 Weiwei Chen
Weiwei Chen Qiang Shu
Qiang Shu Jie Fan
Jie Fan- 1Department of Surgery, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 2The Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou, China
- 3Research and Development, Veterans Affairs Pittsburgh Healthcare System, Pittsburgh, PA, United States
- 4McGowan Institute for Regenerative Medicine, University of Pittsburgh, Pittsburgh, PA, United States
Emerging evidence supports the involvement of nervous system in the regulation of immune responses. Group 2 innate lymphoid cells (ILC2), which function as a crucial bridge between innate and adaptive immunity, are present in large numbers in barrier tissues. Neuropeptides and neurotransmitters have been found to participate in the regulation of ILC2, adding a new dimension to neuroimmunity. However, a comprehensive and detailed overview of the mechanisms of neural regulation of ILC2, associated with previous findings and prospects for future research, is still lacking. In this review, we compile existing information that supports neurons as yet poorly understood regulators of ILC2 in the field of lung innate and adaptive immunity, focusing on neural regulation of the interaction between ILC2 and pulmonary immune cells.
Introduction
The past decade has witnessed an unprecedented interest in the neural modulation of immunity (1–4). The immune barrier consists of innate and adaptive components that adopt different strategies to perceive and respond to pathogen challenge. In the context of innate immunity, innate lymphoid cells (ILCs) have been demonstrated to be a crucial bridge between both immunity branches (5, 6).
ILCs are a heterogeneous family of lymphocytes that lack re-arranged antigen receptors present on B and T cells. In earlier studies, lymphoid tissue inducer (LTi) cells and natural killer (NK) cells were initially identified as the subgroups of the ILCs (7–10). In recent years, more subgroups of the ILCs were discovered and, based on the surface markers, cytokines, and transcription factors, categorized into three major types, ILC1, ILC2 and ILC3 (11, 12). These ILCs groups have distinct phenotypic, developmental, and functional properties. They are the innate counterparts of T lymphocytes: ILC1, ILC2, and ILC3 mirror CD4+ T helper (Th)1, Th2, and Th17 cells, respectively, based on cytokine secretion and transcription factor expression (11, 13, 14). ILC1 consists of conventional NK cells and ILC1s. T-bet, a T-box transcription factor, is indispensable for the differentiation and interferon-gamma (IFN-γ) secretion ability of ILC1. RORα and GATA3 are essential for the development of ILC2, which can be grouped into transient, circulating inflammatory ILC2 (iILC2) and tissue-resident natural ILC2 (nILC2) types (15, 16). ILC3 comprises the classical lymphoid inducer (LTi) cells and LTi-like ILC3 with or without natural cytotoxicity receptors (NCRs), all of which rely on the RORγt (transcription factor) for development and secrete IL-17 and/or IL-22. ILCs protect individuals against infectious agents, response to inflammatory stimuli, and orchestrate lymphoid organogenesis and tissue repair, at various tissues especially mucosal barriers (17–19).
Among all subsets, ILC2 are the center of numerous investigations. They are mainly localized at mucosal barriers, e.g. the small intestine, skin, and lung (19–21). ILC2 are a master regulator of immune and inflammatory responses, but their own regulatory mechanisms remain largely elusive.
ILC2 are activated by host-derived alarmins such as IL-25, IL-33, and thymic stromal lymphopoietin (TSLP), which are expressed during tissue injury (22–24). Once activation takes place, ILC2 release large quantities of cytokines such as IL-4, IL-5, IL-6, IL-9, IL-10, IL-13, IL-17, and amphiregulin (16, 25–28). Furthermore, ILC2 interact with other cells through surface-bound molecules, such as CD80/86, MHC class II, PD-L1, OX40L, and inducible costimulator ligand (ICOS-L), and participate in immune-regulatory functions (29–32). ILC2 play critical roles in the regulation of inflammation, allergic immunity, metabolic homeostasis, parasite rejection, and tissue repair. Dysregulation of ILC2 contributes to inflammatory responses, including allergen-induced lung inflammation (33, 34), airway hyperreactivity (35), and atopic dermatitis (36).
Currently, the nervous system is found to have complex dual functions to quickly stimulate or suppress immune cells to defend the body against various inflammatory responses. There are continuing advances in our knowledge of neural regulation of ILC2, these brilliant results provide a new dimension of immune regulation (37–47). Studies have shown that receptors for norepinephrine, acetylcholine (Ach), neuromedin U (NMU), neuromedin B (NMB), α-Calcitonin Gene-Related Peptide (CGRP), and other neurotransmitters are present on T cells, dendritic cells (DCs), macrophages, ILC2, and other immune cells (19, 37, 38, 44, 48–50), and pattern-recognition receptors (PRRs) and cytokine receptors are distributed on neurons (51–54). Interestingly, immune cells are also able to synthesize and secrete catecholamines, acetylcholine, CGRP, and other neurotransmitters (39, 48, 49, 55). Moreover, ILC subtypes express the nicotinic and muscarinic cholinergic receptor for Ach, β2-adrenergic receptor (β2AR) for epinephrine and norepinephrine, calcitonin receptor-like (CALCRL) for CGRP, neuromedin U receptor 1 (NMUR1) for NMU, neuromedin B receptor (NMBR) for NMB, and VPAC1/2 (vasoactive intestinal peptide receptor) for vasoactive intestinal peptide (VIP) (19, 39, 44, 50, 56). These findings suggest physical machinery for neuro-immune communications. Also, type I cytokines can also influence cells of the center nervous system (CNS) and mediate what is called “sickness behavior” (57, 58). In this review, we highlight existing information that describes neurons as novel regulators of ILC2 in the context of pulmonary innate and adaptive immunity.
Mechanisms Underlying ILC2 Interact with Other Immune Cells
ILC2 function both as initiator of adaptive immunity or as responder to signals produced by B and T cells. Using ILC2-targeted models, investigations have shown multiple mechanisms by which ILC2 regulate innate and adaptive immune system.
Many previous studies showed that “crosstalk” exist between T cells and ILC2 (Figure 1). For instance, ILC2 are the largest group of the cytokine-secreting leukocytes after ovalbumin or HDM treatment (59), and ILC2 activity is essential for the efficient differentiation of Th2 cells (29, 60–63). In Rag2−/− mice, in which T cells and B cells are depleted due to Rag deficiency, the numbers of ILC2 also markedly decreased after helminth infection, indicating that T cells advance the survival of ILC2 (64). Epithelial cells derived cytokines and alarmins activate ILC2, which can be main producer of type 2 cytokines. Moreover, ILC2 can activate CD4+T cells either in the priming phase or during the effector phase since they present major histocompatibility complex class II (MHCII) (65, 66). IL-33–activated ILC2 enhances DCs migration into cancer tissues via C-C motif chemokine ligand 5 (CCL5) and further improve CD8+ T cell-mediated tumor immunity (67). Combination of anti-PD1 checkpoint blockade with rIL33 treatment collaborates to improve anti-tumor immunity by unleashing ILC2 activity (68). Activated ILC2 further facilitate the polarization of the anti-inflammatory M2 macrophages, which in turn stimulate Foxp3 regulatory T cells (Tregs) (69). Tregs are a subpopulation of T cells which modulate the adaptive immune responses through direct cell-cell interactions, as well as through the inhibitory functions of TGF-β and IL-10.
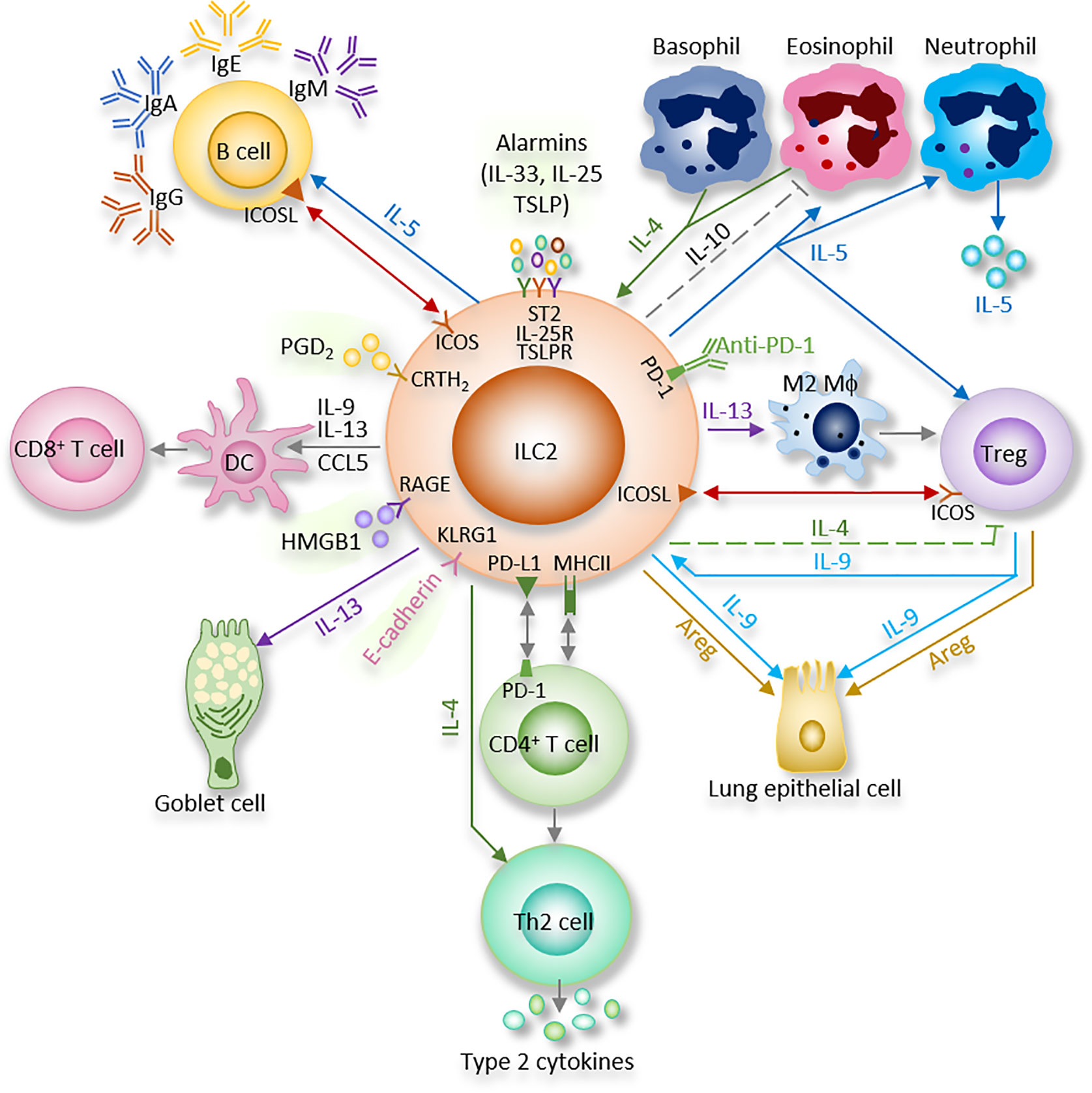
Figure 1 ILC2 interacts with other immune and non-immune cells through a variety of cytokines and cell surface mediators. (1) After activation with alarmins, ILC2 produce type II cytokines and mediators; (2) ILC2 interact with T cells via MHCII, CCL5, PD-1/PD-L1, OX40/OX40L, CD86, CD80, IL-4, IL-5, and IL-13; (3) ILC2 activate B cells to undergo isotype-switching, survival, self-renewal, and secrete antibodies via ICOS/ICOS-L, IL-5; (4) ILC2 stimulate Tregs via IL-5, IL-9, ICOS/ICOS-L, while ILC2-released IL-4 suppress Tregs. Tregs are capable of inhibiting ILC2; (5) ILC2 prime macrophages into a type 2 immune cell phenotype via IL-13; (6) Epithelial cells derived alarmins activate ILC2. ILC2-released IL-9 and Areg protect lung endothelial cells. (7) ILC2 increase eosinophils via IL-5, HMGB1. ILC2 inhibit eosinophils via IL-10. Eosinophil- or basophil-released IL-4 activate ILC2; (8) ILC2 activate DCs via IL-9, IL-13, HMGB1.
Through cytokines and interactions of ICOS with its ligand ICOS-L, ILC2 activate B cell to undergo isotype-switching, survival, and secrete IgG1 and IgE (64, 70, 71). IgE produced by B cells, together with type 2 cytokines released by Th2 cells and ILC2, lead to further activation of smooth muscle contraction, mucus production, granulocyte effector cells, and, which in turn, result in the encapsulation or expulsion of inflammatory stimuli (72). B cell-derived IgE is an important effector of type 2 immunity, and the recognition of allergens by IgE on mast cells is responsible for induction of the cardinal features of classic allergic responses, including anaphylactic shock (70). ILC2-derived IL-5 is an important growth factor that contributes to B1 cell self-renewal (73, 74). ILC2 sorted from mesenteric fat-associated lymphoid clusters are able to increase IgA production by peritoneal B cells in vitro (74).
Multiple indirect (cytokines) and direct (surface-bound molecules) mechanisms are involved in interactions between ILC2 and other cells as summarized below (Figure 1) (29, 75–77).
IL-4
IL-4 is expressed by activated ILC2 (78, 79). ILC2-released IL-4 participates in blocking the expansion of allergen-specific Tregs, thus involving in food allergy (80). ILC2-released IL-4 is also able to polarize TH2 cells during helminth infection (81).
