 Emily F. Cornish
Emily F. Cornish Iva Filipovic
Iva Filipovic Fredrika Åsenius
Fredrika Åsenius David J. Williams
David J. Williams Thomas McDonnell3*
Thomas McDonnell3*- 1Elizabeth Garrett Anderson Institute for Women's Health, University College London, London, United Kingdom
- 2Center for Infectious Medicine, Department of Medicine Huddinge, Karolinska Institute, Stockholm, Sweden
- 3Department of Biochemical Engineering, University College London, London, United Kingdom
Immunological adaptations in pregnancy allow maternal tolerance of the semi-allogeneic fetus but also increase maternal susceptibility to infection. At implantation, the endometrial stroma, glands, arteries and immune cells undergo anatomical and functional transformation to create the decidua, the specialized secretory endometrium of pregnancy. The maternal decidua and the invading fetal trophoblast constitute a dynamic junction that facilitates a complex immunological dialogue between the two. The decidual and peripheral immune systems together assume a pivotal role in regulating the critical balance between tolerance and defense against infection. Throughout pregnancy, this equilibrium is repeatedly subjected to microbial challenge. Acute viral infection in pregnancy is associated with a wide spectrum of adverse consequences for both mother and fetus. Vertical transmission from mother to fetus can cause developmental anomalies, growth restriction, preterm birth and stillbirth, while the mother is predisposed to heightened morbidity and maternal death. A rapid, effective response to invasive pathogens is therefore essential in order to avoid overwhelming maternal infection and consequent fetal compromise. This sentinel response is mediated by the innate immune system: a heritable, highly evolutionarily conserved system comprising physical barriers, antimicrobial peptides (AMP) and a variety of immune cells—principally neutrophils, macrophages, dendritic cells, and natural killer cells—which express pattern-receptors that detect invariant molecular signatures unique to pathogenic micro-organisms. Recognition of these signatures during acute infection triggers signaling cascades that enhance antimicrobial properties such as phagocytosis, secretion of pro-inflammatory cytokines and activation of the complement system. As well as coordinating the initial immune response, macrophages and dendritic cells present microbial antigens to lymphocytes, initiating and influencing the development of specific, long-lasting adaptive immunity. Despite extensive progress in unraveling the immunological adaptations of pregnancy, pregnant women remain particularly susceptible to certain acute viral infections and continue to experience mortality rates equivalent to those observed in pandemics several decades ago. Here, we focus specifically on the pregnancy-induced vulnerabilities in innate immunity that contribute to the disproportionately high maternal mortality observed in the following acute viral infections: Lassa fever, Ebola virus disease (EVD), dengue fever, hepatitis E, influenza, and novel coronavirus infections.
Introduction
Pregnancy creates a unique immunological paradox: the maternal immune system must undergo complex adaptations to permit tolerance of the semi-allogeneic fetus while simultaneously maintaining robust defenses against invasive pathogens. Initial theories of maternal-fetal tolerance proposed that a temporary state of maternal immunosuppression was vital to allow successful implantation and development of a pregnancy (1–3). With the advent of technologies including microscopy, advanced cytometry and single-cell sequencing, these models have been superseded by new data that suggest a tightly regulated balance between inflammatory and tolerogenic states during the “immune chronology” of normal pregnancy (4–8). Longitudinal studies of peripheral, decidual, and amniotic fluid cytokine profiles and immune cell subsets demonstrate that a pro-inflammatory environment predominates during early trophoblast invasion and at parturition, while the second and third trimesters require an anti-inflammatory bias to facilitate fetal growth (5, 8–11). The balance between innate and adaptive immunity shifts in favor of innate mechanisms, particularly in the first trimester; as pregnancy progresses, silencing of chemokine genes inhibits accumulation of effector T cells in the decidua, peripheral B cells are depleted and pregnancy-specific hormones skew B cell polarization toward a tolerogenic IL-10-producing phenotype (12–14).
These changes are orchestrated by the sentinel innate immune cells of the maternal decidua—neutrophils, macrophages, dendritic cells, and natural killer cells—and their molecular interactions with invading fetal trophoblast at the maternal-fetal interface. Activation of decidual innate immunity is crucial in the establishment of a pregnancy-specific immune environment: it recruits additional populations of leukocytes to the decidua, educates adaptive cells to refine appropriate effector and memory responses, and modulates the phenotype and functions of peripheral immune cells (15–18). Dysregulation of this complex bi-directional relationship has been implicated in several obstetric and perinatal complications, including recurrent miscarriage, pre-eclampsia, fetal growth restriction, chorioamnionitis, and preterm birth (19–26).
The corollary of this capacity for immunological tolerance is an increased susceptibility to infection (27–29). The innate cells that mediate maternal-fetal crosstalk and induction of tolerance also constitute the frontier of defense against infection through a wide repertoire of effector mechanisms (30–32). Chief amongst these are the expression of pattern-recognition receptors that detect pathogen-specific molecular signatures and soluble mediators such as the complement system. The World Health Organization estimates that sepsis accounts for 10.7% of maternal deaths globally (33) and there is evidence of a particular vulnerability to acute viral infection. For example, pregnant women suffered disproportionately high mortality rates in the influenza pandemics of 1918, 1957, and 2009 (34–36), and hepatitis E, typically a mild and self-limiting illness, has a 26% case-fatality rate in pregnant women (28). These disparities are likely to arise from a combination of immunological, hormonal, and physiological adaptations that are specific to pregnancy (37–39).
Viral infection in pregnancy carries four distinct risks:
1. Adverse pregnancy outcomes: acute viral infection is consistently associated with a broad spectrum of obstetric complications [reviewed in (40–42)]
2. Acute severe maternal disease with consequent morbidity and/or mortality
3. Vertical transmission to the fetus (during pregnancy), resulting in congenital infection that can cause intrauterine death or permanent disability
4. Perinatal transmission to the fetus (during delivery), which can cause severe neonatal disease.
Although congenital infection is a major public health concern, this article will not cover viruses that cause fetal damage through vertical or horizontal transmission as these have been recently and comprehensively reviewed (42, 43). Instead, we focus on the following six viruses listed in Table 1 that cause acute severe maternal disease, reviewing the innate immune mechanisms and viral evasion strategies that contribute to their effects in pregnancy.
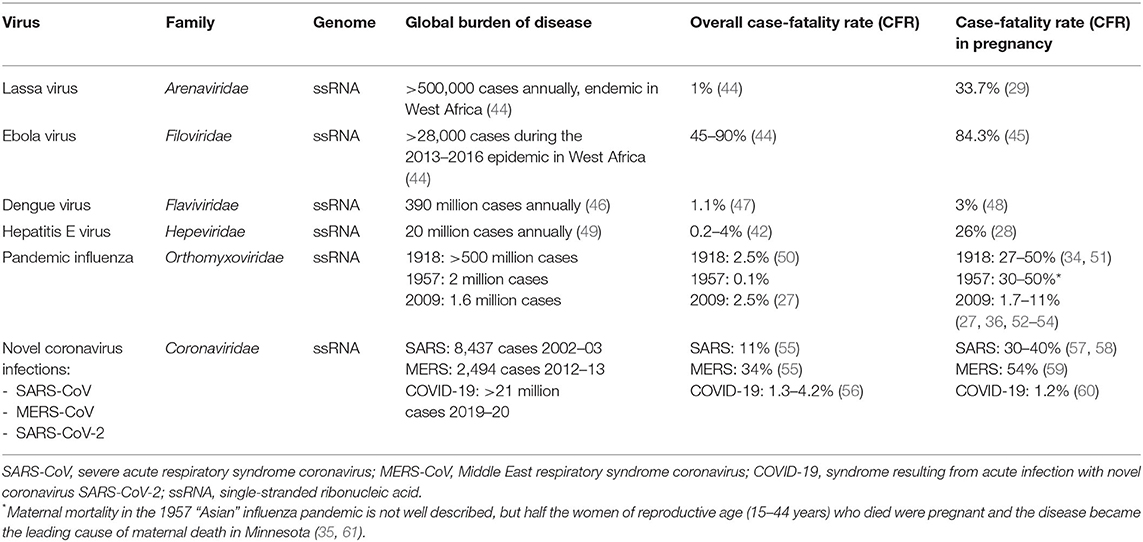
Table 1. RNA viruses that cause severe disease in pregnancy.
Decidual Innate Immunity in Pregnancy: At the Frontier of Maternal-Fetal Tolerance and Infection
Pregnancy is a unique immunological state. Alterations in systemic maternal immunity and cellular dialogue at the maternal-fetal interface combine to maintain tolerance of the fetal allograft while simultaneously preserving the ability to respond to infection. Shortly after conception, rising progesterone levels trigger decidualization, the transformation of the endometrium into a specialized tissue that promotes implantation of the blastocyst (62). Intensive study of the cellular composition of the decidua over recent years has identified diverse immune cell populations, including natural killer (NK) cells, macrophages, dendritic cells (DC) and T cells (63–65) (Figure 1). Interactions between these decidual immune cells and the invading fetal extravillous trophoblast exert a critical influence on subsequent placentation, fetal growth and pregnancy outcome (23, 25, 67–69).
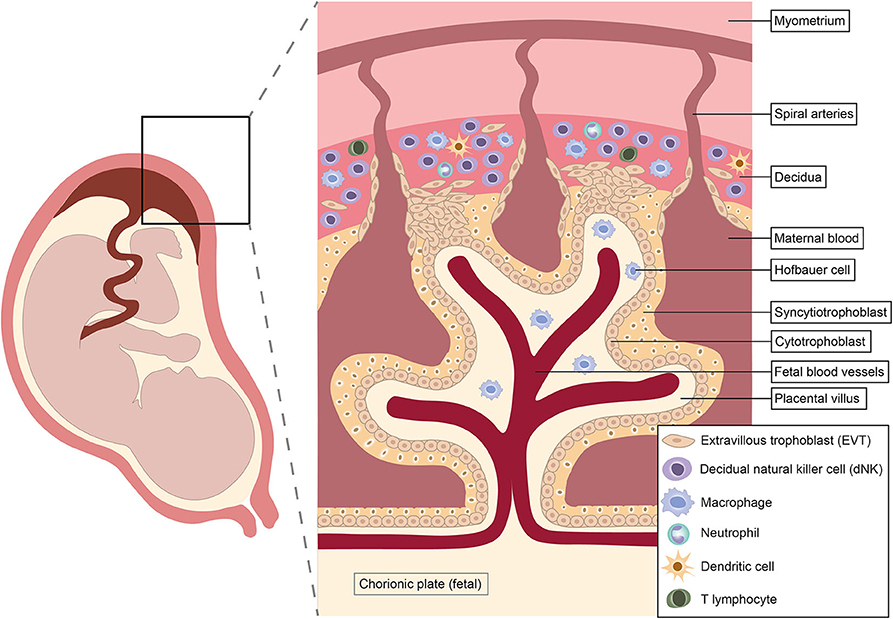
Figure 1. Architecture and immunological repertoire of the maternal-fetal interface. Immune cells shown reflect approximate abundances of different populations in the first and second trimesters of pregnancy. Natural killer cells predominate in early pregnancy, but disappear from mid-gestation onwards (62, 66).
The unique non-classical human leukocyte antigen (HLA) class I molecule HLA-G is exclusively expressed on extravillous trophoblast (EVT) (70, 71). Invading EVT comes into direct contact with maternal cells when it infiltrates through the decidua into the myometrium, remodeling maternal spiral arteries into dilated low-resistance channels that maximize blood flow to the developing feto-placental unit (42).
The EVT is capable of both immune evasion and induction of tolerance due to its unique HLA expression profile, consisting of only the class I molecules HLA-C, HLA-E, and HLA-G. Trophoblast HLA-G undergoes high-affinity binding with leukocyte immunoglobulin-like receptor B1 (LILRB1), an inhibitory receptor widely expressed on decidual antigen-presenting cells. This interaction modulates decidual DC signaling, suppresses production of pro-inflammatory cytokines and inhibits proliferation of maternal T cells. HLA-G therefore provides a critical tolerogenic signal at the maternal-fetal interface (72–74).
The role of decidual innate immune cells in defense against infection is an emerging and rapidly evolving field. Current knowledge is summarized in a recent review by Yockey et al. (75) and relevant aspects of the decidual innate immune response to viral infection will be discussed in detail below. Despite significant advances, our understanding of the recalibration of the innate-adaptive equilibrium in pregnancy remains incomplete. This is due to the practical and ethical difficulties of obtaining decidual tissue samples and the uncertain correlation between peripheral and decidual immune cell activity (6, 25, 76). Renewed focus on the role of the maternal innate immune system in the apparent conflict between fetal tolerance and robust defense against intracellular pathogens will provide further insights into how these two priorities interact in pregnancy. For the purposes of this review, the term “tolerance” refers to the pregnancy-specific state of tolerance of the semi-allogeneic fetus, rather than to the specific T cell phenomenon.
Systemic Innate Immunity in Pregnancy
Systemically, there is a global upregulation of innate immune cells and effector mechanisms in normal pregnancy (6, 77). Complement activity increases compared to the non-pregnant state (78, 79) and there is a substantial rise in circulating phagocytes and type I interferon (IFN)-producing plasmacytoid DC with advancing gestational age (9, 80). Longitudinal studies of serial blood samples from pregnant women show specific enhancement of innate pathways that mediate antiviral immunity: for example, IFN-α-induced STAT1 signaling, a critical response to viral challenge, increases throughout gestation in NK cells, monocytes and myeloid DC (6, 81). This state must be finely calibrated: excessive activation can be associated with tissue damage during response to acute viral infection (9, 77, 82, 83) and adverse obstetric outcomes [such as complement overactivity in antiphospholipid syndrome (84, 85)], while an attenuated immune response could predispose to overwhelming infection. Whether maternal susceptibility to RNA viral infections is due to over- or under-activity of the innate immune system is not yet clear; it is likely that some effector mechanisms are upregulated while others are suppressed. Data specific to individual viruses will be discussed below.
Of all the viruses discussed in this review, influenza has received the most scientific attention with regards to its propensity for pregnancy, but precise mechanisms underlying this vulnerability remain uncertain. While some animal models (86, 87) and a few ex vivo human studies (82, 83) have determined specific innate immune correlates of the increased severity of influenza in pregnancy, much less is known about the other viruses included in this article. This review will synthesize available knowledge on the nature, magnitude, and timing of innate immune responses to viral infection in pregnancy and how these interact with decidual, hormonal and physiological influences.
Molecular Mechanisms of Innate Immunity to Viruses
Human antiviral immunity is a two-step process. Non-specific innate mechanisms are activated immediately, predominate during the first 5–7 days of infection, and are then superseded by T and B cell-mediated antigen-specific adaptive responses. Rapid-onset innate responses may be sufficient to eliminate the virus, but if not, they limit replication during the critical temporal gap between onset of viral challenge and development of adaptive virus-specific cytotoxic lymphocytes, reducing the likelihood of disseminated disease (88).
The innate antiviral response is activated when pattern-recognition receptors (PRRs) detect pathogen-associated molecular patterns (PAMPs), particularly viral nucleic acids. Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs), which are expressed on innate cell membranes and throughout the intracellular compartment, are the primary viral sentinels (89). This PRR-PAMP interaction triggers activation of latent transcription factors that upregulate a vast repertoire of antiviral effector proteins: type I interferons, other pro-inflammatory cytokines such as TNF and IL-1β, chemokines and the antimicrobial peptides (e.g., defensins, cathelicidins, surfactant proteins) (88, 90, 91).
The Interferon Response
Interferons, which are classified according to their cell surface receptors, are grouped into three types. Type I (including IFN-α and IFN-β) and type II (IFN-γ) are produced by virtually all cells, while the more recently discovered type III (IFN-λ) is produced by epithelial and dendritic cells (92). The production of type I IFNs is the hallmark of effective antiviral immunity. Once secreted by virally infected cells, they act in a paracrine manner to induce IFN-stimulated gene (ISG) expression in neighboring cells, creating an antiviral state in the surrounding environment (shown in Figure 2). IFN binding to the type I IFN receptor, IFNAR, leads to receptor endocytosis and activation of the receptor-associated tyrosine kinases Janus kinase I (JAK-1) and tyrosine kinase 2 (TYK2). These in turn activate transcription factors STAT1 and STAT2, which associate with interferon regulatory factor 9 (IRF9). IRF9 is translocated to the nucleus and triggers IFN-stimulated response elements (IRSEs) to upregulate expression of ISG, the definitive effectors of the antiviral response (91–93). Type III IFN can also induce ISG but their effects appear to be limited to sites of epithelial damage at anatomical barriers, exerting a localized antiviral response that is superseded by potent systemic type I IFNs if the infection is not successfully contained (94).
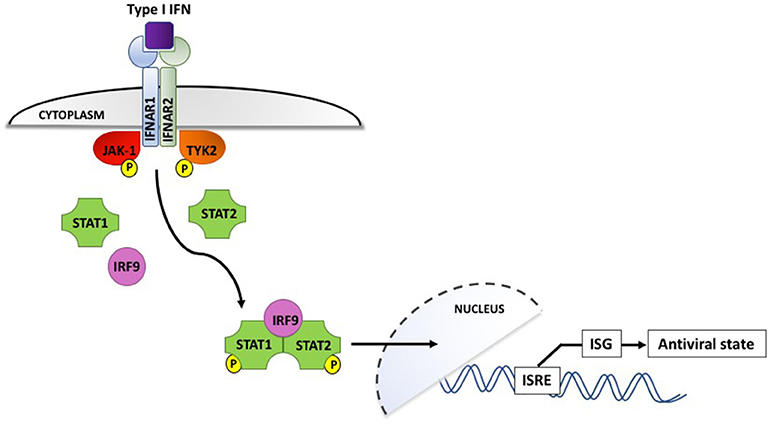
Figure 2. Type I interferon signaling via the IFNAR receptor induces phosphorylation and activation of the JAK-1 and TYK2 tyrosine kinases, which interact with IRF9 to upregulate interferon-stimulated genes and induce an antiviral state in the surrounding cellular microenvironment. IFN, interferon; IFNAR, interferon α/β receptor; IRF9, interferon regulatory factor 9; ISG, interferon-stimulated genes; ISRE, interferon-stimulated response elements; JAK-1, Janus-associated kinase I; STAT1/2, signal transducer and activator of transcription 1/2; TYK2, tyrosine kinase 2. P denotes phosphorylation.
