 Anna S. Świerzko
Anna S. Świerzko Maciej Cedzyński
Maciej Cedzyński- Laboratory of Immunobiology of Infections, Institute of Medical Biology, Polish Academy of Sciences, Łódź, Poland
Lung diseases are among the leading causes of morbidity and mortality. Complement activation may prevent a variety of respiratory infections, but on the other hand, could exacerbate tissue damage or contribute to adverse side effects. In this review, the associations of factors specific for complement activation via the lectin pathway (LP) with infections of the respiratory system, from birth to adulthood, are discussed. The most extensive data concern mannose-binding lectin (MBL) which together with other collectins (collectin-10, collectin-11) and the ficolins (ficolin-1, ficolin-2, ficolin-3) belong to pattern-recognition molecules (PRM) specific for the LP. Those PRM form complexes with MBL-associated serine proteases (MASP-1, MASP-2, MASP-3) and related non-enzymatic factors (MAp19, MAp44). Beside diseases affecting humanity for centuries like tuberculosis or neonatal pneumonia, some recently published data concerning COVID-19 are summarized.
Introduction
Lung diseases are thought to be the 3rd leading cause of morbidity and mortality worldwide. Recently, respiratory infections have been causing more deaths than previously expected due to the pandemic of coronavirus disease 2019 (COVID-19), the etiological agent of which is a betacoronavirus called severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Complement activation may prevent or exacerbate lung injury. Therefore, investigation of the protective and harmful associations of complement factors in diseases of the respiratory system is crucial for understanding pathogenic mechanisms and establishing therapeutic strategies (1).
Activation of complement via the lectin pathway is initiated by several pattern-recognition molecules (PRM), complexed with mannose-binding lectin-associated serine proteases (MASP). These PRM are classified into two lectin families: collectins (mannose-binding lectin, MBL; collectin-10, CL-10 and collectin-11, CL-11) and ficolins (ficolin-1, ficolin-2, ficolin-3). As well as direct elimination of pathogens via complement-dependent lysis, they may act as opsonins and contribute to phagocytosis (Figure 1). Collectins and ficolins are structurally and functionally related. Their molecules are multimers of basic subunits, consisting of three polypeptide chains. Both collectins and ficolins are characterized by four domains: an N-terminal cysteine-rich region, a collagen-like region, an α-helical neck domain and a C-terminal functional domain. The last is a globular carbohydrate-recognition domain (CRD) (in collectins) or a fibrinogen-like (FBG) domain (in ficolins) responsible for ligand recognition and binding [reviewed in (2–4)]. Although it was initially believed that multimeric molecules of all complement-activating collectins and ficolins are built-up from identical polypeptides/subunits, it was demonstrated that collectin-10 (known also as collectin liver-1) and collectin-11 (or collectin kidney-1) may form heterocomplexes termed CL-LK (5–7). Later, Jarlhelt et al. (8) evidenced a similar phenomenon for ficolin-2 (L-ficolin) and ficolin-3 (H-ficolin or Hakata antigen). In both cases, heterocomplexes are suspected to have additional biological relevance (i.e. broader spectrum of ligands) compared with their parent molecules. MBL and CL-LK express a high affinity for D-mannose (D-Man), N-acetyl-D-mannosamine (D-ManNAc), N-acetyl-D-glucosamine (D-GlcNAc), D-fucose (D-Fuc), and L-fucose (L-Fuc) (2–4). Ficolins generally recognize acetylated compounds (not necessarily of carbohydrate nature), including N-acetyl-D-glucosamine, N-acetyl-D-galactosamine (D-GalNAc), and sialic acid but also D-galactose (D-Gal) (3). This wide repertoire enables interaction with numerous glycoconjugates on microbial surfaces like capsular polysaccharides, lipopolysaccharides, exopolysaccharides, fungal mannans or beta-glucans, viral glycoproteins, etc.
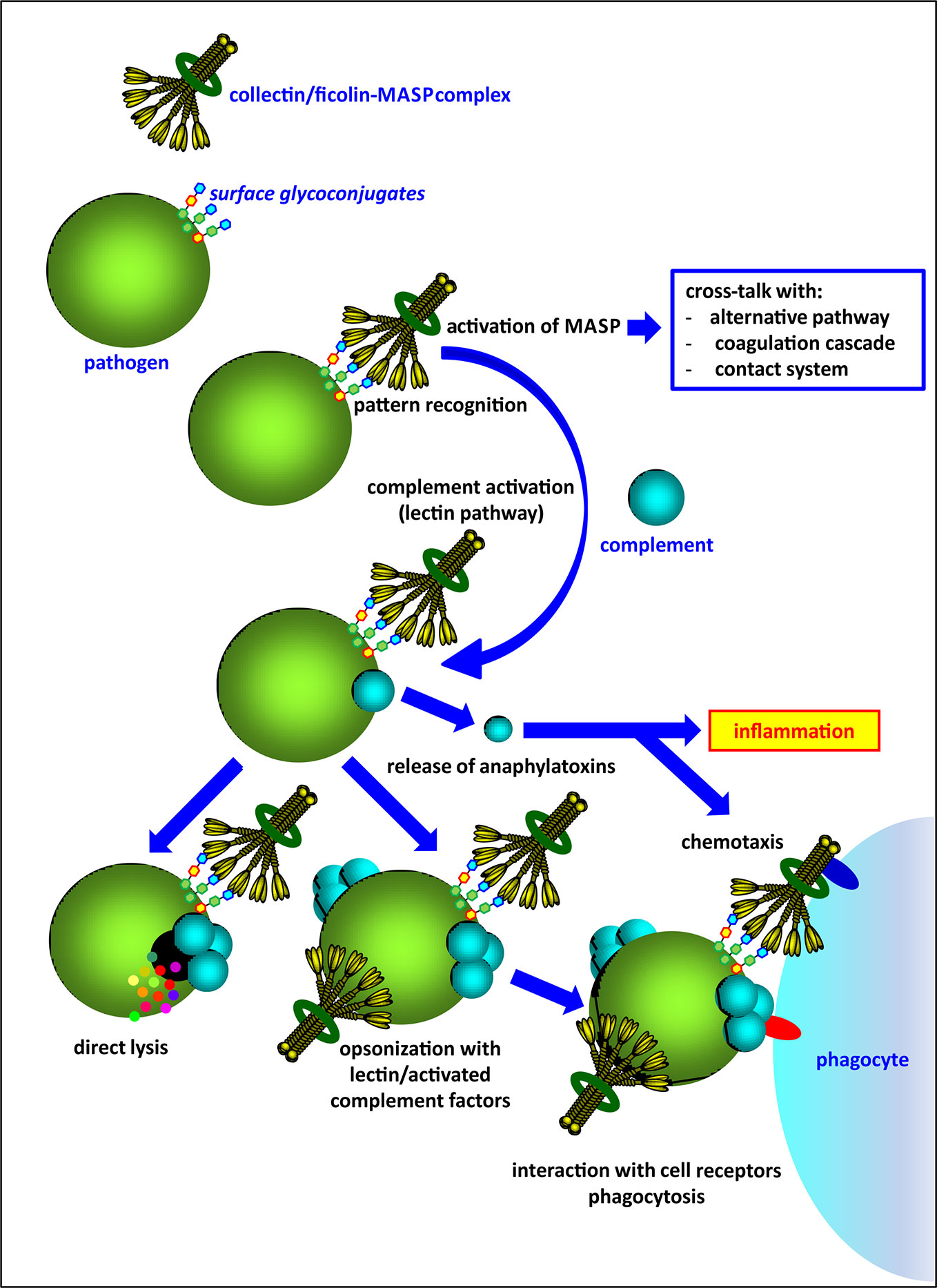
Figure 1 An overview of the activity of complement-activating collectins and ficolins, complexed with associated-serine proteases (MASP).
MBL recognizes a variety of respiratory pathogens, including Staphylococcus aureus, Mycobacterium tuberculosis, M. avium complex, Haemophilus influenzae, Legionella pneumophila, Klebsiella pneumoniae, Mycoplasma pneumoniae, Chlamydia pneumoniae, Ch. psittaci, Ch. trachomatis, Nocardia farcinica, Aspergillus fumigatus, influenza A virus and severe acute respiratory syndrome coronavirus (SARS-CoV-1). Furthermore, it binds to bacteria causing severe infections in cystic fibrosis (CF) patients like Pseudomonas aeruginosa and Burkholderia cepacia [reviewed in (9)] (Table 1). In the case of Streptococcus pneumoniae, efficient binding was originally observed only for non-capsulated strains since capsular polysaccharide abrogates recognition (10).

Table 1 Collectins and ficolins activating complement via the lectin pathway and their interactions with respiratory pathogens.
