 Thomas Daubon
Thomas Daubon Audrey Hemadou2†
Audrey Hemadou2† Maya Saleh
Maya Saleh- 1University of Bordeaux, CNRS, Institut de Biochimie et Génétique Cellulaires (IBGC), UMR 5095, Bordeaux, France
- 2University of Bordeaux, CNRS, ImmunoConcEpT, UMR 5164, Bordeaux, France
- 3Department of Medicine, McGill University, Montreal, QC, Canada
Glioblastoma (GBM) are the most common tumors of the central nervous system and among the deadliest cancers in adults. GBM overall survival has not improved over the last decade despite optimization of therapeutic standard-of-care. While immune checkpoint inhibitors (ICI) have revolutionized cancer care, they unfortunately have little therapeutic success in GBM. Here, we elaborate on normal brain and GBM-associated immune landscapes. We describe the role of microglia and tumor-associated macrophages (TAMs) in immune suppression and highlight the impact of energy metabolism in immune evasion. We also describe the challenges and opportunities of immunotherapies in GBM and discuss new avenues based on harnessing the anti-tumor activity of myeloid cells, vaccines, chimeric antigen receptors (CAR)-T and -NK cells, oncolytic viruses, nanocarriers, and combination therapies.
Preface
The adult human brain is a tissue of vast complexity, composed of multiple cell types defined by their location, function, or molecular characteristics. Five main classes of cerebral cells have been described: neurons, astrocytes, oligodendrocytes, endothelial cells, and microglia. Interactions among these cell types orchestrate the structure and function of the brain in electrical signaling, axonal ensheathing, regulation of blood flow, metabolic coupling and immune surveillance. For instance, astrocytes which are key effectors of the brain's energy metabolism, convert glucose into lactate, which is delivered to neurons and retro-converted into pyruvate to fuel the Krebs cycle (1). The neurovascular unit (NVU), which encompasses the blood-brain barrier (BBB), is a functional physiological unit that regulates the blood/cerebral parenchyma interface. It is composed of endothelial cells, smooth muscle cells, pericytes, astrocytes, microglia and neurons. The NVU governs brain homeostasis, controlling cerebral perfusion and protecting from potential pathogens or toxins present in the blood. The NVU is significantly altered in CNS malignancy, especially in glioblastoma (GBM), which are grade IV malignant glioma that are highly vascularized with dense tortuous and leaky blood vessels, permitting massive immune cell infiltration in the tumor core. GBMs are mainly derived from neural stem cells, differentiating into astrocytic or neuronal lineages. This cancer is one of the deadliest types in humans, with an average survival time of <15 months upon diagnosis. Even with the standard-of-care treatment, consisting of surgical resection when possible, followed by radiation and chemotherapy with the drug Temozolomide (TMZ), the estimated recurrence rate is more than 90%. Recurrence is mostly caused by the regrowth of highly invasive cells that spread out of the tumor core, partially due to its hypoxic and acidic environment (2), and are therefore not removed by surgical resection. The long-standing assumption that GBM tumors were clonal masses with identical molecular characteristics have recently been challenged. Indeed, tumor single cell transcriptomics have identified several GBM cellular states with notable plasticity modulated by the tumor microenvironment (3, 4).
Immune Mechanisms of the Healthy Central Nervous System (CNS)
Prior to delving into the immune landscape and immunosuppressive mechanisms of GBM, we briefly overview the architecture of the CNS immune system under physiological conditions, highlighting its unique lymphatic drainage system, immune cell populations and leukocyte trafficking (Figure 1A). Anatomically, the brain parenchyma is surrounded by the meninges, a series of three membranes under the skull, namely the dura mater, the arachnoid membrane and the pia mater (Figure 1B). The brain bathes in cerebrospinal fluid (CSF), generated at the blood-CSF barrier, by epithelial cells of the choroid plexus, through diffusion, pinocytosis and active transport from arterial blood in fenestrated capillaries (Figure 1C). The CSF flows around the brain four ventricles into the subarachnoid space (SAS) in a unidirectional flux through the action of cilia on the choroid plexus and ependymal cells that line the ventricles. It enters the brain parenchyma through aquaporin 4, water channels on the end-feet of astrocytes surrounding the vasculature, and communicates with the interstitial fluid (ISF) through the glymphatic system, a network of perivascular channels formed by astroglia for waste elimination (5). The CSF is reabsorbed by the venous blood in venous sinuses at arachnoid villi. Such turnover occurs three to twelve times daily suggesting that the CSF is an immunologically active fluid. Indeed, the CSF drains trafficking leukocytes to the deep cervical lymph nodes (DCLNs) via the newly discovered meningeal lymphatic vessels in the dura mater (6, 7), or by channeling along cranial nerves through the cribriform plate to the nasal mucosa where it accesses its afferent lymphatics. The ISF, which carries parenchymal solutes and small soluble antigens but not parenchymal immune cells, reaches the DCLNs by channeling along the tight space of the basement membrane lining the walls of cerebral capillaries and arteries. The blood supply of the brain enters through capillaries and post-capillary venules, that push the pia mater in the SAS to form perivascular spaces (Virchow-Robin spaces). The brain vasculature is ensheathed by the BBB (Figure 1D) formed by endothelial cells connected by complex tight junctions and pericytes in the capillary basement membrane, and surrounded by the pia mater, the subpial space and the glia limitans, a thin membrane barrier at the parenchymal basement membrane formed by astrocyte foot processes.
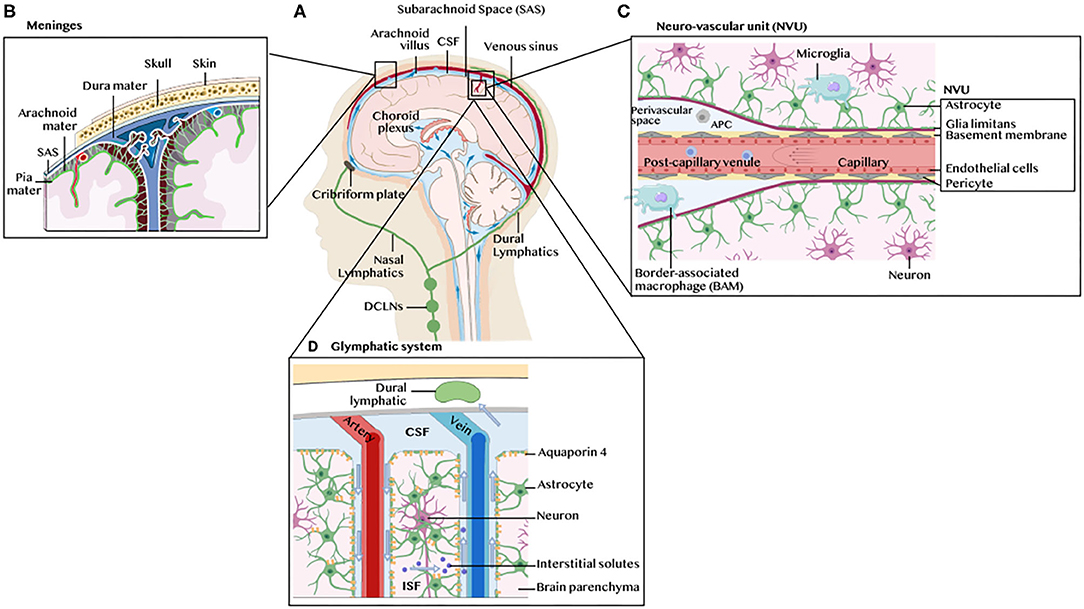
Figure 1. Architecture of the CNS immune system. (A) Schematic illustration of the human brain anatomy namely the brain parenchyma, choroid plexus, ventricles, cerebrospinal fluid (CSF), meninges, dural and nasal lymphatics and the deep cervical lymph nodes (DCLNs). (B) The meninges. These are three membranes that envelop the brain, namely the dura mater, the arachnoid membrane and the pia mater. (C) The neurovascular unit (NVU), blood brain barrier (BBB) and perivascular space. The glia limitans formed by astrocytes end feet ensheath the capillary basement membrane and its pericytes. The perivascular space contains microglia-like perivascular macrophages also dubbed border-associated macrophages (BAMs) and antigen-presenting cells (APC). Microglia are found in the brain parenchyma. (D) The glymphatic system. The CSF enters the brain parenchyma through aquaporin 4, water channels on the end-feet of astrocytes surrounding the vasculature, and communicates with the interstitial fluid (ISF) carrying solutes and small antigens through the glymphatic system, a network of perivascular channels formed by astroglia for waste elimination.