On the other hand, eosinophil- or basophil-released IL-4 is found to affect ILC2 by enhancing ILC2 lineage proliferation, function and stability (82, 83).
IL-5
ILC2 can coordinate adaptive and innate immune functions through IL-5. IL-5 is critical for B cell function (74) and eosinophil homeostasis (Figure 1) (84, 85). When splenic B cells co-culture with mesenteric ILC2, IL-5 from ILC2 plays a pivotal role in the release of IgA from B cells (74). Besides, peripheral ILC2 in pulmonary, peritoneal cavity, and spleen are able to elevate the secretion of IgA, IgE, IgG1, and IgM by B cells in ex vivo co-cultures (86). Furthermore, upon NP-Ficoll (primes for a high affinity IgM anti-NP response) treatment in vivo, IgM produced by B cells is selectively initiated by lung ILC2 in an IL-5–dependent pattern (86). IL-5–producing ILC2 are also essential for the Th2 and Th9 cytokine responses against Trichinella spiralis infection (87). In addition, lung ICOS+ILC2 act a protective factor in a bleomycin model in an IL-5-dependent manner (88). Of note, the timing of IL-5 release by ILC2 seems important for the protective activity (88). Study showed that prostaglandin D2 (PGD2)-chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2) signaling increases ILC2 and its production of IL-5, which promotes Tregs proliferation (89).
Our recent study discovered that high mobility group box 1 (HMGB1, a late mediator of sepsis) signals via receptor for advanced glycation end products (RAGE) to increase lung ILC2 by enhancing ILC2 proliferation and suppressing ILC2 death. The activated ILC2 increase type 2 cytokines production and eosinophil infiltration in the lungs, both of which improve haemorrhagic shock-induced acute lung injury (85). Lung ILC2 activated by IL-33 secrete a large number of IL-5, which further up-regulate neutrophil and its IL-5 production (90).
IL-9
Price et al, reported in detail that ILC2 express IL-9 receptor (78). Using IL-9 reporter and subsequently IL-9 fate mapping mice, two studies delineated the autocrine signaling mechanism of IL-9 in ILC2 that enhances IL-13 and IL-5 release (Figure 1) (66, 91). In papain-induced pulmonary inflammation model, IL-9 was secreted for a short period by ILC2, then ILC2 changed to release IL-13 and IL-5. Furthermore, IL-33, but not IL-25, increased IL-9–producing ILC2 (91). Rauber et al, recently uncovered that IL-9 from ILC2 was necessary for Tregs activation and inflammation resolution in an arthritis model (92). HMGB1-activated ILC2 also secrete IL-9, which increase DCs (93). Moreover, our study discovered that ILC2-released IL-9 protects lung endothelial cells from pyroptosis by suppressing caspase-1 in a septic model (17).
IL-2 released by adaptive immune cells also play a crucial role in the IL-9 expression by ILC2, suggesting again the strong functional link between adaptive and innate lymphoid cells (94). Besides, IL-2 functions as a costimulator to ILC2 and promotes cell proliferation and survival by activating NF-κB pathway and gene transcription through p65 translocation (94). IL-9 and IL-2 work synergistically to direct ILC2 biology, and increased IL-9 production is related to an asthma-like phenotype in humans and mice highlighting the key role of these cytokines (95–98).
IL-13
IL-13 also mediates the interaction between ILC2 and immune and non-immune cells (Figure 1). During infection of helminth in mice, IL-13 released from ILC2 is more abundant than that from Th2 cells for restricting worm expulsion and immune response (23, 64, 78). IL-13 from ILC2 can induce goblet cell hyperplasia as well as mucus secretion (99). Yet the precise mechanism of IL-13 receptors on pulmonary cells at different states remains to be fully elucidated.
Pulmonary IL-13+ ILC2 and CD4+ T cells cooperate to suppress Nippostrongylus brasiliensis (Nb) infection. Immune-damaged larvae have a severe morphological defect that is due to the increase of CD4+ T cells and IL-13+ ILC2, as well as the activation of M2 macrophages (100). Besides, alveolar macrophages can be primed by ILC2-derived IL-13 into a type 2 immune cell phenotype (101). DCs are stimulated by IL-13 to convert to a type 2 chemokine-secreting phenotype. ILC2-derived IL-13 is also able to mediate DCs migration from the lungs to the LNs, thus impacting the differentiation of TH2 cell (102). In addition, the number of IL-13+ ILC2 was reported to be markedly upregulated in patients with uncontrolled asthma, and it was significantly decreased when these patients had their symptoms controlled by treatment, suggesting an important role for ILC2-derived IL-13 in asthma (103). In conclusion, ILC2-derived IL-13 can initiate and affect innate and adaptive type 2 immune responses.
Amphiregulin (Areg)
Areg is a member of the epidermal growth factors (EGF) family and acts via the EGF receptor (EGFR) (104). Both hematopoietic and non-hematopoietic cells in the lung present EGFR (105). ILC2 are a major cellular producer of Areg after activation with IL-33. ILC2-derived Areg is a critical component of effective pulmonary wound healing during influenza infection and restoring epithelial integrity and lung function (20). The initiation of mucus secretion and wound healing can prevent or ameliorate some respiratory diseases, although enhanced and excessive mucus may play a detrimental role in diseases, such as asthma (106, 107). Pulmonary Tregs are also capable of producing Areg without TCR signaling (Figure 1) (108). Thus, innate Areg released by ILC2 and Tregs is of high importance to restore tissue homeostasis and wound healing after pulmonary infection.
ICOS/ICOS-Ligand Interaction
Studies on helminth expulsion revealed ILC2-Tregs crosstalk (109). Tregs and ILC2 colocalize to similar regions within the lung tissues and visceral adipose tissue under homeostatic and inflammatory conditions (Figure 1) (31). Of note, ICOS+ Tregs and ICOS-L+ ILC2 were reported to accumulate in tissues after Nb infection or IL-33 administration, while Tregs accumulation in ICOS-L knock-out mice or after administration with neutralizing monoclonal antibody against ICOS-L was reduced, indicating that ICOS-L+ ILC2 could improve Tregs expansion, thus establishing a pathway for Tregs to cooperate with ILC2 (31). On the other hand, Tregs are capable of inhibiting ILC2 to restrict allergic airway inflammation (110). Besides, ICOS/ICOS-L interaction on ILC2 affects STAT5 signaling, activating ILC2 function and proliferation in an allergic model (111).
PD-1/PD-L1 Interaction
ILC2 constitutively express the checkpoint inhibitor molecule programmed death-ligand 1 (PD-L1), which has been discovered to activate CD4+ Th2 cell responses during type 2 pulmonary responses (Figure 1) (30). Conditional knockout of PD-L1 on ILC2 disrupted cytokine production and early Th2 polarization, resulting in delayed worm expulsion during infection with the gastrointestinal helminth Nb (30). Nevertheless, ILC2 can also express PD-1, which was reported to be upregulated on activated ILC2, and depletion of these PD-1+ ILC2 resolves papain-induced lung inflammation (112).
E-Cadherin/KLRG1 Interaction
E-cadherin, a cell adhesion molecule, interacts with the mature ILC2 marker, killer-cell lectin like receptor G1 (KLRG1). The finding that E-cadherin-KLRG1 ligation on human ILC2 reveals a significant decrease in GATA3 expression and type 2 cytokine (such as IL-5 and IL-13) release and the discovery that E-cadherin expression is suppressed in keratinocytes propose that inhibited E-cadherin expression may activate ILC2, promoting atopic dermatitis (AD) immunopathogenesis (28).
Other Interactions
OX40 ligand (OX40L) expression on ILC2 can be enhanced by IL-33. OX40-OX40L ligation has been reported to increase Th2 cell survival and number (32, 113), thus promoting adaptive immunity (75).
A molecularly distinct subset of lung ILC2 can secrete IL-10 and suppress some pro-inflammatory genes. IL-2, IL-4, IL-27, IL-10, and NMU stimulate IL-10 production from ILC2 and are associated with decreased eosinophil recruitment to the lung, indicating that ILC2 have anti-inflammatory functions similar to Tregs (Figures 1 and 3) (26, 114).
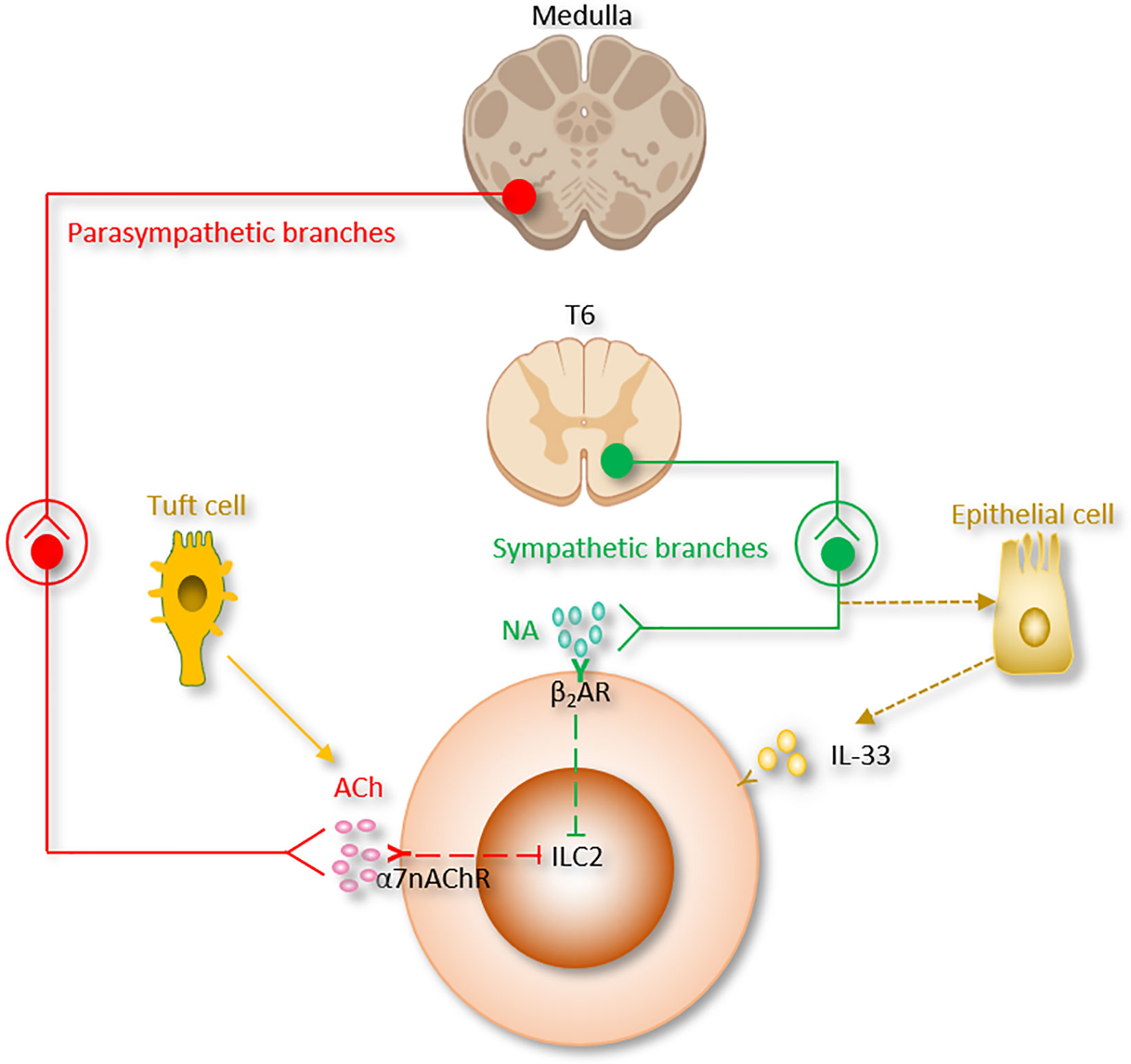
Figure 2 The autonomic nervous system regulation of ILC2 function. Ach, Acetylcholine; α7nAChR, α7 nicotinic acetylcholine receptor; β2AR, β2-adrenergic receptor; NA, Norepinephrine.
