 Yangzhi Qi
Yangzhi Qi Baohui Liu
Baohui Liu Qian Sun
Qian Sun Xiaoxing Xiong
Xiaoxing Xiong Qianxue Chen
Qianxue Chen- Department of Neurosurgery, Renmin Hospital of Wuhan University, Wuhan, China
Glioma is the most malignant primary tumor of the central nervous system and is characterized by an extremely low overall survival. Recent breakthroughs in cancer therapy using immune checkpoint blockade have attracted significant attention. However, despite representing the most promising (immunotherapy) treatment for cancer, the clinical application of immune checkpoint blockade in glioma patients remains challenging due to the “cold phenotype” of glioma and multiple factors inducing resistance, both intrinsic and acquired. Therefore, comprehensive understanding of the tumor microenvironment and the unique immunological status of the brain will be critical for the application of glioma immunotherapy. More sensitive biomarkers to monitor the immune response, as well as combining multiple immunotherapy strategies, may accelerate clinical progress and enable development of effective and safe treatments for glioma patients.
Introduction
In recent years, novel immunotherapies targeting the immune component of the tumor microenvironment have shown great promise for the clinical management of tumors. Among various therapeutic strategies, drugs targeting immune checkpoint molecules are being heralded as a breakthrough in cancer immunotherapy.
Glioma is the most common and deadliest primary brain tumor of the central nervous system (CNS), with a 5-year survival of less than 10%. Glioblastoma multiforme (GBM) accounts for ~50% of glioma cases and is characterized by a 5-year survival rate of less than 5%, corresponding to a grade IV tumor by the World Health Organization (WHO). Unfortunately, the current gold standard of GBM treatment (total resection plus adjuvant radio-chemotherapy) represents only a palliative option for patients, and the median survival after diagnosis is less than 15 months (1).
A striking recent clinical success of checkpoint inhibitors across multiple solid tumors (2, 3) has sparked interest in immune-targeted strategies for glioma treatment. However, the CNS is commonly considered an “immunologically privileged” site as the blood-brain barrier (BBB) inhibits direct contact between the brain and immune system. Considering the unique accessibility and tissue composition of brain, it is therefore not trivial to design effective immunotherapeutic strategies. Herein, we review the unique immunology and tumor microenvironment of the brain. Furthermore, we describe various immune checkpoint blockade strategies, as well as the mechanisms of resistance to immunotherapy.
The CNS Is Immunologically “Unique” Rather Than “Privileged”
The term “immunologically privileged” has been commonly used to describe the failure of the brain to reject heterotopic tissue following transplantation in the past decades. Our understanding of this special characteristic of brain immunology largely originates from experiments by Peter Medawar in the 1940s (4). Although allogeneic tissue transplantation in other areas of the body can lead to immune rejection which continues to the CNS, there is a lack of convincing explanations for the fact that this systemic immune state cannot be initiated from the CNS. Medawar attributed this phenomenon to the lack of lymph nodes and lymphatic vessels in the CNS, which result in the perceived absence of efferent information of the CNS, although this view has been recently disproven (5–12). A series of studies have demonstrated that leukocyte lymphatics exist in the dura sinus and transport antigens from the dura to cervical lymph nodes (9–12). These findings propose an interesting mechanism by which cerebrospinal fluid mediates the immune communication between CNS and circulation via a glial-lymphatic pathway (5–8). Given the existence of an afferent system between the brain and peripheral immune system, many propose that CNS is immunologically “unique” rather than “privileged.”
For the most part, the BBB is responsible for this “unique immunology” of the brain. Structurally, the BBB consist of a bio-membrane between vascular endothelial cells and glial cells. Functionally, the BBB is a dynamic network between circulation and brain that blocks the diffusion of large, hydrophilic molecules or organisms while allowing the influx of small, hydrophobic molecules (13). Except for a few species, such as Neisseria meningitides and Streptococcus pneumoniae which are able to enter the brain circulation via specific mechanisms, the vast majority of blood-borne pathogens are excluded from the brain (14). Given that the CNS is rarely exposed to pathogens, the brain has been believed to only exhibit limited immunity due to poor tolerance of the brain tissue to inflammation. Another unique immunological characteristic of the brain is its resident immune cell population. Originating from myeloid precursors born in the yolk sac, microglial cells (MG) invade the CNS during early embryonic development and serve as the primary resident immune cells (15, 16), while most other immune cell subtypes do not exist in CNS. However, contrary to the previous view that the brain only exhibits limited immunity, recent studies have demonstrated that the systemic immune system is fully involved in the cytotoxic response to CNS antigens (17). After inflammatory stimulation, specific antigens are recognized by MG and presented to activated lymphocytes via the glial-lymphatic pathway, after which a large number of immune cells can easily penetrate the BBB, inducing a strong inflammatory and subsequent immune response (18–20). Despite this, both innate inflammatory and adaptive immune responses have to be tightly regulated as unrestrained inflammation-mediated intracranial hypertension can have serious consequences. Although the concept of immunological privilege of the brain has been overturned, the unique immunological environment of the CNS still represents a significant hurdle for therapies targeting immune checkpoints blockade in the brain.
The Immune Microenvironment of Glioma
The unique brain immunology leads to a particular tumor microenvironment of glioma. A variety of peripheral immune components are present in this glioma microenvironment, including myeloid derived suppressor cells (MDSCs), natural killer cells (NK cells), macrophages, neutrophils, CD4+ helper T cells (Th), CD8+ cytotoxic T lymphocytes (CTLs), and regulatory T (T reg) cells (21, 22), while their infiltration ratio is remarkably low numbers in gliomas compared to other tumors. Furthermore, various tumor-derived cytokines and chemokines reprogram infiltrating immune cells, which causes them to acquire unique functional phenotypes and transform into tumor-associated immune cells. These tumor-associated immune cells can therefore have profound effects on progression, recurrence, and therapeutic resistance of glioma by inducing inflammatory or anti-inflammatory responses (Figure 1).
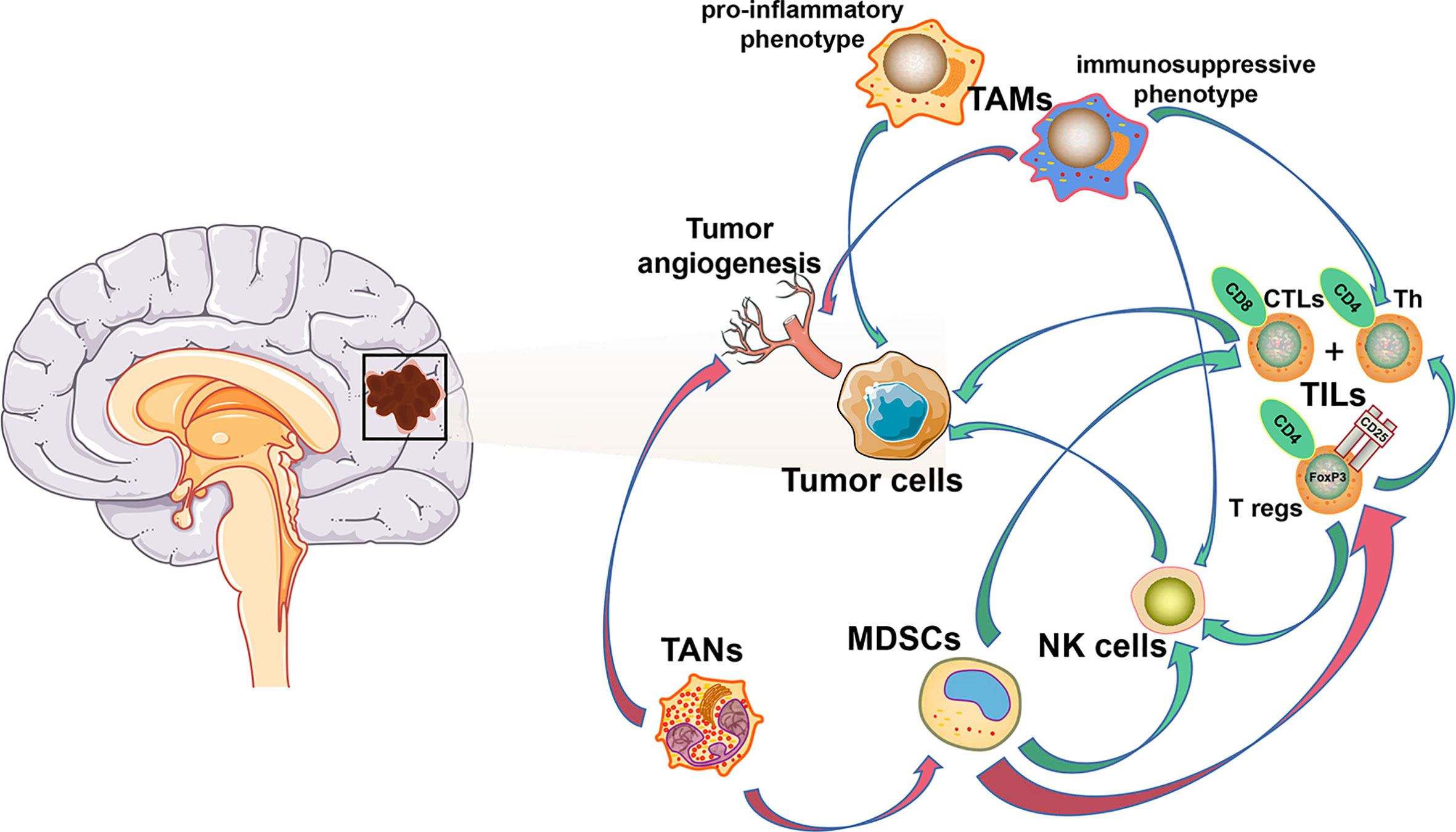
Figure 1 Cellular composition of glioma immune microenvironment. The figure depicts only a general representation of all the cell types that have been reported to be associated with tumor cells in glioma immune microenvironment. Green arrow: down-regulation. Red arrow: up-regulation.