The CNS has long been considered as a site of immune privilege. This was based on earlier findings that transplanted tissue grafts in the brain parenchyma elicit slow adaptive immune responses and are not readily rejected (8), and on the presumed lack of lymphatic vessels. Further, a paucity of innate immune responses to pathogen- or danger-associated molecular patterns (PAMPs and DAMPs) has been reported (9, 10). However, mounting evidence challenge this notion and demonstrate active immunosurveillance in the healthy CNS (11). Together with the discovery of a dural meningeal lymphatic system (6, 7), several studies have shown that unlike the brain parenchyma, the cerebral ventricles elicit immune responses leading to graft rejection (12, 13). Thus, the CNS exhibits compartment-specific immunity regulated by leukocyte entry across endothelial, epithelial and glial cell layers of the blood-brain and blood-CSF barriers. These barriers segregate the parenchyma from the peripheral immune system at steady state while permitting immune communications in the CSF-filled SAS and ventricular space. Such compartmentalization is also reflected by spatially and functionally diverse resident immune cell subsets.
The recent use of high-dimensional single cell approaches [e.g., mass cytometry and single cell RNA sequencing (scRNAseq)] in mice (14) and humans (15), along with intravascular leukocyte tracking and fate mapping systems in reporter mice, has uncovered diverse resident immune cells in the healthy CNS and mapped their localization to different CNS compartments. Microglia, which are derived from a yolk sac progenitor, are found exclusively in the brain parenchyma. A distinct subset of embryonically-derived microglia-like macrophages line the meninges, the choroid plexus and the perivascular spaces, and are dubbed border-associated macrophages (BAMs). Microglia and BAMs make up the bulk of the healthy CNS immune cells accounting for ~80% and ~10% of all CNS steady state leukocytes, respectively. Blood-derived monocytes (Ly6Chi and Ly6Clo), monocyte-derived cells (MdCs), dendritic cells (DCs) and neutrophils are also present in the healthy CNS, albeit at lower frequencies (<3%) (14). T and B cells, innate lymphocytes (ILCs), natural killer (NK), NKT, eosinophils and mast cells are rare (<1%) but also found at steady state. While microglia and BAMs share several surface markers (CD45lo CD11blo F4/80+ CD64+ MeTK+ Cx3CR1+), they differ in the expression of SIGLEC-H, which is typically found on microglia but not on BAMs. In contrast, the latter express CD206, CD38 and CD88. Both subsets potentially act as antigen-presenting cells (APCs), as they can upregulate, in a context-dependent fashion, the expression of CD11c, MHCII and co-stimulatory molecules. For instance, microglia of the white matter express higher levels of MHCII, CD68 and HLA-DR compared to gray matter microglia, and upregulate pro-inflammatory cytokines such as SPP1 (osteopontin) with age (15). There is little evidence that microglia and BAMs migrate to the periphery to prime T cells. Instead they are thought to maintain tissue homeostasis and to locally re-stimulate T cells. On the other hand, brain DCs traffic to the DCLNs using one of two routes: a specific route involving the rostral migratory stream (16), olfactory bulb, cribriform plate, and nasal mucosal lymphatics or via the dural lymphatics (Figure 1A). At steady state, DC trafficking contributes to CNS immune tolerance by inducing regulatory T cells (Treg). Endothelial cells of the meningeal lymphatic vessels are also presumed to maintain brain antigens-reactive T cells in an anergic state (7). Efferent T cells reach the CNS through the choroid plexus or subarachnoid veins and extravasate into the CSF-filled ventricular space and SAS. In the absence of antigen encounter, T cells are eliminated from the CNS by apoptosis or CSF drainage. Cognate antigen recognition on perivascular or leptomeningeal APCs is required for activated T cells to cross the glia limitans into the parenchyma. T cell activation in the brain is often detrimental leading to neuroinflammation and tissue damage. However, this is not always the case, as T cells can mediate neuroprotective effects in response to CNS injury (17).
