 Heather F. Jones
Heather F. Jones Zaki Molvi
Zaki Molvi Martin G. Klatt1
Martin G. Klatt1 Tao Dao
Tao Dao- 1Molecular Pharmacology Program, Memorial Sloan Kettering Cancer Center, New York, NY, United States
- 2Weill Cornell Medicine, New York, NY, United States
- 3Immunology Program, Memorial Sloan Kettering Cancer Center, New York, NY, United States
The use of T cells reactive with intracellular tumor-associated or tumor-specific antigens has been a promising strategy for cancer immunotherapies in the past three decades, but the approach has been constrained by a limited understanding of the T cell receptor’s (TCR) complex functions and specificities. Newer TCR and T cell-based approaches are in development, including engineered adoptive T cells with enhanced TCR affinities, TCR mimic antibodies, and T cell-redirecting bispecific agents. These new therapeutic modalities are exciting opportunities by which TCR recognition can be further exploited for therapeutic benefit. In this review we summarize the development of TCR-based therapeutic strategies and focus on balancing efficacy and potency versus specificity, and hence, possible toxicity, of these powerful therapeutic modalities.
Introduction
Harnessing potent cellular effectors, such as cytotoxic T cells, and soluble molecules of the human immune system has become a successful strategy in the treatment of cancers of a variety of types. While often effective and generally well-tolerated, these effectors are not truly specific for the tumor. Typically, these therapies can either broadly activate cellular effectors, such as with interleukins, interferons, or checkpoint blockade antibodies, or are directed to lineage markers or cell surface differentiation antigens also found on normal cells and tissues. For example, monoclonal antibody (mAb) and chimeric antigen receptor (CAR) T cell therapies have emerged as some of the most successful and important strategies in cancer therapy. However, these modalities are traditionally reactive with a limited repertoire of extracellular antigens. For many cancers, appropriate antigens have not been identified. In contrast, the TCR evolved to detect subtle changes in cellular proteins that can include viral peptides or mutated oncogenic proteins. Thus, TCR-based agents can be directed to the vast majority of truly tumor-specific antigens, or relatively specific tumor-associated proteins, which are derived from intracellular proteins (1–3). Peptides derived from proteins of any subcellular location are presented on the cell surface in the context of major histocompatibility complexes (MHC), known as human leukocyte antigens (HLA) when referring to human MHC, where they are recognized by T cells through their TCRs (3). TCR-based therapies are able to recognize and react to cells expressing these mutated or differentially expressed, cancer-associated proteins presented on MHC. The exploitation of this powerful modality to treat cancer and possibly other serious diseases is dependent on understanding the unique features of their recognition and effector activities, the types of structures that can be developed to take advantage of these functions, and the possible liabilities that these molecules carry.
Immunotherapeutic modalities that take advantage of the TCR’s unique ability to recognize intracellular proteins are both molded by and constrained by key aspects of their structural features and those of their targets, as well as the origins of their antigenic specificity. Critical determinants of success for these agents are (1) the characteristics of the epitope (2); the affinity, avidity, and cellular geometry of the TCR; and (3) the recognition specificity unique to the antigen-TCR interaction. These features of TCRs are markedly divergent from the analogous features of antibodies and must be tackled accordingly to create a successful agent. First, unlike the conformational structure of the molecular targets of antibodies, the potential amino acid sequence epitopes for these TCR agents must be appropriately, expressed, processed, and presented on the cell surface. While peptide presentation on MHC molecules can be predicted in silico, these approaches are inaccurate and ideally, selected epitopes should be validated by using mass spectrometry to verify peptide-MHC presentation and followed by in vitro assays to characterize the functionality of target-specific T cells. Second, although unmodified, native TCRs reactive with peptides in context with their MHC proteins are more likely to yield appropriate specificity and functionality that mimic the actions of an endogenous T cell, as compared to a modified TCR, such native TCRs have orders of magnitude lower affinity than antibodies, which can limit their pharmacologic uses. TCRs may need affinity enhancement to increase the peptide-MHC recognition. In addition, native TCRs, unlike antibodies that operate in solution, cooperate as a collection of molecules along with other proteins in a cell membrane synapse on the T cell that vastly alters their effector functions. Third, TCRs, because of their low affinity and the complex structure of their epitope targets, are far more promiscuous than antibodies; strategies to predict toxicities by determining on-target/off-tumor and off-target antigen recognition of TCR-based agents are essential to ensure TCR agent safety, but such methods are currently in their infancy. There are no marketed drugs in the United States that are based upon the TCR. In this review, we will discuss various approaches to identify, address and overcome these constraints to TCR-based agents in order to advance these innovative drugs to clinical trials (Table 1; Figure 1).

Table 1 TCR-based agents in development.
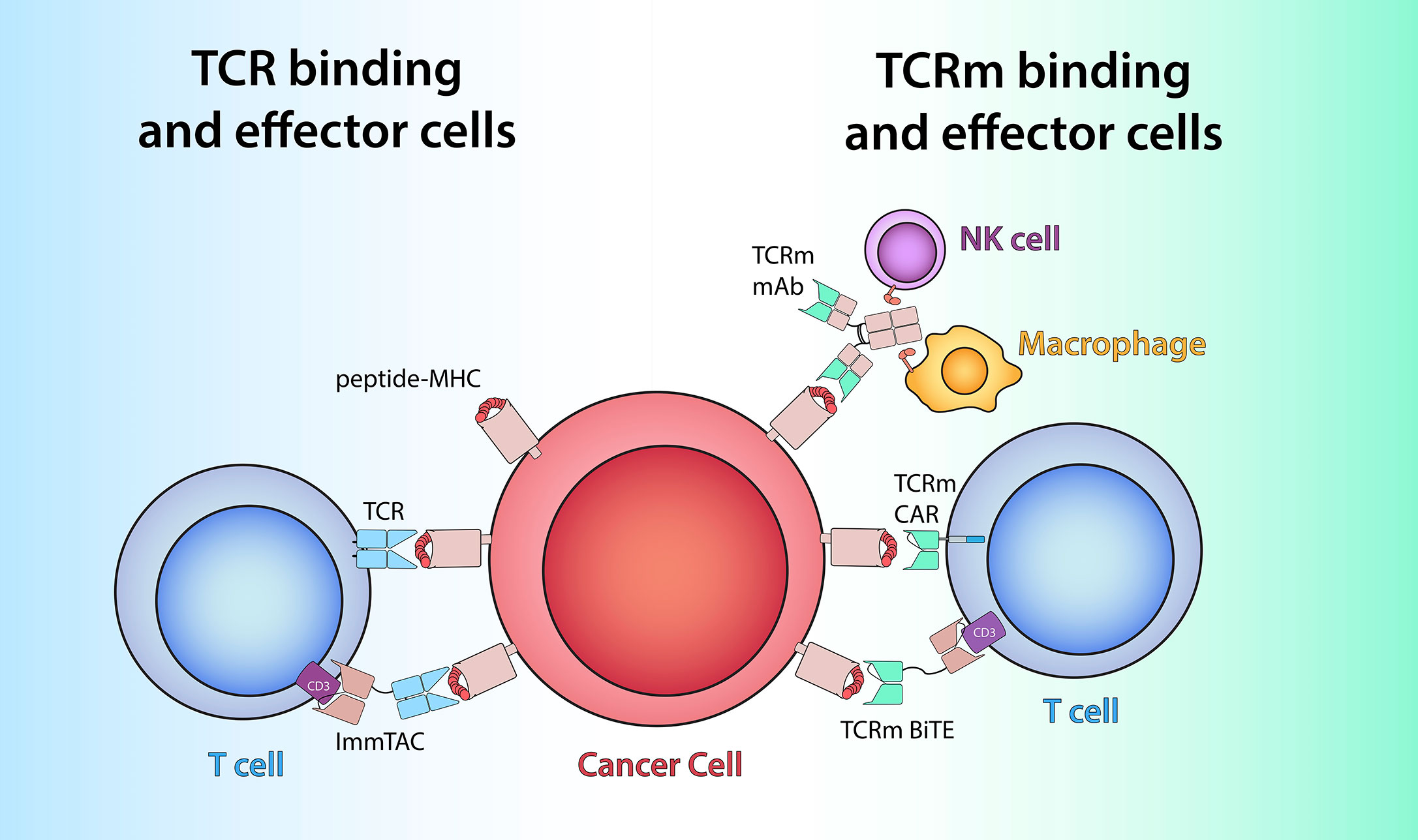
Figure 1 TCR-based therapeutics recognize peptide/MHC antigens (red and pink) on cells by utilizing either TCRs (light blue) or TCRm antigen-binding domains (green). Left: Soluble ImmTAC molecules bind peptide/MHC on cancer cells via alpha/beta TCR heterodimer similar to membrane-bound TCR and redirect the T cells by engaging extracellular CD3-epsilon (purple) via an anti-CD3 scFv. Right: TCRm mAb recognize peptide/MHC complex via its variable region (green) and to engage effector cells such as NK cells and macrophages to elicit Fc-receptor (orange) mediated ADCC or ADCP. TCRm CAR and bispecific mAb leverage TCRm-derived scFv to harness T cell effector function via engagement with intracellular CD3-zeta (blue) or extracellular CD3-epsilon (purple), respectively.
Identification of Targets of TCR-Based Agents
Overall, advancements in screening techniques and engineering now provide multiple approaches and formats to achieve the goal of peptide-MHC recognition to target antigens. However, insufficient processing and presentation of the targeted epitope on the cell surface may limit activity. This underlines the importance of validation of target epitopes to ensure high levels of tumor specificity and efficacy. Ideally this can be achieved in advance by immunoprecipitation of MHC complexes and subsequent mass spectrometry identification of the displayed ligands.
The landscape of targets for TCR therapy of non-viral malignancies is comprised of antigens that demarcate aberrant cells, albeit to a highly variable degree (4). This nuance renders TCR target selection non-trivial. For the purposes of this review, we will divide TCR targets into two broad classes: self-antigens, derived from overexpressed proteins, and neoantigens, which exhibit subtle deviations from self due to the malignant state (5). The common feature between these two is that both are derived from the human genome; however, neoantigens arise directly from genomic deviations caused by the genomic instability of cancer.
The earliest class of cancer antigens known to be recognized by TCRs include self-antigens derived from proteins that are over-presented by MHC in tumor tissue. Classic examples that have been extensively studied are MART-1, Wilms’ tumor-1 (WT1), PR1, MAGE-A3, NY-ESO-1, carcinoembryonic antigen (CEA) and PRAME (6–8). However, an important distinction is that some of these are cancer-associated by means of their lineage-specificity, such as MART-1 and CEA, whereas others are cancer germline antigens, such as NY-ESO-1 and PRAME, that are only expressed in immune privileged sites such as the placenta or testis, but are re-expressed due to genomic instability in tumor cells (9). Lineage associated antigens require careful consideration of on-target/off-tumor effects associated with TCR therapy (7, 10). In contrast, TCRs targeting cancer germline antigens may confer greater tumor-specific recognition, but may be attenuated by escape mechanisms as these are not typically oncogenes critical for tumor survival (5). As this type of tumor-associated antigen has been studied for decades many of the used targets were also confirmed to be presented on the cell surface by MHC ligand isolation and mass spectrometry which renders them bona fide targets (11–13). The growing number of studies utilizing mass spectrometry to verify the presentation of HLA ligands is expected to increase confidence in presented epitopes derived from cancer germline antigens and potentially uncover new epitopes against which new TCR therapeutics can be developed.
More recently neoantigens, peptide antigens that are the result of missense mutations, frameshift mutations, or post-translational modifications, have been exploited as novel targets for TCR-based immunotherapies. However, other types of alterations can produce a neoantigen (14). Neoantigens encoded by missense mutations harbor a nonsynonymous amino acid substitution produced by either a driver or passenger mutation that can be distinguished by T cells by means of an augmented MHC binding affinity or altered TCR recognition. Frameshift mutations produce neoantigens that can be drastically different from wildtype protein-derived peptides, harboring multiple amino acid changes (15). Post-translational amino acid modifications, such as phosphorylation and glycosylation, can exhibit a greatly altered capacity to bind MHC and present a fundamentally different binding moiety to T cells compared to its unmodified variant (16, 17). Careful consideration of a tumor’s biologic properties is also necessary to guide effective target selection. For example, for hematologic malignancies self-antigens such as WT1, PR1, or PRAME, remain among the most useful TCR targets for these tumors rather than rare mutant neoantigens (18–22).
Neoantigens derived from frameshift mutations represent a type of shared neoantigen that is particularly prominent in clear cell renal cell carcinoma (ccRCC) in which neoantigens from common tumor suppressors such as TP53, PTEN, MLL2, MLL3 and ARID1A have been observed (23) and found to be recognized by tumor-infiltrating lymphocytes (TIL) (24). Strikingly, the immunogenic frameshift neoantigens were more distinct from the human proteome than were immunogenic missense mutations, a distinction that has also been made for immunogenic frameshifts in microsatellite instability-high tumors. Because of their high degree of sharing between tumors, immunogenicity, and derivation from driver mutations, frameshift-derived neoantigens may constitute a rational TCR target for tumors with adequate frameshift load.
Post-translationally modified peptides are an emerging class of neoantigens, the discovery of which has been accelerated by recent advances in mass spectrometry (25). Early reports discovered shared phosphorylated peptides (phosphopeptides) presented by tumor cell lines (26, 27) and leukemias (28). Phosphopeptides are an attractive target due to their consistently observed immunogenicity in normal donors, enhanced binding properties, and relationship to aberrant cancer metabolism (16, 28, 29). A recent phosphopeptide vaccine trial in melanoma patients elicited T cell reactivity in patients, suggesting that immune responses to these targets can be generated by tumor-bearing hosts (30). Glycosylated and acetylated peptides have similarly been shown to be immunogenic epitopes presented by tumor cells in similar studies (17, 31, 32). Proteomic data are expected to give depth to the cancer-specific modified peptide repertoire and provide a valuable link between post-translationally modified antigen presentation and tumor metabolism with additional classes of tumor-specific intracellular antigens identified and validated.
Although solid tumors present self-antigens, the efficacy of immune checkpoint blockade in highly mutated solid tumors such as melanoma and non-small cell lung carcinoma (NSCLC) has shifted interest to targeting one of the numerous neoantigens. Correlative studies have repeatedly demonstrated that a critical factor of response to checkpoint blockade is tumor mutational burden, with highly mutated tumors being more likely to respond (33, 34). This paradigm has been further validated by the discovery of T cells that recognize missense-derived neoantigens in responding patients (35). Subsequently, tumor exome sequencing and MHC binding predictions have become an invaluable tool to determine neoantigen load on a patient-by-patient basis (36). However, the accrual of neoantigens as a tumor evolves is unpredictable. Most neoantigens are derived from passenger mutations private to each individual patient or each tumor, rendering scalable TCR targeting infeasible. Furthermore, though some patient specific neoepitopes can be detected by mass spectrometry and their presentation therefore validated, the vast majority of predicted neoepitopes cannot be identified by this technique which further complicates target selection for patient-specific therapeutic approaches (37, 38). Neoantigens derived from shared driver mutations, such as KRAS and PIK3CA missense mutants where presentation has been validated for some HLA alleles (39), might overcome this issue (40–42).
Regulation of Epitope Presentation
One potential disadvantage of targeting peptides presented by MHC molecules is their paucity of density on the cell surface, which may be 100 to 1000-fold lower than other antigens. This can be further exacerbated by down regulation of MHC by cancer cells as a method of immune escape (43). To deal with this issue, the immunopeptidome can be altered by drug or cytokine treatment to either augment expression or investigate the emergence of novel targets. Physiological alterations by interferon gamma (IFNγ) or tumor necrosis factor alpha (TNFα) favor the presentation of longer peptides as well as ligands which preferably bind to HLA-B alleles (44, 45). In addition to upregulating the HLA expression level, IFNγ also can lead to the specific increase in presentation of a given TCR or TCR-mimic monoclonal antibody (TCRm) peptide target through induction of the cytoplasmic immunoproteasome (46–49). This specific induction of ligand presentation could be utilized to render previously unreactive cells targetable (46).
Interestingly, ALK, RET, MEK, and other mitogen-activated protein kinase (MAPK) pathway inhibitors lead not only to a significant increase of HLA complex surface expression, which can help overcome immune escape via downregulation of HLA, but also to a large qualitative change of the displayed peptides (43, 50, 51). Many of which are potentially immunogenic epitopes. Surprisingly, ALK and RET inhibitor treatment can also lead to the presentation of T cell epitopes associated with impaired peptide processing (TEIPPs) which are usually only observed in transporter associated with antigen processing (TAP)-deficient or downregulated cells with low HLA levels. Treatment with the MEK inhibitor trametinib illustrates an example of improved efficacy of a specific TCR-like drug targeting an epitope from the MART-1 protein (50).
The ligandome might be affected to improve presentation of a specific TCR target by use of the proteasome inhibitor carfilzomib, which disfavors presentation of HLA ligands with aromatic C-terminal anchors by altering proteasomal cleavage patterns, as well as the ERAP1 inhibitor, DG013A, which augments presentation of ligands with higher affinity and shorter peptide length by changing endoplasmic peptide trimming (52, 53). Other chemotherapeutic agents, such as gemcitabine can lead to changes through upregulation of HLA-A,B,C complexes and also through immunoproteasome induction (54) or oxaliplatin, which can increase detection by CD4+ T cells through class II peptide presentation (55).
Hypomethylating drugs alone or in combination with histone deacetylase (HDAC) inhibitors are the most effective class of drugs that increase the presentation of specific HLA ligands. This has been most extensively demonstrated in acute myeloid leukemia (AML) for NY-ESO-1, MAGE-A3, and MAGE-A6 (56). Reinduction of cancer germline antigens through reversion of genetic repression marks is also feasible in many solid cancers, e.g. in esophageal squamous cell carcinoma for MAGE-A3 (57), ovarian cancer for protein expressed in prostate, ovary, testis and placenta (POTE) genes (58), mesothelioma for NY-ESO-1, MAGE-A1, MAGE-A3, and XAGE-1b (59) and prostate cancer for NY-ESO-1 (60). Since the presentation of HLA ligands can be altered qualitatively and quantitatively through multiple FDA-approved drugs, therapeutic strategies may benefit from combination therapies of TCR-based agents with HDAC inhibitors and hypomethylating drugs. The synergistic effects of HDAC inhibitors and hypomethylating drugs have been shown to be especially important for immunotherapies as the repressive marks on relevant genes can often only be sufficiently reversed by such a combination treatment (61).
