Contributes to Sex Differences in Macrophage Polarization during Asthma. J Immunol (2017) 199:1573–83. doi: 10.4049/jimmunol.1601975
41. Wang Q, Khan NA, Muthumalage T, Lawyer GR, McDonough SR, Chuang T-D, et al. Dysregulated repair and inflammatory responses by e-cigarette-derived inhaled nicotine and humectant propylene glycol in a sex-dependent manner in mouse lung. FASEB bioAdvances (2019) 1:609–23. doi: 10.1096/fba.2019-00048
42. Yu M, Cao X, Wang X, Xu J, Yang M, Ben K. Migration of mouse antibody-secreting hybridoma cells from blood to genital tract and its regulation by sex hormones are associated with the differential expression patterns of adhesion molecules and chemokines in the tract rather than in the antibody-sec. J Reprod Immunol (2007) 74:78–89. doi: 10.1016/j.jri.2006.09.003
43. Liu J, Morgan M, Hutchison K, Calhoun VD. A study of the influence of sex on genome wide methylation. PloS One (2010) 5:e10028. doi: 10.1371/journal.pone.0010028
44. Duncan CG, Grimm SA, Morgan DL, Bushel PR, Bennett BD, Roberts JD, et al. Dosage compensation and DNA methylation landscape of the X chromosome in mouse liver. Sci Rep (2018) 8:1–17. doi: 10.1038/s41598-018-28356-3
45. Gal-Oz ST, Maier B, Yoshida H, Seddu K, Elbaz N, Czysz C, et al. ImmGen report: sexual dimorphism in the immune system transcriptome. Nat Commun (2019) 10:4295. doi: 10.1038/s41467-019-12348-6
46. Habib P, Dreymueller D, Rösing B, Botung H, Slowik A, Zendedel A, et al. Estrogen serum concentration affects blood immune cell composition and polarization in human females under controlled ovarian stimulation. J Steroid Biochem Mol Biol (2018) 178:340–7. doi: 10.1016/j.jsbmb.2018.02.005
47. Polanczyk M, Zamora A, Subramanian S, Matejuk A, Hess DL, Blankenhorn EP, et al. The protective effect of 17β-estradiol on experimental autoimmune encephalomyelitis is mediated through estrogen receptor-α. Am J Pathol (2003) 163:1599–605. doi: 10.1016/S0002-9440(10)63516-X
48. Skobeloff EM, Spivey WH, Silverman R, Eskin BA, Harchelroad F, Alessi TV. The effect of the menstrual cycle on asthma presentations in the emergency department. Arch Intern Med (1996) 156:1837–40. doi: 10.1001/archinte.156.16.1837
49. Gibon E, Lu L, Goodman SB. Aging, inflammation, stem cells, and bone healing. Stem Cell Res Ther (2016) 7:1–7. doi: 10.1186/s13287-016-0300-9
50. Goodman WA, Garg RR, Reuter BK, Mattioli B, Rissman EF, Pizarro TT. Loss of estrogen-mediated immunoprotection underlies female gender bias in experimental Crohn’s-like ileitis. Mucosal Immunol (2014) 7:1255–65. doi: 10.1038/mi.2014.15
51. Priyanka HP, Krishnan HC, Singh RV, Hima L, ThyagaRajan S. Estrogen modulates in vitro T cell responses in a concentration- and receptor-dependent manner: Effects on intracellular molecular targets and antioxidant enzymes. Mol Immunol (2013) 56:328–39. doi: 10.1016/j.molimm.2013.05.226
52. Lambert KC, Curran EM, Judy BM, Milligan GN, Lubahn DB, Estes DM. Estrogen Receptor α (ERα) Deficiency in Macrophages Results in Increased Stimulation of CD4 + T Cells while 17β-Estradiol Acts through ERα to Increase IL-4 and GATA-3 Expression in CD4 + T Cells Independent of Antigen Presentation. J Immunol (2005) 175:5716–23. doi: 10.4049/jimmunol.175.9.5716
53. Khan D, Ansar Ahmed S. The immune system is a natural target for estrogen action: Opposing effects of estrogen in two prototypical autoimmune diseases. Front Immunol (2016) 6:635. doi: 10.3389/fimmu.2015.00635
54. Kovats S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol (2015) 294:63–9. doi: 10.1016/j.cellimm.2015.01.018
55. Jaillon S, Berthenet K, Garlanda C. Sexual Dimorphism in Innate Immunity. Clin Rev Allergy Immunol (2019) 56:308–21. doi: 10.1007/s12016-017-8648-x
56. Henze L, Schwinge D, Schramm C. The Effects of Androgens on T Cells: Clues to Female Predominance in Autoimmune Liver Diseases? Front Immunol (2020) 11:1567. doi: 10.3389/fimmu.2020.01567
57. Mayne BT, Bianco-Miotto T, Buckberry S, Breen J, Clifton V, Shoubridge C, et al. Large scale gene expression meta-analysis reveals tissue-specific, sex-biased gene expression in humans. Front Genet (2016) 7:183. doi: 10.3389/fgene.2016.00183
58. Cao Y, Wang L, Wang CY, Ye J, Wang Y, Li T, et al. Sex Differences in Correlation with Gene Expression Levels between Ifi200 Family Genes and Four Sets of Immune Disease-Relevant Genes. J Immunol Res (2018) 2018:1–12. doi: 10.1155/2018/1290814
59. Van Nas A, Guhathakurta D, Wang SS, Yehya N, Horvath S, Zhang B, et al. Elucidating the role of gonadal hormones in sexually dimorphic gene coexpression networks. Endocrinology (2009) 150:1235–49. doi: 10.1210/en.2008-0563
60. Wang S, Clarke PAG, Davis R, Mumuni S, Kwabi-Addo B. Sex steroid-induced DNA methylation changes and inflammation response in prostate cancer. Cytokine (2016) 86:110–8. doi: 10.1016/j.cyto.2016.07.006
61. Mamrut S, Avidan N, Staun-Ram E, Ginzburg E, Truffault F, Berrih-Aknin S, et al. Integrative analysis of methylome and transcriptome in human blood identifies extensive sex- and immune cell-specific differentially methylated regions. Epigenetics (2015) 10:943–57. doi: 10.1080/15592294.2015.1084462
62. Fan H, Zhao G, Ren D, Liu F, Dong G, Hou Y. Gender differences of B cell signature related to estrogen-induced IFI44L/BAFF in systemic lupus erythematosus. Immunol Lett (2017) 181:71–8. doi: 10.1016/j.imlet.2016.12.002
63. Fan H, Dong G, Zhao G, Liu F, Yao G, Zhu Y, et al. Gender differences of B cell signature in healthy subjects underlie disparities in incidence and course of SLE related to estrogen. J Immunol Res (2014) 2014:1–17. doi: 10.1155/2014/814598
64. Dunn SE, Ousman SS, Sobel RA, Zuniga L, Baranzini SE, Youssef S, et al. Peroxisome proliferator-activated receptor (PPAR)alpha expression in T cells mediates gender differences in development of T cell-mediated autoimmunity. J Exp Med (2007) 204:321–30. doi: 10.1084/jem.20061839
65. Ji H, Zheng W, Li X, Liu J, Wu X, Zhang MA, et al. Sex-specific T-cell regulation of angiotensin II-dependent hypertension. Hypertens (Dallas Tex 1979) (2014) 64:573–82. doi: 10.1161/HYPERTENSIONAHA.114.03663
66. Zhang MA, Rego D, Moshkova M, Kebir H, Chruscinski A, Nguyen H, et al. Peroxisome proliferator-activated receptor (PPAR)alpha and -gamma regulate IFNgamma and IL-17A production by human T cells in a sex-specific way. Proc Natl Acad Sci USA (2012) 109:9505–10. doi: 10.1073/pnas.1118458109
67. Pace S, Rossi A, Krauth V, Dehm F, Troisi F, Bilancia R, et al. Sex differences in prostaglandin biosynthesis in neutrophils during acute inflammation. Sci Rep (2017) 7:3759. doi: 10.1038/s41598-017-03696-8
68. Prahl M, Jagannathan P, McIntyre TI, Auma A, Wamala S, Nalubega M, et al. Sex Disparity in Cord Blood FoxP3+ CD4 T Regulatory Cells in Infants Exposed to Malaria In Utero. Open Forum Infect Dis (2017) 4:ofx022. doi: 10.1093/ofid/ofx022
69. Muenchhoff M, Goulder PJR. Sex differences in pediatric infectious diseases. J Infect Dis (2014) 209 Suppl:S120–6. doi: 10.1093/infdis/jiu232
70. Astudillo P, Angulo J, Pino K, de Carvalho JB, de Morais GL, Perez S, et al. Correlation between female sex, IL28B genotype, and the clinical severity of bronchiolitis in pediatric patients. Pediatr Res (2020) 87:785–95. doi: 10.1038/s41390-019-0623-1
71. Bongen E, Lucian H, Khatri A, Fragiadakis GK, Bjornson ZB, Nolan GP, et al. Sex Differences in the Blood Transcriptome Identify Robust Changes in Immune Cell Proportions with Aging and Influenza Infection. Cell Rep (2019) 29:1961–1973.e4. doi: 10.1016/j.celrep.2019.10.019
72. Márquez EJ, Chung Ch, Marches R, Rossi RJ, Nehar-Belaid D, Eroglu A, et al. Sexual-dimorphism in human immune system aging. Nat Commun (2020) 11:751. doi: 10.1038/s41467-020-14396-9
73. Voigt EA, Ovsyannikova IG, Kennedy RB, Grill DE, Goergen KM, Schaid DJ, et al. Sex differences in older adults’ immune responses to seasonal influenza vaccination. Front Immunol (2019) 10:180. doi: 10.3389/fimmu.2019.00180
74. Takahashi T, Ellingson MK, Wong P, Israelow B, Lucas C, Klein J, et al. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature (2020). doi: 10.1038/s41586-020-2700-3
75. Jin JM, Bai P, He W, Wu F, Liu XF, Han DM, et al. Gender Differences in Patients With COVID-19: Focus on Severity and Mortality. Front Public Heal (2020) 8:152. doi: 10.3389/fpubh.2020.00152
76. Meng Y, Wu P, Lu W, Liu K, Ma K, Huang L, et al. Sex-specific clinical characteristics and prognosis of coronavirus disease-19 infection in Wuhan, China: A retrospective study of 168 severe patients. PloS Pathog (2020) 16:1–13. doi: 10.1371/journal.ppat.1008520
77. World Health Organization (WHO). Noncommunicable diseases country profiles 2018. Geneva: World Health Organization (WHO) (2018). Available at: https://apps.who.int/iris/handle/10665/274512.
78. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell (2018) 172:162–75.e14. doi: 10.1016/j.cell.2017.12.013
79. Reamon-Buettner SM, Mutschler V, Borlak J. The next innovation cycle in toxicogenomics: Environmental epigenetics. Mutat Res - Rev Mutat Res (2008) 659:158–65. doi: 10.1016/j.mrrev.2008.01.003
80. Aiken CE, Tarry-Adkins JL, Ozanne SE. Transgenerational effects of maternal diet on metabolic and reproductive ageing. Mamm Genome (2016) 27:430–9. doi: 10.1007/s00335-016-9631-1
81. Anderson OS, Sant KE, Dolinoy DC. Nutrition and epigenetics: An interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J Nutr Biochem (2012) 23:853–9. doi: 10.1016/j.jnutbio.2012.03.003
82. McDade TW, Rutherford JN, Adair L, Kuzawa C. Population differences in associations between C-reactive protein concentration and adiposity: Comparison of young adults in the Philippines and the United States. Am J Clin Nutr (2009) 89:1237–45. doi: 10.3945/ajcn.2008.27080
83. Zhang Y, Kutateladze TG. Diet and the epigenome. Nat Commun (2018) 9:9–11. doi: 10.1038/s41467-018-05778-1
84. Marcelino G, Hiane PA, Freitas K de C, Santana LF, Pott A, Donadon JR, et al. Effects of olive oil and its minor components on cardiovascular diseases, inflammation, and gut microbiota. Nutrients (2019) 11:4–6. doi: 10.3390/nu11081826
85. Plastina P, Benincasa C, Perri E, Fazio A, Augimeri G, Poland M, et al. Identification of hydroxytyrosyl oleate, a derivative of hydroxytyrosol with anti-inflammatory properties, in olive oil by-products. Food Chem (2019) 279:105–13. doi: 10.1016/j.foodchem.2018.12.007
86. Visioli F, Wolfram R, Richard D, Abdullah MICB, Crea R. Olive Phenolics increase glutathione levels in healthy volunteers. J Agric Food Chem (2009) 57:1793–6. doi: 10.1021/jf8034429
87. Uusitupa M, Hermansen K, Savolainen MJ, Schwab U, Kolehmainen M, Brader L, et al. Effects of an isocaloric healthy Nordic diet on insulin sensitivity, lipid profile and inflammation markers in metabolic syndrome - a randomized study (SYSDIET). J Intern Med (2013) 274:52–66. doi: 10.1111/joim.12044
88. Adamsson V, Reumark A, Cederholm T, Vessby B, Risérus U, Johansson G. What is a healthy Nordic diet? Foods and nutrients in the NORDIET study. Food Nutr Res (2012) 56:1–9. doi: 10.3402/fnr.v56i0.18189
89. Kanerva N, Kaartinen NE, Rissanen H, Knekt P, Eriksson JG, Sääksjärvi K, et al. Associations of the Baltic Sea diet with cardiometabolic risk factors-a meta-analysis of three Finnish studies. Br J Nutr (2014) 112:616–26. doi: 10.1017/S0007114514001159
90. Kanerva N, Loo BM, Eriksson JG, Leiviskä J, Kaartinen NE, Jula A, et al. Associations of the Baltic Sea diet with obesity-related markers of inflammation. Ann Med (2014) 46:90–6. doi: 10.3109/07853890.2013.870020
91. Poulsen SK, Due A, Jordy AB, Kiens B, Stark KD, Stender S, et al. Health effect of the new nordic diet in adults with increased waist circumference: A 6-mo randomized controlled trial. Am J Clin Nutr (2014) 99:35–45. doi: 10.3945/ajcn.113.069393
92. De Mello VDF, Schwab U, Kolehmainen M, Koenig W, Siloaho M, Poutanen K, et al. A diet high in fatty fish, bilberries and wholegrain products improves markers of endothelial function and inflammation in individuals with impaired glucose metabolism in a randomised controlled trial: The Sysdimet study. Diabetologia (2011) 54:2755–67. doi: 10.1007/s00125-011-2285-3
93. Jobs E, Adamsson V, Larsson A, Jobs M, Nerpin E, Ingelsson E, et al. Influence of a prudent diet on circulating cathepsin S in humans. Nutr J (2014) 13:1–5. doi: 10.1186/1475-2891-13-84
94. Kolehmainen M, Ulven SM, Paananen J, De Mello V, Schwab U, Carlberg C, et al. Healthy Nordic diet downregulates the expression of genes involved in inflammation in subcutaneous adipose tissue in individuals with features of the metabolic syndrome. Am J Clin Nutr (2015) 101:228–39. doi: 10.3945/ajcn.114.092783
95. Myhrstad MCW, de Mello VD, Dahlman I, Kolehmainen M, Paananen J, Rundblad A, et al. Healthy Nordic Diet Modulates the Expression of Genes Related to Mitochondrial Function and Immune Response in Peripheral Blood Mononuclear Cells from Subjects with Metabolic Syndrome–A SYSDIET Sub-Study. Mol Nutr Food Res (2019) 63:1801405. doi: 10.1002/mnfr.201801405
96. Dyerberg J, Bang HO, Stoffersen E, Moncada S, Vane JR. Eicosapentaenoic acid and prevention of thrombosis and atherosclerosis? Lancet (1978) 312:117–9. doi: 10.1016/S0140-6736(78)91505-2
97. Calder PC. Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim Biophys Acta - Mol Cell Biol Lipids (2015) 1851:469–84. doi: 10.1016/j.bbalip.2014.08.010
98. Calder PC. Omega-3 fatty acids and inflammatory processes: From molecules to man. Biochem Soc Trans (2017) 45:1105–15. doi: 10.1042/BST20160474
99. Endo J, Arita M. Cardioprotective mechanism of omega-3 polyunsaturated fatty acids. J Cardiol (2016) 67:22–7. doi: 10.1016/j.jjcc.2015.08.002
100. Matt SM, Johnson RW. Neuro-immune dysfunction during brain aging: new insights in microglial cell regulation. Curr Opin Pharmacol (2016) 26:96–101. doi: 10.1016/j.coph.2015.10.009
101. Hughes DA, Pinder AC, Piper Z, Lund EK. N-3 polyunsaturated fatty acids (PUFA) modulate the expression of functionally associated molecules on human monocytes. Biochem Soc Trans (1995) 23:516–23. doi: 10.1042/bst023303s
102. Hughes DA, Pinder AC. n-3 Polyunsaturated fatty acids inhibit the antigen-presenting function of human monocytes. Am J Clin Nutr (2000) 71:357–60. doi: 10.1093/ajcn/71.1.357s
103. Choi M, Ju J, Suh JS, Park KY, Kim KH. Effects of omega-3-rich harp seal oil on the production of pro-inflammatory cytokines in mouse peritoneal macrophages. Prev Nutr Food Sci (2015) 20:83–7. doi: 10.3746/pnf.2015.20.2.83
104. Sanderson P, Calder PC. Dietary fish oil diminishes lymphocyte adhesion to macrophage and endothelial cell monolayers. Immunology (1998) 94:79–87. doi: 10.1046/j.1365-2567.1998.00474.x
105. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan WQ, et al. GPR120 Is an Omega-3 Fatty Acid Receptor Mediating Potent Anti-inflammatory and Insulin-Sensitizing Effects. Cell (2010) 142:687–98. doi: 10.1016/j.cell.2010.07.041
