 Alexandra Neaga1
Alexandra Neaga1 Cristina Bagacean2,3
Cristina Bagacean2,3 Adrian Tempescul2,3
Adrian Tempescul2,3 Laura Jimbu1
Laura Jimbu1 Oana Mesaros1
Oana Mesaros1 Cristina Blag4
Cristina Blag4 Ciprian Tomuleasa1,5Corina Bocsan6
Ciprian Tomuleasa1,5Corina Bocsan6 Mihaela Gaman7
Mihaela Gaman7 Mihnea Zdrenghea1,5*
Mihnea Zdrenghea1,5*- 1Department of Hematology, Iuliu Haţieganu University of Medicine and Pharmacy, Cluj-Napoca, Romania
- 2Department of Hematology, Brest University Medical School Hospital, Brest, France
- 3U1227 B Lymphocytes and Autoimmunity, University of Brest, INSERM, IBSAM, Brest, France
- 4Department of Pediatrics, Iuliu Hatieganu University of Medicine and Pharmacy, Cluj-Napoca, Romania
- 5Department of Hematology, Ion Chiricuta Oncology Institute, Cluj-Napoca, Romania
- 6Department of Pharmacology, Toxicology and Clinical Pharmacology, Iuliu Haţieganu University of Medicine and Pharmacy, Cluj-Napoca, Romania
- 7Department of Hematology, Carol Davila University of Medicine and Pharmacy, Bucharest, Romania
Acute myeloid leukemia (AML) is an aggressive myeloid malignancy with poor outcomes despite very intensive therapeutic approaches. For the majority of patients which are unfit and treated less intensively, the prognosis is even worse. There has been unspectacular progress in outcome improvement over the last decades and the development of new approaches is of tremendous interest. The tumor microenvironment is credited with an important role in supporting cancer growth, including leukemogenesis. Macrophages are part of the tumor microenvironment and their contribution in this setting is increasingly being deciphered, these cells being credited with a tumor supporting role. Data on macrophage role and polarization in leukemia is scarce. MicroRNAs (miRNAs) have a role in the post-transcriptional regulation of gene expression, by impending translation and promoting degradation of messenger RNAs. They are important modulators of cellular pathways, playing major roles in normal hematopoietic differentiation. miRNA expression is significantly correlated with the prognosis of hematopoietic malignancies, including AML. Oncogenic miRNAs correlate with poor prognosis, while tumor suppressor miRNAs, which inhibit the expression of proto-oncogenes, are correlated with a favorable prognosis. miRNAs are proposed as biomarkers for diagnosis and prognosis and are regarded as therapeutic approaches in many cancers, including AML. miRNAs with epigenetic or modulatory activity, as well as with synergistic activity with chemotherapeutic agents, proved to be promising therapeutic targets in experimental, pre-clinical approaches. The clinical availability of emerging compounds with mimicking or suppressor activity provides the opportunity for future therapeutic targeting of miRNAs. The present paper is focusing on miRNAs which, according to current knowledge, favorably impact on AML outcomes, being regarded as tumor suppressors, and reviews their role in macrophage polarization. We are focusing on miRNA expression in the setting of AML, but data on correlations between miRNA expression and macrophage polarization is mostly coming from studies involving normal tissue.
Introduction
Acute myeloid leukemia (AML), the most common form of acute leukemia in adults, is a heterogeneous myeloid malignancy. It is characterized by an accumulation of immature blast cells due to deficient maturation, uncontrolled proliferation and prolonged survival of a myeloid progenitor. The backbone of intensive treatment approaches is unchanged for almost half a century, and improvements in long-term survival of patients are unimpressive. There is tremendous interest in the development of new therapeutic approaches, including the identification and subsequent validation of new therapeutic targets (1–5).
MicroRNAs (miRNAs) are part of a recently described mechanism of post transcriptional modulation of gene expression. They act inhibiting translation and speeding up degradation of target messenger RNAs by an imperfect pairing with the 3’-untranslated region (3’-UTR) (6, 7).
miRNAs play an important role in regulating normal haematopoiesis: myeloid differentiation, cell cycle, proliferation, apoptosis, and gene methylation (2–4). In hematological malignancies starting from hematopoietic stem and progenitor cells, miRNA could function either as tumor-suppressors or as tumor-promoters/oncomiRs (3–5, 8).
The tumor microenvironment is credited with a critical role in cancer pathogenesis. The involvement of stromal macrophages in solid cancer pathogenesis is currently being deciphered, but data on their role in leukemia is scarce (9).
Tumor Associated Macrophages, Phenotypes, and Regulation Factors
Macrophages are the sentinels of the innate immunity. They were first described in the 1880s by Metchnikoff, who also observed that these peculiar cells recognize, engulf and destroy the pathogens, the senescent cells or other debris, thus describing the phagocytosis process (10). Later, it was discovered that apart from their role in the initiation of inflammatory responses, macrophages are involved in tissue repair and remodeling too (11). There are two types of macrophages: monocyte derived macrophages, that evolve from the blood monocytes, by migrating to tissues and becoming macrophages in inflammatory conditions, and tissue-derived macrophages, that emerge during the gestation period from the yolk sack (12). Both are terminally differentiated, effector cells.
In response to epigenetic, microenvironmental and extrinsic factors, macrophages become activated, acquiring new phenotypes, such as the “classical” pro-inflammatory one and the “alternative” anti-inflammatory one (13, 14). Macrophage polarized activation has profound effects on immune and inflammatory responses, but the understanding of mechanisms involved is still incipient.
There is malleability in phenotype switching by elements of the environment and feedback mechanisms. Excessive inflammation causes cell and tissue destruction, an underlying cause of many diseases, including cancer. Coincident with the activation of Toll-like receptor (TLR) signaling, a feedback of anti-inflammatory pathways and mechanisms start modulating inflammation until tissue repair is complete (15).
As a parallel to Th1 and Th2 cytokines paradigm, the pro-inflammatory, anti-tumoral macrophages are categorized as M1, whilst those with anti-inflammatory, wound healing, and pro-tumorigenic activity, are referred to as M2 macrophages. An imbalance in M1/M2 activation was described in appearance of various diseases, including cancer. Macrophages get polarized towards the M1 phenotype under the effect of lipopolysaccharide (LPS) and Th1 type cytokines, such as INF-γ or TNF-α and finally express on their surface, among other markers, toll like receptor 2 (TLR-2) and TLR-4. The expression of M1 subtype is regulated by transcription factors, such as the signal transducer and activator of transcription 1 and 5 (STAT-1, STAT-5), nuclear factor kB (NF-kB), interferon regulatory factors 3 and 5 (IRF-3, IRF-5), and hypoxia inducible factor-1 alpha (HIF-1α) (15–17). In response to Th2 type cytokines, like IL-4, IL-13, or IL-10, macrophages are balanced towards an M2 phenotype. Expression of the M2 phenotype is regulated by STAT-6, peroxisome proliferator-activated receptors gamma and delta (PPARγ, PPARδ), IRF4, and HIF-2α. Several variants of M2 phenotypes were described, responding to and releasing different types of cytokines: M2a, M2b, M2c, M2d (18, 19).
Despite the fact that M1 and M2 phenotypes are antagonistic, they are also intimately intertwined. The STAT family, the nuclear receptor PPAR-γ, the cAMP responsive element-binding protein (CREB) - CCAAT/enhancer-binding protein (C/EBP) axis, the interferon regulatory factors (IRF), and the NF-κB family all participate in the regulation of macrophage polarization, involving many signaling pathways, including JAK/STAT, JNK, PI3K/AKT, and Notch. The balance between the activation of STAT1 and STAT3/STAT6, tightly regulates M1/M2 polarization and activity. NF-κB p65/p50 and STAT1 activation promotes M1 polarization, while STAT3 and STAT6 activation by IL-4/13 and IL-10 increases M2 polarization. PPARδ and PPARγ control distinct aspects of M2 macrophage activation and oxidative metabolism. KLF4, downstream of STAT6, participates in the promotion of M2 macrophage functions by suppressing the NF-κB/HIF-1α-dependent transcription. IL-4 induces not only the myelocytomatosis viral oncogene homolog (c-Myc), which controls the expression of a subset of M2-associated genes, but also the M2-polarizing IFN regulatory factor (IRF) 4 axis to inhibit IRF5-mediated M1 polarization. IL-10 promotes M2 polarization through the induction of p50 NF-κB homodimer (NF-kB p50/p50), c-Maf, and STAT3 activities. CREB induces IL-10 downstream of p38 activation. The transcription factor CREB is an activator of C/EBPβ expression. C/EBPβ is one among several transcription factors in addition to KLF4, which activates M2 polarization. The CREB-C/EBPβ signaling axis also favours M2 polarization (15, 20).
Tumor Associated Macrophages in AML
When the balance in M1/M2 polarization is broken, the tumor associated macrophages (TAMs) appear, which, as their name suggests, support and stimulate the tumor cells growth. There are substantial differences between the TAMs and LAMs (leukemia associated macrophages) and not all of them are elucidated (21). They share mostly M2 characteristics, as they are able to heal and nurture the tumor. Such macrophages are also present in the tumor microenvironment of AML and there is a paucity of data on their precise phenotype (20). It is also unclear which elements are accountable for changing the status of macrophage polarization, facilitating the growth of malignant cells, instead of restraining it. There is insufficient data about the interaction of stromal cells and leukemic cells, as opposed to the extensive knowledge about the role of macrophages in solid tumors. The bone marrow microenvironment can be altered by the leukemic stem cells (LSCs) in a way that supports the development of LSCs instead of the hematopoietic stem cells (HSCs), ultimately heading to chemoresistance (9). It is important to underline the fact that in vivo activation of macrophages phenotype model in AML is scarce due to complexity and insufficient understanding, but M2-like macrophages are associated with a poorer survival in AML, and the induction of M1 features contributes to extended survival and appears to inhibit the pro-leukemic effect (21).
miRNA Correlation With Macrophage Phenotypes Under Physiologic Conditions
Despite miRNAs being recognized as highly conserved across species, there are a lot of divergent results between human and mouse studies investigating the association of miRNA expression to macrophage phenotypes (22). In the following sections, we chose to refer mostly to human studies.
In vitro studies of polarized macrophages were performed to establish the miRNA profile of M1 versus M2 macrophages. Recent studies suggest that miRNA activity could control macrophage reaction to environmental signals. Macrophages and their precursors can be recruited by tumors to create a nourishing stroma. miRNAs appear to be involved in the regulation of all phases of macrophage development and proliferation, both under normal conditions or in cancer (23). Below we discuss the reported roles of miRNAs in the setting of macrophage polarization towards M1 or M2 phenotype and key regulators known to control macrophage development, recruitment, differentiation, and M1 or M2 polarization.
miR-22-3p was found to be increased in human M2c macrophages (24, 25).
In human MDMs, miR-26a-2* was decreased in both M1 and M2b, and slightly increased in M2c and miR-26a-2 was found in M2c macrophages (20). miR-26a/miR-26b induced a pro-inflammatory M1 polarization by downregulating macrophage colony-stimulating factor (M-CSF) through the PI3K/Akt pathway and IL-10 expression and increasing innate interferons (26).
In human MDMs, miR-29b-1* and miR-29b were found in M1 (20, 24, 25) and in M2b macrophages (20).
