 Dongjuan Song
Dongjuan Song Lijie Lai
Lijie Lai Zhihua Ran
Zhihua Ran- Division of Gastroenterology and Hepatology, Key Laboratory of Gastroenterology and Hepatology, Ministry of Health, Inflammatory Bowel Disease Research Center, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai Institute of Digestive Disease, Shanghai, China
Inflammatory bowel disease (IBD) is characterized by chronic and relapsing inflammatory disorder of the intestine. IBD is associated with complex pathogenesis, and considerable data suggest that innate lymphoid cells contribute to the development and progression of the condition. Group 3 innate lymphoid cells (ILC3s) not only play a protective role in maintaining intestinal homeostasis and gut barrier function, but also a pathogenic role in intestinal inflammation. ILC3s can sense environmental and host-derived signals and combine these cues to modulate cell expansion, migration and function, and transmit information to the broader immune system. Herein, we review current knowledge of how ILC3s can be regulated by dietary nutrients, microbiota and their metabolites, as well as other metabolites. In addition, we describe the phenotypic and functional alterations of ILC3s in IBD and discuss the therapeutic potential of ILC3s in the treatment of IBD.
Introduction
Inflammatory bowel disease (IBD) is a group of chronic inflammatory conditions of the bowel comprising ulcerative colitis (UC) and Crohn’s disease (CD). The increased incidence and prevalence of IBD in recent years poses a significant challenge to society (1, 2). Mounting evidence suggests that genetic background, environmental factors, diet, microbiotic dysbiosis and immune dysregulation contribute to the initiation and progression of IBD (3). Innate lymphoid cells (ILCs) belong to the same family as lymphocytes, however, they lack the rearranged antigen receptors expressed by T cells and B cells and play a central role in immunity, inflammation and gut barrier function (4, 5). The ILC family is classified into three groups: group 1 ILCs including natural killer (NK) cells and ILC1s, group 2 ILCs (ILC2s), and group 3 ILCs (ILC3s). Group 1 ILCs require the transcription factor T-bet and secrete interferon gamma (IFN-γ) upon stimulation with interleukin (IL)-12, IL-15 and IL-18. ILC2s require the transcription factor GATA3 and produce IL-4, IL-5 and IL-13 upon stimulation with IL-25 and IL-33. ILC3s express IL-22, IL-17 and granulocyte macrophage colony-stimulating factor (GM-CSF) when stimulated with IL-23 and IL-1β, which depends on RAR-related orphan receptor gamma t (RORγt) and arylhydrocarbon receptor (AHR) (6, 7). In addition, regulatory ILCs (ILCregs), a novel regulatory subset of ILCs, can produce IL-10 and transforming growth factor β1 (TGF-β1) and help to decrease intestinal inflammation (8).
Single cell analysis of ILC subsets in the small intestine of mice revealed that genes expressed by ILC3s were highly enriched for carbohydrate metabolism and glycolysis, which is different from ILC1s and ILC2s and indicates that each subset of ILC has a unique metabolic profile (9). Another study demonstrated that ILC3 activation relies on the mTOR complex 1(mTORC1)-hypoxia-inducible factor 1α (HIF1α) pathway, which promotes glycolysis and RORγt expression, to promote cellular proliferation as well as IL-22 and IL-17A production. Meanwhile, mTORC1 signaling in ILC3s can activate mitochondrial metabolism and the production of mitochondrial ROS (mROS), which prolongs HIF1α activity, promotes RORγt expression, and ultimately facilitates ILC3 activation. Briefly, ILC3s utilize both glycolysis and mROS production to support effector function (10). ILC3s are enriched in gut mucosal tissue and have a specialized capacity to sense multiple exogenous and endogenous signals, and function as “communication hubs” of the intestinal immune system (11). Indeed, signals from nutrient-derived metabolites, microbiota and microbial metabolites as well as other host metabolites such as 7a,25-dihydroxycholesterol and prostaglandin E2 can be interpreted by ILC3s to regulate proliferation, migration and function of ILC3s as well as their interactions with other cells, which is vital for tissue homeostasis. Dysregulation of ILC3s has been implicated in the pathogenesis of IBD and colorectal cancer (6, 12–14). A better understanding of ILC3s biology in patients with IBD provides valuable insights into potential therapeutic targets. Therefore, it is necessary to evaluate the environmental cues that activate and suppress ILC3s in the gut. In this review, we discuss recent work on how ILC3s are regulated by environmental cues and summarize the involvement of ILC3s in IBD as well as their potential application in IBD therapy.
Overview of Group 3 Innate Lymphoid Cells
ILC3s are a heterogeneous group of cells in humans and mice. Single-cell sequencing analyses of ILCs in human tonsils revealed at least three subsets of ILC3s based on the expression of NKp44, human leucocyte antigen D-related (HLA-DR), and CD62L (15). Furthermore, human NKp44+ILC3s are enriched in barrier tissues such as colon and skin (16). MHCII+ ILC3s with antigen presenting function have also been identified in the colon and small intestine in humans and mice (17, 18). In addition, single-cell sequencing analyses of CD127+ILCs from the small intestinal lamina propria of mice identified five transcriptional states of ILC3s (9). In mice, ILC3s can be divided into CCR6+T-bet- and T-bet+ILC3s (19). CCR6+ILC3s contain fetal lymphoid tissue inducer (LTi) cells, which are indispensable for the organogenesis of secondary lymphoid organs (20). Adult CCR6+LTi-like ILC3s have similar phenotypes to fetal LTi cells and are essential for the development of cryptopatches (CPs) and isolated lymphoid follicles (ILFs) in the gut (21). Although both fetal LTi and adult LTi-like ILC3 express RORγt, they develop from a progenitor distinct from all other ILC subsets (22). T-bet+ILC3s can be further classified on the basis of the expression of natural cytotoxicity receptor (NCR) (NKp46 in mice). T-bet plays a critical role in the differentiation of NCR+ ILC3 from its NCR-ILC3 precursors as well as IFN-r and IL-22 production in NCR+ ILC3 (23, 24).
Regulation of ILC3s by Dietary Nutrients
Recent evidence indicates that dietary vitamins function as key modulators of ILC3s biology (Table 1). For instance, mice fed a diet deficient in vitamin D exhibit reduced abundance of ILC3s and IL-22 secretion by colonic ILC3s, leading to increased susceptibility to Citrobacter rodentium (C. rodentium) infection (25). Consistently, global deletion of vitamin D receptor (VDR) or deficiency in VDR ligand in mice leads to reduced colonic ILC3s and impaired ILC3 response, leading to increased susceptibility to bacterial infection compared with wild-type mice (26). Furthermore, in vitro and in vivo studies revealed that vitamin D/VDR signaling can stimulate colonic ILC3 proliferation, especially LTi cells (26). In contrast, another study reported that VDR knockout (KO) mice had enhanced resistance to bacterial infection due to increased frequencies of ILC3s in the gut and enhanced expression of IL-22 as well as anti-bacterial peptides (39). Of note, an in vitro study revealed that 1α, 25-dihydroxy vitamin D3 (1,25D3), the active form of vitamin D, downregulates the IL-23 receptor pathway in human NKp44+ ILC3s, inhibiting IL-22 and GM-CSF production and inversely enhancing IL-6 production, which encourages ILC3s to maintain an innate-like cytokine expression profile (27). Consequently, these results suggest vitamin D is not only necessary for development and function of ILC3s at steady state, but also can restrain the pro-inflammatory properties of ILC3s.

Table 1 Metabolic regulation of ILC3s by nutrient-derived metabolites.
Similar to vitamin D, lack of vitamin A in the diet results in reduced numbers of ILC3s, IL-22-producing ILCs, CPs and ILFs (29, 30) as well as decreased expression of CCR9 and α4β7 by ILC3s and ILC1s (32) in the small intestine of mice. Furthermore, retinoic acid (RA), a vitamin A metabolite, can induce a homing receptor switch in ILC3s from CCR7 to CCR9 and α4β7 leading to migration of ILC3s to the gut in mice (32) and enhance IL-22 secretion by ILC3s in the mouse small intestine during intestinal inflammation (31). In line with the findings in mice, RA works synergistically with IL-2 to induce integrin α4β7 expression in human ILC3s in vitro. Interestingly, 1,25D3 antagonizes α4β7 expression in human ILC3s induced by RA and IL-2, suggesting the biologically active metabolites of vitamin A and D have antagonistic effects on the expression of integrin in human ILC3s (28). Moreover, RA can accelerate the differentiation of human CD127+ILC1s into IL-22-producing ILC3s in the presence of IL-2, IL-1β and IL-23 (33).
The aryl hydrocarbon receptor (AHR), which is expressed by ILCs and other immune cells, can sense ligands generated from diet, microbiota metabolism, cellular metabolism and environmental pollutants (21). AHR mediates the regulatory effects of dietary and microbial metabolites on ILC3s. AHR-deficient mice exhibit increased vulnerability to colitis and intestinal C. rodentium infection, which is mainly due to the impaired accumulation of ILC3s and IL-22 production in the gut (34, 40, 41) and a decrease in numbers of intraepithelial lymphocytes (IELs) (42). Studies in mice have demonstrated that AHR is indispensable for intestinal ILC3s maintenance and function as well as postnatal development of intestinal lymphoid tissues (34, 40, 41). For instance, AHR promotes the survival of murine intestinal ILC3s, cooperates with RORγt to enhance IL-22 expression and facilitates the expression of IL-7 in the intestine and the expression of IL-7 receptor (IL-7R) by murine ILC3s (40). This is in keeping with the effect of IL-7/IL-7R signaling in the maintenance of ILC3s (43). Moreover, AHR facilitates the expansion of murine CD4-ILC3s through stimulation of cell proliferation, which is induced by AHR-controlled transcription of Kit (34). Additionally, AHR promotes the development of NCR+ILC3s and LTi-like ILC3s by, and partially through activation of Notch signaling (41). Notch signaling has been shown to be essential for adult ILC3s differentiation, but not for fetal LTi development (44). Notably, AHR is also known to block human IL-1R1+ILC3s differentiation into cytolytic NK cells (45).