Cellular Interactions With RNA Viruses
RNA viruses, the focus of this review, are detected by a variety of PRRs. Plasmacytoid dendritic cells (pDC) detect viruses via endosomal TLR7-9 and are responsible for the first wave of type I IFN production, releasing large quantities through the MyD88-IRF7 pathway [reviewed in (95)]. In contrast, macrophages and conventional dendritic cells (cDC) sense viral challenge through RLRs, specifically the cytoplasmic helicases RIG-I and MDA-5. These receptors signal via mitochondrial antiviral-signaling protein (MAVS) and IRF3/7, culminating in a second wave of type I IFN production (91, 95) (Figure 3). The emerging role of NK cells in antiviral immunity and pregnancy, recently reviewed in this journal (66), has provoked controversy and is discussed in more detail below.
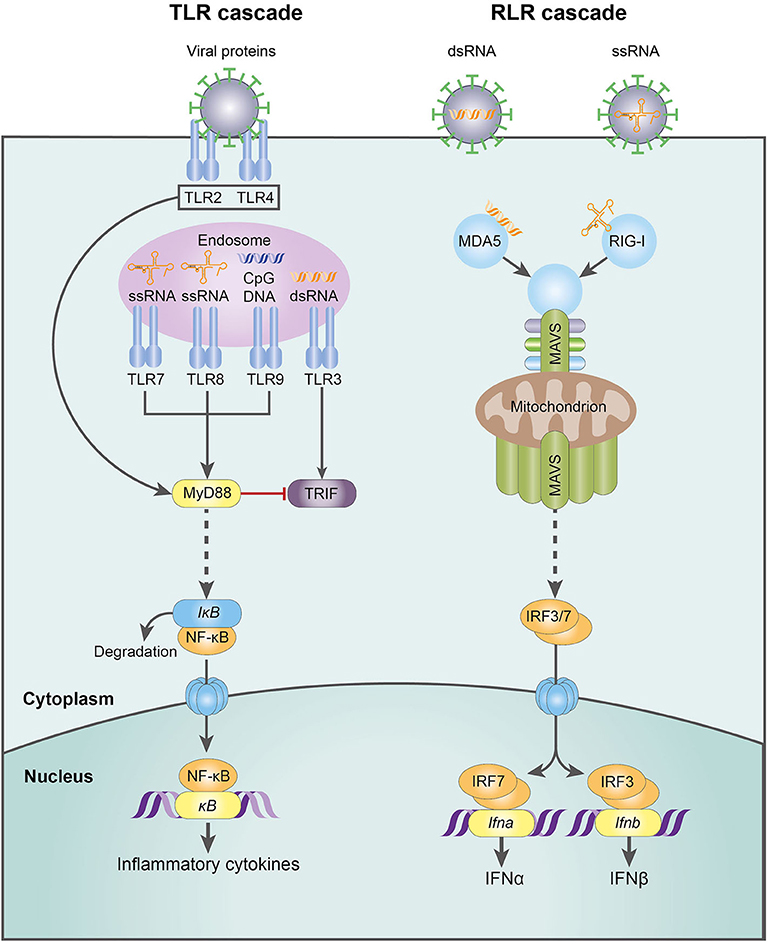
Figure 3. The molecular mechanisms of the TLR- and RLR-mediated innate response to RNA virus infection. CpG DNA, cytosine-guanine oligodeoxynucleotides; dsDNA, double-stranded deoxyribonucleic acid; IFN-α, interferon-α; IFN-β, interferon-β; IkB, inhibitor of NFkB; IRF3, interferon regulatory factor 3; IRF7, interferon regulatory factor 7; MAVS, mitochondrial antiviral-signaling protein; MDA5, melanoma differentiation-associated protein 5; MyD88, myeloid differentiation primary response 88; NFkB, nuclear factor kappa-light-chain-enhancer of activated B cells; RIG-I, retinoic acid-inducible gene I; RLR, RIG-I-like receptor; ssRNA, single-stranded ribonucleic acid; TLR, Toll-like receptor; TRIF, TLR-domain-containing adapter-inducting interferon-β.
Viral Interactions With Programmed Cell Death Pathways
Since viruses depend on host cells for replication, programmed cell death pathways—including apoptosis, necroptosis, and pyroptosis—are a crucial component of antiviral defense. In apoptosis, infected cells undergo an orderly caspase-mediated degradation and are rapidly cleared by surrounding phagocytes (96). This highly regulated disassembly minimizes the release of damage-associated molecular patterns (DAMPs) that could trigger harmful auto-inflammatory responses, but also fails to induce robust antiviral immunity (97).
Necroptosis and pyroptosis differ from apoptosis in that they are powerfully immunogenic. They induce lytic cell death, triggering release of DAMPs and pro-inflammatory cytokines, as detailed in a recent review (98). Although these mechanisms have been implicated in a wide range of autoimmune disorders [reviewed in (99)], they are also a key component of antiviral immunity.
Necroptosis is a caspase-independent process that can be triggered through several different mechanisms, including TNF signaling and TLR detection of viral molecular signatures. These pathways converge on receptor-interacting protein kinase-3 (RIPK3), which phosphorylates and activates the pseudokinase mixed lineage kinase domain-like protein (MLKL). Activated MLKL undergoes conformational changes that expose its pore-forming 4-helical bundle domain, leading to rapid cell lysis (97, 100).
Pyroptosis, a swift and powerfully pro-inflammatory form of programmed cell death, results from activation of the cytosolic NLRP3 inflammasome in virally infected cells. Its caspase-1 effector domain cleaves and activates gasdermin-D, triggering lethal pore formation in the host cell membrane and efflux of pro-inflammatory cytokines (101, 102).
Complement Antiviral Responses
The complement system is another crucial component of antiviral defense. It bridges the innate-adaptive divide through its diverse roles: opsonization and lytic destruction of pathogens, clearance of apoptotic cells and immune complexes, phagocyte chemotaxis and mast cell activation. A highly conserved cascade of over 50 circulating and membrane-bound protein components, it can be activated through three separate pathways: classical, alternative, and lectin. All converge on the formation of C3 convertases, which cleave C3 into active fragments C3a and C3b. Deposition of C3b on cell or pathogen surfaces triggers formation of the C5 convertase, which splits C5 into C5a and C5b, catalyzing the formation of the membrane attack complex (MAC) that penetrates viral membranes to induce lytic destruction (26, 103–105).
Regulation of the Antiviral Response
The pro-inflammatory antiviral response is tightly calibrated: inadequate responses lead to overwhelming infection, while excessive activity causes host tissue damage and is associated with autoimmune disease. A variety of inbuilt negative feedback post-translational modification systems orchestrate spatial and temporal regulation of the antiviral response. For example, ubiquitin-specific peptidase 18 (USP18, also known as ISG43) displaces JAK1 from IFNAR to block type I IFN signaling and also deconjugates ISG15 (one of the most potent inhibitors of viral replication) from its target proteins (91, 106–108).
Such mechanisms are not always sufficient to prevent dysregulation of type I IFN activation, which has been implicated in several auto-inflammatory conditions including systemic lupus erythematosus (91, 109), a disease which shows excessive complement deposition. Similarly, excessive complement activation contributes to acute lung injury in mouse and human studies of influenza and novel coronaviruses, through over-production of the anaphylatoxins C3a and C5a (110–112).
The following sections examine the individual cellular components and effector mechanisms of the innate antiviral response that undergo significant adaptations in pregnancy.
Pregnancy-Induced Modification of Antiviral Immunity
Antimicrobial Peptides
Antimicrobial peptides (AMP) are small molecules that create a microbicidal shield at mucosal surfaces with high pathogen exposure, such as the small intestine, the renal epithelium, and the chorionic membranes. They are secreted by leukocytes and disrupt pathogen membrane integrity, leading to lytic destruction. Three classes of AMP that contribute to antiviral immunity exhibit differential expression in pregnancy: defensins, cathelicidins, and surfactant proteins.
Human defensins are categorized into three families (α, β, and θ). Alpha-defensins are found in neutrophil granules and Paneth cells while beta-defensins are expressed in epithelial cells, including those of the female reproductive tract, and are upregulated by cytokine secretion in response to pathogen-mediated epithelial injury (113, 114).
During pregnancy, the AMPs human-beta-defensin-1 (HBD-1) and HBD-3 are elevated in the amniotic fluid of women who develop preterm labor with coexistent infection (115, 116). HBD1-3 are also expressed in the trophoblast and decidua, suggesting a critical physiological role for defensins in innate immune defenses at the maternal-fetal interface; they also participate in innate-adaptive crosstalk through chemotaxis, recruiting T cells and immature DC (117).
Like defensins, cathelicidins are small AMP secreted by innate cells and mucosal barriers. The sole human cathelicidin, LL-37, is vitamin D-inducible and promotes wound healing, angiogenesis and clearance of cell debris; it can also regulate macrophage and DC responses to pro-inflammatory stimuli (118). LL-37 expression in first-trimester cervicovaginal secretions is significantly higher in women with bacterial vaginosis and incubation of endocervical epithelial cells with LL-37 in vitro induces a pro-inflammatory milieu with enhanced secretion of IL-8 (119). Circulating serum LL-37 is also elevated during pregnancy: LL-37 levels rose consistently in serial samples from Ugandan pregnant women, peaking in the third trimester (120). These studies suggest a role for LL-37 in decidual and systemic pregnancy-specific innate immune defenses.
AMP have been implicated in the innate response to influenza A virus (IAV). LL-37 neutralizes IAV in mouse models by directly damaging viral membranes, whereas surfactant protein-D (SP-D) works by triggering viral aggregation and inhibiting haemagglutinin activity (121–124). In human monocytes, LL-37 and SP-D readily block replication of seasonal IAV, but both had strikingly impaired inhibitory activity against the pandemic H1N1 strain, whereas antiviral activity of a related AMP, H-ficolin, was unchanged (125, 126). The significance of this strain-specific discrepancy in AMP-mediated anti-influenza immunity in pregnancy requires further clarification.
Toll-Like Receptors
The placenta expresses the full repertoire of human TLRs (TLR1-10) (127). Of these, TLR3, TLR7, TLR8, and TLR9 contribute to antiviral immunity: TLR3 binds to double-stranded viral RNA; TLR7 and 8 detect single-stranded RNA viruses; and TLR9, which recognizes unmethylated cytosine-guanine (CpG) motifs in bacterial genomes, can also respond to herpesvirus infection. Antiviral PRR signaling mostly converges on the canonical MyD88 pathway, stimulating activation of NFκB and production of pro-inflammatory cytokines (128, 129). The exceptions are TLR3 and RIG-I, which signal through an alternative MyD88-independent pathway that uses an adapter protein, TRIF, to generate large amounts of IFN-β (130).
TLR expression at the maternal-fetal interface exhibits both temporal and tissue-specific fluctuations in expression levels and functionality (131–134). This suggests a potential contribution to the observed differences in severity of fetal and maternal viral infections in different trimesters (42, 44, 48, 66, 135). In human trophoblast, TLR3 is highly abundant in the first trimester and forms a defensive barrier along the cytotrophoblast with TLR2 and TLR4 (136). TLR3 has dual roles at the placental interface, inhibiting viral replication to protect the developing fetus from vertical transmission but also promoting tolerance through release of indoleamine 2,3-dioxygenase (IDO) (137–139). In the decidua, Duriez and colleagues have demonstrated differential expression of the four antiviral TLRs in decidual macrophages and NK cells, with each cell type producing a distinct cytokine signature in response to TLR7/8 ligation (140). These TLR-mediated responses to viral challenge are at the frontier of the critical balance between tolerance and immunity, which ultimately dictates whether or not a pregnancy will be successful (141).
Complement
In pregnancy, complement activity is increased systemically but suppressed at the maternal-fetal interface (26, 142). While some complement components contribute to normal placentation [C1q, for example, promotes adequate EVT invasion and remodeling of maternal spiral arteries (143, 144)], the majority must be inhibited to ensure successful pregnancy. Synergistic action of regulatory molecules at the placenta impairs formation of the classical and alternative C3 convertases, preventing downstream activation of the C5 convertase and the MAC (104). Failure to suppress these pathways is associated with a wide range of adverse obstetric outcomes, including recurrent miscarriage, fetal growth restriction, preterm birth and pre-eclampsia (26, 104, 145).
The complement system mediates neutralizing antiviral immunity through multiple effector mechanisms, including:
1. MAC-induced pore formation in viral envelopes, leading to lytic destruction;
2. MAC-independent virolysis through deposition of complement components on non-enveloped viruses or direct binding to MBL;
3. Opsonization of virions: this induces aggregation, decreasing the total infectious burden for the host, and may be followed by phagocytosis (103, 146, 147).
However, the diversity of methods for complement-mediated viral attack is mirrored and in some cases surpassed by viral evasion mechanisms, which vary widely according to species and virion structure. For example, Dengue virus non-structural protein 1 is extruded from infected cells and recruits terminal complement inhibitors C4b-binding protein and vitronectin to the cell surface, inhibiting MAC assembly (148, 149); the influenza virus matrix protein 1 inhibits C1q-mediated activation of the classical pathway (150) (Figure 4). These mechanisms have not been characterized in pregnancy and a complete discussion of complement subversion by viruses is beyond the scope of this article, but further detail is available in a recent review (103).
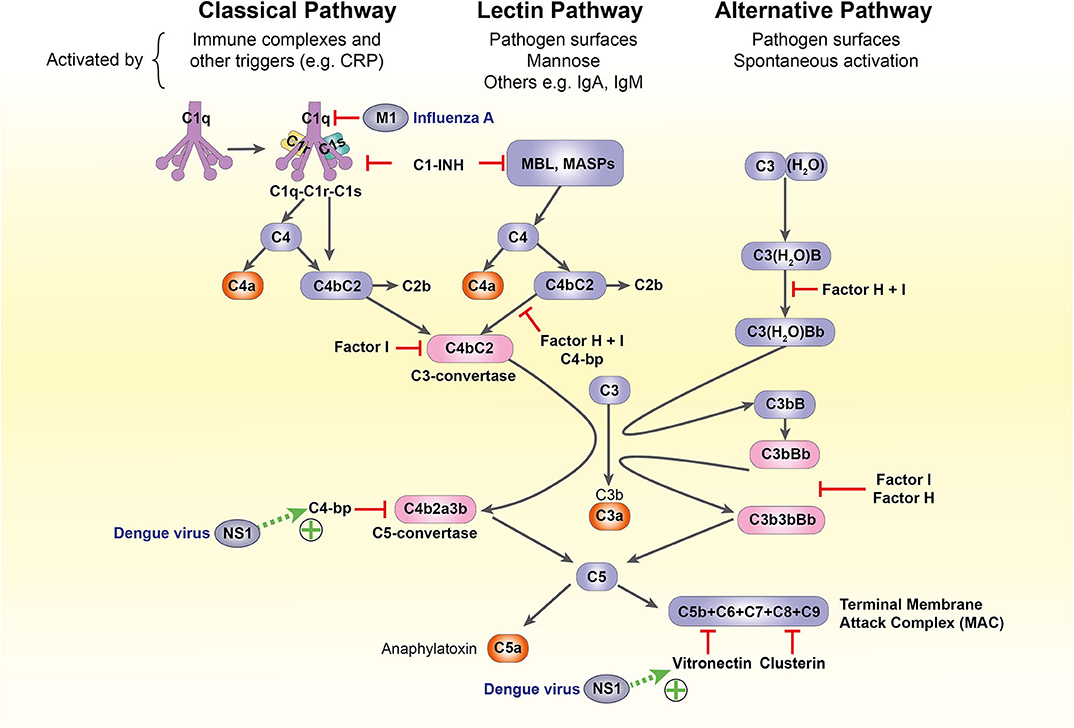
Figure 4. The complement system and its subversion by dengue and influenza A viruses. C1-INH, C1-inhibitor; C4-bp, C4-binding protein; CRP, C-reactive protein; IgA/IgM, immunoglobulin A/M; M1, influenza virus matrix protein 1; MASP, MBL-associated serine protease; MBL, mannose-binding lectin; NS1, non-structural protein 1.
Cellular Innate Immunity
Various cellular mechanisms are exploited by viral infection, a number of which are altered during pregnancy.
Innate Lymphoid Cells
Innate lymphoid cells (ILC), which account for up to 70% of all leukocytes present in first-trimester decidua (62), originate from the common lymphoid progenitor and bridge the innate-adaptive divide. Like other innate cells, they mount rapid responses to infection, lack antigen-specific receptors, and do not exhibit conventional clonal expansion; like T cells, they can modulate adaptive immune responses through production of specific cytokines and regulation of B cell and DC activity. On this basis, ILC have been described as “innate counterparts” of T cells (151). Their abundance at the maternal-fetal interface suggests pivotal roles in both innate immune defense and normal placental development (152). Table 2 summarizes the classification and functions of decidual ILC:
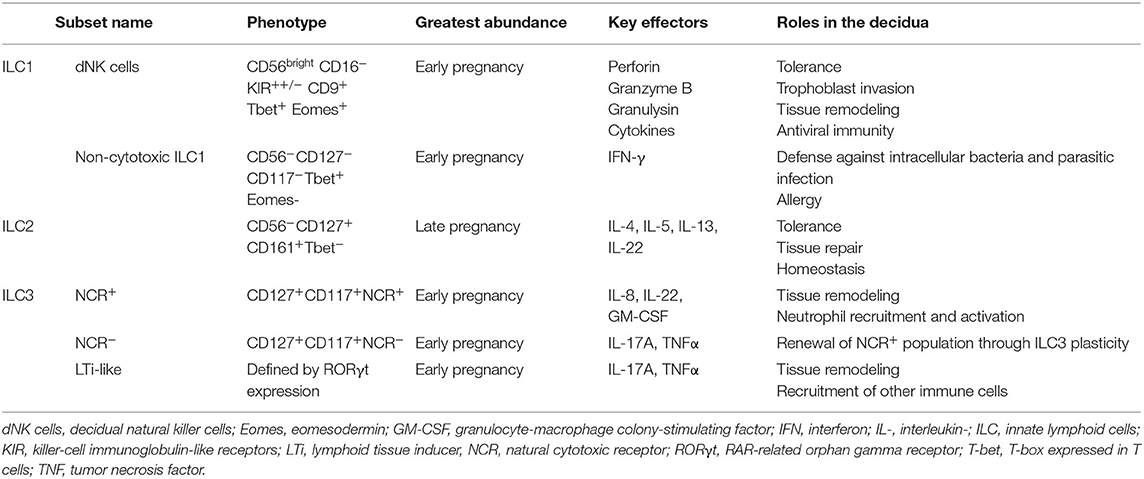
Given the recent discovery of ILC, data on their antiviral properties are still accumulating. In the liver, rapid ILC1-mediated IFN-γ production is essential for early suppression of cytomegalovirus (CMV) viremia (156); in the lungs, production of amphiregulin by ILC2 is critical for restoration of epithelial integrity and lung function following influenza infection, while excessive remodeling can predispose to allergy (157, 158). In pregnancy, investigations have focused on NK cells, both peripheral and decidual.