Collectin-11 interacts with structures exposed on the surfaces of some Gram-positive and Gram-negative bacteria (including respiratory system pathogens: M. tuberculosis, Str. pneumoniae, K. pneumoniae and Ps. aeruginosa), fungi and influenza A virus (6, 11–14) (Table 1). It was also reported to recognize DNA and thought to participate in the response to apoptotic cells, neutrophil extracellular traps and biofilms (15). Moreover, CL-11 was believed to be involved in ischemic injury via recognition of L-fucose and subsequent complement activation at the site of ischemic stress (16, 17). Microbial targets recognized specifically by collectin-10 have not been reported to date; however, as a component of CL-LK, it is believed to extend its specificity and/or modify its affinity (4, 7). Both CL-10 and Cl-11 were demonstrated to contribute to the embryonic development, acting as chemoattractants for the cranial crest nerve cells. A variety of mutations of their genes (COLEC10, and COLEC11, respectively) are associated with the Malpuech, Michels, Mingarelli, and Carnevale (3MC) syndrome, manifested by craniofacial abnormalities [reviewed in (18)].
Ficolins are synthesized by variety of cells (ficolin-1 by neutrophils, monocytes and in bone marrow; ficolin-2 by hepatocytes; ficolin-3 by hepatocytes, alveolar type II pneumocytes and ciliated bronchial cells). They are present in the blood and contribute to the systemic immune response. Ficolin-1 (associated with lung macrophages) and ficolin-3 are furthermore involved locally in the respiratory system [reviewed in (3)].
Ficolin-1, known also as M-ficolin, binds to certain respiratory pathogens, as Staph. aureus, Str. pneumoniae, M. tuberculosis, Ps. aeruginosa, and A. fumigatus (Table 1). It recognizes some pneumococcal capsular polysaccharides [reviewed in (19)]. Ficolin-2 (L-ficolin) targets a relatively broad range of ligands (including bacterial lipoteichoic acids, capsular polysaccharides, fungal 1,3-β-glucans, DNA and elastin) via four binding sites in its FBG domain (20). Like ficolin-1, it is believed to participate in the clearance of late apoptotic cells (21, 22). Staph. aureus, Str. pneumoniae (in both cases encapsulated strains), H. influenzae, M. tuberculosis, Ps. aeruginosa, A. fumigatus, and IAV are among the pathogens of the respiratory system recognized by ficolin-2. Furthermore, ficolin-2 binds to the surface structures of opportunistic Pasteurella pneumotropica [reviewed in (19)] (Table 1). Although the serum concentration of ficolin-3 (H-ficolin, Hakata antigen) was found to be the highest among PRM specific for the lectin pathway [its median level in healthy adults is 20 µg/ml or above (23–25)], few microbial targets have been reported. However, among those few are several respiratory pathogens: A. fumigatus, M. tuberculosis, IAV, and the afore-mentioned Pasteurella pneumotropica [reviewed in (19)] (Table 1). As mentioned, it was recently demonstrated that ficolin-3 and ficolin-2 may form heterocomplexes suspected to have more extensive biological relevance than the corresponding homooligomers (8).
These PRM are able to activate complement via the lectin pathway (LP) after complex formation with proteins of the MASP family, including three enzymes and two related proteins lacking proteolytic activity. As proenzymes, MASP exist as single polypeptide chains. Like C1r and C1s, which are involved in the activation of the classical pathway (CP), they include six domains (from N- to C-termini): CUB1 (C1r/C1s, urchin-epidermal, bone morphogenetic protein), EGF (epidermal growth factor), CUB2, CCP1 (complement control protein), CCP-2, and SP (serine protease, catalytic). When activated, the heavy (H) chain (CUB1–EGF–CUB2–CCP1–CCP2) and the light (L) chain (SP) are created (linked by a disulphide bond). The three N-terminal domains are responsible for MASP dimerization and complex formation with PRM [reviewed in (26)].
MASP-1, MASP-3, and the non-catalytic MBL-associated protein (MAp44 or MAP-1) are encoded by a single (MASP1) gene. MASP-1 undergoes auto-activation upon target recognition by collectins or ficolins. It is able to cleave C2 with low efficiency; therefore it was thought to upregulate LP activation. However, a key role of MASP-1 in MASP-2 activation was established (27). It moreover enables cross-talk with the coagulation and contact systems (28, 29), contributes to activation of platelets (30), endothelial cells, affects endothelial permeability (31–33), and regulates the transcription of complement factor D (34).
MASP-3 was first described to cleave insulin-like growth factor-binding protein-5 (IGFPB-5), regulating activity of insulin-like growth factors hence influencing cell proliferation, differentiation, motility, and survival (35). It was also thought to downregulate LP activation via competitive binding with MASP-2 to PRM. It was however evidenced that its major substrate is pro-factor D. Therefore, MASP-3 is directly involved in activation of complement via the alternative pathway (AP) (36–38). The MAp44 molecule comprises four domains in common with other products of the MASP1 gene (CUB1–EGF–CUB2–CCP1) and 17 specific amino acid (AA) residues. Its biological role has not been established precisely; however, it is supposed to downregulate LP activation (via competition with MASP for formation of complexes with collectins or ficolins) [reviewed in (39)]. Furthermore, MAp44 was found to participate in regulation of cardiac development (40). The afore-mentioned CL-LK interacts with MASP-1 or MASP-3 homodimers (13, 41–43); therefore it may be supposed that neural crest cell migration depends on activity of those complexes. Indeed, a dozen MASP1 mutations have been found associated with 3MC syndrome [reviewed in (18)].
MASP-2 protease and the non-catalytic MBL-associated protein MAp19 (known also as small MBL-associated protein, sMAP) are also products of a single (MASP2) gene. As the first mentioned cleaves C4 and C2, it is crucial for LP activation. Furthermore, it may contribute to the activation of the coagulation cascade, as its substrate is prothrombin [reviewed in (44, 45)]. MASP-2 cleaves kininogen as well; however, it is not associated with bradykinin release (29). Like MASP-1, it is involved in the regulation of transcription of factor D (34). MAp19 consists of CUB1 and EGF domains (shared with MASP-2) and four specific AA residues [reviewed in (44)]. It was thought to downregulate complement activation (via competition with MASP-2 for binding to pattern-recognizing molecules); however, that was not confirmed in a report published by Degn et al. (46).
As well as cross-talk between lectin and alternative pathways and with the coagulation and contact systems, ficolins and MBL were demonstrated to interact with long and/or short pentraxins contributing to the enhancement or regulation of the early immune response [reviewed in (47, 48)]. Previously, pentraxins were known to activate complement via the classical pathway, in association with C1q with no involvement of antibodies (48). The long pentraxin-3 (PTX3) was shown to co-operate with MBL, ficolin-1, and ficolin-2. On the other hand, it can interact with complement regulatory factors [C4-binding protein (C4bp), factor H] and therefore may contribute both to the amplification and modulation of complement activation [reviewed in (49, 50)]. Other (short) pentraxins recognize LP-associated PRM as well: C-reactive protein (CRP) was found to bind to ficolin-1 and -2, while serum amyloid protein (SAP) binds to MBL [reviewed in (47–49)].
Neutrophil extracellular traps (NET) are known to be important factors in host protection. Their release is promoted by complement-dependent opsonization of pathogens, including those invading the respiratory system [like Str. pneumoniae, Staph. aureus, M. tuberculosis, A. fumigatus, IAV, and respiratory syncytial virus (RSV)]. However, a variety of infectious agents, (for example pneumococci and Staph. aureus) developed mechanisms enabling them to escape from NET and promote dissemination from the upper to the lower respiratory tract and beyond (51, 52). Furthermore, when produced excessively, NET may contribute to harmful effects leading to airflow disturbance and chronic inflammation (51). NET-derived extracellular histones act cytotoxically and are mentioned among the main players of tissue damage. Their production may be induced by activation of complement and subsequent inflammatory processes (53). As lectin pathway-associated PRM are constitutively present in the respiratory system (ficolin-3 synthesized by type II pneumocytes and ciliated bronchial cell, ficolin-1 produced by lung macrophages) or are transferred from the bloodstream to the infected sites [MBL and ficolin-2 detected in bronchoalveolar lavage fluid (BALF) from patients suffering from pneumonia or invasive aspergillosis, respectively], they may contribute to excessive inflammation and its detrimental effects. Interestingly, PTX3 (a NET component) is considered to have a protective effect not only by contribution to trapping and killing pathogens but also by interacting with histones and other NET constituents (including myeloperoxidase and azurocidin 1), leading to modulation of the response (53, 54). Therefore, PTX3 appears a molecule enabling both boosting and mitigation of the early antimicrobial response, involved in its complement- and NET-related branches (53, 55). As mentioned above, it is able to amplify activation of the classical and lectin pathways (via interaction with their specific PRM) and downregulate each route (via binding of factor H and C4bp).