106. Zapata-Gonzalez F, Rueda F, Petriz J, Domingo P, Villarroya F, Diaz-Delfin J, et al. Human dendritic cell activities are modulated by the omega-3 fatty acid, docosahexaenoic acid, mainly through PPARγ:RXR heterodimers: comparison with other polyunsaturated fatty acids. J Leukoc Biol (2008) 84:1172–82. doi: 10.1189/jlb.1007688
107. Zhao Y, Joshi-Barve S, Barve S, Chen LH. Eicosapentaenoic Acid Prevents LPS-Induced TNF-α Expression by Preventing NF-κB Activation. J Am Coll Nutr (2004) 23:71–8. doi: 10.1080/07315724.2004.10719345
108. Weldon SM, Mullen AC, Loscher CE, Hurley LA, Roche HM. Docosahexaenoic acid induces an anti-inflammatory profile in lipopolysaccharide-stimulated human THP-1 macrophages more effectively than eicosapentaenoic acid. J Nutr Biochem (2007) 18:250–8. doi: 10.1016/j.jnutbio.2006.04.003
109. Zampelas A. Eicosapentaenoic acid (EPA) from highly concentrated n-3 fatty acid ethyl esters is incorporated into advanced atherosclerotic plaques and higher plaque EPA is associated with decreased plaque inflammation and increased stability. Atherosclerosis (2010) 212:34–5. doi: 10.1016/j.atherosclerosis.2010.06.018
110. Kong W, Yen JH, Ganea D. Docosahexaenoic acid prevents dendritic cell maturation, inhibits antigen-specific Th1/Th17 differentiation and suppresses experimental autoimmune encephalomyelitis. Brain Behav Immun (2011) 25:872–82. doi: 10.1016/j.bbi.2010.09.012
111. Kim W, Khan NA, McMurray DN, Prior IA, Wang N, Chapkin RS. Regulatory activity of polyunsaturated fatty acids in T-cell signaling. Prog Lipid Res (2010) 49:250–61. doi: 10.1016/j.plipres.2010.01.002
112. Rockett BD, Salameh M, Carraway K, Morrison K, Shaikh SR. n-3 PUFA improves fatty acid composition, prevents palmitate-induced apoptosis, and differentially modifies B cell cytokine secretion in vitro and ex vivo. J Lipid Res (2010) 51:1284–97. doi: 10.1194/jlr.M000851
113. Lee TH, Mencia-Huerta JM, Shih C, Corey EJ, Lewis RA, Austen KF. Effects of exogenous arachidonic, eicosapentaenoic, and docosahexaenoic acids on the generation of 5-lipoxygenase pathway products by ionophore-activated human neutrophils. J Clin Invest (1984) 74:1922–33. doi: 10.1172/JCI111612
114. Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature (2007) 447:869–74. doi: 10.1038/nature05877
115. Arita M, Ohira T, Sun Y-P, Elangovan S, Chiang N, Serhan CN. Resolvin E1 Selectively Interacts with Leukotriene B 4 Receptor BLT1 and ChemR23 to Regulate Inflammation. J Immunol (2007) 178:3912–7. doi: 10.4049/jimmunol.178.6.3912
116. Hasturk H, Abdallah R, Kantarci A, Nguyen D, Giordano N, Hamilton J, et al. Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation-induced atherogenesis. Arterioscler Thromb Vasc Biol (2015) 35:1123–33. doi: 10.1161/ATVBAHA.115.305324
117. Streit WJ, Xue QS. Human CNS immune senescence and neurodegeneration. Curr Opin Immunol (2014) 29:93–6. doi: 10.1016/j.coi.2014.05.005
118. Streit WJ. Microglial senescence: does the brain’s immune system have an expiration date? Trends Neurosci (2006) 29:506–10. doi: 10.1016/j.tins.2006.07.001
119. López GH, Ilincheta de Boschero MG, Castagnet PI, Giusto NM. Age-associated changes in the content and fatty acid composition of brain glycerophospholipids. Comp Biochem Physiol – Part B Biochem (1995) 112:331–43. doi: 10.1016/0305-0491(95)00079-8
120. McNamara RK, Liu Y, Jandacek R, Rider T, Tso P. The aging human orbitofrontal cortex: Decreasing polyunsaturated fatty acid composition and associated increases in lipogenic gene expression and stearoyl-CoA desaturase activity. Prostaglandins Leukot Essent Fat Acids (2008) 78:293–304. doi: 10.1016/j.plefa.2008.04.001
121. Joffre C, Grégoire S, De Smedt V, Acar N, Bretillon L, Nadjar A, et al. Modulation of brain PUFA content in different experimental models of mice. Prostaglandins Leukot Essent Fat Acids (2016) 114:1–10. doi: 10.1016/j.plefa.2016.09.003
122. Favrelière S, Perault MC, Huguet F, De Javel D, Bertrand N, Piriou A, et al. DHA-enriched phospholipid diets modulate age-related alterations in rat hippocampus. Neurobiol Aging (2003) 24:233–43. doi: 10.1016/S0197-4580(02)00064-7
123. Little SJ, Lynch MA, Manku M, Nicolaou A. Docosahexaenoic acid-induced changes in phospholipids in cortex of young and aged rats: A lipidomic analysis. Prostaglandins Leukot Essent Fat Acids (2007) 77:155–62. doi: 10.1016/j.plefa.2007.08.009
124. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat Rev Neurosci (2008) 9:46–56. doi: 10.1038/nrn2297
125. Söderberg M, Edlund C, Kristensson K, Dallner G. Fatty acid composition of brain phospholipids in aging and in Alzheimer’s disease. Lipids (1991) 26:421–5. doi: 10.1007/BF02536067
126. Gamoh S, Hashimoto M, Hossain S, Masumura S. Chronic administration of docosahexaenoic acid improves the performance of radial arm maze task in aged rats. Clin Exp Pharmacol Physiol (2001) 28:266–70. doi: 10.1046/j.1440-1681.2001.03437.x
127. Carrié I, Smirnova M, Clément M, De Javel D, Francès H, Bourre JM. Docosahexaenoic acid-rich phospholipid supplementation: Effect on behavior, learning ability, and retinal function in control and n-3 polyunsaturated fatty acid deficient old mice. Nutr Neurosci (2002) 5:43–52. doi: 10.1080/10284150290007074
128. Arsenault D, Julien C, Tremblay C, Calon F. DHA improves cognition and prevents dysfunction of entorhinal cortex neurons in 3xTg-AD mice. PloS One (2011) 6:e17397. doi: 10.1371/journal.pone.0017397
129. Barberger-Gateau P. Association between Mediterranean diet and late-life cognition. JAMA - J Am Med Assoc (2009) 302:2433. doi: 10.1001/jama.2009.1794
130. Tan ZS, Harris WS, Beiser AS, Au R, Himali JJ, Debette S, et al. Red blood cell omega-3 fatty acid levels and markers of accelerated brain aging. Neurology (2012) 78:658–64. doi: 10.1212/WNL.0b013e318249f6a9
131. Titova OE, Sjögren P, Brooks SJ, Kullberg J, Ax E, Kilander L, et al. Dietary intake of eicosapentaenoic and docosahexaenoic acids is linked to gray matter volume and cognitive function in elderly. Age (Omaha) (2013) 35:1495–505. doi: 10.1007/s11357-012-9453-3
132. Whalley LJ, Fox HC, Wahle KW, Starr JM, Deary IJ. Cognitive aging, childhood intelligence, and the use of food supplements: Possible involvement of n-3 fatty acids. Am J Clin Nutr (2004) 80:1650–7. doi: 10.1093/ajcn/80.6.1650