MiR-34a promotes M2 polarization (27).
miR-124 (124-1, 124-2, 124-3) was increased in M2b-polarised cells and seems to be a suitable marker for the M2-like macrophages and a therapeutic target to alter M1/M2 equilibrium (28). miR-124 decreases STAT3 and proinflammatory cytokines, promoting M2 polarization (27, 29).
miR-125a-5p expression was amplified after monocyte polarization under M2b (30) or M1, M2c conditions (24, 25). It has been recently shown that monocyte miR-125a-5p expression is enhanced when cells are polarized toward M2-alternatively activated macrophages with immunoregulatory phenotypes, particularly IL-10-producing M2b (30).
miR-146a was reported to be increased in human M1 polarised macrophages (24, 25). Also, miR-146a suppressed M1 and promoted M2 polarization, enhanced the activation of M2 macrophage and increased the expression of IL-10 through Notch1-dependent mechanism and by increasing PPARγ in murine macrophage RAW264.7 (31).
miR-193b was found increased in M1and in M2a (20, 24, 25). IL-4 treatment increased the accumulation of miR-193b during human monocyte-to-macrophage differentiation (20, 32, 33).
miR-223 promotes M2 polarization (27) via targeting anti-inflammatory SOCS1, CEBP-β, and PPARγ. Rasa1 and Nfat5 are miR-223 targets that are important for PPARγ-dependent macrophage alternative activation. miR-223 also inhibited proinflammatory regulator Pknox1 which regulates macrophage classical activation (34) (Figures 1 and 2).
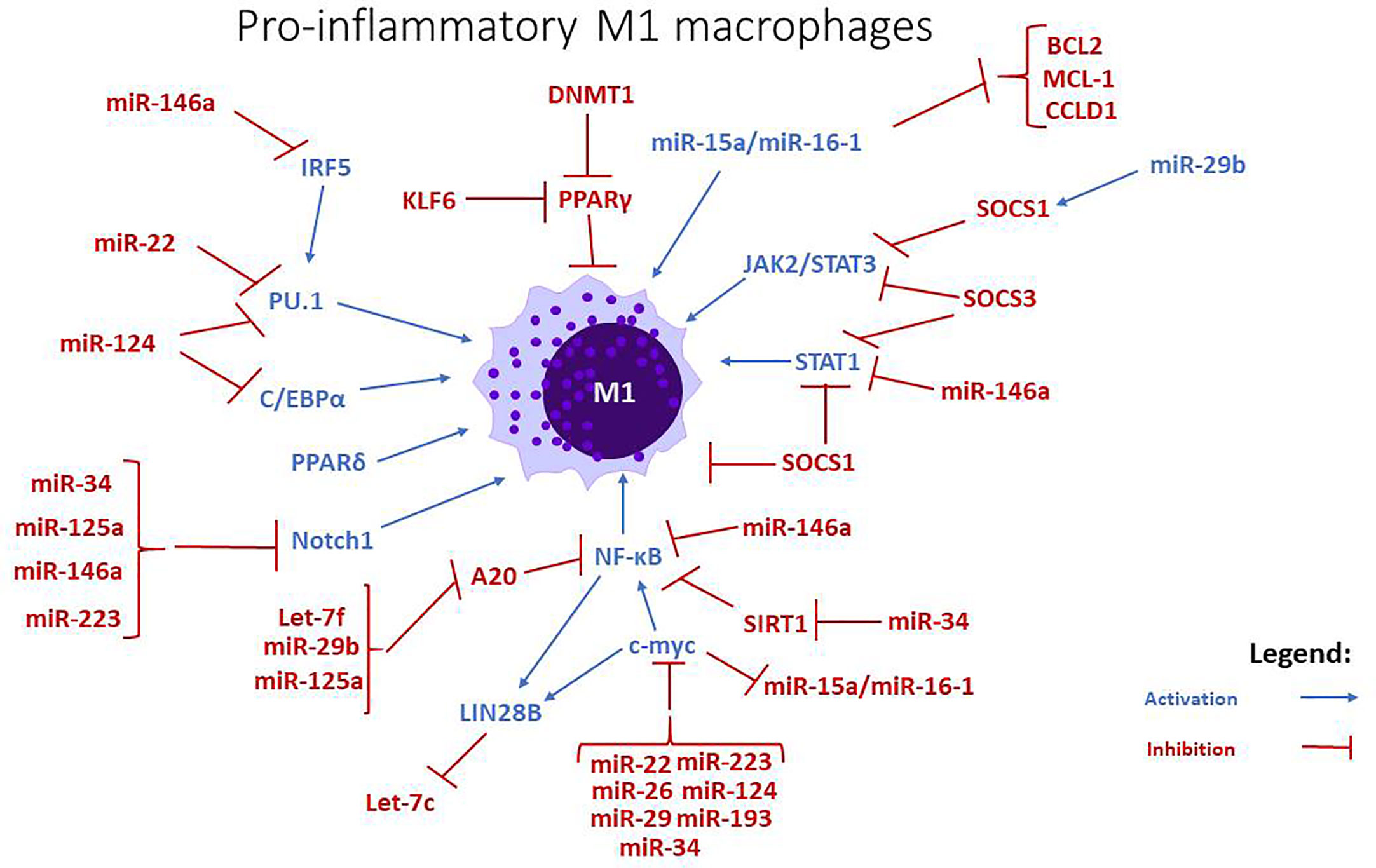
Figure 1 miRNAs and signaling pathways involved in the regulation of pro-inflammatory M1 macrophages. miRNAs like let7-f, miR-15a/-16, miR-22, miR26, miR-29b, stimulate the differentiation towards an M1 phenotype, while miR-34, miR-125a, miR-146a, and miR-223 are negative regulators. The action of miR-193 is uncertain, but some studies imply that it mostly stimulates the polarization towards M2 phenotype.
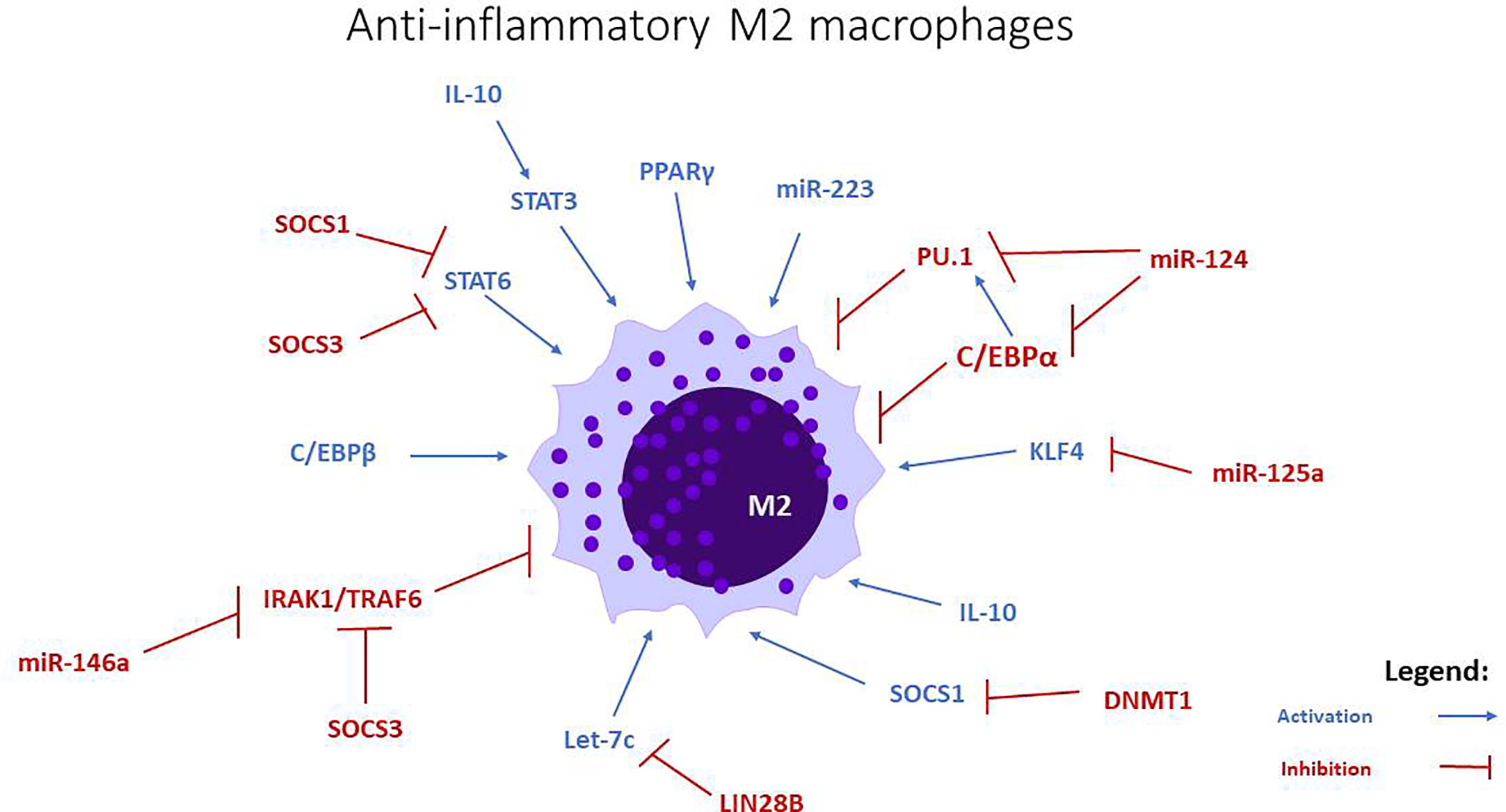
Figure 2 miRNAs involved in M2 anti-inflammatory macrophage polarization. Let-c, miR-34a, miR-124, miR-125a, miR-146a, and miR-223 stimulate the polarization towards this phenotype, through the modulation of transcription factors and intracellular signaling pathways.
The potent anti-inflammatory cytokine IL-10 is important for attenuating the inflammatory response (13, 35). IL-10, via STAT3, suppresses mTOR activity through the induction of an mTOR inhibitor, DDIT4 (35). IL-10-STAT3-DDIT4 is important for the inhibition of mTORC1, and the activation of adenosine monophosphate-activated protein kinase (AMPK) by IL-10 also contributes to mTOR inhibition (35). IL-10 mRNA and protein levels in bone marrow-mononuclear cells (BM‐MNCs) were higher in AML patients as related to healthy volunteers, along with IL-10 receptors and the adhesion molecule E-cadherin (36). IL-10 is capable of up-regulating miRNAs, which contributes to the anti-inflammatory response, like oncomiR miR-146b, and down-regulating those that are highly pro-inflammatory, such as miR-155 or miR-9 (37).
miRNAs Associated With Good Prognosis of AML and Their Potential Therapeutic Targets
Tumor suppressor miRNAs are repressing the expression of genes involved in various oncogenic signaling pathways that are necessary for basic cancer cell survival, as well as cancer stemness, metastasis, and chemoresistance. Epigenetic silencing of tumor suppressor genes frequently occurs due to aberrant DNA hypermethylation in cancer cells. Numerous tumor suppressor miRNAs such as miR-22, miR-29, miR-34a associated with a good prognosis, and long-term survival for AML patients have been found to be decreased in AML (Table 1). In the following section we will focus on the most important tumor suppressor miR, their role in AML and their potential as therapeutic targets.