Peripheral Blood Natural Killer Cells
The majority (90%) of peripheral blood NK (pbNK) cells exhibit a predominantly cytotoxic CD56dim CD16+ phenotype, while the remaining 10% constitute the main cytokine-producing CD56bright CD16− subset (25). Although not antigen-specific, NK cells can utilize the Fc-receptor CD16 to target cells for destruction. Aside from CD16, NK cells integrate signals from an array of germline-encoded activating and inhibitory receptors. Inhibitory signals transmitted through interaction with the ubiquitously expressed HLA class I and the non-classical HLA-E dominate in the healthy human. This balance can be shifted in the case of diseases such as cancer, in which HLA-I and HLA-E may be downregulated, and instead ligands for activating receptors can be induced on transformed cells (e.g., NKG2D ligands) (159).
NK cell receptors can distinguish between “self” and “non-self” through detection of HLA class I and related molecules: their absence can be sensed and triggers degranulation and release of cytotoxic components. Through the same mechanism, viruses, which downregulate HLA-I expression in the cells they infect, become more susceptible to NK-mediated cytotoxicity (76). In a recent study, Le Gars et al. used mass cytometry to compare the responses of isolated pbNK cells from pregnant and non-pregnant women to ex vivo challenge with IAV. In the pregnant cohort, production of IFN-γ by both pbNK subsets was significantly upregulated, as was their capacity to kill influenza-infected monocytes (83). Although robust NK cell activity is usually important for viral clearance, this pregnancy-specific NK enhancement may actually be detrimental: IL-15-deplete mice, who cannot mount NK-mediated responses to influenza, show significantly improved survival compared to controls (160).
Decidual Natural Killer Cells
Decidual NK (dNK) cells are a unique population of CD56bright CD16− KIR++/− cells that expand in the peri-implantation window and remain highly abundant until the end of the second trimester, after which their numbers gradually diminish (66). Despite some phenotypic overlap with pbNK, dNK exhibit a unique set of surface markers and are functionally distinct: their cytotoxic capacity toward allogeneic “non-self” cells, namely the trophoblast with which they are in direct contact, is completely abrogated (62). The lack of dNK cytotoxicity was initially attributed to an attenuation of cytotoxic granule components. However, this has proved wrong: paradoxically, dNK in fact possess equivalent or higher levels of granzyme B, perforin, and granulysin than pbNK (30, 62). The issue is one of translocation: dNK fail to polarize cytotoxic granules to the immunological synapse with target non-self-cells (161). Similarly, it was presumed that these non-cytotoxic dNK would lack the ability to kill virally infected cells. This was disproved when Siewiera et al. demonstrated that dNK isolated from first-trimester decidua rapidly developed into cytotoxic effectors on exposure to CMV-infected autologous decidual fibroblasts, efficiently mobilizing cytolytic apparatus to the immunological synapse and infiltrating CMV+ trophoblastic tissue in culture (31). Importantly, this cytotoxic effect was lost when experiments were repeated with CMV-positive primary EVT, which dNK were unable to kill (162). Whether a similar pattern occurs with other viruses currently remains unproven, and observations specific to CMV should not be extrapolated to other infections.
These findings have led to renewed interest in NK-mediated antiviral immunity and the capacity of viruses to subvert molecular mechanisms of NK activity. In the case of CMV, viral infection can exert unique selective pressures on the pbNK cell compartment, influencing KIR acquisition and triggering clonal expansion of CD57+ populations expressing specific repertoires of the C-type lectin NKG2C (163, 164). However, it is important to emphasize that currently these data refer only to CMV and equivalent effects with other viruses have not been demonstrated. Similarly, expansions of this kind have not been observed among uterine NK cells and it remains uncertain whether the pbNK cell pool (where such proliferation may exist) influences the endometrial or decidual NK cell composition (164). Whether this imprint on NK cells induces long-term immunological memory against repeated viral challenge has proved controversial in humans, where peripheral and lymphoid tissue-resident NK appear to develop memory, as demonstrated in studies of CMV (165), hantavirus (166), and varicella-zoster virus (167). However, an equivalent effect in the uterus remains to be clarified. Overall, the precise role of dNK in antiviral immunity remains controversial and largely unknown.
Neutrophils
Pregnancy induces a physiological neutrophilia both systemically and at the decidua. In pregnant women, neutrophils account for up to 95% of peripheral blood leukocytes, compared to 50–70% outside pregnancy (80, 168). This reflects a state of enhanced innate immune vigilance against infection: neutrophils, the most abundant of all human phagocytes, are recruited to sites of infection or injury by chemokines such as complement C5a and form the first line of defense against microbial challenge. Engulfed micro-organisms are internalized in phagolysosomes and destroyed using a wide range of effector mechanisms including release of serine proteases (e.g., neutrophil elastase, cathepsin G) and production of reactive oxygen species (141, 169). Pathogens that evade phagocytosis can be controlled by the release of neutrophil extracellular traps (NET), large reticular structures containing cytotoxic granule proteins and AMP that sequester and neutralize microbes (170). While the role of NET in defense against bacterial and fungal infections is well-established, their contribution to antiviral immunity has been more difficult to ascertain (171, 172). NET formation may actually be detrimental in viral infections (173), with recent data showing that high levels of NET production exacerbate acute lung injury in influenza (174, 175) and can worsen prognosis in dengue virus infection (176).
The origins of this pregnancy-specific neutrophilia are incompletely understood but are likely to include progesterone-induced upregulation of granulocyte-macrophage colony-stimulating factor (GM-CSF) (177, 178). Mass cytometry analysis of circulating neutrophils in pregnancy by Aghaeepour et al. revealed not only increased abundance but also a hyper-activated state, with progressively enhanced sensitivity to pro-inflammatory stimuli including IL-6 and TNFa (6). However, the same group have also demonstrated a linear increase in “immature-like neutrophil signatures” (denoted by CD10 and CD15 expression) with advancing gestational age, which they suggest could contribute to the increased mortality observed in acute influenza and hepatitis E infections during late pregnancy (179).
Chemokine synthesis in the decidua leads to an influx of innate immune cells. Contact with invading trophoblast during implantation stimulates production of neutrophil chemo-attractants IL-8, CXCL1, and CXCL2 by decidual stromal cells, a process potentiated by the addition of progesterone in vitro (180, 181). However, the existence of a decidual neutrophil population remains controversial. Although one study from 2014 identified neutrophils in second-trimester decidua (182), this has not proved reproducible in subsequent single-cell reconstructions of the maternal-fetal interface (183, 184).
Macrophages
Monocytes are myeloid leukocytes that are released from the bone marrow, mature in the peripheral circulation and infiltrate into tissues, where they can differentiate into specialized populations of macrophages or myeloid DC. As with neutrophils, the monocyte-macrophage lineage expands both systemically and in the uterus during pregnancy (24, 185): macrophages account for up to 25% of the decidual leukocyte population and are outnumbered only by NK cells (80, 141). Macrophages have long been recognized as key phagocytic immune sentinels at mucosal surfaces. At the maternal-fetal interface their roles include antigen presentation to adaptive immune cells, promotion of tolerance through clearance of apoptotic trophoblast debris, secretion of pro-angiogenic factors and direct participation in spiral artery remodeling through the phagocytosis of maternal vascular smooth muscle cells (24).
The diverse functions of macrophages arise from their capacity for polarization into two antagonistic phenotypes, M1 (pro-inflammatory) and M2 (anti-inflammatory) (186). In normal pregnancy, the M1/M2 equilibrium is in constant flux, with M1 predominating during early trophoblast invasion, a shift toward tolerogenic M2 in the second and third trimesters, and a reversion to M1 at the onset of parturition (24). Differential TLR activity in decidual macrophages may contribute to protection of both mother and fetus against infection: stimulation of decidual macrophages with TLR2-4 and 7/8 agonists restricts viral replication and leads to overproduction of IL-10, an anti-inflammatory cytokine that suppresses trophoblast TLR signaling (140, 187).
Pregnancy-specific adaptations in the M1/M2 balance have also been implicated in the response to systemic viral infection. A study comparing the response of peripheral blood monocytes from pregnant and non-pregnant women to in vitro challenge with IAV found that the pro-inflammatory response was exaggerated in pregnant women, although trimester-specific effects were not reported (82). In a mouse model of pandemic H1N1 IAV in pregnancy, mice infected in mid-gestation exhibited higher clinical severity scores and a strong phenotypic shift toward the M2 phenotype in alveolar macrophages obtained from broncho-alveolar lavage samples, compared to those from non-pregnant matched females (87).
Dendritic Cells
Like macrophages, DC are critical for innate-adaptive crosstalk. DC can be broadly divided into two subsets:
1. Myeloid DC (mDC, CD14−CD11c+), which exist in an immature state in the spleen and lymph nodes and can migrate into various tissues where they are associated with Th1-type pro-inflammatory responses
2. Plasmacytoid DC (pDC, CD123+CD11c−), which reside in non-lymphoid tissues and induce Th2-type responses and proliferation of Tregs (25).
Circulating mDC from pregnant women demonstrate a skewed cytokine response to inflammatory stimuli, with increased production of IL-10, programmed death-ligand 1 and CD200, favoring Th2 and Treg polarization over Th1 (6, 188). DC of myeloid origin are recruited into the decidua from a variety of progenitor populations. Decidual DC (dDC) account for <2% of decidual leukocytes, are a highly proliferative and relatively immature population, and are phenotypically distinct from peripheral DC: they display a universal reduction in expression of T cell costimulatory molecules and thus fail to induce specific T cell responses in vitro (189). dDC help to prevent fetal rejection by promoting tolerogenic responses to endocytosed trophoblast antigens, recruit NK cells through production of IL-15 and potentiate decidual angiogenesis via close dialogue with decidual stromal cells (141).
The integral role of pDC in the innate antiviral response to RNA viruses, which centers on their unrivaled capacity for type I IFN production (95), may be further upregulated in pregnancy. In a study comparing pDC isolated from pregnant and non-pregnant women, pDC from pregnant women were more abundant at baseline and demonstrated increased chemokine production (including IFN-inducible protein 10 and CCL4) in response to IAV challenge. The authors note that excessive chemokine release is associated with higher disease severity and mortality from influenza A in vivo (190, 191) and suggest that the exaggerated pDC response they observed could contribute to the disproportionately high fatality rates of influenza in pregnancy (82).
Decidual Stromal Cells
Trophoblast invasion induces dramatic remodeling of the uterine mucosa, transforming the glands, arteries and stroma of the late-secretory-phase endometrium in a progesterone-dependent process known as decidualization (62, 164, 192). The specialized decidual stromal cells (DSC) generated as a result are fundamental for the establishment and homeostasis of the decidual immune system. In particular, DSC are in constant dialogue with dNK and exert critical regulatory influence over their various functions. Progesterone-induced IL-15 production by DSC is essential for the early expansion of dNK (193) and potentiates their cytotoxic degranulation in response to virally infected cells (194). Conversely, DSC-derived IL-33 inhibits dNK perforin and granzyme A synthesis, reduces their cytotoxic capacity and shifts their cytokine effector profile toward the immunosuppressive IL-4, IL-10, and IL-13 (195–197). The observation that T cell chemokine genes are epigenetically silenced in DSC highlights their role in maintaining a tolerogenic environment (12); indeed, they have even shown promise as a novel therapy for acute and chronic graft-versus-host disease in allogeneic stem cell transplant patients (198, 199). Their roles in viral infection remain poorly defined, but recent data show that murine DSC undergo necroptosis in response to transfection with a synthetic analog of viral RNA (200).
It is therefore clear that DSC, instead of merely functioning as a cellular scaffold that hosts the decidual immune system, are vital contributors to its key functions of simultaneous fetal tolerance and immune defense.
Acute Viral Infections That Cause Severe Maternal Disease
Lassa Virus
Lassa virus (LASV) is an enveloped ssRNA arenavirus endemic to West Africa that is acquired through contact with infected rodents (Mastomys natalensis, the multi-mammate rat). It causes regular seasonal outbreaks of Lassa fever, responsible for over 500,000 cases and 5,000 deaths per year. In the general population:
• 80% of patients will be asymptomatic or experience a mild non-specific illness;
• 15–20% require hospital admission due to severe disease (which may manifest as mucosal hemorrhage, hepatitis or multi-organ failure);
• Up to 29% will develop sensorineural hearing loss during their recovery;
However, these figures are strikingly altered in pregnancy. Cohort studies have consistently estimated maternal mortality rates as disproportionately high: 7% in the first two trimesters, rising as high as 87% in the third trimester, with fetal loss rate reported between 75 and 100% (44, 201, 203). The first systematic review of Lassa fever outcomes in pregnancy, published this year, identified just seven studies eligible for inclusion in the meta-analysis. This incorporated a total of 236 women in whom the absolute risk of maternal death was 33.7%, the fetal CFR was 61.5% and the neonatal CFR was 30.2%. Pregnancy conferred a 2.86-fold increase in risk of death (95% confidence interval 1.77–4.63) compared to non-pregnant women of reproductive age (29).
This disparity “underscores the need to prioritize pregnancy” in studies of the immune response to LASV (29). Currently, however, our understanding is almost entirely limited to data from animal models or cultured human cells (29, 204). These have provided insights into the key components of the innate immune response to LASV. The importance of the type I IFN response to LASV has been demonstrated in a non-human primate model: animals who survived viral challenge generated a robust IFN-α response shortly after inoculation, whereas those who died failed to upregulate IFN-α until the terminal phase of illness (205). Given that the type I IFN response to other RNA viruses such as influenza is less effective in pregnancy (206), it is possible that a similar attenuation in the case of LASV infection contributes to the higher mortality in pregnant women.
LASV has also been shown to directly antagonize the type I IFN response through its Z matrix protein, which is common to all arenaviruses, and its nucleoprotein (NP). LASV-NP possesses a unique exonuclease domain that degrades viral dsDNA—usually a potent ligand for RLR—to prevent type I IFN production (207).
In cultured human cells, LASV replicates in DC and macrophages but is remarkably adept at concealing its presence: it does not activate the host cells, induce apoptosis, alter their cell surface expression profile or trigger antigen presentation to T cells. This creates immune-privileged reservoirs that facilitate unchecked LASV replication in early infection, with subsequent systemic spread when the infected cells enter draining lymph nodes (202). It may also be a critical determinant of LASV's pathogenicity—in contrast, infection of macrophages and mDC with Mopeia virus, which shares 75% sequence homology with LASV but is non-pathogenic, leads to rapid activation of both cell types with upregulation of T cell costimulatory molecules and production of pro-inflammatory cytokines including type I IFNs and IL-6 (208–211).
The fact that LASV uses elaborate evasion mechanisms to avoid activation of innate immunity, and that this capacity correlates directly with suppression of adaptive responses, reinforces the role of the innate immune system in defense against Lassa fever. However, no single animal or human study to date has focused specifically on innate anti-LASV responses in pregnancy. The question of why pregnant women experience such high mortality from this disease therefore remains unsolved and should be urgently prioritized in future research.
Ebola Virus
Ebola virus (EBOV) caused over 11,000 deaths in the 2013–2016 outbreak in West Africa and had catastrophic implications for the region's already fragile public health infrastructure. Cases of Ebola virus disease (EVD) were concentrated in Guinea, Sierra Leone and Liberia, which have among the highest maternal mortality ratios in the world, and it is estimated that at least 100 pregnant women died (212–214).
EBOV causes abrupt onset of a febrile illness that can progress to profuse diarrhea, hemorrhage, meningo-encephalitis and hepatic or renal failure (44). Overall CFR is high (45–90%) and if acquired in pregnancy it is almost always lethal for the fetus (213, 215). However, a recent synthesis of available studies demonstrated no significant difference between mortality rates in pregnant vs. non-pregnant women (214). While this may be the case, caution is required on two counts: firstly, as the authors acknowledge, their analysis is limited by the small size, retrospective nature and considerable heterogeneity of included studies; and secondly, while pregnant women may not be intrinsically more vulnerable to EVD, the fact that they are often carers for sick relatives and make frequent visits to health facilities places them at increased risk of acquiring the disease (212).
Like LASV, EBOV targets myeloid DC and macrophages for entry and replication. Both viruses arrest mDC in an immature state that is permissive for viral replication, but their effect on macrophages is different: unlike LASV, EBOV does activate macrophages (44, 216). This allows them to present antigen to T cells, meaning that adaptive responses are at least partially activated in EVD: paradoxically, rather than being protective, this may exacerbate the strong and rapid upregulation of both innate and adaptive immunity that is characteristic of fatal disease (217).
The role of the IFN response in EVD has proved difficult to elucidate. On one hand, EBOV, like LASV, exhibits complex mechanisms that specifically counteract IFN: its VP35 protein contains an inhibitory domain that can scavenge viral dsRNA to prevent it from binding to RIG-I receptors (218) and its VP24 protein blocks nuclear accumulation of phosphorylated STAT1, a critical transcription factor in IFN signaling (219). On the other hand, certain ISGs such as tetherin have been found to actively suppress EBOV replication (220) and the IFN response is massively exaggerated in patients who die of EVD compared to those who survive, suggesting a contribution of host inflammatory response to disease similar to that seen in influenza (217, 221).