The diet provides several exogenous and endogenous AHR ligands or precursors, such as Indole-3-Carbinol (I3C), natural flavonoids, resveratrol, curcumin and tryptophan (46, 47). The absence of dietary AHR ligands increases bacterial load or translocation and aggravates dextran sulfate sodium (DSS)-induced colitis and C. rodentium infection in mice (35, 42). The phenotype of mice fed with phytochemical-free diets mimics that of AHR-deficient mice, which have decreased numbers of ILC3s, insufficient postnatal proliferation of CD4-ILC3s, reduced IL-22 production, and impaired development of intestinal lymphoid follicles (Table 1). The addition of I3C to the diet can counteract these abnormalities (34, 36). Consistently, metabolic clearance of natural AHR ligands by constitutive Cyp1a1 expression in mice contributes to loss of ILC3s and Th17 cells as well as reduced IL-22 production, further increasing vulnerability to C. rodentium infection (48). More importantly, dietary supplementation of AHR ligands or precursors ameliorates DSS-induced colitis and intestinal infection in mice (35, 48–50).
Indeed, several studies showed that ILC3 development can be regulated by maternal nutritional status (Table 1). Maternal levels of dietary retinoids are important for regulation of LTi cells differentiation, for ensuring the correct size of secondary lymphoid organs and for maintenance of immune fitness in adult offspring in mice (20). In addition, a maternal high-fat diet (HFD) has been confirmed to induce the expansion of IL-17-producing NKp46+ILC3s in mice offspring, dependent on the subsequent microbiota alterations (37). Moreover, exposure to TCDD (a ligand of AHR) during pregnancy and lactation in mice led to reduced frequency of colonic ILC3s and decreased expression of RORγt in colonic ILC3s, and increased frequency of colonic NKp46+ILC3 in offspring (38). These results suggest that environment cues in pregnancy can modulate ILC3 biology in offspring.
Taken together, these findings suggest that host nutritional status can regulate the numbers and function of ILC3s, immune response and susceptibility to colitis, thus dietary refinements and nutrient supplementation may be beneficial in alleviating the severity of IBD.
Regulation of ILC3s by Microbiota and Microbial Metabolites
Commensal flora has been demonstrated to promote or repress the function or differentiation of NKp46+ILC3s in mice (51–53). However, the development of murine ILC3s seems to be programmed independent of the gut microbiota (41, 53, 54). Moreover, human ILC3s from tissue exposed to the fecal stream produce more IL-22 compared with ILC3s from that not exposed, indicating that the function of human ILC3s is influenced by microbiota (55). Herein, we review the influence of microbiota and microbial metabolites on ILC3s biology (Figure 1).
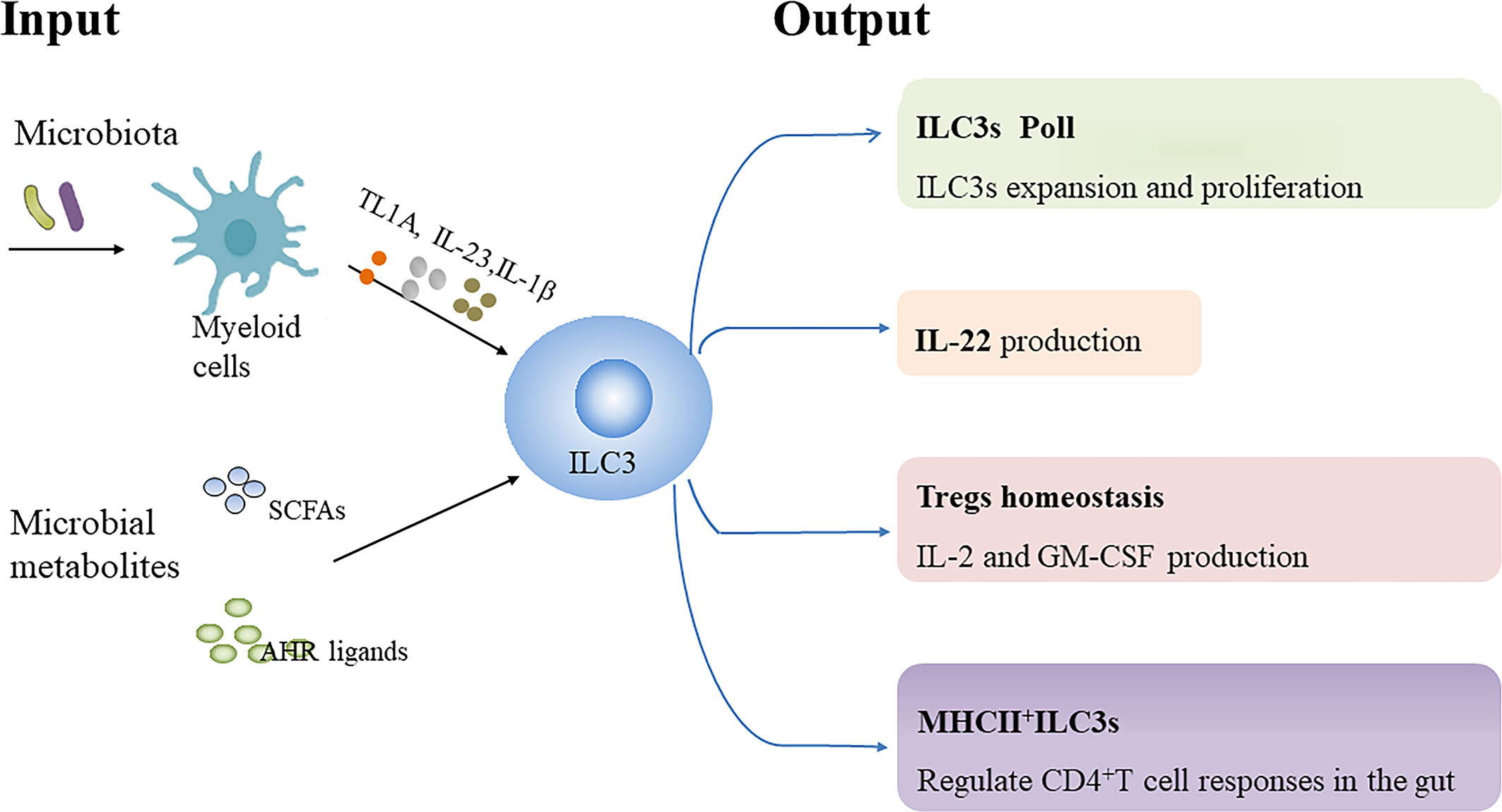
Figure 1 Regulation of ILC3s by microbiota and microbial metabolites. Microbiota-driven IL-1β production by intestinal macrophages enhances IL-2 and granulocyte macrophage colony-stimulating factor (GM-CSF) production by ILC3s, which is essential for regulatory T cells (Tregs) and immunologic homeostasis in the small intestine and colon in mice, respectively. Microbiota-induced IL-23 reduces major histocompatibility complex class II (MHCII) expression in murine NCR-ILC3s, thereby negatively affecting their potential to elicit CD4+T cell responses. TNF-like ligand 1 A (TL1A) cooperates with IL-23 and IL-1β to promote IL-22 production, proliferation and expansion of human ILC3s ex vivo. Additionally, TL1A can enhance IL-22 secretion by murine ILC3s. However, TL1A expression can induce OX40L expression in MHCII+ILC3s, which promote Th1 cells activation in chronic T cell colitis in mice. Collectively, microbial signals can be sensed by myeloid cells to regulate ILC3s biology. In addition, Short-chain fatty acids (SCFAs), the metabolites from gut microbial fermentation of dietary substrates, can regulate murine ILC3s pool as well as their IL-22 production in a receptor-dependent manner. Dietary tryptophan can be metabolized into aryl hydrocarbon receptor (AHR) ligands by commensal microbiota, which promote IL-22 secretion by murine ILC3s.
The production of IL-22 rather than IFN-r by human colonic ILC3s is significantly enhanced following stimulation with commensal or pathogenic bacteria, which may be mediated indirectly by IL-23-and IL-1β-producing CD11c+ myeloid dendritic cells (mDC) and ligation of the NKp44 receptor (56). Additionally, microbiota can stimulate IL-1β production by macrophages through the MyD88 and Nod2 pathway to promote IL-2 production in murine ILC3, which is essential for maintaining Tregs and immunologic homeostasis in the small intestine in mice (57). Moreover, Microbiota-driven IL-1β secretion by intestinal macrophages enhances GM-CSF production by ILC3s, which in turn regulates the frequency and function of macrophages and dendritic cells, helping to maintain colonic Tregs homeostasis (58) (Table 2). Notably, microbiota-induced IL-23 can reduce MHCII expression through mTORC1 and STAT3 signaling in NCR- ILC3s in the small intestine of mice, thereby negatively affecting their potential to induce CD4+ T-cell immune responses (18).Taken together, these findings suggest that microbial signals can be sensed by myeloid cells to regulate ILC3s function.

Table 2 The dichotomous role of ILC3-derived cytokines in the intestinal immune response.