Generation of Therapeutic T Cells
T cells reactive with tumor associated antigens and neoantigens have been found in TILs and peripheral blood lymphocytes (PBL) (62, 63) of cancer patients, as well as in PBLs of healthy donors (64). Such cells, or their TCRs, would be expected to be an appropriate source for effective therapeutic agents. Despite this, many endogenous T cells, including those found within tumors, are still unable to eradicate tumors presenting their cognate antigen. This failure can be attributed in part to the immunosuppressive tumor microenvironment (TME) (3), low affinity of endogenous T cell receptors (TCR) for tumor associated antigens (65, 66), and possibly other factors. Numerous approaches have been employed to generate a more potent anti-tumor T cell response. Tumor-reactive T cells expressing native TCRs can be stimulated in vivo through administration of vaccines, checkpoint blockade inhibitors, or cytokines. Alternatively, reactive T cells can be expanded ex vivo and reinfused for adoptive cell therapy (67). Tumor-reactive T cells can be enriched and used in bulk for treatment, or their individual reactive TCRs can be sequenced and subsequently expressed exogenously in T cells prior to reinfusion.
Cancer Vaccines
Naturally occurring tumor-reactive T cells can be stimulated to boost the anti-tumor T cell response through vaccination with tumor antigens. Vaccines can be peptides (68), DNA or RNA products (69), whole proteins, viruses encoding antigenic peptides (68) or autologous dendritic cells presenting peptide antigens (68, 70). Patients may receive a personalized cancer vaccine, where target peptides are chosen from tumor-specific mutations identified by whole exome sequencing (WES) and filtered through HLA binding prediction (69–71). Some peptides used for vaccination can be modified to further increase their immunogenicity, by substituting peptide residues to result in better binding to HLA molecules. Such “heteroclitic” peptides have been shown to induce stronger T cell responses that cross-react with their native sequences. Characteristic examples are the HLA-A*02:01 restricted peptides, NY-ESO-1 (SLLMWITQC) and WT1 (RMFPNAPYL), where replacing the final amino acid with valine (72), or the first amino acid with tyrosine (73), respectively, increases the HLA binding affinity resulting in increased immunogenicity and enhanced T cell activation. Cancer vaccines have been shown to increase the preexisting anti-tumor T cell response and have proven effective in some patients (68–70, 74–76). Although cancer vaccines are a widely used approach in investigational clinical trials, due to their limited clinical efficacy to date, more potent, passive therapeutic approaches using TCR recognition of tumor antigens have been developed in recent years. This review will focus on these new approaches.
Adoptive T Cell Therapy
Tumor reactive T cells, present either in TILs or PBLs, can be removed from patients for rapid expansion ex vivo, outside of the immunosuppressive TME (77), prior to use for adoptive cell therapy (67). T cells can be stimulated for expansion with resected tumor (67, 78, 79), antibodies targeting CD3 and CD28 (80), or peptide antigens (81, 82). Synthetic peptides of neoepitopes identified by whole exome or RNA sequencing and subsequent HLA binding prediction can be pulsed onto (64, 83–85) or expressed as tandem minigenes (84, 86, 87) on antigen presenting cells, often autologous dendritic cells. For tandem minigenes, mutations are flanked by sequences encoding endogenous amino acids allowing the peptides to be processed and presented on MHC. When using tandem minigenes the reactive peptide and MHC restriction of the respective neoepitope does not have to be identified and it allows multiple antigens to be expressed and presented on the same cell (77, 84, 86, 87). Peptide stimulation has also been employed to generate reactive T cells from the peripheral blood of HLA matched healthy donors (64, 77, 88).
T cells with predefined antigen specificity can be isolated using peptide-MHC multimers (62, 63, 77, 89–94) through either magnetic enrichment or fluorescence activated cell sorting (FACS) when both the antigen and the MHC it binds are known or predicted. Patient specific neoantigens can be predicted based on WES data (94, 95). Multimers have been used to identify antigen specific T cells from patient TILs and PBLs (63, 95) and from healthy donor PBLs (91, 94). In contrast, cell surface biomarkers on T cells can be used to identify reactive T cells without knowledge of the specific antigen or the HLA on which it is presented (96–100). Several biomarkers, including PD-1 (96, 101, 102), LAG-3, TIM-3 (102), OX40 (103), CD137 (84, 97–100, 102–104) and CD107a (105) and cytokine production, such as of IFNγ, indicate the T cell has interacted with its cognate antigen and can be used to isolate T cells (106). Such markers have been used to identify tumor reactive cells from both TILs and PBLs (96). While these approaches have successfully identified tumor reactive T cells, there are limitations. Not all T cells that are multimer positive, and therefore peptide-MHC specific, are able to exert cytotoxic effects against tumor cells expressing these antigens (105). Conversely, multimer staining may not detect all antigen-reactive T cells (107–109). This can be due to decreased TCR surface density or expression of TCRs with low affinity. This can be especially problematic as TCRs reactive with self-antigens and MHC class II antigens tend to have lower affinity for their target. Multimer staining can be enhanced to detect low-affinity TCRs with protein kinase inhibitors, such as dasatinib which decreases TCR downregulation, cross linking antibodies to stabilize multimer binding, anti-coreceptor antibodies, and staining with multimers with more peptide-MHC sites, i.e. dextramers or dodecamers over tetramers (107, 109). Additionally, cytokine production and cytotoxicity are independently regulated, therefor cytokine production does not always correlate with cytotoxic potential (105, 110). These limitations can make it more challenging to accurately identify tumor-specific cytotoxic T cells.
TCR Gene Therapy
Isolated individual reactive TCRs can be transduced and expressed into other T cells, known as TCR-T cells, to broaden therapy to additional patients. Paired TCR alpha and beta chain sequences can be identified from tumor-reactive T cells for subsequent cloning into expression vectors from pooled T cell (111, 112) or single cell (77, 83, 89, 92, 97, 113) sequencing data. Other methods to generate reactive TCRs for subsequent identification circumvent thymic selection to generate high affinity T cells reactive against specific tumor antigens. Immunization of mice expressing human HLA molecules (42, 114) or mice expressing the human TCR repertoire can be used for immunization and isolation of high affinity TCRs (115, 116). High affinity human TCRs with increased activity also can be isolated when human T cells are stimulated ex vivo with tumor antigens on HLA mismatched antigen presenting cells (117–120).
Alternatively, individual TCRs can be affinity enhanced via protein engineering to increase their anti-tumor effects (7, 65, 121, 122). As few as one or two amino acid changes in the complementarity determining regions can increase the affinity of TCRs (8, 65, 114, 123), evident by slower TCR off rates (124). High throughput methods such as phage (124, 125), yeast (126, 127), and T cell display libraries (128, 129), along with somatic hypermutation (130), and in-vitro T cell differentiation (131) have been employed to generate high affinity TCRs, sometimes in conjunction with available structure data (132). While increasing TCR affinity has been shown to increase the effectiveness of the T cell (65, 74, 123), TCRs whose affinities are too high can become less effective (115) and are at higher risk for cross reactivity (74, 115, 123). Drawing any direct correlation between TCR affinity and T cell efficacy can be challenging as affinity itself is determined based on two parameters, the on- and off-rate of TCR binding (133). One model describes T cell responses as requiring long enough dwell time between the TCR and peptide-MHC to stimulate signaling but having a fast enough off-rate to allow for sequential TCR binding and signaling amplification (134, 135). Therefore longer TCR/peptide-MHC half-lives may prevent serial triggering and hamper T cell responses (134). Additionally, mechanisms that decrease the efficacy of T cells have been identified in association with T cells with high affinity TCRs (136). These mechanisms include impaired T cell signaling, upregulation of the inhibitory receptors, such as PD-1, down regulation of costimulatory receptors (135), peripheral deletion, expansion of anergic T cells (136, 137) and TCR down regulation (136).
Finally, T cell therapies can be designed to target patient-specific tumor antigens or public tumor antigens. T cell responses against patient-specific mutated neoantigens have been associated with clinical successes (83, 87, 138) and should be subject to less central tolerance, as such neoantigens are not present in normal tissues (102, 139). Neoantigen-reactive T cells can be highly tumor specific as T cells are able to distinguish between single amino acid changes in peptides, representing either unmutated self or mutant peptide sequences (95). While targeting neoantigens is expected to result in less toxicities (97, 102, 140), finding tumor and patient-specific antigens and reactive TCRs to generate patient specific TCR-T cells is challenging, costly and not currently feasible on a broad scale. Public tumor antigens are not patient or cancer-specific and while they sometimes can be derived from mutant peptides (140, 141), they are often unmutated self-peptides from tumor associated proteins that are minimally or not expressed on normal cells (142, 143). Public antigens have the benefit that the same TCR construct can be used to treat multiple patients (74).
Specificity of TCR-Based Therapies
One the most important questions for clinical application of TCR-based agents is specificity, in order to prevent off-target toxicities. T cells and other TCR-based therapies rely on precise recognition of a short linear peptide sequence, typically 8 to 11 amino acids in length in the groove of a largely structurally constrained HLA class I protein (144). Therefore, the TCR must be able to distinguish between the different antigenic peptides derived from thousands of proteins, which may comprise highly similar amino acid sequences, challenging absolute specificity. The estimated 100 million different TCRs expressed by a human is dwarfed by the number of potential sequence targets in the proteome. Therefore, it is speculated that each TCR can recognize hundreds to thousands of different antigens (145). In this way, TCR promiscuity can be a source of both greater scope of protection, but also significant off-target toxicity. Additionally, native TCRs can have low, micromolar affinity for their cognate target, especially if they are targeting non-mutated peptides, due to thymic selection (65, 66). While increasing affinity, as described above, may increase the anti-tumor effects of the TCR, bypassing thymic selection increases the risk for off-target reactivity and toxicity (122) and hence, a balance between TCR activity and toxicity must be struck. The severe off-target toxicities sometimes seen with TCR therapies has emphasized the need for methods to predict reactive off-target peptides and their cells of origin (65, 142).
On-Target/Off-Tumor Toxicity
TCR-based therapies can lead to autoimmune toxicities caused by on-target/off-tumor responses, which occurs when the target antigen is expressed on normal cells. On-target/off-tumor autoimmune toxicity has been seen in some melanoma patients treated with exogenously expanded TILs, which recognized non-malignant melanocytes, as with T cells reactive against melanocyte differentiation antigens, such as MART-1, and with DMF5 TCR-T cells, specifically reactive with the MART-1 antigen (10, 113, 146). Patients variably experienced uveitis, rash, vitiligo, and hearing loss (147). Interestingly these toxicities are seen with DMF5 TCR T cells, but not with T cells expressing the lower affinity DMF4 TCR (143, 147), reactive with the same MART-1 epitope. The higher affinity TCR, DMF5, is hence both more efficacious and more toxic (147). On-target/off-tumor colitis has additionally been seen with T cells using a TCR developed in an HLA-A*02:01 humanized mouse, affinity enhanced for binding to a CEA epitope presented on HLA-A*02:01, likely due to CEA expression on gastrointestinal cells (7).
Molecular Mimicry and Sequence Similarity Toxicity
Molecular mimicry is when a peptide is able to stimulate TCR reactivity due to structural similarities with the target peptide. An example of molecular mimicry was observed with the affinity enhanced a3a TCR, reactive with a MAGE-A3 peptide on HLA-A*01 (65). While pre-clinical screening showed no evidence of cross reactivity, after TCR-T cell therapy patients died from cardiac failure, which was later attributed to cross reactivity with a peptide from the cardiac protein titin (65). The MAGE-A3 peptide target, EVDPIGHLY, and the Titin peptide target, ESDPIVAQY, differ in four amino acid positions, some of which are in the center of the peptide, the area principally responsible for contact with the TCR (66). Existing methods were unable to predict the cross reactivity preclinically (65, 66). Hence, better methods to predict off-target reactivity for TCRs is an unmet need.
An affinity enhanced ImmTAC designed from the same parent TCR as the a3a TCR was also found to have cross reactivity with the titin peptide. Tissue cross reactivity was still observed with a TCR agent that was more specific to the MAGE-A3 epitope and was attributed to high levels of titin expression on myoblasts (66). Therefore, protein expression is another variable that needs to be considered when testing TCR-based therapies for cross reactivity, as well as affinity and half-life of the TCR/peptide-MHC interaction (66). Additional instances of cross reactivity have been seen due to sequence similarity, as seen with a TCR against a HLA-A*02:01 restricted MAGE-A3 peptide which recognized a peptide unpredicted to be expressed in the brain and led to neurotoxicity including death (114, 142).
Mixed TCR Dimers
If the endogenous TCR alpha or beta chain pairs with an exogenously introduced alpha or beta chain, the resulting TCR, a mixed TCR, could have unknown reactivity with normal peptides (148). These mixed TCRs bypass thymic selection, therefore there is no central tolerance to prevent reactivity with normal tissues (148). To prevent mixed TCR dimer formation, the human constant regions can be interchanged with murine constant regions (122, 147, 149–151), or human TCR alpha and beta constant regions can be interchanged with each other or with the constant regions from gamma-delta T cells, which cannot pair with endogenous alpha-beta chains (152). Additionally, constant regions can be modified to contain cysteines to promote disulfide bond formation and therefor pairing between the alpha and beta chains (85, 148). Other methods to prevent TCR chain mispairing involve transduction of alpha-beta TCRs into gamma-delta T cells (153, 154) and knocking out or down the endogenous TCR chains with clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated protein 9 (Cas9) (155) or small interfering RNA respectively (156–158). TCR mispairing can lead to off-target as well as decreased on-target activity (85, 149). Promoting proper TCR chain pairing increases TCR expression (85, 122, 149), avidity and activity (122).
Predicting Off-Targets of TCR-Based Therapeutic Agents
Prediction of TCR reactivities has proven difficult, as native TCRs are necessarily cross-reactive to enable tissue surveillance. Additionally, TCR reactivity is structurally complex being dependent on the quality and quantity of the expression of the TCR, MHC and presented peptides (1). Steps to detect cross reactive TCRs include screening for reactivity against cell lines known to express the HLA of interest, but not the proteins from which the target peptide is derived (65, 159), as well as HLA mismatched cell lines (159, 160). Reactivity with either of these cells would indicate off-target binding of the TCR and a potential for toxicity. A number of investigations have sought to predict cross-reactivity via structural analyses and predictive algorithms. Single amino acid replacement scans, such as alanine scans (Figure 2A), are often used to identify peptide residues important for TCR recognition (159, 161–163). Alanine scans involve changing every position in a peptide sequence to an alanine, if reactivity is abolished then that particular amino acid residue and its position are considered necessary for TCR reactivity. However, alanine screens do not identify important interactions if the substituted amino acid is similar to alanine. Changing that amino acid to alanine might still result in binding and therefore alanine screens are biased towards identification of TCR interactions with large and charged residues where the substitution to alanine resembles a major change. A second single amino acid replacement scan can be performed to provide a more complete picture of motif residues important for TCR reactivity (65, 160). This motif can subsequently be compared to human peptides (161), to identify putative cross-reactivities (65).