Glioblastoma (GBM) Subtypes and Their Associated Immune Landscapes
In 2007, the WHO graded CNS tumors based on histological criteria (grade II-IV) (18). In 2010, Verhaak et al. used an unsupervised gene expression analysis of 200 GBM and two normal brain samples to identify four GBM subtypes based on molecular signatures (Table 1). These were referred to as neural (NE), proneural (PN), classical (CL) and mesenchymal (MES) (19). The NE subtype, in which the normal brain samples clustered, was characterized by the expression of neuronal gene markers, and was later shown by the same team to be non-tumor specific (20). The PN subtype, associated with the best median patient survival, had two genomic features, PDGFRA alterations and point mutations in IDH1, and was characterized by elevated expression of oligodendrocytic and pro-neural development genes. The CL subtype had high rates of EGFR gene amplification co-occurring with aberrations in the RB pathway. It exhibited high expression of neural precursors and stem cell markers, and elevated expression of effectors of the Notch and sonic hedgehog pathways. The MES subtype, linked to the least favorable outcome, had predominant NF1 gene aberrations and PTEN mutations. As its name implies, it included an epithelial-to-mesenchymal signature indicative of de-differentiated/trans-differentiated tumors. It also had the highest inflammatory signature with a notable upregulation of genes in the TNF and NF-κB pathways. Several studies from the Cancer Genome Atlas (TCGA) project subsequently defined a core of recurrent driver genomic alterations in GBM, involving TP53, RB1, NF1, PDGFRA, EGFR, PTEN, and CTNND2 (21–24). Genetic alterations in IDH1 or IDH2, TERT, and co-deletion of chromosome arms 1p and 19q (1p/19q codel) were rather found in low grade gliomas (LGG; grades II-III) (23, 25). In 2016, the WHO reclassified CNS tumors to integrate molecular information to the diagnosis criteria (26). This classification divided adult gliomas into three groups: (1) oligodendrogliomas, which harbor IDH mutations and 1p/19q codel, (2) astrocytomas, which are IDH mutant but without the 1p/19q codel, and (3) GBM, which are mostly IDH wild-type (WT) (Figure 2). It also introduced histone 3 K27M mutation as a molecular feature of pediatric diffuse midline glioma (27). More recent integration of results from scRNAseq, in vivo single cell lineage tracing and genomic and transcriptomic analyses from TCGA refined the GBM subtypes by identifying four plastic GBM cellular states. These were characterized by six transcriptomic meta-modules and genetic alterations in EGFR, PDGFRA, CDK4, and NF1 (4). Two meta-modules enriched in mesenchymal genes, including hypoxia and glycolysis genes, were referred to as MES1 and MES2, and corresponded to the TCGA-MES subtype in Verhaak et al. (19). An astrocytes-like (AC) module was consistent with the TCGA-CL, and three additional modules referred to as oligodendrocyte progenitor cells-like (OPC) and neural progenitor cells-like (NPC)1 and NPC2, corresponded to the TCGA-PN sub-type (Table 1). Neftel et al. showed, using patient-derived xenografts (PDX) in mice, that tumor cells were able to transit from one cellular state to another, indicative of a plasticity that was modulated by the tumor microenvironment (4).
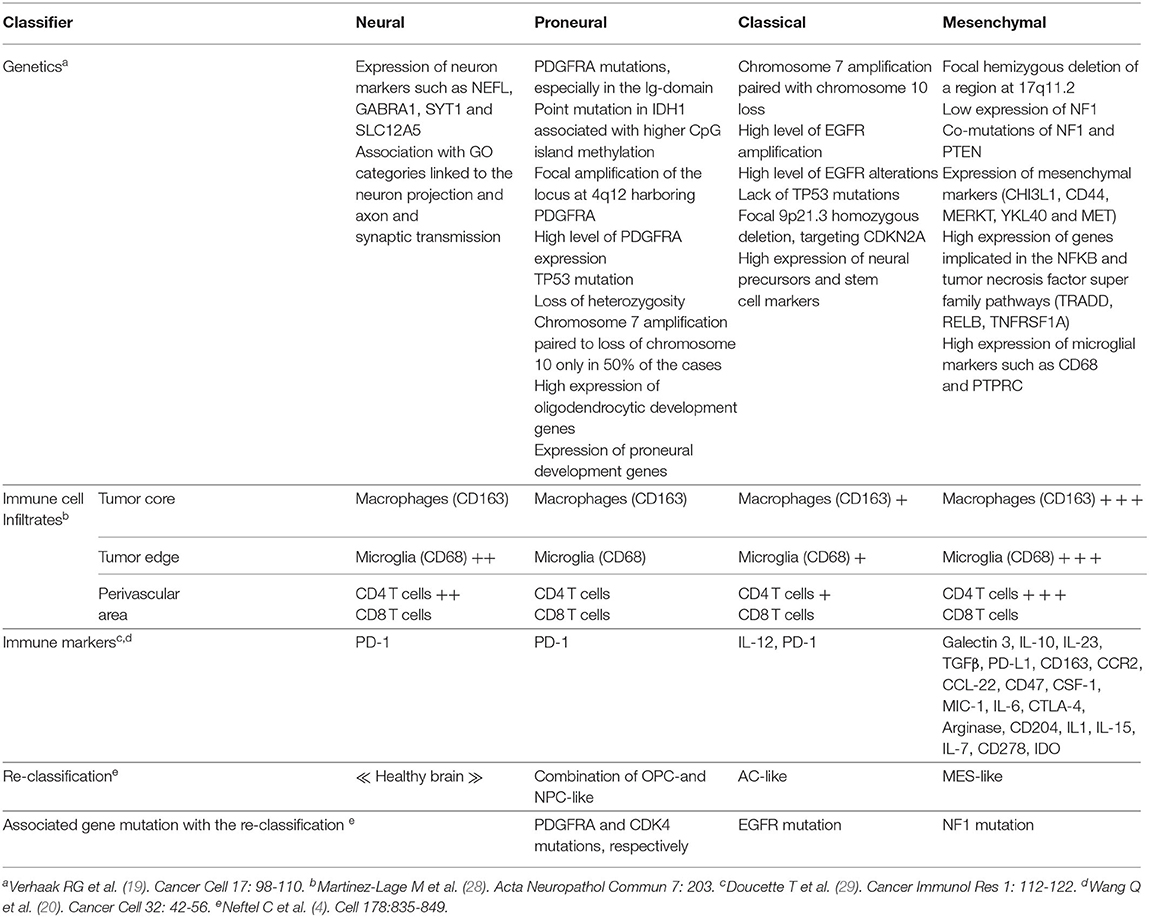
Table 1. GBM molecular classification and associated immune phenotypes.
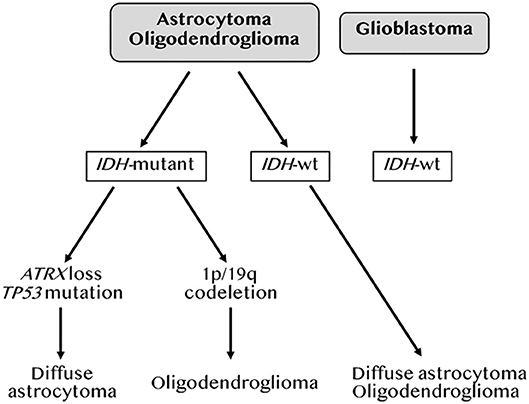
Figure 2. Molecular classification of gliomas. Adapted from the 2016 WHO classification of brain tumors by DeWitt JC et al. (26).
The immune landscape of the GBM subtypes was initially explored by transcriptomics (19, 20, 29). These studies confirmed that the MES subtype exhibited elevated expression of pro-inflammatory mediators together with immunosuppressive factors and immune checkpoints (Table 1). CIBERSORT analysis (30) revealed more TAMs, neutrophils and CD4+ T cells expression signatures in MES, whereas an activated DCs signature was found in CL (20). Analysis of a separate glioma classification system based on IDH1 mutation status and DNA methylation (31) similarly revealed elevated TAMs and neutrophils signatures in one subgroup of IDH1 wild-type (WT) tumors, that was of the MES profile (20). To reassess these findings at the protein level, Martinez-Lage et al. used an automated immunohistochemistry-based analysis of tissue microarray (TMA) from a cohort of 98 patients to define the immune cell counts in each GBM subtype. Microglia and blood-derived TAMs were the most prevalent cells in all four GBM subtypes, but were highest (>80% of all leukocytes) in the MES subtype. Whereas, CD8+ T cell frequencies were similar in all groups, the MES subtype had slightly more CD4+ T cells (~1%) (28).