133. Devore EE, Grodstein F, Van Rooij FJA, Hofman A, Rosner B, Stampfer MJ, et al. Dietary intake of fish and omega-3 fatty acids in relation to long-term dementia risk. Am J Clin Nutr (2009) 90:170–6. doi: 10.3945/ajcn.2008.27037
134. Caligiuri SPB, Aukema HM, Ravandi A, Pierce GN. Elevated levels of pro-inflammatory oxylipins in older subjects are normalized by flaxseed consumption. Exp Gerontol (2014) 59:51–7. doi: 10.1016/j.exger.2014.04.005
135. Rey C, Delpech JC, Madore C, Nadjar A, Greenhalgh AD, Amadieu C, et al. Dietary n-3 long chain PUFA supplementation promotes a pro-resolving oxylipin profile in the brain. Brain Behav Immun (2019) 76:17–27. doi: 10.1016/j.bbi.2018.07.025
136. Oliveira BS, Zunzunegui MV, Quinlan J, Fahmi H, Tu MT, Guerra RO. Systematic review of the association between chronic social stress and telomere length: A life course perspective. Ageing Res Rev (2016) 26:37–52. doi: 10.1016/j.arr.2015.12.006
137. Provençal N, Booij L, Tremblay RE. The developmental origins of chronic physical aggression: Biological pathways triggered by early life adversity. J Exp Biol (2015) 218:123–33. doi: 10.1242/jeb.111401
138. Gu HF, Tang CK, Yang YZ. Psychological stress, immune response, and atherosclerosis. Atherosclerosis (2012) 223:69–77. doi: 10.1016/j.atherosclerosis.2012.01.021
139. Riancho JA, Brennan-Olsen SL. The Epigenome at the Crossroad Between Social Factors, Inflammation, and Osteoporosis Risk. Clin Rev Bone Miner Metab (2017) 15:59–68. doi: 10.1007/s12018-017-9229-5
140. Weaver ICG, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci (2004) 7:847–54. doi: 10.1038/nn1276
141. Klengel T, Pape J, Binder EB, Mehta D. The role of DNA methylation in stress-related psychiatric disorders. Neuropharmacology (2014) 80:115–32. doi: 10.1016/j.neuropharm.2014.01.013
142. Dimitrov S, Lange T, Born J. Selective Mobilization of Cytotoxic Leukocytes by Epinephrine. J Immunol (2010) 184:503–11. doi: 10.4049/jimmunol.0902189
143. Gouin JP, Hantsoo L, Kiecolt-Glaser JK. Immune dysregulation and chronic stress among older adults: a review. Neuroimmunomodulation (2008) 15:251–9. doi: 10.1159/000156468
144. Kiecolt-Glaser JK, Preacher KJ, MacCallum RC, Atkinson C, Malarkey WB, Glaser R. Chronic stress and age-related increases in the proinflammatory cytokine IL-6. Proc Natl Acad Sci USA (2003) 100:9090–5. doi: 10.1073/pnas.1531903100
145. Krohn M, Listing M, Tjahjono G, Reisshauer A, Peters E, Klapp BF, et al. Depression, mood, stress, and Th1/Th2 immune balance in primary breast cancer patients undergoing classical massage therapy. Support Care Cancer (2011) 19:1303–11. doi: 10.1007/s00520-010-0946-2
146. Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-κB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci USA (2010) 107:2669–74. doi: 10.1073/pnas.0910658107
147. Slavich GM, Irwin MR. Social Signal Transduction Theory of Depression. Psychol Bull (2014) 140:774–815. doi: 10.1037/a0035302
148. Hänsel A, Hong S, Cámara RJA, von Känel R. Inflammation as a psychophysiological biomarker in chronic psychosocial stress. Neurosci Biobehav Rev (2010) 35:115–21. doi: 10.1016/j.neubiorev.2009.12.012
149. Powell ND, Bailey MT, Mays JW, Stiner-Jones LM, Hanke ML, Padgett DA, et al. Repeated social defeat activates dendritic cells and enhances Toll-like receptor dependent cytokine secretion. Brain Behav Immun (2009) 23:225–31. doi: 10.1016/j.bbi.2008.09.010
150. Zhu Y, Klomparens EA, Guo S, Geng X. Neuroinflammation caused by mental stress: the effect of chronic restraint stress and acute repeated social defeat stress in mice. Neurol Res (2019) 41:762–9. doi: 10.1080/01616412.2019.1615670
151. Sun P, Wei S, Wei X, Wang J, Zhang Y, Qiao M, et al. Anger Emotional Stress Influences VEGF/VEGFR2 and Its Induced PI3K/AKT/mTOR Signaling Pathway. Neural Plast (2016) 2016:4129015:1-12. doi: 10.1155/2016/4129015
152. López-López AL, Bonilla HJ, Escobar Villanueva M del C, Brianza MP, Vázquez GP, Alarcón FJA. Chronic unpredictable mild stress generates oxidative stress and systemic inflammation in rats. Physiol Behav (2016) 161:15–23. doi: 10.1016/j.physbeh.2016.03.017
153. Fonken LK, Frank MG, Gaudet AD, Maier SF. Stress and aging act through common mechanisms to elicit neuroinflammatory priming. Brain Behav Immun (2018) 73:133–48. doi: 10.1016/j.bbi.2018.07.012
154. Yegorov YE, Poznyak AV, Nikiforov NG, Sobenin IA, Orekhov AN. The link between chronic stress and accelerated aging. Biomedicines (2020) 8:1–14. doi: 10.3390/BIOMEDICINES8070198
155. Boccardi M, Boccardi V. Psychological wellbeing and healthy aging: Focus on telomeres. Geriatr (2019) 4:1–11. doi: 10.3390/geriatrics4010025
156. Epel ES, Prather AA. Stress, Telomeres, and Psychopathology: Toward a Deeper Understanding of a Triad of Early Aging. Annu Rev Clin Psychol (2018) 14:371–97. doi: 10.1146/annurev-clinpsy-032816-045054
157. Prather AA, Gurfein B, Moran P, Daubenmier J, Acree M, Bacchetti P, et al. Tired telomeres: Poor global sleep quality, perceived stress, and telomere length in immune cell subsets in obese men and women. Brain Behav Immun (2015) 47:155–62. doi: 10.1016/j.bbi.2014.12.011
158. Zahs A, Curtis BJ, Waldschmidt TJ, Brown LAS, Gauthier TW, Choudhry MA, et al. Alcohol and epigenetic changes: Summary of the 2011 Alcohol and Immunology Research Interest Group (AIRIG) meeting. Alcohol (2012) 46:783–7. doi: 10.1016/j.alcohol.2012.05.005
159. Mata MM, Napier TC, Graves SM, Mahmood F, Raeisi S, Baum LL. Methamphetamine decreases CD4 T cell frequency and alters pro-inflammatory cytokine production in a model of drug abuse. Eur J Pharmacol (2015) 752:26–33. doi: 10.1016/j.ejphar.2015.02.002
160. Goral J, Karavitis J, Kovacs EJ. Exposure-dependent effects of ethanol on the innate immune system. PMC (2013) 42:140–66. doi: 10.7208/chicago/9780226768618.003.0006
161. Crawford FC, Wood ML, Wilson SE, Mathura VS, Hollen TR, Geall F, et al. Cocaine induced inflammatory response in human neuronal progenitor cells. J Neurochem (2006) 97:662–74. doi: 10.1111/j.1471-4159.2006.03760.x
162. Crews FT, Zou J, Qin L. Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav Immun (2011) 25:S4–S12. doi: 10.1016/j.bbi.2011.03.003