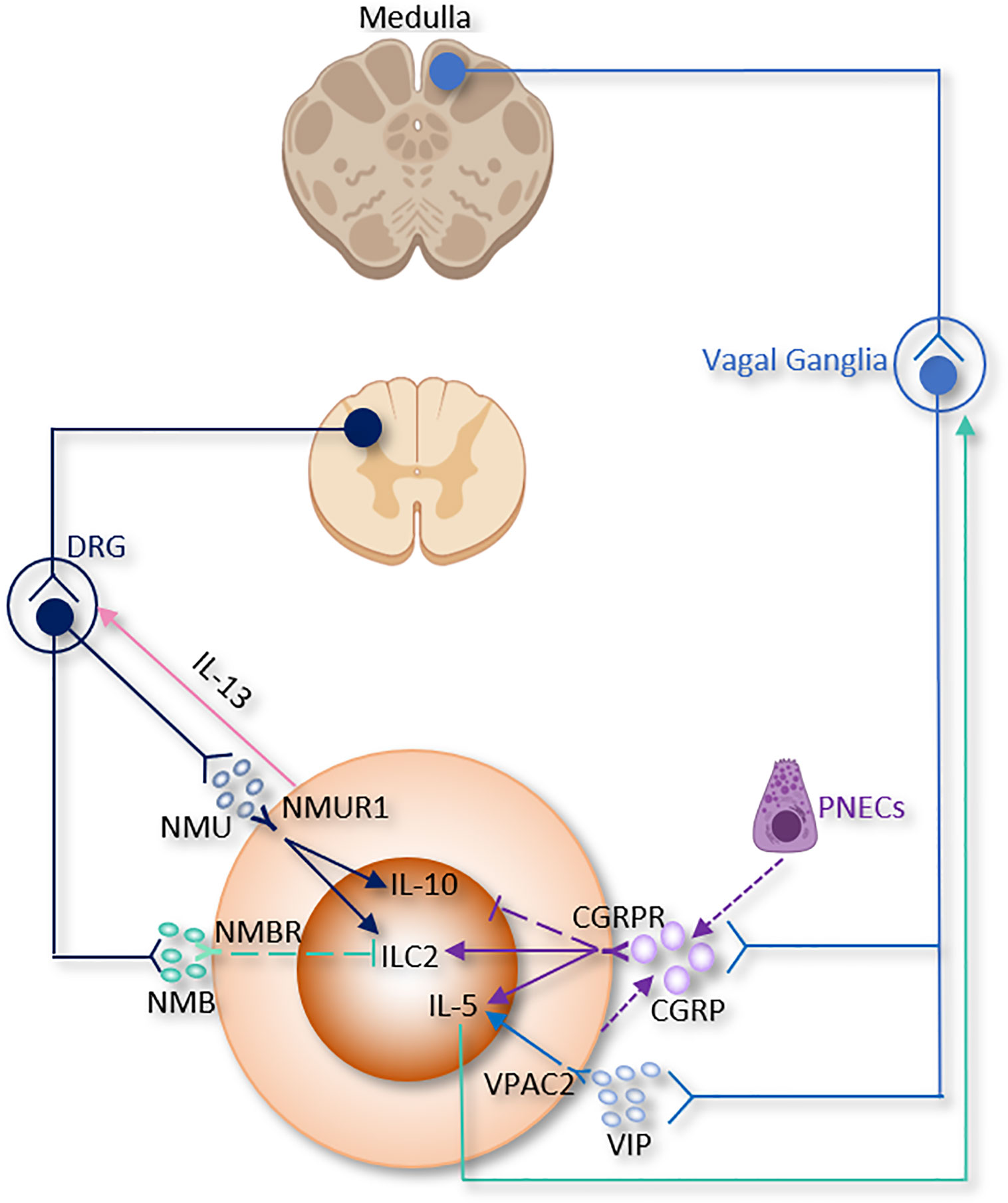
Figure 3 The somatic nervous system regulation of ILC2 function. CGRP, α-calcitonin gene-related peptide; DRG, Dorsal root ganglion; NMB, neuromedin B; NMU, neuromedin U; PNECs, Pulmonary neuroendocrine cells; VIP, Vasoactive Intestinal Peptide.
The expression of CD86, CD80, and MHCII by mouse ILC2 is also involved in ILC2 interactions with CD4+ T cells;
MHCII+ ILC2 can drive the differentiation of naive CD4+ T cells into Th2 in vitro, whereas MHCII-deficient ILC2 upregulate Th2 cell-driven helminth expulsion in vivo (29, 75).
Pulmonary Nervous System and its Regulatory Function
Lung is densely innervated by peripheral nervous system (PNS), which is divided into motor and somatosensory nervous systems (Figure 2).
Motor Nervous System
The motor nervous system consists of the autonomic (sympathetic, parasympathetic, and enteric) and somatic branches.
Autonomic nervous system serves to regulate involuntary functions. Sympathetic branch comes from the upper six thoracic spinal cord segments; the synapse together with the sympathetic ganglia, and postganglionic fibers then innervate the airways. The sympathetic nervous system participates in the body response to stress and regulates bronchodilation and the production of mucous.
The cholinergic parasympathetic branch comes from the vagal nuclei of the medulla; the superior and recurrent laryngeal vagal nerves synapse at the parasympathetic ganglia to innervate the lung (115). The parasympathetic branch is mainly responsible for keeping homeostasis (116). It regulates bronchoconstriction, carbon dioxide and oxygen levels, as well as neural reflexes including coughing.
Somatosensory Nervous System
The somatosensory nervous system delivers sensory stimuli, such as proprioception, touch, and pain. Somatosensory neurons are further divided into pruriceptors and nociceptors responsible for sensing itch-inducing or noxious stimuli, respectively. These neurons are important because their activation is closely related to inflammation and immunity (Figure 2).
All opponents of the PNS play a critical role in orchestrating immunity, inflammation, and tissue repair at host barrier tissues in response to stimuli and stressors. Primary evidence that neurotransmitters may regulate immune responses was that their release from nerves could lead to signaling through the surface receptors of lymphocyte cell (117). Leukocytes have receptors for the neurotransmitters such as dopamine, serotonin and glutamate (117), and also produce neurotransmitters that work as paracrine or autocrine signals (118). The neuronal reflex senses peripheral inflammation and coordinates the host response to injury and/or infection, regulating events within the initiation of inflammation (48, 119). Lung is heavily populated by resident immune cells, such as macrophages, DCs, γδ T cells mast cells and ILCs. It allows fast, integrated reactions to pathogens and noxious stimuli (120).
Sympathetic Nervous System
Sympathetic nervous system helps the center nervous system to control innate immune responses between antiviral and pro-inflammatory actions (Figure 2) (121, 122).
The nerves of the sympathetic nervous system distribute the neurotransmitter catecholamines into tissue microenvironments in which immune response gene transcription occurs, including all lymphoid organs, most musculoskeletal structures and visceral organs, and the vasculature and perivascular tissues (123).
Recent reports have revealed a sympathetic nervous system-mediated steering of innate immune response programs, which include enhanced transcription of pro-inflammatory cytokine genes (such as Il6, tnf, and Il1β) (121, 124) and inhibition of type I IFN-mediated antiviral responses (122).
Stimulation of the sympathetic nervous system has also been shown to change the production and trafficking of innate immune cells, for instance, through the upregulation of myelopoiesis and the mobilization of monocytes, splenic neutrophils, natural killer cells, and haematopoietic stem cells (123). A current study showed that the sympathetic nervous system stimulates IL-33 and then ILC2 in adipose tissue. Cold exposure stimulates IL-33 expression, ILC2 and eosinophils in adipose tissue. Furthermore, sympathetic denervation induced by 6-hydroxydopamine (6-OHDA) cancels this effect (125).
Catecholamines are monoamine neurotransmitters which are mainly released by the postganglionic fibers of the sympathetic nervous system and the chromaffin cells of the adrenal medulla. Included among catecholamines are dopamine, epinephrine (adrenaline), and norepinephrine (noradrenaline) (Table 1 and Figure 2). Release of the epinephrine and norepinephrine from the adrenal glands and adrenergic nerves is part of the fight-or-flight response.
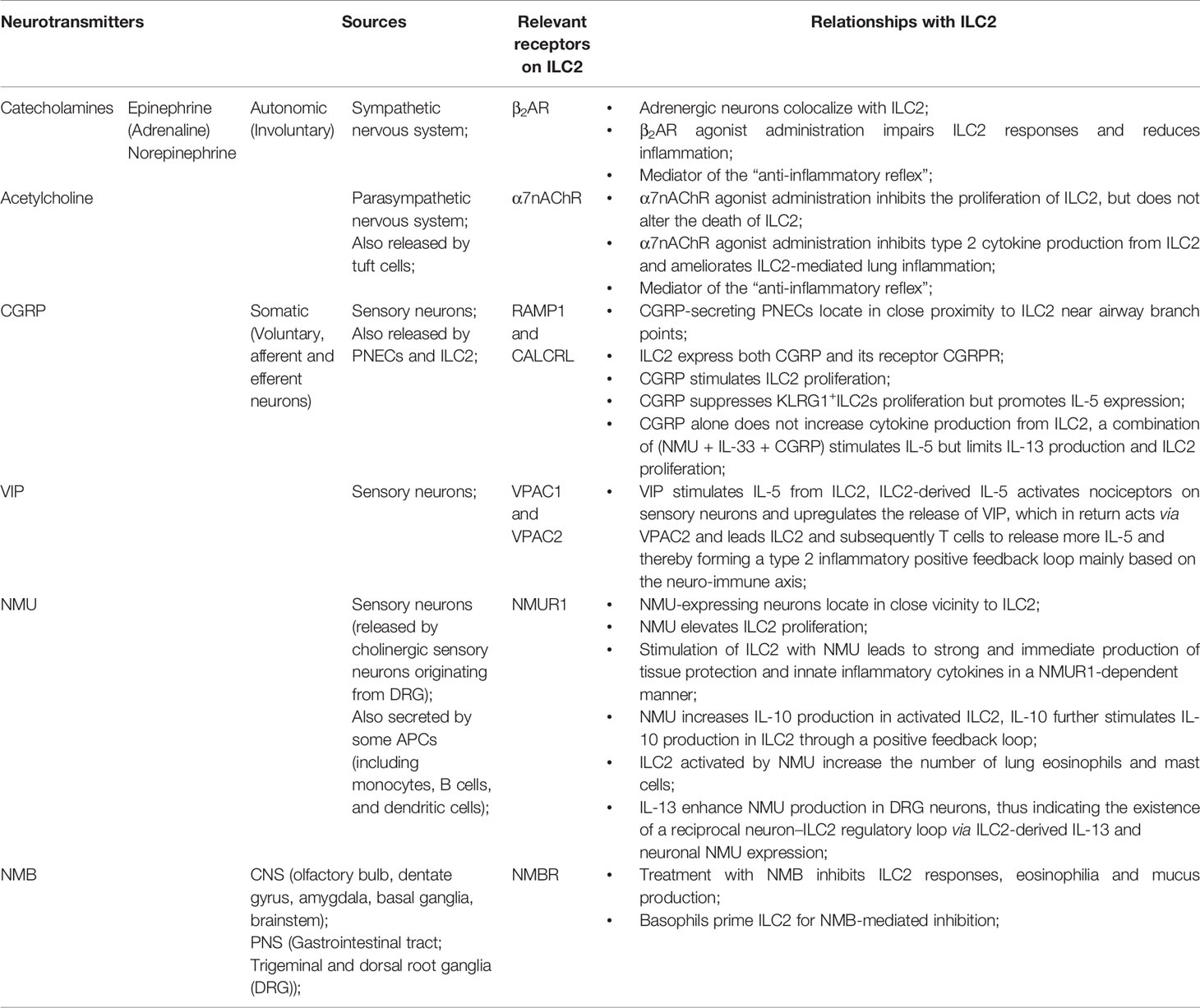
Table 1 Sources, receptors on ILC2, and relationships with ILC2 of several neurotransmitters.
Catecholamines exert their effects via two classes of adrenergic receptors, α and β. Both groups could be functionally divided into subgroups (α1 and α2; β1, β2, and β3).
Norepinephrine regulates leukocyte gene expression through β-adrenergic receptors (123). β-adrenergic receptor is expressed on most immune cells, such as B cells, T cells, and other innate cells (40, 126–128). It was initially thought to regulate adaptive immune responses by suppressing the expression of TH1-type genes, such as Il12β and Ifnγ, and activating the transcription of TH2-type cytokine genes, i.e. Il4 and Il5 (129–131).
Interestingly, β2AR has an inhibitory effect on innate immune responses. β2-agonists suppress cardiodepressant and inflammatory factors, including HMGB1 and TNF. Recently, ILC2 from the lung and the gut-related tissues (small intestinal lamina propria, colonic lamina propria and mesenteric lymph nodes) were found to express β2AR. ILC2 were also shown to colocalize with adrenergic neurons in the mouse intestine (40).
β2AR deficiency led to enhanced ILC2 proliferation and subsequent type 2 cytokine production in lung and intestinal tissues after infection with Nb. Lung eosinophilia was observed following enhanced IL-5 production from ILC2 in β2AR-deficient mice. On the other aspect, β2AR agonist administration disrupted ILC2 responses and suppressed inflammation in vivo. By using conditional β2AR-deficient mice, or by transferring ILC2 progenitors from wild-type mice or β2AR-deficient mice into ILC-deficient mice, the group generated ILC2-specific β2AR-deficient mice and ensured that the β2AR negatively controls ILC2 and type 2 inflammation (40). This study provides another evidence of neuronal regulatory circuit that regulates ILC2-dependent type 2 inflammation. β2AR-agonists are the most effective medications for the treatment of asthma. β2AR-mediated ILC2 regulation could be one of the pathways of β2AR-agonists effect in asthma (33, 38, 44, 110, 132, 133). On the other hand, β2AR is the first adrenergic receptor documented to participate in the “anti-inflammatory reflex” of the parasympathetic system which will be discussed in the next section (Table 1 and Figure 2).
Parasympathetic Nervous System
Parasympathetic and sympathetic systems are usually considered to work in opposition to maintain physiological homeostasis. While current literatures suggested that both branches work together to restrain systemic inflammation in life-threatening illnesses, including arthritis, inflammatory bowel disease, sepsis and endotoxemia (46, 48, 134–138).
Nerve fibers of the parasympathetic nervous system arise from three primary areas: cranial nerves (facial nerve, oculomotor nerve, and glossopharyngeal nerve), vagus nerve, and pelvic splanchnic nerves (three spinal nerves in the sacrum, S2-4).