Tumor necrosis factor superfamily member 15 (TNFSF15) has been identified as a susceptibility gene for CD and is associated with the adaptive immune response (74). The TNFSF15 gene encodes TNF-like ligand 1 A (TL1A) protein, which is the ligand for death domain receptor 3 (DR3). Although TL1A supplementation alone does not increase IL-22 production and proliferation of human ILC3s, TL1A cooperates with IL-23 and IL-1β to promote IL-22 production, proliferation and expansion of human ILC3s ex vivo (55, 75). Colonization of mice with adherent IBD-associated microbiota such as segmented filamentous bacteria (SFB) and adherent-invasive Escherichia coli (AIEC) strain 2A enhance IL-22 production by ILC3s (76, 77). Further evidence revealed firstly that SFB and AIEC strain 2A facilitate TL1A expression from Cx3cr1+ mononuclear phagocytes (MNPs) and secondly that TL1A enhances IL-22 production in murine ILC3s and protects mice against acute colitis, which is dependent on DR3 expressed by ILC3s. Mice with ILC3s-specific DR3 deletion show defective IL-22 secretion by ILC3s and increased susceptibility to DSS-induced colitis (78). Nevertheless, activation of DR3 contributes to the reduced abundance of ILC3s in a GM-CSF-and IL-23-dependent manner, which consequently exacerbates DSS-induced colitis in mice (79). OX40L expression by ILC3s has been demonstrated to be essential for homeostatic expansion of intestinal regulatory T cells (Tregs) in mice, and the expression of OX40L can be increased by TL1A and viral stimulation or inhibited by CD4+ T cells (80). However, TL1A-induced OX40L expression on MHCII+ILC3s promotes the activation of T-BET+ Th1 cells, which is essential for chronic T-cell colitis in mice (78). Additionally, neutralization of TL1A can attenuate α-CD40-induced colitis and DSS-induced chronic colitis in mice (79, 81). Taken together, these data suggest that TL1A/DR3 signaling may help to maintain mucosal homeostasis and protect against acute injury, but play a detrimental role in chronic intestinal inflammation.
Short chain fatty acids (SCFAs), the metabolites from gut microbial fermentation of dietary substrates, exert modulating effects on immune cells and provide energy support for intestinal epithelial cells (IECs) (82). “Metabolite-sensing” free fatty acid receptor 2 (FFAR2/GPR43), also known as a receptor for SCFAs, is expressed by colonic ILC3s. Acetate and propionate (the natural FFAR2 ligands) and synthetic FFAR2 agonists contribute to the proliferation of colonic ILC3s and the production of IL-22 in ILC3s from mice, subsequently contributing to host defenses against DSS-induced colonic injury and C. rodentium infection in mice (83). Further evidence revealed that acetate facilitates IL-22 production in murine ILC3s upon stimulation with IL-1β via enhancement of IL-1 receptor expression in a FFAR2-dependent fashion (84). Of note, butyrate inhibits the number of NKp46+ILC3s as well as their IL-22 production in mouse terminal ileal Peyer’s patches (PPs) through the GPR109a receptor under steady-state conditions, which leads to a reduced frequency of Tregs and antigen-specific immune induction in terminal ileal PPs (85). In accordance with this, Gpr109a-/-Rag1-/- mice developed spontaneous colonic inflammation and had increased ILC3s in the gut relative to Rag1-/- mice especially IL-17-producing ILC3s. Mechanistically, GPR109a suppresses ILC3s through inhibiting microbiota-induced IL-23 production in intestinal dendritic cells to regulate intestinal homeostasis (86). Taken together, the regulation of SCFAs on ILC3 responses may depend on subset, receptor or tissue environment as well as host conditions.
Tryptophan (Trp) metabolites from symbiotic microbiota can promote epithelial barrier function and inhibit the inflammatory response (87–89). Lactobacillus reuteri (L. reuteri) can metabolize Trp into endogenous AHR ligands such as indole-3-aldehyde (IAID) or indole-3-lactic acid under conditions of Trp sufficiency, which promotes IL-22 secretion by ILC3s and induces gut intraepithelial CD4+CD8αα+ T cells in an AHR-dependent manner in mice (87, 90). The microbiota from mice lacking caspase recruitment domain family member 9 (CARD9), a susceptibility gene for IBD, fail to metabolize tryptophan into AHR ligands, resulting in decreased production of IL-22 by ILC3s and Th22 cells, and increased susceptibility in germ-free recipients to colitis (91). Importantly, impaired AHR activation and metabolism of tryptophan by the gut microbiota has been identified in patients with IBD, which is associated with a CARD9 phenotype (91). Administration of some strains of L. reuteri alone or in combination with other Lactobacillus strains has been reported to prevent DSS-induced colitis in mice due to various mechanisms (91–94) and some studies suggest that AHR activation and enhanced IL-22 production play a critical role in the process (91, 92). In addition, it has been observed that rectal administration of L. reuteri can attenuate mucosal inflammation in children with active distal UC (95). Nevertheless, L. reuteri can attenuate immune checkpoint blockade-associated colitis through reducing the population of ILC3s (96).
Apart from bacterial tryptophan metabolism, tryptophan in the gastrointestinal tract can also be metabolized through the kynurenine pathway via the rate-limiting enzyme indoleamine 2,3 dioxygenase-1 (IDO1) and the serotonin pathway via Trp hydroxylase 1 (TpH1) (97). Increased IDO1 expression in the gut negatively correlates with the number of ILC3s as well as IL-17 and TNF-a production by ILC3s during simian immunodeficiency virus infection (98). Interestingly, IDO1-knockout mice had increased numbers of ILC3s in the lungs compared with wild-type mice after paracoccidioides brasiliensis infection (99). Deficiency or inhibition of IDO1 aggravates 2,4,6-trinitrobenzene sulfate (TNBS)-induced colitis in mice (100, 101). In contrast, IDO1-knockout mice are less susceptible to DSS-induced colitis (102). Mice lacking IDO1 had increased abundance of bacteria with tryptophanase activity, which results in the accumulation of microbiota-derived AHR ligands (87, 102). Thus, endogenous Trp metabolism may influence the gut microbiota and bacterial Trp metabolism, thereby regulating the innate lymphoid cells.
Regulation of Other Metabolites
G-Protein-Coupled Receptor 183 (GPR183) and its ligand 7a,25-dihydroxycholesterol (7a,25-OHC), a hydroxylated metabolite of cholesterol, are critical for modulating the distribution of ILC3s, and subsequent interactions between ILC3s and T follicular helper cells (TfH) and B cells (103–105). The GPR183 receptor expressed on LTi-like ILC3s can sense 7a,25-OHC that is produced from fibroblastic stromal cells located in lymphoid structures, which contributes to ILC3s migration to CPs and ILFs and the formation of colonic lymphoid tissues at steady state in mice (103). In addition, GPR183 and 7a,25-OHC regulate the distribution or accumulation of ILC3s in the mesenteric lymph nodes (mLNs), PPs and small intestine of mice (104, 105). ILC3s located within the interfollicular border of mLNs limit TfH-driven B cell responses and IgA production through antigen presentation in the colon at steady state in mice, which is beneficial for the maintenance of host-microbiota mutualism (105). Moreover, GPR183 can protect mice against C. rodentium infection through promoting the enrichment of IL-22-expressing ILC3s in the small intestine of mice (104). However, mice treated with CD40 Ab had enhanced 7a,25-OHC production, which in turn promotes colitis through activating the migration of GPR183+ ILC3s and myeloid cells to inflammatory foci (103). In addition, prostaglandin E2 (PGE2) promotes homeostasis and functionality of murine ILC3s via its receptor EP4, leading to the inhibition of systemic inflammation in mice (106). Adenosine 5′-triphosphate (eATP) and its metabolite adenosine inversely regulate IL-22 secretion from murine ILC3s. Inhibition of NTPDases, which hydrolyzes extracellular eATP into adenosine, can aggravate DSS-induced colitis in mice dependent on reduced frequency of IL-22-producing ILC3s (107).
ILC3s in IBD
Decreased frequency of NKp44+ILC3s has been observed in inflamed tissue from IBD patients compared with non-IBD controls, which was related to disease severity regardless of whether patients were newly diagnosed or had established disease. However, the frequency of ILC1s and ILC2s was increased in newly-diagnosed CD and UC, respectively, and the frequency of ILC1s and ILC2s were both increased in patients with established IBD (12). Reduced frequency of NKp44+ ILC3s in inflamed intestinal tissues from CD patients was accompanied by enrichment of IFN-γ-secreting CD127+ILC1s, indicating that an imbalance between ILC3 and ILC1 may contribute to the pathogenesis of CD (33, 108). Furthermore, the frequency of NKp44+ ILC3s was shown to have an inverse association with the accumulation of IL-17A+ IFN-γ+ and IL-22+IFN-γ+ T cells in inflamed regions of adults with CD (13). In addition, MHCII+ILC3s were significantly reduced in inflamed regions of CD patients compared with non-inflamed regions (13). MHCII+ ILC3s have been reported to mediate negative selection via antigen presentation together with IL-2 withdrawal, leading to cell death of activated commensal bacteria–specific T cells in mice (17). These results suggest that downregulation of MHCII expression may be associated with aberrant immune responses in IBD.
IL-22 functions as a dichotomous cytokine in intestinal inflammation (Table 2). IL-22 deficient mice exhibit aggravated experimental colitis following DSS exposure (109), and IL-22 orchestrates epithelial regeneration, proliferation and glycosylation, the production of mucins and anti-bacterial peptides and protects intestinal stem cells from genotoxic stress (36, 59–61, 63). NKp44+ILC3s produced less IL-22, but acquired the ability to secrete IFN-γ in the inflamed terminal ileum of CD patients compared with unaffected tissue (13). However, IL-22-expressing ILC3s have been confirmed to be responsible for the development of acute innate colitis in mice (62). Foxp3+ Treg cells can attenuate IL-22+ ILC3s-mediated colitis in mice through inhibiting the secretion of IL-23 and IL-1β by Cx3cr1+macrophages (110). Additionally, patients with active mild-to-moderate IBD had increased production of IL-22 in colonic ILC3s compared with controls (55). Serum IL-22 concentrations were markedly increased in CD patients compared with healthy volunteers, and positively correlated with disease activity (111). Furthermore, IL-22 can induce endoplasmic reticulum stress (ER) in colonic epithelial cells, which is functionally important in chronic colitis. Importantly, IL22-responsive transcripts and ER stress response modules were enriched in the colons of patients with IBD compared with non-IBD controls, and the IL22-responsive transcriptional modules positively correlated with the severity of mucosal injury (64). In addition, IL-22 may be implicated in intestinal fibrosis (67, 68). Increased expression of the IL-23/IL-22 axis regulated by mTOR/autophagy signaling in Cx3cr1+MNPs exacerbates fibrosis in the mouse model of TNBS-induced intestinal fibrosis. Interestingly, neutralization of either IL-23 or IL-22 can attenuate the fibrosis reaction. And ILCs, but not T and B cells, are likely participated in intestinal fibrosis (67).