Tumor-Infiltrating Lymphocytes
As the most important component of the immune response in the tumor microenvironment of most solid tumors, tumor-infiltrating lymphocytes (TILs), represented by CD4+ Th, CD8+ CTLs, and CD4+/CD25+/FoxP3+ T reg (23–26), are only present in remarkably low numbers in the CNS compared to other tumor types. CD4+ Th and CD8+ CTL populations increase with tumor malignancy, starting at 39% in WHO grade II tumors, rising to 73% in WHO grade III, and 98% in grade IV (22). Meanwhile, a correlation between increased CD8+ CTL counts and improved patient outcomes has previously been reported (27). T reg cells have a suppressive role in the adaptive immune response and inhibit the proliferation of Th cells and CTLs by secretion of suppressive cytokines (26).
The limited activity and number of TILs in the brain is predominantly caused by the unique immunological status of the brain which encourages only limited immunity in order to prevent an inflammation-mediated intracranial hypertension crisis. In response to tumor-derived inflammatory stimulation, T reg cells secrete anti-inflammatory interleukin-10 (IL-10) and transforming growth factor β (TGF-β) in order to dampen an inflammatory immune response against the tumor (28, 29). In addition to immunosuppressive mechanisms of the CNS, expression of certain genes by the tumor itself also contributes to low levels of TILs. For instance, glioma cells produce a high level of indolamine 2, 3-dioxygenase (IDO) which activates suppressor T cells by depleting tryptophan from the tumor microenvironment (30). Besides, tumor-derived Fas ligand promotes apoptosis of activated T cells and leads to an immune escape of tumor cells by inhibiting dendritic cells and maturation of T cells (31). Overexpression of programmed cell death-ligand 1 (PD-L1) in glioma cells prevents activation of T cells and induces T cell apoptosis via binding to programmed death 1 (PD-1), a well-known inhibitory immune checkpoint molecule (32, 33). Moreover, an overexpression of CTLA-4 mRNA and protein, a strong CD4+ T cell and CD8+ CTL inhibitor, is caused by lack of CD80/86 co-stimulatory molecules (22). Therefore, a comprehensive understanding of tumor heterogeneity and the role of T cells in glioma is of critical importance for the design of therapeutic targets.
Tumor-Associated Macrophages
Tumor-associated macrophages (TAMs) are the major infiltrating immune component in the glioma microenvironment, accounting for ~50% of all immune cells, and have an important role in neoplasia, metastasis, immune escape, and tumor angiogenesis (34, 35). Several studies have reported that a majority of TAMs are derived from circulating monocyte-derived macrophages (MDMs), while the remaining proportion originates from MG (36–38). Immature monocytes migrate to the tumor microenvironment and develop into TAMs following exposure to several cytokines (34, 35). In the glioma microenvironment, tumor- or effector T cell-derived cytokines promote a change in macrophage effector mechanisms on a spectrum between a pro-inflammatory “M1” phenotype with anti-tumor responses, and an immunosuppressive “M2” phenotype with anti-inflammatory responses (39). In the early stages of glioma, TAMs inhibit tumor proliferation via the pro-inflammatory “M1” phenotype, while in advanced glioma, TAMs are predominantly characterized by the “M2” phenotype, which generally induces an immunosuppressive response and immune escape of the tumor. As a special type of TAMs in CNS, MG also exhibit similar plasticity to monocyte-derived macrophages (40).
Studies have demonstrated that large numbers of infiltrating TAMs are closely associated with poor prognosis (41) and the “M2” phenotype has been shown to promote tumor progression via secretion of immunosuppressive cytokines and factors promoting angiogenesis (41, 42). Given this evidence, it seems feasible to block the formation and phenotypic “M2” transformation of TAMs. In mouse models, CSF-1 receptor inhibition with small molecules either blocks the transformation of “M2” phenotype or depletes TAMs (43–46), both of which inhibit glioma progression and invasion. Meanwhile, some other drugs have also been shown to achieve their anti-tumor effect by depleting monocytes that serve as precursors of TAM (47). However, recent studies have expanded our understanding of macrophage polarization (48) and revealed a multifaceted response comprising classical M1 and M2 polarization, including expression changes associated with chronic inflammatory stimuli and exposure to free fatty acids, which is involved in regulation of bone marrow cell function. This indicates that the diverse transcriptional programming of TAMs in glioma extends beyond the simplified view of an “M1” versus “M2” polarization. Thus, despite the fact that both depletion of TAMs and targeting “M2” polarization can represent attractive therapeutic approaches for glioma, a more comprehensive understanding of TAM phenotypes is required for efficient and safe treatments of glioma (43, 49, 50).
Myeloid-Derived Suppressor Cells
Chronic inflammation in the tumor microenvironment is induced by overexpression of pro-inflammatory cytokines, including CSF-1, VEGF, TGF-β, and tumor necrosis factor α (TNF-α) (51, 52). These pro-inflammatory cytokines promote tumor growth, progression, and resistance to immunotherapy by inducing a transformation of immature myeloid cells into myeloid-derived suppressor cells (MDSCs). MDSCs are recruited to peripheral lymphoid organs and the tumor microenvironment from the bone marrow, promoting tumor cell proliferation via various mechanisms, including suppression of cytotoxic NK cell activity, inhibition of the adaptive T cell response, induction of T cell apoptosis and T reg cell proliferation, and secretion of immunosuppressive cytokines (53–56). Therefore, MDSCs contribute to resistance to immunotherapy, and combining treatments targeting MDSCs with other immunotherapies has become a promising therapeutic strategy achieving considerable success (57–61). In glioma, related research has been focusing on strategies that either inhibit the recruitment (targeting of C-C motif chemokine ligand 2, VEGF-A, IL-8, and galectin-1) or the formation of MDSCs from myeloid precursors (targeting of M-CSF, PI3Kγ, TAM-RTKs, and COX-2). Such strategies have shown great promise in preclinical studies (62). As there is increasing evidence that the function of MDSCs is tumor type-dependent, a clear definition of this cell type in glioma remains warranted (49). Transcriptomic characterizations of MDSCs - separately from MG and MDMs – should be carried out to ascertain the suppressive function and mechanisms of differentiation into MDSCs, which could help to evaluate the clinical value of MDSCs-targeted therapies in glioma (63).
Tumor-Associated Neutrophils
Completely contrary to their pro-inflammatory function during infections, neutrophils have been frequently reported to promote tumor progression and metastasis in recent years (64–66). This unique relationship between neutrophils and tumor cells could provide a reasonable explanation for the phenomenon that circulating tumor cells often escape from immune surveillance in breast cancer as neutrophils account for the largest proportion of circulatory leukocytes (66). Besides, current study also indicated that immunosuppressive tumor-associated neutrophils (TANs) or granulocytic MDSCs are enriched in neutrophil-enriched subtypes of triple negative breast cancer and were associated with acquired immune checkpoint blockade resistance (67). In the glioma microenvironment, TANs promote tumor malignancy by mediating angiogenesis (68). Besides, TAN depletion strategies using a Ly6G+ monoclonal antibody have been shown to prolong overall survival in preclinical GBM mouse models (69). However, the mechanisms underlying TAN recruitment to the tumor microenvironment and the role of TANs in tumor progression are not yet comprehensively understood and how the glioma microenvironment heterogeneity affects neutrophil reprogramming still remains to be unraveled.