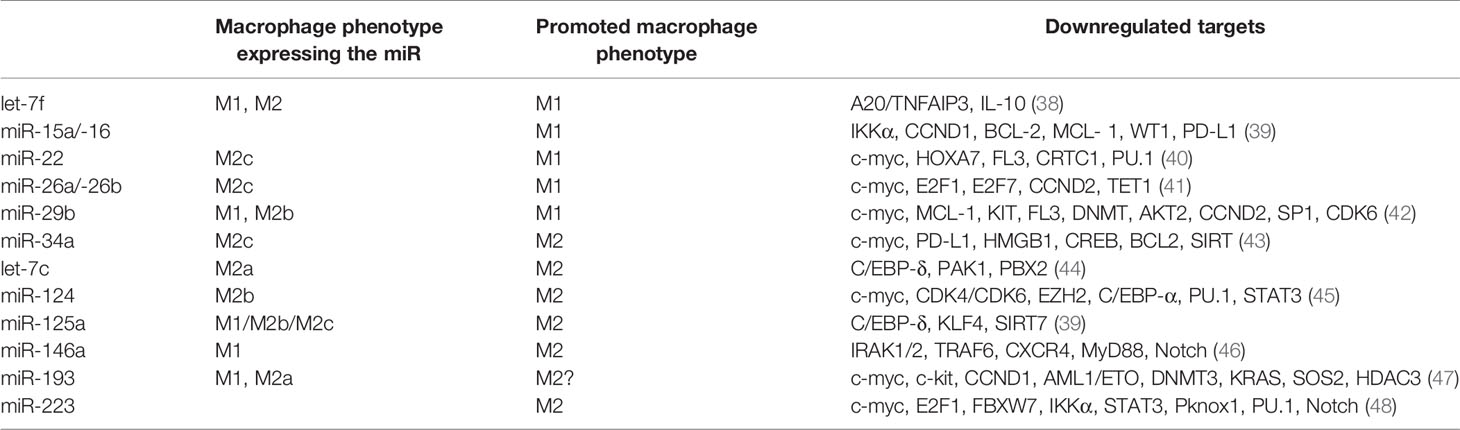
Table 1 Tumor suppressor miRNA targets in AML miR.
Let-7
Let-7 (lethal-7), one of the initial miRNA families to be discovered, is composed of let-7a-1, let-7a-2, let-7a-3, let-7b, let-7c, let-7d, let-7e, let-7f-1, let-7f-2, let-7g, let-7i, and miR-98. It was recently shown that the let-7 family members tend to act as tumor suppressors by supressing oncogenes such as RAS, c-MYC, BCL-XL, NF-kB, and High Mobility Group A2 (HMGA2) in various cancers (49). They also promote differentiation during development. Expression of tumor suppressor let-7 is decreased in a variety of tumors and one of the cause of the global post-transcriptional downregulation of the let-7 miRNA family is the activation of either LIN28A or LIN28B, two highly related RNA binding proteins and proto-oncogenes (49). Elevated levels of LIN28B protein in bone marrow samples independently associate with worse survival in AML patients (50).
let-7c targets C/EBP-δ, a transcriptional factor that plays a significant role in inflammatory reactions (51, 52). Let-7c also targets serine/threonine kinase family member p21-activated kinase (PAK)1 and pre-B cell leukemia transcription factor 2 (PBX2). The upregulation of enhancer of zeste homolog 2 (EZH2) determines the loss of let-7c, PAK1 elevation and human M1 polarization via NIK-IKK-NF-kB signaling (53). PML/RARα-positive blasts from acute promyelocytic leukemia (APL) patients display lower levels of miRNA let-7c than in regular promyelocytes. This expression increases after all-trans-retinoic acid (ATRA) treatment (44). The ectopic expression of let-7c promotes granulocytic differentiation of AML cell lines and blasts. PBX2, a well-known homeodomain protein whose abnormal expression enhances homeobox A9 (HOXA9)-dependent leukemogenesis, is a new let-7c target that may impact the AML phenotype, suggesting that that perturbation of the let-7c-PBX2 pathway may have a beneficial role in AML (44).
let-7f targets A20/TNFAIP3, a feedback inhibitor of the NF-κB pathway that was found in mouse Mycobacterium tuberculosis-infected macrophages (54). let-7f may also target some genes, such as PRKAA2 (AMPK, AMP-activated protein kinase), CCNJL (cyclin J-like), MBD2 (methyl-CpG binding domain protein 2), CYP19A1 (38). However, it was discovered that Let-7f is downregulated in t(11q23) AML versus all other AML patients (39). Dai et al. study reported a reduced expression of let-7f in primary leukemia cells from patients with refractory AML and in adriamycin (ADM)-resistant leukemic cell line, K562/A02 (38). Interestingly, this suggests that let-7f may serve as a tumor suppressor. The enhanced let-7f expression in vitro could make sensitive K562/A02 cells to ADM, showing that down-regulation of let-7f is implicated in the acquisition of drug resistance in AML (38, 39).
miR-15a/miR-16-1
The miR-15a/16-1 cluster is believed to function as a tumor suppressor, because its validated gene targets are important oncogenes. Genes which regulate the cell cycle control cell outcome and induce apoptosis by targeting multiple oncogenes, including BCL2, Cyclin-D1 (CCLD1), Myeloid Cell Leukemia-1 (MCL-1), and others (55, 56). In one study, an important inverse correlation between miR-15a or miR-16-1 expression and Wilms’ tumor 1 (WT1) oncogene protein levels in primary AML blasts was found (55). It was also established that miR-15a was downregulated in patients with t(11q23) versus all other AML patients (39).
Additionally, miR-16 was found to be down-regulated in FLT3-ITD positive murine myeloid FDC-P1 cells and FLT3 inhibition tends to correct this down-regulation, therefore suggesting a tumor suppressor activity in FLT3/ITD-mediated leukemic transformation (57, 58).
Overexpression of the chemokine stromal-derived factor 1 α (SDF-1/CXL12) and its receptor CXCR4 is a hallmark of many hematological malignancies, including AML, and generally correlates with a poor prognosis. CXCR4 is important in the preservation and survival of acute AML blasts in the bone marrow microenvironment. In AML patients, hypoxia induced CXCR4 expression is associated with poor prognosis, not depending on the presence of the mutated FLT3 gene. FLT3-ITD mutations trigger CXCR4 signaling and are associated with increased CXCR4 expression in primary AML cells. However, it was demonstrated that an antagonist of CXCR4, BL-8040, determined the apoptosis of AML cells in vitro and in vivo and induced the robust mobilization of AML blasts from the bone marrow (BM). Apoptosis was mediated by the up-regulation of miR-15a/miR-16-1, resulting in down-regulation of the target antiapoptotic genes BCL2, MCL-1, and cyclin-D1 in MV4-11 and U-937 leukemic cell lines. Interestingly, leukemic cell death was produced directly by the overexpression of miR-15a/miR-16-1. BL-8040-induced apoptosis was also facilitated by the inhibition of survival signals through inhibition of AKT/ERK pathways. Notably, treatment with a BCL2-inhibitor induced apoptosis and combined with the CXCR4 antagonist BL-8040 enhances cell death, making this mechanism a very attractive target (56, 58, 59).
In a recent phase 1b clinical trial, the combination of a second generation CXCR4 antagonist, LY2510924, with idarubicin and cytarabine (IA) was reported to be safe in the relapsed/refractory (R/R) setting. However, LY2510924 at dose of 10 and 20 mg/day suppresses CXCR4 receptor blockade in some, but not all patients. The FDA has allowed a higher dose of 30 mg of LY2510924 in a new clinical trial (60).
Beside the developments mentioned above, there are ongoing clinical trials of miR-16 mimics -TargomiRs, which are minicells (EnGeneIC Dream Vectors) loaded with miR-16-based mimic miR- which successfully completed phase 1 trials and will soon start phase 2 clinical investigation in malignant pleural mesothelioma. Due to these advancements, miR-16 mimics could be evaluated in treating hematological malignancies, including AML (56, 61).
miR-22
miR-22 has an important role in monocytic differentiation in healthy and leukemic cells. miR-22 expression is increased during monocyte/macrophage differentiation of HL-60, THP1 leukemia cells lines, and CD34+ hematopoietic stem/progenitor cells (62).
Studies showed that miR-22 was down-regulated in low miR-188–5p expressers cytogenetically normal acute myeloid leukemia (CN-AML). This fact was associated with longer overall survival (OS) and event free survival (EFS) (63) in de novo AML (40). One of the causes of miR-22-downregulation in AML is TET1/growth factor independent 1 (GFI1)/EZH2/SIN3A-mediated epigenetic repression and/or DNA copy-number loss (40). miR-22 inhibited multiple oncogenes, including CREB-regulated transcription coactivator 1 (CRTC1), homeobox (HOXA)7, FLT3 and MYCBP (a MYC-binding protein), and consequently the CREB and MYC pathways, leading to the inhibition of leukemia progression in vivo (40).
Down-regulation of miR-22 levels and E26 transformation-specific family transcription factor (PU.1) were reported in AML patients (62). miR-22-mediated MECOM (MDS1 and EVI1 complex locus protein EVI1) degradation increased c-Jun levels, which interacts with PU.1 transcription factor to promote monocyte/macrophage differentiation (62). Consequently, the reintroduction of miR-22 inhibited the growth of AML bone marrow blasts, suggesting that the restoration of miR-22 expression or function could have a therapeutic potential to treat AML (62). These studies suggest that miR-22 could be a promising potential marker in diagnosis and treatment monitoring, and a prospective therapeutic target in AML (40, 62).
miR-26a/miR-26b
The initial in vitro and in vivo studies revealed that miR-26a/b can function as an oncogenic miRNA, being associated with unfavourable OS in 53 patients with AML (64). The tumor suppressor ten-eleven translocation (TET) 2, a member of the TET methylcytosine dioxygenase family, is frequently mutated and under extensive miR regulation in hematopoietic malignancies. It was suggested that TET2 gene-targeting miRs, such as miR-26a/b, may play oncogenic roles in hematopoietic cancers (65). However, miR-26 has been incriminated to target cyclin-D2 (CCND2) and cyclin-E2 (CCNE2), consequently inhibiting the cell cycle in malignant cells (66).
The expression of miR-26a was down-regulated in primary blasts from AML patients (41, 67). In myeloid leukemia cells, c-Myc enforces transcriptional repressor E2F1 activity and contributes to increased EZH2 levels (41). During myeloid differentiation of AML cells, miR-26a induction was associated with a decrease in c-Myc and EZH2 levels (41, 67). miR-26a interfering with E2F7 (which promotes cell cycle progression and inhibits monocytic differentiation of AML cells), leads to the inhibition of c-Myc activity and downregulation of the oncogenic miR-17-92 cluster (67).
In another study, miR-26a-5p was also reported to be decreased in AML patients and subsequently the peroxiredoxin III (PrxIII), a reactive oxygen species (ROS) scavenger, was increased (68). Increased levels of PrxIII caused decreases in ROS levels in AML samples, which are required for hematopoietic stem cell differentiation (68).
miR-29b
miR-29b family consists of three different isoforms (miR-29a, miR-29b, miR-29c) and it’s downregulated in different myeloid and lymphoid neoplasms and solid cancers (69). Compared with control groups miR-29 family had lower expression in different subtypes of AML (39, 70–73). miR-29a, miR-29b, and miR-29c were downregulated in PBMCs and bone marrow CD34+ cells in AML patients as opposed to healthy donors (33, 42, 74, 75) miR-29b targeted DNA methyltransferases (DNMTs) DNMT3A and DNMT3B, by reducing the global DNA methylation and specificity protein (SP)1) and DNMT1, by targeting SP1 and enhancing tumor cell chemosensitivity (76, 77). The ectopic transfection of synthetic miR-29b into leukemia cells revealed that miR-29b targets genes connected to apoptosis (MCL-1, TRAF4, and MYBl2), cell cycle (CDK4, CDK6, and CCND2), and cell proliferation (JAK2 and IGF1) (70). The overexpression of miR-29b was inversely correlated to MCL-1 in 45 primary AML samples (39, 70–73).