The production of GM-CSF was elevated in LPMCs from the inflamed mucosa of patients with IBD compared with non-inflamed mucosa and non-IBD controls (112). In addition, the secretion of GM-CSF was also increased by blood ILCs in patients with IBD compared with healthy volunteers (70). Increased production of GM-CSF from ILC3s during colitis mobilizes the migration of ILC3s into adjacent tissue from cryptopatches and promotes inflammatory monocyte accumulation, which mediates the pathogenic role of ILC3s in anti-CD40-induced colitis in mice, and neutralization of GM-CSF can ameliorate intestinal colitis in mice (70, 71) (Table 2). However, GM-CSF gene knockout mice are more susceptible to acute DSS-induced colitis compared with wild-type mice (113). Importantly, elevated levels of GM-CSF auto-antibodies (Ab), which are associated with reduced GM-CSF bioactivity, have been proven to be associated with stricturing/penetrating behavior and higher incidence of intestinal resection in CD patients (114), surgical recurrence in ileal CD patients (115) and disease relapse in IBD patients (116). Importantly, sargramostim (recombinant GM-CSF) is not superior to placebo for inducing clinical improvement or remission in active CD (117). Notably, a recent study revealed the complex role of ILC3-derived GM-CSF in the context of intestinal infection and inflammation. GM-CSF can regulate the activation and polarization of intestinal macrophages in humans and mice, driving the differentiation of pro-inflammatory and microbicidal M1 macrophages, while suppressing wound-healing, pro-fibrotic macrophages (69) (Table 2). Importantly, compared with “non-complicated” biopsies, biopsies from CD patients with complicated disease (stricturing and penetrating behavior) had enrichment of genes that are upregulated in ILC-depleted macrophages, suggesting that ILC3-derived GM-CSF controls the progression of intestinal fibrosis (69). This is consistent with a study that revealed reduced GM-CSF bioactivity was associated with stricturing/penetrating behavior in CD (114). Thus, the role of ILC3s-derived GM-CSF remains to be elucidated.
The frequency of IL-17-expressing CD56− ILC3s was increased in the inflamed ileum and colon of CD patients compared with non-IBD controls, but not patients with UC (118). IL-17A expression by NCR− ILC3s has been demonstrated to drive colitis development in T-bet−/−.Rag2−/− (TRUC) mice (72). Additionally, production of IL-17 and IFN-r in murine ILC3 contributes to colitis development in H. hepaticus-mediated innate colitis in mice, and neutralization of IL-17 or INF-r can significantly attenuate colitis (73). Interestingly, Rora+ ILC3s result in fibrosis mediated by IL-22 and IL-17 production in a salmonella-induced intestinal fibrosis mouse model, and neutralization of IL-17A can attenuate fibrosis, but the effect of neutralized IL-22 expression was not explored in the study (68) (Table 2). However, secukinumab, a human anti-IL-17A monoclonal antibody, failed to show efficacy in CD (119), and this may be due to severe weakening of intestinal epithelial barrier function induced by IL-17 inhibition (120, 121). Additionally, Rag2−/− mice that received T cells from IL17A−/− mice had increased frequency of ILC3, mainly CD4+ILC3 and ILC1s as well as enhanced expression of IL-6 and IL-22, which may partly account for the failure of IL-17A inhibitors in CD (122).
Ectopic or tertiary lymphoid tissues (TLTs) are regarded as ectopic clusters of immune cells in response to chronic non-resolving inflammation, and are a pathologic hallmark of CD (123). TLTs have been observed in the mesenteric creeping fat of patients with CD. Furthermore, the formation of functional LTLs in CD-affected mesentery may be attributed to high local levels of CXCL16, CCL20, CCL21, CXCL13, and CCL19, produced by the mesenteric adipocytes (124). It has been reported that human neuropilin-1(NRP1)+ LTi-like ILC3s were observed in lung tissues from patients with chronic obstructive pulmonary disease, which may participate in ectopic lymphocyte accumulation (125). Additionally, the frequency of human NKp44+ILC3s was significantly reduced in advanced colorectal cancer and non-small cell lung cancer. Furthermore, the accumulation of NKp44+ILC3s may be implicated in the formation of tumor-associated TLTs (126, 127). Studies in mice with IL-7 overexpression provide evidence for the indispensable role of LTi cells in the formation of TLTs (128). However, the formation of TLTs was observed in the intestine of RORγt-deficient mice treated with DSS, indicating that TLTs development seems to be independent of LTi cells (129). Collectively, these findings suggest that future work is required to clarify the role of LTi-like ILC3 in the formation of LTLs in both humans and mice. In addition, although the formation of TLTs aggravates colitis in mice (129), the precise role of TLTs in the pathogenesis of IBD must be further explored, and are reviewed in detail elsewhere (123).
Patients with IBD have an increased risk of developing colorectal cancer (CRC). ILC3s and IL-22 seem to play a pathogenic role in the onset or progression of colorectal cancer (14). ILC3s are vital for IL-23-mediated initiation of gut tumorigenesis (130). IL-22 stimulates STAT3 activation in intestinal epithelial cells to promote cell proliferation, playing a predominant role in the maintenance of tumor development (65, 66). Impaired production of IL-22 by ILC3s and insufficient STAT3 activation thereafter account for the protection of Card9-/- mice from colitis-associated cancer (65). Moreover, NKp46-ILC3s drive the transition from colitis to CRC in Helicobacter hepaticus (Hh)+AOM mice and neutralization of IL-22 can ameliorate established colitis and reduce tumor burden (66). Besides, the absence of IL-22BP, a neutralizing soluble IL-22 receptor, accelerates tumorigenesis in AMO/DSS treated mice (131). However, IL-22 secretion by ILC3s and γδ T cells regulates the DNA damage response (DDR) in colon stem cells and protects them from acquiring dangerous mutations after genotoxic exposure, thus limiting tumorigenesis (36) (Table 2).
Therapeutic Potential of ILC3s in IBD
Anti-TNF treatment has dramatically improved the treatment of IBD over the past two decades, but primary non-response and secondary loss of response are commonly observed (132). Importantly, anti-TNF agents can result in some adverse events in a fraction of patients (133). Thus, safe and effective therapies for IBD are urgently needed.
IL-23 is a heterodimeric cytokine composed of an IL-23-specific P19 subunit and a P40 subunit shared with IL-12. IL-23 responsive ILC3s participate in the pathogenesis of IBD. Ustekinumab, a monoclonal antibody that targets IL-12/IL-23p40, is effective at inducing and sustaining clinical remission in patients with CD, and has shown some evidence of efficacy in UC patients (132). Moreover, IL-23p19 inhibitors including risankizumab, brazikumab, and mirikizumab have been shown to be effective in patients with moderate-to-severe active CD (134, 135) or UC (136) in clinical studies. However, the influence of IL-23 blockade on ILC3s remains to be elucidated. Compared with placebo, brazikumab can significantly reduce the serum levels of IL-22, and CD patients with baseline serum IL-22 concentration ≥15.6 pg/ml are more likely to experience a clinical response or remission at week 8 following treatment with brazikumab compared with patients with low baseline IL-22 concentration (<15.6 pg/ml) (135).
AHR activation may be a potential therapeutic strategy for the treatment of UC. The AHR pathway mediates crosstalk between particular metabolites in the environment and immune cells, which is important for gut barrier protection and mucosal immunity. I3C can prevent TNBS-induced colitis in mice primarily through inducing IL-22 production by ILC3s (137). Furthermore, fecal microbiota transplantation (FMT) and indigo naturalis (IN), a traditional herbal medicine used for UC, can attenuate DSS-induced colitis in mice by up-regulating the expression or activity of AHR (138, 139). Besides, FMT significantly modulates bacterial metabolism of tryptophan indicated by increased levels of indole-3-acetic acid, which is in line with AHR activation in the colon of recipient piglets (140). A randomized controlled clinical trial showed that 8-week treatment with IN was able to induce clinical responses and mucosal healing in patients with UC. However, the long-term administration of IN should be carefully considered in view of potential adverse effects (141). NPD-0414-2 and NPD-0414-24, novel chemical AHR ligands, up-regulate IL-22 and down-regulate IFN-γ transcripts in LPMCs from IBD patients in vitro, which can attenuate TNBS-induced colitis in mice with enhanced expression of IL-22 and reduced expression of IFN-γ in an AHR-dependent manner, without clinical signs of systemic toxicity (142). Of note, PY109, an AHR agonist that has physiochemical drug-likeness properties, ameliorates DSS-induced colitis in mice by promoting the expansion of ILC3s and γδ T cells and expression of IL-22 and IL-17 (143). Collectively, novel AHR agonists with good safety profiles may be effective therapeutic options for the treatment of UC.
Deficiency of Vitamin D is frequently observed in patients with IBD and is associated with increased disease activity and elevated healthcare utilization (144). A prospective study has shown that low serum vitamin D levels (≤ 35 ng/ml) during clinical remission are associated with increased risk of UC relapse (145). In another observational study, CD patients with vitamin D deficiency (25-OH-D concentration < 50 nmol/L) had more relapses during the previous year (146). Importantly, IBD patients with low vitamin D levels who received vitamin D supplements had a significant reduction in their healthcare utilization (144) and correction of 25(OH)D status was associated with reduced risk of CD-related surgery (147). Furthermore, vitamin D status may affect the initial response to TNF inhibitor therapy and IBD patients who had normal vitamin D levels at the initiation of treatment with TNF-α inhibitors had increased odds of remission at 3 months (148). In addition, administration of vitamin A for two months can significantly facilitate clinical remission, clinical response and mucosal healing in UC patients. However, excessive vitamin A supplementation should be avoided due to increased risk of bone fracture and liver toxicity (149). As there are limited data from clinical trials of vitamin D and A in IBD, further studies are needed to conclude whether their administration is clinically effective. In addition, further study to define the optimal levels of vitamin D and A in serum to achieve clinical response is needed.
