 Elisa Eme-Scolan
Elisa Eme-Scolan Samantha J. Dando
Samantha J. Dando- 1École Normale Supérieure de Lyon, Université Claude Bernard Lyon I, Université de Lyon, Lyon, France
- 2Faculty of Health, Centre for Immunology and Infection Control, School of Biomedical Sciences, Queensland University of Technology (QUT), Brisbane, QLD, Australia
Microglia are specialized resident macrophages of the central nervous system (CNS) that have important functions during neurodevelopment, homeostasis and disease. This mini-review provides an overview of the current tools and approaches for studying microglia in vivo. We focus on tools for labeling microglia, highlighting the advantages and limitations of microglia markers/antibodies and reporter mice. We also discuss techniques for imaging microglia in situ, including in vivo live imaging of brain and retinal microglia. Finally, we review microglia depletion approaches and their use to investigate microglial function in CNS homeostasis and disease.
Introduction
The CNS (comprising the brain parenchyma, spinal cord, and neural retina) is populated with specialized resident macrophages called microglia. Microglia are derived from yolk sac progenitors (1) and are long lived cells that are maintained within the CNS through in situ self-renewal (2). Microglia continuously survey their surroundings via highly motile processes (3) and are exquisitely programmed to respond to changes in their microenvironment. In response to injury, infection or inflammation, microglia become “activated” and can shift into numerous functional states to elicit innate immune responses. In addition to performing immune functions, microglia are intimately involved in neurodevelopment and maintaining homeostasis of the healthy CNS. Some of their “non-immune” functions include: phagocytosing apoptotic/dead neural cells and debris (4); supporting neurogenesis, neuronal development and neural circuit assembly (5–7); inducing synapse formation (8); maintaining synaptic structure and function (9); synaptic pruning (10, 11); and maintaining neurons via the formation of somatic junctions (12).
The heterogeneous states of activated microglia exist on a continuum ranging from neuroprotective to neurotoxic/pathogenic (13). There is increasing evidence, largely from animal studies, that uncontrolled activated microglia contribute to the pathogenesis of a range of neurological and ocular diseases, including Alzheimer's disease (AD) (14), multiple sclerosis (15), Parkinson's disease (16), Huntington's disease (17), Amyotrophic Lateral Sclerosis (ALS) (18), neuromyelitis optica (19) and autoimmune uveitis (20). However, protective disease-associated microglia have also been described in AD and ALS (21), and may also exist in retinal degeneration (22). Despite the ongoing debate regarding the protective vs. pathogenic role of microglia, they are clearly involved in a wide range of CNS diseases and display a high level of plasticity.
Microglia are the subject of intense research efforts; however, there are several challenges associated with studying these cells. Challenge 1: microglia cultured in vitro do not recapitulate in vivo microglia in their physiological environment. Although important advances have been made to develop new microglia culture methods, including serum-free culture conditions and iPSC-derived microglia [reviewed in (23–25)], in vitro approaches that reflect microglia within their immune-privileged neural environment are still lacking. Challenge 2: when studying microglia in vivo, manipulation of the CNS (for example, preparing brain slices) can lead to injury and subsequent microglia activation (26), which is a limitation for studying microglia in their physiological state. Challenge 3: microglia share overlapping markers with other myeloid cells (27). It is essential to differentiate microglia from border-associated macrophages (BAMs), which reside within the meninges, choroid plexus and perivascular spaces of the brain, and the choroid that lies adjacent to the retina. Similarly, microglia must also be distinguished from circulating myeloid cells that infiltrate the CNS during neuroinflammation.
In recent years, significant progress has been made to address these challenges by developing new cellular and molecular tools for microglia research. In this mini-review we discuss the current “microglia tool kit” for in vivo research (summarized in Figure 1 and Table 1), and how recently developed approaches can be used to overcome some of the above challenges.
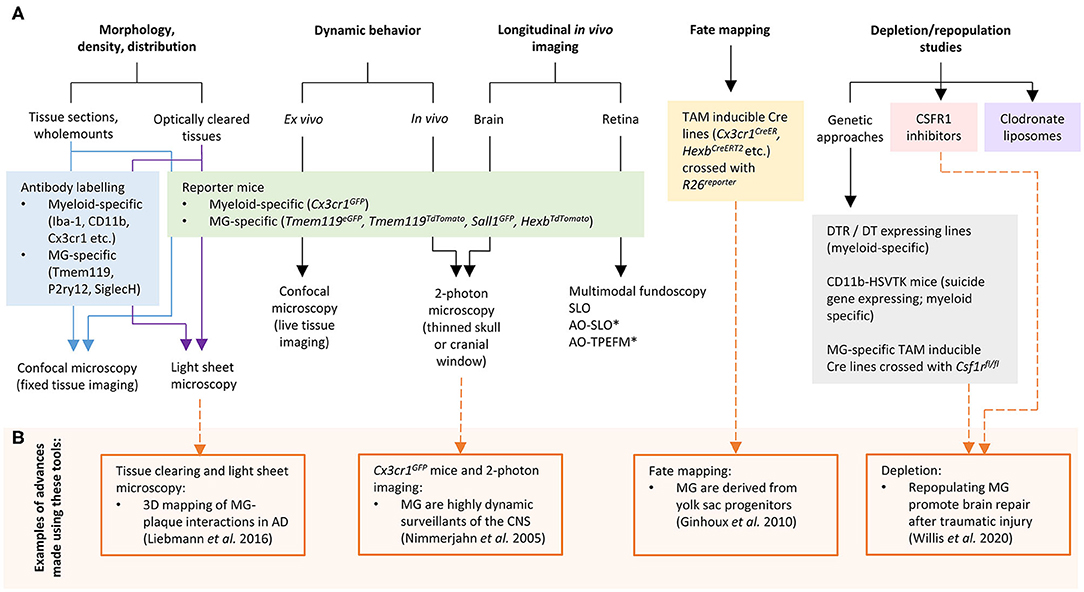
Figure 1. (A) Tools and approaches for studying microglia in vivo. (B) Examples of advances in microglia research that have been made using these tools. AD, Alzheimer's disease; AO, adaptive optics; CSF1R, colony stimulating factor 1 receptor; DT, diphtheria toxin; DTR, diphtheria toxin receptor; MG, microglia; SLO, scanning laser ophthalmoscopy; TPEFM, two-photon excitation fluorescence microscopy. *can also be used for in vivo retinal microglia dynamic behavior studies.