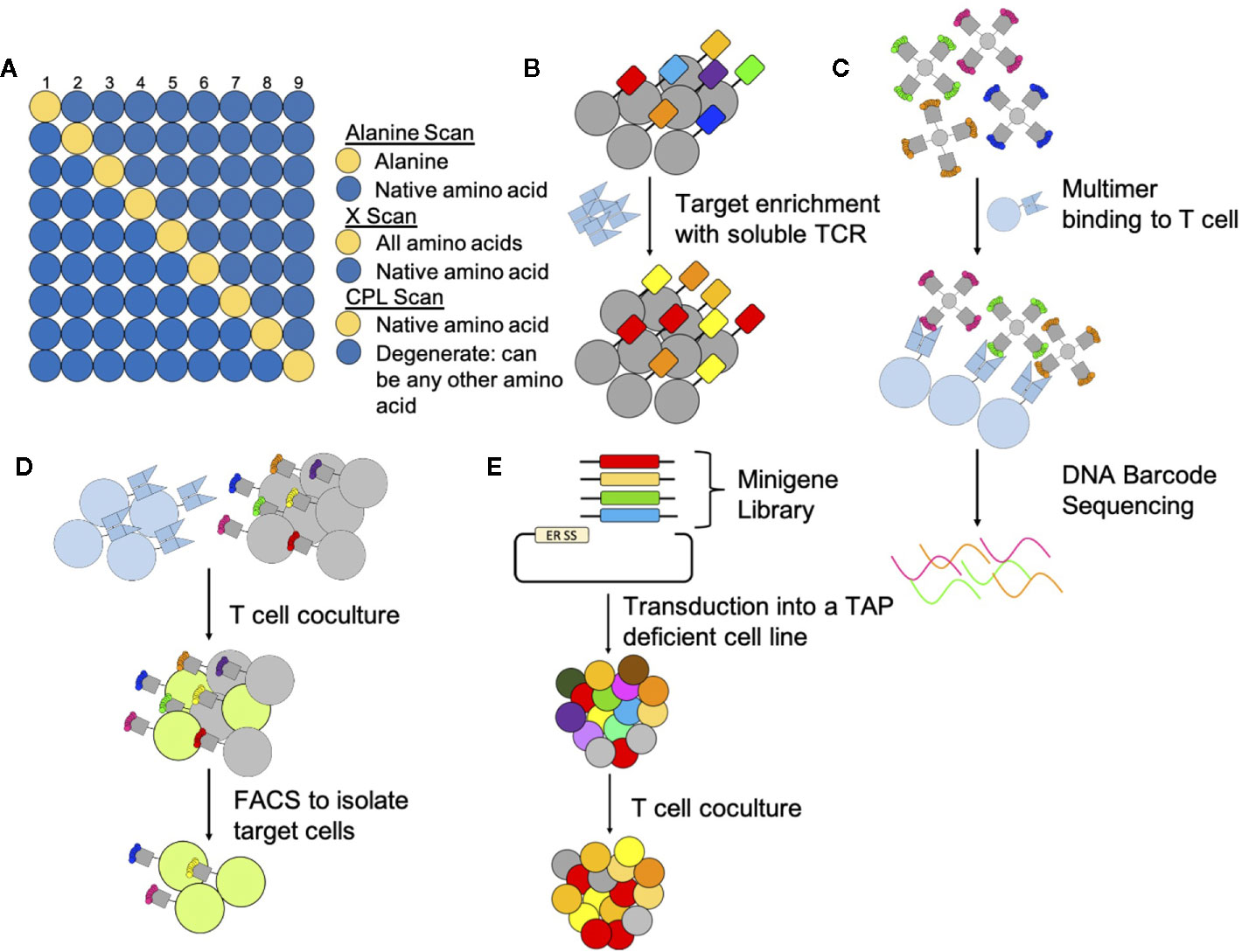
Figure 2 Methods to determine targets and off targets of TCR-based therapeutics. (A) Peptide Scans: Peptide scanning techniques determine which residues are important and in which positions. Alanine scans and x-scans hold all positions in the native peptide (blue) except one (yellow); one position is switched to an alanine for alanine scans or to all other amino acids for X-scans. CPL scans hold one position constant (yellow) and all other positions (blue) can be any combination of amino acids. Peptides are then pulsed onto target cells to assay for TCR reactivity. (B) Yeast Display: Yeast display libraries genetically encode a peptide linked to an MHC. Multiple rounds of selection with soluble TCRs select for peptide-MHC complexes that are recognized by the TCR. (C) Multimers: Multimers can identify T cells that bind specific peptide-MHC complexes. To identify reactive peptide-MHC complexes in a pooled setting multimers are DNA barcoded prior to incubation with and binding to T cells. (D) T scan and SABR: The T Scan method genetically encodes for longer peptides that go through endogenous processing and presentation. Target cells that present peptide-MHC complexes targeted by T cells fluoresce through a granzyme reporter system and are subsequently sorted by FACS. For the SABR method, TCR expressing cells bind to target cells expressing the SABR receptor. After TCR binding, this receptor, which consists of an MHC linked to a CD3z and CD28 domain, signals to an NFAT report system causing target cells to fluoresce. Target cells can then be sorted by FACS. (E) PresentER: The PresentER system genetically encodes for peptides to be presented in MHC complexes on TAP-deficient cells. Target peptides are identified through coculture depletion assays with T cells.
Other amino acid scans screen additional amino acid combinations to determine the TCR binding motif. X-scans (Figure 2A), similar to alanine scans, hold all peptide positions constant except one but change this position to any of the remaining 19 amino acids (162). Combinatorial peptide libraries (CPL) (Figure 2A) are another peptide screening method to determine TCR off-targets which allow additional peptide diversity. CPLs are designed so that one position of a peptide is held constant and the remaining positions are changed to any other amino acid (145, 161, 164). Peptides from CPL scans are screened in subpools (145) for TCR reactivity (161, 165). X-scans and CPLs allow for more potentially cross-reactive peptides to be screened compared to alanine scans providing a more complete picture and ranking of potential TCR reactive peptides (161, 162, 165, 166). In the aforementioned amino acid scans, T cell reactivity can be measured in numerous ways including via cytokine release (145, 161, 162), typically measuring IFNγ, T cell proliferation (164), target cell lysis (164, 166), detection of T cells activation markers and binding of soluble TCRs to peptide-MHC complexes (166).
Computational modeling methods take previously known TCR and peptide-MHC structural and reactivity data to create models to predict peptide-MHC targets (167–169) and cross reactivities (170). These models are based on the premise that TCRs with common targets will have structural and sequence similarities (167, 171). Characteristics used to compare TCRs include length, charge, hydrophobicity and sequence (171). Peptide-MHC complexes have also been compared to assess cross reactivity based on charge and available surface area (170). These methods are primarily limited by the amount of data available for model development and TCR comparison (167, 168).
However, these structural or predictive methods discussed above are neither comprehensive nor fully accurate, as the rules for binding of TCRs to their peptide/MHC sites are still poorly understood. Therefore, more empiric methods have been applied to the problem. Large libraries, where the peptide target is genetically encoded into expression systems have been used to identify TCR targets and off-targets. Display libraries of peptides have been developed for screening in yeast and baculovirus systems (Figure 2B) (172, 173). In these systems the MHC is expressed with the peptide attached by a linker (144, 172–174). For these systems the MHC must fold properly, and the peptide must bind the MHC properly (172). In another library system, known as PresentER (Figure 2E), peptides are directed to be loaded onto the endogenous MHC of mammalian cells through an endoplasmic reticulum signaling sequence. Target peptides are identified after coculture screens with T cells by assaying for peptide dropout via DNA sequencing (175). This system has the advantages of yielding actual peptide-MHC molecules in the context of human cell surface membranes on live cells for both in vitro and in vivo work, as well as allowing functional assays such as recognition and killing of targets to be measured, but is limited in the number of peptides that can be scanned in a single assay to a few tens of thousands, whereas the proteome may contain a million potential epitope sequences that bind to an individual MHC. An additional library screening technique uses signaling and antigen-presenting bifunctional receptors (SABR) (Figure 2D), where the target cell expresses peptides linked to MHC receptors fused to intracellular CD3ζ and CD28 domains. The target cells are identified through fluorescence, as these cells have an NFAT-GFP reporter system which is activated upon signaling from CD3ζ after TCR engagement. The presented target peptides are subsequently identified through sequencing. As with PresentER libraries, SABR libraries are limited in their size. However, SABR libraries can contain up to one million epitopes (176). The previously described libraries genetically encode for short antigenic peptides, the T-scan reporter system (Figure 2D) on the other hand encodes for larger amino acid sequences that need to undergo endogenous processing and presentation (177). Trogocytosis, which describes the facilitated exchange of membrane bound proteins after immune cells and target cells come into close contact, has also been used to identify TCR targets in library screens. This mechanism then allows for the identification of the recognized antigen presented on target cells, that were engaged by a T cell (178). Other library screening methods use DNA-barcoded MHC multimers (Figure 2C). The MHC multimer is screened for binding with a TCR followed by sequencing of the DNA barcode to determine which peptides were able to bind the TCR of interest or to develop recognition motifs to predict additional off targets (163). Such methods may prove useful in the preclinical characterization of TCR reactivity and could be paired with tissue expression data of off-target genes to predict site-specific toxicities.
Current TCR-Based Cellular Agents in Clinical Study
The earliest studies in which TCR-T cells were infused into patients were reported in 2006 (143), an effort that was the culmination of decades of work by Rosenberg and colleagues to characterize the antitumor activity of TILs (179–181). In this early study, TCRs specific for the HLA-A*02:01 presented self-antigens MART-1, gp100, NY-ESO-1, and p53 were transduced into autologous peripheral blood mononuclear cells (PBMC) and infused into melanoma patients. TCRs specific for these antigens are among the most actively studied in TCR gene therapy clinical trials.
Notably, NY-ESO-1-reactive TCRs are being investigated by multiple academic and industry entities worldwide for the treatment of a range of solid and liquid tumors such as melanoma, sarcomas, lung cancers, and multiple myeloma (8, 121, 182, 183). GSK3377794, comprised of autologous T cells transduced with an affinity-matured NY-ESO-1–reactive TCR (184) has reached Phase Ib/II trials testing it in combination with checkpoint inhibitor, anti-PD1, therapy in NSCLC (NCT03709706).
WT1-directed TCRs have also shown promise for treatment of AML (185). WT1 has been designated a highly prioritized antigen (186), and the cytotoxicity of WT1-specific T cells against leukemic cells has been reported by multiple groups (6, 20, 187). WT1-specific T cells can be readily generated from most healthy donors; accordingly, a TCR isolated from a healthy donor could be used without enhancement of its native antigen-binding capability. Moreover, donor-derived Epstein-Barr virus-specific T cells were transduced with the TCR, rather than autologous T cells. This study is notable in several aspects as it utilizes a healthy donor-derived TCR, genetic modification of allogeneic T cells, and provides validation of the preclinical work involved in characterizing WT1 as a leukemia-associated antigen (188, 189). Results from two WT1 TCR Phase I/II trials utilizing autologous T cells are expected to clarify the relationship, if any, between graft versus host disease (GvHD), which has been seen in trials, and WT1-targeting (NCT02550535, NCT01621724, NCT02408016).
A class II-restricted TCR directed against an epitope of MAGE-A3 presented by HLA-DP*04:01/04:02 was shown to be well-tolerated and the CD4 autologous T cells persisted in 17 patients with metastatic cancer in a basket trial (190). Three partial responses in a variety of cancers correlated with T cell persistence of at least one month. Though this TCR was derived from a regulatory T cell clone (191), infused T cells did not appear to differentiate to regulatory T cells (Treg) on the basis of FOXP3 expression. The safety profile of this TCR contrasts that of previous MAGE-A3 TCRs (142, 192), and thus could prove to be an effective therapy with minimal toxicity in a wide range of tumors.
Of particular concern in TCR gene therapy is the safety of affinity enhanced self-antigen TCRs due to potential on and off-target toxicities (10, 123). The TCR targeting an HLA-A*02:01 presented epitope of CEA, described earlier, was found to induce severe colitis in all three colorectal cancer patients tested (7, 193) because of baseline CEA expression in colonic mucosa. An affinity enhanced TCR specific for MAGE-A3 (114) was found to cause severe neurotoxicity due to reactivity with a similar MAGE-A12 epitope (142). Another affinity enhanced TCR to MAGE-A3 caused lethal cardiotoxicity due to recognition of a titin-derived epitope (65, 192). These cases exemplify the critical need for characterizing TCR target recognition before clinical translation.
The safety of neoantigen reactive TCRs appears to be in stark contrast to that of the aforementioned self-antigen reactive TCRs because neoantigens derived from private somatic mutations are theoretically only presented by the tumor. In addition to tumor-selectivity, the potential for acquired resistance to TCR therapies targeting such mutations are expected to be lower in the case of targeting driver mutations. For example, T cells targeting mutant KRAS found in the endogenous TILs of a patient with KRAS-driven metastatic colorectal cancer showed minimal toxicity and all seven of the patient’s lesions initially regressed. Interestingly, at 9 months a lesion escaped by means of downregulating the restricting HLA allele, while maintaining the same KRAS mutation (141). Hence, KRAS driver mutations are not easily mutated into escape variants, but downregulation of HLA can be an alternative mechanism of immune escape. To this end, KRAS mutant-specific TCRs have been generated in HLA transgenic mice and are currently being tested in phase I trials (42) (NCT03190941). Similarly, T cell responses to TP53 hotspot mutations have been found in TILs in different epithelial cancers with multiple HLA allele restrictions (84, 194). The recurrence of TP53 mutations encoding immunogenic neoantigens presents profound opportunities for TCR-based therapy across a variety of solid tumors (195).
Several TCR-transduced cells are being tested in ongoing or recently completed Phase I and II trials (Table 2). New antigens being targeted in these trials include HERV-E and TRAIL-DR4. New TCR modalities are being assessed as well, such as suicide gene-containing T cells that provide a kill-switch and Vγ9Vδ2 TCR-transduced T cells which recognize uncharacterized tumor antigens in an MHC-independent manner.
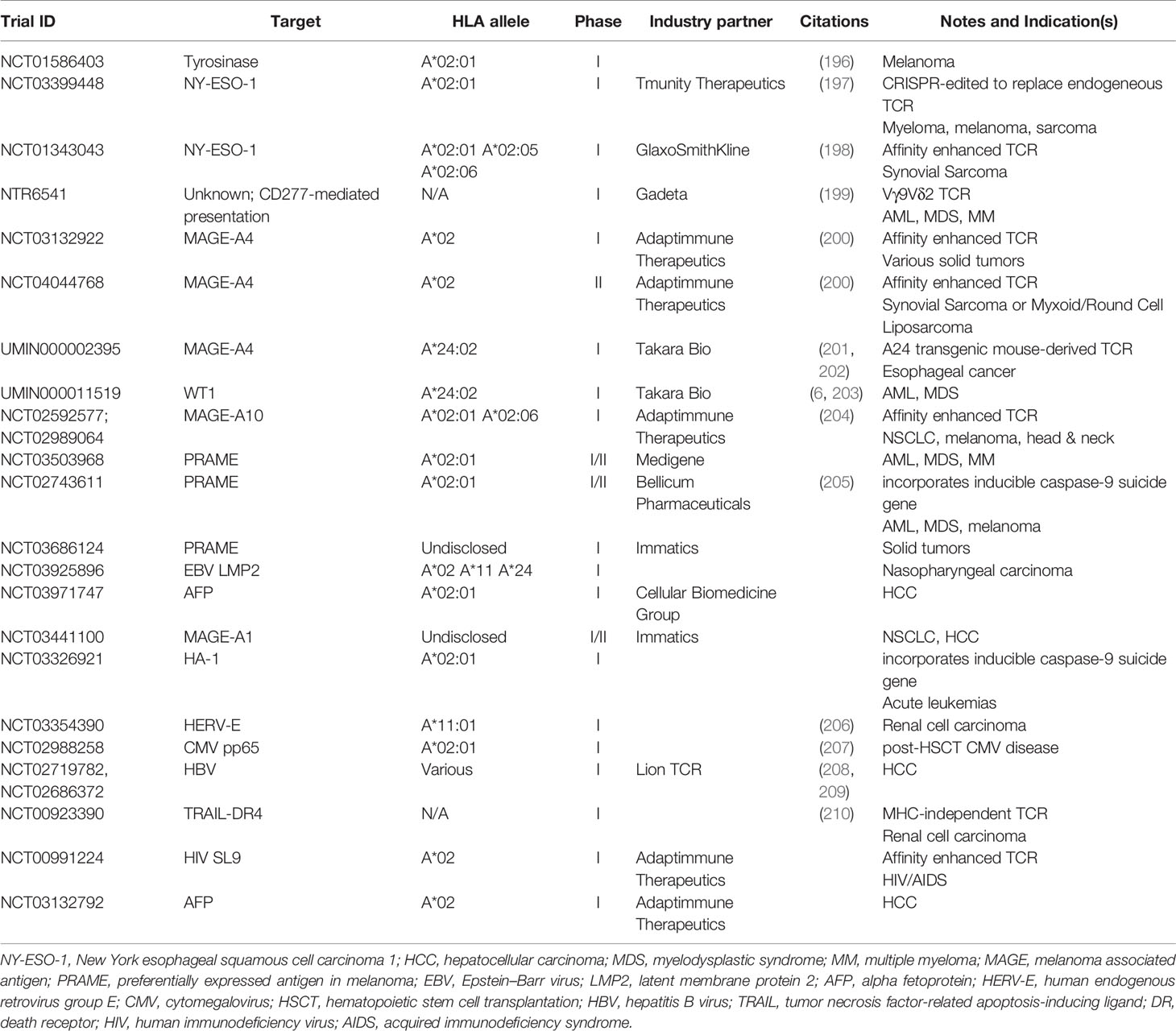
Table 2 Selected TCRs and T cells in ongoing or recently completed clinical trials.
TCR-Based, Non-Cellular Therapies
Current T cell therapies using either TILs or TCR transduced T cells are patient-specific and require TCR gene transduction or expansion of the patient’s T cells in vitro before reinfusion into patients. Such processes have proven to be difficult to translate into a widely available therapy. Several therapeutic modalities (Table 3) have been developed to overcome such limitations and broaden therapeutic options to a wider range of patients. In particular, soluble T cell redirecting biologics based on either TCR or immunoglobulin molecules, in conjunction with redirection of powerful T cell cytotoxicity provides a promising alternative.
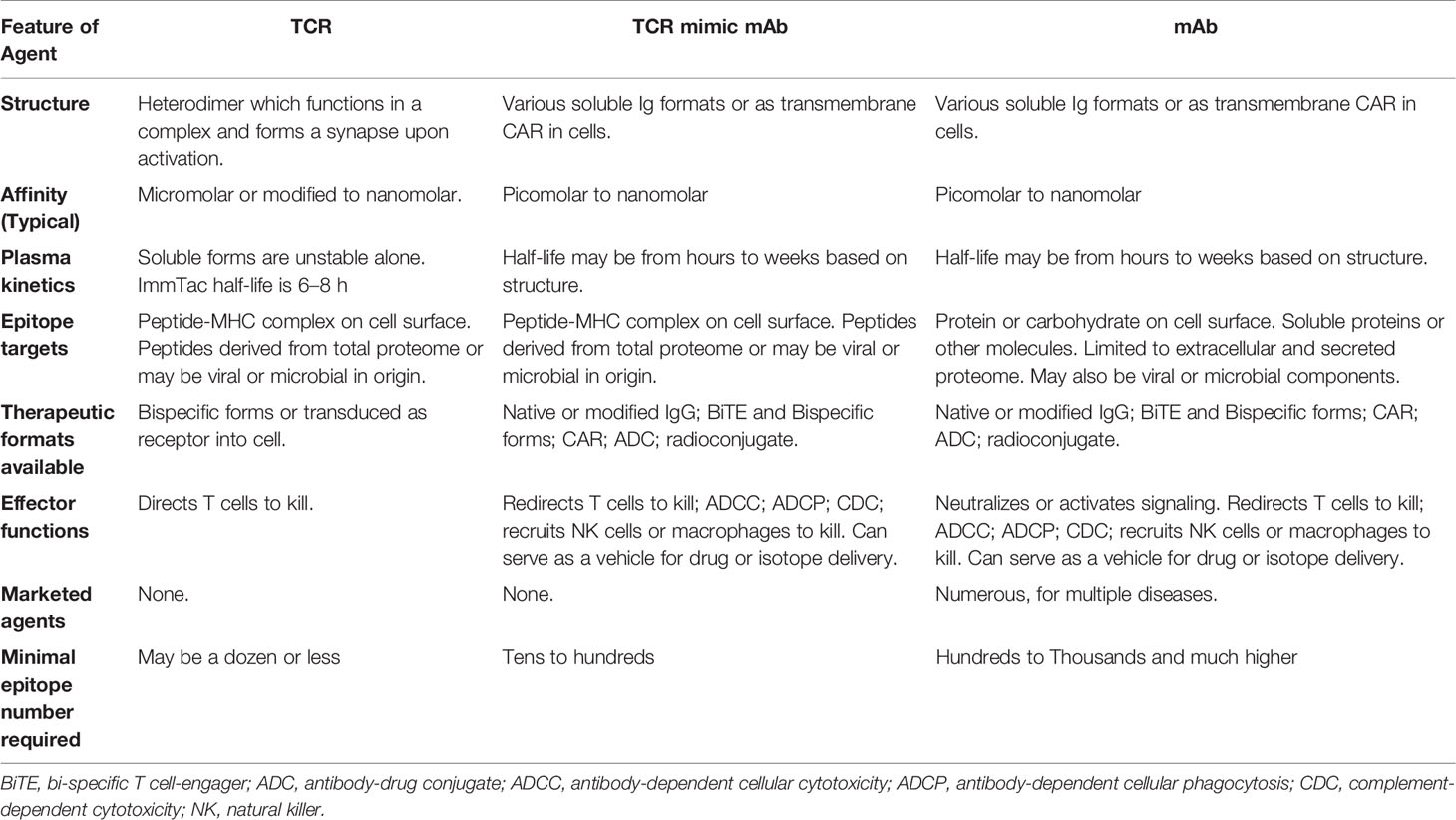
Table 3 Characteristics of TCR, TCRm and traditional mAb.