163. Piechota M, Korostynski M, Solecki W, Gieryk A, Slezak M, Bilecki W, et al. The dissection of transcriptional modules regulated by various drugs of abuse in the mouse striatum. Genome Biol (2010) 11:R48. doi: 10.1186/gb-2010-11-5-r48
164. Renthal W, Maze I, Krishnan V, Covington HE, Xiao G, Kumar A, et al. Histone Deacetylase 5 Epigenetically Controls Behavioral Adaptations to Chronic Emotional Stimuli. Neuron (2007) 56:517–29. doi: 10.1016/j.neuron.2007.09.032
165. Little KY, Ramssen E, Welchko R, Volberg V, Roland CJ, Cassin B. Decreased brain dopamine cell numbers in human cocaine users. Psychiatry Res (2009) 168:173–80. doi: 10.1016/j.psychres.2008.10.034
166. Periyasamy P, Guo ML, Buch S. Cocaine induces astrocytosis through ER stress-mediated activation of autophagy. Autophagy (2016) 12:1310–29. doi: 10.1080/15548627.2016.1183844
167. Fox H, D’Sa C, Kimmerling A, Siedlarz K, Tuit K, Stowe R, et al. Immune system inflammation in cocaine dependent individuals: implications for medications development. Hum Psychopharmacol Clin Exp (2012) 27:156–66. doi: 10.1002/hup
168. Clark KH, Wiley CA, Bradberry CW. Psychostimulant abuse and neuroinflammation: Emerging evidence of their interconnection. Neurotox Res (2013) 23:174–88. doi: 10.1007/s12640-012-9334-7
169. Papageorgiou M, Raza A, Fraser S, Nurgali K, Apostolopoulos V. Methamphetamine and its immune-modulating effects. Maturitas (2019) 121:13–21. doi: 10.1016/j.maturitas.2018.12.003
170. Peerzada H, Gandhi JA, Guimaraes AJ, Nosanchuk JD, Martinez LR. Methamphetamine administration modifies leukocyte proliferation and cytokine production in murine tissues. Immunobiology (2013) 218:1063–8. doi: 10.1016/j.imbio.2013.02.001
171. Quinton M, Yamamoto B. Causes and Consequences of Methamphetamine and MDMA Toxicity. AAPS J (2006) 08:E337. doi: 10.1208/aapsj080238
172. Raineri M, Gonzalez B, Goitia B, Garcia-Rill E, Krasnova IN, Cadet JL, et al. Modafinil Abrogates Methamphetamine-Induced Neuroinflammation and Apoptotic Effects in the Mouse Striatum. PloS One (2012) 7:1–10. doi: 10.1371/journal.pone.0046599
173. Gonçalves J, Baptista S, Martins T, Milhazes N, Borges F, Ribeiro CF, et al. Methamphetamine-induced neuroinflammation and neuronal dysfunction in the mice hippocampus: Preventive effect of indomethacin. Eur J Neurosci (2010) 31:315–26. doi: 10.1111/j.1460-9568.2009.07059.x
174. Chao T, Haney M, Cooper ZD, Vadhan NP, Van Dam NT, Van Snellenberg J, et al. Cognitive function in aging cocaine smokers. J Psychopharmacol (2019) 33:801–10. doi: 10.1177/0269881119849812
175. Chhatre S, Cook R, Mallik E, Jayadevappa R. Trends in substance use admissions among older adults. BMC Health Serv Res (2017) 17:1–8. doi: 10.1186/s12913-017-2538-z
176. Soder HE, Berumen AM, Gomez KE, Green CE, Suchting R, Wardle MC, et al. Elevated neutrophil to lymphocyte ratio in older adults with cocaine use disorder as a marker of chronic inflammation. Clin Psychopharmacol Neurosci (2020) 18:32–40. doi: 10.9758/CPN.2020.18.1.32
177. Hankosky ER, Westbrook SR, Haake RM, Willing J, Raetzman LT, Juraska JM, et al. Age- and sex-dependent effects of methamphetamine on cognitive flexibility and 5-HT2C receptor localization in the orbitofrontal cortex of Sprague-Dawley rats. Behav Brain Res (2018) 349:16–24. doi: 10.1016/j.bbr.2018.04.047
178. Sanvicente-Vieira B, Kommers-Molina J, de Nardi T, Francke I, Grassi-Oliveira R. Crack-cocaine dependence and aging: Effects on working memory. Rev Bras Psiquiatr (2016) 38:58–60. doi: 10.1590/1516-4446-2015-1708
179. Huang C, Wang Q, Wang S, Ren M, Ma R, He Y. Air pollution prevention and control policy in China. Adv Exp Med Biol (2017) 1017:243–61. doi: 10.1007/978-981-10-5657-4_11
180. Saxon A, Diaz-Sanchez D. Air pollution and allergy: You are what you breathe. Nat Immunol (2005) 6:223–6. doi: 10.1038/ni0305-223
181. WHO. WHO Global Ambient Air Quality Database. World Health Organization (2018). Available at: https://www.who.int/airpollution/data/cities/en/#.
182. Shukla A, Bunkar N, Kumar R, Bhargava A, Tiwari R, Chaudhury K, et al. Air pollution associated epigenetic modifications: Transgenerational inheritance and underlying molecular mechanisms. Sci Total Environ (2019) 656:760–77. doi: 10.1016/j.scitotenv.2018.11.381
183. Yang L, Hou XY, Wei Y, Thai P, Chai F. Biomarkers of the health outcomes associated with ambient particulate matter exposure. Sci Total Environ (2017) 579:1446–59. doi: 10.1016/j.scitotenv.2016.11.146
184. Lin CY, Wang CM, Chen ML, Hwang BF. The effects of exposure to air pollution on the development of uterine fibroids. Int J Hyg Environ Health (2019) 222:549–55. doi: 10.1016/j.ijheh.2019.02.004
185. Møller P, Jacobsen NR, Folkmann JK, Danielsen PH, Mikkelsen L, Hemmingsen JG, et al. Role of oxidative damage in toxicity of particulate. Free Radic Res (2010) 44:1–46. doi: 10.3109/10715760903300691
186. Reno AL, Brooks EG, Ameredes BT. Mechanisms of Heightened Airway Sensitivity and Responses to Inhaled SO2 in Asthmatics. Environ Health Insights (2015) 9:13–25. doi: 10.4137/EHI.S15671
187. Zhou W, Tian D, He J, Zhang L, Tang X, Zhang L, et al. Exposure scenario: Another important factor determining the toxic effects of PM2.5 and possible mechanisms involved. Environ Pollut (2017) 226:412–25. doi: 10.1016/j.envpol.2017.04.010
188. Steenhof M, Gosens I, Strak M, Godri KJ, Hoek G, Cassee FR, et al. In vitro toxicity of particulate matter (PM) collected at different sites in the Netherlands is associated with PM composition, size fraction and oxidative potential - the RAPTES project. Part Fibre Toxicol (2011) 8:1–15. doi: 10.1186/1743-8977-8-26
189. Bhargava A, Shukla A, Bunkar N, Shandilya R, Lodhi L, Kumari R, et al. Exposure to ultrafine particulate matter induces NF-KB mediated epigenetic modifications. Environ Pollut (2019) 252:39–50. doi: 10.1016/j.envpol.2019.05.065
190. Pierdominici M, Maselli A, Cecchetti S, Tinari A, Mastrofrancesco A, Alfè M, et al. Diesel exhaust particle exposure in vitro impacts T lymphocyte phenotype and function. Part Fibre Toxicol (2014) 11:1–14. doi: 10.1186/s12989-014-0074-0
191. Ji X, Han M, Yun Y, Li G, Sang N. Acute nitrogen dioxide (NO2) exposure enhances airway inflammation via modulating Th1/Th2 differentiation and activating JAK-STAT pathway. Chemosphere (2015) 120:722–8. doi: 10.1016/j.chemosphere.2014.10.039