However, in some studies, miR-29b has been linked to unfavourable survival rates of AML patients (64, 78). In a cohort of CN-AML, multiple tumor suppressor TET2-targeting miRs, such as miR-29b/-29c and miR-125b, were preferentially overexpressed in TET2-wildtype samples than those with TET2-mutations (65). In vivo expression of TET2-targeting miRNAs generated hematopoietic cell expansion and/or myeloid differentiation bias, and co-expression of TET2 fixed these phenotypes. Cheng et al. suggested that increasing TET2 expression could be a likely approach to combat certain groups of hematopoietic cancers (65).
In another study, miR-29b expression level was inversely related to MLLT11 expression in AML patients and AML patients with low miR-29b and elevated MLLT11 expression had poor OS (71). The decreased miR-29 expression in AML blasts was correlated with increased anti-apoptotic molecule AKT2, cell cycle regulation cyclin-D2, and Myc levels (42). PI3K/AKT pathway is a negative regulator of miR-29b and inversely, miR-29b reduces AKT phosphorylation (42, 79).
The nuclear factor erythroid 2-related factor 2 (NRF2), a transcription factor which protects cells from the oxidative damage, was reported to be constitutively expressed in AML and promoted leukemic cell survival (80). The activation of Nrf2 is correlated with decreased levels of miR-29b and associated with treatment resistance to regular chemotherapy, as well as with reduced survival of cancer patients. The transfection with miR-29b mimic of primary AML samples (either alone or co-treated with standard AML chemotherapy) resulted in increased cell death responsiveness (80).
Another transcription factor, MYC, activated by c-kit, binds and inhibits the miR-29b promoter (81). The decrease in miR-29b expression was correlated with upregulation of SP1 levels, subsequently allowing the formation of the SP1/NF-kB complex. This complex binds regulatory structures to increase c-kit expression and the formation of a complex with histone deacetylase 1 (HDAC1), which promotes the down-regulation of miR-29b expression (81, 82). Remarkably, the proteasome inhibitor bortezomib, acting through a miR-29b-dependent mechanism, downregulated SP1 and decreased global DNA methylation. Consequently, it induced M2 macrophage marker suppressor of cytokine signaling-1 (SOCS-1) promoter demethylation and protein up-regulation, reduced proliferation, and increased apoptosis (83). These promising data suggest that AML patients may benefit from histone-deacetylase inhibitors (HDACI) therapy. Studies of cytogenetic, molecular and in vitro growth features of primary blasts related to HDACI response will deliver understanding into how to select AML patients that are prone to respond to HDACIs (82, 84).
On the other hand, treatment with type I IFN-β supressed miR-29b during human monocyte-to-macrophage differentiation in vitro (33). mir-29 expression can be inhibited by c-Myc, hedgehog signaling, and inflammatory pathways (TLR ligation, activation of NF-kB), commonly activated in cancer (74). C/EBPα is an activator of miR-29a/b (75).
In a study of 53 older AML patients with previously untreated disease, who were not suitable candidates for chemotherapy, or who refused it, higher miR-29b pre-treatment expression was correlated with improved response to decitabine (a DNA methyltransferase inhibitor) and better outcome in AML (85). The HDACI AR-42 increased miR-29b levels and led to downregulation of known miR-29b targets (SP1, DNMT1, DNMT3A, and DNMT3B). Importantly, the anti-leukemic activity in vitro and in vivo of decitabine was increased, when HDACIs were given before decitabine, as decitabine traditionally is given before HDACIs (82). This could be explained by the increased miR-29b levels by HDACI AR-42, which will target DNMT expression, consequently decitabine better inhibiting the activity of remaining DNMT. Alternatively, lower levels of SP1 induced by increased miR-29b expression leads to a reduced transcription of genes, known to promote AML leukemogenesis, such as modified and/or upregulated receptor tyrosine kinases (i.e., FLT3 and KIT) (81, 86).
Conversely, the transferring-conjugated anionic lipopolyplex nanoparticle synthetic miR-29b mimics Tf-NP-miR-29b was shown to downregulate DNMTs, cyclin-dependent kinase (CDK) 6, SP1, KIT, and FLT3, to suppress AML growth, to impair colony formation, and to reduce cell viability in vitro in AML patient samples (87). In addition, priming AML cells with Tf-NP-miR-29b before therapy with decitabine resulted in striking decrease in cell viability and growth, showing improved antileukemic action, compared to in vivo single-agent decitabine treatment (87).
miR-34
miR-34 family, consisting of miR-34a, -34b, and -34c plays a key role as tumor suppressor miR. MiR-34a was the primary recognized tumor suppressor gene which is downregulated in AML (88).
mir-34 exerts its growth inhibition on cancer cells by repressing HDAC1 (89), targeting factors required for G1/S transition (c-MYC, CDK6), anti-apoptotic proteins (Bcl2, sirtuin protein-SIRT1), proteins involved in invasion (c-MET) (90), and induction of sequential down modulating of Erk/Akt activity (91).
Wang et al. reported that PD-L1 was overexpressed in AML samples and that there was an inverse correlation between PD-L1 and miR-34a gene expression (92). PD-L1 is regulated by p53 via miR-34 (93). Overexpression of miR-34a in HL-60 and Kasumi-1 cells blocked PD-L1 expression and reduced PD-L1 surface expression (92). Surface expression of PD-L1 produced by IFN-γ was also reversed by miR-34a. miR-34a transfection diminished the production of PD-L1 downstream of IL-10 and decreased PD-L1-specific T cell apoptosis (92).
The transcription factor, CCAAT enhancer binding protein alpha (C/EBPα) is an activator of miR-34a (3). The CEBPA (which is crucial for granulopoiesis) gene mutations are reported in 8-10% of patients with AML (3). Study of AML samples with CEBPA mutations described a lower expression of miR-34a and increased levels of E2F3, as well as E2F1, a transcriptional target of E2F3. miR-34a influences granulocytic differentiation of AML blast cells with CEBPA mutations. miR-34b was downregulated in patients with t(11q23) AML (39, 88).The high-mobility group box-1 (HMGB1) functions as an anti-apoptotic protein in leukemia, inhibiting cell apoptosis and inducing cell growth. Lower expression of miR-34a correlated with greater expression of HMGB1 in HL-60 and THP-1 cells compared to that in HS-5 cells (43). Higher expression levels of miR-34 and lower expression levels of HMGB1 both considerably promoted apoptosis, inhibited autophagy in HL-60 and THP-1 cells and repressed chemotherapy-induced autophagy by stimulating the autophagy marker LC3 conversion (43). By promoting cell apoptosis and inhibiting autophagy via targeting HMGB1, miR-34a may be a potential promising molecular target for AML therapy. Enhanced expression of miR-34a in TIM3 positive leukemia stem cells (LSC) from AML children inhibits the clone expansion, progression and metastasis of leukemia, suggesting that miR-34a is a key regulator which could be taken into consideration as a novel therapeutic agent against LSC (94).
In addition, mir-34b is a negative regulator of CREB expression in myeloid cell lines by binding directly to the 3′UTR of CREB mRNA. In AML cells, the promoter of miR-34b is highly methylated, leading to miR-34b down-regulation in the majority of patients and, consecutive overexpression of the proto-oncogene CREB (88). Additionally, low miR-34b and high CREB expression levels stimulated abnormal myelopoiesis through CREB-dependent pathways in vitro and in vivo in animal studies, consequently leading to myeloid cell proliferation (88).
miR-34c plays an important role in tumorigenesis, but its role in AML is not well known. A study comparing 122 patients with de novo AML with healthy patients showed downregulated miR-34c in AML in the first group (95). However, in another study low expression of miR-34c is associated with dismal prognosis and poor response to treatment (96).
It is important to point out that miR-34 mimic did not induce side effects in mice (97) and was considered to be the first miR mimics to reach the clinic. Therapeutic delivery of a mimic of naturally occurring miR-34, MRX34, promoted tumor-infiltrating lymphocytes and reduced CD8+PD1+ cells in vivo (93). Furthermore, MRX34 plus radiotherapy increased CD8+ cell numbers more than therapy alone, and miR-34a delivery reduced the numbers of radiation-induced macrophages and T-regulatory cells (93). Still, a clinical trial of MRX34 (NCT01829971) in refractory advanced solid tumors (such as histologically confirmed viral related hepatocellular, lung cancer, melanoma, ovarian, bladder cancer, sarcoma) was stopped by Mirna Therapeutics, following multiple immune-related severe adverse events (5 cases). It remains to see if the miR-34a replacement combined with other anticancer agents could be a valuable therapeutic option (51, 95, 96).
miR-124
In vitro and in vivo studies showed that miR-124 (124-1, 124-2, 124-3) has several molecular targets, including STAT3, proinflammatory cytokines, cyclin-dependent kinase CDK4, CDK6, EZH2, C/EBP-α, and PU.1 (27, 29, 98). miR-124 is a potent tumor suppressor pro-apoptotic miR in several cancers. Downregulation of miR-124 by promoter hypermethylation has been observed in several types of malignancies (99). The promoter of miR-124-1 was homozygously methylated in 15% of AML samples and unmethylated in normal controls (100).
Chen et al. reported that in AML miR-124-1 is underexpressed in 36% of AML patients and is better expressed in the patients with t(15;17) than in others (62% versus 30%) (101). However, there was a trend in AML patients with reduced miR-124-1 to have longer OS and RFS than those without miR-124-1 underexpression (101).
miR-124 expression is inhibited in myelodysplastic syndrome (MDS) and is upregulated in response to epigenetic treatment (EGT) (45, 102). The upregulation of miR-124 expression in response to single-agent EGT with either azacytidine (AZA) or the histone deacetylase inhibitor panobinostat (LBH589) reported in vitro in the HL60 leukemic cell line (45) resulted in inhibition of CDK4, CDK6 and EZH2 expression. Accordingly, combination with EGT led to a significant and additive inhibitory effect. PBMCs from patients which responded to the combination EGT for high risk MDS or AML demonstrated in vitro significant induction of miR-124 and inhibition of CDK4 and CDK6 (and a trend to inhibit EZH2) mRNA expression. These findings suggest miRNA-124 is a potential biomarker of early response to EGT in myeloid malignancies and a possible therapeutic target (45).
miR-125a-5p
miR-125 family consists of miR-125a and miR-125b and regulates hematopoiesis (2). Both miR-125a and miR-125b are overexpressed in different lymphoid and myeloid malignancies (2, 103), but both are also present in solid cancers (104). Different studies suggest that 15%–25% of AML cases overexpress miR-125b. In a knock-in mouse model study, miR-125b was associated with leukemogenesis –via MLL-AF9 – and suppressed apoptosis (2). The highest expression of miR-125a found was in hematopoietic stem cells and expression gradually decreases with differentiation (103). In solid cancers, downregulation of miR-125a is associated with higher invasion, lymph node metastasis and tumor size. In AML low expression of miR-125a is associated with decreased blast count, higher count of leukocytes, platelets, monocytes and promonocytes and with M5 and M3 FAB subtypes (104). The action of both IFN (IFN-β or IFN-γ) and IL-4 consisted of repressing miR-125a-5p accumulation during human monocyte maturation to macrophages (33) and IL-4-dependent repression in alternative activated macrophages occurred via the IL-4Rα-STAT6 signaling pathway (105). miR-125a-5p and miR-125b have been reported to down-regulate oncogenic SIRT7, a member of the sirtuin family of NAD+-dependent histone/protein deacetylase (106).