Immune Mobilizing Monoclonal TCRs Against Cancer (ImmTac)
A new class of bi-specific molecules, ImmTacs, are soluble T cell engagers (sTE), designed to use a TCR specific for a peptide-HLA complex, genetically linked to a single chain variable fragment (scFv) of an anti-CD3 mAb. Structurally, an ImmTac begins with a human TCR or an affinity enhanced TCR. While recombinant soluble TCRs (lacking transmembrane and intracellular parts) should theoretically be an ideal therapeutic vehicle for targeting intracellular tumor antigens, TCRs are inherently unstable in soluble form and tend to form aggregates, which poses a significant technical challenge in developing such molecules as therapeutics (211). To address this issue, ImmTacs are designed to stabilize soluble TCRs through the incorporation of an interchain disulfide bond buried within the core of the TCR. Finally, an anti-CD3 scFv is encoded via a flexible linker to the beta chain of the TCR (212). Once TCRs engage their antigenic peptide-MHC complexes, the anti-CD3 effector arm mediates potent redirection of polyclonal T cells to the target. With this technology, cells expressing fewer than 100 copies of the targeted peptide-MHC complexes can be killed. Since natural TCRs have low, micromolar affinity, ImmTac technology allows increases in TCR affinity up to the subnanomolar range, allowing ImmTacs to target low density peptide-MHC complexes of intracytoplasmic tumor antigens (213). The most extensively studied ImmTac molecule, tebentafusp (IMCgp100), an affinity enhanced TCR specific for a gp100 peptide (a melanocyte differentiation antigen) presented on HLA-A*02 complexes, has demonstrated clinical efficacy as a monotherapy against the gp100-positive uveal melanoma (214, 215). In addition, three other molecules: GSK01 (directed to the cancer germline antigen NY-ESO-1), IMC-C103C (directed to cancer germline antigen MAGE-A4) and IMC-F106C (directed to a cancer testis antigen PRAME) are in phase I/II trials for treatment of multiple myeloma, melanoma and a range of other cancers (216). ImmTacs’ high potency and drug-like soluble format make them easy agents to develop and distribute widely.
TCR-Mimic Monoclonal Antibody (TCRm)
MAb-based therapy has become one of the most successful and important strategies for the treatment of cancer and rheumatologic diseases (217, 218). MAbs are characterized by high target specificity, limited side effects and prolonged half-life in vivo. The intrinsic multifunctional cellular engagement of mAbs include antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), blocking of ligand-based signaling, and direct signaling or inhibition via various pathways. In addition to their intrinsic properties, mAbs can in an antigen-specific manner deliver potent cytotoxic agents such as toxins, drugs, or radionuclides to cancer cells (219). Finally, mAbs can be re-engineered to generate CARs or bi-specific antibodies to redirect the T cells or other effector cells for potent anticancer therapy (220). Commercial therapeutic mAbs are directed to extracellular or cell surface proteins; therefore, the vast majority of intracellular tumor-associated antigens (TAAs) are not addressed by FDA approved mAb therapy (221).
TCRms targeting peptide-MHC complexes, combine TCR recognition of peptide-MHC complexes, with the potency and versatility of mAb drugs. The well-characterized platform of TCRms provides a highly feasible method to target intracellular tumor antigens for a broad range of patients. Increasing advances in library screening technology allow rapid identification and selection of highly specific TCRms to intracellular tumor antigens. A number of mouse and human TCRm antibody fragments such as antigen binding fragments (Fab) or scFvs have been identified (222, 223), as well as several full-length human TCRms, and have been investigated as potential therapeutic agents. A murine hybridoma-generated TCRm (8F4) reactive with the myeloid leukemia antigen PR1-derived epitope (VLQELNVTV) was found to bind HLA-A*02:01 and was later humanized. This TCRm eliminated human AML in xenografts (224) and has now advanced to clinical trials. While active alone, to improve its potency 8F4 was engineered into a bi-specific T cell-engager (BiTE) to redirect polyclonal T cells to PR1-positive leukemias (225). The first fully human TCRm, ESK1, specific for a WT1-derived epitope/HLA-A*02:01 complex, was developed by our group (226). WT1 is expressed in a wide range of human cancers and has been an important target for TCR-based adoptive cell transfer of engineered T cells, as well as peptide, DNA, and dendritic cell vaccines (185, 227–231). WT1 expression encompasses both hematopoietic and solid tumors and therefore, a TCRm targeting this oncoprotein should have a broad application as a therapeutic agent against a variety of human leukemias, myeloma and solid tumors (226, 232).
Our group has described TCRms specific for a cancer germline antigen, PRAME, which is widely expressed in various cancers (recognizing ALYVDSLFFL/HLA-A*02:01) (46), and FOXP3, the hallmark protein for Tregs (recognizing TLIRWAILEA/HLA-A*02:01) (233). The TCRm specific for the FOXP3 epitope is particularly interesting because of the profoundly suppressive role of Tregs in the TME. Strategies of depleting Tregs by mAbs against surface proteins such as CD25, CCR4 and GITR, have not been successful thus far because these molecules are shared between Tregs and other immune effector cells (234–236). Other human TCRms to important cancer-associated and viral targets have been described (Table 4).
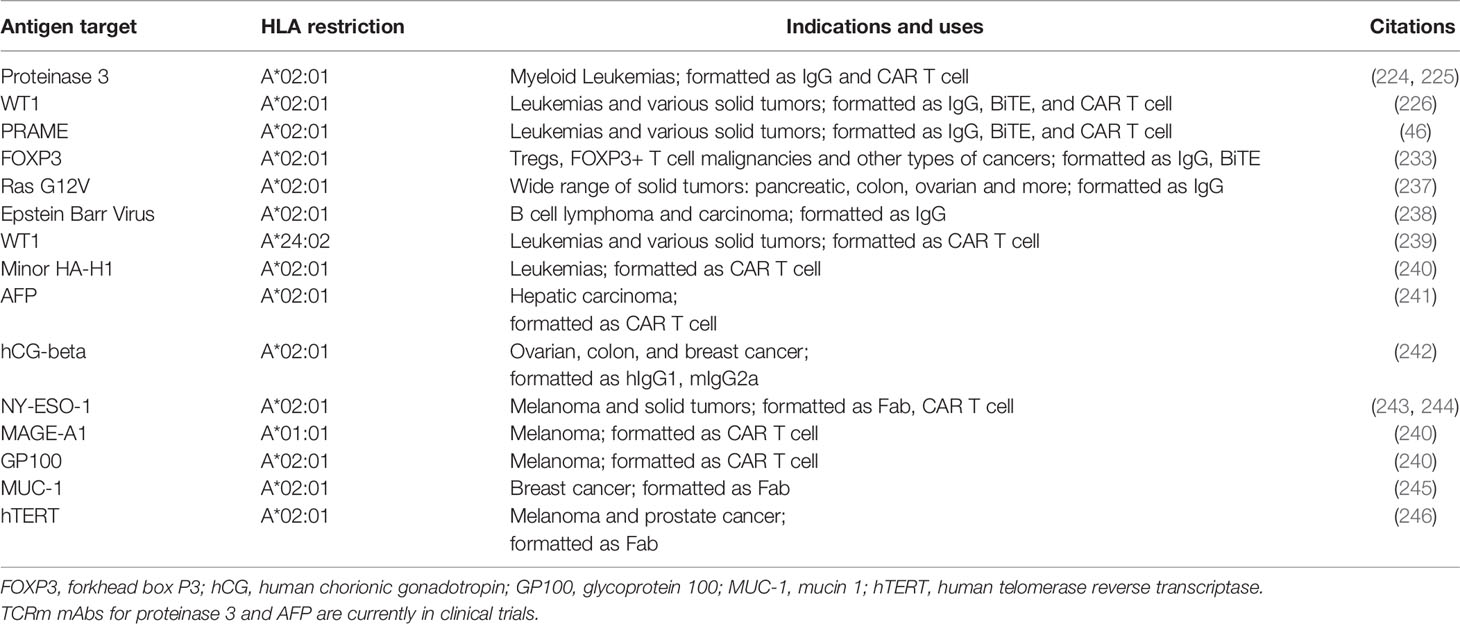
Table 4 Human TCRm reported.
The antibody-based format for TCRm offers the opportunity for optimization via protein engineering strategies to address different needs:
A. Fragment crystallizable (Fc) region modification. The ADCC activity of mAbs can be enhanced 5-to 10-fold by Fc region protein engineering (247) or by modification of Fc-region glycosylation (248, 249).
B. Bispecific mAbs (BsAb). BsAbs are designed to recognize two different epitopes or antigens, and they comprise a large family of molecules, with a wide variety of formats (250). Such bispecific molecules function by recruiting and activating polyclonal T cells or other effector cells. BiTEs are a subtype of BsAb, composed of a scFv specific for tumor antigen on one arm, linked to a scFv for CD3 on the other arm. Such a BiTE molecule functions by recruiting and activating polyclonal T cells at tumor sites, thereby bypassing MHC restriction and co-stimulation, while retaining epitope specificity needed for traditional TCRs (251). The ESK1-BiTE was the first TCRm-based BiTE, which showed superior cytotoxicity than an immunoglobulin form against a wide range of tumor cells expressing WT1 in vitro and in vivo in mice. The ESK1-BiTE also induced robust secondary CD8 T cell responses against other epitopes via epitope spreading (232). Such a mechanism may be important for long-lasting anti-tumor immunity by controlling the outgrowth of tumor cells that have lost the target protein or that have downregulated the primary target during tumor evolution. In addition, as a small molecule, BiTEs may penetrate more easily than CAR T cells into the TME of solid tumors, where it can bridge tumor targets with TILs.
C. TCRm CAR T cells. CAR T cell constructs use immunoglobulin scFvs, recognizing extracellular cell surface protein antigens expressed by cancer cells. The scFv is linked to a transmembrane and intracellular signaling domain generally containing the CD3ζ chain of the TCR complex, as well as costimulatory domains of CD28 or 41BB. Following the clinical success of CD19 CAR T cell therapy in human leukemia (252), many CAR T cells have been developed targeting a variety of cell surface molecules (253). Using TCRms, described above, CAR T cells have been generated against the intracellular tumor antigen WT1, by use of the ESK1 TCRm scFv, thereby opening the door for CAR T cells to enter an entirely new universe of antigens (254). These studies show CAR T cells can be used to target intracellular tumor antigens, in contrast to CAR T cells using traditional scFVs that target extracellular proteins, and thus expand the utility of this platform to include a large majority of cancers. The first CAR T cells expressing a TCRm scFv for alpha fetoprotein (AFP)-HLA-A*02 recently advanced to human trials for the treatment of hepatocellular carcinoma. Currently, more CAR T cells are being developed from TCRms recognizing various epitopes from NY-ESO-1, gp100, and MAGE-A1, in the context of HLA molecules (240).
D. Affinity maturation. Cell surface protein targets of mAbs normally have a high density of typically 10,000 to 1,000,000 molecules per cell. In contrast, intracellular tumor antigens presented as peptide-HLA complexes on the cell surface typically have low densities, often far less than a few hundred molecules (46, 226, 255, 256). Therefore, WT1-specific TCR gene therapy (185) and ImmTac molecules use affinity enhanced TCRs (212). Similarly, in vitro affinity maturation of mAbs, often with phage library technology, has successfully been used to optimize specific mAbs with increased affinity for their targets (257). Most TCRms have been derived using phage display technology and have yielded relatively high affinity TCRms, thereby reducing the need for affinity maturation.
Challenges and Opportunities for Soluble TCR Constructs and TCRm
Soluble TCR-based agents represent novel classes of biologics that make immunotherapy accessible for some of the most interesting and highly tumor specific intracellular antigens and offer pharmacological and manufacturing advantages (Table 3). However, fundamental questions remain to be studied in order to further advance these drugs. First, given the intrinsic nature of TCR recognition of a linear peptide bound to HLAs, cross reactivity to other similar complexes is an important issue in all TCR-based therapies. Second, unlike TCR, TCRm are not natural structures that evolved with thymic selection to recognize peptide-HLA complexes. As antibodies are generally selected on membrane bound soluble proteins or carbohydrate antigens during B cell development in vivo, selection methods using phage or other libraries may introduce unnatural biases and unstable structures in addition to cross-reactivity. TCRm may never completely mimic natural TCR recognition. For example, crystallography studies have shown that the ESK1 Fab primarily interacts with N-terminal residue of the peptide and HLA-A*02:01 (258). An alanine substitution study showed that the TCRm mAb specific for the PRAME peptide-HLA-A*02:01 mainly recognized the C-terminal residues of the peptide (46). In general, TCRs dock onto peptide-HLA complexes using a conserved canonical binding mode, forming a large binding interface between the TCR and peptide-HLA, enabling broader contacts across both peptide backbones and HLA heavy chain. Lessons learned from the early development of TCRm could help optimize screening strategies of phage libraries to select ideal phage clones that more closely mimic TCR recognition. These strategies include selection of mAbs that bind with optimal valency between HLA and peptide, or mAbs that bind to a broader range of amino acid residues in the center of the peptide in a fashion similar to TCRs (259). Additionally, native TCRs tend to have orders of magnitude lower affinities than TCRm or engineered TCRs. The impact this has on activity and specificity, and the importance of affinity in different formats is not well understood. For example, a TCR-T cell or TCRm CAR T cell may need less affinity than a soluble format such as a BiTE due to its multivalent avidity. However, an anti-Ep-CAM BiTE has been shown to form a synapse after engaging its target because of the proximate contacts between effector and target cells (260). A recent study directly compared a scFv specific for NY-ESO-1p157/HLA-A*02:01 complex in BiTE and CAR-T cell formats. The conclusion was that the BiTE and CAR T cells showed a similar functional avidity, assessed by cytokine production and killing activity (261). A study comparing a TCR specific for a WT1-derived peptide/HLA-A*02:01 complex and a TCRm specific for the same complex showed that while the native low-affinity alpha-beta TCR maintained potent cytotoxic activity and specificity, the high-affinity TCR-like antibody CAR T cells exhibited reduced activity and loss of specificity. This TCR-like mAb in a monovalent or bivalent context maintained high specificity, however, when the avidity of this mAb was increased through expression in a CAR T cell format, it exhibited loss of specificity (262). This study suggested that TCRm is less suitable for CAR T cell format than being used as mAb format coupling with more potent drugs. However, function of each mAb depends on the specificity of the particular TCRm used for construction of the CAR T cells. Although this study raised an interesting question, one pair of TCRm vs TCR may not generalize the function of these two formats. Interestingly, a number of TCRms have been converted to a CAR T cell format (Table 4). With a rapidly growing numbers of these new modalities, detailed studies are required to address these fundamental biological questions.
In addition, despite all the advantages that TCR-based, non-cellular therapies offer, they also have certain limitations. Both ImmTacs and BiTEs have a short half-life (4 to 8 h), which requires continued administration of the agents (216, 232). However, this can be overcome by a growing number of engineering technologies.
Summary and Perspective
TCR-based therapies provide a number of unique advantages over other immunotherapies, but also present challenges associated with their structures and the methods used for their generation. On and off-target identifications and toxicity prediction remain problematic. TCR-based agents are a powerful modality on which to create immunotherapies as the TCR is able to target the vast repertoire of cancer associated and mutated proteins found in all subcellular locations (3). The TCR’s unique and valuable recognition properties have been taken advantage of in adoptive cell therapies, where reactive T cells are enriched or T cells are modified to express reactive TCRs, and in non-cellular therapies, which bypass the extensive process of T cell enrichment, modification and expansion, while mimicking the peptide recognition properties of the TCR in the form of soluble TCRs or antibodies. In distinct contrast to traditional antibodies and CAR T cells, TCR-based therapies recognize a short peptide bound to an MHC found on the surface of cells (144). The TCR’s unique ability to recognize intracellular proteins in these complexes both shapes and constrains their functions. Moreover, the origins of their antigenic specificity are dependent on a linear sequence of amino acids that may be shared by other proteins in the proteome. In addition, the complicated and highly regulated process of antigen presentation means that not all peptides are presented, and others are displayed are at insufficient densities to trigger TCR-based recognition or responses. This problem may be overcome in some cases with small molecule drugs or cytokines and such a strategy could be considered for combination therapies. Endogenous TCRs are able to discern between peptides with single amino acid changes (95), enabling precise differentiation between peptides; despite this, TCRs are inherently promiscuous to enable proper immune surveillance, resulting in potential liabilities for their use therapeutically. Compounding this issue is that in silico predictions of peptide presentation on MHC molecules are inaccurate. Ideally, selected epitopes should be validated by using mass spectrometry to verify peptide-MHC presentation, yet these methods are costly and tedious.