192. Muñoz X, Barreiro E, Bustamante V, Lopez-Campos JL, González-Barcala FJ, Cruz MJ. Diesel exhausts particles: Their role in increasing the incidence of asthma. Reviewing the evidence of a causal link. Sci Total Environ (2019) 652:1129–38. doi: 10.1016/j.scitotenv.2018.10.188
193. Leclercq B, Kluza J, Antherieu S, Sotty J, Alleman LY, Perdrix E, et al. Air pollution-derived PM2.5 impairs mitochondrial function in healthy and chronic obstructive pulmonary diseased human bronchial epithelial cells. Environ Pollut (2018) 243:1434–49. doi: 10.1016/j.envpol.2018.09.062
194. Confalonieri M, Salton F, Fabiano F. Acute respiratory distress syndrome. Eur Respir Rev (2017) 26:160116. doi: 10.1183/16000617.0116-2016
195. Gu X, Chu X, Zeng XL, Bao HR, Liu XJ. Effects of PM2.5 exposure on the Notch signaling pathway and immune imbalance in chronic obstructive pulmonary disease. Environ Pollut (2017) 226:163–73. doi: 10.1016/j.envpol.2017.03.070
196. Yao Y, Wang D, Ma H, Li C, Chang X, Low P, et al. The impact on T-regulatory cell related immune responses in rural women exposed to polycyclic aromatic hydrocarbons (PAHs) in household air pollution in Gansu, China: A pilot investigation. Environ Res (2019) 173:306–17. doi: 10.1016/j.envres.2019.03.053
197. Xu Z, Li Z, Liao Z, Gao S, Hua L, Ye X, et al. PM 2.5 induced pulmonary fibrosis in vivo and in vitro. Ecotoxicol Environ Saf (2019) 171:112–21. doi: 10.1016/j.ecoenv.2018.12.061
198. Mi S, Li Z, Yang H, Liu H, Wang J-P, Ma Y, et al. Blocking IL-17A Promotes the Resolution of Pulmonary Inflammation and Fibrosis Via TGF-β1–Dependent and –Independent Mechanisms. J Immunol (2011) 187:3003–14. doi: 10.4049/jimmunol.1004081
199. Li R, Zhang M, Wang Y, Yung KKL, Su R, Li Z, et al. Effects of sub-chronic exposure to atmospheric PM2.5 on fibrosis, inflammation, endoplasmic reticulum stress and apoptosis in the livers of rats. Toxicol Res (Camb) (2018) 7:271–82. doi: 10.1039/c7tx00262a
200. Leclercq B, Platel A, Antherieu S, Alleman LY, Hardy EM, Perdrix E, et al. Genetic and epigenetic alterations in normal and sensitive COPD-diseased human bronchial epithelial cells repeatedly exposed to air pollution-derived PM2.5. Environ Pollut (2017) 230:163–77. doi: 10.1016/j.envpol.2017.06.028
201. Byun HM, Panni T, Motta V, Hou L, Nordio F, Apostoli P, et al. Effects of airborne pollutants on mitochondrial DNA Methylation. Part Fibre Toxicol (2013) 10:1–8. doi: 10.1186/1743-8977-10-18
202. Gruzieva O, Xu CJ, Yousefi P, Relton C, Merid SK, Breton CV, et al. Prenatal particulate air pollution and DNA methylation in newborns: An epigenome-wide meta-analysis. Environ Health Perspect (2019) 127:057012. doi: 10.1289/EHP4522
203. Ding R, Jin Y, Liu X, Ye H, Zhu Z, Zhang Y, et al. Dose- and time- effect responses of DNA methylation and histone H3K9 acetylation changes induced by traffic-related air pollution. Sci Rep (2017) 7:1–9. doi: 10.1038/srep43737
204. Rider CF, Carlsten C. Air pollution and DNA methylation: Effects of exposure in humans. Clin Epigenet (2019) 11:1–15. doi: 10.1186/s13148-019-0713-2
205. Hu J, Yu Y. Epigenetic response profiles into environmental epigenotoxicant screening and health risk assessment: A critical review. Chemosphere (2019) 226:259–72. doi: 10.1016/j.chemosphere.2019.03.096
206. White AJ, Kresovich JK, Keller JP, Xu Z, Kaufman JD, Weinberg CR, et al. Air pollution, particulate matter composition and methylation-based biologic age. Environ Int (2019) 132:105071. doi: 10.1016/j.envint.2019.105071
207. Martens DS, Nawrot TS. Air Pollution Stress and the Aging Phenotype: The Telomere Connection. Curr Environ Heal Rep (2016) 3:258–69. doi: 10.1007/s40572-016-0098-8
208. Wang H, Shen X, Liu J, Wu C, Gao J, Zhang Z, et al. The effect of exposure time and concentration of airborne PM2.5 on lung injury in mice: A transcriptome analysis. Redox Biol (2019) 26:101264. doi: 10.1016/j.redox.2019.101264
209. Partridge L, Deelen J, Slagboom PE. Facing up to the global challenges of ageing. Nature (2018) 561:45–56. doi: 10.1038/s41586-018-0457-8
210. Newgard CB, Sharpless NE, Newgard CB, Sharpless NE. Coming of age: molecular drivers of aging and therapeutic opportunities Find the latest version: Review series introduction Coming of age: molecular drivers of aging and therapeutic opportunities. J Clin Invest (2013) 123:946–50. doi: 10.1172/JCI68833.946
211. Fong JH. Disability incidence and functional decline among older adults with major chronic diseases. BMC Geriatr (2019) 19:323. doi: 10.1186/s12877-019-1348-z
212. Pequeno NPF, Cabral NL de A, Marchioni DM, Lima SCVC, Lyra C de O. Quality of life assessment instruments for adults: a systematic review of population-based studies. Health Qual Life Outcomes (2020) 18:208. doi: 10.1186/s12955-020-01347-7
213. Jakovljevic M, Jakab M, Gerdtham U, McDaid D, Ogura S, Varavikova E, et al. Comparative financing analysis and political economy of noncommunicable diseases. J Med Econ (2019) 22:722–7. doi: 10.1080/13696998.2019.1600523
214. Fulop T, Witkowski JM, Olivieri F, Larbi A. The integration of inflammaging in age-related diseases. Semin Immunol (2018) 40:17–35. doi: 10.1016/j.smim.2018.09.003
215. Cornejo F, Vruwink M, Metz C, Muñoz P, Salgado N, Poblete J, et al. Scavenger Receptor-A deficiency impairs immune response of microglia and astrocytes potentiating Alzheimer’s disease pathophysiology. Brain Behav Immun (2018) 69:336–50. doi: 10.1016/j.bbi.2017.12.007
216. Sanada F, Taniyama Y, Muratsu J, Otsu R, Shimizu H, Rakugi H, et al. Source of Chronic Inflammation in Aging. Front Cardiovasc Med (2018) 5:12. doi: 10.3389/fcvm.2018.00012
217. von Bernhardi R, Eugenín-von Bernhardi L, Eugenín J. Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci (2015) 7:124:124. doi: 10.3389/fnagi.2015.00124
218. Sadighi Akha AA. Aging and the immune system: An overview. J Immunol Methods (2018) 463:21–6. doi: 10.1016/j.jim.2018.08.005
219. Saez-Atienzar S, Masliah E. Cellular senescence and Alzheimer disease: the egg and the chicken scenario. Nat Rev Neurosci (2020) 21:433–44. doi: 10.1038/s41583-020-0325-z