Natural Killer Cells
A variety of mechanisms suppressing the activity of natural killer (NK) cells, the most efficient innate cytotoxic lymphocytes, have been identified during tumor cell progression. Similar to normal cells, glioma cells can inhibit antigen presenting cell (APC)-mediated recognition and NK cell-mediated killing through expression of MHC class I molecules (MHC I) that interact with NK cell immunoglobulin-like receptors (KIRs) (49). Besides, infiltrating NK cells in the glioma microenvironment have been reported to be commonly nonfunctional, largely owing to the combined negative regulatory effect of TAMs, MDSCs, and T reg cells (49, 70).
Immune Checkpoint Blockade Strategy and Inhibitors
There is no doubt that among various immunotherapies, despite that checkpoint blockade might not be the most promising treatment for glioma, it has been the immunotherapy most developed in clinical use. Via a combination of specific antibodies and checkpoint molecules, effector T cells can be reactivated and exert tumor cell cytotoxicity. In the next paragraphs, we describe classical immune checkpoint molecules and their inhibitors (Table 1).
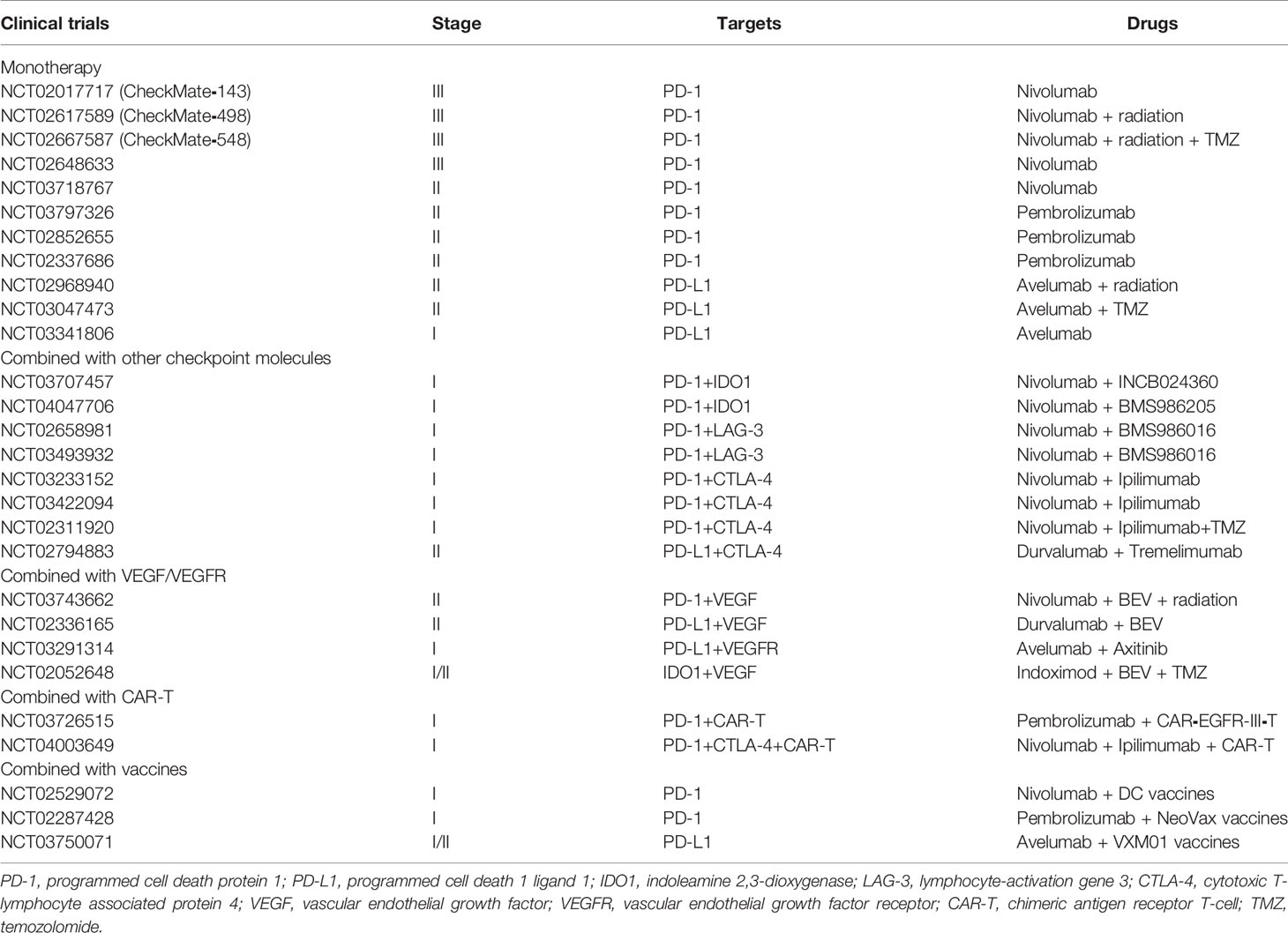
Table 1 Current clinical trials of immune checkpoint blockade.
PD-1/PD-L1
PD-1 and its ligands PD-L1/2 are the most comprehensively studied immune checkpoint molecules to date. PD-1 negatively regulates T cell receptor-mediated signaling transduction pathways and, in combination with PD-L1, inhibits activation and cytotoxic T cell effects and blocks the production of inflammatory factors, resulting in T cell inactivity. Expression of PD-1 on immune cells is tightly regulated. For instance, PD-1 expression appears on the surface of T cells shortly (less than 24 h) after T cell activation and decreases with the elimination or clearance of the antigen (49). Under chronic inflammatory conditions or in cancer, antigens repetitively stimulate CTLs to maintain high levels of PD-1 expression, eventually resulting in T cell exhaustion and depletion. Tumor-expressed PD-L1 is regulated by several mechanisms, including phosphatidylinositol 3-kinase (PI3K) signaling pathway activation and TIL-secreted interferon γ (IFN-γ) (71). In glioma, PD-L1 is predominantly expressed on tumor cells and TAMs and negatively relates to patient outcome (72–74). To date, two anti-PD-1 antibodies (Nivolumab, Pembrolizumab) and three anti-PD-L1 antibodies (Atezolizumab, Avelumab, Durvalumab) have been put into clinical application and have achieved dramatic successes against a variety of solid tumors (75–77). However, they have so far not been approved for clinical treatment of GBM despite numerous preclinical successes reported over the past decades (78–83). For instance, in the preclinical GL261 model, anti-PD-1 treatment success is dosage dependent, with the best outcome reported being a cure rate of 50% (81, 83). Anti-PD-1 monotherapy has been observed to result in an increased ratio of CD8+ CTLs to T reg cells, and enhanced efficacy when combined with radiation and other checkpoint inhibitors (81, 83).
Schalper et al. (84) reported treatment of 30 GBM patients (3 primary, 27 recurrent) with preoperative and postoperative nivolumab (NCT02550249), resulting in increased transcription of chemokines, infiltration of TILs, and diversity of TCR in tumor microenvironment. While no patients with recurrent GBM benefited from treatment as measured by overall survival (OS), two of the three primary GBM patients survived for 33 month and 28 months, respectively. CheckMate⁃143 phase III trial (NCT02017717) found no OS benefit when comparing nivolumab with bevacizumab (anti-VEGFA) in the treatment of recurrent GBM (median OS 9.8 vs. 10.0 months) (85). In the CheckMate⁃498 trial (NCT02617589), newly diagnosed GBM patients with unmethylated O6-methylguanine-DNA methytransferase (MGMT) promoter who received nivolumab plus radiotherapy did not benefit from this treatment compared with radiotherapy plus temozolomide (TMZ) as measured by OS (86). More recently, similarly disappointing results have been reported in the CheckMate-548 study (NCT02667587). Here, newly diagnosed GBM patients with methylated MGMT promoter did not show a PFS benefit with anti-PD-1 treatment; the OS effect is still pending (87). Primary results of a study by Lukas et al. (88) reporting on a clinical trial using atezolizumab, an anti-PD-L1 antibody (NCT01375842), showed that increased CD4+ T cells and IDH mutation indicated better treatment efficiency of atezolizumab.