While manipulating TCR affinity has been shown to increase their effectiveness (65, 74, 123) and would provide agents with better pharmacologic properties, TCRs whose affinities are too high risk cross reactivities (74, 115, 123). Additionally, affinity enhanced, modified and artificial TCR-based therapies do not necessarily adhere to the same binding rules as native TCRs, due to the absence of thymic selection. Therefore, toxicities from TCR-based therapies are of concern and methods to identify potential cross-reactive or off-target toxicities are imperative. Current preclinical strategies to predict toxicities by determining on-target/off-tumor and off-target antigens are largely empiric and are unable to cover the vast potential repertoire of epitopes in the proteome. These failures have led to clinical toxicity in early trials. To avoid toxicity, the targeting of neoantigens may be used, but identifying patient specific tumor antigens and reactive TCRs is challenging, costly and currently not feasible broadly. Alternatively, public tumor antigens offer broader applications, but are rare and may not be absolutely cancer specific.
Despite challenges to development, TCR-based therapies have shown great potential in clinical use, targeting seemly un-targetable intracellular proteins. In distinction to commercial therapeutic mAbs or CAR T cells, which are generally limited to a small number of extracellular or cell surface proteins; TCR-based agents allow, for the first time, access to the vast majority of intracellular tumor associated antigens (TAAs) that are not currently addressed by FDA approved therapies. It is expected that technologies to automate the identification of target and off-target epitopes, and rapid new methods to generate TCRs, as well as new soluble and cell bound structures that take advantage of the unique recognition properties of the TCR, will soon result in a great expansion in these agents to a broader population of patients with cancer and other diseases.
Author Contributions
All authors contributed to the content and revisions of this review and approved the submitted version.
Funding
This work was supported by the National Cancer Institute (F31 CA253995, P01 CA23766, R01 CA55349, R35 CA241894, and P30 CA08748), the Greenberg Fund, the Solomon Fund, the Tudor Fund, the Center for Experimental Therapeutics of MSKCC, and an Individual Research Grant from the German Research Foundation (KL 3118/1-1).
Conflict of Interest
MSKCC has filed for patent protection on behalf of DAS and TD for inventions related to this paper. TD is an advisory board member for Eureka Therapeutics. DAS is an advisor for, or has equity in, Eureka Therapeutics, Oncopep, Sellas, Pfizer, and Iovance,
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Lorenz FKM, Ellinger C, Kieback E, Wilde S, Lietz M, Schendel DJ, et al. Unbiased Identification of T-Cell Receptors Targeting Immunodominant Peptide–MHC Complexes for T-Cell Receptor Immunotherapy. Hum Gene Ther (2017) 28:1158–68. doi: 10.1089/hum.2017.122
2. Harris DT, Hager MV, Smith SN, Cai Q, Stone JD, Kruger P, et al. Comparison of T Cell Activities Mediated by Human TCRs and CARs That Use the Same Recognition Domains. J Immunol (2018) 200:1088–100. doi: 10.4049/jimmunol.1700236
3. Chandran SS, Klebanoff CA. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol Rev (2019) 290:127–47. doi: 10.1111/imr.12772
4. Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med (2016) 22:26–36. doi: 10.1038/nm.4015
5. McGranahan N, Swanton C. Neoantigen quality, not quantity. Sci Transl Med (2019) 11:eaax7918. doi: 10.1126/scitranslmed.aax7918
6. Ohminami H, Yasukawa M, Fujita S. HLA class I-restricted lysis of leukemia cells by a CD8+ cytotoxic T-lymphocyte clone specific for WT1 peptide. Blood (2000) 95:286–93. doi: 10.1182/blood.V95.1.286
7. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan D-AN, Feldman SA, et al. T Cells Targeting Carcinoembryonic Antigen Can Mediate Regression of Metastatic Colorectal Cancer but Induce Severe Transient Colitis. Mol Ther (2011) 19:620–6. doi: 10.1038/mt.2010.272
8. Robbins PF, Kassim SH, Tran TLN, Crystal JS, Morgan RA, Feldman SA, et al. A Pilot Trial Using Lymphocytes Genetically Engineered with an NY-ESO-1-Reactive T-cell Receptor: Long-term Follow-up and Correlates with Response. Clin Cancer Res (2015) 21:1019–27. doi: 10.1158/1078-0432.CCR-14-2708
9. Butterfield LH, Kaufman HL, Marincola FM. Cancer immunotherapy principles and practice. New York, NY: Springer Publishing Company (2017).
10. Yee C, Thompson JA, Roche P, Byrd DR, Lee PP, Piepkorn M, et al. Melanocyte Destruction after Antigen-Specific Immunotherapy of Melanoma. J Exp Med (2000) 192:1637–44. doi: 10.1084/jem.192.11.1637
11. Schuster H, Peper JK, Bösmüller H-C, Röhle K, Backert L, Bilich T, et al. The immunopeptidomic landscape of ovarian carcinomas. Proc Natl Acad Sci (2017) 114:E9942–51. doi: 10.1073/pnas.1707658114
12. Sarkizova S, Klaeger S, Le PM, Li LW, Oliveira G, Keshishian H, et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat Biotechnol (2020) 38:199–209. doi: 10.1038/s41587-019-0322-9
13. Pritchard AL, Hastie ML, Neller M, Gorman JJ, Schmidt CW, Hayward NK. Exploration of peptides bound to MHC class I molecules in melanoma. Pigment Cell Melanoma Res (2015) 28:281–94. doi: 10.1111/pcmr.12357
14. Bräunlein E, Krackhardt AM. Identification and Characterization of Neoantigens As Well As Respective Immune Responses in Cancer Patients. Front Immunol (2017) 8:1702. doi: 10.3389/fimmu.2017.01702
15. Schwitalle Y, Linnebacher M, Ripberger E, Gebert J, Von Knebel Doeberitz M. Immunogenic peptides generated by frameshift mutations in DNA mismatch repair-deficient cancer cells. Cancer Immun (2004) 4:1–10.
16. Mohammed F, Stones DH, Zarling AL, Willcox CR, Shabanowitz J, Cummings KL, et al. The antigenic identity of human class I MHC phosphopeptides is critically dependent upon phosphorylation status. Oncotarget (2017) 8:54160–72. doi: 10.18632/oncotarget.16952
17. Penny S, Malaker S, Steadman L, Trantham P, Bai D, Shabanowitz J, et al. Glycosylated and methylated peptides as neoantigens in leukaemia. Eur J Cancer (2016) 61:S217. doi: 10.1016/S0959-8049(16)61765-3
18. Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med (2013) 5:174ra27. doi: 10.1126/scitranslmed.3004916
19. Alatrash G, Molldrem JJ, Qazilbas MH. Targeting PR1 in myeloid leukemia. Oncotarget (2018) 9:4280–1. doi: 10.18632/oncotarget.23403
20. Doubrovina E, Carpenter T, Pankov D, Selvakumar A, Hasan A, O’Reilly RJ. Mapping of novel peptides of WT-1 and presenting HLA alleles that induce epitope-specific HLA-restricted T cells with cytotoxic activity against WT-1+ leukemias. Blood (2012) 120:1633–46. doi: 10.1182/blood-2011-11-394619
21. Gao L, Bellantuono I, Elsässer A, Marley SB, Gordon MY, Goldman JM, et al. Selective elimination of leukemic CD34+ progenitor cells by cytotoxic T lymphocytes specific for WT1. Blood (2000) 95:2198–203. doi: 10.1182/blood.V95.7.2198
22. Zamora AE, Crawford JC, Allen EK, Guo XZJ, Bakke J, Carter RA, et al. Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8+ T cell responses. Sci Transl Med (2019) 11:eaat8549. doi: 10.1126/scitranslmed.aat8549
23. Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol (2017) 18:1009–21. doi: 10.1016/S1470-2045(17)30516-8
24. Hansen UK, Ramskov S, Bjerregaard AM, Borch A, Andersen R, Draghi A, et al. Tumor-Infiltrating T Cells From Clear Cell Renal Cell Carcinoma Patients Recognize Neoepitopes Derived From Point and Frameshift Mutations. Front Immunol (2020) 11:373. doi: 10.3389/fimmu.2020.00373
25. Abelin JG, Trantham PD, Penny SA, Patterson AM, Ward ST, Hildebrand WH, et al. Complementary IMAC enrichment methods for HLA-associated phosphopeptide identification by mass spectrometry. Nat Protoc (2015) 10:1308–18. doi: 10.1038/nprot.2015.086
26. Zarling AL, Ficarro SB, White FM, Shabanowitz J, Hunt DF, Engelhard VH. Phosphorylated Peptides Are Naturally Processed and Presented by Major Histocompatibility Complex Class I Molecules in Vivo. J Exp Med (2000) 192:1755–62. doi: 10.1084/jem.192.12.1755
27. Zarling AL, Polefrone JM, Evans AM, Mikesh LM, Shabanowitz J, Lewis ST, et al. Identification of class I MHC-associated phosphopeptides as targets for cancer immunotherapy. Proc Natl Acad Sci (2006) 103:14889–94. doi: 10.1073/pnas.0604045103
28. Cobbold M, De La Peña H, Norris A, Polefrone JM, Qian J, English AM, et al. MHC class I-associated phosphopeptides are the targets of memory-like immunity in leukemia. Sci Transl Med (2013) 5:203ra125. doi: 10.1126/scitranslmed.3006061
29. Mohammed F, Cobbold M, Zarling AL, Salim M, Barrett-Wilt GA, Shabanowitz J, et al. Phosphorylation-dependent interaction between antigenic peptides and MHC class I: a molecular basis for the presentation of transformed self. Nat Immunol (2008) 9:1236–43. doi: 10.1038/ni.1660
30. Engelhard VH, Obeng RC, Cummings KL, Petroni GR, Ambakhutwala AL, Chianese-Bullock KA, et al. MHC-restricted phosphopeptide antigens: preclinical validation and first-in-humans clinical trial in participants with high-risk melanoma. J Immunother Cancer (2020) 8:e000262. doi: 10.1136/jitc-2019-000262
31. Malaker SA, Penny SA, Steadman LG, Myers PT, Loke JC, Raghavan M, et al. Identification of glycopeptides as posttranslationally modified neoantigens in Leukemia. Cancer Immunol Res (2017) 5:376–84. doi: 10.1158/2326-6066.CIR-16-0280
32. Sun M, Liu J, Qi J, Tefsen B, Shi Y, Yan J, et al. N α -Terminal Acetylation for T Cell Recognition: Molecular Basis of MHC Class I–Restricted N α -Acetylpeptide Presentation. J Immunol (2014) 192:5509–19. doi: 10.4049/jimmunol.1400199
33. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Sci (80-) (2016) 351:1463–9. doi: 10.1126/science.aaf1490
34. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N Engl J Med (2014) 371:2189–99. doi: 10.1056/NEJMoa1406498
35. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor Exome Analysis Reveals Neoantigen-Specific T-Cell Reactivity in an Ipilimumab-Responsive Melanoma. J Clin Oncol (2013) 31:e439–42. doi: 10.1200/JCO.2012.47.7521
36. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu Y-C, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med (2018) 24:724–30. doi: 10.1038/s41591-018-0040-8
37. Bassani-Sternberg M, Bräunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun (2016) 7:13404. doi: 10.1038/ncomms13404
38. Newey A, Griffiths B, Michaux J, Pak HS, Stevenson BJ, Woolston A, et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class I neoantigen landscape and no increase in neoantigens with interferon or MEK-inhibitor treatment. J Immunother Cancer (2019) 7:309. doi: 10.1186/s40425-019-0769-8
39. Wang Q, Douglass J, Hwang MS, Hsiue EH-C, Mog BJ, Zhang M, et al. Direct Detection and Quantification of Neoantigens. Cancer Immunol Res (2019) 7:1748–54. doi: 10.1158/2326-6066.CIR-19-0107
40. Iiizumi S, Ohtake J, Murakami N, Kouro T, Kawahara M, Isoda F, et al. Identification of novel HLA class II-restricted neoantigens derived from driver mutations. Cancers (Basel) (2019) 11:266. doi: 10.3390/cancers11020266
41. Chandran S, Ma J, Klatt MG, Dündar F, Zumbo P, Femia MR, et al. Abstract CN01-03: T cell receptor gene therapy for a public neoantigen derived from mutated PIK3CA, a dominant driver oncogene in breast and endometrial cancers. In: Mol. Cancer Ther. Philadelphia, PA, USA: American Association for Cancer Research (2019). p. CN01–03. doi: 10.1158/1535-7163.TARG-19-CN01-03
42. Wang QJ, Yu Z, Griffith K, Hanada K-I, Restifo NP, Yang JC. Identification of T-cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol Res (2016) 4:204–14. doi: 10.1158/2326-6066.CIR-15-0188
43. Oh CY, Klatt MG, Bourne C, Dao T, Dacek MM, Brea EJ, et al. ALK and RET Inhibitors Promote HLA Class I Antigen Presentation and Unmask New Antigens within the Tumor Immunopeptidome. Cancer Immunol Res (2019) 7:1984–97. doi: 10.1158/2326-6066.CIR-19-0056
44. Chong C, Marino F, Pak H, Racle J, Daniel RT, Müller M, et al. High-throughput and Sensitive Immunopeptidomics Platform Reveals Profound Interferonγ-Mediated Remodeling of the Human Leukocyte Antigen (HLA) Ligandome. Mol Cell Proteomics (2018) 17:533–48. doi: 10.1074/mcp.TIR117.000383
45. Javitt A, Barnea E, Kramer MP, Wolf-Levy H, Levin Y, Admon A, et al. Pro-inflammatory Cytokines Alter the Immunopeptidome Landscape by Modulation of HLA-B Expression. Front Immunol (2019) 10:141. doi: 10.3389/fimmu.2019.00141
46. Chang AY, Dao T, Gejman RS, Jarvis CA, Scott A, Dubrovsky L, et al. A therapeutic T cell receptor mimic antibody targets tumor-associated PRAME peptide/HLA-I antigens. J Clin Invest (2017) 127:2705–18. doi: 10.1172/JCI92335
47. Soto CM, Stone JD, Chervin AS, Engels B, Schreiber H, Roy EJ, et al. MHC-class I-restricted CD4 T cells: a nanomolar affinity TCR has improved anti-tumor efficacy in vivo compared to the micromolar wild-type TCR. Cancer Immunol Immunother (2013) 62:359–69. doi: 10.1007/s00262-012-1336-z
48. Sun Y, Sijts AJAM, Song M, Janek K, Nussbaum AK, Kral S, et al. Expression of the proteasome activator PA28 rescues the presentation of a cytotoxic T lymphocyte epitope on melanoma cells. Cancer Res (2002) 62:2875–82.
49. Sewell AK, Price DA, Teisserenc H, Booth BL, Gileadi U, Flavin FM, et al. IFN-gamma exposes a cryptic cytotoxic T lymphocyte epitope in HIV-1 reverse transcriptase. J Immunol (1999) 162:7075–9.
50. Lai J, Wang Y, Wu S-S, Ding D, Sun Z-Y, Zhang Y, et al. Elimination of melanoma by sortase A-generated TCR-like antibody-drug conjugates (TL-ADCs) targeting intracellular melanoma antigen MART-1. Biomaterials (2018) 178:158–69. doi: 10.1016/j.biomaterials.2018.06.017
51. Brea EJ, Oh CY, Manchado E, Budhu S, Gejman RS, Mo G, et al. Kinase Regulation of Human MHC Class I Molecule Expression on Cancer Cells. Cancer Immunol Res (2016) 4:936–47. doi: 10.1158/2326-6066.CIR-16-0177
52. Kowalewski DJ, Walz S, Backert L, Schuster H, Kohlbacher O, Weisel K, et al. Carfilzomib alters the HLA-presented peptidome of myeloma cells and impairs presentation of peptides with aromatic C-termini. Blood Cancer J (2016) 6:e411–1. doi: 10.1038/bcj.2016.14
53. Koumantou D, Barnea E, Martin-Esteban A, Maben Z, Papakyriakou A, Mpakali A, et al. Editing the immunopeptidome of melanoma cells using a potent inhibitor of endoplasmic reticulum aminopeptidase 1 (ERAP1). Cancer Immunol Immunother (2019) 68:1245–61. doi: 10.1007/s00262-019-02358-0
54. Gravett AM, Stevanović S, Dalgleish AG, Copier J. Gemcitabine alters the proteasome composition and immunopeptidome of tumour cells. Oncoimmunology (2018) 7:e1438107. doi: 10.1080/2162402X.2018.1438107
55. Galaine J, Turco C, Vauchy C, Royer B, Mercier-Letondal P, Queiroz L, et al. CD4 T cells target colorectal cancer antigens upregulated by oxaliplatin. Int J Cancer (2019) 145:3112–25. doi: 10.1002/ijc.32620
56. Srivastava P, Paluch BE, Matsuzaki J, James SR, Collamat-Lai G, Blagitko-Dorfs N, et al. Induction of cancer testis antigen expression in circulating acute myeloid leukemia blasts following hypomethylating agent monotherapy. Oncotarget (2016) 7:12840–56. doi: 10.18632/oncotarget.7326
57. Shi X, Chen X, Fang B, Ping Y, Qin G, Yue D, et al. Decitabine enhances tumor recognition by T cells through upregulating the MAGE-A3 expression in esophageal carcinoma. BioMed Pharmacother (2019) 112:108632. doi: 10.1016/j.biopha.2019.108632
58. Sharma A, Albahrani M, Zhang W, Kufel CN, James SR, Odunsi K, et al. Epigenetic activation of POTE genes in ovarian cancer. Epigenetics (2019) 14:185–97. doi: 10.1080/15592294.2019.1581590
59. Bensaid D, Blondy T, Deshayes S, Dehame V, Bertrand P, Grégoire M, et al. Assessment of new HDAC inhibitors for immunotherapy of malignant pleural mesothelioma. Clin Epigenet (2018) 10:79. doi: 10.1186/s13148-018-0517-9
60. Heninger E, Krueger TEG, Thiede SM, Sperger JM, Byers BL, Kircher MR, et al. Inducible expression of cancer-testis antigens in human prostate cancer. Oncotarget (2016) 7:84359–74. doi: 10.18632/oncotarget.12711
61. Cameron EE, Bachman KE, Myöhänen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet (1999) 21:103–7. doi: 10.1038/5047
62. Yee C, Savage PA, Lee PP, Davis MM, Greenberg PD. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J Immunol (1999) 162:2227–34.