220. Nardini C, Moreau JF, Gensous N, Ravaioli F, Garagnani P, Bacalini MG. The epigenetics of inflammaging: The contribution of age-related heterochromatin loss and locus-specific remodelling and the modulation by environmental stimuli. Semin Immunol (2018) 40:49–60. doi: 10.1016/j.smim.2018.10.009
221. Topart C, Werner E, Arimondo PB. Wandering along the epigenetic timeline. Clin Epigenet (2020) 12:97. doi: 10.1186/s13148-020-00893-7
222. Khan A, Alsahli M, Rahmani A. Myeloperoxidase as an Active Disease Biomarker: Recent Biochemical and Pathological Perspectives. Med Sci (2018) 6:33. doi: 10.3390/medsci6020033
223. Papaconstantinou J. The Role of Signaling Pathways of Inflammation and Oxidative Stress in Development of Senescence and Aging Phenotypes in Cardiovascular Disease. Cells (2019) 8:1383. doi: 10.3390/cells8111383
224. Moldogazieva NT, Mokhosoev IM, Mel’nikova TI, Porozov YB, Terentiev AA. Oxidative Stress and Advanced Lipoxidation and Glycation End Products (ALEs and AGEs) in Aging and Age-Related Diseases. Oxid Med Cell Longev (2019) 2019:3085756. doi: 10.1155/2019/3085756
225. Fane M, Weeraratna AT. How the ageing microenvironment influences tumour progression. Nat Rev Cancer (2020) 20:89–106. doi: 10.1038/s41568-019-0222-9
226. De Almeida AJPO, De Almeida Rezende MS, Dantas SH, De Lima Silva S, De Oliveira JCPL, De Lourdes Assunção Araújo De Azevedo F, et al. Unveiling the Role of Inflammation and Oxidative Stress on Age-Related Cardiovascular Diseases. Oxid Med Cell Longev (2020) 2020:1–20. doi: 10.1155/2020/1954398
227. Singh SS, Rai SN, Birla H, Zahra W, Rathore AS, Singh SP. NF-κB-Mediated Neuroinflammation in Parkinson’s Disease and Potential Therapeutic Effect of Polyphenols. Neurotox Res (2020) 37:491–507. doi: 10.1007/s12640-019-00147-2
228. Zannas AS, Jia M, Hafner K, Baumert J, Wiechmann T, Pape JC, et al. Epigenetic upregulation of FKBP5 by aging and stress contributes to NF-κB-driven inflammation and cardiovascular risk. Proc Natl Acad Sci USA (2019) 166:11370–9. doi: 10.1073/pnas.1816847116
229. Hammond SL, Bantle CM, Popichak KA, Wright KA, Thompson D, Forero C, et al. NF-κB signaling in astrocytes modulates brain inflammation and neuronal injury following sequential exposure to manganese and MPTP during development and aging. Toxicol Sci (2020) 56:1–38. doi: 10.1093/toxsci/kfaa115
230. Bright F, Werry EL, Dobson-Stone C, Piguet O, Ittner LM, Halliday GM, et al. Neuroinflammation in frontotemporal dementia. Nat Rev Neurol (2019) 15:540–55. doi: 10.1038/s41582-019-0231-z
231. Ventura MT, Casciaro M, Gangemi S, Buquicchio R. Immunosenescence in aging: Between immune cells depletion and cytokines up-regulation. Clin Mol Allergy (2017) 15:1–8. doi: 10.1186/s12948-017-0077-0
232. Declerck K, Vanden Berghe W. Back to the future: Epigenetic clock plasticity towards healthy aging. Mech Ageing Dev (2018) 174:18–29. doi: 10.1016/j.mad.2018.01.002
233. Xu W, Larbi A. Immunity and Inflammation: From Jekyll to Hyde. Exp Gerontol (2018) 107:98–101. doi: 10.1016/j.exger.2017.11.018
234. Hu My, Lin Yy, Zhang Bj, Lu D, Lu Z, Cai W. Update of inflammasome activation in microglia/macrophage in aging and aging-related disease. CNS Neurosci Ther (2019) 25:1299–307. doi: 10.1111/cns.13262
235. Pinti M, Appay V, Campisi J, Frasca D, Fülöp T, Sauce D, et al. Aging of the immune system: Focus on inflammation and vaccination. Eur J Immunol (2016) 46:2286–301. doi: 10.1002/eji.201546178
236. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nat Rev Endocrinol (2018) 14:576–90. doi: 10.1038/s41574-018-0059-4
237. Stevenson AJ, McCartney DL, Hillary RF, Campbell A, Morris SW, Bermingham ML, et al. Characterisation of an inflammation-related epigenetic score and its association with cognitive ability. Clin Epigenet (2020) 12:113. doi: 10.1186/s13148-020-00903-8
238. Tserel L, Kolde R, Limbach M, Tretyakov K, Kasela S, Kisand K, et al. Age-related profiling of DNA methylation in CD8+ T cells reveals changes in immune response and transcriptional regulator genes. Sci Rep (2015) 5:1–11. doi: 10.1038/srep13107
239. Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res (2010) 20:434–9. doi: 10.1101/gr.103101.109
240. Jasiulionis MG. Abnormal epigenetic regulation of immune system during aging. Front Immunol (2018) 9:197. doi: 10.3389/fimmu.2018.00197
241. Messina F, Cecconi F, Rodolfo C. Do You Remember Mitochondria? Front Physiol (2020) 11:271. doi: 10.3389/fphys.2020.00271
242. Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial Turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal (2010) 12:503–35. doi: 10.1089/ars.2009.2598
243. Gowers IR, Walters K, Kiss-Toth E, Read RC, Duff GW, Wilson AG. Age-related loss of CpG methylation in the tumour necrosis factor promoter. Cytokine (2011) 56:792–7. doi: 10.1016/j.cyto.2011.09.009
244. Bradburn S, McPhee J, Bagley L, Carroll M, Slevin M, Al-Shanti N, et al. Dysregulation of C-X-C motif ligand 10 during aging and association with cognitive performance. Neurobiol Aging (2018) 63:54–64. doi: 10.1016/j.neurobiolaging.2017.11.009
245. Mejia-Ramirez E, Florian MC. Understanding intrinsic hematopoietic stem cell aging. Haematologica (2020) 105:22–37. doi: 10.3324/haematol.2018.211342
246. Norum KR. World Health Organization’s Global Strategy on diet, physical activity and health: The process behind the scenes. Scand J Nutr (2005) 49:83–8. doi: 10.1080/11026480510037147
247. Cabral-Santos C, de Lima Junior EA, Fernandes IM da C, Pinto RZ, Rosa-Neto JC, Bishop NC, et al. Interleukin-10 responses from acute exercise in healthy subjects: A systematic review. J Cell Physiol (2019) 234:9956–65. doi: 10.1002/jcp.27920
248. Lancaster GI, Khan Q, Drysdale P, Wallace F, Jeukendrup AE, Drayson MT, et al. The physiological regulation of toll-like receptor expression and function in humans. J Physiol (2005) 563:945–55. doi: 10.1113/jphysiol.2004.081224
249. Oliveira M, Gleeson M. The influence of prolonged cycling on monocyte Toll-like receptor 2 and 4 expression in healthy men. Eur J Appl Physiol (2010) 109:251–7. doi: 10.1007/s00421-009-1350-9
250. Kawanishi N, Yano H, Yokogawa Y, Suzuki K. Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in high-fat-diet-induced obese mice. Exerc Immunol Rev (2010) 16:105–18.