Alternative stratification of GBM based on consensus immunome clusters (CIC) identified two immunologically active GBM clusters (32). These clusters expressed genes associated with cytotoxic T lymphocyte (CTLs) and NK cell activation, such as granzyme B (GZMB) and interferon gamma (IFNG), and genes linked to feedback inhibitory mechanisms including FOXP3, immune checkpoint inhibitors (CTLA-4, PD-1, TIM3, VISTA) and their ligands e.g. PD-L1 and galectin-9 (32). Nevertheless, these CICs did not discriminate patients with respect to survival outcome, potentially due to the low frequencies of CTLs and NKs and the strong immunosuppressive environment mediated by the myeloid compartment. Indeed, GBM tumor-infiltrating lymphocytes (TILs) display an exhausted phenotype (33), and GBM-infiltrating NK cells express reduced levels of activating receptors e.g., NKp30, NKG2D, and DNAX accessory molecule-1 (DNAM-1) (32).
GBM-Associated Myeloid Cells Diversity, Ontogeny and Tumor Geography
Myeloid cells are key determinants of tumor progression and patient outcome in several cancers (34), and are being actively pursued as targets of new immunotherapies (35, 36). The predominance and diversity of myeloid cells in GBM has warranted extensive analysis of their phenotypes and functions in this cancer. This is critical for discriminate therapy, as general targeting of macrophages with inhibition of colony stimulating factor 1 receptor (CSF1R) failed to enhance overall survival in recurrent GBM (37). The use of lineage tracing systems in glioma mouse models revealed distinct GBM-associated myeloid cell ontogeny, i.e., TAMs derived from microglia (MG-TAMs) or from hematopoietic stem cells in the bone marrow (BM-TAMs) (38). RNAseq analysis of these subsets highlighted the impact of ontogeny-imposed chromatin states and tumor cues on their functions in tumor growth and response to therapy. For instance, differential resistance to the anti-angiogenesis therapy bevacizumab was reported to be mediated by BM-TAMs (39). ATAC-seq and transcription factor (TF) landscape analysis identified TFs linked to microglia identity [e.g., MEF2 (40)] in MG-TAMs, whereas BM-TAMs were enriched in TFs involved in monocyte to macrophage differentiation, i.e. RUNX, CEBP, PU.1, IRF4 and STAT3. Notably, a RUNX-induced gene, integrin subunit alpha 4 (Itga4, also known as Cd49d) was identified as a distinguishing cell surface marker between the two TAM subsets in both mice and humans. It is expressed on BM-TAMs but epigenetically suppressed in microglia and MG-TAMs. Further analysis, using three different scRNAseq platforms, uncovered 66 core genes that distinguish the two TAM lineages (41). CX3CR1, which is commonly used to isolate microglia in mice, is not specific to microglia, since monocytes upregulate its expression as they differentiate in tissues. Instead, the purinergic receptor P2RY12 has recently emerged as a new microglia marker. MG-TAMs are therefore CD11b+ CX3CR1+ P2RY12+CD49D− whereas BM-TAMs are CD11b+ CX3CR1+ P2RY12− CD49D+ (41) (Figure 3). Both TAM subsets display a “non-canonical” state, expressing both M1 and M2 markers. However, BM-TAMs exhibit higher expression of immunosuppressive cytokines and effectors of oxidative metabolism, characteristic of the M2 phenotype (41). Collectively, while several studies confirm a critical role of BM-TAMs in GBM, MG-TAMs are not mere bystanders. A recent report, exploring the efficacy and targets of the phagocytosis checkpoint inhibitor anti-CD47, demonstrated that MG-TAMs are important effectors of glioma cell phagocytosis contributing to overall survival of glioma-bearing mice (42).
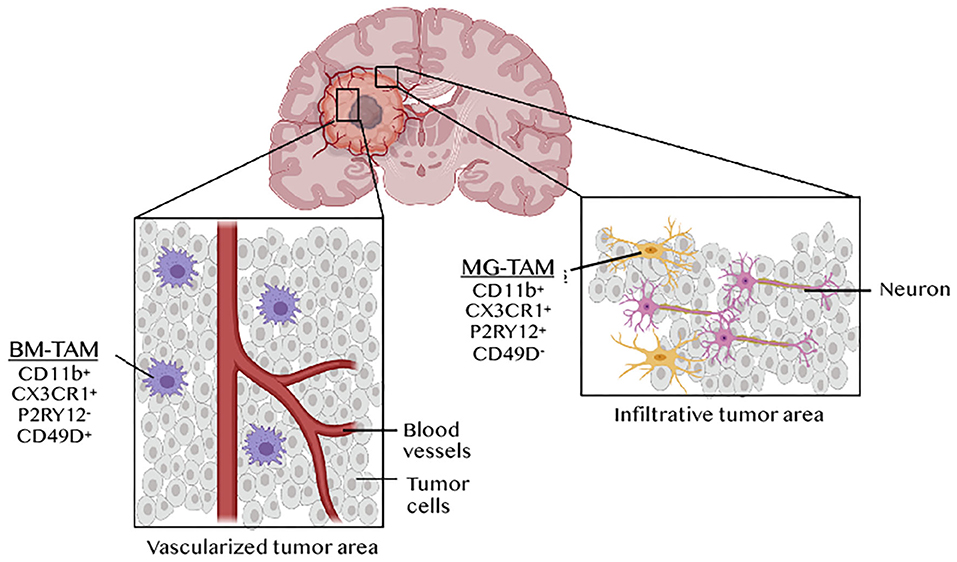
Figure 3. TAM ontogeny and tumor geography in GBM. Tumor-associated macrophages (TAMs) in GBM originates from microglia (MG-TAMs) or from bone marrow-derived monocytes differentiating into macrophages upon recruitment (BM-TAMs). These can be distinguished based on the differential expression of the integrin CD49D on BM-TAMs and of the purinergic receptor P2RY12 on MG-TAMs. BM-TAMs that infiltrate into the tumor core are smaller and less branched than MG-TAMs that are found in the peri-tumoral area.
RNAseq analysis of distinct anatomically defined tumor regions (e.g., leading edge, infiltrating region, necrotic zone, blood vessels etc.) and in situ hybridization for BIN1 (an MG-TAM marker) or TGFBI (a BM-TAM marker), revealed tumor geographic variation in TAM composition. BM-TAMs were enriched near the blood vessels whereas MG-TAMs were found in infiltrated white matter (41). This was confirmed in a glioma model using the Cx3cr1GFP;Ccr2RFP reporter mouse, which showed that BM-TAMs, which constituted 85% of the total TAM population, localized in the perivascular areas of the tumor core, whereas MG-TAMs accounting for 15% of all TAMs, were restricted to the peritumoral area (43) (Figure 3). Besides differential gene expression profiles, these two TAM subsets have different morphological and migratory characteristics, as shown by 2-photon microscopy. MG-TAMs are stationary, larger in size and more branched than BMDM-TAMs that are highly mobile and smaller (44). Clinically, BMDM-TAM infiltration correlates with poor patient survival (28, 41).