CTLA-4
Cytotoxic T-lymphocyte associated protein 4 (CTLA-4) expression on activated T cells or T reg cells was the first identified member of the immunoglobulin superfamily, and also the first immune regulation molecule used in targeted therapy. CTLA-4 inhibits T cell co-stimulatory signaling pathways by combining with ligands CD80 and CD86 expressed on APCs (89). Unlike PD-1, CTLA-4 signaling occurs at the early stages of T cell activation, and CTLA-4 is mainly expressed on T cells of the lymph node (90). In preclinical experiments, anti-CTLA-4 monotherapy prolonged OS in the GL261 syngeneic mouse model (81). Although CTLA-4 blockade strategy results in an increased median survival with 25% cure rate, the response of monotherapy was still considered limited as combined application of anti-PD-1 therapy or radiotherapy can remarkably improve efficacy (81, 90). Reardon et al. (81) also reported that combination of anti-CTLA-4 and anti-PD-1 therapy increased the cure rate to 75%. For further investigation, CTLA-4 blockade as a monotherapy or in combination with anti-PD-1 treatment is therefore currently being tested in a phase III clinical trial in patients with recurrent GBM (NCT02017717).
B7 Family
In recent years, there have been increasing numbers of studies investigating immune checkpoint molecules of the B7 family. In addition to PD-L1 (B7-H1), studies have investigated B7-H3 (CD276), B7-H4, B7-H5 (Vista), B7-H6, and B7-H7 (HHLA2), amongst others. B7-H3 and B7-H7 have a dual function, enabling both co-stimulation and co-inhibition (91). By interacting with specific ligands, these molecules can therefore have different roles in tumor progression. For instance, recent research points out that B7-H3 positively relates to the Toll-like receptor signaling pathway and the poor survival of glioma patients, while it has also been reported to co-stimulate immunological function and be involved in anti-tumor functions (92–95). Similarly, B7-H7 shows the same phenomenon in various solid tumors (96, 97). Inhibiting NK-mediated recognition of B7-H6 is an important mechanism of the tumor immune escape. NK cells eliminate B7-H6-expressing tumor cells either directly via cytotoxicity or indirectly by cytokine secretion, which highlights a role for the tumor-induced “self”-molecule B7-H6 in alerting innate immunity (91). Both B7-H4 and B7-H5 have co-inhibitory functions on the immune system (91), although research on these and other members of the B7 family is still in progress. As the largest immune checkpoint family, the function and mechanisms of B7 family members in glioma remains largely unknown. Thus, a more comprehensive understating of the function of the B7 family in glioma could help to explore more effective therapeutic targets in immunotherapy.
IDO, LAG-3, and TIM-3
Indoleamine 2,3-dioxygenase (IDO) is the key enzyme of the L-tryptophan metabolism via the kynurenine pathway. Although IDO expressed on tumor cells and dendritic cells (DCs) is not a typical checkpoint molecule, it can inhibit T cell activation by modulation of the tryptophan metabolism which has an important role in the function of T cells (98–101). Metabolites of tryptophan also induce T cell apoptosis (101). Besides, an interaction of kynurenine and TGF-β can induce FoxP3 expression in T cells, which results in T reg cell polarization (102, 103). Preclinical models have shown that clinical trials with IDO inhibitors did not meet the expectations (104).
Lymphocyte-activation gene 3 (LAG-3) has four extracellular immunoglobulin superfamily-like domains which bind to MHC II, and is responsible for transmission of inhibitory signals (105). In addition to MHC II, another ligand for LAG-3 is Gal⁃3, which is involved in the inhibition of CD8+ CTLs (106). Tumor-derived antigens induce LAG-3 overexpression and thereby lead to the depletion of CD8+ CTLs (107). Research in mouse xenografts revealed that co-targeting of PD-1 and LAG-3 on TILs can limit tumor growth, which is likely superior to a single inhibitory mechanism (108, 109). Given this finding, recent trials have focused on anti-PD-1 and anti-LAG-3 combination therapies rather than monotherapies. However, the vast majority of this research in still in preclinical stages.
T cell immunoglobulin domain and mucin domain protein-3 (TIM-3) is expressed on CD4+ and CD8+ T cells, monocytes, and macrophages (110). TIM-3 regulates T cell depletion and is involved in tumor immunosuppression and immune escape via binding to its ligand Gal-9 (110). Clinical trials reported that GBM patients with overexpression of TIM-3 have higher tumor malignancy, a lower quality of life, and worse prognosis (111, 112).
Although several checkpoint-related molecules have been discovered, there have been none as influential as PD-1 and CTLA-4, and the efficiency of the vast majority of checkpoint inhibitors in glioma remains doubtful. While single checkpoint inhibition is the standard of care in many tumor entities, checkpoint molecules cooperate or antagonize each other in tumor progression, making it difficult for a single checkpoint inhibitor to play a decisive role in systemic immunity. Therefore, combination of checkpoint inhibitors seems to be more efficient than monotherapy.
Lessons From Clinical Failures
There is no doubt that immunotherapy holds promise for the treatment of glioma. However, even promising preclinical data are rarely translated into clinical success in glioma. Two factors complicate the clinical translation for glioma treatment. Firstly, glioma has a “cold tumor” phenotype, which is associated with a poor response to immunotherapy. Owing to the unique environment of CNS, even after inhibiting checkpoint molecules to induce T cell responses against glioma, antigen-specific TILs remain at relatively low levels. Second, current preclinical models have only limited capacity to reflect the real tumor heterogeneity of glioma. Generally, GBM can be classified into four subtypes: classical, proneural, neural, and mesenchymal, with high heterogeneity between each subtype (113, 114). There are remarkable differences in gene expression among these four subtypes, which suggests that targeting checkpoint molecules therapies may only be effective for some subpopulations expressing specific genes, but not for other subpopulations. These two factors interact to form resistance mechanisms at all phases of the antitumor immune response: intrinsic resistance prevents the initiation of a response; adaptive resistance deactivates tumor-infiltrating immune cells; and acquired resistance protects the tumor from elimination in the face of attack by the immune system. Even though dramatic immune responses have been observed in preclinical models using a variety of immunotherapy strategies, patients rarely benefit from these treatments, owing to the extensive immunosuppressive mechanisms of glioma (115, 116). However, these mechanisms render glioma a valuable model for studying how resistance allows tumors to escape immunotherapy.
Intrinsic Resistance
Intrinsic tumor resistance can be classified into three groups: patient-intrinsic factors (including sex, age, and HLA genotype), tumor-intrinsic factors (including the host immune system and tumor-associated stroma), and environmental factors (117–119). Among them, tumor-intrinsic factors, relating to the genetic, transcriptional or functional profile of the tumor cells, are the main determinants of response and resistance (116).
Several studies have demonstrated that tumors can prevent immune responses by not expressing high-quality neoantigens, and they can furthermore rapidly suppress immune responses by expressing multiple immune checkpoint ligands and immunosuppressive cytokines (115, 116). Meanwhile, even with sufficient antigenicity, sensitivity to immune checkpoint blockade can be disrupted by tumorintrinsic genetic defects in the IFN γ signaling pathway and antigen presentation (120–122). A disruption in anti-tumor response to the IFN γ signaling pathway can inhibit the Janus kinase (JAK) and (STAT) signaling pathway, downregulating PD-L1 expression, and making anti-PD-1 treatment ineffective. Besides, the WNT–β-catenin signaling pathway has been confirmed to prevent an anti-tumor immune response by inhibiting dendritic cells and promoting the immunosuppressive cytokine IL-10 (123–126). Meanwhile, the MAPK signaling pathway also contributes to tumor immune escape by upregulation of the expression of the immunosuppressive cytokines IL6 and IL10 (127).
To date, the heterogeneity of glioma is still considered the basis for its resistance to a variety of treatments. For instance, the most extensively studied neoantigen, EGFR variant III, is a truncated EGFR neoantigen with expression in 19% of newly diagnosed GBM patients, of which 11% exhibit high levels of expression (128). Although nearly 82% of recurrent tumors do not express EGFR variant III, the vast majority of mesenchymal subtypes shows overexpression of EGFR variant III (129, 130). This characteristic makes it difficult to stably express specific antigens to induce a durable anti-tumor immune response. Besides, despite the fact that adjuvant radio-chemotherapy can enhance the efficiency of checkpoint blockade strategies, what cannot be ignored is that radio-chemotherapy has well-documented immunosuppressive functions inducing other resistance mechanisms rather than tumor-intrinsic resistance to immunotherapy, which further reduces the immune responses of the CNS (131).