63. Ren L, Leisegang M, Deng B, Matsuda T, Kiyotani K, Kato T, et al. Identification of neoantigen-specific T cells and their targets: implications for immunotherapy of head and neck squamous cell carcinoma. Oncoimmunology (2019) 8:1–10. doi: 10.1080/2162402X.2019.1568813
64. Tsai V, Southwood S, Sidney J, Sakaguchi K, Kawakami Y, Appella E, et al. Identification of subdominant CTL epitopes of the GP100 melanoma-associated tumor antigen by primary in vitro immunization with peptide-pulsed dendritic cells. J Immunol (1997) 158:1796–802.
65. Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a Titin-Derived HLA-A1-Presented Peptide as a Cross-Reactive Target for Engineered MAGE A3-Directed T Cells. Sci Transl Med (2013) 5:197ra103–197ra103. doi: 10.1126/scitranslmed.3006034
66. Raman MCC, Rizkallah PJ, Simmons R, Donnellan Z, Dukes J, Bossi G, et al. Direct molecular mimicry enables off-target cardiovascular toxicity by an enhanced affinity TCR designed for cancer immunotherapy. Sci Rep (2016) 6:18851. doi: 10.1038/srep18851
67. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Sci (80-) (2015) 348:62–8. doi: 10.1126/science.aaa4967
68. Connerotte T, Van Pel A, Godelaine D, Tartour E, Schuler-Thurner B, Lucas S, et al. Functions of Anti-MAGE T-Cells Induced in Melanoma Patients under Different Vaccination Modalities. Cancer Res (2008) 68:3931–40. doi: 10.1158/0008-5472.CAN-07-5898
69. Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature (2017) 547:222–6. doi: 10.1038/nature23003
70. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Sci (80-) (2015) 348:803–8. doi: 10.1126/science.aaa3828
71. Robbins PF, Lu Y-C, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med (2013) 19:747–52. doi: 10.1038/nm.3161
72. Chen J-L, Stewart-Jones G, Bossi G, Lissin NM, Wooldridge L, Choi EML, et al. Structural and kinetic basis for heightened immunogenicity of T cell vaccines. J Exp Med (2005) 201:1243–55. doi: 10.1084/jem.20042323
73. Pinilla-Ibarz J, May RJ, Korontsvit T, Gomez M, Kappel B, Zakhaleva V, et al. Improved human T-cell responses against synthetic HLA-0201 analog peptides derived from the WT1 oncoprotein. Leukemia (2006) 20:2025–33. doi: 10.1038/sj.leu.2404380
74. Bethune MT, Li X-H, Yu J, McLaughlin J, Cheng D, Mathis C, et al. Isolation and characterization of NY-ESO-1–specific T cell receptors restricted on various MHC molecules. Proc Natl Acad Sci (2018) 115:E10702–11. doi: 10.1073/pnas.1810653115
75. Weber J, Sondak VK, Scotland R, Phillip R, Wang F, Rubio V, et al. Granulocyte-macrophage-colony-stimulating factor added to a multipeptide vaccine for resected Stage II melanoma. Cancer (2003) 97:186–200. doi: 10.1002/cncr.11045
76. Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med (1998) 4:321–7. doi: 10.1038/nm0398-321
77. Ali M, Foldvari Z, Giannakopoulou E, Böschen M-L, Strønen E, Yang W, et al. Induction of neoantigen-reactive T cells from healthy donors. Nat Protoc (2019) 14:1926–43. doi: 10.1038/s41596-019-0170-6
78. Meng Q, Valentini D, Rao M, Moro CF, Paraschoudi G, Jäger E, et al. Neoepitope targets of tumour-infiltrating lymphocytes from patients with pancreatic cancer. Br J Cancer (2019) 120:97–108. doi: 10.1038/s41416-018-0262-z
79. Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of Tumor-Infiltrating Lymphocyte Cultures for Use in Adoptive Transfer Therapy for Melanoma Patients. J Immunother (2003) 26:332–42. doi: 10.1097/00002371-200307000-00005
80. Owens GL, Price MJ, Cheadle EJ, Hawkins RE, Gilham DE, Edmondson RJ. Ex vivo expanded tumour-infiltrating lymphocytes from ovarian cancer patients release anti-tumour cytokines in response to autologous primary ovarian cancer cells. Cancer Immunol Immunother (2018) 67:1519–31. doi: 10.1007/s00262-018-2211-3
81. Celis E, Tsai V, Crimi C, DeMars R, Wentworth PA, Chesnut RW, et al. Induction of anti-tumor cytotoxic T lymphocytes in normal humans using primary cultures and synthetic peptide epitopes. Proc Natl Acad Sci (1994) 91:2105–9. doi: 10.1073/pnas.91.6.2105
82. Rivoltini L, Kawakami Y, Sakaguchi K, Southwood S, Sette A, Robbins PF, et al. Induction of tumor-reactive CTL from peripheral blood and tumor-infiltrating lymphocytes of melanoma patients by in vitro stimulation with an immunodominant peptide of the human melanoma antigen MART-1. J Immunol (1995) 154:2257–65.
83. Lu Y-C, Zheng Z, Robbins PF, Tran E, Prickett TD, Gartner JJ, et al. An Efficient Single-Cell RNA-Seq Approach to Identify Neoantigen-Specific T Cell Receptors. Mol Ther (2018) 26:379–89. doi: 10.1016/j.ymthe.2017.10.018
84. Malekzadeh P, Pasetto A, Robbins PF, Parkhurst MR, Paria BC, Jia L, et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J Clin Invest (2019) 129:1109–14. doi: 10.1172/JCI123791
85. Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced Antitumor Activity of T Cells Engineered to Express T-Cell Receptors with a Second Disulfide Bond. Cancer Res (2007) 67:3898–903. doi: 10.1158/0008-5472.CAN-06-3986
86. Tran E, Turcotte S, Gros A, Robbins PF, Lu Y-C, Dudley ME, et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient with Epithelial Cancer. Sci (80-) (2014) 344:641–5. doi: 10.1126/science.1251102
87. Lu Y-C, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, et al. Efficient Identification of Mutated Cancer Antigens Recognized by T Cells Associated with Durable Tumor Regressions. Clin Cancer Res (2014) 20:3401–10. doi: 10.1158/1078-0432.CCR-14-0433
88. Stronen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Sci (80-) (2016) 352:1337–41. doi: 10.1126/science.aaf2288
89. Kobayashi E, Mizukoshi E, Kishi H, Ozawa T, Hamana H, Nagai T, et al. A new cloning and expression system yields and validates TCRs from blood lymphocytes of patients with cancer within 10 days. Nat Med (2013) 19:1542–6. doi: 10.1038/nm.3358
90. Altman JD, Moss PAH, Goulder PJR, Barouch DH, McHeyzer-Williams MG, Bell JI, et al. Phenotypic Analysis of Antigen-Specific T Lymphocytes. Sci (80-) (1996) 274:94–6. doi: 10.1126/science.274.5284.94
91. Kato T, Matsuda T, Ikeda Y, Park J-H, Leisegang M, Yoshimura S, et al. Effective screening of T cells recognizing neoantigens and construction of T-cell receptor-engineered T cells. Oncotarget (2018) 9:11009–19. doi: 10.18632/oncotarget.24232
92. Dössinger G, Bunse M, Bet J, Albrecht J, Paszkiewicz PJ, Weißbrich B, et al. MHC Multimer-Guided and Cell Culture-Independent Isolation of Functional T Cell Receptors from Single Cells Facilitates TCR Identification for Immunotherapy. PLoS One (2013) 8:e61384. doi: 10.1371/journal.pone.0061384
93. Romero P, Rod Dunbar P, Valmori D, Pittet M, Ogg GS, Rimoldi D, et al. Ex Vivo Staining of Metastatic Lymph Nodes by Class I Major Histocompatibility Complex Tetramers Reveals High Numbers of Antigen-experienced Tumor-specific Cytolytic T Lymphocytes. J Exp Med (1998) 188:1641–50. doi: 10.1084/jem.188.9.1641
94. Matsuda T, Leisegang M, Park J-H, Ren L, Kato T, Ikeda Y, et al. Induction of Neoantigen-Specific Cytotoxic T Cells and Construction of T-cell Receptor–Engineered T Cells for Ovarian Cancer. Clin Cancer Res (2018) 24:5357–67. doi: 10.1158/1078-0432.CCR-18-0142
95. Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest (2015) 125:3981–91. doi: 10.1172/JCI82416
96. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med (2016) 22:433–8. doi: 10.1038/nm.4051
97. Parkhurst M, Gros A, Pasetto A, Prickett T, Crystal JS, Robbins P, et al. Isolation of T-cell receptors specifically reactive with mutated tumor-associated antigens from tumor-infiltrating lymphocytes based on CD137 expression. Clin Cancer Res (2017) 23:2491–505. doi: 10.1158/1078-0432.CCR-16-2680
98. Wolfl M, Kuball J, Ho WY, Nguyen H, Manley TJ, Bleakley M, et al. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood (2007) 110:201–10. doi: 10.1182/blood-2006-11-056168
99. Ye Q, Song D-G, Poussin M, Yamamoto T, Best A, Li C, et al. CD137 Accurately Identifies and Enriches for Naturally Occurring Tumor-Reactive T Cells in Tumor. Clin Cancer Res (2014) 20:44–55. doi: 10.1158/1078-0432.CCR-13-0945
100. Wölfl M, Kuball J, Eyrich M, Schlegel PG, Greenberg PD. Use of CD137 to study the full repertoire of CD8+ T cells without the need to know epitope specificities. Cytom Part A (2008) 73A:1043–9. doi: 10.1002/cyto.a.20594
101. Inozume T, Hanada K, Wang QJ, Ahmadzadeh M, Wunderlich JR, Rosenberg SA, et al. Selection of CD8+PD-1+ Lymphocytes in Fresh Human Melanomas Enriches for Tumor-reactive T Cells. J Immunother (2010) 33:956–64. doi: 10.1097/CJI.0b013e3181fad2b0
102. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest (2014) 124:2246–59. doi: 10.1172/JCI73639
103. Malekzadeh P, Yossef R, Cafri G, Paria BC, Lowery FJ, Jafferji M, et al. Antigen Experienced T Cells from Peripheral Blood Recognize p53 Neoantigens. Clin Cancer Res (2020) 26:1267–76. doi: 10.1158/1078-0432.CCR-19-1874
104. Tan Q, Zhang C, Yang W, Liu Y, Heyilimu P, Feng D, et al. Isolation of T cell receptor specifically reactive with autologous tumour cells from tumour-infiltrating lymphocytes and construction of T cell receptor engineered T cells for esophageal squamous cell carcinoma. J Immunother Cancer (2019) 7:232. doi: 10.1186/s40425-019-0709-7
105. Rubio V, Stuge TB, Singh N, Betts MR, Weber JS, Roederer M, et al. Ex vivo identification, isolation and analysis of tumor-cytolytic T cells. Nat Med (2003) 9:1377–82. doi: 10.1038/nm942
106. Becker C, Pohla H, Frankenberger B, Schüler T, Assenmacher M, Schendel DJ, et al. Adoptive tumor therapy with T lymphocytes enriched through an IFN-γ capture assay. Nat Med (2001) 7:1159–62. doi: 10.1038/nm1001-1159
107. Rius C, Attaf M, Tungatt K, Bianchi V, Legut M, Bovay A, et al. Peptide–MHC Class I Tetramers Can Fail To Detect Relevant Functional T Cell Clonotypes and Underestimate Antigen-Reactive T Cell Populations. J Immunol (2018) 200:2263–79. doi: 10.4049/jimmunol.1700242
108. Laugel B, van den Berg HA, Gostick E, Cole DK, Wooldridge L, Boulter J, et al. Different T Cell Receptor Affinity Thresholds and CD8 Coreceptor Dependence Govern Cytotoxic T Lymphocyte Activation and Tetramer Binding Properties. J Biol Chem (2007) 282:23799–810. doi: 10.1074/jbc.M700976200
109. Dolton G, Lissina A, Skowera A, Ladell K, Tungatt K, Jones E, et al. Comparison of peptide–major histocompatibility complex tetramers and dextramers for the identification of antigen-specific T cells. Clin Exp Immunol (2014) 177:47–63. doi: 10.1111/cei.12339
110. Snyder JE, Bowers WJ, Livingstone AM, Lee FE-H, Federoff HJ, Mosmann TR. Measuring the frequency of mouse and human cytotoxic T cells by the Lysispot assay: independent regulation of cytokine secretion and short-term killing. Nat Med (2003) 9:231–6. doi: 10.1038/nm821
111. Linnemann C, Heemskerk B, Kvistborg P, Kluin RJC, Bolotin DA, Chen X, et al. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat Med (2013) 19:1534–41. doi: 10.1038/nm.3359
112. Howie B, Sherwood AM, Berkebile AD, Berka J, Emerson RO, Williamson DW, et al. High-throughput pairing of T cell receptor α and β sequences. Sci Transl Med (2015) 7:301ra131. doi: 10.1126/scitranslmed.aac5624
113. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science (2002) 298:850–4. doi: 10.1126/science.1076514
114. Chinnasamy N, Wargo JA, Yu Z, Rao M, Frankel TL, Riley JP, et al. A TCR Targeting the HLA-A*0201–Restricted Epitope of MAGE-A3 Recognizes Multiple Epitopes of the MAGE-A Antigen Superfamily in Several Types of Cancer. J Immunol (2011) 186:685–96. doi: 10.4049/jimmunol.1001775
115. Obenaus M, Leitão C, Leisegang M, Chen X, Gavvovidis I, van der Bruggen P, et al. Identification of human T-cell receptors with optimal affinity to cancer antigens using antigen-negative humanized mice. Nat Biotechnol (2015) 33:402–7. doi: 10.1038/nbt.3147
116. Li LP, Lampert JC, Chen X, Leitao C, Popović J, Müller W, et al. Transgenic mice with a diverse human T cell antigen receptor repertoire. Nat Med (2010) 16:1029–34. doi: 10.1038/nm.2197
117. Wilde S, Sommermeyer D, Frankenberger B, Schiemann M, Milosevic S, Spranger S, et al. Dendritic cells pulsed with RNA encoding allogeneic MHC and antigen induce T cells with superior antitumor activity and higher TCR functional avidity. Blood (2009) 114:2131–9. doi: 10.1182/blood-2009-03-209387
118. Münz C, Obst R, Osen W, Stevanović S, Rammensee HG. Alloreactivity as a source of high avidity peptide-specific human CTL. J Immunol (1999) 162:25–34.