In a study of 122 newly diagnosed AML samples Garzon et al. reported that miR-125a is downregulated (39) compared to healthy donors. In another AML study (without a healthy control group), miR-125a expression was downregulated in CN-AML. miR-125a expression was most decreased in AML with favourable and intermediate prognostic and associated with decreased survival (104). Ufkin et al. demonstrated in their study on leukemia NB4 cells that miR-125a is transcriptionally suppressed by methylation, and decitabine increased both precursor and mature miR-125a (107).
Mahmud et al. suggested the possible use of the tyrosine kinase inhibitor erlotinib in a subset of adult AML patients that had high epidermal growth factor receptor (EGFR) and EGFR phosphorylation levels relative to normal CD34+ cells measured by RPPA. These studies suggest that ErbB inhibitors are promising therapeutic agents for treating miR-125a-low AML or AML patients that have high levels of epidermal growth factor receptor (EGFR) (107). Treatment with either ectopic expression of miR-125a or inhibition of ErbB via Mubritinib, a selective inhibitor of ErbB2 phosphorylation, restored miR-125a expression in human acute promyelocytic leukemia NB4 cells with low miR-125a levels. As a result, it causes decreased cell proliferation, cell cycle progression and enhanced apoptosis (33, 39, 87, 105, 107).
miR-146a
The family consists of miR-146a and miR-146, which negatively mediates inflammation. Its expression, especially the one of miR-146a, is NF-kB-dependent and it is induced by TLR-ligands and pro-inflammatory cytokines, like TNFα and IL-1β (108–110). As mentioned above, through negative feedback, miR-146a and miR-146b possess anti-inflammatory effect, preventing immune overreaction and malignant transformation, miR-146a being negatively correlated with genes involved in the inflammatory response, such as CXCR4, TLR4, CCl23, Ltb4r, LYZ, IL8RB, MyD88 (111, 112). First of all, the CXCR4 expression in normal and leukemic monocytic cells is under the post-transcriptional control of miR-146a. HIF-1α stimulates the miR-146a expression, which lowers the level of CXCR4, while low expression of miR-146a in AML was correlated with high expression of CXCR4, which through his ligand CXCL-12, stimulates the cell harboring in the microenvironment of the BM and was found to be overexpressed in 25%–30% of AML cases (46, 113). However, it has been pointed out the fact that the hypoxia-mediated regulation of microRNA-146a is absent in acute monocytic leukemia, thus enhancing the expression of CXCR4 and providing the leukemic cells with chemoresistance (46).
Apart from mediating the CXCR4 expression, miR-146a appears to debilitate the effect of NF-κB, by repressing TNF receptor-associated factor 6 (TRAF6) and IL-1 receptor-associated kinase 1 (IRAK-1), key adaptor proteins of the NF-kB (108, 114). It was suggested that down-regulation of miR-146a could promote the progression of AML through TRAF6-mediated induction of NF-κB (114).
In one study of 53 patients with AML, miR-146a expression in bone marrow specimens was inversely correlated with OS (64). However, when compared to the control lot, miR-146a was downregulated in CD34 bone marrow cells isolated from patients with AML with a normal karyotype and even more decreased in patients who presented chromosome 5q deletion (del[5q]) as compared to controls. The 5q deletion is one of the most frequent cytogenetic anomaly found in AML and myelodysplastic syndrome. Re-expression of miR-146a inhibits survival and growth of leukemic cells, suggesting a role in the pathogenesis of AML (115, 116).
It was reported that reduced miR-146a expression in chromosome 5q deletion high-risk (HR) MDS (HR MDS)/AML patients and miR-146a-/- hematopoietic stem/progenitor cells (HSPCs) resulted in TRAF6/NF-kB activation. The increased survival and proliferation of HSPCs from miR-146alow HR MDS/AML is sustained by a neighboring haploid gene, SQSTM1 (p62), necessary for TRAF6-mediated NF-kB signaling. The disintegration of p62-TRAF6 complex resulted in cell-cycle blockade and apoptosis of MDS/AML cells. However, TRAF6 is needed for hematopoietic stem cell homeostasis in order to maintain a minimum level of IKKβ/NF-κB (115).
Human miR-146a and TRAF-interacting protein with forkhead-associated domain B (TIFAB) reside on chromosome 5q and are co-deleted in ∼80% of del(5q) MDS and AML. miR-146a inhibits TRAF6 protein expression and reduces its mRNA levels and TIFAB inhibits TRAF6 by destabilizing its protein (116, 117). The combined deletion of miR-146a and TIFAB, which are genes associated with del(5q) MDS/AML, resulted in the interdependent augmentation of TRAF6 expression and hematopoietic dysfunction (117).
An intimate link between high miR-146a-5p expression and better overall survival in 21 types of solid cancer was suggested by the first meta-analysis of the prognostic value of miR-146a-5p expression in diverse cancers. No correlation was found between miR-146a and AML, but only one study was included in this meta-analysis. More studies are needed to elucidate the role of miR-146a in AML (64).
miR-193
miR-193 is a suppressor miR important in controlling the self-renewal and the expansion of HSCs and progenitor cells by directly suppressing the expression of target genes, such as oncoprotein AML1/ETO, DNMT3a, HDAC3, c-kit, cyclin-D1. It also increases indirectly the activity of tumor suppressor PTEN, which negatively regulates PI3K activity (32, 55).
miR-193a levels were oppositely coordinated with the ones of c-kit proto-oncogene levels measured in 27 primary AML samples and 9 leukemia cell lines (47). The reconstitution of the miR-193a expression in AML cells containing c-kit mutation and/or overexpression, generated a significant cutback in c-kit expression at the RNA, as well as the protein levels, the inhibition of cell growth, and an augmentation of apoptosis and granulocytic differentiation. The inhibitor of DNA methylation 5-azacytidine-induced c-kit proto-oncogene reduction and it could be blocked to a certain degree by a miR-193a inhibitor, that will result in the cancellation of the antiproliferative and the pro-apoptotic effects of 5-azacytidine. These data suggest that miR-193a holds an important role in the formation of AML cells, bringing into the light the hope of a new therapeutic approach by upregulating miR-193a expression for c-kit-positive AML (55). miR-193a and PTEN were identified as targets for AML1/ETO and supported their role in the differentiation block of myeloid precursors in t(8;21) leukemias (32). High levels of miR-193a levels will induce G1 arrest, apoptosis, and will restore the differentiation of the leukemic cells differentiation (83). Li et al. associated the downregulation of miR-193a-3p with fusion protein AML1/ETO expressed in hematopoietic cells from patients diagnosed with t(8;21) AML (32).
The high c-kit expression was associated with the downregulation of miR-193a, which was found in patients with t(11q23)and in cytogenetically normal AML (CN-AML) (39, 118). RUNX1 was also up-regulated in CN-AML patients, as well as gene sets that regulate the cellular cycle and proliferation, particularly c-kit, FLT3, MYCN, MYB, MYC, and CDK6, RUNX1high were associated with poorer OS and EFS of CN-AML patients (118).
More recently miR-193b-3p levels in bone marrow cells were reported to be decreased in adult patients with CN-AML, especially in cases with FLT3 mutations, and the values were higher in the t(15;17) good prognosis subgroup compared to all AML patients (there was no healthy control group). Low expression of miR-193b-3p correlated with significantly worse OS and EFS in adult, as well as in paediatric patients (119). In vitro, miR-193b interfered with cell-cycle progression, differentiation and viability of leukemia cell lines and primary AML samples, by targeting key regulators of MAPK signaling (KIT, KRAS, SOS2) and CCND1 (119, 120). The authors suggest that due to its blocking effect on the entire MAPK signaling cascade, the miR-193b agonists would be highly efficient in AML (119).
miR-223
miR-223 has been reported to be both, an oncogenic regulator of the tumor progression and a tumor suppressor gene, but in AML it seems to act as the later and to be downregulated (48, 121).
miR-223 acts normally as an inhibitor regulator of granulocytic proliferation, along with C/EBPα, E2F1, and PU.1 (2, 122, 123) transcription factors, taking part in an autoregulatory negative feedback. During human monocyte-macrophage differentiation (under GM-CSF- or M-CSF-treatment), the expression of the miR-223 was found to be considerably decreased, which led to higher expression of the serine-threonine kinase IKKα (but not IKKβ or IKKγ) in macrophages, which further induced higher amounts of p52. In the context of decreased expression of RelB, a transcription factor found in untreated macrophages, high p52 expression blocked the basal transcription of both canonical and noncanonical NF-κB target genes (124).
It seems that miR-223 is a direct transcriptional target of the acute myeloid leukamia-specific fusion oncoprotein RUNX1/RUNX1T1 (AML1/ETO) (125). miR-223 levels are lower in t(8;21) AML patients than other AMLs. AML blast treatment with miR-223 oligonucleotides, RUNX1/RUNX1T1 inhibitors, or even hypomethylating agents, has shown to increase miR-223 levels and restore cell differentiation (125).
In another study, miR-223 expression was measured in blasts from 115 AML patients, which led to the correlation of low miR-223 expression levels with worse outcome, while higher miR-223 levels were associated with favourable prognosis (121). Furthermore, miR-223 was hierarchically expressed in AML subpopulations, with lower expression in leukemic stem cell-containing fractions (121).
The miR-223 expression was downregulated in mononuclear cells from bone marrow aspirate samples from patients with AML as compared to healthy subjects (48). In vitro experiments on AML cells pointed out that miR-223 inhibits cell proliferation and stimulates cell apoptosis and also the fact that miR-223 target, F-box and WD repeat domain containing 7 (FBXW7), binds proteins such as c-myc, Jun, cyclin-E, and Notch leading to their proteasomal degradations (48).
Conclusions
The onset and progression of malignancy in AML is not only dependant on intrinsic factors, but also on the tumor microenvironment, and macrophages thus play a crucial role in this setting. Tumors are invaded with by large amounts of macrophages expressing several active miRs, and their interfering with TAM activity could reprogram their phenotypes. The interest in the role of miRNA in cancer, including acute myeloid leukemia has seen a recent surge. In this review we focused on miRNA which, due to their tumor suppressor activity, could play a favourable role in adult AML. The available preclinical data suggest that these miRNA could be useful in deciphering the pathogenesis of AML and contribute to the diagnosis and prognositic assessment of AML. To validate the beneficial effect of the suppressor miRNA mimics, clinical trials are expected. The succesful future approach could be a combination of miRNA mimics with inhibitors of oncogenic miRNA and chemotherapy.
Author Contributions
All authors contributed to conception and design of the study. AN drafted the manuscript. AT, CBa, OM, LJ, CBl, CT, CBo and MG researched and wrote sections of the review. MZ co-ordinated the team and reviewed the sections and final article. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors CT. The handling editor declared a shared affiliation with several of the authors AN, CT, LJ, OM, CLB, CB, MZ at time of review.