The West African epidemic revealed an unexpected tropism for the placenta: women who survived EVD in early pregnancy with complete resolution of viremia and no fetal loss were found to have unusually high rates of miscarriage and stillbirth weeks or months later, with abundant EBOV RNA detected in placental and fetal tissues (222, 223). Immuno-histochemistry analysis of placentas from EBOV-positive mothers shows accumulation of EBOV antigen within the intervillous space, where it co-localizes with a population of atypical maternal macrophages, and in extravillous trophoblast (224). This may reflect shared dependence on specific endocytic mechanisms: EBOV entry into target cells depends on the Niemann-Pick cholesterol transporter protein Niemann-Pick C1 (NPC-1), a protein that is also expressed on the syncytiotrophoblast (225, 226). These findings raise the suspicion of an EBOV predilection for pregnancy, even if the maternal death rate is comparable to that of the non-pregnant population, and support a rationale for close follow-up of female survivors (214).
Dengue Virus
The arboviruses are a group of over 100 arthropod-borne RNA viruses including dengue, Zika, Chikungunya, and yellow fever viruses. They constitute a major threat to global public health: 90% of pregnant women live in areas with either endemic or epidemic transmission of arboviruses (47).
Dengue, a spherical enveloped flavivirus with four ssRNA serotypes (DENV1-4), is the most abundant of the arboviruses and occupies two separate environmental niches: a sylvatic cycle, in which DENV circulates between arthropod vectors and non-human primate reservoirs, and an urban cycle, in which humans and Aedes mosquitoes are the only hosts. Following inoculation, the majority of people remain asymptomatic, but a minority will develop an acute febrile illness accompanied by severe headache, retro-orbital pain, arthralgia, and a rash. Approximately 1% develop severe dengue, a potentially lethal manifestation of disease that results from a sudden increase in systemic vascular permeability and can cause shock, profound thrombocytopenia, hemorrhage, and multi-organ failure. Mortality in severe dengue can be reduced from over 20 to <1% with good supportive care, but there are no specific antiviral therapies with proven benefit (227).
Quantifying the risks associated with DENV infection in pregnancy has proved particularly challenging due to the high proportion of asymptomatic cases, the lack of large prospective studies with comparison groups, the inevitable reporting bias in small case series and the inherent difficulties in accurate diagnosis of acute dengue fever (dengue IgM cross-reacts with other flaviviruses and coinfections are common) (48). Machado and colleagues were the first to definitively establish the association between pregnancy and severe dengue. They analyzed all 151,604 cases of suspected DENV in Rio de Janeiro from 2007 to 2008, compared outcomes in 99 pregnant women to 447 matched non-pregnant women of reproductive age, and found an increased risk of severe dengue in the pregnant women (odds ratio 3.38) with a trend toward higher mortality (3 vs. 1.1%) (47). A 2018 study added to this by investigating correlates of severe maternal disease in French Guiana: they showed an 8.6-fold increase in the risk of postpartum hemorrhage in the presence of severe dengue (228). Whether or not dengue increases the risk of adverse fetal and perinatal outcomes (specifically miscarriage, stillbirth, preterm birth, and low birthweight) remains controversial, with two meta-analyses from 2016 and 2017 reporting conflicting results (229, 230).
The origins of this increased susceptibility to severe dengue in pregnancy remain unknown but are likely to include placental tropism, innate immune adaptations and the physiological increase in vascular permeability that occurs in normal pregnancy, which may both delay diagnosis and exacerbate severe dengue (40). An immunohistochemical analysis of placentas from women with dengue in pregnancy showed accumulation of DENV antigen in the trophoblast and decidua in 92% (22/24) of cases. Microscopy revealed hypoxic changes (including villous edema and infarction) and an unusual observation of sickled erythrocytes in the intervillous space—this occurred despite no history of maternal sickle cell disease and was correlated with maternal death, suggesting that the virus may be able to influence erythrocyte biology in pregnancy (231, 232).
DENV can infect and deplete human megakaryocytes, suppressing their capacity to produce platelets and causing dengue-induced thrombocytopenia. Campbell et al. found that DENV leads to marked upregulation of interferon-induced transmembrane protein 3 (IFITM3) on platelets with corresponding release of type I IFNs, and that the highest levels of IFITM3 expression correlated with mildest disease (233, 234). These mechanisms are yet to be studied in pregnancy, but it is conceivable that enhanced DENV-mediated megakaryocyte depletion or a failure to upregulate platelet IFITM3 could contribute to the higher rates of hemorrhagic complications during pregnancy.
Type I IFN production is central to the innate anti-DENV response, although no specific studies have been conducted in pregnancy. DENV triggers type I IFNs through binding with various PRRs (RIG-I, endosomal TLR3, and endosomal TLR7) and is detected by the mannose-binding lectin complex. This leads to deposition of Cb4 and C2a on the virion surface, formation of the C3 convertase and activation of complement-mediated virolysis, although the virus can subvert this through its non-structural protein NS1 as described above. RNA interference and activation of apoptosis in infected cells are also important contributors to DENV defense, as reviewed by Uno and Ross (96). NS1 also appears to be a critical regulator of the DC and NK-mediated response to dengue infection. Sentinel cutaneous DC detect invading DENV and rapidly recruit NK cells through contact-dependent IFN-mediated upregulation of adhesion molecules, and the NK are key for viral suppression: in humanized mice, depletion of NK cells exacerbates DENV viremia and thrombocytopenia (235).
Although mechanistic insights into DENV pathogenicity with plausible relevance to pregnancy continue to emerge—for example, the observation that vitamin D supplementation reduces cultured human DC susceptibility to DENV2 through downregulation of TLR3, TLR7, and TLR9 signaling (236)—these findings cannot be extrapolated to the pregnant population and dedicated studies are urgently needed.
Hepatitis E
The hepatitis E virus (HEV) is a small, non-enveloped ssRNA hepevirus that causes over 20 million infections annually. Of its four genotypes, HEV-1 and 2 cause human disease and are spread by feco-oral transmission through contaminated water supply, entering the body via enterocytes and replicating in the liver. This route accounts for the wide geographical variation in disease burden of hepatitis E, which is concentrated in areas of poor sanitation (42, 49). In the general population, hepatitis E is asymptomatic in the vast majority of cases, but still causes 3.3 million symptomatic infections (usually mild, self-limiting and clinically indistinguishable from hepatitis A) and 56,000–70,000 deaths per year, a mortality rate of <0.5% (42).
However, outcomes are much worse for pregnant women: HEV mortality can exceed 50%, particularly if acquired in the third trimester. Several large cohort studies from India have demonstrated that pregnant women with HEV are both more likely to develop fulminant hepatic failure (FHF) and more likely to die from it (237–240). A 2019 systematic review including a total of 1,338 pregnant women with hepatitis E showed a 45% risk of fulminant hepatic failure, a median maternal CFR of 26% and a median fetal CFR of 33%. Other obstetric complications were not consistently reported in the included articles but data from four studies suggest an increased risk of postpartum hemorrhage, ranging from 13.6 to 30% (28). There are no proven drug therapies for hepatitis E (ribavirin, which has shown equivocal benefits in small case series, is contraindicated in pregnancy) and the only available vaccine for prevention of hepatitis E is not manufactured or licensed outside China (28).
Like the other viruses discussed in this review, there is therefore a global imperative to identify the factors that confer increased susceptibility to fatal HEV infection in pregnancy. Common themes emerge: the mechanisms are poorly characterized, difficult to recapitulate through in vitro models, influenced by hormonal factors and likely to arise from dysregulation of immune homeostasis at the maternal-fetal interface (241). Studies examining peripheral blood cells and circulating inflammatory mediators from women with HEV-induced FHF have implicated excessive Th2 switching, impaired NFkB-mediated liver regeneration, oxidative stress, and coagulopathy in its pathogenesis; these are reviewed by Perez-Gracia et al. (242), although no convincing individual candidate has emerged. The innate immune response to HEV is also unclear, although it appears to be crucial for prevention of severe disease. In 2015, a comparison of macrophages and DC from pregnant patients with HEV-induced FHF, non-fulminant acute HEV, and healthy pregnant controls found that macrophages from the women who developed fulminant liver disease had significantly impaired phagocytic capacity, with reduced TLR3 and TLR9 expression impeding MyD88-mediated IFN production (239).
Importantly, although the life cycle of HEV remains enigmatic, extrahepatic replication has been demonstrated in both the placenta (241, 243) and, more recently, in cultured human endometrial stromal cells (244). Gouilly et al. (241) showed that HEV-1 causes severe necrotic tissue injury in both decidua and trophoblast, significantly reduces both tissues' ability to produce type III IFNs and distorts the cytokine secretome of cultured DSC, upregulating potent pro-inflammatory mediators including IL-6 and the chemokines CCL-3 and CCL-4. As the authors suggest, these widespread disruptions to the architecture and homeostasis of the maternal-fetal interface are likely to contribute to viral dissemination, adverse obstetric outcomes and increased disease severity in pregnancy.
Influenza—Seasonal and Pandemic
The Orthomyxoviridae family includes three species capable of causing human disease: influenza viruses A, B, and C. Types A and B account for the majority of seasonal influenza, which causes approximately 389,000 deaths per year (245). They have an unusual segmented genome with eight negative-sense RNA strands and, unlike the other viruses discussed in this review, replicate in the nucleus. The pleiomorphic IAV virion consists of a host-derived lipid envelope displaying embedded surface glycoproteins haemagglutinin (HA) and neuraminidase (NA), which account for its antigenicity and are used to classify the different IAV serotypes (e.g., H1N1).
Major influenza pandemics over the last century have consistently shown disproportionately high mortality rates in pregnant women, mostly recently the H1N1 outbreak in 2009 (34–36). A 2011 meta-analysis including a total of 3,110 pregnant women who developed H1N1 showed that rates of hospital admission (52.3%), requirement for intensive care (23.3%) and death (4.1%, of which two thirds occurred in the third trimester) were all significantly increased in pregnancy. Despite only 1% of the susceptible population being pregnant, these women accounted for 5.7% of all deaths in the pooled analysis (52). Although maternal mortality outside a pandemic setting is low, these data prompted the WHO to recommend seasonal influenza vaccination for all pregnant women (52, 246). A subsequent larger systematic review and individual participant data meta-analysis (including 36,498 women of reproductive age) confirmed a 6.8-fold higher risk of requiring hospital admission in pregnant women with H1N1, but did not find any evidence of an increased risk of death (27, 247).
Like Ebola, whether maternal mortality from IAV is actually disproportionately increased remains unclear, but disease is certainly more severe in pregnant women. Pregnant women appear to develop particularly severe lung injury and are also at higher risk of extrapulmonary complications. In a study reporting autopsy findings of 21 Brazilian patients who died of H1N1-related acute respiratory failure, the single pregnant woman in the cohort had the most severe pulmonary disease, with widespread necrotising bronchiolitis, diffuse alveolar damage and significant upregulation of TLR3, IFN-γ, and granzyme B-producing cells in the airway epithelium, suggesting excessive activation of the innate immune response (248).
Mouse models have shown that disrupted TLR signaling is an important component of IAV pathogenesis. Following cell entry, IAV virions are internalized into endosomes, in which the low pH induces a conformational shift in HA resulting in release of the nucleocapsid protein into the cytosol. This process is essential for viral replication but also stimulates the innate immune response: in pDC isolated from mouse spleens, endosomal sensing of IAV ssRNA by TLR7 led to massive IFN-α release, a response that was completely abrogated in TLR7−/− mice (129). TLR3-deficient mice also show a muted response to IAV infection, with significant reductions in pro-inflammatory cytokine expression and CD8+ T cell recruitment to the broncho-alveolar space. However, despite higher viremic burden, this actually confers a survival advantage compared to control TLR3-competent mice (249). In purified human alveolar epithelial cells, RIG-I and TLR3 are the primary IAV sensors and trigger a type III-predominant IFN response (250). As with the other RNA viruses discussed here, IAV has a sophisticated array of innate evasion mechanisms, which are reviewed by Kikkert (251). IAV can also induce necroptosis of infected cells, leading to potent neutrophil recruitment and exacerbated lung injury. Mice deficient in MLKL, a critical mediator of necroptosis, exhibit improved survival in response to a lethal IAV challenge (252).
Pregnancy appears to suppress the systemic type I and type III IFN responses to IAV. A 2012 study showed that peripheral blood mononuclear cells from pregnant women exhibited a 10-fold reduction in the expression of protein kinase receptor, an ISG that is stimulated early in the antiviral response, compared to matched female controls in response to H1N1 IAV challenge. This effect was partially reversed by vaccination, although still not completely restored to non-pregnant levels (206). Some studies have also shown an IAV-induced upregulation of T cell costimulatory markers on pDC, hypothesizing that the virus may break the physiological attenuation of DC maturation in pregnancy and lead to exaggerated immune responses and tissue damage; conversely, others suggest that a failure to activate lung DC and recruit virus-specific CD8 T cells to the airway epithelium could contribute to the inability to control the virus in pregnancy (253, 254). Whether the severe disease observed in pregnancy results from a failure to generate robust innate responses or a harmful virus-induced disruption of the tolerogenic state remains to be clarified.
The role of hormones in modulating lung physiology and the response to IAV has been extensively studied. Progesterone, which is essential for the maintenance of pregnancy, is a muscle relaxant that causes physiological airway dilatation. It also affects the innate immune system, preventing NK cell degranulation and upregulating neutrophil-attractant chemokines in the respiratory epithelium. It interacts closely with the prostaglandins PGE2, which increases vascular permeability in the lungs and downregulates effector mechanisms in neutrophils and macrophages, and PGF2a, a potent broncho- and vasoconstrictor (86). In animal studies, female mice treated with exogenous progesterone are protected against severe IAV disease through increased expression of amphiregulin, which promotes tissue repair in the lungs and improves survival (255). However, the immunoregulatory role of progesterone appears to be strain- and exposure-specific: in another study, pre-treatment of female mice with progesterone protected them against severe disease during initial H1N1 exposure but reduced their survival during subsequent challenge with H3N2 (256). Influenza infection in pregnant mice dysregulates the progesterone-prostaglandin axis, leading to bronchoconstriction, an alveolar influx of neutrophils and respiratory failure (86). The implications of these findings in the severity of influenza in pregnancy are discussed further in a recent review in this journal by Littauer and Skountzou (38).
Novel Coronaviruses
Coronaviruses, which were first isolated from humans in the 1960s, cause frequent mild upper respiratory tract illnesses on a large scale but are not usually associated with significant disease. However, in the last 20 years, three zoonotic novel coronaviruses capable of causing severe pneumonia have emerged: severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV) and COVID-19 (SARS-CoV-2). Global experiences of the first two, which disproportionately affected pregnant women, have caused significant apprehension about the consequences of the third, which remains subject to intensive and ongoing research (55, 257). Given that pneumonia is the leading non-obstetric infectious cause of maternal death, vigilance is required with any emerging respiratory virus and pregnant women should be prioritized in efforts to anticipate and mitigate their effects (258).
SARS-CoV caused over 8,400 cases in 29 countries in the outbreak of 2002–2003. The 916 reported deaths corresponded to a global CFR of ~11%. Studies describing its effects in pregnant women, although small, are strongly suggestive of a higher burden of mortality and morbidity than in the general population. Wong et al. summarized outcomes in 12 pregnant women hospitalized due to confirmed SARS-CoV in Hong Kong in 2003: 50% were admitted to intensive care due to hypoxia (vs. 20% of non-pregnant patients), 33% required mechanical ventilation and 25% died. Associated complications included disseminated intravascular coagulation, renal failure and superimposed bacterial pneumonia. Of the 7 who presented in the first trimester, 5 had a spontaneous miscarriage; and of the 5 who presented beyond 24 weeks' gestation, 80% delivered preterm. Surviving neonates did not develop SARS and there was no evidence of vertical transmission, although placental histopathology did show other features indicative of maternal hypoxemia. The authors note that the use of ribavirin in 11/12 cases (following careful counseling about potential teratogenicity) may have exacerbated the high observed rate of first-trimester miscarriage (57). A case-control study, also from Hong Kong, confirmed the increased severity of disease in pregnancy by comparing 10 pregnant to 40 non-pregnant women with SARS at the same hospital: death (30 vs. 0%), intensive care admission (60 vs. 17.5%) and renal failure were all significantly more common in the pregnant group (58).
MERS-CoV, which emerged in 2012, caused 2494 confirmed cases and 858 deaths, with a CFR of 34.4%. The higher mortality rate has been attributed to both poor infection control, facilitating rapid propagation in healthcare settings, and an inherently more aggressive clinical course, with faster progression to respiratory failure. The clinical spectrum was broader than in SARS, with immunocompetent adults often reporting mild or moderate symptoms while deaths were more concentrated in those who were immunosuppressed, pregnant or had major medical comorbidities (55, 259). A 2019 synthesis of 11 pregnant women with MERS-CoV found that 54% required intensive care and 27% died. Although this was not elevated compared to the CFR in the general population, these numbers are too small to provide confident assurance that pregnant women are not at increased risk from MERS-CoV. As with SARS, no episodes of vertical transmission were documented, although umbilical cord and neonatal blood sampling were not universally performed (59).
The spread of COVID-19 (SARS-CoV-2) has vastly exceeded that of SARS and MERS, with over 21 million cases and 760,000 deaths at the time of writing (260). However, its mortality rate is considerably lower than the two previous novel coronavirus outbreaks, as is the proportion of patients who develop severe disease (pooled estimate 18.0%) (261).
In the early stages of its escalation, experience from the SARS-CoV and MERS-CoV outbreaks prompted some national health policy departments to recommend increased caution in pregnant women. However, accumulating data are reassuring on this front: a systematic review of 2,567 affected pregnancies published in July showed a 7% intensive care unit admission rate with maternal mortality of approximately 1% (262) and a population-level analysis of 427 pregnant women hospitalized with COVID-19 in the UK showed no evidence of an increased risk of severe disease compared to the general hospital population (60). In contrast to SARS-CoV and MERS-CoV, vertical transmission appears to occur in a small proportion of cases of SARS-CoV-2: in a recent meta-analysis, 3.2% of neonates tested positive on nasopharyngeal swabs and 3.7% had positive serology, based on IgM positivity (263), however, caution is required given that the validity of IgM serology tests can be compromised by cross-reactivity. These findings are supported by the recent systematic confirmation of transplacental transmission of SARS-CoV-2 following maternal infection at 35 weeks' gestation (264).