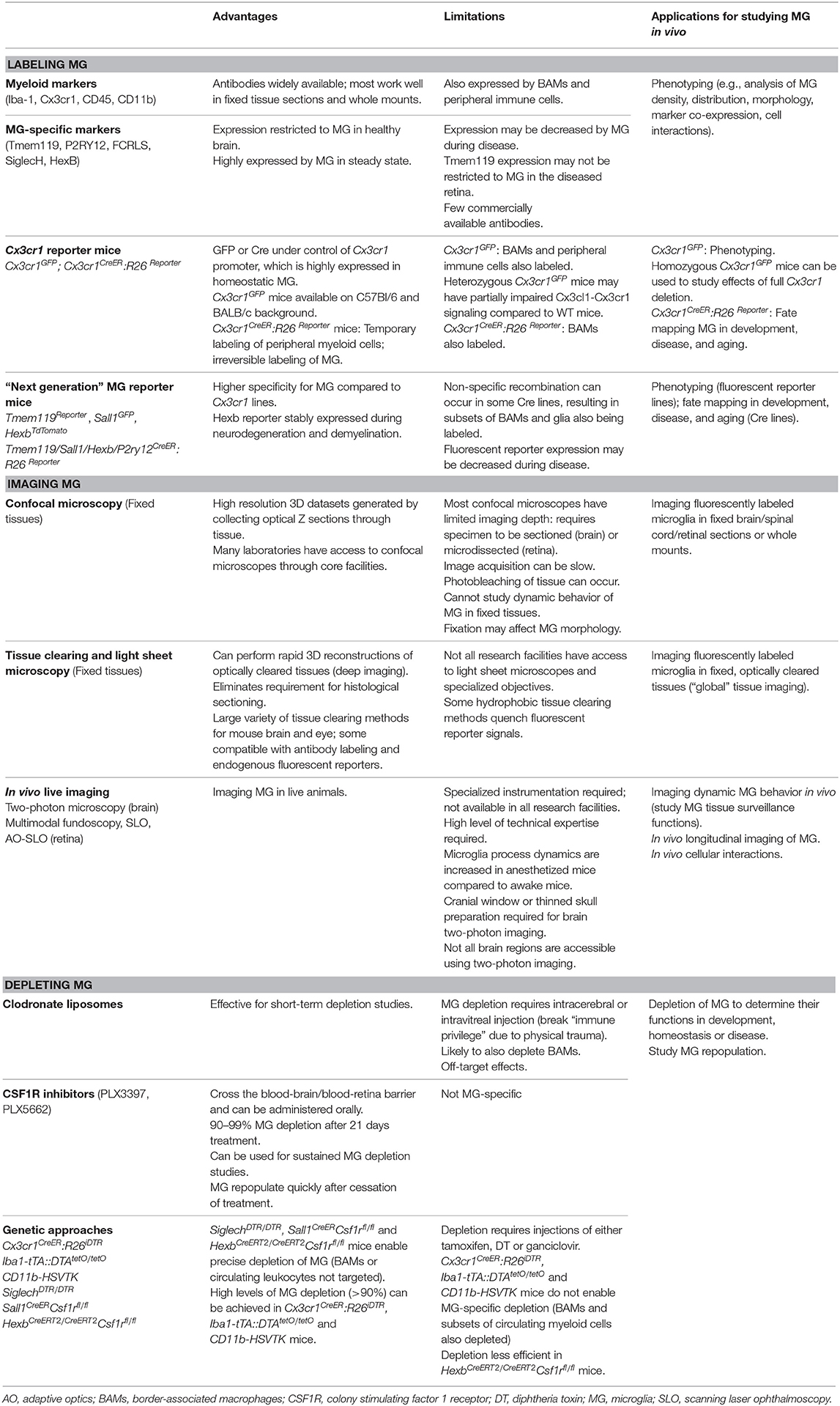
Table 1. Advantages, limitations, and applications of tools to study microglia in vivo.
Tools for Labeling Microglia in the CNS
Markers and Antibodies
Immunohistochemistry and flow cytometry are common techniques in neuroimmunology. Traditionally, Iba-1 antibodies have been used to label/stain microglia using immunohistochemistry; however, Iba-1 is also expressed by BAMs and subsets of peripheral myeloid cells (28, 29). During neuroinflammation, peripheral Iba-1+ myeloid cells that invade the CNS adopt a similar morphology to activated microglia (30), therefore microglia cannot be definitively distinguished from infiltrating leukocytes based solely on Iba-1 expression. Microglia also express several other markers that are common to BAMs and peripheral myeloid cells, including CD45, CD11b, CD68, Cx3cr1, F4/80, and CSF1R (31). Using flow cytometry, it is possible to distinguish microglia from other leukocytes based on their unique expression profile of selected surface markers. For example, microglia in the healthy CNS express low/intermediate levels of CD45, whereas BAMs and peripheral immune cells are CD45hi. Whether microglia retain low/intermediate CD45 expression during neuroinflammation is debated and appears to be disease- and CNS region-dependent. O'Koren et al. reported that retinal microglia retain a CD45lo phenotype in a light injury model and can be distinguished from infiltrating myeloid cells based on their unique CD45lo CD11clo F4/80lo I-A/I-E− signature (32). In contrast, Plemel et al. demonstrated that spinal cord microglia increased CD45 expression in a demyelination model and suggested that classical markers (such as CD45 and Cx3cr1) are less sensitive for distinguishing activated microglia from infiltrating myeloid cells in this model (33). Therefore, more recent studies have focused on identifying microglia-specific markers that can reliably distinguish microglia from other leukocytes during health and disease.
Bulk RNA-seq studies have identified several highly expressed genes that constitute the “microglia homeostatic signature” (28, 34–36). These signature genes, including Tmem119, P2ry12, Olfml3, Hexb, Fcrls, Siglech, Tgfbr1, Gpr34, Sall1, and others, have been the basis for the development of a range of new tools for microglia research in recent years, including microglia-specific antibodies. Tmem119 antibodies were developed by Bennett et al. for specific labeling of microglia using immunohistochemistry and flow cytometry (37). Tmem119 is expressed by mouse and human microglia but not BAMs or peripheral immune cells, and commercially available Tmem119 antibodies are now widely used. Butovsky et al. generated antibodies to P2ry12 and FCRLS, and showed that these distinguished mouse microglia from infiltrating myeloid cells (34). Furthermore, Konishi et al. reported that SiglecH antibodies specifically labeled microglia in the mouse brain but not BAMs or infiltrating monocytes (38). Microglia signature genes have also recently been used as targets for single molecule fluorescence in situ hybridization (smFISH) using RNAscope assays. Hammond et al. used smFISH (alongside single cell RNA-seq) to study microglia throughout the mouse lifespan and demonstrated that (i) microglia universally expressed FCRLS and (ii) diverse subtypes of microglia with unique spatial gene expression signatures exist in the developing, aged, and injured mouse brain (39).