References
1. Khanal N, Upadhyay Banskota S, Bhatt VR. Novel treatment paradigms in acute myeloid leukemia. Clin Pharmacol Ther (2020) 108(3):506–14. doi: 10.1002/cpt.1962
2. Pulikkan JA, Dengler V, Peramangalam PS, Peer Zada AA, Muller-Tidow C, Bohlander SK, et al. Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood (2010) 115(9):1768–78. doi: 10.1182/blood-2009-08-240101
3. Pulikkan JA, Peramangalam PS, Dengler V, Ho PA, Preudhomme C, Meshinchi S, et al. C/EBPalpha regulated microRNA-34a targets E2F3 during granulopoiesis and is down-regulated in AML with CEBPA mutations. Blood (2010) 116(25):5638–49. doi: 10.1182/blood-2010-04-281600
4. Katzerke C, Madan V, Gerloff D, Brauer-Hartmann D, Hartmann JU, Wurm AA, et al. Transcription factor C/EBPalpha-induced microRNA-30c inactivates Notch1 during granulopoiesis and is downregulated in acute myeloid leukemia. Blood (2013) 122(14):2433–42. doi: 10.1182/blood-2012-12-472183
5. O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci U S A (2009) 106(17):7113–8. doi: 10.1073/pnas.0902636106
6. O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol (2010) 10(2):111–22. doi: 10.1038/nri2708
7. Elton TS, Selemon H, Elton SM, Parinandi NL. Regulation of the MIR155 host gene in physiological and pathological processes. Gene (2013) 532(1):1–12. doi: 10.1016/j.gene.2012.12.009
8. Marcucci G, Maharry KS, Metzeler KH, Volinia S, Wu YZ, Mrozek K, et al. Clinical role of microRNAs in cytogenetically normal acute myeloid leukemia: miR-155 upregulation independently identifies high-risk patients. J Clin Oncol (2013) 31(17):2086–93. doi: 10.1200/JCO.2012.45.6228
9. Al-Matary YS, Botezatu L, Opalka B, Hones JM, Lams RF, Thivakaran A, et al. Acute myeloid leukemia cells polarize macrophages towards a leukemia supporting state in a Growth factor independence 1 dependent manner. Haematologica (2016) 101(10):1216–27. doi: 10.3324/haematol.2016.143180
10. Merien F. A journey with Elie Metchnikoff: from innate cell mechanisms in infectious diseases to quantum biology. Front Public Health (2016) 4:125. doi: 10.3389/fpubh.2016.00125
11. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity (2016) 44(3):450–62. doi: 10.1016/j.immuni.2016.02.015
12. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity (2014) 41(1):21–35. doi: 10.1016/j.immuni.2014.06.013
13. Zdrenghea MT, Makrinioti H, Muresan A, Johnston SL, Stanciu LA. The role of macrophage IL-10/innate IFN interplay during virus-induced asthma. Rev Med Virol (2015) 25(1):33–49. doi: 10.1002/rmv.1817
14. Feketea G, Bocsan CI, Popescu C, Gaman M, Stanciu LA, Zdrenghea MT. A Review of Macrophage MicroRNAs’ Role in Human Asthma. Cells (2019) 8(5):420. doi: 10.3390/cells8050420
15. Murray PJ, Smale ST. Restraint of inflammatory signaling by interdependent strata of negative regulatory pathways. Nat Immunol (2012) 13(10):916–24. doi: 10.1038/ni.2391
16. Wang Y, Han C-c, Cui D, Li Y, Ma Y, Wei W. Is macrophage polarization important in rheumatoid arthritis? Int Immunopharmacol (2017) 50:345–52. doi: 10.1016/j.intimp.2017.07.019
17. Murray PJ. Macrophage polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
18. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1 (LPS+) vs. classically and M2 (LPS–) vs. alternatively activated macrophages. Front Immunol (2019) 10:1084. doi: 10.3389/fimmu.2019.01084
19. Rőszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm (2015) 2015:1–16. doi: 10.1155/2015/816460
20. Graff JW, Dickson AM, Clay G, McCaffrey AP, Wilson ME. Identifying functional microRNAs in macrophages with polarized phenotypes. J Biol Chem (2012) 287(26):21816–25. doi: 10.1074/jbc.M111.327031
21. Yang X, Feng W, Wang R, Yang F, Wang L, Chen S, et al. Repolarizing heterogeneous leukemia-associated macrophages with more M1 characteristics eliminates their pro-leukemic effects. Oncoimmunology (2018) 7(4):e1412910. doi: 10.1080/2162402X.2017.1412910
22. Fayyad-Kazan H, Bitar N, Najar M, Lewalle P, Fayyad-Kazan M, Badran R, et al. Circulating miR-150 and miR-342 in plasma are novel potential biomarkers for acute myeloid leukemia. J Trans Med (2013) 11(1):1–10. doi: 10.1186/1479-5876-11-31
23. Squadrito ML, Etzrodt M, De Palma M, Pittet MJ. MicroRNA-mediated control of macrophages and its implications for cancer. Trends Immunol (2013) 34(7):350–9. doi: 10.1016/j.it.2013.02.003
24. Cobos Jimenez V, Bradley EJ, Willemsen AM, van Kampen AH, Baas F, Kootstra NA. Next-generation sequencing of microRNAs uncovers expression signatures in polarized macrophages. Physiol Genomics (2014) 46(3):91–103. doi: 10.1152/physiolgenomics.00140.2013
25. Cobos Jimenez V, Willemsen AM, Bradley EJ, Baas F, van Kampen AH, Kootstra NA. Next-generation sequencing of microRNAs in primary human polarized macrophages. Genom Data (2014) 2:181–3. doi: 10.1016/j.gdata.2014.06.019
26. Chai ZT, Zhu XD, Ao JY, Wang WQ, Gao DM, Kong J, et al. microRNA-26a suppresses recruitment of macrophages by down-regulating macrophage colony-stimulating factor expression through the PI3K/Akt pathway in hepatocellular carcinoma. J Hematol Oncol (2015) 8:56. doi: 10.1186/s13045-015-0150-4
27. Essandoh K, Li Y, Huo J, Fan GC. Mirna-Mediated Macrophage Polarization and its Potential Role in the Regulation of Inflammatory Response. Shock (2016) 46:122. doi: 10.1097/SHK.0000000000000604
28. Veremeyko T, Siddiqui S, Sotnikov I, Yung A, Ponomarev ED. IL-4/IL-13-dependent and independent expression of miR-124 and its contribution to M2 phenotype of monocytic cells in normal conditions and during allergic inflammation. PLoS One (2013) 8(12):e81774. doi: 10.1371/journal.pone.0081774
29. Wang X, Li Y, Dai Y, Liu Q, Ning S, Liu J, et al. Sulforaphane improves chemotherapy efficacy by targeting cancer stem cell-like properties via the miR-124/IL-6R/STAT3 axis. Sci Rep (2016) 6:36796. doi: 10.1038/srep36796
30. Schulert GS, Fall N, Harley JB, Shen N, Lovell DJ, Thornton S, et al. Monocyte MicroRNA Expression in Active Systemic Juvenile Idiopathic Arthritis Implicates MicroRNA-125a-5p in Polarized Monocyte Phenotypes. Arthritis Rheumatol (2016) 68(9):2300–13. doi: 10.1002/art.39694
31. Huang C, Liu XJ, QunZhou, Xie J, Ma TT, Meng XM, et al. MiR-146a modulates macrophage polarization by inhibiting Notch1 pathway in RAW264.7 macrophages. Int Immunopharmacol (2016) 32:46–54. doi: 10.1016/j.intimp.2016.01.009
32. Li Y, Gao L, Luo X, Wang L, Gao X, Wang W, et al. Epigenetic silencing of microRNA-193a contributes to leukemogenesis in t(8;21) acute myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood (2013) 121(3):499–509. doi: 10.1182/blood-2012-07-444729
33. Eigsti RL, Sudan B, Wilson ME, Graff JW. Regulation of activation-associated microRNA accumulation rates during monocyte-to-macrophage differentiation. J Biol Chem (2014) 289(41):28433–47. doi: 10.1074/jbc.M114.599316
34. Ying W, Tseng A, Chang RC, Morin A, Brehm T, Triff K, et al. MicroRNA-223 is a crucial mediator of PPARgamma-regulated alternative macrophage activation. J Clin Invest (2015) 125(11):4149–59. doi: 10.1172/JCI81656
35. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science (2017) 356(6337):513–9. doi: 10.1126/science.aal3535
36. Nishioka C, Ikezoe T, Pan B, Xu K, Yokoyama A. MicroRNA-9 plays a role in interleukin-10-mediated expression of E-cadherin in acute myelogenous leukemia cells. Cancer Sci (2017) 108(4):685–95. doi: 10.1111/cas.13170
37. Peng L, Zhang H, Hao Y, Xu F, Yang J, Zhang R, et al. Reprogramming macrophage orientation by microRNA 146b targeting transcription factor IRF5. EBioMedicine (2016) 14:837–96. doi: 10.1016/j.ebiom.2016.10.041
38. Dai CW, Bai QW, Zhang GS, Cao YX, Shen JK, Pei MF, et al. MicroRNA let-7f is down-regulated in patients with refractory acute myeloid leukemia and is involved in chemotherapy resistance of adriamycin-resistant leukemic cells. Leuk Lymphoma (2014) 55(7):1645–8. doi: 10.3109/10428194.2013.847936
39. Garzon R, Volinia S, Liu CG, Fernandez-Cymering C, Palumbo T, Pichiorri F, et al. MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood (2008) 111(6):3183–9. doi: 10.1182/blood-2007-07-098749
40. Jiang X, Hu C, Arnovitz S, Bugno J, Yu M, Zuo Z, et al. miR-22 has a potent anti-tumour role with therapeutic potential in acute myeloid leukaemia. Nat Commun (2016) 7:11452. doi: 10.1038/ncomms11452
41. Salvatori B, Iosue I, Djodji Damas N, Mangiavacchi A, Chiaretti S, Messina M, et al. Critical Role of c-Myc in Acute Myeloid Leukemia Involving Direct Regulation of miR-26a and Histone Methyltransferase EZH2. Genes Cancer (2011) 2(5):585–92. doi: 10.1177/1947601911416357
42. Gong JN, Yu J, Lin HS, Zhang XH, Yin XL, Xiao Z, et al. The role, mechanism and potentially therapeutic application of microRNA-29 family in acute myeloid leukemia. Cell Death Differ (2014) 21(1):100–12. doi: 10.1038/cdd.2013.133
43. Liu L, Ren W, Chen K. MiR-34a Promotes Apoptosis and Inhibits Autophagy by Targeting HMGB1 in Acute Myeloid Leukemia Cells. Cell Physiol Biochem (2017) 41(5):1981–92. doi: 10.1159/000475277
44. Pelosi A, Careccia S, Lulli V, Romania P, Marziali G, Testa U, et al. miRNA let-7c promotes granulocytic differentiation in acute myeloid leukemia. Oncogene (2013) 32(31):3648–54. doi: 10.1038/onc.2012.398
45. Liu H, Pattie P, Chandrasekara S, Spencer A, Dear AE. Epigenetic regulation of miRNA-124 and multiple downstream targets is associated with treatment response in myeloid malignancies. Oncol Lett (2016) 12(3):2175–80. doi: 10.3892/ol.2016.4912
46. Spinello I, Quaranta MT, Paolillo R, Pelosi E, Cerio AM, Saulle E, et al. Differential hypoxic regulation of the microRNA-146a/CXCR4 pathway in normal and leukemic monocytic cells: impact on response to chemotherapy. Haematologica (2015) 100(9):1160–71. doi: 10.3324/haematol.2014.120295
47. Gao XN, Lin J, Li YH, Gao L, Wang XR, Wang W, et al. MicroRNA-193a represses c-kit expression and functions as a methylation-silenced tumor suppressor in acute myeloid leukemia. Oncogene (2011) 30(31):3416–28. doi: 10.1038/onc.2011.62
48. Xiao Y, Su C, Deng T. miR-223 decreases cell proliferation and enhances cell apoptosis in acute myeloid leukemia via targeting FBXW7. Oncol Lett (2016) 12(5):3531–6. doi: 10.3892/ol.2016.5115
49. Rowe RG, Wang LD, Coma S, Han A, Mathieu R, Pearson DS, et al. Developmental regulation of myeloerythroid progenitor function by the Lin28b-let-7-Hmga2 axis. J Exp Med (2016) 213(8):1497–512. doi: 10.1084/jem.20151912