Lectin Pathway-Associated Molecules in Respiratory Infections: From Newborn to Teenager
MBL was first believed to be protective against infections especially in infants and children aged 5–18 months (“window of vulnerability”) (56). However, numerous reports demonstrated associations of MBL deficiency with an increased susceptibility to various infections in newborns, older children/adolescents and adults (the last age group is discussed below).
Inherited lack of functional MBL is conferred by LXPA/O (referred to also as LXA/O or XA/O) and O/O genotypes. They correspond to six single nucleotide polymorphisms (SNPs) of the MBL2 gene. Two of them are localized in the promoter region: −550 G>C (rs11003125, commonly called H/L) and −221 C>G (rs7096206, Y/X); one to the 5′-untranslated region (+4 C>T, rs7095891, P>Q). The coding region (exon 1) polymorphisms: +223 C>T (Arg52Cys, rs5030737), +230 G>A (Gly54Asp, rs1800450) and +239 G>A (Gly57Glu, rs1800451) are called A>D, A>B and A>C, respectively (D, B, and C variants are collectively designated O). Promoter SNPs affect the gene expression level (and thus MBL concentration in serum), while D/B/C alleles are related to markedly diminished ability to opsonize microbial cells and to activate complement. Those structural mutations lead to impaired oligomerization of subunits and diminished complex formation with associated serine proteases. Moreover, an increased sensitivity to endogenous metalloproteases is associated with shorter MBL half-life resulting in a lower serum level. As strong linkage disequilibria exist between the afore-mentioned polymorphisms, only seven haplotypes are commonly observed: HYPA, LYPA, LYQA, LXPA, HYPD, LYPB, and LYQC. Additionally, several rare variants (HXPA, LYPD, HYPB, LYQB) have been found in various populations [reviewed in (2)].
An adverse influence of MBL deficiency on risk of perinatal pneumonia, especially in premature babies was evidenced in several studies (57–60). A similar relationship was found for low ficolin-2 concentration in cord serum (58, 59). In contrast, babies with confirmed infections (mainly pneumonias) had higher ficolin-1 levels compared with newborns with no infections before leaving hospital (59).
Koch et al. (42) found that low MBL enhanced susceptibility to acute (mainly viral) infections of the respiratory system in infants aged 6–17 months supporting the “window of vulnerability” hypothesis. They suggested that in the younger (up to 5 months) population such an effect is moderated by maternal antibodies while in older (18–23 months) children, MBL dysfunction modifies disease course rather than influences infection risk itself (42). However, an important role of MBL in protection from pediatric respiratory infections of various etiologies was demonstrated also by Garred et al. (61) and Summerfield et al. (62). They found a relationship between O/O homozygosity and recurrent and/or severe pneumonias (61, 62). Our data indicated that insufficiency of MBL is associated with recurrence of infections of the respiratory system in children, especially when accompanied with other defects of the humoral response (63, 64). Furthermore, in two patients, the rare MASP-2 deficiency [related to +359 A>G (Asp120Gly or Asp105Gly in mature protein, rs72550870) homozygous mutation of the MASP2 gene] was found. One of them suffered from recurrent pneumonias (63), while another had frequent infections of the upper respiratory tract and skin abscesses (64). The +359 A>G mutation, affecting the CUB1 domain, precludes formation of complexes with collectins and ficolins and therefore lectin pathway activation.
Low concentrations of ficolin-2 were also reported to enhance susceptibility to respiratory infections in children and teenagers (age range 1–16 years, mean 8.9), however, in the context of allergic rhinitis/asthma only. That association was not found in patients without allergic diseases. Based on those data, it was supposed that ficolin-2 may be protective against pathogens exacerbating allergic inflammation in the lung (64, 65). Ruskamp et al. (66) did not find any associations with nine FCN2 gene SNP (located in promoter, introns, and coding region) or two intronic FCN3 polymorphisms with respiratory system infections in a large cohort of children aged up to 4 years. It should be however, stressed that 6% of individuals only had frequent (>3/year) episodes. In our investigations (63–65), recurrence was defined as at least two pneumonias or serious sinus infections (requiring hospitalization) or at least eight upper respiratory tract infections within 12 months. Therefore the data published are not fully comparable.
Regarding the role of ficolin-1 in pediatric pneumonia, Elkoumi et al. (67) recently reported an association of (−144 C>A (rs10117466) SNP, localized in the promoter region of the FCN1 gene. They found a higher frequency of the A/A genotype (and higher ficolin-1 serum levels) in children aged <5 years suffering from severe disease compared with controls and concluded that this protein may contribute to an enhanced inflammatory response resulting in a poorer outcome.
Bronchiectasis is an outcome of recurrent infections and may occur with or without cystic fibrosis (CF). The only study on children with non-CF bronchiectasis has been published recently (68). This study obtained negative results for MBL2 alleles and haplotypes, while serum MBL concentrations were not determined.
Lectin Pathway-Associated Molecules in Respiratory Infections: Adult
Roy et al. (69) reported MBL2 O/O genotypes to be associated with a high risk of pneumonia and invasive pneumococcal disease in adults. However, this relationship was not confirmed by Kronborg et al. (70). Later, Gomi et al. (71) evidenced higher frequency of the MBL2 B variant in Japanese patients suffering from recurrent respiratory infections compared with healthy controls. They furthermore evidenced the presence of mannose-binding lectin in bronchoalveolar lavage fluid from persons with active infection. Later, the presence of this lectin in BALF during infection was confirmed by Fidler et al. (72).
Collectin-11 and ficolin-2 (but not MBL) were shown to activate complement upon recognition of surface structures of Streptococcus pneumoniae (14). Consequently, Garcia-Laorden et al. (73) found no impact of MBL deficiency on the risk of development of community-acquired pneumonia (CAP) (commonly caused by pneumococci) or invasive pneumococcal disease. However, earlier the same authors postulated that MBL deficiency is associated with higher disease severity (including developing sepsis and multiorgan failure) and its fatal outcome (74), while Chalmers et al. (75) found no impact of low MBL serum concentration on CAP incidence or 1-month mortality. Ficolin-2 recognizes pneumococcal lipoteichoic acid, some capsular polysaccharides of pathogenic serotypes and pneumolysin (the major toxin of those bacteria) (10, 76–78). It was moreover suspected that the low invasiveness of Str. pneumoniae 11A serotype is related to recognition of its capsular polysaccharide by ficolin-2 (79). Nevertheless, Chapman et al. (80) found no relationship between FCN2 gene polymorphisms and invasive pneumococcal disease. Chalmers et al. (75) noticed an association of very low ficolin-2 serum levels (<1,200 ng/ml) with higher risk of 30-day mortality in CAP patients. The data discussed above suggest that ficolin-2 may be protective against infections with some Str. pneumoniae strains, depending on a variety of factors, including bacterial serotype and the FCN2 genotype/ficolin-2 serum concentration in the host. Moreover, MASP-2 primary deficiency was reported to be associated with severe pneumococcal pneumonia and to modify the course of cystic fibrosis (43, 81, 82).
Interestingly, van Kempen et al. (83) reported that MBL2 genotypes conferring high gene expression levels (YA/YA, YA/XA) predispose to CAP caused by intracellular pathogens (Coxiella burnetii, Legionella spp., Chlamydia spp. Mycoplasma pneumoniae), supposedly by the contribution of MBL to enhanced phagocytosis. However, low MBL-dependent complement activity was earlier shown to be a risk factor for legionnaires’ disease (84). Furthermore, +6424 G>T (rs7851696) FCN2 gene polymorphism minor allele (related to low ficolin-2 serum concentration) was observed to be a risk factor for C. burnetii pneumonia (83).
A few cases of the rare ficolin-3 deficiency associated with +1637 C>delC (rs28357092) variant allele homozygosity have been reported. In one of them, it was found associated with numerous severe/recurrent infections, including those affecting the respiratory system. The adult patient suffered from recurrent lower respiratory tract diseases from early childhood. As a young adult, he was hospitalized due to bilateral frontal cerebral abscesses caused by non-hemolytic streptococci and several times due to pneumonia (H. influenzae and Ps. aeruginosa were identified as aetiological agents). Furthermore, severe bronchiectasis, pulmonary fibrosis, and obstructive lung disease were diagnosed (85).
Haerynck et al. (86) found an association of the A variant [related to MASP1 gene +1851 G>A (rs3821805) SNP, localized in exon 12, encoding for MASP-3 SP domain) with earlier onset of chronic Ps. aeruginosa colonization in cystic fibrosis patients. Interestingly, the presence of the A allele does not affect the protein sequence (Leu617Leu). It was therefore supposed it may influence mRNA splicing, its stability, structure, and protein folding (86).