The development of microglia-specific antibodies is useful for labeling these cells during homeostatic conditions. However, these markers may not be reliable for the identification of microglia during development and disease. For example, embryonic microglia do not express Tmem119, and expression of this protein by all microglia does not occur until postnatal day 14 in mice (37). This limits the use of Tmem119 antibodies during development and early postnatal stages. Furthermore, Tmem119 immunoreactivity may not be restricted to microglia in the diseased retina, as Su et al. reported that other cell types (such as Müller cells) may become Tmem119+ in a choroidal neovascularization model (40). Although further studies are required to validate these findings in other experimental models, Tmem119 antibodies should be used cautiously in the diseased retina. Moreover, microglial homeostatic genes including Tmem119, P2ry12, and Siglech are reported to be downregulated in CNS diseases (21, 41, 42), which may further limit their use. This is likely to be disease-dependent however, as some studies have shown stable expression of microglia homeostatic genes during disease (43). Given the range of markers and antibodies that can be used to identify microglia, the choice of targets needs to be carefully considered for each research question.
Reporter Mice
The fractalkine receptor gene Cx3cr1 is the basis of several reporter mouse lines that are widely used in microglia research. In Cx3cr1GFP mice, microglia express GFP under the control of the Cx3cr1 promoter (44). Homeostatic microglia express high levels of Cx3cr1 and are therefore strongly GFP+, which is extremely useful for identifying microglia within tissues. One limitation of these mice is that they are either heterozygous (Cx3cr1GFP/+) or deficient (Cx3cr1GFP/GFP) for Cx3cr1, and therefore neuron-microglia communication (mediated by Cx3cl1-Cx3cr1 signaling) is impaired compared to wild type mice, which can lead to reduced cognitive and synaptic function (45). Another important caveat is that Cx3cr1 is also expressed by BAMs, peripheral monocytes, DCs and NK cells (44), therefore GFP expression is not restricted to microglia. In contrast, tamoxifen inducible Cx3cr1CreER mice crossed with R26Reporter mice are a useful tool to distinguish microglia and CNS-infiltrating myeloid cells (32, 46). In these mice, CreER is constitutively expressed under the control of the Cx3cr1 promoter, whereas R26Reporter expression is dependent on Cre recombination. Upon tamoxifen administration, Cre is activated in Cx3cr1 expressing cells and a stop codon controlling R26Reporter expression is excised, resulting in Cx3cr1+ cells becoming R26Reporter+ (i.e., both tissue resident macrophages and circulating myeloid cells express R26Reporter). Due to the temporary activation of Cre and the turnover of circulating myeloid cells, several weeks after tamoxifen induction blood-borne myeloid cells lose R26Reporterexpression. In contrast, R26Reporter labeling is irreversible in long-lived tissue resident macrophages, including microglia (32, 46). However, BAMs are also long-lived resident cells that retain expression of the tamoxifen-induced reporter using this Cx3cr1CreER approach (47), and additional means are required to distinguish microglia from BAMs.
Recently described reporter mice have taken advantage of microglia-specific signature genes. Tmem119eGFP (48), Tmem119TdTomato (49), Sall1GFP (50, 51), and HexbTdTomato (52) mice are knock-in strains in which expression of fluorescent reporter proteins is largely restricted to microglia. Although Tmem119, Sall1, and other microglia signature genes are typically downregulated during CNS disease, mouse microglia were recently reported to maintain stable Hexb expression in models of neurodegeneration and demyelination, suggesting that HexbTdTomato mice may be suitable for consistent bright labeling of microglia in both homeostasis and CNS disease (52). In addition to the Cx3cr1CreER mice described above, several other tamoxifen inducible Cre lines now also exist for microglia fate mapping and genetic manipulation. These include Tmem119CreERT2 (48), Sall1CreER (50), HexbCreERT2 (52) and P2ry12CreER (53). Although these mouse strains have a much higher specificity for microglia compared to Cx3cr1CreER mice, they still have some limitations. For example, Tmem119CreERT2 and P2ry12CreER mice are not 100% microglia specific as recombination also occurs in some BAM subsets after tamoxifen administration. In addition, recombination is detected in glial cells in Sall1CreER mice (52, 54). Of the available tamoxifen inducible Cre lines, HexbCreERT2 mice are reported to have the highest microglia specificity, although not all microglia are targeted (brain region dependent) and recombination also occurs in a very small percentage of perivascular macrophages (52). Despite these limitations, these “next generation” reporter mice are an excellent resource for the neuroimmunology research community.
Techniques for Imaging Microglia In Situ
Confocal Microscopy of Fixed and Fresh Tissue
The ability to label microglia using the above approaches enables researchers to visualize these cells in situ. Confocal laser scanning microscopy is frequently used to image fluorescently labeled microglia in tissue sections (fixed), retinal wholemounts (fixed or fresh) and organotypic brain slices (fresh) to investigate microglial density, morphology, distribution, cellular interactions and dynamic behavior. However, there are limitations to these approaches. Fixation obviously preserves the tissue but makes it impossible to study the dynamism of microglia. Fixation procedures can also have effects on microglia morphology (55), which may confound some experiments. On the other hand, the dynamic behavior of microglia can be observed in fresh retinal wholemounts (56) and brain slices (57) but as tissues are placed in artificial medium for live imaging, microglia are likely to sense perturbations within their environment. In addition, the process of preparing brain slices and retinal wholemounts for live imaging can cause physical trauma. Therefore, microglia observed using live ex vivo approaches may not recapitulate their in vivo physiological counterparts.
Tissue Clearing and Light Sheet Microscopy
Tissue clearing techniques coupled with light sheet microscopy can be used to visualize microglia within intact transparent CNS tissues. Tissue clearing methods [reviewed in detail in (58)] make tissues transparent, resulting in minimal light scattering and enabling deep imaging and 3D reconstruction of tissues (including brains and eyes). This allows unbiased global investigation of tissues and eliminates the requirement to perform histological sectioning, which is a major advantage of this technique. A range of tissue clearing methods (including hydrophobic, hydrophilic and hydrogel-based methods) can be used to “clear” fixed CNS tissues (58, 59). Depending on the clearing method, tissues can be labeled with antibodies, nanobodies and lectins prior to clearing for visualization of cells and structures of interest. For example, Liebmann et al. used iDISCO clearing and Iba-1 immunolabeling to reconstruct the 3D interactions between brain microglia, vasculature and amyloid-β plaques in a mouse model of Alzheimer's disease (60). Some hydrophobic tissue clearing methods [such as 3DISCO (61)] rapidly quench fluorescent reporter protein signals; however, several hydrophilic and hydrogel-based clearing approaches effectively preserve endogenous fluorescence and can be used to image microglia in fluorescent reporter mice (described above). For instance, Xu et al. developed the FACT tissue clearing method and applied this to Cx3cr1YFP mouse brains for deep imaging of YFP+ microglia (62). Using this method, the authors detected fluorescent signal to a Z depth of ~800 μm although the signal intensity was markedly diminished after ~550 μm (62).