Adaptive Resistance
The discovery that tumors can counter attacks of the immune system by usurping mechanisms that normally prevent autoimmunity is one of the most impactful findings in the history of oncology. Although immune checkpoint molecules may be expressed in various tumors at “baseline,” a remarkable increase of their expression levels can be observed under immunological stimulation (132). Thus, immune checkpoint blockade can trigger strong anti-tumor response. In spite of the durable clinical responses that PD-1 and CTLA-4 blockade strategies have achieved in several advanced tumors, it is undeniable a large proportion of patients do not benefit from checkpoint blockade (132). One explanation for this is that TILs can exhibit severe exhaustion, similar to that observed in chronic viral infections (133). However, while the degree of immune exhaustion in GBM is severe, it does not appear to be singularly so, as other tumors that respond poorly to checkpoint inhibitors use similar adaptive resistance mechanisms (115, 134). Another explanation is that checkpoint molecules with similar mechanisms can compensate for each other. For instance, upregulation of the alternative checkpoint molecule TIM-3 has been observed in tumors resisting PD-1 blockade (135). Downregulation of one immune checkpoint generally upregulates alternative immune checkpoints, eventually leading to the durable immunosuppression and a resistance to the blockade. Given this mechanism, current clinical trials focus on overcoming adaptive resistance of PD-1 and CTLA-4 blockade strategies by targeting alternative immune checkpoints.
Acquired Resistance
Acquired resistance of tumor generally refers to the genetic alternations caused by immunological pressure (115). For instance, in non-small cell lung cancer (NSCLCs) and melanoma, significant downregulation of targeted antigens has been observed in tumor infiltrating region, resulting in the failure of immune targeted therapy (136, 137). Perhaps therapies that overcome intrinsic resistance mechanisms will also render acquired resistance inconsequential by generating a diverse repertoire of T cell clones targeting high-quality targeted antigens that rapidly eliminate a tumor before acquired resistance emerges. However, the exact effects of acquired resistance on malignant glioma remain unknown, as the low response and persistence of treatments in glioma have been considered as an important intrinsic resistance mechanism. In contrast, recent research reported 66 recurrent GBM patients who received PD-1 blockade therapy (138). Among them, 17 patients were identified as responders based on brain imaging and profiling of resected tissue. Tumors in responders were found to be enriched for alterations in the mitogen-activated protein kinase pathway and exhibited branched patterns of evolution, while non-responding tumors more frequently had mutations in the gene encoding PTEN and non-clonal evolution patterns (138). Notably, responders had a significantly longer OS than non-responders (14.3 vs. 10.1 months) (138). Given the heterogeneity of GBM mentioned above, in addition to intrinsic resistance, acquired resistance seems to play an important role in resistance to checkpoint blockade.
Hypoxia in the Glioblastoma Microenvironment
To date, research has mainly focused on the “seed’s” response to therapy (i.e., tumor cells themselves), while the problem of “poor soil” (the tumor microenvironment) is often ignored. Herein, we further explored the role of hypoxia in the tumor immune microenvironment. Accumulating evidence indicates that hypoxia may protect tumors from immune responses through various mechanisms, including by inhibition of NK and CTL cell activity, promotion of immunosuppressive cytokines, and by enhancing immunosuppressive cells (T reg cells, TAMs, and neutrophils) (139).
CTLs and NK Cells
There are an increasing number of studies investigating the effects hypoxia on immune cells. For instance, IL-2, an important growth factor for T and NK cells with a pivotal role in the regulation of the host’s immune response, has been reported to be exquisitely sensitive to changes in oxygen tension (140). Hypoxia can cause a prolonged reduction in IL-2 mRNA expression and inhibit NK cell and CTL activity. Meanwhile, hypoxia has also been shown to reduce the ability of NK cells to release IFNγ, TNFα, GM-CSF, CCL3, and CCL5 (139, 141). In patients with a high risk of hypoxia, CTLs and NK cells appeared to be in resting status rather than active (139), revealing that hypoxia might lead to a state of immune suppression.
Suppressive Immune Cells and Cytokines
Hypoxia is thought to play a key role in TAM polarization. It can promote the “M2” phenotype and contribute to tumor growth, immune suppression, and tumor angiogenesis (142–144). In a bioinformatic study assessing the polarization of cells in the tumor immune microenvironment, T reg cells, neutrophils, and TAMs with an “M2” phenotype increased remarkably under hypoxia (139). Besides, hypoxia also promotes the expression of TGF-β and IL-10, two well-established suppressive cytokines (139, 142, 145).
Future Directions
The extensive immunosuppressive mechanisms in “seed” (including tumor heterogeneity and alteration of checkpoint molecules) and “soil” (hypoxia in tumor microenvironment) complicates the treatment of glioma and explains why promising preclinical data had rarely been translated into clinical success. Given this, individualized treatment and real-time monitoring of treatment response are essential.
Biomarkers
Predicting and monitoring patient responses to treatment have become an urgent requirement for the clinical development of immunotherapies. Tumor tissue biopsies remain the gold standard for diagnosis, but its application is not suitable for response monitoring. Complex and changeable signals on MRI furthermore challenge the differentiation of glioma recurrence from pseudoprogression and radiation brain necrosis. Thus, the availability of biomarkers has greatly enhanced oncological practices and is now the basis of precision medicine for many cancers. However, suitable biomarkers for immunotherapies of glioma are still unknown. Recently, studies have reported that anti-PD-1 therapy results in upregulation of T cell- and IFN γ-related gene expression in immune cells, as well as downregulation of cell-cycle-related gene expression within tumor cells (84, 138, 146). Anti-PD-1 therapy seems to result in different responses in tumors with specific genetic alternation, including increased clonal expansion of T cells, decreased expression of PD-1 in peripheral T cells, and decreased monocytes in circulation (84, 138, 146). Liquid biopsies are anticipated to become a successful strategy for biomarker response monitoring in glioma. For instance, tumor mutation burden (TMB) based on detection of circulating tumor DNA shows a high correlation with anti PD-1 response (147). Meanwhile, current studies also indicated that circulating tumor cells (CTCs) of glioma offer unique advantages for non-invasive monitoring of tumor progression which could furthermore identify pseudoprogression and radiation necrosis (148, 149). Taken together, an efficient biomarker can not only help to choose individualized treatment, but also timely reflect when patients develop resistance to adjust the treatment.
Combined Drug Therapy
Immunotherapy resistance of glioma is a result of multiple factors: intrinsic resistance and adaptive resistance in the early stages of treatment, and acquired resistance over the period of therapy mediated by genetic alternations. Owing to unique resistance mechanisms, monotherapy of checkpoint inhibitors for glioma does not seem to induce durable anti-tumor responses. Thus, combined drug therapy, to some extent, may show advantages and higher efficacy. For instance, in preclinical model, anti-PD-1 combined with anti-TIM-3 synergistically improved survival (135). Furthermore, the combination of immune checkpoint blockade and anti-tumor-associated immune cells (TAMs, MDSCs) also holds promise for the treatment of glioma. Therefore, a more comprehensive understanding of immune cell roles in the tumor microenvironment, as well as specific biomarkers for functional immune cell types and tumor response, may be necessary for individualized treatment of patients with glioma in the era of precision medicine.
Author Contributions
YQ and BL contributed equally to this article. XX and QC designed this study. YQ, QS and BL performed the data collection and collation. All the authors were involved in the analysis and interpretation of data. YQ wrote the paper, with the help of the coauthors. XX and QC reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin (2010) 60(3):166–93. doi: 10.3322/caac.20069
2. Emens LA, Ascierto PA, Darcy PK, Demaria S, Eggermont AMM, Redmond WL, et al. Cancer immunotherapy: Opportunities and challenges in the rapidly evolving clinical landscape. Eur J Cancer (2017) 81:116–29. doi: 10.1016/j.ejca.2017.01.035
3. Lipson EJ, Forde PM, Hammers HJ, Emens LA, Taube JM, Topalian SL. Antagonists of PD-1 and PD-L1 in Cancer Treatment. Semin Oncol (2015) 42(4):587–600. doi: 10.1053/j.seminoncol.2015.05.013