Numerous studies have investigated the role of lectin pathway factors in susceptibility to pulmonary tuberculosis. The majority of them concerned MBL. As revealed in several meta-analyses, the role of MBL in that disease is unclear and inconsistent, apparently reflecting differences in study design, ethnicity and number of patients tested. Although it is generally believed that MBL2 O alleles may contribute to enhanced susceptibility to TB, at least in some populations (87–89), certain data suggest diverse associations for D, B, and C variants. Tong et al. (90) concluded that minor alleles in codons 52 (D) and 54 (B) elevate the risk of developing disease while that in codon 57 (C) has a protective effect (in Chinese population). In contrast, Mandal et al. (91) postulated an unfavorable effect of the C allele and a beneficial effect of the D allele. The protective role of ficolin-2 from tuberculosis (caused by M. tuberculosis) and pulmonary infection with M. avium complex was reported by Luo et al. (92) and Kobayashi et al. (93), respectively.
Recently, we reported a significantly higher frequency of MBL2 O variants among adults diagnosed with pulmonary tuberculosis (PTB), compared with controls. Furthermore, investigation of the FCN1 gene −542 G>A polymorphism revealed a higher incidence of G/G homozygosity among patients. The median concentration of MBL in serum was significantly higher in the disease group, despite afore-mentioned genetic association (94). Similarly, median ficolin-1 level was higher in patients (94), although the FCN1 G allele at position −542 was earlier reported to be associated with the opposite effect (95). In contrast, ficolin-3 concentrations were generally lower among persons with confirmed infection. Furthermore, a high potential of ficolin-1 to differentiate between PTB patients and controls was noted; therefore, it was suggested to be considered a supplementary marker of active tuberculosis. Furthermore, two patients were found to be MASP-2-deficient (94).
The MBL2 D allele was suggested to contribute to higher susceptibility to pulmonary chronic necrotizing aspergillosis in British adults (96). Furthermore, Bidula et al. (97) observed significantly higher BALF ficolin-3 concentrations in patients with confirmed pulmonary A. fumigatus infection compared with controls and considered this lectin to be a promising disease marker. Interestingly, both ficolin-2 and pentraxin-3 recognize A. fumigatus independently; however they were shown to recruit each other to the pathogen surface in a calcium-dependent and synergistic manner (98). That leads to the cross-talk between classical and lectin pathways and amplification of ficolin-2-dependent complement activation (98). As ficolin-2 was detected in BALF from patients suffering from invasive aspergillosis (99), it was supposed that it may enhance complement-mediated phagocytosis (48). Furthermore, PTX3 deficiency in donors was found associated with enhanced risk of that disease in recipients of allogeneic hematopoietic stem cell transplants (100) supporting the conclusion that the cross-talk between PTX3 and ficolin-2 may boost anti-fungal response in the lung (50).
Due to interaction with hemagglutinin and neuraminidase, MBL is able to neutralize influenza A virus (IAV) in a complement-independent way (101–103). However, no role for MBL in protection from the disease has been reported to date. Regarding other respiratory viral infections, low MBL concentration in serum was reported to be associated with higher risk and more severe course of respiratory syncytial virus (RSV) disease in children (104). Furthermore, the MBL2 B variant was associated with the severe acute respiratory syndrome (SARS) (105, 106). On the other hand, Yuan et al. (107) found no relationship with any MBL2 SNP in SARS.
There is also some evidence that MBL insufficiency (defined genetically or at the protein level) affects the course and severity of CF-associated bronchiectasis, although not manifested clinically in childhood (108). Similar results were obtained with non-CF bronchiectasis adult patients (109) although not confirmed by another group (110).
Possible Associations of Complement Activation via the Lectin Pathway With COVID-19
The current pandemic of coronavirus disease 2019 (COVID-19) is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Non-glycosylated SARS-CoV-2 nucleocapsid protein (like corresponding components of SARS-CoV-1 and MERS-CoV) was demonstrated to activate complement via the lectin pathway (111). Data reported by Magro et al. (112) evidenced depositions of MASP-2, co-localized with C4d and SARS-CoV-2 spike glycoprotein (S-gp) in septal capillaries and interalveolar septa of the lungs from COVID-19 non-survivors. Recently, recombinant MBL has been mooted as a therapeutic agent, acting via inhibition of binding of S-gp to the angiotensin converting enzyme 2 (ACE2) cell receptor and by promoting phagocytosis (113).
The RNA sequence of the better-studied SARS-CoV-1 is highly homologous to that of SARS-CoV-2, and both viruses recognize the same human receptor. Ip et al. (105) reported that MBL bound to SARS-CoV-1 and enhanced C4 deposition on the viral surface. They also found that this lectin is able to inhibit the infectivity of SARS-CoV-1 in fetal rhesus monkey kidney cells. In addition, MBL seems to recognize SARS-CoV-1 S-gp which results in preventing infection (114). However, complement activation may also be detrimental. Based on data from a murine model, Gralinski et al. (115) suggested complement activation contributed to acute respiratory distress syndrome (ARDS) associated with SARS-CoV-1 infection: C3−/− mice had reduced lung neutrophilia and less severe systemic inflammatory response.
Similar findings are emerging for SARS-CoV-2 and COVID-19. Complement activation may provoke a “cytokine storm” leading to ARDS and organ failure. That supposition was first supported by promising data from patients treated with complement inhibitors (116–119). Polycarpou et al. (120) suggested that targeting complement might contribute to reducing COVID-19 systemic complications (multiorgan failure, coagulopathy), mediated by lectin pathway activation. Therefore, inhibition of the complement system at the levels of C3 or C5a/C5aR is considered a possible therapeutic option (121, 122).
Matricardi et al. (123) proposed an intriguing model explaining the course of SARS-CoV-2 infection. They suggested an efficient local innate immune response (MBL, natural antibodies etc.) may eliminate the pathogen. However, when the virus replicates and spreads, it induces a strong adaptive response (with involvement of specific IgM and IgG) leading to severe inflammation with involvement of complement and coagulation cascades, resulting in a “cytokine storm” (123). Thus, complement activation seems to be beneficial at the early stage of infection but it may be severely harmful at a later stage. It should be stressed that MBL-dependent complement activation may be associated with adverse effects as well, leading to the amplification of an excessive response. Eriksson et al. (124) reported that MBL contributed to pathological thrombosis and coagulopathy (but not other organ dysfunction or intensity of inflammation) in critically ill COVID-19 patients. In contrast, Holter et al. (125) did not find much difference in MBL concentrations in plasma between COVID-19 patients and controls although a transient increase of its level (at days 3–5 after hospital admission) was noted. It should be however stressed that narsoplimab, specifically targeting MASP-2, has recently been demonstrated to be a promising therapeutic agent, reducing detrimental effects of complement activation and giving no adverse reactions itself (126).
Concluding Remarks
The findings reviewed here detail a variety of associations between factors specific for complement activation via the lectin pathway and infections of the respiratory system, from birth to adulthood. However, they are not entirely consistent (especially those concerning pulmonary tuberculosis) and therefore have to be considered in relation to ethnic, geographical and social backgrounds, study design, number of patients recruited, etc. Nowadays, extensive investigations concerning the lectin pathway (and complement in general) associations with pandemic SARS-CoV-2 infection have to be considered crucial as they may result in elaboration of efficient treatment strategies.
Author Contributions
MC and AŚ conceptualized the background of this review, collected, and selected literature to be discussed. MC wrote draft manuscript. AŚ reviewed the draft version. MC prepared the submitted version. All authors contributed to the article and approved the submitted version.
Funding
This work was partially supported by National Science Centre, Poland, grant UMO-2015/17/B/NZ6/04250 and Institute of Medical Biology, Polish Academy of Sciences.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are very grateful to Dr David C. Kilpatrick for critical reading of the manuscript and helpful discussion.
References
1. Pandya PH, Wilkes DS. Complement system in lung disease. Am J Respir Cell Mol Biol (2014) 51:467–73. doi: 10.1165/rcmb.2013-0485TR
2. Cedzyński M, Kilpatrick DC, Świerzko AS. Mannose-binding lectin. In: Barnum S, Schein T, editors. The Complement Factsbook, 2nd edition. London, UK: Academic Press, Elsevier (2018). p. 33–43.
3. Matsushita M, Barnum S, Schein T. Ficolins. In: The Complement Factsbook, 2nd edition. London, UK: Academic Press, Elsevier (2018). p. 45–56.
4. Ohtani K, Wakamiya N. The collectins. In: Barnum S, Schein T, editors. The Complement Factsbook, 2nd edition. London, UK: Academic Press, Elsevier (2018). p. 57–65.