Unlike the brain, the eye is naturally transparent. However, the retinal pigment epithelium, the interface between the retinal photoreceptors and the choroid, is highly melanized (darkly pigmented), which remains a challenge for standard clearing methods. Specialized clearing techniques including EyeCi (63), DEEP-Clear (64), and EyeDISCO (65) were developed to address this issue and enable light sheet imaging of retinas within intact eyeballs. However, to the best of our knowledge these techniques have not yet been used for retinal microglia studies.
In vivo Live Imaging of Microglia
Two-photon microscopy is a powerful technique that allows unparalleled imaging of microglia in live animals. Time-lapse in vivo two-photon imaging of Cx3cr1GFP mouse brains led to the seminal discovery that microglia are highly dynamic surveillants that constantly extend and retract their processes during homeostasis (3). Several studies have since used two-photon imaging to study microglial dynamism in CNS disease models and aging (66–68). Most two-photon microglia imaging studies are performed on mice under general anesthesia through either a cranial window or thinned skull preparation [reviewed in detail in (69)]. The major advantage of this approach is that it is performed in live animals and therefore the cells are observed within their physiological environment. However, there are some important limitations. Firstly, microglial arbor, surveillance territory and process dynamics are increased in anesthetized mice compared to awake mice (70, 71) and methods to image microglia in awake mice may be required for some studies. Secondly, cranial window surgery and skull thinning procedures can induce CNS damage and subsequent microglial activation (69). Thirdly, not all brain regions are accessible using this approach. It remains difficult to image deep brain regions, therefore most two-photon studies are performed on cortical microglia. Using HexbTdTomato mice, Masuda et al. demonstrated that it is possible to perform time lapse in vivo two-photon imaging of microglia to a depth of 500 μm from the surface of the brain (52).
Specialized tools are available for in vivo live imaging of retinal microglia. Multimodal fundoscopy and scanning laser ophthalmoscopy (SLO) enable longitudinal studies of microglia in fluorescent reporter mice (72, 73) but these methods do not have sufficient resolution for examination of microglia fine process morphology or dynamic behavior, nor do they provide any depth information. Adaptive optics (AO) is a recent advancement that corrects optical aberrations and significantly improves resolution. When combined with SLO, AO-SLO enables microglia within distinct retinal layers to be resolved in vivo (74). Miller et al. demonstrated that AO-SLO can be used to quantify the 3D distribution, morphology and dynamism of microglia within individual layers of the Cx3cr1GFP mouse retina (75). More recently, Qin et al. developed an adaptive optics two-photon excitation fluorescence microscopy (AO-TPEFM) system that provides further improvements in the resolution of in vivo retinal microglia imaging (76). Combined with microglia-specific fluorescent reporter mice, AO-SLO and AO-TPEFM imaging will be useful tools to examine in vivo longitudinal changes in microglia in retinal diseases.
In vivo microglia imaging in humans remains challenging due to a lack of specific tools but positron emission tomography (PET) combined with 18-kDa translocator protein (TPSO)-binding radioligands is used to non-specifically assess microglial activation and neuroinflammation in patients in vivo (77, 78). The use of this clinical neuroimaging technique in multiple sclerosis patients has been reviewed previously (77) and will not be discussed here.
Tools for Depleting Microglia
Clodronate Liposomes
To attribute in vivo functions to microglia, depletion studies are often required. One approach to deplete microglia in vivo involves clodronate liposomes. Clodronate liposomes are phagocytosed by macrophages and subsequently released into the cytosol, where clodronate inhibits mitochondrial ADP/ATP translocase and induces macrophage apoptosis (79). To deplete microglia, clodronate liposomes are injected intracerebrally (80, 81) or intravitreally (82) to bypass the blood-brain/blood-retina barrier. This approach is effective for short-term microglia depletion; however, intracerebral injections can induce physical trauma and break “immune privilege” of the CNS (83). Moreover, off-target effects may occur (84), and control liposomes (that do not contain clodronate) have been shown to non-specifically activate microglia (85). Intracerebral delivery of clodronate liposomes is also likely to deplete BAMs (in particular, perivascular macrophages), although the specificity of depletion following intracerebral administration of clodronate liposomes has not been investigated to the best of our knowledge. In contrast, intraventricular injection of clodronate liposomes results in selective depletion of BAMs and not microglia (86).
CSF1R Inhibitors
CSF1R inhibitors are effective for microglia depletion, as adult microglia depend on CSF1R for survival (87). Unlike CSF1R antibodies and clodronate liposomes, CSF1R inhibitors are small molecules that readily cross the blood-brain/blood-retina barrier and can be administered orally, either by oral gavage or mixed within standard rodent chow. PLX3397 and PLX5662 are the most commonly used CSF1R inhibitors for microglia depletion as these have high potency (PLX3397) (87) and high brain penetrance (PLX5662) (88). CSF1R inhibitors are effective to achieve long-term, sustained microglial depletion in vivo. However, this approach does not eliminate 100% of microglia. For example, PLX3397 depletes 90–99% of microglia from the mouse brain after 21 days of treatment (89). Microglial repopulation begins after CSF1R inhibitor treatment ceases, and it is thought that repopulating microglia are generated from the few microglial cells that are not eliminated by the treatment (72). In the mouse retina, repopulated microglia display similar dynamism to endogenous microglia, suggesting that repopulated microglia restore homeostatic tissue surveillance (72). Interestingly, the adult mouse brain only has capacity for a single microglia repopulation event, as following repeated PLX3397 depletion cycles microglia fail to repopulate the brain (90). Using PLX3397 depletion, Spangenberg et al. reported that microglial elimination improved contextual memory deficits in a mouse model (5xFAD) of Alzheimer's disease (91). Further studies demonstrated that sustained PLX5662 microglia depletion impaired plaque formation in 5xFAD mice (88, 92). As such, CSF1R inhibitors are being investigated as a potential treatment option for CNS diseases (93). However, studies involving CSF1R depletion strategies must be interpreted cautiously, as CSF1R inhibitors also likely target BAMs and some peripheral immune populations (93).