The parasympathetic nervous system mainly utilizes acetylcholine (ACh) as its neurotransmitter (Table 1 and Figure 2). Tuft cells, capable of secreting the ILC2 activator IL-25, also secrete Ach (139, 140). The ACh has two kinds of receptors, the nicotinic and muscarinic cholinergic receptors. α7 nicotinic acetylcholine receptor (α7nAChR), one of the nicotinic acetylcholine receptors, is expressed by ILC2 at steady state, and this expression is further increased following alarmin-induced activation such as IL-33 (44). The α7nAChR is also present on B cells (141), T cells (142, 143), macrophages (144) and ILC3 (41).
α7nAChR-/- mice were more susceptible to severe lung injury and higher mortality than α7nAChR+/+ mice. Increased α7nAChR+ alveolar neutrophils and macrophages were observed in the mice injured lungs. The immunomodulatory cholinergic α7nAChR pathway of alveolar neutrophils and macrophages alleviated E. coli- and LPS-induced acute lung injury by inhibiting transalveolar neutrophil migration and chemokine production.
It was reported that the expression of HMGB1 protein was suppressed by α7nAChR agonist nicotine and the survival of post-sepsis acute lung injury was improved (145). In addition, administration of α7nAChR agonist inhibits type 2 cytokine production from ILC2 and ameliorates ILC2-mediated lung inflammation induced via IL-33 stimulation or Alternaria alternata inhalation. Mechanistically, α7nAChR agonist is reported to inhibit cellular markers for proliferation in ILC2 (Ki67, NF-κB and GATA3 signaling pathways) (27, 44, 146, 147) (Table 1). These findings indicate that α7nAChR may be a potential therapeutic target for acute lung injury (120).
On the other aspect, Bcl-2, an anti-apoptotic factor of ILC2, was unchanged by α7nAChR agonist treatment. These studies indicate that parasympathetic nervous system modulates ILC2 proliferation, but not death. The suppressive effects of α7nAChR on ILC2 may serve as the mechanism underlying the observed reduced pulmonary allergic inflammation induced by nicotine treatment (148). Moreover, in cancer immunity, nicotine treatment stimulates tumor growth by suppressing apoptosis and promoting cell proliferation (149–152).
Anti-Inflammatory Reflex
The vagus nerve is the main parasympathetic nerve connecting between the central nervous system and peripheral organs (128, 153). Pharmacological or electrical stimulation of the vagus nerve can restrain the systemic inflammation response, organ damage, and mortality in different experimental hemorrhage and resuscitation (43), pancreatitis (154), ischemia and reperfusion (43, 155, 156), colitis (157), endotoxemia (42, 155, 158, 159) and sepsis (145, 160).
Mechanically, the motor and sensory vagus nerve form a complex neural reflex circuit termed the “anti-inflammatory reflex” which control spleen cytokine production through splenic nerve (42, 161–166). ACh released by the vagus nerve in the celiac mesenteric ganglia stimulates postsynaptic α7nAChR of splenic nerve (46). The adrenergic splenic nerve release norepinephrine to activate a discrete subset of spleen lymphocytes via β2AR. Activated lymphocytes then release Ach (48, 167). Lymphocyte-derived Ach downregulates macrophage cytokine release, and switches them toward a tissue-protective, M2 anti-inflammatory phenotype. α7nAChR mediates Ach-induced signal transduction in macrophages and monocytes (144). α7nAChR inhibits the inflammasome activity (168), enhances the JAK2-STAT3 pathway (169), stabilizes mitochondrial membranes, and suppresses the nuclear translocation of NF-κB (119, 145, 168–170).
The eventual influence of inflammatory reflex on the spleen is the inhibition of cytokine release by spleen macrophages, which produce over 90% of the IL-1 and TNF during acute endotoxemia (159, 167). The anti-inflammatory reflex is a special instance of a functional network between the efferent parasympathetic vagus nerve, the splenic nerve (termed sympathetic) and T cells relaying neural signals. In depicting this cooperation, the use of the classical sympathetic-versus-parasympathetic neuronal designation should be modified.
Indirect studies suggested that anti-inflammatory reflex is involved in the regulation of ILC. Dalli et al, reported that dissection of the right vagus downregulated the number of peritoneal ILC3 and changed peritoneal macrophage responses. Right vagotomy led to decreased peritoneal levels of pro-resolving mediators, which include the protective immunoresolvent protectin conjugates in tissue regeneration 1 (PCTR1), as well as increased inflammation-initiating eicosanoids. Ach restored the PCTR1 production from ILC3. Treatment of PCTR1 or ILC3 repaired tissue and ameliorated E. coli infections in vagotomized mice (41). Another group studied the regulation of ILC2 using coding α7nAChR (Chrna7) knockout mice, pulmonary C fibre (PCF, which releases ACh and neuropeptides) degeneration mice, and vagotomized mice. Knockout of Chrna7 enhanced resident ILC2s and trafficking iILC2s in the lung, worsened allergic inflammation. However, PCF degeneration and vagotomy significantly reduced these two types of ILC2 and attenuated asthma responses (171). Although there is no direct evidence suggests that “anti-inflammatory reflex” regulates ILC2, it is promising to investigate since the number of ILC2 is significantly increased in spleen during inflammation or infection (172).
In sum, parasympathetic nervous system participates in the pathogenesis of various diseases, with a different role in each disease.
Sensory Neurons
The lung is innervated by a dense network of sensory neurons that mainly comes from vagal afferents whose cell bodies reside in the vagal ganglia (jugular and nodose ganglia); while other sensory nerve innervation originates from the dorsal root ganglion (DRG) (Figure 3) (115, 173–175). Nociceptive receptors are richly expressed in sensory neurons endings, which are abundantly present in the lung parenchyma and near the airways; this poises them to act as the first wall for host defense and these neurons interact directly with inflammatory stimuli such as ATP, pathogens, allergens, protons, heat, mechanical injury and chemical irritants like immune cells such as APCs, macrophages, and other phagocytes (116, 176–178).
Sensory neuronal action potentials evoked by this interaction are then transmitted into the CNS within milliseconds of the detection of inflammation or invasion. This action-potential signaling mechanism is significantly faster than immune cell. Once activated, nociceptive receptors induce coughing, pain and bronchoconstriction (173, 178–180). Neuropeptides emanating from nociceptor nerve terminals also participate in the nociceptors crosstalk with immune cells (173, 181–183).
Excitation of nociceptors increases the release of multiple neuropeptides, such as substance P, VIP and CGRP which regulate both innate and adaptive immune cells (184) (Table 1 and Figure 3).
CGRP
CGRP, encoded by Calca, is a member of the calcitonin family peptides that not only secreted by peripheral nociceptive neurons but also found in central neurons (45, 185). CGRP binds to a heteromeric receptor composed of a receptor activity-modifying protein (RAMP1) and a G-protein coupled receptor termed CALCRL. CGRP via these receptors stimulates AC, which results in cAMP and PKA pathway activation and leads to the phosphorylation of several downstream pathways including NOS, MAPK, and CREB pathways (186).
In skin bacterial infection, lymph node hypertrophy and TNF-α production are found to be suppressed by CGRP (183, 187). CGRP levels markedly increased in the bronchoalveolar lavage fluid (BALF) after Staphylococcus aureus infection. S. aureus also increases cultured neuronal production of CGRP in vitro. CGRP could alleviate the symptoms of S. aureus-induced pneumonia by suppressing TNF-α, CXCL1, γδ T cells and neutrophils (188).
A recent study discovered the relationship between CGRP and pulmonary neuroendocrine cells (PNECs), which comprise ~1% of the airway cell population (189, 190). PNECs (locate in close proximity to ILC2 near airway branch points) could secrete CGRP, which aggravates allergen-induced asthma in mice by stimulating ILC2 proliferation and the secretion of IL-5 from ILC2 (133). On the other hand, Il5hiILC2 produce both CGRP and its receptor CGRPR following Nb infection. CGRP treatment alone does not increase cytokine production from ILC2, a combination of neuromedin U (NMU) + IL-33 with CGRP stimulates IL-5 but limits IL-13 production and ILC2 proliferation. Worm expulsion and ILC2 responses are augmented without CGRP signaling (39). Interestingly, Xu et al, reported that OVA-induced inflammation increased the expression of Calca in KLRG1+ILC2. CGRP suppressed KLRG1+ILC2s proliferation but promoted IL-5 expression (191). Collectively, these paradoxes point to both pro- and anti-inflammatory properties of CGRP on immune responses in the lung and warrant further investigation (Table 1 and Figure 3).
VIP
The neuropeptide VIP also involves in the regulation of ILC2. It has been firstly characterized as a polypeptide isolated from the small intestine with multiple impacts on different systems such as respiratory and cardiovascular systems (192). VIP can be perceived by VIP receptor type 1 (VPAC1) or VIP receptor type 2 (VPAC2), which are differentially regulated according to cell type and activity conditions (193, 194). Similar to CGRP, VIP enhances the AC/cAMP/PKA pathway and phospholipase C, which causes the accumulation of intracellular Ca2+ (195).
Of note, pulmonary and intestinal ILC2 express VPAC1 and VPAC2 and produce IL-5 when they are incubated with IL-7 and VIP- or VPAC2-specific agonist (84). Talbot et al, discovered a critical relationship between ILC2, VIP, T cells, and nociceptive neurons (182). Reciprocally, ILC2-derived IL-5 activates nociceptors on afferent neurons and upregulates the release of VIP, which in return acts via VPAC2 and leads ILC2 and subsequently T cells to release more IL-5 and thereby forming a type 2 inflammatory positive feedback loop mainly based on the neuro-immune axis (Table 1 and Figure 3) (182). Since the levels of blood eosinophils and type 2 cytokine release from ILC2 are regulated by circadian rhythm and food intake, this suggests that VIP might influence blood eosinophils via upregulation of ILC2 (84).
NMU
NMU is a neuropeptide mainly released by cholinergic sensory neurons originating from DRG, but not parasympathetic neurons in the vagal ganglion (196). The initial biological functions ascribed to NMU were food intake and body weight reduction, smooth muscle contraction of the uterus, pronociceptive effects promotion and circadian rhythm regulation (197, 198). In addition, NMU is also occasionally secreted by some APCs, including monocytes, B cells, and dendritic cells (199). Thus, it is suggested to play an important role in the regulation of adaptive and innate immunity. In an allergen-induced asthma model, airway eosinophilia was shown to decrease in nmu-/- mice. NMU directly stimulated extracellular/signal-regulated kinase phosphorylation and Ca2+ mobilization. NMU also induced cell adhesion to components of the extracellular matrix, and chemotaxis in vitro (200).
Recent studies reported that NMU from lamina propria play a regulatory role in mice type 2 innate immunity through binding to the neuromedin U receptor 1 (Nmur1), which is selectively enriched in ILC2. Consistent with this idea, NMU-expressing neurons have been discovered in close vicinity to ILC2 in the lungs (19, 37, 38). Lung ILC2 present NMUR1 at steady state and upon IL-25 stimulation, however, NMUR1 was inhibited upon IL-33 exposure (38).
In a mice model of worm infection in the lungs and intestine, stimulation of ILC2 with NMU led to strong and immediate production of tissue protection and innate inflammatory cytokines in a NMUR1-dependent manner, thereby alleviating worm burden (37).
In a model of airway allergy, ILC2 were activated by NMU in vitro, and in vivo co-treatment of NMU with IL-25 significantly increased lung histopathology. Disruption of NMU-NMUR1 pathway decreased ILC2 number and effector function, and changed transcriptional programs following in vivo allergen exposure (38). NMU elevates pulmonary ILC2 proliferation and a selectively potent secretion of innate IL-5, IL-13, and amphiregulin (19, 37, 38). Furthermore, ILC2 activated by NMU increase the number of lung eosinophils and mast cells, thus alleviating antihelminth responses (19, 37, 38). Interestingly, IL-13 enhance NMU production in DRG neurons, thus indicating the existence of a reciprocal neuron–ILC2 regulatory loop via ILC2-derived IL-13 and neuronal NMU expression (38). IL-10, primarily secreted by Tregs, was increased by NMU in activated intestinal ILC2. IL-10 further stimulated IL-10 production in ILC2 through a positive feedback loop (Table 1 and Figure 3) (114). These findings suggested that NMU treatment enhance inflammation-induced damage in the lungs and pointed to a double-edged sword of NMU-NMUR1 signaling.
NMB
NMB belongs to the neuromedin family that includes NMB, NMC, NMK, NML, NMN, NMU and NMS (201, 202). It is expressed widely in the CNS (olfactory bulb, dentate gyrus, amygdala, basal ganglia, and brainstem) and the PNS (gastrointestinal tract, trigeminal and dorsal root ganglia (DRG)) (203, 204).
NMB controls cell growth, blood glucose, body temperature, emotion, energy homeostasis, exocrine and endocrine secretion, food intake, grooming and scratching, nociception and smooth muscle contraction. Inclan-Rico, Juan M et al, found that administration of NMB suppressed ILC2 responses via NMU receptor (NMBR), eosinophilia and mucus production after Nb infection in the lung. In consistent with in vivo results, in vitro treatment of NMB inhibited the growth of sorted ILC2 (Table 1 and Figure 3). Of note, ILC2 sorted from basophil-depleted mice were unchanged by NMB stimulation, indicating that basophils are indispensable for the inhibitory effects of NMB on ILC2 (50).
Promising Directions for Research on the Neural Regulation of ILC2 in the Lung
The discoveries of neural control of ILC2 have added a new dimension to neuroimmunity. All previously known findings of ILC2 in the lung could be re-examined from this perspective.