TAM Recruitment and Immunosuppressive Mechanisms in GBM
Interleukin (IL)-6, produced by vascular endothelial cells and TAMs, has been implicated in several pro-tumoral processes in GBM: (1) it contributes to the disruption of the BBB by downregulating intercellular tight junction proteins on endothelial cells (45). Concordantly, endothelial cell-specific deletion of IL-6 prevented glioma growth and improved mouse survival (46); (2) it reinforces GBM metabolic dependence on aerobic glycolysis (47), as discussed below; and (3) it promotes the recruitment of macrophages through the induction of CCL5/CXCL5 and favors their alternative activation through PPARγ/HIF-2α signaling (46). The CCL2-CCR2 pathway is equally important for BM-TAM recruitment. Glioma cells instruct this pathway through indoleamine 2,3-deoxygenase (IDO)-dependent production of kynurenine (KYN), a metabolite that triggers CCR2 upregulation through aryl hydrocarbon receptor (AHR). Myeloid-specific deletion of AHR in mice blunted BM-TAMs glioma infiltration. In humans, the KYN-AHR pathway is upregulated in GBM and is associated with an unfavorable outcome (48). A direct correlate has been established between loss of PTEN and BM-TAM recruitment via lysyl oxidase (LOX), a macrophage chemoattractant that signals through the β1 integrin (ITGB1)-PYK2 pathway. Concordantly, YAP1, LOX and β1 integrin are elevated in GBM, and are associated with reduced overall survival. LOX-elicited TAMs infiltrate the tumor microenvironment and support glioma growth via SPP1 (osteopontin), which inhibits glioma cell apoptosis, promotes angiogenesis and sustains the TAM tolerogenic phenotype by signaling through the Integrin αvβ5 (49, 50).
GBM and other brain tumors are notorious for eliciting local and systemic immunosuppression, mediated in great part by TAMs. TAM-derived TGFβ was initially considered as a key inducer of systemic immune tolerance (51). However, targeting this immunosuppressive cytokine alone did not impact the survival of mice bearing brain tumors (52), implicating additional mechanisms. The expression of PD-L1 on circulating monocytes and BM-TAMs might similarly trigger systemic immunosuppression, through a feed forward mechanism involving IL-10 (53). Beyond soluble immunosuppressive cytokines, direct cell-cell contacts, e.g., through PD-L1 (54), tolerogenic HLA molecules (55) and the apoptosis-inducing receptor Fas (56) contribute to immune escape. A recent study reported a role of tumor-associated glycosylation in local and systemic immunosuppression (57). This was mediated through a direct interaction between O-linked glycans on glioma cells with their receptor, Macrophage Galactose-type Lectin (MGL), on TAMs leading to immunosuppression signaling. Of note, the current GBM standard of care often prescribes dexamethasone to alleviate cerebral edema. This immunosuppressive corticosteroid further contributes to the GBM immunosuppressive environment, interfering with anti-tumor immunity and presenting a challenge for the future of immunotherapies in this cancer.
Metabolic Remodeling of the GBM Tumor Microenvironment
Hypoxia and necrosis are well-known features of GBM. HIF-1α, stabilized by the inhibition of prolyl hydroxylase (PHD) activity in hypoxia, is a transcription factor that modifies the expression of thousands of genes, notably effectors of glycolysis and lactic fermentation. The expression of glucose transporters (GLUT1), glycolytic enzymes (PDK1, Hexokinase or PKM2), and lactate dehydrogenase A (LDHA) help in replenishing NAD+ to support the glycolytic process. Monocarboxylate transporter (MCT)4 expression is also increased following stabilization of HIF-1α, leading to passive release of lactate out of the cells (58). Production of H+ happens during glycolysis, lactic fermentation, but also during respiration when CO2 is hydrated into HCO3− and H+ ions by carbonic anhydrases (CAs). H+ ions efflux from the cytoplasm via H+ ATPases and Na+/H+ exchangers (NHEs) leads to a decrease in the extracellular pHe. Tumor acidosis promotes cancer cell invasion through cytoskeletal remodeling, but also by modulating the activity of immune cells in the tumor microenvironment. For instance, LDHA-mediated production of lactic acid was shown to blunt the cytotoxic activity of CTLs and NK cells in melanoma through inhibition of NFAT expression (59). This supports previous findings demonstrating that lactate accumulation in T cells, due to decreased efflux via MCT1 (which controls lactate shuttling in a gradient dependent manner), blunted CTL activity (60). TAMs reinforce GBM metabolic shift to aerobic glycolysis through IL-6 that enhances the activity of phosphoglycerate kinase 1 (PGK1) by promoting its phosphorylation (47).
Glioma cells also display a high dependence on amino acid metabolism accompanied by an elevated uptake of branched chain amino acids (BCAA). Through the overexpression of branched chain amino acid transaminase 1 (BCAT1), glioma tumors excrete elevated levels of branched-chain ketoacids (BCKA) through MCT1. Which influx into TAMs and blunt their phagocytic activity (61). GBM TAMs were also shown to drive T cell dysfunction through elevated expression of the ectonucleosidase CD39 that, together with CD73, induces the production of the immunosuppressive metabolite adenosine (48).
The Future of Immunotherapies in GBM
Immune Checkpoint Inhibitors (ICI)
Immune checkpoint inhibitors (ICI) targeting the PD-1 or CTLA-4 pathways have revolutionized cancer therapy in the last decade. However, they have had little clinical benefit in GBM, at the least in the adjuvant setting. The recently published results of the open-label, randomized, phase 3 trial CheckMate-143, which evaluated nivolumab vs. bevacizumab in patients with recurrent GBM were disappointing, as there was no significant difference in median overall survival (mOS) between the two arms (62). The two ongoing phase 3 trials CheckMate-498 and CheckMate-548 evaluating the use of nivolumab in patients with newly-diagnosed GBM, either methylguanine methyltransferase (MGMT)-unmethylated or MGMT-methylated, also failed to meet their primary endpoints, according to an update by Bristol-Myers Squibb. In the neoadjuvant setting, the results are controversial. The anti-PD-1 nivolumab, administered as a neoadjuvant, did not impact patient survival in resectable GBM in a phase 2 clinical trial (63). In contrast, another study reported a survival benefit of the anti-PD-1 pembrolizumab in 35 patients with recurrent and resectable GBM (64). Collectively, the dismal results of ICI in GBM may be due to the poor immunogenicity of GBM tumors. In 2017, the FDA approved the use of the anti-PD-1 pembrolizumab in solid tumors with microsatellite instability high (MSI-H) or mismatch repair deficiency (dMMR) tumors. This year, it further approved the use of pembrolizumab for the treatment of adult and pediatric patients with non-resectable or metastatic tumor mutation burden-high (TMB-H) solid tumors. dMMR gliomas are rare (65), but earlier results from two case reports showed a response to pembrolizumab in one pediatric (66) and one adult (67) patients. Despite these promising results, a recent study reported that PD-1 blockade did not impact mOS in hypermutated gliomas, consistent with an observed lack of TILs in these cancers (68). However, another study reported significant clinical and radiological responses of nivolumab in two young siblings with biallelic mismatch repair deficiency (66), suggesting that ICI therapy might benefit pediatric GBM with high mutational burden [e.g., with MSH6 mutations (69)]. It is plausible that treatments that increase mutational burden might synergize with ICI, as has been shown in other cancers (70). Nanoscale immunoconjugates (NICs), which deliver ICIs, covalently attached on a natural biopolymer scaffold, across the BBB using transferrin receptor (TfR)-mediated transcytosis, or via angiopep-2 (AP-2)- LDLR-related protein 1 (LRP1), were shown to outperform free ICIs in increasing TILs and improving survival in a murine glioma model (71). However, this remains to be tested in patients. Alternative immunotherapies for GBM are being explored. These are primarily focused on vaccines, chimeric antigen receptors (CAR)-T cells, oncolytic viruses and strategies that harness the anti-tumor activity of myeloid cells or the use of adipose stem/stromal cells (ASC) and stromal vascular fraction (SVF) injected in the surgical cavity [reviewed in Bateman et al. (72)].