119. Sadovnikova E, Stauss HJ. Peptide-specific cytotoxic T lymphocytes restricted by nonself major histocompatibility complex class I molecules: Reagents for tumor immunotherapy. Proc Natl Acad Sci (1996) 93:13114–8. doi: 10.1073/pnas.93.23.13114
120. Falkenburg WJJ, Melenhorst JJ, van de Meent M, Kester MGD, Hombrink P, Heemskerk MHM, et al. Allogeneic HLA-A*02–Restricted WT1-Specific T Cells from Mismatched Donors Are Highly Reactive but Show Off-Target Promiscuity. J Immunol (2011) 187:2824–33. doi: 10.4049/jimmunol.1100852
121. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor Regression in Patients With Metastatic Synovial Cell Sarcoma and Melanoma Using Genetically Engineered Lymphocytes Reactive With NY-ESO-1. J Clin Oncol (2011) 29:917–24. doi: 10.1200/JCO.2010.32.2537
122. Johnson LA, Heemskerk B, Powell DJ, Cohen CJ, Morgan RA, Dudley ME, et al. Gene Transfer of Tumor-Reactive TCR Confers Both High Avidity and Tumor Reactivity to Nonreactive Peripheral Blood Mononuclear Cells and Tumor-Infiltrating Lymphocytes. J Immunol (2006) 177:6548–59. doi: 10.4049/jimmunol.177.9.6548
123. Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, et al. Single and Dual Amino Acid Substitutions in TCR CDRs Can Enhance Antigen-Specific T Cell Functions. J Immunol (2008) 180:6116–31. doi: 10.4049/jimmunol.180.9.6116
124. Li Y, Moysey R, Molloy PE, Vuidepot A-L, Mahon T, Baston E, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol (2005) 23:349–54. doi: 10.1038/nbt1070
125. Dunn SM, Rizkallah PJ, Baston E, Mahon T, Cameron B, Moysey R, et al. Directed evolution of human T cell receptor CDR2 residues by phage display dramatically enhances affinity for cognate peptide-MHC without increasing apparent cross-reactivity. Protein Sci (2006) 15:710–21. doi: 10.1110/ps.051936406
126. Holler PD, Holman PO, Shusta EV, O’Herrin S, Wittrup KD, Kranz DM. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc Natl Acad Sci (2000) 97:5387–92. doi: 10.1073/pnas.080078297
127. Chlewicki LK, Holler PD, Monti BC, Clutter MR, Kranz DM. High-affinity, Peptide-specific T Cell Receptors can be Generated by Mutations in CDR1, CDR2 or CDR3. J Mol Biol (2005) 346:223–39. doi: 10.1016/j.jmb.2004.11.057
128. Malecek K, Zhong S, McGary K, Yu C, Huang K, Johnson LA, et al. Engineering improved T cell receptors using an alanine-scan guided T cell display selection system. J Immunol Methods (2013) 392:1–11. doi: 10.1016/j.jim.2013.02.018
129. Chervin AS, Aggen DH, Raseman JM, Kranz DM. Engineering higher affinity T cell receptors using a T cell display system. J Immunol Methods (2008) 339:175–84. doi: 10.1016/j.jim.2008.09.016
130. Bassan D, Gozlan YM, Sharbi-Yunger A, Tzehoval E, Eisenbach L. Optimizing T-cell receptor avidity with somatic hypermutation. Int J Cancer (2019) 145:2816–26. doi: 10.1002/ijc.32612
131. Schmitt TM, Aggen DH, Ishida-Tsubota K, Ochsenreither S, Kranz DM, Greenberg PD. Generation of higher affinity T cell receptors by antigen-driven differentiation of progenitor T cells in vitro. Nat Biotechnol (2017) 35:1188–95. doi: 10.1038/nbt.4004
132. Malecek K, Grigoryan A, Zhong S, Gu WJ, Johnson LA, Rosenberg SA, et al. Specific Increase in Potency via Structure-Based Design of a TCR. J Immunol (2014) 193:2587–99. doi: 10.4049/jimmunol.1302344
133. Gálvez J, Gálvez JJ, García-Peñarrubia P. Is TCR/pMHC Affinity a Good Estimate of the T-cell Response? An Answer Based on Predictions From 12 Phenotypic Models. Front Immunol (2019) 10:349. doi: 10.3389/fimmu.2019.00349
134. Irving M, Zoete V, Hebeisen M, Schmid D, Baumgartner P, Guillaume P, et al. Interplay between T Cell Receptor Binding Kinetics and the Level of Cognate Peptide Presented by Major Histocompatibility Complexes Governs CD8 + T Cell Responsiveness. J Biol Chem (2012) 287:23068–78. doi: 10.1074/jbc.M112.357673
135. Hebeisen M, Baitsch L, Presotto D, Baumgaertner P, Romero P, Michielin O, et al. SHP-1 phosphatase activity counteracts increased T cell receptor affinity. J Clin Invest (2013) 123:1044–56. doi: 10.1172/JCI65325
136. Duong MN, Erdes E, Hebeisen M, Rufer N. Chronic TCR-MHC (self)-interactions limit the functional potential of TCR affinity-increased CD8 T lymphocytes. J Immunother Cancer (2019) 7:284. doi: 10.1186/s40425-019-0773-z
137. McMahan RH, McWilliams JA, Jordan KR, Dow SW, Wilson DB, Slansky JE. Relating TCR-peptide-MHC affinity to immunogenicity for the design of tumor vaccines. J Clin Invest (2006) 116:2543–51. doi: 10.1172/JCI26936
138. Prickett TD, Crystal JS, Cohen CJ, Pasetto A, Parkhurst MR, Gartner JJ, et al. Durable Complete Response from Metastatic Melanoma after Transfer of Autologous T Cells Recognizing 10 Mutated Tumor Antigens. Cancer Immunol Res (2016) 4:669–78. doi: 10.1158/2326-6066.CIR-15-0215
139. Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, et al. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci (2005) 102:16013–8. doi: 10.1073/pnas.0500090102
140. Linard B, Bézieau S, Benlalam H, Labarrière N, Guilloux Y, Diez E, et al. A ras- Mutated Peptide Targeted by CTL Infiltrating a Human Melanoma Lesion. J Immunol (2002) 168:4802–8. doi: 10.4049/jimmunol.168.9.4802
141. Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
142. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother (2013) 36:133–51. doi: 10.1097/CJI.0b013e3182829903
143. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Sci (80-) (2006) 314:126–9. doi: 10.1126/science.1129003
144. Gee MH, Han A, Lofgren SM, Beausang JF, Mendoza JL, Birnbaum ME, et al. Antigen Identification for Orphan T Cell Receptors Expressed on Tumor-Infiltrating Lymphocytes. Cell (2018) 172:549–63.e16. doi: 10.1016/j.cell.2017.11.043
145. Wooldridge L, Laugel B, Ekeruche J, Clement M, van den Berg HA, Price DA, et al. CD8 Controls T Cell Cross-Reactivity. J Immunol (2010) 185:4625–32. doi: 10.4049/jimmunol.1001480
146. Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive Cell Transfer Therapy Following Non-Myeloablative but Lymphodepleting Chemotherapy for the Treatment of Patients With Refractory Metastatic Melanoma. J Clin Oncol (2005) 23:2346–57. doi: 10.1200/JCO.2005.00.240
147. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood (2009) 114:535–46. doi: 10.1182/blood-2009-03-211714
148. van Loenen MM, de Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, et al. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci (2010) 107:10972–7. doi: 10.1073/pnas.1005802107
149. Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced Antitumor Activity of Murine-Human Hybrid T-Cell Receptor (TCR) in Human Lymphocytes Is Associated with Improved Pairing and TCR/CD3 Stability. Cancer Res (2006) 66:8878–86. doi: 10.1158/0008-5472.CAN-06-1450
150. Hart DP, Xue S-A, Thomas S, Cesco-Gaspere M, Tranter A, Willcox B, et al. Retroviral transfer of a dominant TCR prevents surface expression of a large proportion of the endogenous TCR repertoire in human T cells. Gene Ther (2008) 15:625–31. doi: 10.1038/sj.gt.3303078
151. Bialer G, Horovitz-Fried M, Ya’acobi S, Morgan RA, Cohen CJ. Selected Murine Residues Endow Human TCR with Enhanced Tumor Recognition. J Immunol (2010) 184:6232–41. doi: 10.4049/jimmunol.0902047
152. Bethune MT, Gee MH, Bunse M, Lee MS, Gschweng EH, Pagadala MS, et al. Domain-swapped T cell receptors improve the safety of TCR gene therapy. Elife (2016) 5:e19095. doi: 10.7554/eLife.19095
153. van der Veken LT, Coccoris M, Swart E, Falkenburg JHF, Schumacher TN, Heemskerk MHM. αβ T Cell Receptor Transfer to γδ T Cells Generates Functional Effector Cells without Mixed TCR Dimers In Vivo. J Immunol (2009) 182:164–70. doi: 10.4049/jimmunol.182.1.164
154. van der Veken LT, Hagedoorn RS, van Loenen MM, Willemze R, Falkenburg JHF, Heemskerk MHM. αβ T-Cell Receptor Engineered γδ T Cells Mediate Effective Antileukemic Reactivity. Cancer Res (2006) 66:3331–7. doi: 10.1158/0008-5472.CAN-05-4190
155. Legut M, Dolton G, Mian AA, Ottmann OG, Sewell AK. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood (2018) 131:311–22. doi: 10.1182/blood-2017-05-787598
156. Okamoto S, Mineno J, Ikeda H, Fujiwara H, Yasukawa M, Shiku H, et al. Improved Expression and Reactivity of Transduced Tumor-Specific TCRs in Human Lymphocytes by Specific Silencing of Endogenous TCR. Cancer Res (2009) 69:9003–11. doi: 10.1158/0008-5472.CAN-09-1450
157. Okamoto S, Amaishi Y, Goto Y, Ikeda H, Fujiwara H, Kuzushima K, et al. A Promising Vector for TCR Gene Therapy: Differential Effect of siRNA, 2A Peptide, and Disulfide Bond on the Introduced TCR Expression. Mol Ther Nucleic Acids (2012) 1:e63. doi: 10.1038/mtna.2012.52
158. Campillo-Davo D, Fujiki F, Van den Bergh JMJ, De Reu H, Smits ELJM, Goossens H, et al. Efficient and Non-genotoxic RNA-Based Engineering of Human T Cells Using Tumor-Specific T Cell Receptors With Minimal TCR Mispairing. Front Immunol (2018) 9:2503. doi: 10.3389/fimmu.2018.02503
159. Sharma P, Harris DT, Stone JD, Kranz DM. T-cell Receptors Engineered De Novo for Peptide Specificity Can Mediate Optimal T-cell Activity without Self Cross-Reactivity. Cancer Immunol Res (2019) 7:2025–35. doi: 10.1158/2326-6066.CIR-19-0035
160. Audehm S, Glaser M, Pecoraro M, Bräunlein E, Mall S, Klar R, et al. Key Features Relevant to Select Antigens and TCR From the MHC-Mismatched Repertoire to Treat Cancer. Front Immunol (2019) 10:1485. doi: 10.3389/fimmu.2019.01485
161. Bijen HM, van der Steen DM, Hagedoorn RS, Wouters AK, Wooldridge L, Falkenburg JHF, et al. Preclinical Strategies to Identify Off-Target Toxicity of High-Affinity TCRs. Mol Ther (2018) 26:1206–14. doi: 10.1016/j.ymthe.2018.02.017
162. Harper J, Adams KJ, Bossi G, Wright DE, Stacey AR, Bedke N, et al. An approved in vitro approach to preclinical safety and efficacy evaluation of engineered T cell receptor anti-CD3 bispecific (ImmTAC) molecules. PloS One (2018) 13:e0205491. doi: 10.1371/journal.pone.0205491
163. Bentzen AK, Such L, Jensen KK, Marquard AM, Jessen LE, Miller NJ, et al. T cell receptor fingerprinting enables in-depth characterization of the interactions governing recognition of peptide–MHC complexes. Nat Biotechnol (2018) 36:1191–6. doi: 10.1038/nbt.4303
164. Schaubert KL, Price DA, Salkowitz JR, Sewell AK, Sidney J, Asher TE, et al. Generation of robust CD8+ T-cell responses against subdominant epitopes in conserved regions of HIV-1 by repertoire mining with mimotopes. Eur J Immunol (2010) 40:1950–62. doi: 10.1002/eji.200940079
165. Zhao Y, Gran B, Pinilla C, Markovic-Plese S, Hemmer B, Tzou A, et al. Combinatorial Peptide Libraries and Biometric Score Matrices Permit the Quantitative Analysis of Specific and Degenerate Interactions Between Clonotypic TCR and MHC Peptide Ligands. J Immunol (2001) 167:2130–41. doi: 10.4049/jimmunol.167.4.2130
166. Karapetyan AR, Chaipan C, Winkelbach K, Wimberger S, Jeong JS, Joshi B, et al. TCR Fingerprinting and Off-Target Peptide Identification. Front Immunol (2019) 10:2501. doi: 10.3389/fimmu.2019.02501
167. Lanzarotti E, Marcatili P, Nielsen M. T-Cell Receptor Cognate Target Prediction Based on Paired α and β Chain Sequence and Structural CDR Loop Similarities. Front Immunol (2019) 10:2080. doi: 10.3389/fimmu.2019.02080
168. Lanzarotti E, Marcatili P, Nielsen M. Identification of the cognate peptide-MHC target of T cell receptors using molecular modeling and force field scoring. Mol Immunol (2018) 94:91–7. doi: 10.1016/j.molimm.2017.12.019
169. Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F, et al. Identifying specificity groups in the T cell receptor repertoire. Nature (2017) 547:94–8. doi: 10.1038/nature22976
170. Mendes MFA, Antunes DA, Rigo MM, Sinigaglia M, Vieira GF. Improved structural method for T-cell cross-reactivity prediction. Mol Immunol (2015) 67:303–10. doi: 10.1016/j.molimm.2015.06.017
171. Dash P, Fiore-Gartland AJ, Hertz T, Wang GC, Sharma S, Souquette A, et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature (2017) 547:89–93. doi: 10.1038/nature22383
172. Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J, et al. Deconstructing the Peptide-MHC Specificity of T Cell Recognition. Cell (2014) 157:1073–87. doi: 10.1016/j.cell.2014.03.047
173. Crawford F, Jordan KR, Stadinski B, Wang Y, Huseby E, Marrack P, et al. Use of baculovirus MHC/peptide display libraries to characterize T-cell receptor ligands. Immunol Rev (2006) 210:156–70. doi: 10.1111/j.0105-2896.2006.00365.x
174. Adams JJ, Narayanan S, Birnbaum ME, Sidhu SS, Blevins SJ, Gee MH, et al. Structural interplay between germline interactions and adaptive recognition determines the bandwidth of TCR-peptide-MHC cross-reactivity. Nat Immunol (2016) 17:87–94. doi: 10.1038/ni.3310
175. Gejman RS, Jones HF, Klatt MG, Chang AY, Oh CY, Chandran SS, et al. Identification of the Targets of T-cell Receptor Therapeutic Agents and Cells by Use of a High-Throughput Genetic Platform. Cancer Immunol Res (2020) 8:672–84. doi: 10.1158/2326-6066.CIR-19-0745
176. Joglekar AV, Leonard MT, Jeppson JD, Swift M, Li G, Wong S, et al. T cell antigen discovery via signaling and antigen-presenting bifunctional receptors. Nat Methods (2019) 16:191–8. doi: 10.1038/s41592-018-0304-8
177. Kula T, Dezfulian MH, Wang CI, Abdelfattah NS, Hartman ZC, Wucherpfennig KW, et al. T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes. Cell (2019) 178:1016–28.e13. doi: 10.1016/j.cell.2019.07.009
178. Li G, Bethune MT, Wong S, Joglekar AV, Leonard MT, Wang JK, et al. T cell antigen discovery via trogocytosis. Nat Methods (2019) 16:183–90. doi: 10.1038/s41592-018-0305-7
179. Karpati RM, Banks SM, Malissen B, Rosenberg SA, Sheard MA, Weber JS, et al. Phenotypic characterization of murine tumor-infiltrating T lymphocytes. J Immunol (1991) 146:2043–51.
180. Grimm EA, Mazumder A, Zhang HZ, Rosenberg SA. Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. J Exp Med (1982) 155:1823–41. doi: 10.1084/jem.155.6.1823
181. Mazumder A, Grimm EA, Zhang HZ, Rosenberg SA. Lysis of fresh human solid tumors by autologous lymphocytes activated in vitro with lectins. Cancer Res (1982) 42:913–8.