4. Medawar PB. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol (1948) 29(1):58–69.
5. Goldmann J, Kwidzinski E, Brandt C, Mahlo J, Richter D, Bechmann I. T cells traffic from brain to cervical lymph nodes via the cribroid plate and the nasal mucosa. J Leukoc Biol (2006) 80(4):797–801. doi: 10.1189/jlb.0306176
6. Bradbury MW, Westrop RJ. Factors influencing exit of substances from cerebrospinal fluid into deep cervical lymph of the rabbit. J Physiol (1983) 339:519–34. doi: 10.1113/jphysiol.1983.sp014731
7. Cserr HF, Knopf PM. Cervical lymphatics, the blood-brain barrier and the immunoreactivity of the brain: a new view. Immunol Today (1992) 13(12):507–12. doi: 10.1016/0167-5699(92)90027-5
8. Widner H, Jönsson BA, Hallstadius L, Wingårdh K, Strand SE, Johansson BB. Scintigraphic method to quantify the passage from brain parenchyma to the deep cervical lymph nodes in rats. Eur J Nucl Med (1987) 13(9):456–61. doi: 10.1007/BF00281860
9. Eide PK, Vatnehol SAS, Emblem KE, Ringstad G. Magnetic resonance imaging provides evidence of glymphatic drainage from human brain to cervical lymph nodes. Sci Rep (2018) 8(1):7194. doi: 10.1038/s41598-018-25666-4
10. Louveau A, Herz J, Alme MN, Salvador AF, Dong MQ, Viar KE, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci (2018) 21(10):1380–91. doi: 10.1038/s41593-018-0227-9
11. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med (2015) 212(7):991–9. doi: 10.1084/jem.20142290
12. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523(7560):337–41. doi: 10.1038/nature14432
13. Bauer HC, Krizbai IA, Bauer H, Traweger A. “You Shall Not Pass”-tight junctions of the blood brain barrier. Front Neurosci (2014) 8:392. doi: 10.3389/fnins.2014.00392
14. Coureuil M, Lécuyer H, Bourdoulous S, Nassif X. A journey into the brain: insight into how bacterial pathogens cross blood-brain barriers. Nat Rev Microbiol (2017) 15(3):149–59. doi: 10.1038/nrmicro.2016.178
15. Hutter G, Theruvath J, Graef CM, Zhang M, Schoen MK, Manz EM, et al. Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc Natl Acad Sci U.S.A. (2019) 116(3):997–1006. doi: 10.1073/pnas.1721434116
16. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330(6005):841–5. doi: 10.1126/science.1194637
17. Klein RS, Izikson L, Means T, Gibson HD, Lin E, Sobel RA, et al. IFN-inducible protein 10/CXC chemokine ligand 10-independent induction of experimental autoimmune encephalomyelitis. J Immunol (Baltimore Md 1950) (2004) 172(1):550–9. doi: 10.4049/jimmunol.172.1.550
18. Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev (2006) 213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x
19. Masson F, Calzascia T, Di Berardino-Besson W, de Tribolet N, Dietrich PY, Walker PR. Brain microenvironment promotes the final functional maturation of tumor-specific effector CD8+ T cells. J Immunol (Baltimore Md 1950) (2007) 179(2):845–53. doi: 10.4049/jimmunol.179.2.845
20. Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol (2016) 53(2):1181–94. doi: 10.1007/s12035-014-9070-5
21. Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W, et al. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin Cancer Res (2008) 14(16):5166–72. doi: 10.1158/1078-0432.CCR-08-0320
22. Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab Investigat J Tech Methods Pathol (2017) 97(5):498–518. doi: 10.1038/labinvest.2017.19
23. Waziri A. Glioblastoma-derived mechanisms of systemic immunosuppression. Neurosurg Clin N Am (2010) 21(1):31–42. doi: 10.1016/j.nec.2009.08.005
24. Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-β. Clin Cancer Res (2011) 17(13):4296–308. doi: 10.1158/1078-0432.CCR-10-2557
25. Alexiou GA, Vartholomatos G, Karamoutsios A, Batistatou A, Kyritsis AP, Voulgaris S. Circulating progenitor cells: a comparison of patients with glioblastoma or meningioma. Acta Neurol Belg (2013) 113(1):7–11. doi: 10.1007/s13760-012-0097-y
26. Wainwright DA, Dey M, Chang A, Lesniak MS. Targeting Tregs in Malignant Brain Cancer: Overcoming IDO. Front Immunol (2013) 4:116. doi: 10.3389/fimmu.2013.00116
27. Zhang W, Wu S, Guo K, Hu Z, Peng J, Li J. Correlation and clinical significance of LC3, CD68+ microglia, CD4+ T lymphocytes, and CD8+ T lymphocytes in gliomas. Clin Neurol Neurosurg (2018) 168:167–74. doi: 10.1016/j.clineuro.2018.02.044
28. Gong D, Shi W, Yi SJ, Chen H, Groffen J, Heisterkamp N. TGFβ signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol (2012) 13:31. doi: 10.1186/1471-2172-13-31
29. Vitkovic L, Maeda S, Sternberg E. Anti-inflammatory cytokines: expression and action in the brain. Neuroimmunomodulation (2001) 9(6):295–312. doi: 10.1159/000059387
30. Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res (2012) 18(22):6110–21. doi: 10.1158/1078-0432.CCR-12-2130
31. Jansen T, Tyler B, Mankowski JL, Recinos VR, Pradilla G, Legnani F, et al. FasL gene knock-down therapy enhances the antiglioma immune response. Neuro Oncol (2010) 12(5):482–9. doi: 10.1093/neuonc/nop052
32. Perng P, Lim M. Immunosuppressive Mechanisms of Malignant Gliomas: Parallels at Non-CNS Sites. Front Oncol (2015) 5:153. doi: 10.3389/fonc.2015.00153
33. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity (2007) 27(1):111–22. doi: 10.1016/j.immuni.2007.05.016
34. de Groot AE, Pienta KJ. Epigenetic control of macrophage polarization: implications for targeting tumor-associated macrophages. Oncotarget (2018) 9(29):20908–27. doi: 10.18632/oncotarget.24556
35. Liu Y, Cao X. The origin and function of tumor-associated macrophages. Cell Mol Immunol (2015) 12(1):1–4. doi: 10.1038/cmi.2014.83
36. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol (2015) 17(2):170–82. doi: 10.1038/ncb3090
37. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep (2016) 17(9):2445–59. doi: 10.1016/j.celrep.2016.10.052
38. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res (2017) 77(9):2266–78. doi: 10.1158/0008-5472.CAN-16-2310
39. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
40. Walker DG, Lue LF. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther (2015) 7(1):56. doi: 10.1186/s13195-015-0139-9
41. Roesch S, Rapp C, Dettling S, Herold-Mende C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int J Mol Sci (2018) 19(2):436. doi: 10.3390/ijms19020436
42. da Fonseca AC, Badie B. Microglia and macrophages in malignant gliomas: recent discoveries and implications for promising therapies. Clin Dev Immunol (2013) 2013:264124. doi: 10.1155/2013/264124
43. Yan D, Kowal J, Akkari L, Schuhmacher AJ, Huse JT, West BL, et al. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene (2017) 36(43):6049–58. doi: 10.1038/onc.2017.261
44. Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science (2016) 352(6288):aad3018. doi: 10.1126/science.aad3018
45. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med (2013) 19(10):1264–72. doi: 10.1038/nm.3337
46. Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med (Cambridge Mass) (2012) 18(1):519–27. doi: 10.2119/molmed.2011.00217
47. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell (2013) 23(2):249–62. doi: 10.1016/j.ccr.2013.01.008
48. Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell (2020) 181(7):1643–60.e17. doi: 10.1016/j.cell.2020.05.007
49. Boussiotis VA, Charest A. Immunotherapies for malignant glioma. Oncogene (2018) 37(9):1121–41. doi: 10.1038/s41388-017-0024-z
50. Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer (2020) 20(1):12–25. doi: 10.1038/s41568-019-0224-7
51. Meirow Y, Kanterman J, Baniyash M. Paving the Road to Tumor Development and Spreading: Myeloid-Derived Suppressor Cells are Ruling the Fate. Front Immunol (2015) 6:523. doi: 10.3389/fimmu.2015.00523
52. Umansky V, Blattner C, Gebhardt C, Utikal J. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines (Basel) (2016) 4(4):36. doi: 10.3390/vaccines4040036
53. Sceneay J, Chow MT, Chen A, Halse HM, Wong CS, Andrews DM, et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res (2012) 72(16):3906–11. doi: 10.1158/0008-5472.CAN-11-3873
54. Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res (2012) 72(6):1384–94. doi: 10.1158/0008-5472.CAN-11-2905