Previous studies on the lung ILC2 have been mainly performed in models of allergic disease (205–208), helminth infection (16, 50, 209, 210), and septic lung injury (17, 85, 211, 212). Multiple neural pathways have been reported to be involved in these disease models (4, 213, 214). Although recent studies have found some clues, the relationship between nervous system and ILC2 still remains contradictory and inconclusive. For example, CGRP shows opposite effects on different ILC2 subtypes. NMU is able to enhance the pro-inflammatory function of ILC2 as well as its anti-inflammatory function. Does the nervous system or neural mediators play ameliorating or worsening roles in these diseases by regulating ILC2? If future studies can prove this hypothesis, then ILC2 will not only be a bridge between innate and adaptive immunity, but also between the nervous system and the immune system.
The second promising research direction will be the effects of neural-regulated ILC2 on nervous system. Type I cytokines and their receptors (such as IL-1, IL-6, and TNFs) are expressed widely in CNS cells and are important for the development and function of the CNS (58). However, the impacts of ILC2-released type II cytokines and mediators on nervous system remains to be elucidated. Currently, a reciprocal DRG–ILC2 regulatory loop via ILC2-derived IL-13 and neuronal NMU expression has been found (38). Besides, ILC2-derived IL-5 activates nociceptors on afferent neurons and upregulates the release of VIP, which in return, acting via VPAC2 leads to ILC2 and T cells to release more IL-5 and, thereby, forming a type 2 inflammatory positive feedback loop (182). It would be important to explore unidentified neuron-ILC2 positive/negative regulatory loops.
The third area of interest will be the neural regulation of ILC1 and ILC3. Recent studies have shown that three ILCs are functionally plastic. For instance, plastic iILC2 can coproduce both type-2 cytokines and the ILC3-characteristic cytokine (IL-17A) (215). Under certain conditions, c-Kit+ILC2 can convert to ILC3-like cells (216). Besides, IL-12 and IL-18 converted ILC2 into ILC1 in patients with chronic obstructive pulmonary disease (COPD) (217). Does neuromodulation affect the interconversion of ILC2 and two other cell subtypes? If these plastic properties can be elucidated, we can gain a comprehensive understanding of the relationship between ILC as a cell type and the nervous system.
Least but not last, these results have great therapeutic implications for precision medicine. For example, NMUR1 is selectively expressed by ILC2, while receptors for classical ILC2 activators, i.e. IL-25, IL-33, and TSLP, are widely expressed by various cell types (19, 37, 38). Meanwhile, researchers have developed many methods to selectively stimulate and inhibit neurons (218, 219). Combining these advances will allow us to identify more effective clinical targets.
Conclusion
Emerging evidence from in vivo animal models, human studies, and in vitro experiments indicates that neuropeptides and neurotransmitters released from various neurons and non-neuronal cells are critical in regulation of immune responses in different tissues including the lung. This review article provides a comprehensive overview of the effects of novel neural mediators and pathways on ILC2 and underlying mechanisms as well as the insights into the direct and indirect interactions between ILC2 and other immune cells, highlighting ILC2 as the bridge between innate and adaptive immunity. However, the research in neuro-immune area is, in general, in a premature status, and numerous questions remain to be addressed. For examples, the most of signaling pathways that mediate neural regulation of ILC2 are yet clear; and the mechanisms, by which ILC2 selectively respond to neutral and non-neural signaling need to be elucidated as well. In addition, translational and clinical investigations are required to promote the application resulted from the studies in this area.
Author Contributions
WC collected the data and drafted the manuscript. WC, QS, and JF conceived and designed the study. JF reviewed and finalized the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the USA National Institutes of Health Grant R01-HL-079669 (JF), USA National Institutes of Health Grant R01HL076179 (JF), USA National Institutes of Health Grant R01-HL-139547 (JF), VA Merit Award 1I01BX002729 (JF), and VA BLR&D Award 1IK6BX004211 (JF).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
α7nAChR, α7 nicotinic acetylcholine receptor; Ach, Acetylcholine; AD, Atopic Dermatitis; Areg, Amphiregulin; CGRP, α-Calcitonin Gene-Related Peptide; CRTH2, Chemoattractant Receptor-homologous molecule expressed on Th2 cells; DCs, Dendritic Cells; PGD2, Prostaglandin D2; EGF, Epidermal Growth Factors; EGFR, EGF Receptor; HMGB1, High Mobility Group Box 1; ICOS, Inducible Costimulator; ILCs, Innate Lymphoid Cells; ILC2, Group 2 Innate Lymphoid Cells; KLRG1, Killer-cell Lectin Like Receptor G1; NMU, Neuromedin U; NMB, Neuromedin B; Nb, Nippostrongylus brasiliensis; PCTR1, Protectin Conjugates in Tissue Regeneration 1; PD-L1, Programmed Death-Ligand 1; PNECs, Pulmonary Neuroendocrine cells; PNS, Peripheral Nervous System; RAGE, Receptor for Advanced Glycation End products; RAMP1, Receptor Activity-Modifying Protein; TLRs, Toll-Like Receptors; Tregs, Regulatory T Cells; TSLP, Thymic Stromal Lymphopoietin; VIP, Vasoactive Intestinal Peptide.
References
1. Chavan SS, Tracey KJ. Essential Neuroscience in Immunology. J Immunol (2017) 198:3389–97. doi: 10.4049/jimmunol.1601613
2. Irwin MR, Cole SW. Reciprocal regulation of the neural and innate immune systems. Nat Rev Immunol (2011) 11:625–32. doi: 10.1038/nri3042
3. Hoffman C, Aballay A. Role of neurons in the control of immune defense. Curr Opin Immunol (2019) 60:30–6. doi: 10.1016/j.coi.2019.04.005
4. Zhai Q, Lai D, Cui P, Zhou R, Chen Q, Hou J, et al. Selective Activation of Basal Forebrain Cholinergic Neurons Attenuates Polymicrobial Sepsis-Induced Inflammation via the Cholinergic Anti-Inflammatory Pathway. Crit Care Med (2017) 45:e1075–82. doi: 10.1097/CCM.0000000000002646
5. Zhong C, Zhu J. Transcriptional regulators dictate innate lymphoid cell fates. Protein Cell (2017) 8:242–54. doi: 10.1007/s13238-017-0369-7
6. Schuijs MJ, Halim TYF. Group 2 innate lymphocytes at the interface between innate and adaptive immunity. Ann N Y Acad Sci (2018) 1417:87–103. doi: 10.1111/nyas.13604
7. Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol (1975) 5:112–7. doi: 10.1002/eji.1830050208
8. Pross HF, Jondal M. Cytotoxic lymphocytes from normal donors. A functional marker ofhuman non-T lymphocytes. Clin Exp Immunol (1975) 21:226–35. PMID: 810282
9. Kelly KA, Scollay R. Seeding of neonatal lymph nodes by T cells and identification of a novel population of CD3-CD4+ cells. Eur J Immunol (1992) 22:329–34. doi: 10.1002/eji.1830220207
10. Mebius RE, Rennert P, Weissman IL. Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity (1997) 7:493–504. doi: 10.1016/S1074-7613(00)80371-4
11. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13:145–9. doi: 10.1038/nri3365
12. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate Lymphoid Cells: 10 Years On. Cell (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
13. Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science (2015) 348:aaa6566. doi: 10.1126/science.aaa6566
14. Artis D, Spits H. The biology of innate lymphoid cells. Nature (2015) 517:293–301. doi: 10.1038/nature14189
15. Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, et al. Transcription factor RORalpha is critical for nuocyte development. Nat Immunol (2012) 13:229–36. doi: 10.1038/ni.2208
16. Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat Immunol (2015) 16:161–9. doi: 10.1038/ni.3078
17. Lai D, Tang J, Chen L, Fan EK, Scott MJ, Li Y, et al. Group 2 innate lymphoid cells protect lung endothelial cells from pyroptosis in sepsis. Cell Death Dis (2018) 9:369. doi: 10.1038/s41419-018-0412-5
18. Hammad H, Lambrecht BN. Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity (2015) 43:29–40. doi: 10.1016/j.immuni.2015.07.007
19. Klose CSN, Mahlakõiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature (2017) 549:282–6. doi: 10.1038/nature23676
20. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol (2011) 12:1045–54. doi: 10.1038/ni.2131
21. Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity (2012) 36:451–63. doi: 10.1016/j.immuni.2011.12.020
22. Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity (2001) 15:985–95. doi: 10.1016/S1074-7613(01)00243-6
23. Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med (2006) 203:1105–16. doi: 10.1084/jem.20051615
24. Cording S, Medvedovic J, Aychek T, Eberl G. Innate lymphoid cells in defense, immunopathology and immunotherapy. Nat Immunol (2016) 17:755–7. doi: 10.1038/ni.3448
25. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D, et al. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A (2015) 112:10762–7. doi: 10.1073/pnas.1509070112
26. Seehus CR, Kadavallore A, Torre B, Yeckes AR, Wang Y, Tang J, et al. Alternative activation generates IL-10 producing type 2 innatelymphoid cells. Nat Commun (2017) 8:1900. doi: 10.1038/s41467-017-02023-z
27. Mjosberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, et al. The transcription factor GATA3 is essential for the function ofhuman type 2 innate lymphoid cells. Immunity (2012) 37:649–59. doi: 10.1016/j.immuni.2012.08.015
28. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells inatopic dermatitis. J Exp Med (2013) 210:2939–50. doi: 10.1084/jem.20130351
29. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells andCD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity (2014) 41:283–95. doi: 10.1016/j.immuni.2014.06.016
30. Schwartz C, Khan AR, Floudas A, Saunders SP, Hams E, Rodewald HR, et al. ILC2s regulate adaptive Th2 cell functions via PD-L1 checkpointcontrol. J Exp Med (2017) 214:2507–21. doi: doi: 10.1084/jem.20170051
31. Molofsky AB, Van Gool F, Liang HE, Van Dyken SJ, Nussbaum JC, Lee J, et al. Interleukin-33 and Interferon-γ Counter-Regulate Group 2 Innate Lymphoid Cell Activation during Immune Perturbation. Immunity (2015) 43:161–74. doi: 10.1016/j.immuni.2015.05.019
32. Webb GJ, Hirschfield GM, Lane PJ. OX40, OX40L and Autoimmunity: a Comprehensive Review. Clin Rev Allergy Immunol (2016) 50:312–32. doi: 10.1007/s12016-015-8498-3
33. Chang YJ, DeKruyff RH, Umetsu DT. The role of type 2 innate lymphoid cells in asthma. J Leukoc Biol (2013) 94:933–40. doi: 10.1189/jlb.0313127
34. Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, et al. Innate IL-13-producing nuocytes arise during allergic lunginflammation and contribute to airways hyperreactivity. J Allergy ClinImmunol (2012) 129:191–198.e191-194. doi: 10.1016/j.jaci.2011.09.041
35. Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, et al. Innate lymphoid cells responding to IL-33 mediate airwayhyperreactivity independently of adaptive immunity. J Allergy Clin Immunol (2012) 129:216–227.e211-216. doi: 10.1016/j.jaci.2011.10.036
36. Imai Y, Yasuda K, Sakaguchi Y, Haneda T, Mizutani H, Yoshimoto T, et al. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc Natl Acad Sci U S A (2013) 110:13921–6. doi: 10.1073/pnas.1307321110
37. Cardoso V, Chesné J, Ribeiro H, García-Cassani B, Carvalho T, Bouchery T, et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature (2017) 549:277–81. doi: 10.1038/nature23469
38. Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature (2017) 549:351–6. doi: 10.1038/nature24029
39. Nagashima H, Mahlakõiv T, Shih HY, Davis FP, Meylan F, Huang Y, et al. Neuropeptide CGRP Limits Group 2 Innate Lymphoid Cell Responses andConstrains Type 2 Inflammation. Immunity (2019) 51:682-95.e6. doi: 10.1016/j.immuni.2019.06.009
40. Moriyama S, Brestoff JR, Flamar AL, Moeller JB, Klose CSN, Rankin LC, et al. β2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science (2018) 359:1056–61. doi: 10.1126/science.aan4829
41. Dalli J, Colas RA, Arnardottir H, Serhan CN. Vagal Regulation of Group 3 Innate Lymphoid Cells and the Immunoresolvent PCTR1 Controls Infection Resolution. Immunity (2017) 46:92–105. doi: 10.1016/j.immuni.2016.12.009
42. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature (2000) 405:458–62. doi: 10.1038/35013070
43. Cai B, Chen F, Ji Y, Kiss L, de Jonge WJ, Conejero-Goldberg C, et al. Alpha7 cholinergic-agonist prevents systemic inflammation and improves survival during resuscitation. J Cell Mol Med (2009) 13:3774–85. doi: 10.1111/j.1582-4934.2008.00550.x
44. Galle-Treger L, Suzuki Y, Patel N, Sankaranarayanan I, Aron JL, Maazi H, et al. Nicotinic acetylcholine receptor agonist attenuates ILC2-dependent airway hyperreactivity. Nat Commun (2016) 7:13202. doi: 10.1038/ncomms13202
45. Holzmann B. Modulation of immune responses by the neuropeptide CGRP. Amino Acids (2013) 45:1–7. doi: 10.1007/s00726-011-1161-2