251. Gleeson M, Bishop NC, Stensel DJ, Lindley MR, Mastana SS, Nimmo MA. The anti-inflammatory effects of exercise: Mechanisms and implications for the prevention and treatment of disease. Nat Rev Immunol (2011) 11:607–10. doi: 10.1038/nri3041
252. Zanchi NE, Lira FS, De Siqueira Filho MA, Rosa JC, De Oliveira Carvalho CR, Seelaender M, et al. Chronic low frequency/low volume resistance training reduces pro-inflammatory cytokine protein levels and TLR4 mRNA in rat skeletal muscle. Eur J Appl Physiol (2010) 109:1095–102. doi: 10.1007/s00421-010-1456-0
253. Ma Y, He M, Qiang L. Exercise therapy downregulates the overexpression of TLR4, TLR2, MyD88 and NF-κB after cerebral ischemia in rats. Int J Mol Sci (2013) 14:3718–33. doi: 10.3390/ijms14023718
254. Kang EB, Koo JH, Jang YC, Yang CH, Lee Y, Cosio-Lima LM, et al. Neuroprotective Effects of Endurance Exercise Against High-Fat Diet-Induced Hippocampal Neuroinflammation. J Neuroendocrinol (2016) 28:1–10. doi: 10.1111/jne.12385
255. Van Der Vaart H, Postma DS, Timens W, Ten Hacken NHT. Acute effects of cigarette smoke on inflammation and oxidative stress: A review. Thorax (2004) 59:713–21. doi: 10.1136/thx.2003.012468
256. Krüger K, Seimetz M, Ringseis R, Wilhelm J, Pichl A, Couturier A, et al. Exercise training reverses inflammation and muscle wasting after tobacco smoke exposure. Am J Physiol - Regul Integr Comp Physiol (2018) 314:R366–76. doi: 10.1152/ajpregu.00316.2017
257. Lambert CP, Wright NR, Finck BN, Villareal DT. Exercise but not diet-induced weight loss decreases skeletal muscle inflammatory gene expression in frail obese elderly persons. J Appl Physiol (2008) 105:473–8. doi: 10.1152/japplphysiol.00006.2008
258. Rodriguez-Miguelez P, Fernandez-Gonzalo R, Almar M, Mejías Y, Rivas A, de Paz JA, et al. Role of Toll-like receptor 2 and 4 signaling pathways on the inflammatory response to resistance training in elderly subjects. Age (Omaha) (2014) 36:9734. doi: 10.1007/s11357-014-9734-0
259. Deckx N, Wens I, Nuyts AH, Hens N, De Winter BY, Koppen G, et al. 12 Weeks of Combined Endurance and Resistance Training Reduces Innate Markers of Inflammation in a Randomized Controlled Clinical Trial in Patients With Multiple Sclerosis. Mediators Inflammation (2016) 2016:1–13. doi: 10.1155/2016/6789276
260. Cristi-Montero C, Sánchez-Collado P, Veneroso C, Cuevas MJ, González-Gallego J. Efecto del ejercicio agudo sobre la expresión del receptor tipo Toll-4 y los mecanismos inflamatorios en corazón de rata. Rev Med Chil (2012) 140:1282–8. doi: 10.4067/S0034-98872012001000007
261. Balan M, Locke M. Acute exercise activates myocardial nuclear factor kappa B. Cell Stress Chaperones (2011) 16:105–11. doi: 10.1007/s12192-010-0217-7
262. Rosa JC, Lira FS, Eguchi R, Pimentel GD, Venâncio DP, Cunha CA, et al. Exhaustive exercise increases inflammatory response via toll like receptor-4 and NF-κBp65 pathway in rat adipose tissue. J Cell Physiol (2011) 226:1604–7. doi: 10.1002/jcp.22490
263. Barcelos RP, Bresciani G, Rodriguez-Miguelez P, Cuevas MJ, Soares FAA, Barbosa NV, et al. Diclofenac pretreatment effects on the toll-like receptor 4/nuclear factor kappa B-mediated inflammatory response to eccentric exercise in rat liver. Life Sci (2016) 148:247–53. doi: 10.1016/j.lfs.2016.02.006
264. Barcelos RP, Bresciani G, Cuevas MJ, Martínez-Flórez S, Soares FAA, González-Gallego J. Diclofenac pretreatment modulates exercise-induced inflammation in skeletal muscle of rats through the TLR4/NF-κB pathway. Appl Physiol Nutr Metab (2017) 42:757–64. doi: 10.1139/apnm-2016-0593
265. Bruunsgaard H, Galbo H, Halkjaer-Kristensen J, Johansen TL, MacLean DA, Pedersen BK. Exercise-induced increase in serum inferleukin-6 in humans is related to muscle damage. J Physiol (1997) 499:833–41. doi: 10.1113/jphysiol.1997.sp021972
266. Chiang J, Shen YC, Wang YH, Hou YC, Chen CC, Liao JF, et al. Honokiol protects rats against eccentric exercise-induced skeletal muscle damage by inhibiting NF-κB induced oxidative stress and inflammation. Eur J Pharmacol (2009) 610:119–27. doi: 10.1016/j.ejphar.2009.03.035
267. Neubauer O, Sabapathy S, Lazarus R, Jowett JBM, Desbrow B, Peake JM, et al. Transcriptome analysis of neutrophils after endurance exercise reveals novel signaling mechanisms in the immune response to physiological stress. J Appl Physiol (2013) 114:1677–88. doi: 10.1152/japplphysiol.00143.2013
268. Horsburgh S, Robson-Ansley P, Adams R, Smith C. Exercise and inflammation-related epigenetic modifications: Focus on DNA methylation. Exerc Immunol Rev (2015) 21:26–41.
269. Hunter DJ, James L, Hussey B, Wadley AJ, Lindley MR, Mastana SS. Impact of aerobic exercise and fatty acid supplementation on global and gene-specific DNA methylation. Epigenetics (2019) 14:294–309. doi: 10.1080/15592294.2019.1582276
270. Ferioli M, Zauli G, Maiorano P, Milani D, Mirandola P, Neri LM. Role of physical exercise in the regulation of epigenetic mechanisms in inflammation, cancer, neurodegenerative diseases, and aging process. J Cell Physiol (2019) 234:14852–64. doi: 10.1002/jcp.28304
271. Ntanasis-Stathopoulos J, Tzanninis JG, Philippou A, Koutsilieris M. Epigenetic regulation on gene expression induced by physical exercise. J Musculoskelet Neuronal Interact (2013) 13:133–46.
272. Taniguchi S, Sagara J. Regulatory molecules involved in inflammasome formation with special reference to a key mediator protein, ASC. Semin Immunopathol (2007) 29:231–8. doi: 10.1007/s00281-007-0082-3
273. Nakajima K, Takeoka M, Mori M, Hashimoto S, Sakurai A, Nose H, et al. Exercise effects on methylation of ASC gene. Int J Sports Med (2010) 31:671–5. doi: 10.1055/s-0029-1246140
274. Zhang Y, Hashimoto S, Fujii C, Hida S, Ito K, Matsumura T, et al. NFκB2 Gene as a Novel Candidate that Epigenetically Responds to Interval Walking Training. Int J Sports Med (2015) 36:769–75. doi: 10.1055/s-0035-1547221
275. Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev (2007) 128:92–105. doi: 10.1016/j.mad.2006.11.016
276. Von Bernhardi R, Cornejo F, Parada GE, Eugenín J. Role of TGF β signaling in the pathogenesis of Alzheimer’s disease. Front Cell Neurosci (2015) 9:1–21. doi: 10.3389/fncel.2015.00426
277. Cornejo F, Von Bernhardi R. Age-Dependent Changes in the Activation and Regulation of Microglia. Adv Exp Med Biol (2016) 949:205–26. doi: 10.1007/978-3-319-40764-7
278. Butcher SK, Lord JM. Stress responses and innate immunity: Aging as a contributory factor. Aging Cell (2004) 3:151–60. doi: 10.1111/j.1474-9728.2004.00103.x
279. Cannizzo ES, Clement CC, Sahu R, Follo C, Santambrogio L. Oxidative stress, inflamm-aging and immunosenescence. J Proteomics (2011) 74:2313–23. doi: 10.1016/j.jprot.2011.06.005
280. Olivieri F, Albertini MC, Orciani M, Ceka A, Cricca M, Procopio AD, et al. DNA damage response (DDR) and senescence: shuttled inflamma-miRNAs on the stage of inflamm-aging. Oncotarget (2015) 6:35509–21. doi: 10.18632/oncotarget.5899
281. Jones DL, Rando TA. Emerging models and paradigms for stem cell ageing. Nat Publ Gr (2011) 13:506–12. doi: 10.1038/ncb0511-506
282. Dupuis TFG, Le SBA, Bourgade PK, Witkowski EFJM, Larbi GPA. From inflamm-aging to immune-paralysis: a slippery slope during aging for immune-adaptation. Biogerontology (2015) 17:147–57. doi: 10.1007/s10522-015-9615-7
283. Calabrese V, Santoro A, Monti D, Crupi R, Di Paola R, Latteri S, et al. Aging and Parkinson’s Disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic Biol Med (2018) 115:80–91. doi: 10.1016/j.freeradbiomed.2017.10.379
Keywords: pollution, oxidative stress, nutrition, immune system, gender, exercise, epigenetic changes, drug abuse
Citation: Bachmann MC, Bellalta S, Basoalto R, Gómez-Valenzuela F, Jalil Y, Lépez M, Matamoros A and von Bernhardi R (2020) The Challenge by Multiple Environmental and Biological Factors Induce Inflammation in Aging: Their Role in the Promotion of Chronic Disease. Front. Immunol. 11:570083. doi: 10.3389/fimmu.2020.570083
Received: 06 June 2020; Accepted: 24 September 2020;
Published: 14 October 2020.
Edited by:
Fulvio D’Acquisto, University of Roehampton London, United KingdomReviewed by:
Christine Noel Metz, Feinstein Institute for Medical Research, United StatesLuciele Guerra Minuzzi, State University of Campinas, Brazil
Copyright © 2020 Bachmann, Bellalta, Basoalto, Gómez-Valenzuela, Jalil, Lépez, Matamoros and von Bernhardi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rommy von Bernhardi, cnZvbmJAbWVkLnB1Yy5jbA==
†These authors have contributed equally to this work