5. Henriksen ML, Madsen KL, Skjoedt K, Hansen S. Calcium-sensitive immunoaffinity chromatography: gentle and highly specific retrieval of a scarce plasma antigen, collectin-LK (CL-LK). J Immunol Methods (2014) 413:25–31. doi: 10.1016/j.jim.2014.07.006
6. Hansen SW, Ohtani K, Roy N, Wakamiya N. The collectins CL-L1, CL-K1 and CL-P1, and their roles in complement and innate immunity. Immunobiology (2016) 221:1058–67. doi: 10.1016/j.imbio.2016.05.012
7. Hansen SWK, Aagaard JB, Bjerrum KB, Hejbol EK, Nielsen O, Schroder HD, et al. CL-L1 and CL-K1 exhibit widespread tissue distribution with high and co-localized expression in secretory epithelia and mucosa. Front Immunol (2018) 9:1757. doi: 10.3389/fimmu.2018.01757
8. Jarlhelt I, Pilely K, Clausen JB, Skjoedt MO, Bayarri-Olmos R, Garred P. Circulating ficolin-2 and ficolin-3 form heterocomplexes. J Immunol (2020) 204:1919–28. doi: 10.4049/jimmunol.1900694
9. Eisen DP. Mannose-binding lectin deficiency and respiratory tract infection. J Innate Immun (2010) 2:114–22. doi: 10.1159/000228159
10. Krarup A, Skov Sorensen UB, Matsushita M, Jensenius JC, Thiel S. Effect of capsulation of opportunistic pathogenic bacteria on binding of the pattern recognition molecules mannan-binding lectin, L-ficolin, and H-ficolin. Infect Immun (2005) 73:1052–60. doi: 10.1128/IAI.73.2.1052-1060.2005
11. Keshi H, Sakamoto T, Kawai T, Ohtani K, Katoh T, Jang SJ, et al. Identification and characterization of a novel human collectin CL-K1. Mol Immunol (2006) 50:1001–13. doi: 10.1111/j.1348-0421.2006.tb03868.x
12. Hansen S, Selman L, Palaniyar N, Ziegler K, Brandt J, Kliem A, et al. Collectin 11 (CL-11, CL-K1) is a MASP-1/3-associated plasma collectin with microbial-binding activity. J Immunol (2010) 185:6096–104. doi: 10.4049/jimmunol.1002185
13. Ma YJ, Skjoedt MO, Garred P. Collectin-11/MASP complex formation triggers activation of the lectin complement pathway - the fifth lectin pathway initiation complex. J Innate Immun (2013) 5:242–50. doi: 10.1159/000345356
14. Fidler KJ, Hilliard TN, Bush A, Johnson M, Alton E, Geddes D, et al. Mannose-binding lectin is present in the infected airway: a possible pulmonary defence mechanism. Thorax (2009) 64:150–5. doi: 10.1136/thx.2008.100073
15. Henriksen ML, Brandt J, Iyer SS, Thielens NM, Hansen S. Characterization of the interaction between collectin 11 (CL-11, CL-K1) and nucleic acids. Mol Immunol (2013) 56:757–67. doi: 10.1016/j.molimm.2013.07.011
16. Farrar CA, Tran D, Li K, Wu W, Peng Q, Schwaeble W, et al. Collectin-11 detects stress-induced L-fucose pattern to trigger renal epithelial injury. J Clin Invest (2016) 126:1911–25. doi: 10.1172/JCI83000
17. Nauser CL, Howard MC, Fanelli G, Farrar CA, Sacks S. Collectin-11 (CL-11) is a major sentinel at epithelial surfaces and key pattern recognition molecule in complement-mediated ischaemic injury. Front Immunol (2018) 9:2023. doi: 10.3389/fimmu.2018.02023
18. Gajek G, Świerzko AS, Cedzyński M. Association of polymorphisms of MASP1/3, COLEC10, and COLEC11 genes with 3MC syndrome. Int J Mol Sci (2020) 21(15):5483. doi: 10.3390/ijms21155483
19. Bidula S, Sexton DW, Schelenz S. Ficolins and the recognition of pathogenic microorganisms: an overview of the innate immune response and contribution of single nucleotide polymorphisms. J Immunol Res (2019) 2019:3205072. doi: 10.1155/2019/3205072
20. Garlatti V, Belloy N, Martin L, Lacroix M, Matsushita M, Endo Y, et al. Structural insights into the innate immune recognition specificities of L- and H-ficolins. EMBO J (2007) 26:623–33. doi: 10.1038/sj.emboj.7601500
21. Kuraya M, Ming Z, Liu X, Matsushita M, Fujita T. Specific binding of L-ficolin and H-ficolin to apoptotic cells leads to complement activation. Immunobiology (2005) 209:689–97. doi: 10.1016/j.imbio.2004.11.001
22. Jensen ML, Honore C, Hummelshoj T, Hansen BE, Madsen HO, Garred P. Ficolin-2 recognizes DNA and participates in the clearance of dying host cells. Mol Immunol (2007) 44:856–65. doi: 10.1016/j.molimm.2006.04.002
23. Troldborg A, Hansen A, Hansen SWK, Jensenius JC, Stengaard-Pedersen K, Thiel S. Lectin complement pathway proteins in healthy individuals. Clin Exp Immunol (2017) 188:138–47. doi: 10.1111/cei.12909
24. Świerzko AS, Michalski M, Sokołowska A, Nowicki M, Szala-Poździej A, Eppa Ł, et al. Associations of ficolins with haematological malignancies in patients receiving high-dose chemotherapy and autologous haematopoietic stem cell transplantations (auto-HSCT). Front Immunol (2020) 10:3097. doi: 10.3389/fimmu.2019.03097
25. Sokołowska A, Świerzko AS, Gajek G, Gołos A, Michalski M, Nowicki M, et al. Associations of ficolins and mannose-binding lectin with acute myeloid leukaemia in adults. Sci Rep (2020) 10:1056.1. doi: 10.1038/s41598-020-67516-2
26. Kjaer TR, Le LTM, Pedersen JS, Sander B, Golas MM, Jensenius JC, et al. Structural insights into the initiating complex of the lectin pathway of complement activation. Structure (2015) 23:342–51. doi: 10.1016/j.str.2014.10.024
27. Degn SE, Jensen L, Hansen AG, Duman D, Tekin M, Jensenius JC, et al. Mannan-binding lectin-associated serine protease (MASP)-1 is crucial for lectin pathway activation in human serum, whereas neither MASP-1 nor MASP-3 is required for alternative pathway function. J Immunol (2012) 189:3957–69. doi: 10.4049/jimmunol.1201736
28. Dobo J, Harmat V, Beinrohr L, Sebestyen E, Zavodszky P, Gal P. MASP-1, a promiscuous complement protease: structure of its catalytic region reveals the basis of its broad specificity. J Immunol (2009) 183:1207–14. doi: 10.4049/jimmunol.0901141
29. Dobo J, Major B, Kekesi KA, Szabo I, Megyeri M, Hajela K, et al. Cleavage of kininogen and subsequent bradykinin release by the complement component: mannose-binding lectin-associated serine protease (MASP)-1. PLoS One (2011) 6(5):e20036. doi: 10.1371/journal.pone.0020036
30. Megyeri M, Mako V, Beinrohr L, Doleschall Z, Prohaszka Z, Cervenak L, et al. Complement protease MASP-1 activates human endothelial cells: PAR4 activation is a link between complement and endothelial function. J Immunol (2009) (183):3409–16. doi: 10.4049/jimmunol.0900879
31. Jani PK, Kajdacsi E, Megyeri M, Dobo J, Doleschall Z, Futosi K, et al. MASP-1 induces a unique cytokine pattern in endothelial cells: a novel link between complement system and neutrophil granulocytes. PloS One (2014) 9(1):e87104. doi: 10.1371/journal.pone.0087104
32. Schwaner E, Nemeth Z, Jani PK, Kajdacsi E, Debreczeni ML, Doleschall Z, et al. Transcriptome analysis of inflammation-related gene expression in endothelial cells activated by complement MASP-1. Sci Rep (2017) 7:10462. doi: 10.1038/s41598-017-09058-8
33. Debreczeni ML, Nemeth Z, Kajdacsi E, Schwaner E, Mako V, Masszi A, et al. MASP-1 increases endothelial permeability. Front Immunol (2019) 10:991. doi: 10.3389/fimmu.2019.00991
34. Holers MV, Borodovsky A, Scheinman RI, Ho N, Ramos Ramirez J, Dobo J, et al. Key components of the complement lectin pathway are not only required for the development of inflammatory arthritis but also regulate the transcription of factor D. Front Immunol (2020) 11:201. doi: 10.3389/fimmu.2020.00201
35. Cortesio CL, Jiang W. Mannan-binding lectin-associated serine protease 3 cleaves synthetic peptides and insulin-like growth factor-binding protein 5. Arch Biochem Biophys (2006) 449:164–70. doi: 10.1016/j.abb.2006.02.006