Conclusions
ILC3s function as “communication hubs”, which respond to environmental cues and propagate signals to the broader immune system. Herein, we highlighted the dependence of ILC3s on dietary metabolites such as vitamin D, vitamin A as well as its metabolite RA and AHR ligands, microbiota and microbial metabolites such as SCFAs and microbial tryptophan metabolites. In addition, other metabolites such as PGE2 and 7a,25-OHC can also be sensed by ILC3s and give rise to functional outputs. Moreover, maternal nutritional status can modulate ILC3s biology in offspring. Group 3 innate lymphoid cells maintain mucosal homeostasis dependent on moderate production of IL-22, IL-17 and GM-CSF in the steady state. However, excessive production may contribute to the progression of IBD and colorectal cancer. Importantly, IL-22 and IL-17 produced by ILC3s may be involved in the development of intestinal fibrosis. Targeting ILC3s hold promise for treating IBD. Antibodies targeting IL-23-specific P19 or P40 have shown clinical efficacy. In addition, AHR agonist with good safety profiles may be effective treatments for UC. Importantly, nutritional interventions and dietary modifications should always be considered in patients with IBD.
Author Contributions
DS drafted the initial manuscript. ZR provided critical feedback. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (grant 81670497).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet (Lond Engl) (2018) 390(10114):2769–78. doi: 10.1016/S0140-6736(17)32448-0
2. Ananthakrishnan AN, Kaplan GG, Ng SC. Changing Global Epidemiology of Inflammatory Bowel Diseases-Sustaining Healthcare Delivery into the 21st Century. Clin Gastroenterol Hepatol (2020) 18(6):1252–60. doi: 10.1016/j.cgh.2020.01.028
3. Zhang Y-Z, Li Y-Y. Inflammatory bowel disease: pathogenesis. World J Gastroenterol (2014) 20(1):91–9. doi: 10.3748/wjg.v20.i1.91
4. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate Lymphoid Cells: 10 Years On. Cell (2018) 174(5):1054–66. doi: 10.1016/j.cell.2018.07.017
5. Fan H, Wang A, Wang Y, Sun Y, Han J, Chen W, et al. Innate Lymphoid Cells: Regulators of Gut Barrier Function and Immune Homeostasis. J Immunol Res (2019) 2019:2525984. doi: 10.1155/2019/2525984
6. Zeng B, Shi S, Ashworth G, Dong C, Liu J, Xing F. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis (2019) 10(4):315. doi: 10.1038/s41419-019-1540-2
7. Ebbo M, Crinier A, Vély F, Vivier E. Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol (2017) 17(11):665–78. doi: 10.1038/nri.2017.86
8. Wang S, Xia P, Chen Y, Qu Y, Xiong Z, Ye B, et al. Regulatory Innate Lymphoid Cells Control Innate Intestinal Inflammation. Cell (2017) 171(1):201–16.e218. doi: 10.1016/j.cell.2017.07.027
9. Gury-BenAri M, Thaiss CA, Serafini N, Winter DR, Giladi A, Lara-Astiaso D, et al. The Spectrum and Regulatory Landscape of Intestinal Innate Lymphoid Cells Are Shaped by the Microbiome. Cell (2016) 166(5):1231–46.e13. doi: 10.1016/j.cell.2016.07.043
10. Di Luccia B, Gilfillan S, Cella M, Colonna M, Huang SC. ILC3s integrate glycolysis and mitochondrial production of reactive oxygen species to fulfill activation demands. J Exp Med (2019) 216(10):2231–41. doi: 10.1084/jem.20180549
11. Withers DR, Hepworth MR. Group 3 Innate Lymphoid Cells: Communications Hubs of the Intestinal Immune System. Front Immunol (2017) 8:1298. doi: 10.3389/fimmu.2017.01298
12. Forkel M, van Tol S, Hoog C, Michaelsson J, Almer S, Mjosberg J. Distinct Alterations in the Composition of Mucosal Innate Lymphoid Cells in Newly Diagnosed and Established Crohn’s Disease and Ulcerative Colitis. J Crohn’s Colitis (2019) 13(1):67–78. doi: 10.1093/ecco-jcc/jjy119
13. Li J, Doty AL, Tang Y, Berrie D, Iqbal A, Tan SA, et al. Enrichment of IL-17A(+) IFN-γ(+) and IL-22(+) IFN-γ(+) T cell subsets is associated with reduction of NKp44(+) ILC3s in the terminal ileum of Crohn’s disease patients. Clin Exp Immunol (2017) 190(1):143–53. doi: 10.1111/cei.12996
14. Atreya I, Kindermann M, Wirtz S. Innate lymphoid cells in intestinal cancer development. Semin Immunol (2019) 41:101267. doi: 10.1016/j.smim.2019.02.001
15. Björklund ÅK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, et al. The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol (2016) 17(4):451–60. doi: 10.1038/ni.3368
16. Simoni Y, Fehlings M, Kløverpris HN, McGovern N, Koo SL, Loh CY, et al. Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity (2017) 46(1):148–61. doi: 10.1016/j.immuni.2016.11.005
17. Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science (New York NY) (2015) 348(6238):1031–5. doi: 10.1126/science.aaa4812
18. Lehmann FM, von Burg N, Ivanek R, Teufel C, Horvath E, Peter A, et al. Microbiota-induced tissue signals regulate ILC3-mediated antigen presentation. Nat Commun (2020) 11(1):1794. doi: 10.1038/s41467-020-15612-2
19. Artis D, Spits H. The biology of innate lymphoid cells. Nature (2015) 517(7534):293–301. doi: 10.1038/nature14189
20. van de Pavert SA, Ferreira M, Domingues RG, Ribeiro H, Molenaar R, Moreira-Santos L, et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature (2014) 508(7494):123–7. doi: 10.1038/nature13158
21. Li S, Bostick JW, Zhou L. Regulation of Innate Lymphoid Cells by Aryl Hydrocarbon Receptor. Front Immunol (2018) 8:1909. doi: 10.3389/fimmu.2017.01909
22. Montaldo E, Juelke K, Romagnani C. Group 3 innate lymphoid cells (ILC3s): Origin, differentiation, and plasticity in humans and mice. Eur J Immunol (2015) 45(8):2171–82. doi: 10.1002/eji.201545598
23. Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, et al. A T-bet gradient controls the fate and function of CCR6-RORγt+ innate lymphoid cells. Nature (2013) 494(7436):261–5. doi: 10.1038/nature11813
24. Sciumé G, Hirahara K, Takahashi H, Laurence A, Villarino AV, Singleton KL, et al. Distinct requirements for T-bet in gut innate lymphoid cells. J Exp Med (2012) 209(13):2331–8. doi: 10.1084/jem.20122097
25. Lin Y-D, Arora J, Diehl K, Bora SA, Cantorna MT. Vitamin D Is Required for ILC3 Derived IL-22 and Protection From Citrobacter rodentium Infection. Front Immunol (2019) 10:1. doi: 10.3389/fimmu.2019.00001
26. He L, Zhou M, Li YC. Vitamin D/Vitamin D Receptor Signaling Is Required for Normal Development and Function of Group 3 Innate Lymphoid Cells in the Gut. iScience (2019) 17:119–31. doi: 10.1016/j.isci.2019.06.026
27. Konya V, Czarnewski P, Forkel M, Rao A, Kokkinou E, Villablanca EJ, et al. Vitamin D downregulates the IL-23 receptor pathway in human mucosal group 3 innate lymphoid cells. J Allergy Clin Immunol (2018) 141(1):279–92. doi: 10.1016/j.jaci.2017.01.045
28. Ruiter B, Patil SU, Shreffler WG. Vitamins A and D have antagonistic effects on expression of effector cytokines and gut-homing integrin in human innate lymphoid cells. Clin Exp Allergy (2015) 45(7):1214–25. doi: 10.1111/cea.12568
29. Goverse G, Labao-Almeida C, Ferreira M, Molenaar R, Wahlen S, Konijn T, et al. Vitamin A Controls the Presence of RORγ+ Innate Lymphoid Cells and Lymphoid Tissue in the Small Intestine. J Immunol (Baltimore Md 1950) (2016) 196(12):5148–55. doi: 10.4049/jimmunol.1501106
30. Spencer SP, Wilhelm C, Yang Q, Hall JA, Bouladoux N, Boyd A, et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science (New York NY) (2014) 343(6169):432–7. doi: 10.1126/science.1247606
31. Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, et al. Retinoic acid expression associates with enhanced IL-22 production by γδ T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med (2013) 210(6):1117–24. doi: 10.1084/jem.20121588
32. Kim MH, Taparowsky EJ, Kim CH. Retinoic Acid Differentially Regulates the Migration of Innate Lymphoid Cell Subsets to the Gut. Immunity (2015) 43(1):107–19. doi: 10.1016/j.immuni.2015.06.009
33. Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and -23 Control Plasticity of CD127(+) Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity (2015) 43(1):146–60. doi: 10.1016/j.immuni.2015.06.019
34. Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science (New York NY) (2011) 334(6062):1561–5. doi: 10.1126/science.1214914
35. Julliard W, De Wolfe TJ, Fechner JH, Safdar N, Agni R, Mezrich JD. Amelioration of Clostridium difficile Infection in Mice by Dietary Supplementation With Indole-3-carbinol. Ann Surg (2017) 265(6):1183–91. doi: 10.1097/SLA.0000000000001830