The exponential transmission of COVID-19 has prompted renewed efforts to identify integral components of the immune response to coronaviruses. While innate host defenses rely on the same cardinal mechanisms as for the other RNA viruses discussed—namely, the interferon response—coronaviruses appear to have evolved a particularly diverse repertoire of innate immune evasion strategies. Inhaled coronaviruses enter pneumocytes and macrophages in the upper respiratory tract and replicate in the cytosol, hijacking host intracellular membranes to create “replication organelles” (RO) that facilitate viral replication while simultaneously shielding the RNA within double-membrane vesicles to prevent detection by intracellular PRRs such as RIG-I (265).
Coronaviruses go to great lengths to avoid recognition: their non-structural proteins even exhibit endonuclease activity, allowing them to degrade their own RNA and forestall activation of PAMP-mediated antiviral signaling (266). As well as these evasion strategies, coronaviruses can also directly antagonize the innate immune response: the SARS-CoV non-structural protein-1 (NSP-1) binds to the 40S subunit of host ribosomes and induces “translational shutoff,” bringing all cellular expression of antiviral effector proteins to a halt (267). This particularly intricate capacity of CoV to both elude and suppress innate effector mechanisms may contribute to their pandemic potential. However, no studies thus far have been specifically designed to investigate the disparities between individual coronaviruses in terms of their effects on maternal disease and vertical transmission in pregnancy.
Challenges and Future Developments
Determining exactly how innate immune mechanisms and corresponding viral evasion strategies contribute to the disproportionately severe disease observed in pregnancy is an ongoing problem. Challenges range from the technical and ethical (e.g., the difficulty of accessing human decidual tissue and replicating human pregnancy in animal models) to the global, with the ever-present threat of widespread outbreaks caused by novel viruses with pandemic potential. Emerging viruses, epitomized by the current COVID-19 pandemic and the ongoing Ebola outbreak in North Kivu, highlight the vulnerability and inequity in the global health infrastructure. However, there is also a considerable threat from resurgence of previously controlled viruses: the global re-emergence of measles since 2016, including in several countries where transmission had previously been eradicated, has caused thousands of deaths and poses a major risk to pregnant women (268). A 2017 UK case report described a patient who required emergency Cesarean section and extracorporeal membrane oxygenation for deteriorating respiratory failure at 32 weeks' gestation (269). Control of preventable viral diseases like measles depends on high rates of vaccination coverage, which is compromised by conflict, migration, and persistent belief in the discredited association with autism (268).
The ability to protect vulnerable groups from viral infection depends partly on public health measures such as vaccination and vector control but also on our understanding of the biological correlates of this vulnerability. The advent of technologies such as mass cytometry and single-cell RNA sequencing have already shown promise (270), offering unparalleled insights into the cellular architecture of the maternal-fetal interface in early pregnancy (183, 271, 272). These, along with the recent development of human trophoblast organoids that replicate the placental secretome in vitro, may transform our ability to study the effects of viral infection in pregnancy (273).
Conclusion
The RNA viruses discussed in this article share many overlapping features in their clinical manifestations and their interactions with the maternal immune system (Figure 5). Common themes include placental tropism, an association with adverse obstetric outcomes and the importance of a tightly regulated interferon response, which is reflected in the evolution of diverse viral IFN evasion mechanisms. Innate immune cells are uniquely positioned at the maternal-fetal interface and orchestrate the balance between the conflicting immunological priorities of pregnancy: tolerance of the fetal allograft and defense of both mother and fetus against invasive pathogens.
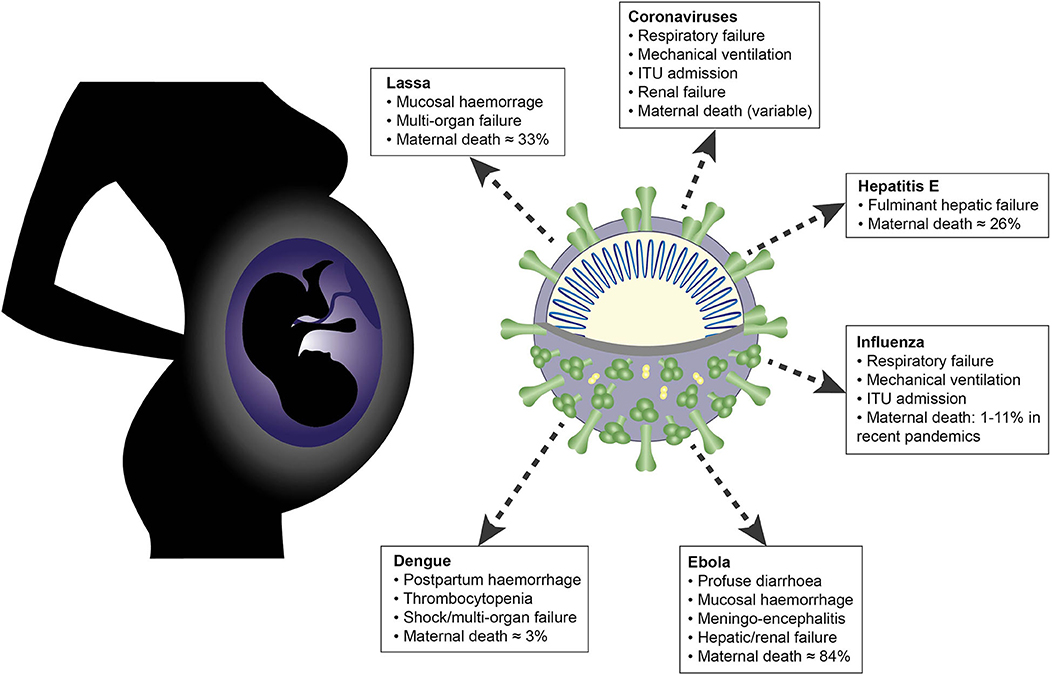
Figure 5. Schematic summarizing effects of maternal infection with the following RNA viruses during pregnancy: Lassa, Ebola, dengue, hepatitis E, influenza and novel coronaviruses.
The disproportionate rates of severe disease and mortality observed in pregnant women with Lassa, Ebola, dengue, hepatitis E, influenza and certain novel coronavirus infections cannot be attributed purely to immunological adaptations: social and behavioral factors also contribute (212). However, it is only through research that is tailored toward these pregnancy-specific factors that the outcomes will improve. In particular, as recently and eloquently argued by Gomes et al. (274) and Schwartz and Graham (55), the systematic exclusion of pregnant women from the design of vaccine trials for viral illnesses will perpetuate and exacerbate the problem. Addressing the gaps in our knowledge of innate immunity in pregnancy is an urgent priority.
Author Contributions
EC and TM conceived the article. EC wrote the first draft of the manuscript. TM wrote sections of the manuscript, provided critical revisions, and edited the text. FÅ generated all the figures for the manuscript. DW and IF provided critical revisions of the manuscript and edited the text. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Medawar PB. Some immunological and endocrinological problems raised by the evolution of viviparity in vertebrates. Symp Soc Exp Biol. (1953) 44:320–38.
2. Raghupathy R. Th1-type immunity is incompatible with successful pregnancy. Immunol Today. (1997) 18:478–82. doi: 10.1016/S0167-5699(97)01127-4
3. Billington WD. The immunological problem of pregnancy: 50 years with the hope of progress. A tribute to Peter Medawar. J Reprod Immunol. (2003) 60:1–11. doi: 10.1016/S0165-0378(03)00083-4
4. Luppi P, Haluszczak C, Betters D, Richard CA, Trucco M, DeLoia JA. Monocytes are progressively activated in the circulation of pregnant women. J Leukoc Biol. (2002) 72:874–84. doi: 10.1034/j.1600-0897.2002.1o041.x
5. Kraus TA, Sperling RS, Engel SM, Lo Y, Kellerman L, Singh T, et al. Peripheral blood cytokine profiling during pregnancy and post-partum periods. Am J Reprod Immunol. (2010) 64:411–26. doi: 10.1111/j.1600-0897.2010.00889.x
6. Aghaeepour N, Ganio EA, McIlwain D, Tsai AS, Tingle M, Van Gassen S, et al. An immune clock of human pregnancy. Sci Immunol. (2017) 2:eaan2946. doi: 10.1126/sciimmunol.aan2946
7. Gomez-Lopez N, Romero R, Hassan SS, Bhatti G, Berry SM, Kusanovic JP, et al. The cellular transcriptome in the maternal circulation during normal pregnancy: a longitudinal study. Front Immunol. (2019) 10:2863. doi: 10.3389/fimmu.2019.02863
8. Forger F, Villiger PM. Immunological adaptations in pregnancy that modulate rheumatoid arthritis disease activity. Nat Rev Rheumatol. (2020) 16:113–22. doi: 10.1038/s41584-019-0351-2
9. Kraus TA, Engel SM, Sperling RS, Kellerman L, Lo Y, Wallenstein S, et al. Characterizing the pregnancy immune phenotype: results of the viral immunity and pregnancy (VIP) study. J Clin Immunol. (2012) 32:300–11. doi: 10.1007/s10875-011-9627-2
10. Graham C, Chooniedass R, Stefura WP, Becker AB, Sears MR, Turvey SE, et al. In vivo immune signatures of healthy human pregnancy: Inherently inflammatory or anti-inflammatory? PLoS ONE. (2017) 12:e0177813. doi: 10.1371/journal.pone.0177813
11. Gomez-Lopez N, Romero R, Xu Y, Miller D, Leng Y, Panaitescu B, et al. The immunophenotype of amniotic fluid leukocytes in normal and complicated pregnancies. Am J Reprod Immunol. (2018) 79:e12827. doi: 10.1111/aji.12827
12. Nancy P, Tagliani E, Tay CS, Asp P, Levy DE, Erlebacher A. Chemokine gene silencing in decidual stromal cells limits T cell access to the maternal-fetal interface. Science. (2012) 336:1317–21. doi: 10.1126/science.1220030
13. Fettke F, Schumacher A, Canellada A, Toledo N, Bekeredjian-Ding I, Bondt A, et al. Maternal and fetal mechanisms of B cell regulation during pregnancy: human chorionic gonadotropin stimulates B cells to produce IL-10 while alpha-fetoprotein drives them into apoptosis. Front Immunol. (2016) 7:495. doi: 10.3389/fimmu.2016.00495
14. Lima J, Cambridge G, Vilas-Boas A, Martins C, Borrego LM, Leandro M. Serum markers of B-cell activation in pregnancy during late gestation, delivery, and the postpartum period. Am J Reprod Immunol. (2019) 81:e13090. doi: 10.1111/aji.13090
15. Holmlund U, Cebers G, Dahlfors AR, Sandstedt B, Bremme K, Ekstrom ES, et al. Expression and regulation of the pattern recognition receptors Toll-like receptor-2 and Toll-like receptor-4 in the human placenta. Immunology. (2002) 107:145–51. doi: 10.1046/j.1365-2567.2002.01491.x
16. Fest S, Aldo PB, Abrahams VM, Visintin I, Alvero A, Chen R, et al. Trophoblast-macrophage interactions: a regulatory network for the protection of pregnancy. Am J Reprod Immunol. (2007) 57:55–66. doi: 10.1111/j.1600-0897.2006.00446.x
17. Kwan M, Hazan A, Zhang J, Jones RL, Harris LK, Whittle W, et al. Dynamic changes in maternal decidual leukocyte populations from first to second trimester gestation. Placenta. (2014) 35:1027–34. doi: 10.1016/j.placenta.2014.09.018
18. Sharkey AM, Xiong S, Kennedy PR, Gardner L, Farrell LE, Chazara O, et al. Tissue-specific education of decidual NK cells. J Immunol. (2015) 195:3026–32. doi: 10.4049/jimmunol.1501229
19. Sacks GP, Studena K, Sargent K, Redman CW. Normal pregnancy and preeclampsia both produce inflammatory changes in peripheral blood leukocytes akin to those of sepsis. Am J Obstet Gynecol. (1998) 179:80–6. doi: 10.1016/S0002-9378(98)70254-6
20. Redman CW, Sacks GP, Sargent IL. Preeclampsia: an excessive maternal inflammatory response to pregnancy. Am J Obstet Gynecol. (1999) 180(2 Pt 1):499–506. doi: 10.1016/S0002-9378(99)70239-5
21. Naccasha N, Gervasi MT, Chaiworapongsa T, Berman S, Yoon BH, Maymon E, et al. Phenotypic and metabolic characteristics of monocytes and granulocytes in normal pregnancy and maternal infection. Am J Obstet Gynecol. (2001) 185:1118–23. doi: 10.1067/mob.2001.117682
22. Sugiura-Ogasawara M, Nozawa K, Nakanishi T, Hattori Y, Ozaki Y. Complement as a predictor of further miscarriage in couples with recurrent miscarriages. Hum Reprod. (2006) 21:2711–4. doi: 10.1093/humrep/del229
23. Hiby SE, Apps R, Sharkey AM, Farrell LE, Gardner L, Mulder A, et al. Maternal activating KIRs protect against human reproductive failure mediated by fetal HLA-C2. J Clin Invest. (2010) 120:4102–10. doi: 10.1172/JCI43998
24. Yao Y, Xu XH, Jin L. Macrophage polarization in physiological and pathological pregnancy. Front Immunol. (2019) 10:792. doi: 10.3389/fimmu.2019.00792
25. Yang F, Zheng Q, Jin L. Dynamic function and composition changes of immune cells during normal and pathological pregnancy at the maternal-fetal interface. Front Immunol. (2019) 10:2317. doi: 10.3389/fimmu.2019.02317
26. Pierik E, Prins JR, van Goor H, Dekker GA, Daha MR, Seelen MAJ, et al. Dysregulation of complement activation and placental dysfunction: a potential target to treat preeclampsia? Front Immunol. (2019) 10:3098. doi: 10.3389/fimmu.2019.03098
27. Mertz D, Geraci J, Winkup J, Gessner BD, Ortiz JR, Loeb M. Pregnancy as a risk factor for severe outcomes from influenza virus infection: a systematic review and meta-analysis of observational studies. Vaccine. (2017) 35:521–8. doi: 10.1016/j.vaccine.2016.12.012
28. Berglov A, Hallager S, Weis N. Hepatitis E during pregnancy: maternal and foetal case-fatality rates and adverse outcomes-a systematic review. J Viral Hepat. (2019) 26:1240–8. doi: 10.1111/jvh.13129
29. Kayem ND, Benson C, Aye CYL, Barker S, Tome M, Kennedy S, et al. Lassa fever in pregnancy: a systematic review and meta-analysis. Trans R Soc Trop Med Hyg. (2020) 114:385–96. doi: 10.1093/trstmh/traa011
30. Koopman LA, Kopcow HD, Rybalov B, Boyson JE, Orange JS, Schatz F, et al. Human decidual natural killer cells are a unique NK cell subset with immunomodulatory potential. J Exp Med. (2003) 198:1201–12. doi: 10.1084/jem.20030305
31. Siewiera J, El Costa H, Tabiasco J, Berrebi A, Cartron G, Le Bouteiller P, et al. Human cytomegalovirus infection elicits new decidual natural killer cell effector functions. PLoS Pathog. (2013) 9:e1003257. doi: 10.1371/journal.ppat.1003257
32. Zhang D, Ren L, Zhao M, Yang C, Liu X, Zhang H, et al. Role of Tim-3 in decidual macrophage functional polarization during abnormal pregnancy with toxoplasma gondii infection. Front Immunol. (2019) 10:1550. doi: 10.3389/fimmu.2019.01550
33. Say L, Chou D, Gemmill A, Tuncalp O, Moller AB, Daniels J, et al. Global causes of maternal death: a WHO systematic analysis. Lancet Glob Health. (2014) 2:e323–33. doi: 10.1016/S2214-109X(14)70227-X
34. Harris JW. Influenza occurring in pregnant women. JAMA. (1919) 72:978–80. doi: 10.1001/jama.1919.02610140008002
35. Freeman DW, Barno A. Deaths from Asian influenza associated with pregnancy. Am J Obstet Gynecol. (1959) 78:1172–5. doi: 10.1016/0002-9378(59)90570-8
36. Siston AM, Rasmussen SA, Honein MA, Fry AM, Seib K, Callaghan WM, et al. Pandemic 2009 influenza A(H1N1) virus illness among pregnant women in the United States. JAMA. (2010) 303:1517–25. doi: 10.1001/jama.2010.479
37. Moffett A, Chazara O, Colucci F. Maternal allo-recognition of the fetus. Fertil Steril. (2017) 107:1269–72. doi: 10.1016/j.fertnstert.2017.05.001
38. Littauer EQ, Skountzou I. hormonal regulation of physiology, innate immunity and antibody response to H1N1 influenza virus infection during pregnancy. Front Immunol. (2018) 9:2455. doi: 10.3389/fimmu.2018.02455
39. Shah NM, Imami N, Johnson MR. Progesterone modulation of pregnancy-related immune responses. Front Immunol. (2018) 9:1293. doi: 10.3389/fimmu.2018.01293
40. Charlier C, Beaudoin MC, Couderc T, Lortholary O, Lecuit M. Arboviruses and pregnancy: maternal, fetal, neonatal effects. Lancet Child Adolesc Health. (2017) 1:134–46. doi: 10.1016/S2352-4642(17)30021-4
41. Racicot K, Mor G. Risks associated with viral infections during pregnancy. J Clin Invest. (2017) 127:1591–9. doi: 10.1172/JCI87490
42. Pereira L. Congenital viral infection: traversing the uterine-placental interface. Annu Rev Virol. (2018) 5:273–99. doi: 10.1146/annurev-virology-092917-043236
43. Rogan SC, Beigi RH. Treatment of viral infections during pregnancy. Clin Perinatol. (2019) 46:235–56. doi: 10.1016/j.clp.2019.02.009
44. Prescott JB, Marzi A, Safronetz D, Robertson SJ, Feldmann H, Best SM. Immunobiology of Ebola and Lassa virus infections. Nat Rev Immunol. (2017) 17:195–207. doi: 10.1038/nri.2016.138
45. Garba I, Dattijo LM, Habib AG. Ebola virus disease and pregnancy outcome: a review of the literature. Trop J Obstet Gynaecol. (2017) 34:6–10. doi: 10.4103/TJOG.TJOG_3_17
46. Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, et al. The global distribution and burden of dengue. Nature. (2013) 496:504–7. doi: 10.1038/nature12060
47. Machado CR, Machado ES, Rohloff RD, Azevedo M, Campos DP, de Oliveira RB, et al. Is pregnancy associated with severe dengue? A review of data from the Rio de Janeiro surveillance information system. PLoS Negl Trop Dis. (2013) 7:e2217. doi: 10.1371/journal.pntd.0002217
48. Carles G. What are the true consequences of dengue during pregnancy? Lancet Infect Dis. (2016) 16:765–6. doi: 10.1016/S1473-3099(16)00130-4
49. Dagnew M, Belachew A, Tiruneh M, Moges F. Hepatitis E virus infection among pregnant women in Africa: systematic review and meta-analysis. BMC Infect Dis. (2019) 19:519. doi: 10.1186/s12879-019-4125-x
50. Li FC, Choi BC, Sly T, Pak AW. Finding the real case-fatality rate of H5N1 avian influenza. J Epidemiol Community Health. (2008) 62:555–9. doi: 10.1136/jech.2007.064030
51. Woolston WJC DO. Epidemic pneumonia (Spanish influenza) in pregnant: effect in one hundred and one cases. JAMA. (1918) 71:1898–9. doi: 10.1001/jama.1918.02600490030008
52. Mosby LG, Rasmussen SA, Jamieson DJ. 2009 pandemic influenza A (H1N1) in pregnancy: a systematic review of the literature. Am J Obstet Gynecol. (2011) 205:10–8. doi: 10.1016/j.ajog.2010.12.033
53. ANZIC Influenza Investigators and the Australasian Maternity Outcomes Surveillance System. Critical illness due to 2009 A/H1N1 influenza in pregnant and postpartum women: population based cohort study. BMJ. (2010) 340:c1279. doi: 10.1136/bmj.c1279
54. Yates L, Pierce M, Stephens S, Mill AC, Spark P, Kurinczuk JJ, et al. Influenza A/H1N1v in pregnancy: an investigation of the characteristics and management of affected women and the relationship to pregnancy outcomes for mother and infant. Health Technol Assess. (2010) 14:109–82. doi: 10.3310/hta14340-02
55. Schwartz DA, Graham AL. Potential maternal and infant outcomes from (Wuhan) coronavirus 2019-nCoV infecting pregnant women: lessons from SARS, MERS, and other human coronavirus infections. Viruses. (2020) 12:194. doi: 10.3390/v12020194
56. He W, Yi GY, Zhu Y. Estimation of the basic reproduction number, average incubation time, asymptomatic infection rate, and case fatality rate for COVID-19: meta-analysis and sensitivity analysis. J Med Virol. (2020). doi: 10.1002/jmv.26041. [Epub ahead of print].