55. Yan HH, Pickup M, Pang Y, Gorska AE, Li Z, Chytil A, et al. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res (2010) 70(15):6139–49. doi: 10.1158/0008-5472.CAN-10-0706
56. Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol (2015) 98(6):913–22. doi: 10.1189/jlb.4RI0515-204R
57. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U.S.A. (2014) 111(32):11774–9. doi: 10.1073/pnas.1410626111
58. Christmas BJ, Rafie CI, Hopkins AC, Scott BA, Ma HS, Cruz KA, et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol Res (2018) 6(12):1561–77. doi: 10.1158/2326-6066.CIR-18-0070
59. Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin Cancer Res (2017) 23(17):5187–201. doi: 10.1158/1078-0432.CCR-17-0741
60. Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight (2019) 4(7):e126853. doi: 10.1172/jci.insight.126853
61. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell (2016) 29(6):832–45. doi: 10.1016/j.ccell.2016.04.014
62. Ding AS, Routkevitch D, Jackson C, Lim M. Targeting Myeloid Cells in Combination Treatments for Glioma and Other Tumors. Front Immunol (2019) 10:1715. doi: 10.3389/fimmu.2019.01715
63. Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells (2020) 9(3):561. doi: 10.3390/cells9030561
64. Khan S, Mittal S, McGee K, Alfaro-Munoz KD, Majd N, Balasubramaniyan V, et al. Role of Neutrophils and Myeloid-Derived Suppressor Cells in Glioma Progression and Treatment Resistance. Int J Mol Sci (2020) 21(6):1954. doi: 10.3390/ijms21061954
65. Garrido-Navas C, de Miguel-Perez D, Exposito-Hernandez J, Bayarri C, Amezcua V, Ortigosa A, et al. Cooperative and Escaping Mechanisms between Circulating Tumor Cells and Blood Constituents. Cells (2019) 8(11):1382. doi: 10.3390/cells8111382
66. Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature (2019) 566(7745):553–7. doi: 10.1038/s41586-019-0915-y
67. Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol (2019) 21(9):1113–26. doi: 10.1038/s41556-019-0373-7
68. Fujita M, Scheurer ME, Decker SA, McDonald HA, Kohanbash G, Kastenhuber ER, et al. Role of type 1 IFNs in antiglioma immunosurveillance–using mouse studies to guide examination of novel prognostic markers in humans. Clin Cancer Res (2010) 16(13):3409–19. doi: 10.1158/1078-0432.CCR-10-0644
69. Liang J, Piao Y, Holmes L, Fuller GN, Henry V, Tiao N, et al. Neutrophils promote the malignant glioma phenotype through S100A4. Clin Cancer Res (2014) 20(1):187–98. doi: 10.1158/1078-0432.CCR-13-1279
70. Poli A, Wang J, Domingues O, Planagumà J, Yan T, Rygh CB, et al. Targeting glioblastoma with NK cells and mAb against NG2/CSPG4 prolongs animal survival. Oncotarget (2013) 4(9):1527–46. doi: 10.18632/oncotarget.1291
71. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
72. Heiland DH, Haaker G, Delev D, Mercas B, Masalha W, Heynckes S, et al. Comprehensive analysis of PD-L1 expression in glioblastoma multiforme. Oncotarget (2017) 8(26):42214–25. doi: 10.18632/oncotarget.15031
73. Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol (2016) 18(2):195–205. doi: 10.1093/neuonc/nov172
74. Liu S, Wang Z, Wang Y, Fan X, Zhang C, Ma W, et al. PD-1 related transcriptome profile and clinical outcome in diffuse gliomas. Oncoimmunology (2018) 7(2):e1382792. doi: 10.1080/2162402X.2017.1382792
75. Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol (2015) 16(4):375–84. doi: 10.1016/S1470-2045(15)70076-8
76. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
77. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. New Engl J Med (2013) 369(2):134–44. doi: 10.1056/NEJMoa1305133
78. Agarwalla P, Barnard Z, Fecci P, Dranoff G, Curry WT Jr. Sequential immunotherapy by vaccination with GM-CSF-expressing glioma cells and CTLA-4 blockade effectively treats established murine intracranial tumors. J Immunother (Hagerstown Md 1997) (2012) 35(5):385–9. doi: 10.1097/CJI.0b013e3182562d59
79. Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res (2007) 13(7):2158–67. doi: 10.1158/1078-0432.CCR-06-2070
80. Antonios JP, Soto H, Everson RG, Orpilla J, Moughon D, Shin N, et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight (2016) 1(10):e87059. doi: 10.1172/jci.insight.87059
81. Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol Res (2016) 4(2):124–35. doi: 10.1158/2326-6066.CIR-15-0151
82. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res (2014) 20(20):5290–301. doi: 10.1158/1078-0432.CCR-14-0514
83. Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys (2013) 86(2):343–9. doi: 10.1016/j.ijrobp.2012.12.025
84. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med (2019) 25(3):470–6. doi: 10.1038/s41591-018-0339-5
85. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol (2020) 6(7):1003–10.. doi: 10.1001/jamaoncol.2020.1024
86. Bristol⁃Myers Squibb. Announces Phase 3 CheckMate⁃498 study did not meet primary endpoint of overall survival with opdivo (nivolumab) plus radiation in patients with newly diagnosed MGMT⁃unmethylated glioblastoma multiforme. Princeton, NJ: Bristol⁃Myers Squibb Company (2019). 2019⁃11-04.
87. Bristol⁃Myers Squibb. Provides Update on Phase 3 Opdivo (Nivolumab) CheckMate ⁃ 548 trial in patients with newly diagnosed MGMT ⁃ methylated glioblastoma multiforme. Princeton, NJ: Bristol⁃Myers Squibb Company (2019). 2019⁃11⁃ 04.
88. Lukas RV, Rodon J, Becker K, Wong ET, Shih K, Touat M, et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J Neuro-Oncol (2018) 140(2):317–28. doi: 10.1007/s11060-018-2955-9
89. Fong B, Jin R, Wang X, Safaee M, Lisiero DN, Yang I, et al. Monitoring of regulatory T cell frequencies and expression of CTLA-4 on T cells, before and after DC vaccination, can predict survival in GBM patients. PloS One (2012) 7(4):e32614. doi: 10.1371/journal.pone.0032614
90. Belcaid Z, Phallen JA, Zeng J, See AP, Mathios D, Gottschalk C, et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PloS One (2014) 9(7):e101764. doi: 10.1371/journal.pone.0101764
91. Ni L, Dong C. New B7 Family Checkpoints in Human Cancers. Mol Cancer Ther (2017) 16(7):1203–11. doi: 10.1158/1535-7163.MCT-16-0761
92. Wang Z, Wang Z, Zhang C, Liu X, Li G, Liu S, et al. Genetic and clinical characterization of B7-H3 (CD276) expression and epigenetic regulation in diffuse brain glioma. Cancer Sci (2018) 109(9):2697–705. doi: 10.1111/cas.13744
93. Zhou Z, Luther N, Ibrahim GM, Hawkins C, Vibhakar R, Handler MH, et al. B7-H3, a potential therapeutic target, is expressed in diffuse intrinsic pontine glioma. J Neuro-Oncol (2013) 111(3):257–64. doi: 10.1007/s11060-012-1021-2
94. Wu CP, Jiang JT, Tan M, Zhu YB, Ji M, Xu KF, et al. Relationship between co-stimulatory molecule B7-H3 expression and gastric carcinoma histology and prognosis. World J Gastroenterol (2006) 12(3):457–9. doi: 10.3748/wjg.v12.i3.457
95. Loos M, Hedderich DM, Ottenhausen M, Giese NA, Laschinger M, Esposito I, et al. Expression of the costimulatory molecule B7-H3 is associated with prolonged survival in human pancreatic cancer. BMC Cancer (2009) 9:463. doi: 10.1186/1471-2407-9-463
96. Qi Y, Deng G, Xu P, Zhang H, Yuan F, Geng R, et al. HHLA2 is a novel prognostic predictor and potential therapeutic target in malignant glioma. Oncol Rep (2019) 42(6):2309–22. doi: 10.3892/or.2019.7343
97. Yan H, Qiu W, Koehne de Gonzalez AK, Wei JS, Tu M, Xi CH, et al. HHLA2 is a novel immune checkpoint protein in pancreatic ductal adenocarcinoma and predicts post-surgical survival. Cancer Lett (2019) 442:333–40. doi: 10.1016/j.canlet.2018.11.007
98. Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother CII (2014) 63(7):721–35. doi: 10.1007/s00262-014-1549-4
99. Godin-Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Cancer Res (2011) 17(22):6985–91. doi: 10.1158/1078-0432.CCR-11-1331
100. Löb S, Königsrainer A, Rammensee HG, Opelz G, Terness P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: can we see the wood for the trees? Nat Rev Cancer (2009) 9(6):445–52. doi: 10.1038/nrc2639
101. Mbongue JC, Nicholas DA, Torrez TW, Kim NS, Firek AF, Langridge WH. The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity. Vaccines (Basel) (2015) 3(3):703–29. doi: 10.3390/vaccines3030703