50. Zhou J, Chan ZL, Bi C, Lu X, Chong PS, Chooi JY, et al. LIN28B Activation by PRL-3 Promotes Leukemogenesis and a Stem Cell-like Transcriptional Program in AML. Mol Cancer Res (2017) 15(3):294–303. doi: 10.1158/1541-7786.MCR-16-0275-T
51. Banerjee S, Xie N, Cui H, Tan Z, Yang S, Icyuz M, et al. MicroRNA let-7c regulates macrophage polarization. J Immunol (2013) 190(12):6542–9. doi: 10.4049/jimmunol.1202496
52. Lu L, McCurdy S, Huang S, Zhu X, Peplowska K, Tiirikainen M, et al. Time Series miRNA-mRNA integrated analysis reveals critical miRNAs and targets in macrophage polarization. Sci Rep (2016) 6:37446. doi: 10.1038/srep37446
53. Zhang W, Liu H, Liu W, Liu Y, Xu J. Polycomb-mediated loss of microRNA let-7c determines inflammatory macrophage polarization via PAK1-dependent NF-kappaB pathway. Cell Death Differ (2015) 22(2):287–97. doi: 10.1038/cdd.2014.142
54. Kumar M, Sahu SK, Kumar R, Subuddhi A, Maji RK, Jana K, et al. MicroRNA let-7 modulates the immune response to Mycobacterium tuberculosis infection via control of A20, an inhibitor of the NF-kappaB pathway. Cell Host Microbe (2015) 17(3):345–56. doi: 10.1016/j.chom.2015.01.007
55. Gao SM, Xing CY, Chen CQ, Lin SS, Dong PH, Yu FJ. miR-15a and miR-16-1 inhibit the proliferation of leukemic cells by down-regulating WT1 protein level. J Exp Clin Cancer Res (2011) 30:110. doi: 10.1186/1756-9966-30-110
56. Abraham M, Klein S, Bulvik B, Wald H, Weiss ID, Olam D, et al. The CXCR4 inhibitor BL-8040 induces the apoptosis of AML blasts by down-regulating ERK BCL-2, MCL-1 and cyclin-D1 via altered miR-15a/16-1 expression. Leukemia (2017) 31:2336–46. doi: 10.1038/leu.2017.82
57. Kim KT, Carroll AP, Mashkani B, Cairns MJ, Small D, Scott RJ. MicroRNA-16 is down-regulated in mutated FLT3 expressing murine myeloid FDC-P1 cells and interacts with Pim-1. PLoS One (2012) 7(9):e44546. doi: 10.1371/journal.pone.0044546
58. Borthakur G, Nagler A, Ofran Y, Rowe JM, Altman JK, Frankfurt O, et al. BL-8040, a peptidic CXCR4 antagonist, induces leukemia cell death and specific leukemia cell mobilization in relapsed/refractory acute myeloid leukemia patients in an ongoing phase IIa clinical trial. Am Soc Hematol Washington DC (2014) 124:950. doi: 10.1182/blood.V124.21.950.950
59. Abraham M, Pereg Y, Bulvik B, Klein S, Mishalian I, Wald H, et al. Single dose of the CXCR4 antagonist BL-8040 induces rapid mobilization for the collection of human CD34+ cells in healthy volunteers. Clin Cancer Res (2017) 23(22):6790–801. doi: 10.1158/1078-0432.CCR-16-2919
60. Cho B-S, Zeng Z, Mu H, Wang Z, Konoplev S, McQueen T, et al. Antileukemia activity of the novel peptidic CXCR4 antagonist LY2510924 as monotherapy and in combination with chemotherapy. Blood (2015) 126(2):222–32. doi: 10.1182/blood-2015-02-628677
61. van Zandwijk N, Pavlakis N, Kao SC, Linton A, Boyer MJ, Clarke S, et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: a first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol (2017) 18(10):1386–96. doi: 10.1016/S1470-2045(17)30621-6
62. Shen C, Chen MT, Zhang XH, Yin XL, Ning HM, Su R, et al. The PU.1-Modulated MicroRNA-22 Is a Regulator of Monocyte/Macrophage Differentiation and Acute Myeloid Leukemia. PLoS Genet (2016) 12(9):e1006259. doi: 10.1371/journal.pgen.1006259
63. Jinlong S, Lin F, Yonghui L, Li Y, Weidong W. Identification of let-7a-2-3p or/and miR-188-5p as prognostic biomarkers in cytogenetically normal acute myeloid leukemia. PLoS One (2015) 10(2):e0118099. doi: 10.1371/journal.pone.0118099
64. Wang Y, Li Z, He C, Wang D, Yuan X, Chen J, et al. MicroRNAs expression signatures are associated with lineage and survival in acute leukemias. Blood Cells Mol Dis (2010) 44(3):191–7. doi: 10.1016/j.bcmd.2009.12.010
65. Cheng J, Guo S, Chen S, Mastriano SJ, Liu C, D’Alessio AC, et al. An extensive network of TET2-targeting MicroRNAs regulates malignant hematopoiesis. Cell Rep (2013) 5(2):471–81. doi: 10.1016/j.celrep.2013.08.050
66. Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell (2009) 137(6):1005–17. doi: 10.1016/j.cell.2009.04.021
67. Salvatori B, Iosue I, Mangiavacchi A, Loddo G, Padula F, Chiaretti S, et al. The microRNA-26a target E2F7 sustains cell proliferation and inhibits monocytic differentiation of acute myeloid leukemia cells. Cell Death Dis (2012) 3:e413. doi: 10.1038/cddis.2012.151
68. Jiang W, Min J, Sui X, Qian Y, Liu Y, Liu Z, et al. MicroRNA-26a-5p and microRNA-23b-3p up-regulate peroxiredoxin III in acute myeloid leukemia. Leuk Lymphoma (2015) 56(2):460–71. doi: 10.3109/10428194.2014.924115
69. Ngankeu A, Ranganathan P, Havelange V, Nicolet D, Volinia S, Powell BL, et al. Discovery and functional implications of a miR-29b-1/miR-29a cluster polymorphism in acute myeloid leukemia. Oncotarget (2018) 9(4):4354. doi: 10.18632/oncotarget.23150
70. Garzon R, Heaphy CE, Havelange V, Fabbri M, Volinia S, Tsao T, et al. MicroRNA 29b functions in acute myeloid leukemia. Blood (2009) 114(26):5331–41. doi: 10.1182/blood-2009-03-211938
71. Xiong Y, Li Z, Ji M, Tan AC, Bemis J, Tse JV, et al. MIR29B regulates expression of MLLT11 (AF1Q), an MLL fusion partner, and low MIR29B expression associates with adverse cytogenetics and poor overall survival in AML. Br J Haematol (2011) 153(6):753–7. doi: 10.1111/j.1365-2141.2011.08662.x
72. Wang F, Wang XS, Yang GH, Zhai PF, Xiao Z, Xia LY, et al. miR-29a and miR-142-3p downregulation and diagnostic implication in human acute myeloid leukemia. Mol Biol Rep (2012) 39(3):2713–22. doi: 10.1007/s11033-011-1026-5
73. Xu L, Xu Y, Jing Z, Wang X, Zha X, Zeng C, et al. Altered expression pattern of miR-29a, miR-29b and the target genes in myeloid leukemia. Exp Hematol Oncol (2014) 3:17. doi: 10.1186/2162-3619-3-17
74. Mott JL, Kurita S, Cazanave SC, Bronk SF, Werneburg NW, Fernandez-Zapico ME. Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-kappaB. J Cell Biochem (2010) 110(5):1155–64. doi: 10.1002/jcb.22630
75. Eyholzer M, Schmid S, Wilkens L, Mueller BU, Pabst T. The tumour-suppressive miR-29a/b1 cluster is regulated by CEBPA and blocked in human AML. Br J Cancer (2010) 103(2):275–84. doi: 10.1038/sj.bjc.6605751
76. Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood (2009) 113(25):6411–8. doi: 10.1182/blood-2008-07-170589
77. Amodio N, Rossi M, Raimondi L, Pitari MR, Botta C, Tagliaferri P, et al. miR-29s: a family of epi-miRNAs with therapeutic implications in hematologic malignancies. Oncotarget (2015) 6(15):12837–61. doi: 10.18632/oncotarget.3805
78. Bhise NS, Chauhan L, Shin M, Cao X, Pounds S, Lamba V, et al. MicroRNA-mRNA Pairs Associated with Outcome in AML: From In Vitro Cell-Based Studies to AML Patients. Front Pharmacol (2015) 6:324. doi: 10.3389/fphar.2015.00324
79. Amodio N, Di Martino MT, Foresta U, Leone E, Lionetti M, Leotta M, et al. miR-29b sensitizes multiple myeloma cells to bortezomib-induced apoptosis through the activation of a feedback loop with the transcription factor Sp1. Cell Death Dis (2012) 3:e436. doi: 10.1038/cddis.2012.175
80. Shah NM, Zaitseva L, Bowles KM, MacEwan DJ, Rushworth SA. NRF2-driven miR-125B1 and miR-29B1 transcriptional regulation controls a novel anti-apoptotic miRNA regulatory network for AML survival. Cell Death Differ (2015) 22(4):654–64. doi: 10.1038/cdd.2014.152
81. Liu S, Wu LC, Pang J, Santhanam R, Schwind S, Wu YZ, et al. Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell (2010) 17(4):333–47. doi: 10.1016/j.ccr.2010.03.008
82. Mims A, Walker AR, Huang X, Sun J, Wang H, Santhanam R, et al. Increased anti-leukemic activity of decitabine via AR-42-induced upregulation of miR-29b: a novel epigenetic-targeting approach in acute myeloid leukemia. Leukemia (2013) 27(4):871–8. doi: 10.1038/leu.2012.342
83. Amodio N, Bellizzi D, Leotta M, Raimondi L, Biamonte L, D’Aquila P, et al. miR-29b induces SOCS-1 expression by promoter demethylation and negatively regulates migration of multiple myeloma and endothelial cells. Cell Cycle (2013) 12(23):3650–62. doi: 10.4161/cc.26585
84. Stapnes C, Ryningen A, Hatfield K, Oyan AM, Eide GE, Corbascio M, et al. Functional characteristics and gene expression profiles of primary acute myeloid leukaemia cells identify patient subgroups that differ in susceptibility to histone deacetylase inhibitors. Int J Oncol (2007) 31(6):1529–38. doi: 10.3892/ijo.31.6.1529
85. Blum W, Garzon R, Klisovic RB, Schwind S, Walker A, Geyer S, et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci U S A (2010) 107(16):7473–8. doi: 10.1073/pnas.1002650107
86. Blum W, Schwind S, Tarighat SS, Geyer S, Eisfeld AK, Whitman S, et al. Clinical and pharmacodynamic activity of bortezomib and decitabine in acute myeloid leukemia. Blood (2012) 119(25):6025–31. doi: 10.1182/blood-2012-03-413898
87. Huang X, Schwind S, Yu B, Santhanam R, Wang H, Hoellerbauer P, et al. Targeted delivery of microRNA-29b by transferrin-conjugated anionic lipopolyplex nanoparticles: a novel therapeutic strategy in acute myeloid leukemia. Clin Cancer Res (2013) 19(9):2355–67. doi: 10.1158/1078-0432.CCR-12-3191
88. Pigazzi M, Manara E, Bresolin S, Tregnago C, Beghin A, Baron E, et al. MicroRNA-34b promoter hypermethylation induces CREB overexpression and contributes to myeloid transformation. Haematologica (2013) 98(4):602–10. doi: 10.3324/haematol.2012.070664
89. Zhao J, Lammers P, Torrance CJ, Bader AG. TP53-independent function of miR-34a via HDAC1 and p21(CIP1/WAF1.). Mol Ther (2013) 21(9):1678–86. doi: 10.1038/mt.2013.148
90. Hermeking H. The mir-34 family in cancer and apoptosis. Cell Death Differ (2010) 17(2):193–9. doi: 10.1038/cdd.2009.56
91. Di Martino MT, Campani V, Misso G, Gallo Cantafio ME, Gulla A, Foresta U, et al. In vivo activity of miR-34a mimics delivered by stable nucleic acid lipid particles (SNALPs) against multiple myeloma. PLoS One (2014) 9(2):e90005. doi: 10.1371/journal.pone.0090005
92. Wang X, Li J, Dong K, Lin F, Long M, Ouyang Y, et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signall (2015) 27(3):443–52. doi: 10.1016/j.cellsig.2014.12.003
93. Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst (2016) 108(1):1–9. doi: 10.1093/jnci/djv303
94. Wang S, Wang T, Li MZ, Cheng XL, Li XL. Expression of microRNA miR-34a inhibits leukemia stem cells and its metastasis. Eur Rev Med Pharmacol Sci (2016) 20(13):2878–83.