57. Wong SF, Chow KM, Leung TN, Ng WF, Ng TK, Shek CC, et al. Pregnancy and perinatal outcomes of women with severe acute respiratory syndrome. Am J Obstet Gynecol. (2004) 191:292–7. doi: 10.1016/j.ajog.2003.11.019
58. Lam CM, Wong SF, Leung TN, Chow KM, Yu WC, Wong TY, et al. A case-controlled study comparing clinical course and outcomes of pregnant and non-pregnant women with severe acute respiratory syndrome. BJOG. (2004) 111:771–4. doi: 10.1111/j.1471-0528.2004.00199.x
59. Alfaraj SH, Al-Tawfiq JA, Memish ZA. Middle east respiratory syndrome coronavirus (MERS-CoV) infection during pregnancy: report of two cases & review of the literature. J Microbiol Immunol Infect. (2019) 52:501–3. doi: 10.1016/j.jmii.2018.04.005
60. Knight M, Bunch K, Vousden N, Morris E, Simpson N, Gale C, et al. Characteristics and outcomes of pregnant women admitted to hospital with confirmed SARS-CoV-2 infection in UK: national population based cohort study. BMJ. (2020) 369:m2107. doi: 10.1136/bmj.m2107
61. Greenberg M, Jacobziner H, Pakter J, Weisl BA. Maternal mortality in the epidemic of Asian influenza, New York City, 1957. Am J Obstet Gynecol. (1958) 76:897–902. doi: 10.1016/0002-9378(58)90027-9
62. Huhn O, Ivarsson MA, Gardner L, Hollinshead M, Stinchcombe JC, Chen P, et al. Distinctive phenotypes and functions of innate lymphoid cells in human decidua during early pregnancy. Nat Commun. (2020) 11:381. doi: 10.1038/s41467-019-14123-z
63. Starkey PM, Sargent IL, Redman CW. Cell populations in human early pregnancy decidua: characterization and isolation of large granular lymphocytes by flow cytometry. Immunology. (1988) 65:129–34.
64. Ferry BL, Starkey PM, Sargent IL, Watt GM, Jackson M, Redman CW. Cell populations in the human early pregnancy decidua: natural killer activity and response to interleukin-2 of CD56-positive large granular lymphocytes. Immunology. (1990) 70:446–52.
65. Vince GS, Starkey PM, Jackson MC, Sargent IL, Redman CW. Flow cytometric characterisation of cell populations in human pregnancy decidua and isolation of decidual macrophages. J Immunol Methods. (1990) 132:181–9. doi: 10.1016/0022-1759(90)90028-T
66. Jabrane-Ferrat N. Features of human decidual NK cells in healthy pregnancy and during viral infection. Front Immunol. (2019) 10:1397. doi: 10.3389/fimmu.2019.01397
67. Hiby SE, Walker JJ, O'Shaughnessy KM, Redman CW, Carrington M, Trowsdale J, et al. Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J Exp Med. (2004) 200:957–65. doi: 10.1084/jem.20041214
68. Hiby SE, Apps R, Chazara O, Farrell LE, Magnus P, Trogstad L, et al. Maternal KIR in combination with paternal HLA-C2 regulate human birth weight. J Immunol. (2014) 192:5069–73. doi: 10.4049/jimmunol.1400577
69. Papuchova H, Meissner TB, Li Q, Strominger JL, Tilburgs T. The dual role of HLA-C in tolerance and immunity at the maternal-fetal interface. Front Immunol. (2019) 10:2730. doi: 10.3389/fimmu.2019.02730
70. Orr HT, Bach FH, Ploegh HL, Strominger JL, Kavathas P, DeMars R. Use of HLA loss mutants to analyse the structure of the human major histocompatibility complex. Nature. (1982) 296:454–6. doi: 10.1038/296454a0
71. Kovats S, Main EK, Librach C, Stubblebine M, Fisher SJ, DeMars R. A class I antigen, HLA-G, expressed in human trophoblasts. Science. (1990) 248:220–3. doi: 10.1126/science.2326636
72. Apps R, Gardner L, Sharkey AM, Holmes N, Moffett A. A homodimeric complex of HLA-G on normal trophoblast cells modulates antigen-presenting cells via LILRB1. Eur J Immunol. (2007) 37:1924–37. doi: 10.1002/eji.200737089
73. Apps R, Gardner L, Moffett A. A critical look at HLA-G. Trends Immunol. (2008) 29:313–21. doi: 10.1016/j.it.2008.02.012
74. Gregori S, Amodio G, Quattrone F, Panina-Bordignon P. HLA-G orchestrates the early interaction of human trophoblasts with the maternal niche. Front Immunol. (2015) 6:128. doi: 10.3389/fimmu.2015.00128
75. Yockey LJ, Lucas C, Iwasaki A. Contributions of maternal and fetal antiviral immunity in congenital disease. Science. (2020) 368:608–12. doi: 10.1126/science.aaz1960
76. Crespo AC, van der Zwan A, Ramalho-Santos J, Strominger JL, Tilburgs T. Cytotoxic potential of decidual NK cells and CD8+ T cells awakened by infections. J Reprod Immunol. (2017) 119:85–90. doi: 10.1016/j.jri.2016.08.001
77. Kay AW, Fukuyama J, Aziz N, Dekker CL, Mackey S, Swan GE, et al. Enhanced natural killer-cell and T-cell responses to influenza A virus during pregnancy. Proc Natl Acad Sci USA. (2014) 111:14506–11. doi: 10.1073/pnas.1416569111
78. Richani K, Soto E, Romero R, Espinoza J, Chaiworapongsa T, Nien JK, et al. Normal pregnancy is characterized by systemic activation of the complement system. J Matern Fetal Neonatal Med. (2005) 17:239–45. doi: 10.1080/14767050500072722
79. Derzsy Z, Prohaszka Z, Rigo J Jr, Fust G, Molvarec A. Activation of the complement system in normal pregnancy and preeclampsia. Mol Immunol. (2010) 47:1500–6. doi: 10.1016/j.molimm.2010.01.021
80. Lurie S, Rahamim E, Piper I, Golan A, Sadan O. Total and differential leukocyte counts percentiles in normal pregnancy. Eur J Obstet Gynecol Reprod Biol. (2008) 136:16–9. doi: 10.1016/j.ejogrb.2006.12.013
81. Chen K, Liu J, Liu S, Xia M, Zhang X, Han D, et al. Methyltransferase SETD2-mediated methylation of STAT1 is critical for interferon antiviral activity. Cell. (2017) 170:492–506.e14. doi: 10.1016/j.cell.2017.06.042
82. Le Gars M, Kay AW, Bayless NL, Aziz N, Dekker CL, Swan GE, et al. Increased proinflammatory responses of monocytes and plasmacytoid dendritic cells to influenza A virus infection during pregnancy. J Infect Dis. (2016) 214:1666–71. doi: 10.1093/infdis/jiw448
83. Le Gars M, Seiler C, Kay AW, Bayless NL, Starosvetsky E, Moore L, et al. Pregnancy-induced alterations in NK cell phenotype and function. Front Immunol. (2019) 10:2469. doi: 10.3389/fimmu.2019.02469
84. Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest. (2003) 112:1644–54. doi: 10.1172/JCI200318817
85. Kim MY, Guerra MM, Kaplowitz E, Laskin CA, Petri M, Branch DW, et al. Complement activation predicts adverse pregnancy outcome in patients with systemic lupus erythematosus and/or antiphospholipid antibodies. Ann Rheum Dis. (2018) 77:549–55. doi: 10.1136/annrheumdis-2017-212224
86. Littauer EQ, Esser ES, Antao OQ, Vassilieva EV, Compans RW, Skountzou I. H1N1 influenza virus infection results in adverse pregnancy outcomes by disrupting tissue-specific hormonal regulation. PLoS Pathog. (2017) 13:e1006757. doi: 10.1371/journal.ppat.1006757
87. Lauzon-Joset JF, Scott NM, Mincham KT, Stumbles PA, Holt PG, Strickland DH. Pregnancy induces a steady-state shift in alveolar macrophage M1/M2 phenotype that is associated with a heightened severity of influenza virus infection: mechanistic insight using mouse models. J Infect Dis. (2019) 219:1823–1. doi: 10.1093/infdis/jiy732
88. Yang Q, Shu HB. Deciphering the pathways to antiviral innate immunity and inflammation. Adv Immunol. (2020) 145:1–36. doi: 10.1016/bs.ai.2019.11.001
89. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
90. Yan N, Chen ZJ. Intrinsic antiviral immunity. Nat Immunol. (2012) 13:214–22. doi: 10.1038/ni.2229
91. Reich NC. Too much of a good thing: detrimental effects of interferon. Semin Immunol. (2019) 43:101282. doi: 10.1016/j.smim.2019.101282
92. Crosse KM, Monson EA, Beard MR, Helbig KJ. Interferon-stimulated genes as enhancers of antiviral innate immune signaling. J Innate Immun. (2018) 10:85–93. doi: 10.1159/000484258
93. Lee AJ, Ashkar AA. The dual nature of type I and type II interferons. Front Immunol. (2018) 9:2061. doi: 10.3389/fimmu.2018.02061
94. Lazear HM, Schoggins JW, Diamond MS. shared and distinct functions of type I and Type III interferons. Immunity. (2019) 50:907–23. doi: 10.1016/j.immuni.2019.03.025
95. Ali S, Mann-Nuttel R, Schulze A, Richter L, Alferink J, Scheu S. Sources of type I interferons in infectious immunity: plasmacytoid dendritic cells not always in the Driver's Seat. Front Immunol. (2019) 10:778. doi: 10.3389/fimmu.2019.00778
96. Uno N, Ross TM. Dengue virus and the host innate immune response. Emerg Microbes Infect. (2018) 7:167. doi: 10.1038/s41426-018-0168-0
97. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. (2015) 517:311–20. doi: 10.1038/nature14191
98. Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. (2019) 26:99–114. doi: 10.1038/s41418-018-0212-6
99. Linkermann A, Green DR. Necroptosis. N Engl J Med. (2014) 370:455–65. doi: 10.1056/NEJMra1310050
100. Wang Y, Hao Q, Florence JM, Jung BG, Kurdowska AK, Samten B, et al. Influenza virus infection induces ZBP1 expression and necroptosis in mouse lungs. Front Cell Infect Microbiol. (2019) 9:286. doi: 10.3389/fcimb.2019.00286
101. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
102. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
103. Agrawal P, Nawadkar R, Ojha H, Kumar J, Sahu A. Complement evasion strategies of viruses: an overview. Front Microbiol. (2017) 8:1117. doi: 10.3389/fmicb.2017.01117
104. Regal JF, Burwick RM, Fleming SD. The complement system and preeclampsia. Curr Hypertens Rep. (2017) 19:87. doi: 10.1007/s11906-017-0784-4
105. Rattan A, Pawar SD, Nawadkar R, Kulkarni N, Lal G, Mullick J, et al. Synergy between the classical and alternative pathways of complement is essential for conferring effective protection against the pandemic influenza A(H1N1) 2009 virus infection. PLoS Pathog. (2017) 13:e1006248. doi: 10.1371/journal.ppat.1006248
106. Malakhova OA, Yan M, Malakhov MP, Yuan Y, Ritchie KJ, Kim KI, et al. Protein ISGylation modulates the JAK-STAT signaling pathway. Genes Dev. (2003) 17:455–60. doi: 10.1101/gad.1056303
107. Marafioti T, Paterson JC, Ballabio E, Reichard KK, Tedoldi S, Hollowood K, et al. Novel markers of normal and neoplastic human plasmacytoid dendritic cells. Blood. (2008) 111:3778–92. doi: 10.1182/blood-2007-10-117531
108. Honke N, Shaabani N, Zhang DE, Hardt C, Lang KS. Multiple functions of USP18. Cell Death Dis. (2016) 7:e2444. doi: 10.1038/cddis.2016.326
109. Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci. (2011) 1238:91–8. doi: 10.1111/j.1749-6632.2011.06220.x
110. Sun S, Zhao G, Liu C, Wu X, Guo Y, Yu H, et al. Inhibition of complement activation alleviates acute lung injury induced by highly pathogenic avian influenza H5N1 virus infection. Am J Respir Cell Mol Biol. (2013) 49:221–30. doi: 10.1165/rcmb.2012-0428OC
111. Wilk CM. Coronaviruses hijack the complement system. Nat Rev Immunol. (2020) 20:350. doi: 10.1038/s41577-020-0314-5
112. Gao T, Hu M, Zhang X, Li H, Zhu L, Liu H, et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv. (2020). doi: 10.1101/2020.03.29.20041962
113. Schaefer TM, Fahey JV, Wright JA, Wira CR. Innate immunity in the human female reproductive tract: antiviral response of uterine epithelial cells to the TLR3 agonist poly(I:C). J Immunol. (2005) 174:992–1002. doi: 10.4049/jimmunol.174.2.992
114. Klotman ME, Chang TL. Defensins in innate antiviral immunity. Nat Rev Immunol. (2006) 6:447–56. doi: 10.1038/nri1860
115. Varrey A, Romero R, Panaitescu B, Miller D, Chaiworapongsa T, Patwardhan M, et al. Human beta-defensin-1: a natural antimicrobial peptide present in amniotic fluid that is increased in spontaneous preterm labor with intra-amniotic infection. Am J Reprod Immunol. (2018) 80:e13031. doi: 10.1111/aji.13031
116. Para R, Romero R, Miller D, Panaitescu B, Varrey A, Chaiworapongsa T, et al. Human beta-defensin-3 participates in intra-amniotic host defense in women with labor at term, spontaneous preterm labor and intact membranes, and preterm prelabor rupture of membranes. J Matern Fetal Neonatal Med. (2019) 32:1–16. doi: 10.1080/14767058.2019.1597047
117. King AE, Kelly RW, Sallenave JM, Bocking AD, Challis JR. Innate immune defences in the human uterus during pregnancy. Placenta. (2007) 28:1099–106. doi: 10.1016/j.placenta.2007.06.002
118. Tecle T, Tripathi S, Hartshorn KL. Review: defensins and cathelicidins in lung immunity. Innate Immun. (2010) 16:151–9. doi: 10.1177/1753425910365734
119. Frew L, Makieva S, McKinlay AT, McHugh BJ, Doust A, Norman JE, et al. Human cathelicidin production by the cervix. PLoS ONE. (2014) 9:e103434. doi: 10.1371/journal.pone.0103434
120. Ramos NL, Sekikubo M, Kironde F, Mirembe F, Saaf M, Brauner A. The impact of vitamin D on the innate immune response to uropathogenic Escherichia coli during pregnancy. Clin Microbiol Infect. (2015) 21:482 e1–7. doi: 10.1016/j.cmi.2014.12.010
121. Hartshorn KL. Role of surfactant protein A and D (SP-A and SP-D) in human antiviral host defense. Front Biosci. (2010) 2:527–46. doi: 10.2741/s83
122. Tripathi S, Tecle T, Verma A, Crouch E, White M, Hartshorn KL. The human cathelicidin LL-37 inhibits influenza A viruses through a mechanism distinct from that of surfactant protein D or defensins. J Gen Virol. (2013) 94(Pt 1):40–9. doi: 10.1099/vir.0.045013-0
123. Hsieh IN, De Luna X, White MR, Hartshorn KL. The role and molecular mechanism of action of surfactant protein D in innate host defense against influenza A virus. Front Immunol. (2018) 9:1368. doi: 10.3389/fimmu.2018.01368
124. Parsons LM, An Y, Qi L, White MR, van der Woude R, Hartshorn KL, et al. Influenza virus hemagglutinins H2, H5, H6, and H11 are not targets of pulmonary surfactant protein D: N-glycan subtypes in host-pathogen interactions. J Virol. (2020) 94:e01951–19. doi: 10.1128/JVI.01951-19
125. Tripathi S, Wang G, White M, Qi L, Taubenberger J, Hartshorn KL. Antiviral activity of the human cathelicidin, LL-37, and derived peptides on seasonal and pandemic influenza A viruses. PLoS ONE. (2015) 10:e0124706. doi: 10.1371/journal.pone.0124706
126. White MR, Tripathi S, Verma A, Kingma P, Takahashi K, Jensenius J, et al. Collectins, H-ficolin and LL-37 reduce influence viral replication in human monocytes and modulate virus-induced cytokine production. Innate Immun. (2017) 23:77–88. doi: 10.1177/1753425916678470
127. Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. (2002) 168:554–61. doi: 10.4049/jimmunol.168.2.554
128. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. (2004) 303:1526–9. doi: 10.1126/science.1093620
129. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. (2004) 303:1529–31. doi: 10.1126/science.1093616
130. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. (2003) 301:640–3. doi: 10.1126/science.1087262
131. Beijar EC, Mallard C, Powell TL. Expression and subcellular localization of TLR-4 in term and first trimester human placenta. Placenta. (2006) 27:322–6. doi: 10.1016/j.placenta.2004.12.012
132. Krikun G, Lockwood CJ, Abrahams VM, Mor G, Paidas M, Guller S. Expression of Toll-like receptors in the human decidua. Histol Histopathol. (2007) 22:847–54. doi: 10.14670/HH-22.847