36. Gronke K, Hernandez PP, Zimmermann J, Klose CSN, Kofoed-Branzk M, Guendel F, et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature (2019) 566(7743):249–53. doi: 10.1038/s41586-019-0899-7
37. Babu ST, Niu X, Raetz M, Savani RC, Hooper LV, Mirpuri J. Maternal high-fat diet results in microbiota-dependent expansion of ILC3s in mice offspring. JCI Insight (2018) 3(19):e99223. doi: 10.1172/jci.insight.99223
38. Li Y, Xie HQ, Zhang W, Wei Y, Sha R, Xu L, et al. Type 3 innate lymphoid cells are altered in colons of C57BL/6 mice with dioxin exposure. Sci Total Environ (2019) 662:639–45. doi: 10.1016/j.scitotenv.2019.01.139
39. Chen J, Waddell A, Lin YD, Cantorna MT. Dysbiosis caused by vitamin D receptor deficiency confers colonization resistance to Citrobacter rodentium through modulation of innate lymphoid cells. Mucosal Immunol (2015) 8(3):618–26. doi: 10.1038/mi.2014.94
40. Qiu J, Heller JJ, Guo X, Chen Z-ME, Fish K, Fu Y-X, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity (2012) 36(1):92–104. doi: 10.1016/j.immuni.2011.11.011
41. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol (2011) 13(2):144–51. doi: 10.1038/ni.2187
42. Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell (2011) 147(3):629–40. doi: 10.1016/j.cell.2011.09.025
43. Sheikh A, Abraham N. Interleukin-7 Receptor Alpha in Innate Lymphoid Cells: More Than a Marker. Front Immunol (2019) 10:2897. doi: 10.3389/fimmu.2019.02897
44. Possot C, Schmutz S, Chea S, Boucontet L, Louise A, Cumano A, et al. Notch signaling is necessary for adult, but not fetal, development of RORgammat(+) innate lymphoid cells. Nat Immunol (2011) 12(10):949–58. doi: 10.1038/ni.2105
45. Hughes T, Briercheck EL, Freud AG, Trotta R, McClory S, Scoville SD, et al. The transcription Factor AHR prevents the differentiation of a stage 3 innate lymphoid cell subset to natural killer cells. Cell Rep (2014) 8(1):150–62. doi: 10.1016/j.celrep.2014.05.042
46. Lamas B, Natividad JM, Sokol H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol (2018) 11(4):1024–38. doi: 10.1038/s41385-018-0019-2
47. Veldhoen M, Ferreira C. Influence of nutrient-derived metabolites on lymphocyte immunity. Nat Med (2015) 21(7):709–18. doi: 10.1038/nm.3894
48. Schiering C, Wincent E, Metidji A, Iseppon A, Li Y, Potocnik AJ, et al. Feedback control of AHR signalling regulates intestinal immunity. Nature (2017) 542(7640):242–5. doi: 10.1038/nature21080
49. Islam J, Sato S, Watanabe K, Watanabe T, Ardiansyah, Hirahara K, et al. Dietary tryptophan alleviates dextran sodium sulfate-induced colitis through aryl hydrocarbon receptor in mice. J Nutr Biochem (2017) 42:43–50. doi: 10.1016/j.jnutbio.2016.12.019
50. Hubbard TD, Murray IA, Nichols RG, Cassel K, Podolsky M, Kuzu G, et al. Dietary Broccoli Impacts Microbial Community Structure and Attenuates Chemically Induced Colitis in Mice in an Ah receptor dependent manner. J Funct Foods (2017) 37:685–98. doi: 10.1016/j.jff.2017.08.038
51. Satoh-Takayama N, Vosshenrich CAJ, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity (2008) 29(6):958–70. doi: 10.1016/j.immuni.2008.11.001
52. Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat Immunol (2009) 10(1):83–91. doi: 10.1038/ni.1684
53. Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Berard M, Kleinschek M, et al. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol (2011) 12(4):320–6. doi: 10.1038/ni.2002
54. Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, et al. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science (New York NY) (2010) 330(6004):665–9. doi: 10.1126/science.1194597
55. Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med (2014) 211(8):1571–83. doi: 10.1084/jem.20140678
56. Castleman MJ, Dillon SM, Purba CM, Cogswell AC, Kibbie JJ, McCarter MD, et al. Commensal and Pathogenic Bacteria Indirectly Induce IL-22 but Not IFNγ Production From Human Colonic ILC3s via Multiple Mechanisms. Front Immunol (2019) 10:649. doi: 10.3389/fimmu.2019.00649
57. Zhou L, Chu C, Teng F, Bessman NJ, Goc J, Santosa EK, et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature (2019) 568(7752):405–9. doi: 10.1038/s41586-019-1082-x
58. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science (New York NY) (2014) 343(6178):1249288. doi: 10.1126/science.1249288
59. Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature (2015) 528(7583):560–4. doi: 10.1038/nature16460
60. Moniruzzaman M, Wang R, Jeet V, McGuckin MA, Hasnain SZ. Interleukin (IL)-22 from IL-20 Subfamily of Cytokines Induces Colonic Epithelial Cell Proliferation Predominantly through ERK1/2 Pathway. Int J Mol Sci (2019) 20(14):3468. doi: 10.3390/ijms20143468
61. Goto Y, Obata T, Kunisawa J, Sato S, Ivanov II, Lamichhane A. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science (2014) 345(6202):1254009. doi: 10.1126/science.1254009
62. Eken A, Singh AK, Treuting PM, Oukka M. IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol (2014) 7(1):143–54. doi: 10.1038/mi.2013.33
63. Mizoguchi A, Yano A, Himuro H, Ezaki Y, Sadanaga T, Mizoguchi E. Clinical importance of IL-22 cascade in IBD. J Gastroenterol (2018) 53(4):465–74. doi: 10.1007/s00535-017-1401-7
64. Powell N, Pantazi E, Pavlidis P, Tsakmaki A, Li K, Yang F, et al. Interleukin-22 orchestrates a pathological endoplasmic reticulum stress response transcriptional programme in colonic epithelial cells. Gut (2020) 69(3):578–90. doi: 10.1136/gutjnl-2019-318483
65. Bergmann H, Roth S, Pechloff K, Kiss EA, Kuhn S, Heikenwalder M, et al. Card9-dependent IL-1β regulates IL-22 production from group 3 innate lymphoid cells and promotes colitis-associated cancer. Eur J Immunol (2017) 47(8):1342–53. doi: 10.1002/eji.201646765
66. Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med (2013) 210(5):917–31. doi: 10.1084/jem.20122308
67. Mathur R, Alam MM, Zhao X-F, Liao Y, Shen J, Morgan S, et al. Induction of autophagy in Cx3cr1(+) mononuclear cells limits IL-23/IL-22 axis-mediated intestinal fibrosis. Mucosal Immunol (2019) 12(3):612–23. doi: 10.1038/s41385-019-0146-4
68. Lo BC, Gold MJ, Hughes MR, Antignano F, Valdez Y, Zaph C, et al. The orphan nuclear receptor ROR alpha and group 3 innate lymphoid cells drive fibrosis in a mouse model of Crohn’s disease. Sci Immunol (2016) 1(3):eaaf8864. doi: 10.1126/sciimmunol.aaf8864
69. Castro-Dopico T, Fleming A, Dennison TW, Ferdinand JR, Harcourt K, Stewart BJ, et al. GM-CSF Calibrates Macrophage Defense and Wound Healing Programs during Intestinal Infection and Inflammation. Cell Rep (2020) 32(1):107857. doi: 10.1016/j.celrep.2020.107857
70. Pearson C, Thornton EE, McKenzie B, Schaupp A-L, Huskens N, Griseri T, et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife (2016) 5:e10066. doi: 10.7554/eLife.10066
71. Song C, Lee JS, Gilfillan S, Robinette ML, Newberry RD, Stappenbeck TS, et al. Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. J Exp Med (2015) 212(11):1869–82. doi: 10.1084/jem.20151403
72. Ermann J, Staton T, Glickman JN, de Waal Malefyt R, Glimcher LH. Nod/Ripk2 signaling in dendritic cells activates IL-17A-secreting innate lymphoid cells and drives colitis in T-bet-/-.Rag2-/- (TRUC) mice. Proc Natl Acad Sci U S A (2014) 111(25):E2559–66. doi: 10.1073/pnas.1408540111
73. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature (2010) 464(7293):1371–5. doi: 10.1038/nature08949
74. Park SC, Jeen YT. Genetic Studies of Inflammatory Bowel Disease-Focusing on Asian Patients. Cells (2019) 8(5):404. doi: 10.3390/cells8050404
75. Ahn YO, Weeres MA, Neulen ML, Choi J, Kang SH, Heo DS, et al. Human group3 innate lymphoid cells express DR3 and respond to TL1A with enhanced IL-22 production and IL-2-dependent proliferation. Eur J Immunol (2015) 45(8):2335–42. doi: 10.1002/eji.201445213
76. Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, et al. An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell (2015) 163(2):381–93. doi: 10.1016/j.cell.2015.08.061
77. Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, et al. IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci Trans Med (2017) 9(376):eaaf9655. doi: 10.1126/scitranslmed.aaf9655
78. Castellanos JG, Woo V, Viladomiu M, Putzel G, Lima S, Diehl GE, et al. Microbiota-Induced TNF-like Ligand 1A Drives Group 3 Innate Lymphoid Cell-Mediated Barrier Protection and Intestinal T Cell Activation during Colitis. Immunity (2018) 49(6):1077–89.e5. doi: 10.1016/j.immuni.2018.10.014
79. Li J, Shi W, Sun H, Ji Y, Chen Y, Guo X. Activation of DR3 signaling causes loss of ILC3s and exacerbates intestinal inflammation. Nat Commun (2019) 10(1):3371. doi: 10.1038/s41467-019-11304-8