Genetic Approaches
Microglia depletion can also be achieved with genetic approaches. In Cx3cr1CreER:R26iDTR mice, microglia and other long-lived resident immune cells (but not circulating myeloid cells) express the diphtheria toxin receptor (DTR) following tamoxifen administration. Upon exposure to diphtheria toxin (DT), ~99% of microglia are depleted within 1 day, and low numbers of microglia are maintained in the CNS for up to 7 days post-DT (46). Iba1-tTA::DTAtetO/tetO mice also take advantage of a DT system. In these mice, selective expression of DT occurs in Iba-1+ cells after withdrawal of doxycycline. This results in ~90% depletion of microglia (94); however, Iba-1+ BAMs and peripheral myeloid cells are also likely to be targeted in these mice. Depletion systems based on the CD11b promoter also broadly target myeloid cells and are not microglia specific. In CD11b-HSVTK mice, expression of the suicide gene HSVTK is driven by the CD11b promoter. After administration of the drug ganciclovir, HSVTK converts ganciclovir to a toxic compound, which induces apoptosis of CD11b+ cells. Intracerebroventricular infusion of ganciclovir achieves high levels (<90%) of microglia depletion; however, this is short-lived as microglia robustly repopulate the CNS within 2 weeks of ganciclovir treatment (95).
Genetic depletion systems targeting microglia-specific genes offer a more precise approach to microglia elimination without affecting BAMs and circulating myeloid cells. Using SiglechDTR/DTR mice, Konishi et al. reported that ~80–85% of Iba-1+ CD206− microglia were depleted in the cortex and area postrema following intraperitoneal DT injection. In contrast, the numbers of perivascular macrophages, meningeal macrophages and infiltrating monocytes following nerve injury were unaffected (38). Tamoxifen inducible Cre lines also exist for specific microglia depletion, including Sall1CreERCsf1rfl/fl and HexbCreERT2/CreERT2Csf1rfl/fl mice. In these strains, the conditional Csf1r allele is deleted by tamoxifen inducible Cre expressed from the Sall1 or Hexb locus (50, 52). Whilst these strains specifically target microglia (and not BAMs or circulating myeloid cells), recombination appears to be less efficient that other genetic depletion systems. For example, in HexbCreERT2/CreERT2Csf1rfl/fl mice only 60% of microglia are depleted after tamoxifen injection (52). Moreover, in Sall1CreERCsf1rfl/fl mice small numbers of Sall1+ CD45− cells in the adult kidney and liver are also depleted (50). Whilst tamoxifen-induced depletion of these cells in adult mice does not result in overt inflammation in the serum, spleen, liver, and kidneys (50), it is possible there may be other effects in these peripheral organs.
Conclusion
Researchers have previously applied traditional immunological approaches to study microglia the CNS. Thanks to rapid advances in the neuroimmunology field, we now have a suite of tools and techniques for microglia research. The discovery of microglia-specific signature genes has elevated the choice of markers, antibodies and reporter mice for studying these cells in vivo. However, further research to identify microglia-specific markers that are stably expressed during neuroinflammation is required. The combination of microglia-specific labeling, powerful in vivo imaging, and precise depletion approaches is already accelerating discoveries relating to microglial biology. These tools provide opportunities to investigate microglial function and phenotype in vivo, and to understand the spatial and disease-dependent heterogeneity of these multitasking, multifunctional cells.
Author Contributions
EE-S and SD conceived and researched data for the article and wrote the review. All authors contributed to the article and approved the submitted version.
Funding
SD was supported by an Australian Research Council DECRA (DE180101075).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Antoine Corbin and Dr. Rejane Rua for insightful discussions and comments on early drafts of the manuscript, and the Master de Biologie of the École Normale Supérieure de Lyon for their support and guidance.
References
1. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
2. Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FMV. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. (2007) 10:1538–43. doi: 10.1038/nn2014
3. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
4. Márquez-Ropero M, Benito E, Plaza-Zabala A, Sierra A. Microglial corpse clearance: lessons from macrophages. Front Immunol. (2020) 11:506. doi: 10.3389/fimmu.2020.00506
5. Wallace J, Lord J, Dissing-Olesen L, Stevens B, Murthy VN. Microglial depletion disrupts normal functional development of adult-born neurons in the olfactory bulb. eLife. (2020) 9:e50531. doi: 10.7554/eLife.50531
6. Squarzoni P, Oller G, Hoeffel G, Pont-Lezica L, Rostaing P, Low D, et al. Microglia modulate wiring of the embryonic forebrain. Cell Rep. (2014) 8:1271–9. doi: 10.1016/j.celrep.2014.07.042
7. Diaz-Aparicio I, Paris I, Sierra-Torre V, Plaza-Zabala A, Rodríguez-Iglesias N, Márquez-Ropero M, et al. Microglia actively remodel adult hippocampal neurogenesis through the phagocytosis secretome. J Neurosci. (2020) 40:1453–82. doi: 10.1523/JNEUROSCI.0993-19.2019
8. Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, et al. Microglia contact induces synapse formation in developing somatosensory cortex. Nat Commun. (2016) 7:12540. doi: 10.1038/ncomms12540
9. Wang X, Zhao L, Zhang J, Fariss RN, Ma W, Kretschmer F, et al. Requirement for microglia for the maintenance of synaptic function and integrity in the mature retina. J Neurosci. (2016) 36:2827–42. doi: 10.1523/JNEUROSCI.3575-15.2016
10. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. (2011) 333:1456–8. doi: 10.1126/science.1202529
11. Tremblay M-È, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLOS Biol. (2010) 8:e1000527. doi: 10.1371/journal.pbio.1000527
12. Cserép C, Pósfai B, Lénárt N, Fekete R, László ZI, Lele Z, et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science. (2020) 367:528–37. doi: 10.1126/science.aax6752
13. Hellwig S, Heinrich A, Biber K. The brain's best friend: microglial neurotoxicity revisited. Front Cell Neurosci. (2013) 7:71. doi: 10.3389/fncel.2013.00071
14. Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med. (2019) 216:2546–61. doi: 10.1084/jem.20190980