36. Dobo J, Szakacs D, Oroszlan G, Kortvely E, Kiss B, Boros E, et al. MASP-3 is the exclusive pro-factor D activator in resting blood: the lectin and the alternative complement pathways are fundamentally linked. Sci Rep (2016) 6:31877. doi: 10.1038/srep31877
37. Pihl R, Jensen L, Hansen AG, Thogersen IB, Andres S, Dagnes-Hansen F, et al. Analysis of factor D isoforms in Malpuech-Michels-Mingarelli-Carnevale patients highlights the role of MASP-3 as a maturase in the alternative pathway of complement. J Immunol (2017) 199:2158–70. doi: 10.4049/jimmunol.1700518
38. Hayashi M, Machida T, Ishida Y, Ogata Y, Omori T, Takasumi M, et al. Cutting edge: role of MASP-3 in the physiological activation of factor D of the alternative complement pathway. J Immunol (2019) 203:1411–6. doi: 10.4049/jimmunol.1900605
39. Yongqing T, Drentin N, Duncan RC, Wijeyewickrema LC, Pike RN. Mannose-binding lectin serine proteases and associated proteins of the lectin pathway of complement: two genes, five proteins and many functions? Biochim Biophys Acta (2012) 1824:253–62. doi: 10.1016/j.bbapap.2011.05.021
40. Mortensen SA, Skov LL, Kjaer-Sorensen K, Hansen AG, Hansen S, Dagnaes-Hansen F, et al. Endogenous natural complement inhibitor regulates cardiac development. J Immunol (2017) 198:3118–26. doi: 10.4049/jimmunol.1601958
41. Girija UV, Furze CM, Gingras AR, Yoshizaki T, Ohtani K, Marshall JE, et al. Molecular basis of sugar recognition by collectin-K1 and the effects of mutations associated with 3MC syndrome. BMC Biol (2015) 13:1–14. doi: 10.1186/s12915-015-0136-2
42. Koch A, Melbye M, Sorensen P, Homoe P, Madsen HO, Molbak K, et al. Acute respiratory tract infections and mannose-binding lectin insufficiency during early childhood. JAMA (2001) 285:1316–21. doi: 10.1001/jama.285.10.1316
43. Thiel S, Steffensen R, Christensen IJ, Ip WK, Lau YL, Reason IJ, et al. Deficiency of mannan-binding lectin associated serine protease-2 due to missense polymorphisms. Genes Immun (2007) 8:154–63. doi: 10.1038/sj.gene.6364373
44. Garred P, Genster N, Pilely K, Bayarri-Olmos R, Rosbjerg A, Ma YJ, et al. A journey through the lectin pathway of complement-MBL and beyond. Immunol Rev (2016) 274:74–97. doi: 10.1111/imr.12468
45. Pihl R, Jensenius JC, Thiel S. MASP-2. In: Barnum S, Schein T, editors. The Complement Factsbook, 2nd edition. London, UK: Academic Press, Elsevier (2018). p. 79–87.
46. Degn SE, Thiel S, Nielsen O, Hansen AG, Steffensen R, Jensenius JC. MAp19, the alternative splice product of the MASP2 gene. J Immunol Methods (2011) 373:89–101. doi: 10.1016/j.jim.2011.08.006
47. Garred P, Genster N, Pilely K, Bayarri-Olmos R, Rosbjerg A, Ma YJ, et al. A journey through the lectin pathway of complement – MBL and beyond. Immunol Rev (2016) 274:74–97. doi: 10.1111/imr.12468
48. Ma YJ, Lee BL, Garred P. An overview of synergy and crosstalk between pentraxins and collectins/ficolins: their functional relevance in complement activation. Exp Mol Med (2017) 49:e230. doi: 10.1038/emm.2017.51
49. Doni A, Garlanda C, Bottazzi B, Meri S, Garred P, Mantovani A. Interactions of the humoral pattern recognition molecule PTX3 with the complement system. Immunobiology (2012) 217:1122–8. doi: 10.1016/j.imbio.2012.07.004
50. Ma YJ, Garred P. Pentraxins in complement activation and regulation. Front Immunol (2018) 9:3046. doi: 10.3389/fimmu.2018.03046
51. Delgado-Rizo V, Martinez-Guzman MA, Iniguez-Gutierrez L, Garcia-Orozco A, Alvardo-Navarro A, Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol (2017) 8:81. doi: 10.3389/fimmu.2017.00081
52. Palmer LJ, Damgaard C, Holstrup P, Nierlsen CH. Influence of complement on neutrophil extracellular trap release induced by bacteria. J Periodont Res (2016) 51:70–6. doi: 10.1111/jre.12284
53. Daigo K, Takamatsu Y, Hamakubo T. The protective effect against extracellular histones afforded by long-pentraxin PTX3 as a regulator of NETs. Front Immunol (2016) 7:344. doi: 10.3389/fimmu.2016.00344
54. Daigo K, Yamaguchi N, Kawamura T, Matsubara K, Jiang S, Ohashi R, et al. The proteomic profile of circulating pentraxin 3 (PTX3) complex in sepsis demonstrates the interaction with azurocidin 1 and other components of neutrophil extracellular traps. Mol Cell Proteomics (2012) 11(6):M111.015073. doi: 10.1074/mcp.M111.015073
55. Daigo K, Hamakubo T. Host-protective effect of circulating pentraxin 3 (PTX3) and complex formation with neutrophil extracellular traps. Front Immunol (2012) 3:378. doi: 10.3389/fimmu.2012.00378
56. Super M, Thiel S, Lu J, Levinsky RJ, Turner MW. Association of low levels of mannan-binding protein with a common defect of opsonisation. Lancet (1989) 2(8674):1236–9. doi: 10.1016/S0140-6736(89)91849-7
57. Frakking FN, Brouwer N, van Eijkelenburg NK, Merkus MP, Kuijpers TW, Offringa M, et al. Low mannose-binding lectin (MBL) levels in neonates with pneumonia and sepsis. Clin Exp Immunol (2007) 150:255–62. doi: 10.1111/j.1365-2249.2007.03479.x
58. Swierzko AST, Atkinson APM, Cedzynski M, MacDonald SL, Szala A, Domzalska-Popadiuk I, et al. Two factors of the lectin pathway of complement, L-ficolin and mannan-binding lectin, and their associations with prematurity, low birthweight and infections in a large cohort of Polish neonates. Mol Immunol (2009) 46:551–8. doi: 10.1016/j.molimm.2008.07.025
59. Świerzko AS, Szala-Poździej A, Kilpatrick DC, Sobociński M, Chojnacka K, Sokołowska A, et al. Components of the lectin pathway of complement activation in paediatric patients of intensive care units. Immunobiology (2016) 221:657–69. doi: 10.1016/j.imbio.2016.01.003
60. Ozkan H, Koksal N, Cetinkaya M, Kilic S, Celebi S, Oral B, et al. Serum mannose-binding lectin (MBL) gene polymorphism and low MBL levels are associated with neonatal sepsis and pneumonia. J Perinatol (2012) 32:210–7. doi: 10.1038/jp.2011.79
61. Garred P, Madsen HO, Hofmann B, Svejgaard A. Increased frequency of homozygosity of abnormal mannan-binding protein alleles in patients with suspected immunodeficiency. Lancet (1995) 346:941–3. doi: 10.1016/S0140-6736(95)91559-1
62. Summerfield JA, Sumiya M, Levin M, Turner MW. Association of mutations in mannose binding protein gene with childhood infection in consecutive hospital series. BMJ (1997) 314:1229–32. doi: 10.1136/bmj.314.7089.1229
63. Cedzyński M, Szemraj J, Świerzko AST, Bąk-Romaniszyn L, Banasik M, Zeman K, et al. Mannan-binding lectin (MBL) insufficiency in children with recurrent infections of the respiratory system. Clin Exp Immunol (2004) 136:304–11. doi: 10.1111/j.1365-2249.2004.02453.x
64. Cedzyński M, Atkinson APM, Świerzko AST, MacDonald SL, Szala A, Zeman K, et al. L-ficolin (ficolin-2) insufficiency is associated with combined allergic and infectious respiratory disease in children. Mol Immunol (2009) 47:415–9. doi: 10.1016/j.molimm.2009.08.028
65. Atkinson APM, Cedzyński M, Szemraj J, Świerzko AST, Bąk-Romaniszyn L, Banasik M, et al. L-ficolin in children with recurrent respiratory infections. Clin Exp Immunol (2004) 138:517–20. doi: 10.1111/j.1365-2249.2004.02634.x
66. Ruskamp JM, Hoekstra MO, Postma DS, Kerkhof M, Bottema RW, Koppelman GH, et al. Exploring the role of polymorphisms in ficolin genes in respiratory tract infections in children. Clin Exp Immunol (2008) 155:433–40. doi: 10.1111/j.1365-2249.2008.03844.x