46. Vida G, Peña G, Deitch EA, Ulloa L. α7-cholinergic receptor mediates vagal induction of splenic norepinephrine. J Immunol (2011) 186:4340–6. doi: 10.4049/jimmunol.1003722
47. Vida G, Peña G, Kanashiro A, Thompson-Bonilla Mdel R, Palange D, Deitch EA, et al. β2-Adrenoreceptors of regulatory lymphocytes are essential for vagal neuromodulation of the innate immune system. FASEB J (2011) 25:4476–85. doi: 10.1096/fj.11-191007
48. Rosas-Ballina M, Olofsson PS, Ochani M, Valdés-Ferrer SI, Levine YA, Reardon C, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science (2011) 334:98–101. doi: 10.1126/science.1209985
49. Kawashima K, Fujii T, Moriwaki Y, Misawa H. Critical roles of acetylcholine and the muscarinic and nicotinic acetylcholine receptors in the regulation of immune function. Life Sci (2012) 91:1027–32. doi: 10.1016/j.lfs.2012.05.006
50. Inclan-Rico JM, Ponessa JJ, Valero-Pacheco N, Hernandez CM, Sy CB, Lemenze AD, et al. Basophils prime group 2 innate lymphoid cells for neuropeptide-mediated inhibition. Nat Immunol (2020) 21:1181–93. doi: 10.1038/s41590-020-0753-y
51. Hosoi T, Okuma Y, Matsuda T, Nomura Y. Novel pathway for LPS-induced afferent vagus nerve activation: possible role of nodose ganglion. Auton Neurosci (2005) 120:104–7. doi: 10.1016/j.autneu.2004.11.012
52. de Lartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE. Diet-induced obesity leads to the development of leptin resistance in vagal afferent neurons. Am J Physiol Endocrinol Metab (2011) 301:E187–195. doi: 10.1152/ajpendo.00056.2011
53. Ma F, Zhang L, Westlund KN. Reactive oxygen species mediate TNFR1 increase after TRPV1 activation in mouse DRG neurons. Mol Pain (2009) 5:31. doi: 10.1186/1744-8069-5-31
54. Li M, Shi J, Tang JR, Chen D, Ai B, Chen J, et al. Effects of complete Freund’s adjuvant on immunohistochemical distribution of IL-1beta and IL-1R I in neurons and glia cells of dorsal root ganglion. Acta Pharmacol Sin (2005) 26:192–8. doi: 10.1111/j.1745-7254.2005.00522.x
55. Cosentino M, Bombelli R, Ferrari M, Marino F, Rasini E, Maestroni GJ, et al. HPLC-ED measurement of endogenous catecholamines in human immune cells and hematopoietic cell lines. Life Sci (2000) 68:283–95. doi: 10.1016/S0024-3205(00)00937-1
56. Quatrini L, Vivier E, Ugolini S. Neuroendocrine regulation of innate lymphoid cells. Immunol Rev (2018) 286:120–36. doi: 10.1111/imr.12707
57. Schedlowski M, Engler H, Grigoleit JS. Endotoxin-induced experimental systemic inflammation in humans: a model to disentangle immune-to-brain communication. Brain Behav Immun (2014) 35:1–8. doi: 10.1016/j.bbi.2013.09.015
58. Mousa A, Bakhiet M. Role of cytokine signaling during nervous system development. Int J Mol Sci (2013) 14:13931–57. doi: 10.3390/ijms140713931
59. Klein Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, et al. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol (2012) 42:1106–16. doi: 10.1002/eji.201142018
60. Van Dyken SJ, Nussbaum JC, Lee J, Molofsky AB, Liang HE, Pollack JL, et al. A tissue checkpoint regulates type 2 immunity. Nat Immunol (2016) 17:1381–7. doi: 10.1038/ni.3582
61. Guo L, Huang Y, Chen X, Hu-Li J, Urban JF Jr, Paul WE, et al. Innate immunological function of TH2 cells in vivo. Nat Immunol (2015) 16:1051–9. doi: 10.1038/ni.3244
62. Halim TY, MacLaren A, Romanish MT, Gold MJ, McNagny KM, Takei F, et al. Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity (2012) 37:463–74. doi: 10.1016/j.immuni.2012.06.012
63. Gold MJ, et al. Group 2 innate lymphoid cells facilitate sensitization to local, but not systemic, TH2-inducing allergen exposures. J Allergy Clin Immunol (2014) 133:1142–8. doi: 10.1016/j.jaci.2014.02.033
64. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature (2010) 464:1367–70. doi: 10.1038/nature08900
65. Saenz SA, Siracusa MC, Perrigoue JG, Spencer SP, Urban JF Jr, Tocker JE, et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature (2010) 464:1362–6. doi: 10.1038/nature08901
66. Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, et al. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol (2011) 12:1071–7. doi: 10.1038/ni.2133
67. Mattiola I, Diefenbach A. Enabling anti-tumor immunity by unleashing ILC2. Cell Res (2020) 30:461–2. doi: 10.1038/s41422-020-0330-9
68. Moral JA, Leung J, Rojas LA, Ruan J, Zhao J, Sethna Z, et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature (2020) 579:130–5. doi: 10.1038/s41586-020-2015-4
69. Besnard AG, Guabiraba R, Niedbala W, Palomo J, Reverchon F, Shaw TN, et al. IL-33-mediated protection against experimental cerebral malaria is linked to induction of type 2 innate lymphoid cells, M2 macrophages and regulatory T cells. PLoS Pathog (2015) 11:e1004607. doi: 10.1371/journal.ppat.1004607
70. Licona-Limon P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nat Immunol (2013) 14:536–42. doi: 10.1038/ni.2617
71. Fukuoka A, Futatsugi-Yumikura S, Takahashi S, Kazama H, Iyoda T, Yoshimoto T, et al. Identification of a novel type 2 innate immunocyte with the ability to enhance IgE production. Int Immunol (2013) 25:373–82. doi: 10.1093/intimm/dxs160
72. Moriyama S, Artis D. Neuronal regulation of group 2 innate lymphoid cells and type 2 inflammation. Adv Immunol (2019) 143:1–9. doi: 10.1016/bs.ai.2019.08.001
73. Erickson LD, Foy TM, Waldschmidt TJ. Murine B1 B cells require IL-5 for optimal T cell-dependent activation. J Immunol (2001) 166:1531–9. doi: 10.4049/jimmunol.166.3.1531
74. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature (2010) 463:540–4. doi: 10.1038/nature08636
75. Drake LY, Iijima K, Kita H. Group 2 innate lymphoid cells and CD4+ T cells cooperate to mediate type 2 immune response in mice. Allergy (2014) 69:1300–7. doi: 10.1111/all.12446
76. Mirchandani AS, Besnard AG, Yip E, Scott C, Bain CC, Cerovic V, et al. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J Immunol (2014) 192:2442–8. doi: 10.4049/jimmunol.1300974
77. von Burg N, Turchinovich G, Finke D. Maintenance of Immune Homeostasis through ILC/T Cell Interactions. Front Immunol (2015) 6:416. doi: 10.3389/fimmu.2015.00416
78. Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci U S A (2010) 107:11489–94. doi: 10.1073/pnas.1003988107
79. Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol (2013) 132:205–13. doi: 10.1016/j.jaci.2013.03.048
80. Noval Rivas M, Burton OT, Oettgen HC, Chatila T. IL-4 production by group 2 innate lymphoid cells promotes foodallergy by blocking regulatory T-cell function. J Allergy Clin Immunol (2016) 138:801–811.e809. doi: 10.1016/j.jaci.2016.02.030
81. Pelly VS, Kannan Y, Coomes SM, Entwistle LJ, Rückerl D, Seddon B, et al. IL-4-producing ILC2s are required for the differentiation of T. Mucosal Immunol (2016) 9:1407–17. doi: 10.1038/mi.2016.4
82. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1β, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol (2016) 17:636–45. doi: 10.1038/ni.3444
83. Motomura Y, Morita H, Moro K, Nakae S, Artis D, Endo TA, et al. Basophil-derived interleukin-4 controls the function of natural helper cells, a member of ILC2s, in lung inflammation. Immunity (2014) 40:758–71. doi: 10.1016/j.immuni.2014.04.013
84. Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature (2013) 502:245–8. doi: 10.1038/nature12526
85. Zhang K, Jin Y, Lai D, Wang J, Wang Y, Wu X, et al. RAGE-induced ILC2 expansion in acute lung injury due to haemorrhagic shock. Thorax (2020) 75:209–19. doi: 10.1136/thoraxjnl-2019-213613
86. Drake LY, Iijima K, Bartemes K, Kita H. Group 2 Innate Lymphoid Cells Promote an Early Antibody Response to a Respiratory Antigen in Mice. J Immunol (2016) 197:1335–42. doi: 10.4049/jimmunol.1502669
87. Angkasekwinai P, Sodthawon W, Jeerawattanawart S, Hansakon A, Pattanapanyasat K, Wang YH, et al. ILC2s activated by IL-25 promote antigen-specific Th2 and Th9 functions that contribute to the control of Trichinella spiralis infection. PLoS One (2017) 12:e0184684. doi: 10.1371/journal.pone.0184684
88. Hrusch CL, Manns ST, Bryazka D, Casaos J, Bonham CA, Jaffery MR, et al. ICOS protects against mortality from acute lung injury through activation of IL-5. Mucosal Immunol (2018) 11:61–70. doi: 10.1038/mi.2017.42
89. Wu L, Lin Q, Ma Z, Chowdhury FA, Mazumder MHH, Du W. Mesenchymal PGD2 activates an ILC2-Treg axis to promoteproliferation of normal and malignant HSPCs. Leukemia (2020) 360:eaan8546. doi: 10.1038/s41375-020-0843-8
90. Xu J, Guardado J, Hoffman R, Xu H, Namas R, Vodovotz Y, et al. IL33-mediated ILC2 activation and neutrophil IL5 production in the lung response after severe trauma: A reverse translation study from a human cohort to a mouse trauma model. PLoS Med (2017) 14:e1002365. doi: 10.1371/journal.pmed.1002365
91. Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, et al. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med (2013) 210:2951–65. doi: 10.1084/jem.20130071
92. Rauber S, Luber M, Weber S, Maul L, Soare A, Wohlfahrt T, et al. Resolution of inflammation by interleukin-9-producing type 2 innatelymphoid cells. Nat Med (2017) 23:938–44. doi: 10.1038/nm.4373
93. Wan J, Huang L, Ji X, Yao S, Hamed Abdelaziz M, Cai W, et al. HMGB1-induced ILC2s activate dendritic cells by producing IL-9 in asthmatic mouse model. Cell Immunol (2020) 352:104085. doi: 10.1016/j.cellimm.2020.104085
94. Roediger B, Kyle R, Tay SS, Mitchell AJ, Bolton HA, Guy TV, et al. IL-2 is a critical regulator of group 2 innate lymphoid cellfunction during pulmonary inflammation. J Allergy Clin Immunol (2015) 136:1653–1663.e1657. doi: 10.1016/j.jaci.2015.03.043
95. Erpenbeck VJ, Hohlfeld JM, Volkmann B, Hagenberg A, Geldmacher H, Braun A, et al. Segmental allergen challenge in patients with atopic asthma leads to increased IL-9 expression in bronchoalveolar lavage fluid lymphocytes. J Allergy Clin Immunol (2003) 111:1319–27. doi: 10.1067/mai.2003.1485
96. Temann UA, Geba GP, Rankin JA, Flavell RA. Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J Exp Med (1998) 188:1307–20. doi: 10.1084/jem.188.7.1307
97. Temann UA, Laouar Y, Eynon EE, Homer R, Flavell RA. IL9 leads to airway inflammation by inducing IL13 expression in airway epithelial cells. Int Immunol (2007) 19:1–10. doi: 10.1093/intimm/dxl117
98. Shimbara A, Christodoulopoulos P, Soussi-Gounni A, Olivenstein R, Nakamura Y, Levitt RC, et al. IL-9 and its receptor in allergic and nonallergic lung disease: increased expression in asthma. J Allergy Clin Immunol (2000) 105:108–15. doi: 10.1016/S0091-6749(00)90185-4
99. Sugita K, Steer CA, Martinez-Gonzalez I, Altunbulakli C, Morita H, Castro-Giner F, et al. Type 2 innate lymphoid cells disrupt bronchial epithelial barrierintegrity by targeting tight junctions through IL-13 in asthmatic patients. J Allergy Clin Immunol (2018) 141:300–310.e311. doi: 10.1016/j.jaci.2017.02.038
100. Bouchery T, Kyle R, Camberis M, Shepherd A, Filbey K, Smith A, et al. ILC2s and T cells cooperate to ensure maintenance of M2 macrophages for lung immunity against hookworms. Nat Commun (2015) 6:6970. doi: 10.1038/ncomms7970
101. Saluzzo S, Gorki AD, Rana BMJ, Martins R, Scanlon S, Starkl P, et al. First-Breath-Induced Type 2 Pathways Shape the Lung Immune Environment. Cell Rep (2017) 18:1893–905. doi: 10.1016/j.celrep.2017.01.071
102. Halim TY, Steer CA, Mathä L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity (2014) 40:425–35. doi: 10.1016/j.immuni.2014.01.011
103. Jia Y, Fang X, Zhu X, Bai C, Zhu L, Jin M, et al. IL-13. Am J Respir Cell Mol Biol (2016) 55:675–83. doi: 10.1165/rcmb.2016-0099OC
104. Zaiss DMW, Gause WC, Osborne LC, Artis D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity (2015) 42:216–26. doi: 10.1016/j.immuni.2015.01.020
105. Zaiss DM, van Loosdregt J, Gorlani A, Bekker CP, Gröne A, Sibilia M, et al. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity (2013) 38:275–84. doi: 10.1016/j.immuni.2012.09.023
106. Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol (2010) 298:L715–731. doi: 10.1152/ajplung.00361.2009
107. Puddicombe SM, Polosa R, Richter A, Krishna MT, Howarth PH, Holgate ST, et al. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J (2000) 14:1362–74. doi: 10.1096/fj.14.10.1362
108. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell (2015) 162:1078–89. doi: 10.1016/j.cell.2015.08.021
109. Smith KA, Filbey KJ, Reynolds LA, Hewitson JP, Harcus Y, Boon L, et al. Low-level regulatory T-cell activity is essential for functional type-2 effector immunity to expel gastrointestinal helminths. Mucosal Immunol (2016) 9:428–43. doi: 10.1038/mi.2015.73
110. Rigas D, Lewis G, Aron JL, Wang B, Banie H, Sankaranarayanan I, et al. Type 2 innate lymphoid cell suppression by regulatory T cellsattenuates airway hyperreactivity and requires inducible T-cell costimulator-inducible T-cell costimulator ligand interaction. J Allergy Clin Immunol (2017) 139:1468–1477.e1462. doi: 10.1016/j.jaci.2016.08.034
111. Maazi H, Patel N, Sankaranarayanan I, Suzuki Y, Rigas D, Soroosh P, et al. ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity (2015) 42:538–51. doi: 10.1016/j.immuni.2015.02.007