15. Voet S, Prinz M, van Loo G. Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol Med. (2019) 25:112–23. doi: 10.1016/j.molmed.2018.11.005
16. Guo M, Wang J, Zhao Y, Feng Y, Han S, Dong Q, et al. Microglial exosomes facilitate α-synuclein transmission in Parkinson's disease. Brain. (2020) 143:1476–97. doi: 10.1093/brain/awaa090
17. Crapser JD, Ochaba J, Soni N, Reidling JC, Thompson LM, Green KN. Microglial depletion prevents extracellular matrix changes and striatal volume reduction in a model of Huntington's disease. Brain. (2020) 143:266–88. doi: 10.1093/brain/awz363
18. Christoforidou E, Joilin G, Hafezparast M. Potential of activated microglia as a source of dysregulated extracellular microRNAs contributing to neurodegeneration in amyotrophic lateral sclerosis. J Neuroinflamm. (2020) 17:135. doi: 10.1186/s12974-020-01822-4
19. Chen T, Lennon VA, Liu YU, Bosco DB, Li Y, Yi M-H, et al. Astrocyte-microglia interaction drives evolving neuromyelitis optica lesion. J Clin Invest. (2020) 130:4025–38. doi: 10.1172/JCI134816
20. Okunuki Y, Mukai R, Nakao T, Tabor SJ, Butovsky O, Dana R, et al. Retinal microglia initiate neuroinflammation in ocular autoimmunity. Proc Natl Acad Sci USA. (2019) 116:9989–98. doi: 10.1073/pnas.1820387116
21. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell. (2017) 169:1276–90.e17. doi: 10.1016/j.cell.2017.05.018
22. Yu C, Roubeix C, Sennlaub F, Saban DR. Microglia versus monocytes: distinct roles in degenerative diseases of the retina. Trends Neurosci. (2020) 43:433–49. doi: 10.1016/j.tins.2020.03.012
23. Guttenplan KA, Liddelow SA. Astrocytes and microglia: models and tools. J Exp Med. (2019) 216:71–83. doi: 10.1084/jem.20180200
24. Timmerman R, Burm SM, Bajramovic JJ. An overview of in vitro methods to study microglia. Front Cell Neurosci. (2018) 12:242. doi: 10.3389/fncel.2018.00242
25. Hasselmann J, Blurton-Jones M. Human iPSC-derived microglia: a growing toolset to study the brain's innate immune cells. Glia. (2020) 68:721–39. doi: 10.1002/glia.23781
26. Stence N, Waite M, Dailey ME. Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia. (2001) 33:256–66. doi: 10.1002/1098-1136(200103)33:3<256::AID-GLIA1024>3.0.CO;2-J
27. McMenamin PG, Saban DR, Dando SJ. Immune cells in the retina and choroid: two different tissue environments that require different defenses and surveillance. Prog Retin Eye Res. (2019) 70:85–98. doi: 10.1016/j.preteyeres.2018.12.002
28. Chiu IM, Morimoto ETA, Goodarzi H, Liao JT, O'Keeffe S, Phatnani HP, et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. (2013) 4:385–401. doi: 10.1016/j.celrep.2013.06.018
29. Dando SJ, Kazanis R, Chinnery HR, McMenamin PG. Regional and functional heterogeneity of antigen presenting cells in the mouse brain and meninges. Glia. (2019) 67:935–49. doi: 10.1002/glia.23581
30. Miró-Mur F, Pérez-de-Puig I, Ferrer-Ferrer M, Urra X, Justicia C, Chamorro A, et al. Immature monocytes recruited to the ischemic mouse brain differentiate into macrophages with features of alternative activation. Brain Behav Immun. (2016) 53:18–33. doi: 10.1016/j.bbi.2015.08.010
31. Dando SJ, Naranjo Golborne C, Chinnery HR, Ruitenberg MJ, McMenamin PG. A case of mistaken identity: CD11c-eYFP + cells in the normal mouse brain parenchyma and neural retina display the phenotype of microglia, not dendritic cells. Glia. (2016) 64:1331–49. doi: 10.1002/glia.23005
32. O'Koren EG, Mathew R, Saban DR. Fate mapping reveals that microglia and recruited monocyte-derived macrophages are definitively distinguishable by phenotype in the retina. Sci Rep. (2016) 6:20636. doi: 10.1038/srep20636
33. Plemel JR, Stratton JA, Michaels NJ, Rawji KS, Zhang E, Sinha S, et al. Microglia response following acute demyelination is heterogeneous and limits infiltrating macrophage dispersion. Sci Adv. (2020) 6:eaay6324. doi: 10.1126/sciadv.aay6324
34. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. (2014) 17:131–43. doi: 10.1038/nn.3599
35. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. (2012) 13:1118–28. doi: 10.1038/ni.2419
36. Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang L, Means TK, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. (2013) 16:1896–905. doi: 10.1038/nn.3554
37. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci USA. (2016) 113:E1738–46. doi: 10.1073/pnas.1525528113
38. Konishi H, Kobayashi M, Kunisawa T, Imai K, Sayo A, Malissen B, et al. Siglec-H is a microglia-specific marker that discriminates microglia from CNS-associated macrophages and CNS-infiltrating monocytes. Glia. (2017) 65:1927–43. doi: 10.1002/glia.23204
39. Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. (2019) 50:253–71.e6. doi: 10.1016/j.immuni.2018.11.004
40. Su N, März S, Plagemann T, Cao J, Schnittler H-J, Eter N, et al. Occurrence of transmembrane protein 119 in the retina is not restricted to the microglia: an immunohistochemical study. Trans Vis Sci Tech. (2019) 8:29. doi: 10.1167/tvst.8.6.29
41. Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun. (2015) 3:31. doi: 10.1186/s40478-015-0203-5
42. Butovsky O, Weiner HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. (2018) 19:622–635. doi: 10.1038/s41583-018-0057-5
43. van der Poel M, Ulas T, Mizee MR, Hsiao C-C, Miedema SSM, Adelia, et al. Transcriptional profiling of human microglia reveals grey–white matter heterogeneity and multiple sclerosis-associated changes. Nat Commun. (2019) 10:1139. doi: 10.1038/s41467-019-08976-7
44. Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, et al. Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. (2000) 20:4106–14. doi: 10.1128/MCB.20.11.4106-4114.2000
45. Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci. (2011) 31:16241–50. doi: 10.1523/JNEUROSCI.3667-11.2011
46. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, Lafaille JJ, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. (2013) 155:1596–609. doi: 10.1016/j.cell.2013.11.030
47. Goldmann T, Wieghofer P, Jordão MJC, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. (2016) 17:797–805. doi: 10.1038/ni.3423
48. Kaiser T, Feng G. Tmem119-EGFP and Tmem119-CreERT2 transgenic mice for labeling and manipulating microglia. eNeuro. (2019) 6:ENEURO.0448-18.2019. doi: 10.1523/ENEURO.0448-18.2019
49. Ruan C, Sun L, Kroshilina A, Beckers L, De Jager P, Bradshaw EM, et al. A novel Tmem119-tdTomato reporter mouse model for studying microglia in the central nervous system. Brain Behav Immun. (2020) 83:180–91. doi: 10.1016/j.bbi.2019.10.009
50. Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol. (2016) 17:1397–406. doi: 10.1038/ni.3585
51. Takasato M, Osafune K, Matsumoto Y, Kataoka Y, Yoshida N, Meguro H, et al. Identification of kidney mesenchymal genes by a combination of microarray analysis and Sall1-GFP knockin mice. Mech Dev. (2004) 121:547–57. doi: 10.1016/j.mod.2004.04.007
52. Masuda T, Amann L, Sankowski R, Staszewski O, Lenz M, d'Errico P, et al. Novel Hexb-based tools for studying microglia in the CNS. Nat Immunol. (2020) 21:802–15. doi: 10.1038/s41590-020-0774-6
53. McKinsey GL, Lizama CO, Keown-Lang AE, Niu A, Santander N, Larpthaveesarp A, et al. A new genetic strategy for targeting microglia in development and disease. eLife. (2020) 9:e54590. doi: 10.7554/eLife.54590
54. Chappell-Maor L, Kolesnikov M, Grozovski J, Kim J-S, Shemer A, Haimon Z, et al. Comparative analysis of CreER transgenic mice for the study of brain macrophages – a case study. Immunology. (2019) 50:353–62. doi: 10.1101/725218
55. Cătălin B, Stopper L, Bălşeanu T-A, Scheller A. The in situ morphology of microglia is highly sensitive to the mode of tissue fixation. J Chem Neuroanat. (2017) 86:59–66. doi: 10.1016/j.jchemneu.2017.08.007
56. Lee JE, Liang KJ, Fariss RN, Wong WT. Ex vivo dynamic imaging of retinal microglia using time-lapse confocal microscopy. Invest Ophthalmol Vis Sci. (2008) 49:4169–76. doi: 10.1167/iovs.08-2076
57. Dailey ME, Eyo U, Fuller L, Hass J, Kurpius D. Imaging microglia in brain slices and slice cultures. Cold Spring Harbor Protoc. (2013) 2013:pdb.prot079483-pdb.prot079483. doi: 10.1101/pdb.prot079483
58. Ueda HR, Ertürk A, Chung K, Gradinaru V, Chédotal A, Tomancak P, et al. Tissue clearing and its applications in neuroscience. Nat Rev Neurosci. (2020) 21:61–79. doi: 10.1038/s41583-019-0250-1
59. Wan P, Zhu J, Xu J, Li Y, Yu T, Zhu D. Evaluation of seven optical clearing methods in mouse brain. NPH. (2018) 5:035007. doi: 10.1117/1.NPh.5.3.035007
60. Liebmann T, Renier N, Bettayeb K, Greengard P, Tessier-Lavigne M, Flajolet M. Three-dimensional study of Alzheimer's disease hallmarks using the iDISCO clearing method. Cell Rep. (2016) 16:1138–52. doi: 10.1016/j.celrep.2016.06.060
61. Ertürk A, Becker K, Jährling N, Mauch CP, Hojer CD, Egen JG, et al. Three-dimensional imaging of solvent-cleared organs using 3DISCO. Nat Protoc. (2012) 7:1983–95. doi: 10.1038/nprot.2012.119
62. Xu N, Tamadon A, Liu Y, Ma T, Leak RK, Chen J, et al. Fast free-of-acrylamide clearing tissue (FACT)—an optimized new protocol for rapid, high-resolution imaging of three-dimensional brain tissue. Sci Rep. (2017) 7:9895 doi: 10.1038/s41598-017-10204-5
63. Henning Y, Osadnik C, Malkemper EP. EyeCi: optical clearing and imaging of immunolabeled mouse eyes using light-sheet fluorescence microscopy. Exp Eye Res. (2019) 180:137–45. doi: 10.1016/j.exer.2018.12.001
64. Pende M, Vadiwala K, Schmidbaur H, Stockinger AW, Murawala P, Saghafi S, et al. A versatile depigmentation, clearing, and labeling method for exploring nervous system diversity. Sci Adv. (2020) 6:aba0365. doi: 10.1126/sciadv.aba0365
65. Vigouroux RJ, Cesar Q, Chédotal A, Nguyen-Ba-Charvet KT. Revisiting the role of Dcc in visual system development with a novel eye clearing method. eLife. (2020) 9:e51275. doi: 10.7554/eLife.51275
66. Bayerl SH, Niesner R, Cseresnyes Z, Radbruch H, Pohlan J, Brandenburg S, et al. Time lapse in vivo microscopy reveals distinct dynamics of microglia-tumor environment interactions—a new role for the tumor perivascular space as highway for trafficking microglia. Glia. (2016) 64:1210–26. doi: 10.1002/glia.22994
67. Drost N, Houtman J, Cseresnyés Z, Niesner R, Rinnenthal J-L, Miller KR, et al. The Amyloid-beta rich CNS environment alters myeloid cell functionality independent of their origin. Sci Rep. (2020) 10:7152. doi: 10.1038/s41598-020-63989-3
68. Füger P, Hefendehl JK, Veeraraghavalu K, Wendeln A-C, Schlosser C, Obermüller U, et al. Microglia turnover with aging and in an Alzheimer's model via long-term in vivo single-cell imaging. Nat Neurosci. (2017) 20:1371–6. doi: 10.1038/nn.4631
69. Hierro-Bujalance C, Bacskai BJ, Garcia-Alloza M. In vivo imaging of microglia with multiphoton microscopy. Front Aging Neurosci. (2018) 10:218. doi: 10.3389/fnagi.2018.00218
70. Liu YU, Ying Y, Li Y, Eyo UB, Chen T, Zheng J, et al. Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nat Neurosci. (2019) 22:1771–81. doi: 10.1038/s41593-019-0511-3
71. Stowell RD, Sipe GO, Dawes RP, Batchelor HN, Lordy KA, Whitelaw BS, et al. Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nat Neurosci. (2019) 22:1782–92. doi: 10.1038/s41593-019-0514-0
72. Zhang Y, Zhao L, Wang X, Ma W, Lazere A, Qian H, et al. Repopulating retinal microglia restore endogenous organization and function under CX3CL1-CX3CR1 regulation. Sci Adv. (2018) 4:eaap8492. doi: 10.1126/sciadv.aap8492
73. Chen X, Kezic JM, Forrester JV, Goldberg GL, Wicks IP, Bernard CC, et al. In vivo multi-modal imaging of experimental autoimmune uveoretinitis in transgenic reporter mice reveals the dynamic nature of inflammatory changes during disease progression. J Neuroinflamm. (2015) 12:17. doi: 10.1186/s12974-015-0235-6
74. Zawadzki RJ, Zhang P, Zam A, Miller EB, Goswami M, Wang X, et al. Adaptive-optics SLO imaging combined with widefield OCT and SLO enables precise 3D localization of fluorescent cells in the mouse retina. Biomed Opt Exp. (2015) 6:2191–210. doi: 10.1364/BOE.6.002191
75. Miller EB, Zhang P, Ching K, Pugh EN, Burns ME. in vivo imaging reveals transient microglia recruitment and functional recovery of photoreceptor signaling after injury. Proc Natl Acad Sci USA. (2019) 116:16603–12. doi: 10.1073/pnas.1903336116
76. Qin Z, He S, Yang C, Yung JS-Y, Chen C, Leung CK-S, et al. Adaptive optics two-photon microscopy enables near-diffraction-limited and functional retinal imaging in vivo. Light Sci Appl. (2020) 9:79. doi: 10.1038/s41377-020-0317-9
77. Airas L, Nylund M, Rissanen E. Evaluation of microglial activation in multiple sclerosis patients using positron emission tomography. Front Neurol. (2018) 9:181. doi: 10.3389/fneur.2018.00181
78. Conen S, Gregory CJ, Hinz R, Smallman R, Corsi-Zuelli F, Deakin B, et al. Neuroinflammation as measured by positron emission tomography in patients with recent onset and established schizophrenia: implications for immune pathogenesis. Mol Psychiatry. (2020). doi: 10.1038/s41380-020-0829-y. [Epub ahead of print].