Vaccines
In the vaccine arena, three phase 3 clinical trials have been completed with different outcomes. ACT IV, a phase III trial evaluating Rindopepimut (also known as CDX-110), a 13-amino acid peptide vaccine targeting EGFRvIII, a constitutively active mutant form of EGFR expressed in ~30% of GBM patients, in combination with TMZ was terminated for futility, as no significant difference in mOS was observed in patients with newly-diagnosed GBM (73). The failure of this approach might be due to heterogenous expression of EGFRvIII within the tumor or loss of its expression leading to clonal outgrowth of resistant cells. A second phase III trial that evaluated an autologous tumor lysate-pulsed DC vaccine (DCVax®-L) in combination with TMZ showed some clinical benefit, reporting longer progression free survival (PFS) and mOS in patients with recurrent GBM (74). However, this is a logistically complicated approach as it requires personalization, apheresis, and DC expansion prior to administration back into patients. A third phase III trial conducted in Japan using personalized peptide vaccination for HLA-24+ recurrent GBM did not meet the primary nor the secondary endpoints (75). More recently, two phase I/Ib trials reported beneficial effects of personalized peptide vaccines. The first, the Glioma Actively Personalized Vaccine Consortium (GAPVAC), employed two sets of personalized peptide vaccines designed according to patients tumor mutations, transcriptomic and immuno-peptidomic profiles, and showed that these vaccines were able to elicit sustained CD8+ T cell and CD4+ Th1 responses against neoantigens (76). The second, which employed a pool of synthetic long peptides mimicking neoantigens, also reported the generation of poly-functional neoantigen-specific CD4+ T cells and CD8+ T cells in the periphery and enhanced infiltration of TILs (77). Together, these trials indicate that vaccine approaches are feasible as they elicit anti-tumor immune responses but whether this will translate into clinical benefit, as a monotherapy, requires additional testing.
CAR-T and CAR-NK Cells
CAR-T cells are patients-derived T cells engineered to express a CAR, which consists of the antigen-recognition region of an antibody fused in tandem with the cytoplasmic domains of the T cell receptor chain CD3ζ and costimulatory receptors (e.g., CD28 and/or 4-1BB). Currently, approved CAR-T cells target CD19 in B cell malignancies. The challenges of this therapy include the identification of tumor-specific or tumor-associated antigens, especially important in solid tumors, circumventing antigen loss, and countering the exhaustion of transferred CAR-T cells, among others. Several trials and pre-clinical studies have been conducted using CAR-T cells in GBM. The first was a case report that used an IL13Rα2-CAR-T cells in one patient. The CAR-T was delivered through repeated infusions in the resected tumor cavity followed by infusions in the ventricular system. This regimen led to the regression of all cranial and spinal tumors accompanied by a notable immune activity in the CSF (78). A first-in-human study including 10 patients with recurrent GBM followed. This study evaluated EGFRvIII-CAR-T cells injected intravenously. While the CAR-T cells expanded in the blood and trafficked to the tumor, the antigen was lost in 5 out 7 patients and the tumor microenvironment exhibited elevated expression of inhibitory molecules and a high frequency of Treg cells (79). Improvement of CAR-T therapy requires the identification of a tumor-associated antigen expressed stably throughout tumor growth and with limited heterogeneity. Chondroitin sulfate proteoglycan 4 (CSPG4) was found to fit this criterion. It is highly expressed in 67% of GBM cells and is sustained by TNF derived from microglia. Intracranial delivery of CSPG4-CAR-T cells was effective in vivo in nude mice transplanted with CSPG4-expressing glioma cells or neurospheres (80). Transgenic expression of cytokines, such as IL-15, was also demonstrated as a mean to improve anti-glioma activity of CAR-T cells, as shown with IL13Rα2-CAR-T cells (81). Since the final CAR-T cell product is a mix of CD4+ CAR-T and CD8+ CAR-T cells, another mean to refine this approach is to characterize the T cell subset that mediates anti-tumor activity. Using orthotopic GBM mouse models and IL13Rα2-CAR-T cells, the CD4+ CAR-T cell subset was found to be more effective than the CD8+ CAR-T cells, which were rapidly exhausted (82). Co-expression of the IL-8 receptor, CXCR1/CXCR2, was found to enhance CAR-T cells trafficking and persistence in the tumor in a glioma mouse model (83). Engineering EGFRvIII-CAR-T cells to co-express a bispecific T-cell engager (BiTE) against wild-type EGFR was demonstrated to ameliorate this therapy by countering the heterogeneity of EGFRvIII expression (84). A CAR-engineered NK cell targeting both WT EGFR and EGFRvIII mutant, NK-92-EGFR-CAR, was similarly efficient in targeting and killing GBM cells in mice engrafted with patients' mesenchymal GBM stem cells (85). Additional CAR target antigens in GBM include B7-H3 (86, 87), HER2 (88–90) and EphA2 (91), as demonstrated in preclinical studies, and in a phase I dose escalation clinical trial using a HER2-CAR (92). Interestingly, generation of a tri-cistronic transgene encoding three CAR molecules against HER2, EphA2 and IL13Rα2, dubbed universal CAR-T (UCAR), was shown to overcome interpatient heterogeneity and target 100% of tumor cells (93). Another approach to overcome problems of tumor heterogeneity and antigen escape, is a new CAR design employing a toxin as the targeting entity was developed and tested in a murine model of glioma. This is based on GBM cells' affinity to bind chlorotoxin (CLTX) by matrix metalloproteinase-2. (CLTX)-CAR-T cells efficiently limited tumor growth in the absence of off-target effects (94).