67. Elkoumi MA, Abdellatif SH, Mohamed FY, Sherif AH, Elashkar SSA, Saleh RM, et al. Ficolin-1 gene (FCN1) -144 C/A polymorphism is associated with adverse outcome of severe pneumonia in the under-five Egyptian children: A multicenter study. Pediatr Pulmonol (2020) 55:1175–83. doi: 10.1002/ppul.24719
68. Dogru D, Polat SE, Tan C, Tezcan I, Yalcin SS, Utine E, et al. Impact of mannose-binding lectin 2 gene polymorphisms on disease severity in noncystic fibrosis bronchiectasis in children. Pediatr Pulmonol (2020) 55:1190–8. doi: 10.1002/ppul.24711
69. Roy S, Knox K, Segal S, Griffiths D, Moore CE, Welsh KI, et al. MBL genotype and risk of invasive pneumococcal disease: a case-control study. Lancet (2002) 359:1569–73. doi: 10.1016/S0140-6736(02)08516-1
70. Kronborg G, Weis N, Madsen HO, Pedersen SS, Wejse C, Nielsen H, et al. Variant mannose-binding lectin alleles are not associated with susceptibility to or outcome of invasive pneumococcal infection in randomly included patients. J Infect Dis (2002) 185:1517–20. doi: 10.1086/340216
71. Gomi K, Tokue Y, Kobayashi T, Takahashi T, Watanabe A, Fujita T, et al. Mannose-binding lectin gene polymorphism is a modulating factor in repeated respiratory infections. Chest (2004) 126:95–9. doi: 10.1378/chest.126.1.95
72. Ali YM, Lynch NJ, Haleem KS, Fujita T, Endo Y, Hansen S, et al. The lectin pathway of complement activation is a critical component of the innate immune response to pneumococcal infection. PLoS Pathog (2012) 8(7):e1002793. doi: 10.1371/journal.ppat.1002793
73. Garcia-Laorden MI, Rodriguez de Castro F, Sole-Violan J, Payeras A, Briones ML, Borderias L, et al. The role of mannose-binding lectin in pneumococcal infection. Eur Respir J (2013) 41:131–9. doi: 10.1183/09031936.00174111
74. Garcia-Laorden MI, Sole-Violan J, Rodriguez de Castro F, Aspa J, Briones ML, Garcia-Saavedra A, et al. Mannose-binding lectin and mannose-binding lectin-associated serine protease 2 in susceptibility, severity, and outcome of pneumonia in adults. J Allergy Clin Immunol (2008) 122:368–74. doi: 10.1016/j.jaci.2008.05.037
75. Chalmers JD, Fleming GB, Rutherford J, Matsushita M, Kilpatrick DC, Hill AT. Serum ficolin-2 in hospitalised patients with community-acquired pneumonia. Inflammation (2014) 37:1635–41. doi: 10.1007/s10753-014-9891-4
76. Lynch NJ, Roscher S, Hartung T, Morath S, Matsushita M, Maennel DN, et al. L-ficolin specifically binds to lipoteichoic acid, a cell wall constituent of gram-positive bacteria, and activates the lectin pathway of complement. J Immunol (2004) 172:1198–202. doi: 10.4049/jimmunol.172.2.1198
77. Vassal-Stermann E, Lacroix M, Gout E, Laffly E, Pedersen CM, Martin L, et al. Human L-ficolin recognizes phosphocholine moieties of pneumococcal teichoic acid. J Immunol (2014) 193:5699–708. doi: 10.4049/jimmunol.1400127
78. Ali YM, Kenawy HI, Muhammad A, Sim RB, Andrew PW, Schwaeble WJ. Human L-ficolin, a recognition molecule of the lectin activation pathway of complement, activates complement by binding to pneumolysin, the major toxin of Streptococcus pneumoniae. PLoS One (2013) 8(12):e82583. doi: 10.1371/journal.pone.0082583
79. Brady AM, Calix JJ, Yu J, Geno KA, Cutter GR, Nahm MH. Low invasiveness of pneumococcal serotype 11A is linked to ficolin-2 recognition of O-acetylated capsule epitopes and lectin complement pathway activation. J Infect Dis (2014) 210:1155–65. doi: 10.1093/infdis/jiu195
80. Chapman SJ, Vannberg FO, Khor CC, Segal S, Moore CE, Knox K, et al. Functional polymorphisms in the FCN2 gene are not associated with invasive pneumococcal disease. Mol Immunol (2007) 44:3267–70. doi: 10.1016/j.molimm.2006.04.013
81. Stengaard-Pedersen K, Thiel S, Gadjeva M, Moller-Kristensen M, Sorensen R, Jensen LT, et al. Inherited deficiency of mannan-binding lectin associated serine protease 2. N Engl J Med (2003) 349:554–60. doi: 10.1056/NEJMoa022836
82. Olesen HV, Jensenius JC, Steffensen R, Thiel S, Schiotz PO. The mannan-binding lectin pathway and lung disease in cystic fibrosis - disfunction of mannan-binding lectin-associated serine protease 2 (MASP-2) may be a major modifier. Clin Immunol (2006) 121:324–31. doi: 10.1016/j.clim.2006.08.014
83. van Kempen G, Meijvis S, Endeman H, Vlaminckx B, Meek B, de Jong B, et al. Mannose-binding lectin and L-ficolin polymorphisms in patients with community-acquired pneumonia caused by intracellular pathogens. Immunology (2017) 151:81–8. doi: 10.1111/imm.12705
84. Eisen DP, Stubbs J, Spilsbury D, Carnie J, Leydon J, Howden BP. Low mannose-binding lectin complement activation function is associated with predisposition to Legionnaires’ disease. Clin Exp Immunol (2007) 149:97–102. doi: 10.1111/j.1365-2249.2007.03390.x
85. Munthe-Fog L, Hummelshoj T, Honore C, Madsen HO, Permin H, Garred P. Immunodeficiency associated with FCN3 mutation and ficolin-3 deficiency. N Engl J Med (2009) 360:2637–44. doi: 10.1056/NEJMoa0900381
86. Haerynck F, Van Steen K, Cattaert T, Loeys B, Van Daele S, Schelstraete P, et al. Polymorphisms in the lectin pathway genes as a possible cause of early chronic Pseudomonas aeruginosa colonization in cystic fibrosis patients. Hum Immunol (2012) 73:1175–83. doi: 10.1016/j.humimm.2012.08.010
87. Denholm JT, McBryde ES, Eisen DP. Mannose-binding lectin and susceptibility to tuberculosis: a meta-analysis. Clin Exp Immunol (2010) 162:84–90. doi: 10.1111/j.1365-2249.2010.04221.x
88. Wu YJ, Yang X, Chen TD, Zhang ZX, You YZ, Fan ZD. Genetic polymorphisms of exon 1 of MBL2 contribute to tuberculosis risk especially in Asian populations: an updated meta-analysis of 26 studies. Infect Drug Resist (2018) 11:1237–48. doi: 10.2147/IDR.S168465
89. Cao Y, Wang X, Cao Z, Wu C, Wu D, Cheng X. Genetic polymorphisms of MBL2 and tuberculosis susceptibility: a meta-analysis of 22 case-control studies. Arch Med Sci (2018) 14:1212–32. doi: 10.5114/aoms.2017.65319
90. Tong X, Wan Q, Li Z, Liu S, Huang J, Wu M, et al. Association between the mannose-binding lectin (MBL)-2 gene variants and serum MBL with pulmonary tuberculosis: An update meta-analysis and systematic review. Microb Pathog (2019) 132:374–80. doi: 10.1016/j.micpath.2019.04.023
91. Mandal RK, Khan MA, Hussain A, Dar SA, Aloufi S, Jawed A, et al. Association of MBL2 gene polymorphisms with pulmonary tuberculosis susceptibility: trial sequence meta-analysis as evidence. Infect Drug Resist (2019) 12:185–210. doi: 10.2147/IDR.S188980
92. Luo F, Sun X, Wang Y, Wang Q, Wu Y, Pan Q, et al. Ficolin-2 defends against virulent Mycobacteria tuberculosis infection in vivo, and its insufficiency is associated with infection in humans. PLoS One (2013) 8(9):e73859. doi: 10.1371/journal.pone.0073859
93. Kobayashi T, Kuronuma K, Saito A, Ikeda K, Ariki S, Saitou A, et al. Insufficient serum L-ficolin is associated with disease presence and extent of pulmonary Mycobacterium avium complex disease. Resp Res (2019) 20(1):224. doi: 10.1186/s12931-019-1185-9
94. Sokołowska A, Świerzko AS, Szala-Poździej A, Augustynowicz-Kopeć E, Kozińska M, Niemiec T, et al. Selected factors of the innate immunity in Polish patients suffering from pulmonary tuberculosis. Immunobiology (2020) 225:151905. doi: 10.1016/j.imbio.2020.151905