80. Deng T, Suo C, Chang J, Yang R, Li J, Cai T, et al. ILC3-derived OX40L is essential for homeostasis of intestinal Tregs in immunodeficient mice. Cell Mol Immunol (2020) 17(2):163–77. doi: 10.1038/s41423-019-0200-x
81. Takedatsu H, Michelsen KS, Wei B, Landers CJ, Thomas LS, Dhall D, et al. TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T-helper 1 and T-helper 17 activation. Gastroenterology (2008) 135(2):552–67. doi: 10.1053/j.gastro.2008.04.037
82. Russo E, Giudici F, Fiorindi C, Ficari F, Scaringi S, Amedei A. Immunomodulating Activity and Therapeutic Effects of Short Chain Fatty Acids and Tryptophan Post-biotics in Inflammatory Bowel Disease. Front Immunol (2019) 10:2754. doi: 10.3389/fimmu.2019.02754
83. Chun E, Lavoie S, Fonseca-Pereira D, Bae S, Michaud M, Hoveyda HR, et al. Metabolite-Sensing Receptor Ffar2 Regulates Colonic Group 3 Innate Lymphoid Cells and Gut Immunity. Immunity (2019) 51(5):871–84.e876. doi: 10.1016/j.immuni.2019.09.014
84. Fachi JL, Sécca C, Rodrigues PB, Mato FCPD, Di Luccia B, Felipe JDS, et al. Acetate coordinates neutrophil and ILC3 responses against C. difficile through FFAR2. J Exp Med (2020) 217(3):e20190489. doi: 10.1084/jem.20190489
85. Kim S-H, Cho B-H, Kiyono H, Jang Y-S. Microbiota-derived butyrate suppresses group 3 innate lymphoid cells in terminal ileal Peyer’s patches. Sci Rep (2017) 7(1):3980. doi: 10.1038/s41598-017-02729-6
86. Bhatt B, Zeng P, Zhu H, Sivaprakasam S, Li S, Xiao H. Gpr109a Limits Microbiota-Induced IL-23 Production To Constrain ILC3-Mediated Colonic Inflammation. J Immunol (2018) 200(8):2905–14. doi: 10.4049/jimmunol.1701625
87. Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity (2013) 39(2):372–85. doi: 10.1016/j.immuni.2013.08.003
88. Wlodarska M, Luo C, Kolde R, d’Hennezel E, Annand JW, Heim CE, et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe (2017) 22(1):25–37.e6. doi: 10.1016/j.chom.2017.06.007
89. Venkatesh M, Mukherjee S, Wang H, Li H, Sun K, Benechet AP, et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity (2014) 41(2):296–310. doi: 10.1016/j.immuni.2014.06.014
90. Cervantes-Barragan L, Chai JN, Tianero MD, Di Luccia B, Ahern PP, Merriman J, et al. Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8alphaalpha(+) T cells. (2017) Science 357(6353):806–10. doi: 10.1126/science.aah5825
91. Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med (2016) 22(6):598–605. doi: 10.1038/nm.4102
92. Hou Q, Ye L, Liu H, Huang L, Yang Q, Turner JR, et al. Lactobacillus accelerates ISCs regeneration to protect the integrity of intestinal mucosa through activation of STAT3 signaling pathway induced by LPLs secretion of IL-22. Cell Death Differ (2018) 25(9):1657–70. doi: 10.1038/s41418-018-0070-2
93. Ahl D, Liu H, Schreiber O, Roos S, Phillipson M, Holm L. Lactobacillus reuteri increases mucus thickness and ameliorates dextran sulphate sodium-induced colitis in mice. Acta Physiol (Oxford Engl) (2016) 217(4):300–10. doi: 10.1111/apha.12695
94. Sun M-C, Zhang F-C, Yin X, Cheng B-J, Zhao C-H, Wang Y-L, et al. Lactobacillus reuteri F-9-35 Prevents DSS-Induced Colitis by Inhibiting Proinflammatory Gene Expression and Restoring the Gut Microbiota in Mice. J Food Sci (2018) 83(10):2645–52. doi: 10.1111/1750-3841.14326
95. Oliva S, Di Nardo G, Ferrari F, Mallardo S, Rossi P, Patrizi G, et al. Randomised clinical trial: the effectiveness of Lactobacillus reuteri ATCC 55730 rectal enema in children with active distal ulcerative colitis. Aliment Pharmacol Ther (2012) 35(3):327–34. doi: 10.1111/j.1365-2036.2011.04939.x
96. Wang T, Zheng N, Luo Q, Jiang L, He B, Yuan X, et al. Probiotics Lactobacillus reuteri Abrogates Immune Checkpoint Blockade-Associated Colitis by Inhibiting Group 3 Innate Lymphoid Cells. Front Immunol (2019) 10:1235. doi: 10.3389/fimmu.2019.01235
97. Agus A, Planchais J, Sokol H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe (2018) 23(6):716–24. doi: 10.1016/j.chom.2018.05.003
98. Reeves RK, Rajakumar PA, Evans TI, Connole M, Gillis J, Wong FE, et al. Gut inflammation and indoleamine deoxygenase inhibit IL-17 production and promote cytotoxic potential in NKp44+ mucosal NK cells during SIV infection. Blood (2011) 118(12):3321–30. doi: 10.1182/blood-2011-04-347260
99. de Araújo EF, Feriotti C, Galdino NADL, Preite NW, Calich VLG, Loures FV. The IDO-AhR Axis Controls Th17/Treg Immunity in a Pulmonary Model of Fungal Infection. Front Immunol (2017) 8:880. doi: 10.3389/fimmu.2017.00880
100. Takamatsu M, Hirata A, Ohtaki H, Hoshi M, Hatano Y, Tomita H, et al. IDO1 plays an immunosuppressive role in 2,4,6-trinitrobenzene sulfate-induced colitis in mice. J Immunol (2013) 191(6):3057–64. doi: 10.4049/jimmunol.1203306
101. Gurtner GJ, Newberry RD, Schloemann SR, McDonald KG, Stenson WF. Inhibition of indoleamine 2,3-dioxygenase augments trinitrobenzene sulfonic acid colitis in mice. Gastroenterology (2003) 125(6):1762–73. doi: 10.1053/j.gastro.2003.08.031
102. Shin JH, Lee YK, Shon WJ, Kim B, Jeon CO, Cho JY, et al. Gut microorganisms and their metabolites modulate the severity of acute colitis in a tryptophan metabolism-dependent manner. Eur J Nutr (2020). doi: 10.1007/s00394-020-02194-4
103. Emgard J, Kammoun H, Garcia-Cassani B, Chesne J, Parigi SM, Jacob JM, et al. Oxysterol Sensing through the Receptor GPR183 Promotes the Lymphoid-Tissue-Inducing Function of Innate Lymphoid Cells and Colonic Inflammation. Immunity (2018) 48(1):120–32.e8. doi: 10.1016/j.immuni.2017.11.020
104. Chu C, Moriyama S, Li Z, Zhou L, Flamar AL, Klose CSN, et al. Anti-microbial Functions of Group 3 Innate Lymphoid Cells in Gut-Associated Lymphoid Tissues Are Regulated by G-Protein-Coupled Receptor 183. Cell Rep (2018) 23(13):3750–8. doi: 10.1016/j.celrep.2018.05.099
105. Melo-Gonzalez F, Kammoun H, Evren E, Dutton EE, Papadopoulou M, Bradford BM, et al. Antigen-presenting ILC3 regulate T cell-dependent IgA responses to colonic mucosal bacteria. J Exp Med (2019) 216(4):728–42. doi: 10.1084/jem.20180871
106. Duffin R, O’Connor RA, Crittenden S, Forster T, Yu C, Zheng X, et al. Prostaglandin E₂ constrains systemic inflammation through an innate lymphoid cell-IL-22 axis. Science (New York NY) (2016) 351(6279):1333–8. doi: 10.1126/science.aad9903
107. Crittenden S, Cheyne A, Adams A, Forster T, Robb CT, Felton J, et al. Purine metabolism controls innate lymphoid cell function and protects against intestinal injury. Immunol Cell Biol (2018) 96(10):1049–59. doi: 10.1111/imcb.12167
108. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol (2013) 14(3):221–9. doi: 10.1038/ni.2534
109. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity (2008) 29(6):947–57. doi: 10.1016/j.immuni.2008.11.003
110. Bauché D, Joyce-Shaikh B, Jain R, Grein J, Ku KS, Blumenschein WM, et al. LAG3(+) Regulatory T Cells Restrain Interleukin-23-Producing CX3CR1(+) Gut-Resident Macrophages during Group 3 Innate Lymphoid Cell-Driven Colitis. Immunity (2018) 49(2):342–52.e5. doi: 10.1016/j.immuni.2018.07.007
111. Schmechel S, Konrad A, Diegelmann J, Glas J, Wetzke M, Paschos E, et al. Linking genetic susceptibility to Crohn’s disease with Th17 cell function: IL-22 serum levels are increased in Crohn’s disease and correlate with disease activity and IL23R genotype status. Inflamm Bowel Dis (2008) 14(2):204–12. doi: 10.1002/ibd.20315
112. Noguchi M, Hiwatashi N, Liu ZX, Toyota T. Increased secretion of granulocyte-macrophage colony-stimulating factor in mucosal lesions of inflammatory bowel disease. Digestion (2001) 63(Suppl 1):32–6. doi: 10.1159/000051908
113. Xu Y, Hunt NH, Bao S. The role of granulocyte macrophage-colony-stimulating factor in acute intestinal inflammation. Cell Res (2008) 18(12):1220–9. doi: 10.1038/cr.2008.310
114. Gathungu G, Kim MO, Ferguson JP, Sharma Y, Zhang W, Ng SM, et al. Granulocyte-macrophage colony-stimulating factor autoantibodies: a marker of aggressive Crohn’s disease. Inflamm Bowel Dis (2013) 19(8):1671–80. doi: 10.1097/MIB.0b013e318281f506
115. Gathungu G, Zhang Y, Tian X, Bonkowski E, Rowehl L, Krumsiek J, et al. Impaired granulocyte-macrophage colony-stimulating factor bioactivity accelerates surgical recurrence in ileal Crohn’s disease. World J Gastroenterol (2018) 24(5):623–30. doi: 10.3748/wjg.v24.i5.623