Oncolytic Viruses
Oncolytic viruses (OV) constitute an interesting therapeutic approach in GBM, as besides their lytic activity, they might overcome GBM immunosuppression by stimulating innate immunity. Several types of OVs have been tested including replication-competent viruses such as polio and measles viruses, Herpes simplex viruses (HSV), adenoviruses and retroviruses. Notably, recombinant non-pathogenic polio-rhinovirus chimera (PVSRIPO), which binds the poliovirus receptor CD155 on cancer cells, was evaluated in 61 GBM patients via intra-tumoral injection and was effective in 21% patients who survived past 36 months (95). Replication-deficient adenoviruses, e.g., aglatimagene besadenovec, have also been used as vectors to deliver tumoricidal genes such as the HSV thymidine kinase that converts ganciclovir into a toxic nucleotide analog that poisons infected dividing cells. Two phase II clinical trials evaluated this Adv-tk viro-immunotherapy in GBM and reported improved PFS and OS (96, 97). An oncolytic HSV expressing E-cadherin, a ligand for the inhibitory NK receptor KLRG1, resulted in a better outcome in a glioma mouse model, by inhibiting NK cells and permitting viral spread (98). More recently, a Zika OV was shown to specifically target GBM stem cells (GSCs) rather than neural precursor cells, through a SOX2-Integrin αvβ5 Axis (99), suggesting a potentially superior anti-tumoral activity for brain tumor therapy. A triple combination of anti-CTLA-4, anti-PD-1 and a recombinant oncolytic HSV expressing mouse IL-12 (G47Δ-mIL12) cured most mice in two glioma models. CD4+ T cells, CD8+ T cells and M1 macrophages mediated this response, highlighting the need for combinatory approaches in future trials (100).
Macrophage-Based Immunotherapies
Additional promising strategies for GBM immunotherapy include harnessing the anti-tumor activity of myeloid and NK cells. Targeting the phagocytosis checkpoint CD47 using a humanized anti-CD47 antibody, Hu5F9-G4, has shown promise in a glioma PDX mouse model of five aggressive pediatric brain cancers (101). Furthermore, anti-CD47 in combination with TMZ was shown to enhance phagocytosis and promote cytotoxic CD8+ T cell priming by stimulating antigen cross-presentation through cGAS-STING activation (102). Members of the Let-7 micro-RNA family have also been used as a therapeutic tool in a mouse glioma model; they boosted microglial anti-tumor activity by stimulating TLR7 (103). Alternatively, blocking TAM recruitment or polarization has also shown some efficacy in preclinical models. A 4-1BB–osteopontin (OPN) bi-specific aptamer for instance increased median survival by neutralizing macrophage infiltration while co-stimulating effector T cell activity (50). Di-mannose nanocarriers that bind the mannose receptor CD206 on M2 macrophages, used to deliver in vitro-transcribed mRNA encoding M1-polarizing transcription factors, were shown to reprogram TAMs and improve survival in different cancer models (ovarian, lung metastasis) including GBM (104).
Perspectives
There is a significant need to develop novel GBM immunotherapies. To date, more than 70 clinical trials with the terms GBM and immunotherapy are found in the clinicaltrials.gov webpage, of which 7 are phase III, 31 phase II and 37 phase I trials (Table 2). These trials explore the various strategies described above notably personalized vaccines, adoptive cell transfer therapy and combinations. It is our hope that this endeavor will soon impact patients' lives (Figure 4).
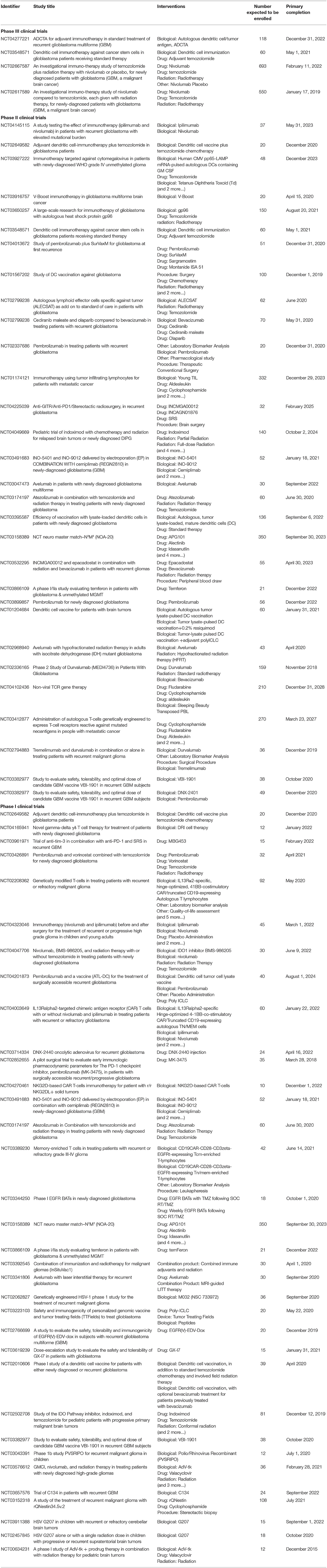
Table 2. Clinical trials of immunotherapies for GBM.
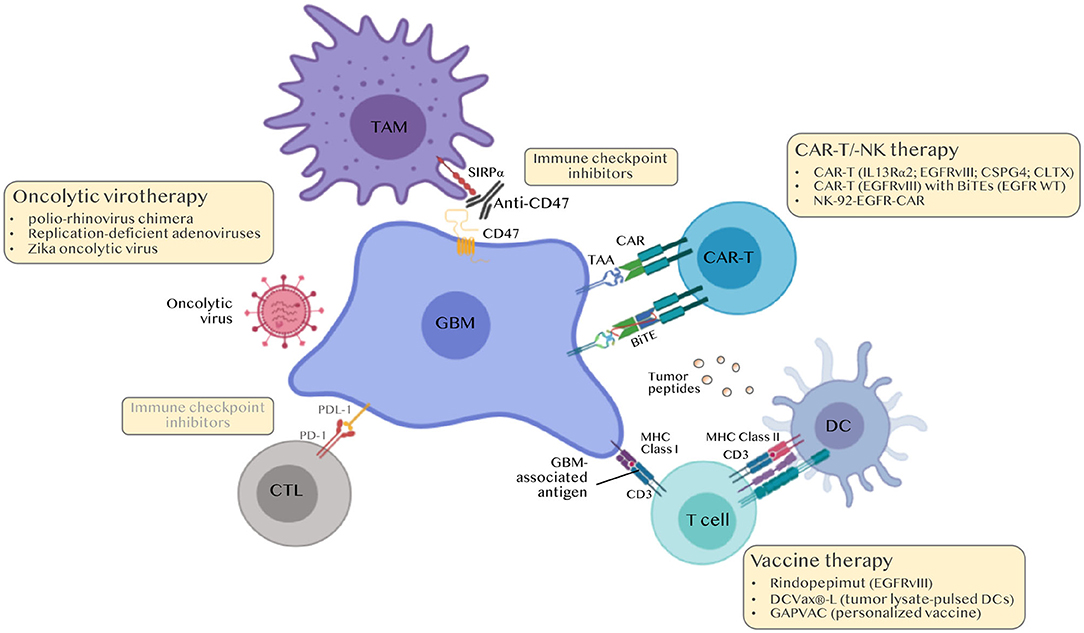
Figure 4. Immunotherapies for the treatment of GBM. Classical immune checkpoint inhibitors (ICI) i.e., anti-PD-1/PDL-1 and anti-CTLA4 were ineffective in GBM. Current approaches include modulating TAMs (anti-CD47 to boost phagocytosis, nano-immunoconjugates to modulate TAM phenotype, aptamers to inhibit TAM recruitment), personalized peptide vaccines, chimeric antigen receptor (CAR)-T and CAR-NK cell approaches and oncolytic viruses. BiTEs, Bi-specific T-cell engagers.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
MS was funded by the ARC Foundation, IDEX Bordeaux and the New Aquitaine Region. TD was funded by ARC Foundation, Ligue Contre le Cancer, SIRIC BRIO.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Machler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, Von Faber-Castell A, et al. In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab. (2016) 23:94–102. doi: 10.1016/j.cmet.2015.10.010
2. Daubon T, Leon C, Clarke K, Andrique L, Salabert L, Darbo E, et al. Deciphering the complex role of thrombospondin-1 in glioblastoma development. Nat Commun. (2019) 10:1146. doi: 10.1038/s41467-019-08480-y
3. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. (2014) 344:1396–401. doi: 10.1126/science.1254257
4. Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. (2019) 178:835–49 e821. doi: 10.1016/j.cell.2019.06.024
5. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. (2012) 4:147ra111. doi: 10.1126/scitranslmed.3003748
6. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. (2015) 212:991–9. doi: 10.1084/jem.20142290
7. Louveau A, Harris TH, Kipnis J. Revisiting the mechanisms of CNS immune privilege. Trends Immunol. (2015) 36:569–77. doi: 10.1016/j.it.2015.08.006