95. Ammitzboll CG, Kjaer TR, Steffensen R, Stengaard-Pedersen K, Nielsen HJ, Thiel S, et al. Non-synonymous polymorphisms in the FCN1 gene determine ligand-binding ability and serum levels of M-ficolin. PLoS One (2012) 7(11):e50585. doi: 10.1371/journal.pone.0050585
96. Crosdale DJ, Poulton KV, Ollier WE, Thomson W, Denning DW. Mannose-binding lectin gene polymorphism as a susceptibility factor for chronic necrotizing pulmonary aspergillosis. J Infect Dis (2001) 184:653–6. doi: 10.1086/322791
97. Bidula S, Sexton DW, Yates M, Abdolrasouli A, Shah A, Wallis R, et al. H-ficolin binds Aspergillus fumigatus leading to activation of the lectin complement pathway and modulation of lung epithelial immune responses. Immunology (2015) 146:281–91. doi: 10.1111/imm.12501
98. Ma YJ, Doni A, Hummelshoj T, Honore C, Bastone A, Mantovani A, et al. Synergy between ficolin-2 and pentraxin 3 boosts innate immune recognition and complement deposition. J Biol Chem (2009) 284:28263–75. doi: 10.1074/jbc.M109.009225
99. Bidula S, Sexton DW, Abdolrasouli A, Shah A, Reed A, Armstrong-James D, et al. The serum opsonin ficolin-2 is detected in lungs of human transplant recipients following fungal infections and modulates inflammation and killing of Aspergillus fumigatus. J Infect Dis (2015) 212:234–46. doi: 10.1093/infdis/jiv027
100. Cunha C, Aversa F, Lacerda JF, Busca A, Kurzai O, Grube M, et al. Genetic PTX3 deficiency and aspergillosis in stem-cell transplantation. N Engl J Med (2014) 370:421–32. doi: 10.1056/NEJMoa1211161
101. Malhotra R, Haurum JS, Thiel S, Sim RB. Binding of human collectins (SP-A and MBP) to influenza virus. Biochem J (1994) 304:455–61. doi: 10.1042/bj3040455
102. Kase T, Suzuki Y, Kawai T, Sakamoto T, Ohtani K, Eda S, et al. Human mannan-binding lectin inhibits the infection of influenza A virus without complement. Immunology (1999) 97:385–92. doi: 10.1046/j.1365-2567.1999.00781.x
103. Thielens NM, Tacnet-Delorme P, Arlaud GJ. Interaction of C1q and mannan-binding lectin with viruses. Immunobiology (2002) 205:563–74. doi: 10.1078/0171-2985-00155
104. Ribeiro LZ, Tripp RA, Rossi LM, Palma PV, Yokosawa J, Mantese OC, et al. Serum mannose binding lectin levels are linked with respiratory syncytial virus (RSV) disease. J Clin Immunol (2008) 28:166–73. doi: 10.1007/s10875-007-9141-8
105. Ip WK, Chan KH, Law HK, Tso GH, Kong EK, Wong WH, et al. Mannose-binding lectin in severe acute respiratory syndrome coronavirus infection. J Infect Dis (2005) 1910:1697–704. doi: 10.1086/429631
106. Zhang H, Zhou G, Zhi L, Yang H, Zhai Y, Dong X, et al. Association between mannose-binding lectin gene polymorphisms and susceptibility to severe acute respiratory syndrome coronavirus infection. J Infect Dis (2005) 192:1355–61. doi: 10.1086/491479
107. Yuan FF, Tanner J, Chan PKS, Biffin S, Dyer WB, Geczy AF, et al. Influence of FcγRIIA and MBL polymorphisms on severe acute respiratory syndrome. Tissue Antigens (2005) 66:291–6. doi: 10.1111/j.1399-0039.2005.00476.x
108. Chalmers JD, Fleming GB, Hill AT, Kilpatrick DC. Impact of mannose-binding lectin insufficiency on the course of cystic fibrosis: a review and meta-analysis. Glycobiology (2011) 21:271–82. doi: 10.1093/glycob/cwq161
109. Chalmers JD, McHugh BJ, Doherty C, Smith MP, Govan JR, Kilpatrick DC, et al. Mannose-binding lectin deficiency and disease severity in non-cystic fibrosis bronchiectasis: a prospective study. Lancet Respir Med (2013) 3:224–32. doi: 10.1016/S2213-2600(13)70001-8
110. Macfarlane JG, Jary H, Hester KLM, McAlinden P, Wake J, Small T, et al. Low serum mannose-binding lectin level is not associated with disease severity in non-cystic fibrosis bronchiectasis. Innate Immun (2012) 18:782–92. doi: 10.1177/1753425912440472
111. Gao T, Hu M, Zhang X, Li H, Zhu L, Liu H, et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. MedRxiv (2020) 2020.03.29.20041962. doi: 10.1101/2020.03.29.20041962
112. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
113. Chatterjee SK, Saha S, Munoz MNM. Molecular pathogenesis, immunopathogenesis and novel therapeutic strategy against COVID-19. Front Mol Biosci (2020) 7:196. doi: 10.3389/fmolb.2020.00196
114. Zhou Y, Lu K, Pfefferle S, Bertram S, Glowacka I, Drosten C, et al. A single asparagine-linked glycosylation site of the severe acute respiratory syndrome coronavirus spike glycoprotein facilitates inhibition by mannose-binding lectin through multiple mechanisms. J Virol (2010) 84:8753–64. doi: 10.1128/JVI.00554-10
115. Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio (2018) 9:e01753–18. doi: 10.1128/mBio.01753-18
116. Mastaglio S, Ruggeri A, Risitano AM, Angelillo P, Yancopolou D, Mastellos DC, et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol (2020) 215:108450. doi: 10.1016/j.clim.2020.108450
117. Risitano AM, Mastellos DC, Huber-Lang M, Yancopolou D, Garlanda C, Ciceri F, et al. Complement as a target in COVID-19? Nat Rev Immunol (2020) 20:343–4. doi: 10.1038/s41577-020-0320-7
118. Diurno F, Numis FG, Porta G, Cirillo, Maddaluno S, Ragozzino A, et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur Rev Med Pharmacol Sci (2020) 24:4040–7. doi: 10.26355/eurrev_202004_20875
119. Song W-C, FitzGerald GA. COVID-19, microangiopathy, hemostatic activation, and complement. J Clin Invest (2020) 130:3950–3. doi: 10.1172/JCI140183
120. Polycarpou A, Howard M, Farrar CA, Greenlaw R, Fanelli G, Wallis R, et al. Rationale for targeting complement in COVID-19. EMBO Mol Med (2020) 12:e12642. doi: 10.15252/emmm.202012642
121. Chauhan AJ, Wiffen LJ, Brown TP. COVID-19: A collision of complement, coagulation and inflammatory pathways. J Thromb Haemost (2020) 18:2110–7. doi: 10.1111/jth.14981
122. Stahel PF, Barnum SR. Complement inhibition in coronavirus disease (COVID)-19: a neglected therapeutic option. Front Immunol (2020) 11:1661. doi: 10.3389/fimmu.2020.01661
123. Matricardi PM, Dal Negro RW, Nisini R. The first, holistic model of COVID-19: implications for prevention, diagnosis, and public health measures. Pediatr Allergy Immunol (2020) 31:454–70. doi: 10.1111/pai.13271
124. Eriksson O, Hulstrom M, Persson B, Lipcsey M, Nilsson Ekdahl K, Nilsson B, et al. Mannose-binding lectin is associated with thrombosis and coagulopathy in critically ill COVID-19 patients. Thromb Haemost (2020). doi: 10.1055/s-0040-1715835
125. Holter JC, Pischke SE, de Boer E, Lind A, Jenum S, Holten AR, et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc Natl Acad Sci U S A (2020) 117:25018–25. doi: 10.1073/pnas.2010540117
Keywords: collectin, complement, ficolin, MASP, mannose-binding lectin, respiratory infection, MBL
Citation: Świerzko AS and Cedzyński M (2020) The Influence of the Lectin Pathway of Complement Activation on Infections of the Respiratory System. Front. Immunol. 11:585243. doi: 10.3389/fimmu.2020.585243
Received: 20 July 2020; Accepted: 01 October 2020;
Published: 21 October 2020.
Edited by:
Mihály Józsi, Eötvös Loránd University, HungaryReviewed by:
Péter Gál, Hungarian Academy of Sciences (MTA), HungaryYing Jie Ma, Copenhagen University Hospital, Denmark
Copyright © 2020 Świerzko and Cedzyński. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maciej Cedzyński, bWNlZHp5bnNraUBjYm0ucGFuLnBs