133. Patni S, Wynen LP, Seager AL, Morgan G, White JO, Thornton CA. Expression and activity of Toll-like receptors 1–9 in the human term placenta and changes associated with labor at term. Biol Reprod. (2009) 80:243–8. doi: 10.1095/biolreprod.108.069252
134. Patni S, Bryant AH, Wynen LP, Seager AL, Morgan G, Thornton CA. Functional activity but not gene expression of toll-like receptors is decreased in the preterm versus term human placenta. Placenta. (2015) 36:1031–8. doi: 10.1016/j.placenta.2015.06.017
135. Silasi M, Cardenas I, Kwon JY, Racicot K, Aldo P, Mor G. Viral infections during pregnancy. Am J Reprod Immunol. (2015) 73:199–213. doi: 10.1111/aji.12355
136. Pudney J, He X, Masheeb Z, Kindelberger DW, Kuohung W, Ingalls RR. Differential expression of toll-like receptors in the human placenta across early gestation. Placenta. (2016) 46:1–10. doi: 10.1016/j.placenta.2016.07.005
137. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. (1998) 281:1191–3. doi: 10.1126/science.281.5380.1191
138. Wang B, Koga K, Osuga Y, Cardenas I, Izumi G, Takamura M, et al. Toll-like receptor-3 ligation-induced indoleamine 2, 3-dioxygenase expression in human trophoblasts. Endocrinology. (2011) 152:4984–92. doi: 10.1210/en.2011-0278
139. Koga K, Izumi G, Mor G, Fujii T, Osuga Y. Toll-like receptors at the maternal-fetal interface in normal pregnancy and pregnancy complications. Am J Reprod Immunol. (2014) 72:192–205. doi: 10.1111/aji.12258
140. Duriez M, Quillay H, Madec Y, El Costa H, Cannou C, Marlin R, et al. Human decidual macrophages and NK cells differentially express Toll-like receptors and display distinct cytokine profiles upon TLR stimulation. Front Microbiol. (2014) 5:316. doi: 10.3389/fmicb.2014.00316
141. Olmos-Ortiz A, Flores-Espinosa P, Mancilla-Herrera I, Vega-Sanchez R, Diaz L, Zaga-Clavellina V. Innate immune cells and toll-like receptor-dependent responses at the maternal-fetal interface. Int J Mol Sci. (2019) 20:3654. doi: 10.3390/ijms20153654
142. Girardi G, Lingo JJ, Fleming SD, Regal JF. Essential role of complement in pregnancy: from implantation to parturition and beyond. Front Immunol. (2020) 11:1681. doi: 10.3389/fimmu.2020.01681
143. Denny KJ, Woodruff TM, Taylor SM, Callaway LK. Complement in pregnancy: a delicate balance. Am J Reprod Immunol. (2013) 69:3–11. doi: 10.1111/aji.12000
144. Agostinis C, Tedesco F, Bulla R. Alternative functions of the complement protein C1q at embryo implantation site. J Reprod Immunol. (2017) 119:74–80. doi: 10.1016/j.jri.2016.09.001
145. Girardi G. Complement activation, a threat to pregnancy. Semin Immunopathol. (2018) 40:103–11. doi: 10.1007/s00281-017-0645-x
146. Ip WK, Chan KH, Law HK, Tso GH, Kong EK, Wong WH, et al. Mannose-binding lectin in severe acute respiratory syndrome coronavirus infection. J Infect Dis. (2005) 191:1697–704. doi: 10.1086/429631
147. Tam JC, Bidgood SR, McEwan WA, James LC. Intracellular sensing of complement C3 activates cell autonomous immunity. Science. (2014) 345:1256070. doi: 10.1126/science.1256070
148. Avirutnan P, Hauhart RE, Somnuke P, Blom AM, Diamond MS, Atkinson JP. Binding of flavivirus nonstructural protein NS1 to C4b binding protein modulates complement activation. J Immunol. (2011) 187:424–33. doi: 10.4049/jimmunol.1100750
149. Conde JN, da Silva EM, Allonso D, Coelho DR, Andrade IDS, de Medeiros LN, et al. Inhibition of the membrane attack complex by dengue virus NS1 through interaction with vitronectin and terminal complement proteins. J Virol. (2016) 90:9570–81. doi: 10.1128/JVI.00912-16
150. Zhang J, Li G, Liu X, Wang Z, Liu W, Ye X. Influenza A virus M1 blocks the classical complement pathway through interacting with C1qA. J Gen Virol. (2009) 90(Pt 11):2751–8. doi: 10.1099/vir.0.014316-0
151. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
152. Miller D, Motomura K, Garcia-Flores V, Romero R, Gomez-Lopez N. Innate lymphoid cells in the maternal and fetal compartments. Front Immunol. (2018) 9:2396. doi: 10.3389/fimmu.2018.02396
153. Vacca P, Montaldo E, Croxatto D, Loiacono F, Canegallo F, Venturini PL, et al. Identification of diverse innate lymphoid cells in human decidua. Mucosal Immunol. (2015) 8:254–64. doi: 10.1038/mi.2014.63
154. Croxatto D, Micheletti A, Montaldo E, Orecchia P, Loiacono F, Canegallo F, et al. Group 3 innate lymphoid cells regulate neutrophil migration and function in human decidua. Mucosal Immunol. (2016) 9:1372–83. doi: 10.1038/mi.2016.10
155. Vacca P, Vitale C, Munari E, Cassatella MA, Mingari MC, Moretta L. Human innate lymphoid cells: their functional and cellular interactions in decidua. Front Immunol. (2018) 9:1897. doi: 10.3389/fimmu.2018.01897
156. Weizman OE, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C. ILC1 confer early host protection at initial sites of viral infection. Cell. (2017) 171:795–808.e12. doi: 10.1016/j.cell.2017.09.052
157. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. (2011) 12:1045–54. doi: 10.1038/ni.2131
158. Monticelli LA, Sonnenberg GF, Artis D. Innate lymphoid cells: critical regulators of allergic inflammation and tissue repair in the lung. Curr Opin Immunol. (2012) 24:284–9. doi: 10.1016/j.coi.2012.03.012
159. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. (2012) 12:239–52. doi: 10.1038/nri3174
160. Abdul-Careem MF, Mian MF, Yue G, Gillgrass A, Chenoweth MJ, Barra NG, et al. Critical role of natural killer cells in lung immunopathology during influenza infection in mice. J Infect Dis. (2012) 206:167–77. doi: 10.1093/infdis/jis340
161. Kopcow HD, Allan DS, Chen X, Rybalov B, Andzelm MM, Ge B, et al. Human decidual NK cells form immature activating synapses and are not cytotoxic. Proc Natl Acad Sci USA. (2005) 102:15563–8. doi: 10.1073/pnas.0507835102
162. Crespo AC, Strominger JL, Tilburgs T. Expression of KIR2DS1 by decidual natural killer cells increases their ability to control placental HCMV infection. Proc Natl Acad Sci USA. (2016) 113:15072–7. doi: 10.1073/pnas.1617927114
163. Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol. (2011) 89:216–24. doi: 10.1038/icb.2010.78
164. Ivarsson MA, Stiglund N, Marquardt N, Westgren M, Gidlof S, Bjorkstrom NK. Composition and dynamics of the uterine NK cell KIR repertoire in menstrual blood. Mucosal Immunol. (2017) 10:322–31. doi: 10.1038/mi.2016.50
165. Lopez-Verges S, Milush JM, Schwartz BS, Pando MJ, Jarjoura J, York VA, et al. Expansion of a unique CD57(+)NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci USA. (2011) 108:14725–32. doi: 10.1073/pnas.1110900108
166. Bjorkstrom NK, Lindgren T, Stoltz M, Fauriat C, Braun M, Evander M, et al. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J Exp Med. (2011) 208:13–21. doi: 10.1084/jem.20100762
167. Nikzad R, Angelo LS, Aviles-Padilla K, Le DT, Singh VK, Bimler L, et al. Human natural killer cells mediate adaptive immunity to viral antigens. Sci Immunol. (2019) 4:eaat8116. doi: 10.1126/sciimmunol.aat8116
168. Mandala WL, Gondwe EN, Molyneux ME, MacLennan JM, MacLennan CA. Leukocyte counts and lymphocyte subsets in relation to pregnancy and HIV infection in Malawian women. Am J Reprod Immunol. (2017) 78:e1267. doi: 10.1111/aji.12678
169. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev. (2010) 62:726–59. doi: 10.1124/pr.110.002733
170. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
171. Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe. (2012) 12:109–16. doi: 10.1016/j.chom.2012.05.015
172. Schonrich G, Raftery MJ. Neutrophil extracellular traps go viral. Front Immunol. (2016) 7:366. doi: 10.3389/fimmu.2016.00366
173. Jenne CN, Kubes P. Virus-induced NETs—critical component of host defense or pathogenic mediator? PLoS Pathog. (2015) 11:e1004546. doi: 10.1371/journal.ppat.1004546
174. Pillai PS, Molony RD, Martinod K, Dong H, Pang IK, Tal MC, et al. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science. (2016) 352:463–6. doi: 10.1126/science.aaf3926
175. Zhu L, Liu L, Zhang Y, Pu L, Liu J, Li X, et al. High level of neutrophil extracellular traps correlates with poor prognosis of severe influenza A infection. J Infect Dis. (2018) 217:428–37. doi: 10.1093/infdis/jix475
176. Sung PS, Huang TF, Hsieh SL. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat Commun. (2019) 10:2402. doi: 10.1038/s41467-019-10360-4
177. Perricone R, De Carolis C, Giacomelli R, Guarino MD, De Sanctis G, Fontana L. GM-CSF and pregnancy: evidence of significantly reduced blood concentrations in unexplained recurrent abortion efficiently reverted by intravenous immunoglobulin treatment. Am J Reprod Immunol. (2003) 50:232–7. doi: 10.1034/j.1600-0897.2003.00083.x
178. Robertson SA. GM-CSF regulation of embryo development and pregnancy. Cytokine Growth Factor Rev. (2007) 18:287–98. doi: 10.1016/j.cytogfr.2007.04.008
179. Blazkova J, Gupta S, Liu Y, Gaudilliere B, Ganio EA, Bolen CR, et al. Multicenter systems analysis of human blood reveals immature neutrophils in males and during pregnancy. J Immunol. (2017) 198:2479–88. doi: 10.4049/jimmunol.1601855
180. Caballero-Campo P, Dominguez F, Coloma J, Meseguer M, Remohi J, Pellicer A, et al. Hormonal and embryonic regulation of chemokines IL-8, MCP-1 and RANTES in the human endometrium during the window of implantation. Mol Hum Reprod. (2002) 8:375–84. doi: 10.1093/molehr/8.4.375
181. Popovici RM, Betzler NK, Krause MS, Luo M, Jauckus J, Germeyer A, et al. Gene expression profiling of human endometrial-trophoblast interaction in a coculture model. Endocrinology. (2006) 147:5662–75. doi: 10.1210/en.2006-0916
182. Amsalem H, Kwan M, Hazan A, Zhang J, Jones RL, Whittle W, et al. Identification of a novel neutrophil population: proangiogenic granulocytes in second-trimester human decidua. J Immunol. (2014) 193:3070–9. doi: 10.4049/jimmunol.1303117
183. Vento-Tormo R, Efremova M, Botting RA, Turco MY, Vento-Tormo M, Meyer KB, et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature. (2018) 563:347–53. doi: 10.1038/s41586-018-0698-6
184. Suryawanshi H, Morozov P, Straus A, Sahasrabudhe N, Max KEA, Garzia A, et al. A single-cell survey of the human first-trimester placenta and decidua. Sci Adv. (2018) 4:eaau4788. doi: 10.1126/sciadv.aau4788
185. Faas MM, de Vos P. Maternal monocytes in pregnancy and preeclampsia in humans and in rats. J Reprod Immunol. (2017) 119:91–7. doi: 10.1016/j.jri.2016.06.009
186. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. (2014) 5:614. doi: 10.3389/fimmu.2014.00614
187. El Costa H, Quillay H, Marlin R, Cannou C, Duriez M, Benjelloun F, et al. The local environment orchestrates mucosal decidual macrophage differentiation and substantially inhibits HIV-1 replication. Mucosal Immunol. (2016) 9:634–46. doi: 10.1038/mi.2015.87
188. Bachy V, Williams DJ, Ibrahim MA. Altered dendritic cell function in normal pregnancy. J Reprod Immunol. (2008) 78:11–21. doi: 10.1016/j.jri.2007.09.004
189. Kammerer U, Eggert AO, Kapp M, McLellan AD, Geijtenbeek TB, Dietl J, et al. Unique appearance of proliferating antigen-presenting cells expressing DC-SIGN (CD209) in the decidua of early human pregnancy. Am J Pathol. (2003) 162:887–96. doi: 10.1016/S0002-9440(10)63884-9
190. de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med. (2006) 12:1203–7. doi: 10.1038/nm1477
191. To KK, Hung IF, Li IW, Lee KL, Koo CK, Yan WW, et al. Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clin Infect Dis. (2010) 50:850–9. doi: 10.1086/650581
192. Ramathal CY, Bagchi IC, Taylor RN, Bagchi MK. Endometrial decidualization: of mice and men. Semin Reprod Med. (2010) 28:17–26. doi: 10.1055/s-0029-1242989
193. Okada S, Okada H, Sanezumi M, Nakajima T, Yasuda K, Kanzaki H. Expression of interleukin-15 in human endometrium and decidua. Mol Hum Reprod. (2000) 6:75–80. doi: 10.1093/molehr/6.1.75
194. Tilburgs T, Evans JH, Crespo AC, Strominger JL. The HLA-G cycle provides for both NK tolerance and immunity at the maternal-fetal interface. Proc Natl Acad Sci USA. (2015) 112:13312–7. doi: 10.1073/pnas.1517724112
195. Croxatto D, Vacca P, Canegallo F, Conte R, Venturini PL, Moretta L, et al. Stromal cells from human decidua exert a strong inhibitory effect on NK cell function and dendritic cell differentiation. PLoS ONE. (2014) 9:e89006. doi: 10.1371/journal.pone.0089006
196. Hu WT, Huang LL, Li MQ, Jin LP, Li DJ, Zhu XY. Decidual stromal cell-derived IL-33 contributes to Th2 bias and inhibits decidual NK cell cytotoxicity through NF-kappaB signaling in human early pregnancy. J Reprod Immunol. (2015) 109:52–65. doi: 10.1016/j.jri.2015.01.004
197. Vinketova K, Mourdjeva M, Oreshkova T. Human decidual stromal cells as a component of the implantation niche and a modulator of maternal immunity. J Pregnancy. (2016) 2016:8689436. doi: 10.1155/2016/8689436
198. Erkers T, Kaipe H, Nava S, Mollden P, Gustafsson B, Axelsson R, et al. Treatment of severe chronic graft-versus-host disease with decidual stromal cells and tracing with (111)indium radiolabeling. Stem Cells Dev. (2015) 24:253–63. doi: 10.1089/scd.2014.0265
199. Ringden O, Baygan A, Remberger M, Gustafsson B, Winiarski J, Khoein B, et al. Placenta-derived decidua stromal cells for treatment of severe acute graft-versus-host disease. Stem Cells Transl Med. (2018) 7:325–31. doi: 10.1002/sctm.17-0167
200. Yu SX, Zhou FH, Chen W, Jiang GM, Du CT, Hu GQ, et al. Decidual stromal cell necroptosis contributes to polyinosinic-polycytidylic acid-triggered abnormal murine pregnancy. Front Immunol. (2017) 8:916. doi: 10.3389/fimmu.2017.00916
201. Ogbu O, Ajuluchukwu E, Uneke CJ. Lassa fever in West African sub-region: an overview. J Vector Borne Dis. (2007) 44:1–11. Available online at: http://www.mrcindia.org/journal/issues/441001.pdf
202. Brisse ME, Ly H. Hemorrhagic fever-causing arenaviruses: lethal pathogens and potent immune suppressors. Front Immunol. (2019) 10:372. doi: 10.3389/fimmu.2019.00372
203. Price ME, Fisher-Hoch SP, Craven RB, McCormick JB. A prospective study of maternal and fetal outcome in acute Lassa fever infection during pregnancy. BMJ. (1988) 297:584–7. doi: 10.1136/bmj.297.6648.584
204. Smith DR, Holbrook MR, Gowen BB. Animal models of viral hemorrhagic fever. Antiviral Res. (2014) 112:59–79. doi: 10.1016/j.antiviral.2014.10.001
205. Baize S, Marianneau P, Loth P, Reynard S, Journeaux A, Chevallier M, et al. Early and strong immune responses are associated with control of viral replication and recovery in lassa virus-infected cynomolgus monkeys. J Virol. (2009) 83:5890–903. doi: 10.1128/JVI.01948-08
206. Forbes RL, Wark PA, Murphy VE, Gibson PG. Pregnant women have attenuated innate interferon responses to 2009 pandemic influenza A virus subtype H1N1. J Infect Dis. (2012) 206:646–53. doi: 10.1093/infdis/jis377
207. Hastie KM, King LB, Zandonatti MA, Saphire EO. Structural basis for the dsRNA specificity of the Lassa virus NP exonuclease. PLoS ONE. (2012) 7:e44211. doi: 10.1371/journal.pone.0044211