79. Lehenkari PP, Kellinsalmi M, Näpänkangas JP, Ylitalo KV, Mönkkönen J, Rogers MJ, et al. Further insight into mechanism of action of clodronate: inhibition of mitochondrial ADP/ATP translocase by a nonhydrolyzable, adenine-containing metabolite. Mol Pharmacol. (2002) 61:1255–62. doi: 10.1124/mol.61.5.1255
80. Faustino JV, Wang X, Johnson CE, Klibanov A, Derugin N, Wendland MF, et al. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci. (2011) 31:12992–3001. doi: 10.1523/JNEUROSCI.2102-11.2011
81. Andreou KE, Soto MS, Allen D, Economopoulos V, de Bernardi A, Larkin JR, et al. Anti-inflammatory microglia/macrophages as a potential therapeutic target in brain metastasis. Front Oncol. (2017) 7:251. doi: 10.3389/fonc.2017.00251
82. Todd L, Palazzo I, Suarez L, Liu X, Volkov L, Hoang TV, et al. Reactive microglia and IL1β/IL-1R1-signaling mediate neuroprotection in excitotoxin-damaged mouse retina. J Neuroinflamm. (2019) 16:118. doi: 10.1186/s12974-019-1505-5
83. Forrester JV, McMenamin PG, Dando SJ. CNS infection and immune privilege. Nat Rev Neurosci. (2018) 19:655–71. doi: 10.1038/s41583-018-0070-8
84. Han X, Li Q, Lan X, EL-Mufti L, Ren H, Wang J. Microglial depletion with clodronate liposomes increases proinflammatory cytokine levels, induces astrocyte activation, and damages blood vessel integrity. Mol Neurobiol. (2019) 56:6184–96. doi: 10.1007/s12035-019-1502-9
85. Zelinka CP, Scott MA, Volkov L, Fischer AJ. The reactivity, distribution and abundance of Non-Astrocytic Inner Retinal Glial (NIRG) cells are regulated by microglia, acute damage, and IGF1. PLoS ONE. (2012) 7:e44477. doi: 10.1371/journal.pone.0044477
86. Polfliet MMJ, Goede PH, van Kesteren-Hendrikx EML, van Rooijen N, Dijkstra CD, van den Berg TK. A method for the selective depletion of perivascular and meningeal macrophages in the central nervous system. J Neuroimmunol. (2001) 116:188–95. doi: 10.1016/S0165-5728(01)00282-X
87. Elmore MRP, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. (2014) 82:380–97. doi: 10.1016/j.neuron.2014.02.040
88. Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer's disease model. Nat Commun. (2019) 10:1–21. doi: 10.1038/s41467-019-11674-z
89. Han J, Harris RA, Zhang X-M. An updated assessment of microglia depletion: current concepts and future directions. Mol Brain. (2017) 10:25. doi: 10.1186/s13041-017-0307-x
90. Najafi AR, Crapser J, Jiang S, Ng W, Mortazavi A, West BL, et al. A limited capacity for microglial repopulation in the adult brain. Glia. (2018) 66:2385–96. doi: 10.1002/glia.23477
91. Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MRP, Blurton-Jones M, et al. Eliminating microglia in Alzheimer's mice prevents neuronal loss without modulating amyloid-β pathology. Brain. (2016) 139:1265–81. doi: 10.1093/brain/aww016
92. Casali BT, MacPherson KP, Reed-Geaghan EG, Landreth GE. Microglia depletion rapidly and reversibly alters amyloid pathology by modification of plaque compaction and morphologies. Neurobiol Dis. (2020) 142:104956. doi: 10.1016/j.nbd.2020.104956
93. Han J, Zhu K, Zhang X-M, Harris RA. Enforced microglial depletion and repopulation as a promising strategy for the treatment of neurological disorders. Glia. (2019) 67:217–31. doi: 10.1002/glia.23529
94. Takeda A, Shinozaki Y, Kashiwagi K, Ohno N, Eto K, Wake H, et al. Microglia mediate non-cell-autonomous cell death of retinal ganglion cells. Glia. (2018) 66:2366–84. doi: 10.1002/glia.23475
Keywords: microglia, central nervous system, brain, retina, reporter mice, microglia homeostatic genes, microglia imaging, microglia depletion
Citation: Eme-Scolan E and Dando SJ (2020) Tools and Approaches for Studying Microglia In vivo. Front. Immunol. 11:583647. doi: 10.3389/fimmu.2020.583647
Received: 15 July 2020; Accepted: 24 August 2020;
Published: 07 October 2020.
Edited by:
Amanda Sierra, Achucarro Basque Center for Neuroscience, SpainReviewed by:
Souvarish Sarkar, Brigham and Women's Hospital and Harvard Medical School, United StatesRishein Gupta, University of Texas at San Antonio, United States
Copyright © 2020 Eme-Scolan and Dando. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samantha J. Dando, c2FtYW50aGEuZGFuZG9AcXV0LmVkdS5hdQ==