112. Yu Y, Tsang JC, Wang C, Clare S, Wang J, Chen X, et al. Single-cell RNA-seq identifies a PD-1. Nature (2016) 539:102–6. doi: 10.1038/nature20105
113. Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu Rev Immunol (2010) 28:57–78. doi: 10.1146/annurev-immunol-030409-101243
114. Bando JK, Gilfillan S, Di Luccia B, Fachi JL, Sécca C, Cella M, et al. ILC2s are the predominant source of intestinal ILC-derived IL-10. J Exp Med (2020) 217(2):e20191520. doi: 10.1084/jem.20191520
115. Belvisi MG. Overview of the innervation of the lung. Curr Opin Pharmacol (2002) 2:211–5. doi: 10.1016/S1471-4892(02)00145-5
116. McMahon SB, La Russa F, Bennett DL. Crosstalk between the nociceptive and immune systems in host defence and disease. Nat Rev Neurosci (2015) 16:389–402. doi: 10.1038/nrn3946
117. Franco R, Schoneveld OJ, Pappa A, Panayiotidis M II. The central role of glutathione in the pathophysiology of human diseases. Arch Physiol Biochem (2007) 113:234–58. doi: 10.1080/13813450701661198
118. Papa I, Saliba D, Ponzoni M, Bustamante S, Canete PF, Gonzalez-Figueroa P, et al. TFH-derived dopamine accelerates productive synapses in germinal centres. Nature (2017) 547:318–23. doi: 10.1038/nature23013
119. Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex–linking immunity and metabolism. Nat Rev Endocrinol (2012) 8:743–54. doi: 10.1038/nrendo.2012.189
120. Su X, Matthay MA, Malik AB. Requisite role of the cholinergic alpha7 nicotinic acetylcholine receptor pathway in suppressing Gram-negative sepsis-induced acute lung inflammatory injury. J Immunol (2010) 184:401–10. doi: 10.4049/jimmunol.0901808
121. Cole SW, Arevalo JM, Takahashi R, Sloan EK, Lutgendorf SK, Sood AK, et al. Computational identification of gene-social environment interaction at the human IL6 locus. Proc Natl Acad Sci U S A (2010) 107:5681–6. doi: 10.1073/pnas.0911515107
122. Collado-Hidalgo A, Sung C, Cole S. Adrenergic inhibition of innate anti-viral response: PKA blockade of Type I interferon gene transcription mediates catecholamine support for HIV-1 replication. Brain Behav Immun (2006) 20:552–63. doi: 10.1016/j.bbi.2006.01.005
123. Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987-2007). Brain Behav Immun (2007) 21:736–45. doi: 10.1016/j.bbi.2007.03.008
124. Grebe KM, Takeda K, Hickman HD, Bailey AL, Embry AC, Bennink JR, et al. Cutting edge: Sympathetic nervous system increases proinflammatory cytokines and exacerbates influenza A virus pathogenesis. J Immunol (2010) 184:540–4. doi: 10.4049/jimmunol.0903395
125. Ding X, Luo Y, Zhang X, Zheng H, Yang X, Yang X, et al. IL-33-driven ILC2/eosinophil axis in fat is induced by sympathetic tone and suppressed by obesity. J Endocrinol (2016) 231:35–48. doi: 10.1530/JOE-16-0229
126. Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve–an integrative interface between twosupersystems: the brain and the immune system. Pharmacol Rev(2000) 52:595–638.
127. Muller PA, Koscsó B, Rajani GM, Stevanovic K, Berres ML, Hashimoto D, et al. Crosstalk between Muscularis Macrophages and Enteric Neurons Regulates Gastrointestinal Motility. Cell (2014) 158:1210. doi: 10.1016/j.cell.2014.08.002
128. Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol (2006) 6:318–28. doi: 10.1038/nri1810
129. Lee HJ, Takemoto N, Kurata H, Kamogawa Y, Miyatake S, O'Garra A, et al. GATA-3 induces T helper cell type 2 (Th2) cytokine expression and chromatin remodeling in committed Th1 cells. J Exp Med (2000) 192:105–15. doi: 10.1084/jem.192.1.105
130. Panina-Bordignon P, Mazzeo D, Lucia PD, D'Ambrosio D, Lang R, Fabbri L, et al. Beta2-agonists prevent Th1 development by selective inhibition of interleukin 12. J Clin Invest (1997) 100:1513–9. doi: 10.1172/JCI119674
131. Cole SW, Korin YD, Fahey JL, Zack JA. Norepinephrine accelerates HIV replication via protein kinase A-dependent effects on cytokine production. J Immunol (1998) 161:610–6.
132. Cai T, Qiu J, Ji Y, Li W, Ding Z, Suo C, et al. IL-17-producing ST2. J Allergy ClinImmunol (2019) 143:229–244.e229. doi: 10.1073/pnas.0900591106
133. Sui P, Wiesner DL, Xu J, Zhang Y, Lee J, Van Dyken S, et al. Pulmonary neuroendocrine cells amplify allergic asthma responses. Science (2018) 360:eaan8546. doi: 10.1126/science.aan8546
134. Pavlov VA, Ochani M, Gallowitsch-Puerta M, Ochani K, Huston JM, Czura CJ, et al. Central muscarinic cholinergic regulation of the systemic inflammatory response during endotoxemia. Proc Natl Acad Sci U S A (2006) 103:5219–23. doi: 10.1073/pnas.0600506103
135. Rosas-Ballina M, Valdés-Ferrer SI, Dancho ME, Ochani M, Katz D, Cheng KF, et al. Xanomeline suppresses excessive pro-inflammatory cytokine responses through neural signal-mediated pathways and improves survival in lethal inflammation. Brain Behav Immun (2015) 44:19–27. doi: 10.1016/j.bbi.2014.07.010
136. Ji H, Rabbi MF, Labis B, Pavlov VA, Tracey KJ, Ghia JE. Central cholinergic activation of a vagus nerve-to-spleen circuit alleviates experimental colitis. Mucosal Immunol (2014) 7:335–47. doi: 10.1038/mi.2013.52
137. Miksa M, Das P, Zhou M, Wu R, Dong W, Ji Y, et al. Pivotal role of the alpha(2A)-adrenoceptor in producing inflammation and organ injury in a rat model of sepsis. PLoS One (2009) 4:e5504. doi: 10.1371/journal.pone.0005504
138. Levine YA, Koopman FA, Faltys M, Caravaca A, Bendele A, Zitnik R, et al. Neurostimulation of the cholinergic anti-inflammatory pathway ameliorates disease in rat collagen-induced arthritis. PLoS One (2014) 9:e104530. doi: 10.1371/journal.pone.0104530
139. von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature (2016) 529:221–5. doi: 10.1038/nature16161
140. Schneider C, O’Leary CE, Locksley RM. Regulation of immune responses by tuft cells. Nat Rev Immunol (2019) 19:584–93. doi: 10.1038/s41577-019-0176-x
141. Skok MV, Grailhe R, Agenes F, Changeux JP. The role of nicotinic receptors in B-lymphocyte development and activation. Life Sci (2007) 80:2334–6. doi: 10.1016/j.lfs.2007.02.005
142. Nizri E, Hamra-Amitay Y, Sicsic C, Lavon I, Brenner T. Anti-inflammatory properties of cholinergic up-regulation: A new role for acetylcholinesterase inhibitors. Neuropharmacology (2006) 50:540–7. doi: 10.1016/j.neuropharm.2005.10.013
143. Nizri E, Irony-Tur-Sinai M, Lory O, Orr-Urtreger A, Lavi E, Brenner T, et al. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J Immunol (2009) 183:6681–8. doi: 10.4049/jimmunol.0902212
144. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature (2003) 421:384–8. doi: 10.1038/nature01339
145. Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med (2004) 10:1216–21. doi: 10.1038/nm1124
146. Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity (2012) 37:634–48. doi: 10.1016/j.immuni.2012.06.020
147. Klein Wolterink RG, Serafini N, van Nimwegen M, Vosshenrich CA, de Bruijn MJ, Fonseca Pereira D, et al. Essential, dose-dependent role for the transcription factor Gata3 in the development of IL-5+ and IL-13+ type 2 innate lymphoid cells. Proc Natl Acad Sci U S A (2013) 110:10240–5. doi: 10.1073/pnas.1217158110
148. Mishra NC, Rir-Sima-Ah J, Langley RJ, Singh SP, Peña-Philippides JC, Koga T, et al. Nicotine primarily suppresses lung Th2 but not goblet cell and muscle cell responses to allergens. J Immunol (2008) 180:7655–63. doi: 10.4049/jimmunol.180.11.7655
149. Cesario A, Russo P, Nastrucci C, Granone P. Is alpha7-nAChR a possible target for lung cancer and malignant pleural mesothelioma treatment? Curr Drug Targets (2012) 13:688–94. doi: 10.2174/138945012800398900
150. West KA, Brognard J, Clark AS, Linnoila IR, Yang X, Swain SM, et al. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Invest (2003) 111:81–90. doi: 10.1172/JCI200316147
151. Dasgupta P, Kinkade R, Joshi B, Decook C, Haura E, Chellappan S, et al. Nicotine inhibits apoptosis induced by chemotherapeutic drugs by up-regulating XIAP and survivin. Proc Natl Acad Sci U S A (2006) 103:6332–7. doi: 10.1073/pnas.0509313103
152. Schaal C, Chellappan SP. Nicotine-mediated cell proliferation and tumor progression in smoking-related cancers. Mol Cancer Res (2014) 12:14–23. doi: 10.1158/1541-7786.MCR-13-0541
153. Steinman L. Elaborate interactions between the immune and nervous systems. Nat Immunol (2004) 5:575–81. doi: 10.1038/ni1078
154. van Westerloo DJ, Giebelen IA, Florquin S, Bruno MJ, Larosa GJ, Ulloa L, et al. The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology (2006) 130:1822–30. doi: 10.1053/j.gastro.2006.02.022
155. Bernik TR, Friedman SG, Ochani M, DiRaimo R, Susarla S, Czura CJ, et al. Cholinergic antiinflammatory pathway inhibition of tumor necrosis factor during ischemia reperfusion. J Vasc Surg (2002) 36:1231–6. doi: 10.1067/mva.2002.129643
156. Altavilla D, Guarini S, Bitto A, Mioni C, Giuliani D, Bigiani A, et al. Activation of the cholinergic anti-inflammatory pathway reduces NF-kappab activation, blunts TNF-alpha production, and protects againts splanchic artery occlusion shock. Shock (2006) 25:500–6. doi: 10.1097/01.shk.0000209539.91553.82
157. Pullan RD, Rhodes J, Ganesh S, Mani V, Morris JS, Williams GT, et al. Transdermal nicotine for active ulcerative colitis. N Engl J Med (1994) 330:811–5. doi: 10.1056/NEJM199403243301202
158. Bernik TR, Friedman SG, Ochani M, DiRaimo R, Ulloa LR, Yang H, et al. Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med (2002) 195:781–8. doi: 10.1084/jem.20011714
159. Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med (2006) 203:1623–8. doi: 10.1084/jem.20052362
160. van Westerloo DJ, Giebelen IA, Florquin S, Daalhuisen J, Bruno MJ, de Vos AF, et al. The cholinergic anti-inflammatory pathway regulates the host response during septic peritonitis. J Infect Dis (2005) 191:2138–48. doi: 10.1086/430323
161. Pavlov VA, Wang H, Czura CJ, Friedman SG, Tracey KJ. The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Mol Med (2003) 9:125–34. doi: 10.1007/BF03402177
162. Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest (2007) 117:289–96. doi: 10.1172/JCI30555
165. Andersson U, Tracey KJ. Reflex principles of immunological homeostasis. Annu Rev Immunol (2012) 30:313–35. doi: 10.1146/annurev-immunol-020711-075015
166. Oke SL, Tracey KJ. From CNI-1493 to the immunological homunculus: physiology of the inflammatory reflex. J Leukoc Biol (2008) 83:512–7. doi: 10.1189/jlb.0607363
167. Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, et al. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A (2008) 105:11008–13. doi: 10.1073/pnas.0803237105
168. Lu B, Kwan K, Levine YA, Olofsson PS, Yang H, Li J, et al. alpha7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol Med (2014) 20:350–8. doi: 10.2119/molmed.2013.00117
169. de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol (2005) 6:844–51. doi: 10.1038/ni1229
170. Guarini S, Altavilla D, Cainazzo MM, Giuliani D, Bigiani A, Marini H, et al. Efferent vagal fibre stimulation blunts nuclear factor-kappaB activation and protects against hypovolemic hemorrhagic shock. Circulation (2003) 107:1189–94. doi: 10.1161/01.CIR.0000050627.90734.ED
171. Feng X, Li L, Feng J, He W, Li N, Shi T, et al. Vagal-α7nAChR signalling attenuates allergic asthma responses and facilitates asthma tolerance by regulating inflammatory group 2 innate lymphoid cells. Immunol Cell Biol (2020). doi: 10.1111/imcb.12400
172. Zhou L. Striking similarity: GATA-3 regulates ILC2 and Th2 cells. Immunity (2012) 37:589–91. doi: 10.1016/j.immuni.2012.10.002
173. Trankner D, Hahne N, Sugino K, Hoon MA, Zuker C. Population of sensory neurons essential for asthmatic hyperreactivity of inflamed airways. Proc Natl Acad Sci U S A (2014) 111:11515–20. doi: 10.1073/pnas.1411032111
174. Chang RB, Strochlic DE, Williams EK, Umans BD, Liberles SD. Vagal Sensory Neuron Subtypes that Differentially Control Breathing. Cell (2015) 161:622–33. doi: 10.1016/j.cell.2015.03.022
175. Mazzone SB, Undem BJ. Vagal Afferent Innervation of the Airways in Health and Disease. Physiol Rev (2016) 96:975–1024. doi: 10.1152/physrev.00039.2015
176. Kollarik M, Ru F, Brozmanova M. Vagal afferent nerves with the properties of nociceptors. Auton Neurosci (2010) 153:12–20. doi: 10.1016/j.autneu.2009.08.001
177. Ni D, Gu Q, Hu HZ, Gao N, Zhu MX, Lee LY, et al. Thermal sensitivity of isolated vagal pulmonary sensory neurons: role of transient receptor potential vanilloid receptors. Am J Physiol Regul Integr Comp Physiol (2006) 291:R541–550. doi: 10.1152/ajpregu.00016.2006
178. Basbaum A II, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell (2009) 139:267–84. doi: 10.1016/j.cell.2009.09.028
179. Canning BJ, Mori N, Mazzone SB. Vagal afferent nerves regulating the cough reflex. Respir Physiol Neurobiol (2006) 152:223–42. doi: 10.1016/j.resp.2006.03.001
180. Dubin AE, Patapoutian A. Nociceptors: the sensors of the pain pathway. J Clin Invest (2010) 120:3760–72. doi: 10.1172/JCI42843
181. Caceres A I, Brackmann M, Elia MD, Bessac BF, del Camino D, D'Amours M, et al. A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma. Proc Natl Acad Sci U S A (2009) 106:9099–104.