208. Baize S, Kaplon J, Faure C, Pannetier D, Georges-Courbot MC, Deubel V. Lassa virus infection of human dendritic cells and macrophages is productive but fails to activate cells. J Immunol. (2004) 172:2861–9. doi: 10.4049/jimmunol.172.5.2861
209. Pannetier D, Faure C, Georges-Courbot MC, Deubel V, Baize S. Human macrophages, but not dendritic cells, are activated and produce alpha/beta interferons in response to Mopeia virus infection. J Virol. (2004) 78:10516–24. doi: 10.1128/JVI.78.19.10516-10524.2004
210. Pannetier D, Reynard S, Russier M, Journeaux A, Tordo N, Deubel V, et al. Human dendritic cells infected with the nonpathogenic Mopeia virus induce stronger T-cell responses than those infected with Lassa virus. J Virol. (2011) 85:8293–306. doi: 10.1128/JVI.02120-10
211. Schaeffer J, Carnec X, Reynard S, Mateo M, Picard C, Pietrosemoli N, et al. Lassa virus activates myeloid dendritic cells but suppresses their ability to stimulate T cells. PLoS Pathog. (2018) 14:e1007430. doi: 10.1371/journal.ppat.1007430
212. Black BO, Caluwaerts S, Achar J. Ebola viral disease and pregnancy. Obstet Med. (2015) 8:108–13. doi: 10.1177/1753495X15597354
213. Bebell LM, Oduyebo T, Riley LE. Ebola virus disease and pregnancy: a review of the current knowledge of Ebola virus pathogenesis, maternal, neonatal outcomes. Birth Defects Res. (2017) 109:353–62. doi: 10.1002/bdra.23558
214. Henwood PC, Bebell LM, Roshania R, Wolfman V, Mallow M, Kalyanpur A, et al. Ebola virus disease and pregnancy: a retrospective cohort study of patients managed at 5 Ebola treatment units in West Africa. Clin Infect Dis. (2017) 65:292–9. doi: 10.1093/cid/cix290
215. Mupapa K, Mukundu W, Bwaka MA, Kipasa M, De Roo A, Kuvula K, et al. Ebola hemorrhagic fever and pregnancy. J Infect Dis. (1999) 179(Suppl, 1):S11–2. doi: 10.1086/514289
216. Stroher U, West E, Bugany H, Klenk HD, Schnittler HJ, Feldmann H. Infection and activation of monocytes by Marburg and Ebola viruses. J Virol. (2001) 75:11025–33. doi: 10.1128/JVI.75.22.11025-11033.2001
217. Villinger F, Rollin PE, Brar SS, Chikkala NF, Winter J, Sundstrom JB, et al. Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. J Infect Dis. (1999) 179(Suppl. 1):S188–91. doi: 10.1086/514283
218. Leung DW, Prins KC, Borek DM, Farahbakhsh M, Tufariello JM, Ramanan P, et al. Structural basis for dsRNA recognition and interferon antagonism by Ebola VP35. Nat Struct Mol Biol. (2010) 17:165–72. doi: 10.1038/nsmb.1765
219. Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, et al. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol. (2006) 80:5156–67. doi: 10.1128/JVI.02349-05
220. Jouvenet N, Neil SJ, Zhadina M, Zang T, Kratovac Z, Lee Y, et al. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol. (2009) 83:1837–44. doi: 10.1128/JVI.02211-08
221. Raj RS, Bonney EA, Phillippe M. Influenza, immune system, and pregnancy. Reprod Sci. (2014) 21:1434–51. doi: 10.1177/1933719114537720
222. Bower H, Grass JE, Veltus E, Brault A, Campbell S, Basile AJ, et al. Delivery of an ebola virus-positive stillborn infant in a rural community health center, sierra leone, 2015. Am J Trop Med Hyg. (2016) 94:417–9. doi: 10.4269/ajtmh.15-0619
223. Fallah MP, Skrip LA, Dahn BT, Nyenswah TG, Flumo H, Glayweon M, et al. Pregnancy outcomes in Liberian women who conceived after recovery from Ebola virus disease. Lancet Glob Health. (2016) 4:e678–9. doi: 10.1016/S2214-109X(16)30147-4
224. Muehlenbachs A, de la Rosa Vazquez O, Bausch DG, Schafer IJ, Paddock CD, Nyakio JP, et al. Ebola virus disease in pregnancy: clinical, histopathologic, immunohistochemical findings. J Infect Dis. (2017) 215:64–9. doi: 10.1093/infdis/jiw206
225. Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. (2011) 477:340–3. doi: 10.1038/nature10348
226. Olgun NS. Viral infections in pregnancy: a focus on ebola virus. Curr Pharm Des. (2018) 24:993–8. doi: 10.2174/1381612824666180130121946
227. World Health Organization. World Health Organization Fact Sheet: Dengue and Severe Dengue. Geneva: World Health Organization (2020).
228. Basurko C, Everhard S, Matheus S, Restrepo M, Hilderal H, Lambert V, et al. A prospective matched study on symptomatic dengue in pregnancy. PLoS ONE. (2018) 13:e0202005. doi: 10.1371/journal.pone.0202005
229. Paixao ES, Teixeira MG, Costa M, Rodrigues LC. Dengue during pregnancy and adverse fetal outcomes: a systematic review and meta-analysis. Lancet Infect Dis. (2016) 16:857–65. doi: 10.1016/S1473-3099(16)00088-8
230. Xiong YQ, Mo Y, Shi TL, Zhu L, Chen Q. Dengue virus infection during pregnancy increased the risk of adverse fetal outcomes? An updated meta-analysis. J Clin Virol. (2017) 94:42–9. doi: 10.1016/j.jcv.2017.07.008
231. Ribeiro CF, Silami VG, Brasil P, Nogueira RM. Sickle-cell erythrocytes in the placentas of dengue-infected women. Int J Infect Dis. (2012) 16:e72. doi: 10.1016/j.ijid.2011.09.005
232. Ribeiro CF, Lopes VGS, Brasil P, Pires ARC, Rohloff R, Nogueira RMR. Dengue infection in pregnancy and its impact on the placenta. Int J Infect Dis. (2017) 55:109–12. doi: 10.1016/j.ijid.2017.01.002
233. Vogt MB, Lahon A, Arya RP, Spencer Clinton JL, Rico-Hesse R. Dengue viruses infect human megakaryocytes, with probable clinical consequences. PLoS Negl Trop Dis. (2019) 13:e0007837. doi: 10.1371/journal.pntd.0007837
234. Campbell RA, Schwertz H, Hottz ED, Rowley JW, Manne BK, Washington AV, et al. Human megakaryocytes possess intrinsic antiviral immunity through regulated induction of IFITM3. Blood. (2019) 133:2013–26. doi: 10.1182/blood-2018-09-873984
235. Costa VV, Ye W, Chen Q, Teixeira MM, Preiser P, Ooi EE, et al. Dengue virus-infected dendritic cells, but not monocytes, activate natural killer cells through a contact-dependent mechanism involving adhesion molecules. mBio. (2017) 8:e00741–17. doi: 10.1128/mBio.00741-17
236. Martinez-Moreno J, Hernandez JC, Urcuqui-Inchima S. Effect of high doses of vitamin D supplementation on dengue virus replication, Toll-like receptor expression, and cytokine profiles on dendritic cells. Mol Cell Biochem. (2020) 464:169–80. doi: 10.1007/s11010-019-03658-w
237. Jilani N, Das BC, Husain SA, Baweja UK, Chattopadhya D, Gupta RK, et al. Hepatitis E virus infection and fulminant hepatic failure during pregnancy. J Gastroenterol Hepatol. (2007) 22:676–82. doi: 10.1111/j.1440-1746.2007.04913.x
238. Shalimar, Acharya SK. Hepatitis e and acute liver failure in pregnancy. J Clin Exp Hepatol. (2013) 3:213–24. doi: 10.1016/j.jceh.2013.08.009
239. Sehgal R, Patra S, David P, Vyas A, Khanam A, Hissar S, et al. Impaired monocyte-macrophage functions and defective Toll-like receptor signaling in hepatitis E virus-infected pregnant women with acute liver failure. Hepatology. (2015) 62:1683–96. doi: 10.1002/hep.28143
240. Karna R, Hazam RK, Borkakoti J, Kumar A, Kar P. A 5-year single-center experience of hepatitis E virus infection during pregnancy. J Clin Exp Hepatol. (2020) 10:135–8. doi: 10.1016/j.jceh.2019.09.003
241. Gouilly J, Chen Q, Siewiera J, Cartron G, Levy C, Dubois M, et al. Genotype specific pathogenicity of hepatitis E virus at the human maternal-fetal interface. Nat Commun. (2018) 9:4748. doi: 10.1038/s41467-018-07200-2
242. Perez-Gracia MT, Suay-Garcia B, Mateos-Lindemann ML. Hepatitis E and pregnancy: current state. Rev Med Virol. (2017) 27:e1929. doi: 10.1002/rmv.1929
243. Bose PD, Das BC, Hazam RK, Kumar A, Medhi S, Kar P. Evidence of extrahepatic replication of hepatitis E virus in human placenta. J Gen Virol. (2014) 95(Pt 6):1266–71. doi: 10.1099/vir.0.063602-0
244. El-Mokhtar MA, Othman ER, Khashbah MY, Ismael A, Ghaliony MA, Seddik MI, et al. evidence of the extrahepatic replication of hepatitis e virus in human endometrial stromal cells. Pathogens. (2020) 9:245. doi: 10.3390/pathogens9040295
245. Paget J, Spreeuwenberg P, Charu V, Taylor RJ, Iuliano AD, Bresee J, et al. Global mortality associated with seasonal influenza epidemics: new burden estimates and predictors from the GLaMOR Project. J Glob Health. (2019) 9:020421. doi: 10.7189/jogh.09.020421
246. Lapinsky SE. Critical illness as a result of influenza A/H1N1 infection in pregnancy. BMJ. (2010) 340:c1235. doi: 10.1136/bmj.c1235
247. Mertz D, Lo CK, Lytvyn L, Ortiz JR, Loeb M, Flurisk I. Pregnancy as a risk factor for severe influenza infection: an individual participant data meta-analysis. BMC Infect Dis. (2019) 19:683. doi: 10.1186/s12879-019-4318-3
248. Mauad T, Hajjar LA, Callegari GD, da Silva LF, Schout D, Galas FR, et al. Lung pathology in fatal novel human influenza A (H1N1) infection. Am J Respir Crit Care Med. (2010) 181:72–9. doi: 10.1164/rccm.200909-1420OC
249. Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, et al. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. (2006) 2:e53. doi: 10.1371/journal.ppat.0020053
250. Wu W, Zhang W, Duggan ES, Booth JL, Zou MH, Metcalf JP. RIG-I and TLR3 are both required for maximum interferon induction by influenza virus in human lung alveolar epithelial cells. Virology. (2015) 482:181–8. doi: 10.1016/j.virol.2015.03.048
251. Kikkert M. Innate immune evasion by human respiratory RNA viruses. J Innate Immun. (2020) 12:4–20. doi: 10.1159/000503030
252. Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, et al. Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell. (2020) 180:1115–29.e13. doi: 10.1016/j.cell.2020.02.050
253. Lambrecht BN, Hammad H. Lung dendritic cells in respiratory viral infection and asthma: from protection to immunopathology. Annu Rev Immunol. (2012) 30:243–70. doi: 10.1146/annurev-immunol-020711-075021
254. Vanders RL, Murphy VE, Gibson PG, Hansbro PM, Wark PA. CD8 T cells and dendritic cells: key players in the attenuated maternal immune response to influenza infection. J Reprod Immunol. (2015) 107:1–9. doi: 10.1016/j.jri.2014.09.051
255. Hall OJ, Limjunyawong N, Vermillion MS, Robinson DP, Wohlgemuth N, Pekosz A, et al. Progesterone-based therapy protects against influenza by promoting lung repair and recovery in females. PLoS Pathog. (2016) 12:e1005840. doi: 10.1371/journal.ppat.1005840
256. Hall OJ, Nachbagauer R, Vermillion MS, Fink AL, Phuong V, Krammer F, et al. Progesterone-based contraceptives reduce adaptive immune responses and protection against sequential influenza A virus infections. J Virol. (2017) 91:e02160–16. doi: 10.1128/JVI.02160-16
257. Schwartz DA. An analysis of 38 pregnant women with COVID-19, their newborn infants, and maternal-fetal transmission of SARS-CoV-2: maternal coronavirus infections and pregnancy outcomes. Arch Pathol Lab Med. (2020) 144:799–805. doi: 10.5858/arpa.2020-0901-SA
258. Mehta N, Chen K, Hardy E, Powrie R. Respiratory disease in pregnancy. Best Pract Res Clin Obstet Gynaecol. (2015) 29:598–611. doi: 10.1016/j.bpobgyn.2015.04.005
259. Hui DS. Epidemic and emerging coronaviruses (severe acute respiratory syndrome and middle east respiratory syndrome). Clin Chest Med. (2017) 38:71–86. doi: 10.1016/j.ccm.2016.11.007
260. World Health Organization. Coronavirus Disease (COVID-19) Situation Report-−209. Geneva: World Health Organization (2020).
261. Hu Y, Sun J, Dai Z, Deng H, Li X, Huang Q, et al. Prevalence and severity of corona virus disease 2019 (COVID-19): a systematic review and meta-analysis. J Clin Virol. (2020) 127:104371. doi: 10.1016/j.jcv.2020.104371
262. Khalil A, Kalafat E, Benlioglu C, O'Brien P, Morris E, Draycott T, et al. SARS-CoV-2 infection in pregnancy: a systematic review and meta-analysis of clinical features and pregnancy outcomes. EClinicalMedicine. (2020). 25:100446. doi: 10.1016/j.eclinm.2020.100446
263. Kotlyar A, Grechukhina O, Chen A, Popkhadze S, Grimshaw A, Tal O, et al. Vertical transmission of COVID-19: a systematic review and meta-analysis. Am J Obstet Gynecol. (2020). doi: 10.1016/j.ajog.2020.07.049. [Epub ahead of print].
264. Vivanti AJ, Vauloup-Fellous C, Prevot S, Zupan V, Suffee C, Do Cao J, et al. Transplacental transmission of SARS-CoV-2 infection. Nat Commun. (2020) 11:3572. doi: 10.1038/s41467-020-17436-6
265. Romero-Brey I, Bartenschlager R. Membranous replication factories induced by plus-strand RNA viruses. Viruses. (2014) 6:2826–57. doi: 10.3390/v6072826
266. Kindler E, Gil-Cruz C, Spanier J, Li Y, Wilhelm J, Rabouw HH, et al. Early endonuclease-mediated evasion of RNA sensing ensures efficient coronavirus replication. PLoS Pathog. (2017) 13:e1006195. doi: 10.1371/journal.ppat.1006195
267. Tanaka T, Kamitani W, DeDiego ML, Enjuanes L, Matsuura Y. Severe acute respiratory syndrome coronavirus nsp1 facilitates efficient propagation in cells through a specific translational shutoff of host mRNA. J Virol. (2012) 86:11128–37. doi: 10.1128/JVI.01700-12
268. Paules CI, Marston HD, Fauci AS. Measles in 2019 - going backward. N Engl J Med. (2019) 380:2185–7. doi: 10.1056/NEJMp1905099
269. Bansal J, Hameed A. Measles in pregnancy. BMJ Case Rep. (2019) 12:e228781. doi: 10.1136/bcr-2018-228781
270. Pique-Regi R, Romero R, Tarca AL, Sendler ED, Xu Y, Garcia-Flores V, et al. Single cell transcriptional signatures of the human placenta in term and preterm parturition. Elife. (2019) 8:52004. doi: 10.7554/eLife.52004
271. Stevenson DK, Wong RJ, Aghaeepour N, Maric I, Angst MS, Contrepois K, et al. Towards personalized medicine in maternal and child health: integrating biologic and social determinants. Pediatr Res. (2020). doi: 10.1038/s41390-020-0981-8. [Epub ahead of print].
272. Toothaker JM, Presicce P, Cappelletti M, Stras SF, McCourt CC, Chougnet CA, et al. Immune cells in the placental villi contribute to intra-amniotic inflammation. Front Immunol. (2020) 11:866. doi: 10.3389/fimmu.2020.00866
273. Turco MY, Gardner L, Kay RG, Hamilton RS, Prater M, Hollinshead MS, et al. Trophoblast organoids as a model for maternal-fetal interactions during human placentation. Nature. (2018) 564:263–7. doi: 10.1038/s41586-018-0753-3
Keywords: pregnancy, innate antiviral immunity, Lassa virus, Ebola virus, dengue virus, hepatitis E, influenza virus, emerging coronavirus
Citation: Cornish EF, Filipovic I, Åsenius F, Williams DJ and McDonnell T (2020) Innate Immune Responses to Acute Viral Infection During Pregnancy. Front. Immunol. 11:572567. doi: 10.3389/fimmu.2020.572567
Received: 14 June 2020; Accepted: 31 August 2020;
Published: 30 September 2020.
Edited by:
Sherry Fleming, Kansas State University, United StatesReviewed by:
Nardhy Gomez-Lopez, Wayne State University, United StatesBoris Michael Hartmann, Österreichische Agentur für Gesundheit und Ernährungssicherheit, Austria
Copyright © 2020 Cornish, Filipovic, Åsenius, Williams and McDonnell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas McDonnell, dGhvbWFzLm1jZG9ubmVsbC4xMUB1Y2wuYWMudWs=