95. Yang DQ, Zhou JD, Wang YX, Deng ZQ, Yang J, Yao DM, et al. Low miR-34c expression is associated with poor outcome in de novo acute myeloid leukemia. Int J Lab Hematol (2017) 39(1):42–50. doi: 10.1111/ijlh.12566
96. Peng D, Wang H, Li L, Ma X, Chen Y, Zhou H, et al. miR-34c-5p promotes eradication of acute myeloid leukemia stem cells by inducing senescence through selective RAB27B targeting to inhibit exosome shedding. Leukemia (2018) 32(5):1180–8. doi: 10.1038/s41375-018-0015-2
97. Bader AG. miR-34 - a microRNA replacement therapy is headed to the clinic. Front Genet (2012) 3:120. doi: 10.3389/fgene.2012.00120
98. Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat Med (2011) 17(1):64–70. doi: 10.1038/nm.2266
99. Gambari R, Brognara E, Spandidos DA, Fabbri E. Targeting oncomiRNAs and mimicking tumor suppressor miRNAs: Nuew trends in the development of miRNA therapeutic strategies in oncology (Review). Int J Oncol (2016) 49:5–32. doi: 10.3892/ijo.2016.3503
100. Wong KY, So CC, Loong F, Chung LP, Lam WW, Liang R, et al. Epigenetic inactivation of the miR-124-1 in haematological malignancies. PLoS One (2011) 6(4):e19027. doi: 10.1371/journal.pone.0019027
101. Chen XX, Lin J, Qian J, Qian W, Yang J, Ma JC, et al. Dysregulation of miR-124-1 predicts favorable prognosis in acute myeloid leukemia. Clin Biochem (2014) 47(1-2):63–6. doi: 10.1016/j.clinbiochem.2013.09.020
102. Marcucci G, Mrózek K, Radmacher MD, Garzon R, Bloomfield CD. The prognostic and functional role of microRNAs in acute myeloid leukemia. Blood J Am Soc Hematol (2011) 117(4):1121–9. doi: 10.1182/blood-2010-09-191312
103. Ganan-Gomez I, Wei Y, Yang H, Pierce S, Bueso-Ramos C, Calin G, et al. Overexpression of miR-125a in myelodysplastic syndrome CD34+ cells modulates NF-κB activation and enhances erythroid differentiation arrest. PLoS One (2014) 9(4):e93404. doi: 10.1371/journal.pone.0093404
104. Yang M, Tang X, Wang Z, Wu X, Tang D, Wang D. miR-125 inhibits colorectal cancer proliferation and invasion by targeting TAZ. Biosci Rep (2019) 39(12):1–10. doi: 10.1042/BSR20190193
105. Czimmerer Z, Varga T, Kiss M, Vazquez CO, Doan-Xuan QM, Ruckerl D, et al. The IL-4/STAT6 signaling axis establishes a conserved microRNA signature in human and mouse macrophages regulating cell survival via miR-342-3p. Genome Med (2016) 8(1):63. doi: 10.1186/s13073-016-0315-y
106. Kim JK, Noh JH, Jung KH, Eun JW, Bae HJ, Kim MG, et al. Sirtuin7 oncogenic potential in human hepatocellular carcinoma and its regulation by the tumor suppressors MiR-125a-5p and MiR-125b. Hepatology (2013) 57(3):1055–67. doi: 10.1002/hep.26101
107. Ufkin ML, Peterson S, Yang X, Driscoll H, Duarte C, Sathyanarayana P. miR-125a regulates cell cycle, proliferation, and apoptosis by targeting the ErbB pathway in acute myeloid leukemia. Leuk Res (2014) 38(3):402–10. doi: 10.1016/j.leukres.2013.12.021
108. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A (2006) 103(33):12481–6. doi: 10.1073/pnas.0605298103
109. So AY, Zhao JL, Baltimore D. The Yin and Yang of microRNAs: leukemia and immunity. Immunol Rev (2013) 253(1):129–45. doi: 10.1111/imr.12043
110. Curtale G, Mirolo M, Renzi TA, Rossato M, Bazzoni F, Locati M. Negative regulation of Toll-like receptor 4 signaling by IL-10-dependent microRNA-146b. Proc Natl Acad Sci U S A (2013) 110(28):11499–504. doi: 10.1073/pnas.1219852110
111. Havelange V, Stauffer N, Heaphy CC, Volinia S, Andreeff M, Marcucci G, et al. Functional implications of microRNAs in acute myeloid leukemia by integrating microRNA and messenger RNA expression profiling. Cancer (2011) 117(20):4696–706. doi: 10.1002/cncr.26096
112. Croasdell A, Sime PJ, Phipps RP. Resolvin D2 decreases TLR4 expression to mediate resolution in human monocytes. FASEB J (2016) 30(9):3181–93. doi: 10.1096/fj.201600375R
113. Spinello I, Quaranta MT, Riccioni R, Riti V, Pasquini L, Boe A, et al. MicroRNA-146a and AMD3100, two ways to control CXCR4 expression in acute myeloid leukemias. Blood Cancer J (2011) 1(6):e26. doi: 10.1038/bcj.2011.24
114. Magilnick N, Reyes EY, Wang W-L, Vonderfecht SL, Gohda J, Inoue J-i, et al. miR-146a–Traf6 regulatory axis controls autoimmunity and myelopoiesis, but is dispensable for hematopoietic stem cell homeostasis and tumor suppression. Proc Natl Acad Sci (2017) 114(34):E7140–E9. doi: 10.1073/pnas.1706833114
115. Fang J, Barker B, Bolanos L, Liu X, Jerez A, Makishima H, et al. Myeloid malignancies with chromosome 5q deletions acquire a dependency on an intrachromosomal NF-kappaB gene network. Cell Rep (2014) 8(5):1328–38. doi: 10.1016/j.celrep.2014.07.062
116. Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat Med (2010) 16(1):49–58. doi: 10.1038/nm.2054
117. Varney ME, Niederkorn M, Konno H, Matsumura T, Gohda J, Yoshida N, et al. Loss of Tifab, a del(5q) MDS gene, alters hematopoiesis through derepression of Toll-like receptor-TRAF6 signaling. J Exp Med (2015) 212(11):1967–85. doi: 10.1084/jem.20141898
118. Fu L, Fu H, Tian L, Xu K, Hu K, Wang J, et al. High expression of RUNX1 is associated with poorer outcomes in cytogenetically normal acute myeloid leukemia. Oncotarget (2016) 7(13):15828–39. doi: 10.18632/oncotarget.7489
119. Bhayadia R, Krowiorz K, Haetscher N, Jammal R, Emmrich S, Obulkasim A, et al. Endogenous tumor suppressor microRNA-193b: therapeutic and prognostic value in acute myeloid leukemia. J Clin Oncol (2018) 36(10):1007–16. doi: 10.1200/JCO.2017.75.2204
120. Liu Y, Cheng Z, Pang Y, Cui L, Qian T, Quan L, et al. Role of microRNAs, circRNAs and long noncoding RNAs in acute myeloid leukemia. J Hematol Oncol (2019) 12(1):51. doi: 10.1186/s13045-019-0734-5
121. Gentner B, Pochert N, Rouhi A, Boccalatte F, Plati T, Berg T, et al. MicroRNA-223 dose levels fine tune proliferation and differentiation in human cord blood progenitors and acute myeloid leukemia. Exp Hematol (2015) 43(10):858–68.e7. doi: 10.1016/j.exphem.2015.05.018
122. Garzon R, Pichiorri F, Palumbo T, Visentini M, Aqeilan R, Cimmino A, et al. MicroRNA gene expression during retinoic acid-induced differentiation of human acute promyelocytic leukemia. Oncogene (2007) 26(28):4148–57. doi: 10.1038/sj.onc.1210186
123. Brauer-Hartmann D, Hartmann JU, Wurm AA, Gerloff D, Katzerke C, Verga Falzacappa MV, et al. PML/RARalpha-Regulated miR-181a/b Cluster Targets the Tumor Suppressor RASSF1A in Acute Promyelocytic Leukemia. Cancer Res (2015) 75(16):3411–24. doi: 10.1158/0008-5472.CAN-14-3521
124. Li T, Morgan MJ, Choksi S, Zhang Y, Kim Y-S, Liu Z-g. MicroRNAs modulate the noncanonical transcription factor NF-[kappa]B pathway by regulating expression of the kinase IKK[alpha] during macrophage differentiation. Nat Immunol (2010) 11(9):799–805. doi: 10.1038/ni.1918
Keywords: acute myeloid leukemia, non-coding RNAs, macrophages (M1/M2), macrophage polarization, tumor suppressor microRNA
Citation: Neaga A, Bagacean C, Tempescul A, Jimbu L, Mesaros O, Blag C, Tomuleasa C, Bocsan C, Gaman M and Zdrenghea M (2021) MicroRNAs Associated With a Good Prognosis of Acute Myeloid Leukemia and Their Effect on Macrophage Polarization. Front. Immunol. 11:582915. doi: 10.3389/fimmu.2020.582915
Received: 14 July 2020; Accepted: 24 November 2020;
Published: 15 January 2021.
Edited by:
Ioana Berindan Neagoe, Iuliu Haţieganu University of Medicine and Pharmacy, RomaniaReviewed by:
Nithyananda Thorenoor, Pennsylvania State University, United StatesFlorian Kuchenbauer, University of British Columbia, Canada
Liren Qian, Sixth Medical Center of PLA General Hospital, China
Copyright © 2021 Neaga, Bagacean, Tempescul, Jimbu, Mesaros, Blag, Tomuleasa, Bocsan, Gaman and Zdrenghea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mihnea Zdrenghea, bXpkcmVuZ2hlYUB1bWZjbHVqLnJv