182. Talbot S, Abdulnour RE, Burkett PR, Lee S, Cronin SJ, Pascal MA, et al. Silencing Nociceptor Neurons Reduces Allergic Airway Inflammation. Neuron (2015) 87:341–54. doi: 10.1016/j.neuron.2015.06.007
183. Pinho-Ribeiro FA, Verri WA, Chiu IM. Nociceptor Sensory Neuron-Immune Interactions in Pain and Inflammation. Trends Immunol (2017) 38:5–19. doi: 10.1016/j.it.2016.10.001
184. Blake KJ, Jiang XR, Chiu IM. Neuronal Regulation of Immunity in the Skin and Lungs. Trends Neurosci (2019) 42:537–51. doi: 10.1016/j.tins.2019.05.005
185. Russell FA, King R, Smillie SJ, Kodji X, Brain SD. Calcitonin gene-related peptide: physiology and pathophysiology. Physiol Rev (2014) 94:1099–142. doi: 10.1152/physrev.00034.2013
186. Walker CS, Conner AC, Poyner DR, Hay DL. Regulation of signal transduction by calcitonin gene-related peptide receptors. Trends Pharmacol Sci (2010) 31:476–83. doi: 10.1016/j.tips.2010.06.006
187. Chiu IM, Heesters BA, Ghasemlou N, Von Hehn CA, Zhao F, Tran J, et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature (2013) 501:52–7. doi: 10.1038/nature12479
188. Baral P, Umans BD, Li L, Wallrapp A, Bist M, Kirschbaum T, et al. Nociceptor sensory neurons suppress neutrophil and γδ T cell responses in bacterial lung infections and lethal pneumonia. Nat Med (2018) 24:417–26. doi: 10.1038/nm.4501
189. Kuo CS, Krasnow MA. Formation of a Neurosensory Organ by Epithelial Cell Slithering. Cell (2015) 163:394–405. doi: 10.1016/j.cell.2015.09.021
190. Noguchi M, Sumiyama K, Morimoto M. Directed Migration of Pulmonary Neuroendocrine Cells toward Airway Branches Organizes the Stereotypic Location of Neuroepithelial Bodies. Cell Rep (2015) 13:2679–86. doi: 10.1016/j.celrep.2015.11.058
191. Xu H, Ding J, Porter CBM, Wallrapp A, Tabaka M, Ma S, et al. Transcriptional Atlas of Intestinal Immune Cells Reveals thatNeuropeptide α-CGRP Modulates Group 2 Innate Lymphoid Cell Responses. Immunity (2019) 51:696–708.e699. doi: 10.1016/j.immuni.2019.09.004
192. Said S II, Mutt V. Polypeptide with broad biological activity: isolation from small intestine. Science (1970) 169:1217–8. doi: 10.1126/science.169.3951.1217
193. Lara-Marquez M, O’Dorisio M, O’Dorisio T, Shah M, Karacay B. Selective gene expression and activation-dependent regulation of vasoactive intestinal peptide receptor type 1 and type 2 in human T cells. J Immunol (2001) 166:2522–30. doi: 10.4049/jimmunol.166.4.2522
194. Calvo JR, Pozo D, Guerrero JM. Functional and molecular characterization of VIP receptors and signal transduction in human and rodent immune systems. Adv Neuroimmunol (1996) 6:39–47. doi: 10.1016/S0960-5428(96)00005-8
195. Dickson L, Finlayson K. VPAC and PAC receptors: From ligands to function. Pharmacol Ther (2009) 121:294–316. doi: 10.1016/j.pharmthera.2008.11.006
196. Moriyama M, Furue H, Katafuchi T, Teranishi H, Sato T, Kano T, et al. Presynaptic modulation by neuromedin U of sensory synaptic transmission in rat spinal dorsal horn neurones. J Physiol (2004) 559:707–13. doi: 10.1113/jphysiol.2004.070110
197. Howard AD, Wang R, Pong SS, Mellin TN, Strack A, Guan XM, et al. Identification of receptors for neuromedin U and its role in feeding. Nature (2000) 406:70–4. doi: 10.1038/35017610
198. Nakahara K, Kojima M, Hanada R, Egi Y, Ida T, Miyazato M, et al. Neuromedin U is involved in nociceptive reflexes and adaptation to environmental stimuli in mice. Biochem Biophys Res Commun (2004) 323:615–20. doi: 10.1016/j.bbrc.2004.08.136
199. Hedrick JA, Morse K, Shan L, Qiao X, Pang L, Wang S, et al. Identification of a human gastrointestinal tract and immune system receptor for the peptide neuromedin U. Mol Pharmacol (2000) 58:870–5. doi: 10.1124/mol.58.4.870
200. Moriyama M, Fukuyama S, Inoue H, Matsumoto T, Sato T, Tanaka K, et al. The neuropeptide neuromedin U activates eosinophils and is involved in allergen-induced eosinophilia. Am J Physiol Lung Cell Mol Physiol (2006) 290:L971–977. doi: 10.1152/ajplung.00345.2005
201. Ohki-Hamazaki H, Neuromedin B. Prog Neurobiol (2000) 62:297–312. doi: 10.1016/S0301-0082(00)00004-6
202. Gajjar S, Patel BM. Neuromedin: An insight into its types, receptors and therapeutic opportunities. Pharmacol Rep (2017) 69:438–47. doi: 10.1016/j.pharep.2017.01.009
203. Krane IM, Naylor SL, Helin-Davis D, Chin WW, Spindel ER. Molecular cloning of cDNAs encoding the human bombesin-like peptideneuromedin B. Chromosomal localization and comparison to cDNAs encoding its amphibian homolog ranatensin. J Biol Chem (1988) 263:13317–23.
204. Wada E, Way J, Lebacq-Verheyden AM, Battey JF, Neuromedin B. and gastrin-releasing peptide mRNAs are differentially distributed in the rat nervous system. J Neurosci (1990) 10:2917–30. doi: 10.1523/JNEUROSCI.10-09-02917.1990
205. Khumalo J, Kirstein F, Hadebe S, Brombacher F. IL-4Rα signalling in CD4+CD25+FoxP3+ T regulatory cells restrain airway inflammation via limiting local tissue IL-33. JCI Insight (2020) 5:136206. doi: 10.1172/jci.insight.136206
206. Howard E, Lewis G, Galle-Treger L, Hurrell BP, Helou DG, Shafiei-Jahani P, et al. IL-10 production by ILC2s requires Blimp-1 and cMaf, modulatescellular metabolism and ameliorates airway hyperreactivity. J Allergy ClinImmunol (2020) 6:S0091-6749(20)31234-3. doi: 10.1016/j.jaci.2020.08.024
207. Xiao Q, He J, Lei A, Xu H, Zhang L, Zhou P, et al. PPARγ enhances ILC2 function during allergic airwayinflammation via transcription regulation of ST2. Mucosal Immunol (2020). doi: 10.1038/s41385-020-00339-6
208. Helou DG, Shafiei-Jahani P, Lo R, Howard E, Hurrell BP, Galle-Treger L, et al. PD-1 pathway regulates ILC2 metabolism and PD-1 agonist treatmentameliorates airway hyperreactivity. Nat Commun (2020) 11:3998. doi: 10.1038/s41467-020-17813-1
209. Miller MM, Patel PS, Bao K, Danhorn T, O'Connor BP, Reinhardt RL, et al. BATF acts as an essential regulator of IL-25-responsive migratoryILC2 cell fate and function. Sci Immunol (2020) 5(43):eaay3994. doi: 10.1126/sciimmunol.aay3994
210. Huang Y, Mao K, Chen X, Sun MA, Kawabe T, Li W, et al. S1P-dependent interorgan trafficking of group 2 innate lymphoidcells supports host defense. Science (2018) 359:114–9. doi: 10.1126/science.aam5809
211. Akama Y, Park EJ, Satoh-Takayama N, Gaowa A, Ito A, Kawamoto E, et al. Sepsis Induces Deregulation of IL-13 Production and PD-1 Expression in Lung Group 2 Innate Lymphoid Cells. Shock (2020). doi: 10.1097/SHK.0000000000001647
212. Xu H, Xu J, Xu L, Jin S, Turnquist HR, Hoffman R, et al. Interleukin-33 contributes to ILC2 activation and early inflammation-associated lung injury during abdominal sepsis. Immunol Cell Biol (2018) 96:935–47. doi: 10.1111/imcb.12159
213. Wang W, Cohen JA, Wallrapp A, Trieu KG, Barrios J, Shao F, et al. Age-Related Dopaminergic Innervation Augments T Helper 2-TypeAllergic Inflammation in the Postnatal Lung. Immunity (2019) 51:1102–1118.e1107. doi: 10.1016/j.immuni.2019.10.002
214. O’Leary CE, Schneider C, Locksley RM. Tuft Cells-Systemically Dispersed Sensory Epithelia Integrating Immune and Neural Circuitry. Annu Rev Immunol (2019) 37:47–72. doi: 10.1146/annurev-immunol-042718-041505
215. Zhang K, Xu X, Pasha MA, Siebel CW, Costello A, Haczku A, et al. Cutting Edge: Notch Signaling Promotes the Plasticity of Group-2 Innate Lymphoid Cells. J Immunol (2017) 198:1798–803. doi: 10.4049/jimmunol.1601421
216. Bernink JH, Ohne Y, Teunissen MBM, Wang J, Wu J, Krabbendam L, et al. c-Kit-positive ILC2s exhibit an ILC3-like signature that may contribute to IL-17-mediated pathologies. Nat Immunol (2019) 20:992–1003. doi: 10.1038/s41590-019-0423-0
217. Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol (2016) 17:626–35. doi: 10.1038/ni.3443
218. Wiegert JS, Mahn M, Prigge M, Printz Y, Yizhar O. Silencing Neurons: Tools, Applications, and Experimental Constraints. Neuron (2017) 95:504–29. doi: 10.1016/j.neuron.2017.06.050
Keywords: neuroimmunity, adaptive immunity, innate immunity, lung, group 2 innate lymphoid cells (ILC2)
Citation: Chen W, Shu Q and Fan J (2020) Neural Regulation of Interactions Between Group 2 Innate Lymphoid Cells and Pulmonary Immune Cells. Front. Immunol. 11:576929. doi: 10.3389/fimmu.2020.576929
Received: 27 June 2020; Accepted: 05 October 2020;
Published: 29 October 2020.
Edited by:
Maureen Ann Cox, University of Oklahoma Health Sciences Center, United StatesReviewed by:
Chaofeng Han, Second Military Medical University, ChinaCyril Seillet, Walter and Eliza Hall Institute of Medical Research, Australia
Copyright © 2020 Chen, Shu and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Fan, amlmN0BwaXR0LmVkdQ==