182. Xia Y, Tian X, Wang J, Qiao D, Liu X, Xiao L, et al. Treatment of metastatic non−small cell lung cancer with NY−ESO−1 specific TCR engineered−T cells in a phase I clinical trial: A case report. Oncol Lett (2018) 16:6998–7007. doi: 10.3892/ol.2018.9534
183. D’Angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 c259 T Cells in Synovial Sarcoma. Cancer Discovery (2018) 8:944–57. doi: 10.1158/2159-8290.CD-17-1417
184. Nishihori T, Kaufman JL, Hoffman JE, Blouch K, Pandit S, Butler E, et al. Open-Label Pilot Study of Genetically Engineered NY-ESO-1 Specific T Cells (GSK3377794) Alone or in Combination with Pembrolizumab in Relapsed and Refractory Multiple Myeloma. Blood (2019) 134:3134–4. doi: 10.1182/blood-2019-128059
185. Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med (2019) 25:1064–72. doi: 10.1038/s41591-019-0472-9
186. Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The Prioritization of Cancer Antigens: A National Cancer Institute Pilot Project for the Acceleration of Translational Research. Clin Cancer Res (2009) 15:5323–37. doi: 10.1158/1078-0432.CCR-09-0737
187. Xue S-A, Gao L, Hart D, Gillmore R, Qasim W, Thrasher A, et al. Elimination of human leukemia cells in NOD/SCID mice by WT1-TCR gene–transduced human T cells. Blood (2005) 106:3062–7. doi: 10.1182/blood-2005-01-0146
188. Koehne G, Kosuri S, Doubrovina ES, Orlando E, Giralt SA, Jungbluth AA, et al. Targeting Wilms’ Tumor 1 Protein Following CD34-Selected Allografts By Adoptive Transfer of Donor-Derived CTLs in Patients with Advanced Multiple Myeloma. Biol Blood Marrow Transplant (2016) 22:S141. doi: 10.1016/j.bbmt.2015.11.480
189. Tyler EM, Jungbluth AA, O’Reilly RJ, Koehne G. WT1-specific T-cell responses in high-risk multiple myeloma patients undergoing allogeneic T cell-depleted hematopoietic stem cell transplantation and donor lymphocyte infusions. Blood (2013) 121:308–17. doi: 10.1182/blood-2012-06-435040
190. Lu Y-C, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, et al. Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II–Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J Clin Oncol (2017) 35:3322–9. doi: 10.1200/JCO.2017.74.5463
191. Yao X, Lu Y-C, Parker LL, Li YF, El-Gamil M, Black MA, et al. Isolation and Characterization of an HLA-DPB1*04. J Immunother (2016) 39:191–201. doi: 10.1097/CJI.0000000000000123
192. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood (2013) 122:863–71. doi: 10.1182/blood-2013-03-490565
193. Parkhurst MR, Joo J, Riley JP, Yu Z, Li Y, Robbins PF, et al. Characterization of Genetically Modified T-Cell Receptors that Recognize the CEA:691-699 Peptide in the Context of HLA-A2.1 on Human Colorectal Cancer Cells. Clin Cancer Res (2009) 15:169–80. doi: 10.1158/1078-0432.CCR-08-1638
194. Deniger DC, Pasetto A, Robbins PF, Gartner JJ, Prickett TD, Paria BC, et al. T-cell responses to TP53 “Hotspot” Mutations and unique neoantigens expressed by human ovarian cancers. Clin Cancer Res (2018) 24:5562–73. doi: 10.1158/1078-0432.CCR-18-0573
195. Baugh EH, Ke H, Levine AJ, Bonneau RA, Chan CS. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ (2018) 25:154–60. doi: 10.1038/cdd.2017.180
196. Moore T, Wagner CR, Scurti GM, Hutchens KA, Godellas C, Clark AL, et al. Clinical and immunologic evaluation of three metastatic melanoma patients treated with autologous melanoma-reactive TCR-transduced T cells. Cancer Immunol Immunother (2018) 67:311–25. doi: 10.1007/s00262-017-2073-0
197. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Sci (80 ) (2020) 367:eaba7365. doi: 10.1126/science.aba7365
198. Ramachandran I, Lowther DE, Dryer-Minnerly R, Wang R, Fayngerts S, Nunez D, et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J Immunother Cancer (2019) 7:276. doi: 10.1186/s40425-019-0762-2
199. Johanna I, Straetemans T, Heijhuurs S, Aarts-Riemens T, Norell H, Bongiovanni L, et al. Evaluating in vivo efficacy - Toxicity profile of TEG001 in humanized mice xenografts against primary human AML disease and healthy hematopoietic cells. J Immunother Cancer (2019) 7:1–13. doi: 10.1186/s40425-019-0558-4
200. Sanderson JP, Crowley DJ, Wiedermann GE, Quinn LL, Crossland KL, Tunbridge HM, et al. Preclinical evaluation of an affinity-enhanced MAGE-A4-specific T-cell receptor for adoptive T-cell therapy. Oncoimmunology (2020) 9:1682381. doi: 10.1080/2162402X.2019.1682381
201. Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, et al. Adoptive transfer of MAGE-A4 T-cell receptor gene-transduced lymphocytes in patients with recurrent esophageal cancer. Clin Cancer Res (2015) 21:2268–77. doi: 10.1158/1078-0432.CCR-14-1559
202. Miyahara Y, Naota H, Wang L, Hiasa A, Goto M, Watanabe M, et al. Determination of cellularly processed HLA-A2402-restricted novel CTL epitopes derived from two cancer germ line genes, MAGE-A4 and SAGE. Clin Cancer Res (2005) 11:5581–9. doi: 10.1158/1078-0432.CCR-04-2585
203. Tawara I, Kageyama S, Miyahara Y, Fujiwara H, Nishida T, Akatsuka Y, et al. Safety and persistence of WT1-specific T-cell receptor gene–transduced lymphocytes in patients with AML and MDS. Blood (2017) 130:1985–94. doi: 10.1182/blood-2017-06-791202
204. Lam VK, Hong DS, Heymach JV, Blumenschein G, Butler MO, Johnson M, et al. Safety and anti-tumor effects of MAGE-A10c796 TCR T-cells in two clinical trials. Ann Oncol (2018) 29:viii731. doi: 10.1093/annonc/mdy424.048
205. Amir AL, van der Steen DM, van Loenen MM, Hagedoorn RS, de Boer R, Kester MDG, et al. PRAME-Specific Allo-HLA-Restricted T Cells with Potent Antitumor Reactivity Useful for Therapeutic T-Cell Receptor Gene Transfer. Clin Cancer Res (2011) 17:5615–25. doi: 10.1158/1078-0432.CCR-11-1066
206. Takahashi Y, Harashima N, Kajigaya S, Yokoyama H, Cherkasova E, McCoy JP, et al. Regression of human kidney cancer following allogeneic stem cell transplantation is associated with recognition of an HERV-E antigen by T cells. J Clin Invest (2008) 118:1099–109. doi: 10.1172/JCI34409
207. Xue S-A, Gao L, Ahmadi M, Ghorashian S, Barros RD, Pospori C, et al. Human MHC Class I-restricted high avidity CD4 + T cells generated by co-transfer of TCR and CD8 mediate efficient tumor rejection in vivo. Oncoimmunology (2013) 2:e22590. doi: 10.4161/onci.22590
208. Koh S, Shimasaki N, Suwanarusk R, Ho ZZ, Chia A, Banu N, et al. A Practical Approach to Immunotherapy of Hepatocellular Carcinoma Using T Cells Redirected Against Hepatitis B Virus. Mol Ther Nucleic Acids (2013) 2:e114. doi: 10.1038/mtna.2013.43
209. Qasim W, Brunetto M, Gehring AJ, Xue SA, Schurich A, Khakpoor A, et al. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J Hepatol (2015) 62:486–91. doi: 10.1016/j.jhep.2014.10.001
210. Hanada K, Wang QJ, Inozume T, Yang JC. Molecular identification of an MHC-independent ligand recognized by a human α/β T-cell receptor. Blood (2011) 117:4816–25. doi: 10.1182/blood-2010-11-317743
211. Lunde E, Loset GA, Bogen B, Sandlie I. Stabilizing mutations increase secretion of functional soluble TCR-Ig fusion proteins. BMC Biotechnol (2010) 10:61. doi: 10.1186/1472-6750-10-61
212. Liddy N, Bossi G, Adams KJ, Lissina A, Mahon TM, Hassan NJ, et al. Monoclonal TCR-redirected tumor cell killing. Nat Med (2012) 18:980–7. doi: 10.1038/nm.2764
213. Hickman ES, Lomax ME, Jakobsen BK. Antigen Selection for Enhanced Affinity T-Cell Receptor–Based Cancer Therapies. J Biomol Screen (2016) 21:769–85. doi: 10.1177/1087057116637837
214. Connelly S. Novel Immunotherapy Tebentafusp Granted Fast Track Designation for Metastatic Uveal Melanoma (2019). Available at: https://www.targetedonc.com/view/novel-immunotherapy-tebentafusp-granted-fast-track-designation-for-metastatic-uveal-melanoma (Accessed May 17, 2020).
215. Middleton MR, McAlpine C, Woodcock VK, Corrie P, Infante JR, Steven NM, et al. Tebentafusp, A TCR/Anti-CD3 Bispecific Fusion Protein Targeting gp100, Potently Activated Antitumor Immune Responses in Patients with Metastatic Melanoma. Clin Cancer Res (2020) 26:5869–78. doi: 10.1158/1078-0432.CCR-20-1247
216. Lowe KL, Cole D, Kenefeck R, OKelly I, Lepore M, Jakobsen BK. Novel TCR-based biologics: mobilising T cells to warm ‘cold’ tumours. Cancer Treat Rev (2019) 77:35–43. doi: 10.1016/j.ctrv.2019.06.001
217. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer (2012) 12:278–87. doi: 10.1038/nrc3236
218. Murphy G, Isenberg DA. New therapies for systemic lupus erythematosus — past imperfect, future tense. Nat Rev Rheumatol (2019) 15:403–12. doi: 10.1038/s41584-019-0235-5
219. Scott AM, Allison JP, Wolchok JD. Monoclonal antibodies in cancer therapy. Cancer Immun (2012) 12:14.
220. Veomett N, Dao T, Scheinberg DA. Therapeutic antibodies to intracellular targets in cancer therapy. Expert Opin Biol Ther (2013) 13:1485–8. doi: 10.1517/14712598.2013.833602
221. Dao T, Liu C, Scheinberg DA. Approaching untargetable tumor-associated antigens with antibodies. Oncoimmunology (2013) 2:e24678. doi: 10.4161/onci.24678
222. Dahan R, Reiter Y. T-cell-receptor-like antibodies – generation, function and applications. Expert Rev Mol Med (2012) 14:e6. doi: 10.1017/erm.2012.2
223. Denkberg G, Cohen CJ, Lev A, Chames P, Hoogenboom HR, Reiter Y. Direct visualization of distinct T cell epitopes derived from a melanoma tumor-associated antigen by using human recombinant antibodies with MHC- restricted T cell receptor-like specificity. Proc Natl Acad Sci (2002) 99:9421–6. doi: 10.1073/pnas.132285699
224. Sergeeva A, Alatrash G, He H, Ruisaard K, Lu S, Wygant J, et al. An anti–PR1/HLA-A2 T-cell receptor–like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood (2011) 117:4262–72. doi: 10.1182/blood-2010-07-299248
225. Herrmann AC, Im JS, Pareek S, Ruiz-Vasquez W, Lu S, Sergeeva A, et al. A Novel T-Cell Engaging Bi-specific Antibody Targeting the Leukemia Antigen PR1/HLA-A2. Front Immunol (2019) 9:3153. doi: 10.3389/fimmu.2018.03153
226. Dao T, Yan S, Veomett N, Pankov D, Zhou L, Korontsvit T, et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl Med (2013) 5:176ra33. doi: 10.1126/scitranslmed.3005661
227. Anguille S, Van de Velde AL, Smits EL, Van Tendeloo VF, Juliusson G, Cools N, et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood (2017) 130:1713–21. doi: 10.1182/blood-2017-04-780155
228. May RJ, Dao T, Pinilla-Ibarz J, Korontsvit T, Zakhaleva V, Zhang RH, et al. Peptide Epitopes from the Wilms’ Tumor 1 Oncoprotein Stimulate CD4+ and CD8+ T Cells That Recognize and Kill Human Malignant Mesothelioma Tumor Cells. Clin Cancer Res (2007) 13:4547–55. doi: 10.1158/1078-0432.CCR-07-0708
229. Oka Y, Tsuboi A, Taguchi T, Osaki T, Kyo T, Nakajima H, et al. Induction of WT1 (Wilms’ tumor gene)-specific cytotoxic T lymphocytes by WT1 peptide vaccine and the resultant cancer regression. Proc Natl Acad Sci (2004) 101:13885–90. doi: 10.1073/pnas.0405884101
230. Rezvani K, Yong ASM, Mielke S, Savani BN, Musse L, Superata J, et al. Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood (2008) 111:236–42. doi: 10.1182/blood-2007-08-108241
231. Keilholz U, Letsch A, Busse A, Asemissen AM, Bauer S, Blau IW, et al. A clinical and immunologic phase 2 trial of Wilms tumor gene product 1 (WT1) peptide vaccination in patients with AML and MDS. Blood (2009) 113:6541–8. doi: 10.1182/blood-2009-02-202598
232. Dao T, Pankov D, Scott A, Korontsvit T, Zakhaleva V, Xu Y, et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol (2015) 33:1079–86. doi: 10.1038/nbt.3349
233. Dao T, Mun SS, Scott AC, Jarvis CA, Korontsvit T, Yang Z, et al. Depleting T regulatory cells by targeting intracellular Foxp3 with a TCR mimic antibody. Oncoimmunology (2019) 8:e1570778. doi: 10.1080/2162402X.2019.1570778
234. Attia P, Maker AV, Haworth LR, Rogers-Freezer L, Rosenberg SA. Inability of a Fusion Protein of IL-2 and Diphtheria Toxin (Denileukin Diftitox, DAB389IL-2, ONTAK) to Eliminate Regulatory T Lymphocytes in Patients With Melanoma. J Immunother (2005) 28:582–92. doi: 10.1097/01.cji.0000175468.19742.10
235. Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci (2013) 110:17945–50. doi: 10.1073/pnas.1316796110
236. Schaer DA, Murphy JT, Wolchok JD. Modulation of GITR for cancer immunotherapy. Curr Opin Immunol (2012) 24:217–24. doi: 10.1016/j.coi.2011.12.011
237. Inaguma Y, Akahori Y, Murayama Y, Shiraishi K, Tsuzuki-Iba S, Endoh A, et al. Construction and molecular characterization of a T-cell receptor-like antibody and CAR-T cells specific for minor histocompatibility antigen HA-1H. Gene Ther (2014) 21:575–84. doi: 10.1038/gt.2014.30
238. Ahmed M, Lopez-Albaitero A, Pankov D, Santich BH, Liu H, Yan S, et al. TCR-mimic bispecific antibodies targeting LMP2A show potent activity against EBV malignancies. JCI Insight (2018) 3:e97805. doi: 10.1172/jci.insight.97805
239. Akahori Y, Wang L, Yoneyama M, Seo N, Okumura S, Miyahara Y, et al. Antitumor activity of CAR-T cells targeting the intracellular oncoprotein WT1 can be enhanced by vaccination. Blood (2018) 132:1134–45. doi: 10.1182/blood-2017-08-802926
240. Akatsuka Y. TCR-Like CAR-T Cells Targeting MHC-Bound Minor Histocompatibility Antigens. Front Immunol (2020) 11:257. doi: 10.3389/fimmu.2020.00257
241. Liu H, Xu Y, Xiang J, Long L, Green S, Yang Z, et al. Targeting Alpha-Fetoprotein (AFP)–MHC Complex with CAR T-Cell Therapy for Liver Cancer. Clin Cancer Res (2017) 23:478–88. doi: 10.1158/1078-0432.CCR-16-1203
242. Verma B, Neethling FA, Caseltine S, Fabrizio G, Largo S, Duty JA, et al. TCR Mimic Monoclonal Antibody Targets a Specific Peptide/HLA Class I Complex and Significantly Impedes Tumor Growth In Vivo Using Breast Cancer Models. J Immunol (2010) 184:2156–65. doi: 10.4049/jimmunol.0902414
243. Stewart-Jones G, Wadle A, Hombach A, Shenderov E, Held G, Fischer E, et al. Rational development of high-affinity T-cell receptor-like antibodies. Proc Natl Acad Sci (2009) 106:5784–8. doi: 10.1073/pnas.0901425106
244. Held G, Matsuo M, Epel M, Gnjatic S, Ritter G, Lee SY, et al. Dissecting cytotoxic T cell responses towards the NY-ESO-1 protein by peptide/MHC-specific antibody fragments. Eur J Immunol (2004) 34:2919–29. doi: 10.1002/eji.200425297
245. Cohen CJ, Hoffmann N, Farago M, Hoogenboom HR, Eisenbach L, Reiter Y. Direct detection and quantitation of a distinct T-cell epitope derived from tumor-specific epithelial cell-associated mucin using human recombinant antibodies endowed with the antigen-specific, major histocompatibility complex-restricted specificity of T cells. Cancer Res (2002) 62:5835–44.
246. Lev A, Denkberg G, Cohen CJ, Tzukerman M, Skorecki KL, Chames P, et al. Isolation and characterization of human recombinant antibodies endowed with the antigen-specific, major histocompatibility complex-restricted specificity of T cells directed toward the widely expressed tumor T-cell epitopes of the telomerase catalytic sub. Cancer Res (2002) 62:3184–94.
247. Desjarlais JR, Lazar GA, Zhukovsky EA, Chu SY. Optimizing engagement of the immune system by anti-tumor antibodies: an engineer’s perspective. Drug Discovery Today (2007) 12:898–910. doi: 10.1016/j.drudis.2007.08.009
248. Jefferis R. Glycosylation of Recombinant Antibody Therapeutics. Biotechnol Prog (2008) 21:11–6. doi: 10.1021/bp040016j
249. Veomett N, Dao T, Liu H, Xiang J, Pankov D, Dubrovsky L, et al. Therapeutic Efficacy of an Fc-Enhanced TCR-like Antibody to the Intracellular WT1 Oncoprotein. Clin Cancer Res (2014) 20:4036–46. doi: 10.1158/1078-0432.CCR-13-2756
250. Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discovery (2019) 18:585–608. doi: 10.1038/s41573-019-0028-1
251. Nagorsen D, Baeuerle PA. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp Cell Res (2011) 317:1255–60. doi: 10.1016/j.yexcr.2011.03.010
252. Hill JA, Li D, Hay KA, Green ML, Cherian S, Chen X, et al. Infectious complications of CD19-targeted chimeric antigen receptor–modified T-cell immunotherapy. Blood (2018) 131:121–30. doi: 10.1182/blood-2017-07-793760
253. Martinez M, Moon EK. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front Immunol (2019) 10:128. doi: 10.3389/fimmu.2019.00128
254. Rafiq S, Purdon TJ, Daniyan AF, Koneru M, Dao T, Liu C, et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia (2017) 31:1788–97. doi: 10.1038/leu.2016.373
255. Riemer AB, Keskin DB, Zhang G, Handley M, Anderson KS, Brusic V, et al. A Conserved E7-derived Cytotoxic T Lymphocyte Epitope Expressed on Human Papillomavirus 16-transformed HLA-A2 + Epithelial Cancers. J Biol Chem (2010) 285:29608–22. doi: 10.1074/jbc.M110.126722
256. Sykulev Y, Joo M, Vturina I, Tsomides TJ, Eisen HN. Evidence that a Single Peptide–MHC Complex on a Target Cell Can Elicit a Cytolytic T Cell Response. Immunity (1996) 4:565–71. doi: 10.1016/S1074-7613(00)80483-5
257. Lu R-M, Hwang Y-C, Liu I-J, Lee C-C, Tsai H-Z, Li H-J, et al. Development of therapeutic antibodies for the treatment of diseases. J BioMed Sci (2020) 27:1. doi: 10.1186/s12929-019-0592-z
258. Ataie N, Xiang J, Cheng N, Brea EJ, Lu W, Scheinberg DA, et al. Structure of a TCR-Mimic Antibody with Target Predicts Pharmacogenetics. J Mol Biol (2016) 428:194–205. doi: 10.1016/j.jmb.2015.12.002
259. Chang AY, Gejman RS, Brea EJ, Oh CY, Mathias MD, Pankov D, et al. Opportunities and challenges for TCR mimic antibodies in cancer therapy. Expert Opin Biol Ther (2016) 16:979–87. doi: 10.1080/14712598.2016.1176138
260. Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol (2006) 43:763–71. doi: 10.1016/j.molimm.2005.03.007
261. Maruta M, Ochi T, Tanimoto K, Asai H, Saitou T, Fujiwara H, et al. Direct comparison of target-reactivity and cross-reactivity induced by CAR- and BiTE-redirected T cells for the development of antibody-based T-cell therapy. Sci Rep (2019) 9:13293. doi: 10.1038/s41598-019-49834-2
Keywords: T cell receptor, bispecific T cell engager, cross-reactivity, tumor infiltrating lymphocytes, immune mobilizing monoclonal T cell receptors against cancer, peptide- major histocompatibility complexes, T cell receptor mimic monoclonal antibody, T cell receptor-T cell
Citation: Jones HF, Molvi Z, Klatt MG, Dao T and Scheinberg DA (2021) Empirical and Rational Design of T Cell Receptor-Based Immunotherapies. Front. Immunol. 11:585385. doi: 10.3389/fimmu.2020.585385
Received: 20 July 2020; Accepted: 04 December 2020;
Published: 25 January 2021.
Edited by:
Francisco Martin, Andalusian Autonomous Government of Genomics and Oncological Research (GENYO), SpainReviewed by:
Estefania Garcia-Guerrero, Fundación Pública Andaluza para la Gestión de la Investigación en Salud de Sevilla (FISEVI), SpainZwi Berneman, University of Antwerp, Belgium
Copyright © 2021 Jones, Molvi, Klatt, Dao and Scheinberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David A. Scheinberg, c2NoZWluYmRAbXNrY2Mub3Jn