116. Däbritz J, Bonkowski E, Chalk C, Trapnell BC, Langhorst J, Denson LA, et al. Granulocyte macrophage colony-stimulating factor auto-antibodies and disease relapse in inflammatory bowel disease. Am J Gastroenterol (2013) 108(12):1901–10. doi: 10.1038/ajg.2013.360
117. Roth L, MacDonald JK, McDonald JWD, Chande N. Sargramostim (GM-CSF) for induction of remission in Crohn’s disease: a cochrane inflammatory bowel disease and functional bowel disorders systematic review of randomized trials. Inflamm Bowel Dis (2012) 18(7):1333–9. doi: 10.1002/ibd.22973
118. Geremia A, Arancibia-Cárcamo CV, Fleming MPP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med (2011) 208(6):1127–33. doi: 10.1084/jem.20101712
119. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PDR, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut (2012) 61(12):1693–700. doi: 10.1136/gutjnl-2011-301668
120. Maxwell JR, Zhang Y, Brown WA, Smith CL, Byrne FR, Fiorino M, et al. Differential Roles for Interleukin-23 and Interleukin-17 in Intestinal Immunoregulation. Immunity (2015) 43(4):739–50. doi: 10.1016/j.immuni.2015.08.019
121. Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity (2015) 43(4):727–38. doi: 10.1016/j.immuni.2015.09.003
122. Park CH, Lee AR, Ahn SB, Eun CS, Han DS. Role of innate lymphoid cells in chronic colitis during anti-IL-17A therapy. Sci Rep (2020) 10(1):297. doi: 10.1038/s41598-019-57233-w
123. McNamee EN, Rivera-Nieves J. Ectopic Tertiary Lymphoid Tissue in Inflammatory Bowel Disease: Protective or Provocateur? Front Immunol (2016) 7:308. doi: 10.3389/fimmu.2016.00308
124. Guedj K, Abitbol Y, Cazals-Hatem D, Morvan M, Maggiori L, Panis Y, et al. Adipocytes orchestrate the formation of tertiary lymphoid organs in the creeping fat of Crohn’s disease affected mesentery. J Autoimmun (2019) 103:102281. doi: 10.1016/j.jaut.2019.05.009
125. Shikhagaie MM, Björklund ÅK, Mjösberg J, Erjefält JS, Cornelissen AS, Ros XR, et al. Neuropilin-1 Is Expressed on Lymphoid Tissue Residing LTi-like Group 3 Innate Lymphoid Cells and Associated with Ectopic Lymphoid Aggregates. Cell Rep (2017) 18(7):1761–73. doi: 10.1016/j.celrep.2017.01.063
126. Ikeda A, Ogino T, Kayama H, Okuzaki D, Nishimura J, Fujino S, et al. Human NKp44(+) Group 3 Innate Lymphoid Cells Associate with Tumor-Associated Tertiary Lymphoid Structures in Colorectal Cancer. Cancer Immunol Res (2020) 8(6):724–31. doi: 10.1158/2326-6066.CIR-19-0775
127. Carrega P, Loiacono F, Di Carlo E, Scaramuccia A, Mora M, Conte R, et al. NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat Commun (2015) 6:8280. doi: 10.1038/ncomms9280
128. Meier D, Bornmann C, Chappaz S, Schmutz S, Otten LA, Ceredig R, et al. Ectopic lymphoid-organ development occurs through interleukin 7-mediated enhanced survival of lymphoid-tissue-inducer cells. Immunity (2007) 26(5):643–54. doi: 10.1016/j.immuni.2007.04.009
129. Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. J Exp Med (2011) 208(1):125–34. doi: 10.1084/jem.20100052
130. Chan IH, Jain R, Tessmer MS, Gorman D, Mangadu R, Sathe M, et al. Interleukin-23 is sufficient to induce rapid de novo gut tumorigenesis, independent of carcinogens, through activation of innate lymphoid cells. Mucosal Immunol (2014) 7(4):842–56. doi: 10.1038/mi.2013.101
131. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature (2012) 491(7423):259–63. doi: 10.1038/nature11535
132. Wong U, Cross RK. Expert opinion on interleukin-12/23 and interleukin-23 antagonists as potential therapeutic options for the treatment of inflammatory bowel disease. Expert Opin Invest Drugs (2019) 28(5):473–9. doi: 10.1080/13543784.2019.1597053
133. Shivaji UN, Sharratt CL, Thomas T, Smith SCL, Iacucci M, Moran GW, et al. Review article: managing the adverse events caused by anti-TNF therapy in inflammatory bowel disease. Aliment Pharmacol Ther (2019) 49(6):664–80. doi: 10.1111/apt.15097
134. Feagan BG, Sandborn WJ, D’Haens G, Panés J, Kaser A, Ferrante M, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet (Lond Engl) (2017) 389(10080):1699–709. doi: 10.1016/S0140-6736(17)30570-6
135. Sands BE, Chen J, Feagan BG, Penney M, Rees WA, Danese S, et al. Efficacy and Safety of MEDI2070, an Antibody Against Interleukin 23, in Patients With Moderate to Severe Crohn’s Disease: A Phase 2a Study. Gastroenterology (2017) 153(1):77–86.e6. doi: 10.1053/j.gastro.2017.03.049
136. Sandborn WJ, Ferrante M, Bhandari BR, Berliba E, Feagan BG, Hibi T, et al. Efficacy and Safety of Mirikizumab in a Randomized Phase 2 Study of Patients With Ulcerative Colitis. Gastroenterology (2020) 158(3):537–49.e10. doi: 10.1053/j.gastro.2019.08.043
137. Busbee PB, Menzel L, Alrafas HR, Dopkins N, Becker W, Miranda K, et al. Indole-3-carbinol prevents colitis and associated microbial dysbiosis in an IL-22-dependent manner. JCI Insight (2020) 5(1):e127551. doi: 10.1172/jci.insight.127551
138. Wei YL, Chen YQ, Gong H, Li N, Wu KQ, Hu W, et al. Fecal Microbiota Transplantation Ameliorates Experimentally Induced Colitis in Mice by Upregulating AhR. Front Microbiol (2018) 9:1921. doi: 10.3389/fmicb.2018.01921
139. Kawai S, Iijima H, Shinzaki S, Hiyama S, Yamaguchi T, Araki M, et al. Indigo Naturalis ameliorates murine dextran sodium sulfate-induced colitis via aryl hydrocarbon receptor activation. J Gastroenterol (2017) 52(8):904–19. doi: 10.1007/s00535-016-1292-z
140. Geng S, Cheng S, Li Y, Wen Z, Ma X, Jiang X, et al. Faecal Microbiota Transplantation Reduces Susceptibility to Epithelial Injury and Modulates Tryptophan Metabolism of the Microbial Community in a Piglet Model. J Crohn’s Colitis (2018) 12(11):1359–74. doi: 10.1093/ecco-jcc/jjy103
141. Naganuma M, Sugimoto S, Mitsuyama K, Kobayashi T, Yoshimura N, Ohi H, et al. Efficacy of Indigo Naturalis in a Multicenter Randomized Controlled Trial of Patients With Ulcerative Colitis. Gastroenterology (2018) 154(4):935–47. doi: 10.1053/j.gastro.2017.11.024
142. Marafini I, Di Fusco D, Dinallo V, Franzè E, Stolfi C, Sica G, et al. NPD-0414-2 and NPD-0414-24, Two Chemical Entities Designed as Aryl Hydrocarbon Receptor (AhR) Ligands, Inhibit Gut Inflammatory Signals. Front Pharmacol (2019) 10:380. doi: 10.3389/fphar.2019.00380
143. Chen J, Haller CA, Jernigan FE, Koerner SK, Wong DJ, Wang Y, et al. Modulation of lymphocyte-mediated tissue repair by rational design of heterocyclic aryl hydrocarbon receptor agonists. Sci Adv (2020) 6(3):eaay8230. doi: 10.1126/sciadv.aay8230
144. Kabbani TA, Koutroubakis IE, Schoen RE, Ramos-Rivers C, Shah N, Swoger J, et al. Association of Vitamin D Level With Clinical Status in Inflammatory Bowel Disease: A 5-Year Longitudinal Study. Am J Gastroenterol (2016) 111(5):712–9. doi: 10.1038/ajg.2016.53
145. Gubatan J, Mitsuhashi S, Zenlea T, Rosenberg L, Robson S, Moss AC. Low Serum Vitamin D During Remission Increases Risk of Clinical Relapse in Patients With Ulcerative Colitis. Clin Gastroenterol Hepatol (2017) 15(2):240–6.e1. doi: 10.1016/j.cgh.2016.05.035
146. Frigstad SO, Høivik M, Jahnsen J, Dahl SR, Cvancarova M, Grimstad T, et al. Vitamin D deficiency in inflammatory bowel disease: prevalence and predictors in a Norwegian outpatient population. Scand J Gastroenterol (2017) 52(1):100–6. doi: 10.1080/00365521.2016.1233577
147. Ananthakrishnan AN, Cagan A, Gainer VS, Cai T, Cheng S-C, Savova G, et al. Normalization of plasma 25-hydroxy vitamin D is associated with reduced risk of surgery in Crohn’s disease. Inflamm Bowel Dis (2013) 19(9):1921–7. doi: 10.1097/MIB.0b013e3182902ad9
148. Winter RW, Collins E, Cao B, Carrellas M, Crowell AM, Korzenik JR. Higher 25-hydroxyvitamin D levels are associated with greater odds of remission with anti-tumour necrosis factor-α medications among patients with inflammatory bowel diseases. Aliment Pharmacol Ther (2017) 45(5):653–9. doi: 10.1111/apt.13936
Keywords: group 3 innate lymphoid cells, immunometabolism, intestinal inflammation, inflammatory bowel disease, therapeutics
Citation: Song D, Lai L and Ran Z (2020) Metabolic Regulation of Group 3 Innate Lymphoid Cells and Their Role in Inflammatory Bowel Disease. Front. Immunol. 11:580467. doi: 10.3389/fimmu.2020.580467
Received: 06 July 2020; Accepted: 30 September 2020;
Published: 26 October 2020.
Edited by:
Hergen Spits, University of Amsterdam, NetherlandsReviewed by:
Guido Ferlazzo, University of Messina, ItalyJochem Bernink, Hubrecht Institute (KNAW), Netherlands
Copyright © 2020 Song, Lai and Ran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhihua Ran, emhpaHVhcmFuMTk2MkAxMjYuY29t