102. Mellor AL, Munn DH. Physiologic control of the functional status of Foxp3+ regulatory T cells. J Immunol (Baltimore Md 1950) (2011) 186(8):4535–40. doi: 10.4049/jimmunol.1002937
103. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol (Baltimore Md 1950) (2010) 185(6):3190–8. doi: 10.4049/jimmunol.0903670
104. Hanihara M, Kawataki T, Oh-Oka K, Mitsuka K, Nakao A, Kinouchi H. Synergistic antitumor effect with indoleamine 2,3-dioxygenase inhibition and temozolomide in a murine glioma model. J Neurosurg (2016) 124(6):1594–601. doi: 10.3171/2015.5.JNS141901
105. Workman CJ, Vignali DA. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur J Immunol (2003) 33(4):970–9. doi: 10.1002/eji.200323382
106. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol Res (2015) 3(4):412–23. doi: 10.1158/2326-6066.CIR-14-0150
107. Andrews LP, Marciscano AE, Drake CG, Vignali DA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev (2017) 276(1):80–96. doi: 10.1111/imr.12519
108. Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget (2015) 6(29):27359–77. doi: 10.18632/oncotarget.4751
109. Jing W, Gershan JA, Weber J, Tlomak D, McOlash L, Sabatos-Peyton C, et al. Combined immune checkpoint protein blockade and low dose whole body irradiation as immunotherapy for myeloma. J Immunother Cancer (2015) 3(1):2. doi: 10.1186/s40425-014-0043-z
110. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev (2017) 276(1):97–111. doi: 10.1111/imr.12520
111. Han S, Feng S, Xu L, Shi W, Wang X, Wang H, et al. Tim-3 on peripheral CD4⁺ and CD8⁺ T cells is involved in the development of glioma. DNA Cell Biol (2014) 33(4):245–50. doi: 10.1089/dna.2013.2306
112. Li G, Wang Z, Zhang C, Liu X, Cai J, Wang Z, et al. Molecular and clinical characterization of TIM-3 in glioma through 1,024 samples. Oncoimmunology (2017) 6(8):e1328339. doi: 10.1080/2162402X.2017.1328339
113. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17(1):98–110. doi: 10.1016/j.ccr.2009.12.020
114. The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455(7216):1061–8. doi: 10.1038/nature07385
115. Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol (2019) 20(9):1100–9. doi: 10.1038/s41590-019-0433-y
116. Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol (2020) 20(1):25–39. doi: 10.1038/s41577-019-0218-4
117. Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science (2018) 359(6375):582–7. doi: 10.1126/science.aao4572
118. Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity (2016) 44(6):1255–69. doi: 10.1016/j.immuni.2016.06.001
119. Fessler J, Matson V, Gajewski TF. Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer (2019) 7(1):108. doi: 10.1186/s40425-019-0574-4
120. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science (2013) 340(6129):202–7. doi: 10.1126/science.1235208
121. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science (2013) 340(6129):207–11. doi: 10.1126/science.1235214
122. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell (2016) 167(6):1540–54.e12. doi: 10.1016/j.cell.2016.11.022
123. Yaguchi T, Goto Y, Kido K, Mochimaru H, Sakurai T, Tsukamoto N, et al. Immune suppression and resistance mediated by constitutive activation of Wnt/β-catenin signaling in human melanoma cells. J Immunol (Baltimore Md 1950) (2012) 189(5):2110–7. doi: 10.4049/jimmunol.1102282
124. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature (2015) 523(7559):231–5. doi: 10.1038/nature14404
125. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell (2017) 31(5):711–23.e4. doi: 10.1016/j.ccell.2017.04.003
126. Holtzhausen A, Zhao F, Evans KS, Tsutsui M, Orabona C, Tyler DS, et al. Melanoma-Derived Wnt5a Promotes Local Dendritic-Cell Expression of IDO and Immunotolerance: Opportunities for Pharmacologic Enhancement of Immunotherapy. Cancer Immunol Res (2015) 3(9):1082–95. doi: 10.1158/2326-6066.CIR-14-0167
127. Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med (2006) 203(7):1651–6. doi: 10.1084/jem.20051848
128. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155(2):462–77. doi: 10.1016/j.cell.2013.09.034
129. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol (2010) 28(31):4722–9. doi: 10.1200/JCO.2010.28.6963
130. Wood MD, Reis GF, Reuss DE, Phillips JJ. Protein Analysis of Glioblastoma Primary and Posttreatment Pairs Suggests a Mesenchymal Shift at Recurrence. J Neuropathol Exp Neurol (2016) 75(10):925–35. doi: 10.1093/jnen/nlw068
131. Seifert L, Werba G, Tiwari S, Giao Ly NN, Nguy S, Alothman S, et al. Radiation Therapy Induces Macrophages to Suppress T-Cell Responses Against Pancreatic Tumors in Mice. Gastroenterology (2016) 150(7):1659–72.e5. doi: 10.1053/j.gastro.2016.02.070
132. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27(4):450–61. doi: 10.1016/j.ccell.2015.03.001
133. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin Cancer Res (2018) 24(17):4175–86. doi: 10.1158/1078-0432.CCR-17-1846
134. Bauer C, Kühnemuth B, Duewell P, Ormanns S, Gress T, Schnurr M. Prevailing over T cell exhaustion: New developments in the immunotherapy of pancreatic cancer. Cancer Lett (2016) 381(1):259–68. doi: 10.1016/j.canlet.2016.02.057
135. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
136. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discovery (2017) 7(3):264–76. doi: 10.1158/1538-7445.AM2017-NG01
137. Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature (2019) 567(7749):479–85. doi: 10.1038/s41586-019-1032-7
138. Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med (2019) 25(3):462–9. doi: 10.1038/s41591-019-0349-y
139. Lin W, Wu S, Chen X, Ye Y, Weng Y, Pan Y, et al. Characterization of Hypoxia Signature to Evaluate the Tumor Immune Microenvironment and Predict Prognosis in Glioma Groups. Front Oncol (2020) 10:796. doi: 10.3389/fonc.2020.00796
140. Zuckerberg AL, Goldberg LI, Lederman HM. Effects of hypoxia on interleukin-2 mRNA expression by T lymphocytes. Crit Care Med (1994) 22(2):197–203. doi: 10.1097/00003246-199402000-00008
141. Parodi M, Raggi F, Cangelosi D, Manzini C, Balsamo M, Blengio F, et al. Hypoxia Modifies the Transcriptome of Human NK Cells, Modulates Their Immunoregulatory Profile, and Influences NK Cell Subset Migration. Front Immunol (2018) 9:2358. doi: 10.3389/fimmu.2018.02358
142. Guo X, Xue H, Shao Q, Wang J, Guo X, Chen X, et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget (2016) 7(49):80521–42. doi: 10.18632/oncotarget.11825
143. Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell (2013) 24(6):695–709. doi: 10.1016/j.ccr.2013.11.007
144. Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Investigation (2016) 126(10):3672–9. doi: 10.1172/JCI84427
145. Dennis KL, Blatner NR, Gounari F, Khazaie K. Current status of interleukin-10 and regulatory T-cells in cancer. Curr Opin Oncol (2013) 25(6):637–45. doi: 10.1097/CCO.0000000000000006
146. Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med (2019) 25(3):477–86. doi: 10.1038/s41591-018-0337-7
147. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Sci (N Y NY) (2018) 362(6411):eaar3593. doi: 10.1126/science.aar3593
148. Macarthur KM, Kao GD, Chandrasekaran S, Alonso-Basanta M, Chapman C, Lustig RA, et al. Detection of brain tumor cells in the peripheral blood by a telomerase promoter-based assay. Cancer Res (2014) 74(8):2152–9. doi: 10.1158/0008-5472.CAN-13-0813
Keywords: brain immunology, glioma microenvironment, immune checkpoint blockade, immunotherapy resistance, immune-response monitoring biomarker
Citation: Qi Y, Liu B, Sun Q, Xiong X and Chen Q (2020) Immune Checkpoint Targeted Therapy in Glioma: Status and Hopes. Front. Immunol. 11:578877. doi: 10.3389/fimmu.2020.578877
Received: 01 July 2020; Accepted: 29 October 2020;
Published: 27 November 2020.
Edited by:
Katy Rezvani, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
William K. Decker, Baylor College of Medicine, United StatesFranziska Eckert, Tübingen University Hospital, Germany
Copyright © 2020 Qi, Liu, Sun, Xiong and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoxing Xiong, eGlhb3hpbmd4aW9uZ0B3aHUuZWR1LmNu; Qianxue Chen, Y2hlbnF4NjY2QHdodS5lZHUuY24=
†These authors have contributed equally to this work