8. Medawar PB. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol. (1948) 29:58–69.
9. Andersson PB, Perry VH, Gordon S. The acute inflammatory response to lipopolysaccharide in CNS parenchyma differs from that in other body tissues. Neuroscience. (1992) 48:169–86. doi: 10.1016/0306-4522(92)90347-5
10. Locatelli G, Wortge S, Buch T, Ingold B, Frommer F, Sobottka B, et al. Primary oligodendrocyte death does not elicit anti-CNS immunity. Nat Neurosci. (2012) 15:543–50. doi: 10.1038/nn.3062
11. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. (2012) 122:1164–71. doi: 10.1172/JCI58644
12. Mason DW, Charlton HM, Jones AJ, Lavy CB, Puklavec M, Simmonds SJ. The fate of allogeneic and xenogeneic neuronal tissue transplanted into the third ventricle of rodents. Neuroscience. (1986) 19:685–94. doi: 10.1016/0306-4522(86)90292-7
13. Nicholas MK, Antel JP, Stefansson K, Arnason BG. Rejection of fetal neocortical neural transplants by H-2 incompatible mice. J Immunol. (1987) 139:2275–83.
14. Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity. (2018) 48:599. doi: 10.1016/j.immuni.2018.01.011
15. Sankowski R, Bottcher C, Masuda T, Geirsdottir L, Sagar Sindram E, Seredenina T, et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat Neurosci. (2019) 22:2098–110. doi: 10.1038/s41593-019-0532-y
16. Mohammad MG, Tsai VW, Ruitenberg MJ, Hassanpour M, Li H, Hart PH, et al. Immune cell trafficking from the brain maintains CNS immune tolerance. J Clin Invest. (2014) 124:1228–41. doi: 10.1172/JCI71544
17. Walsh JT, Hendrix S, Boato F, Smirnov I, Zheng J, Lukens JR, et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J Clin Invest. (2015) 125:2547. doi: 10.1172/JCI76210
18. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. (2007) 114:97–109. doi: 10.1007/s00401-007-0243-4
19. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. (2010) 17:98–110. doi: 10.1016/j.ccr.2009.12.020
20. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. (2017) 32:42–56 e46. doi: 10.1016/j.ccell.2017.06.003
21. Brennan CW, Verhaak RG, Mckenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. (2013) 155:462–77. doi: 10.1016/j.cell.2013.09.034
22. Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. (2013) 45:1141–9. doi: 10.1038/ng.2734
23. Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med. (2015) 372:2499–508. doi: 10.1056/NEJMoa1407279
24. Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. (2015) 47:458–68. doi: 10.1038/ng.3273
25. Cancer Genome Atlas Research N, Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. (2015) 372:2481–98. doi: 10.1056/NEJMoa1402121
26. Dewitt JC, Mock A, Louis DN. The 2016 WHO classification of central nervous system tumors: what neurologists need to know. Curr Opin Neurol. (2017) 30:643–9. doi: 10.1097/WCO.0000000000000490
27. Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. (2012) 124:439–47. doi: 10.1007/s00401-012-0998-0
28. Martinez-Lage M, Lynch TM, Bi Y, Cocito C, Way GP, Pal S, et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol Commun. (2019) 7:203. doi: 10.1186/s40478-019-0803-6
29. Doucette T, Rao G, Rao A, Shen L, Aldape K, Wei J, et al. Immune heterogeneity of glioblastoma subtypes: extrapolation from the cancer genome atlas. Cancer Immunol Res. (2013) 1:112–22. doi: 10.1158/2326-6066.CIR-13-0028
30. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. (2015) 12:453–7. doi: 10.1038/nmeth.3337
31. Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. (2016) 164:550–63. doi: 10.1016/j.cell.2015.12.028
32. Close HJ, Stead LF, Nsengimana J, Reilly KA, Droop A, Wurdak H, et al. Expression profiling of single cells and patient cohorts identifies multiple immunosuppressive pathways and an altered NK cell phenotype in glioblastoma. Clin Exp Immunol. (2020) 200:33–44. doi: 10.1111/cei.13403
33. Mohme M, Schliffke S, Maire CL, Runger A, Glau L, Mende KC, et al. Immunophenotyping of newly diagnosed and recurrent glioblastoma defines distinct immune exhaustion profiles in peripheral and tumor-infiltrating lymphocytes. Clin Cancer Res. (2018) 24:4187–200. doi: 10.1158/1078-0432.CCR-17-2617
34. Denardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. (2019) 19:369–82. doi: 10.1038/s41577-019-0127-6
35. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. (2018) 17:887–904. doi: 10.1038/nrd.2018.169
36. Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer. (2019) 19:568–86. doi: 10.1038/s41568-019-0183-z
37. Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. (2016) 18:557–64. doi: 10.1093/neuonc/nov245
38. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. (2016) 17:2445–59. doi: 10.1016/j.celrep.2016.10.052
39. Castro BA, Flanigan P, Jahangiri A, Hoffman D, Chen W, Kuang R, et al. Macrophage migration inhibitory factor downregulation: a novel mechanism of resistance to anti-angiogenic therapy. Oncogene. (2017) 36:3749–59. doi: 10.1038/onc.2017.1
40. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. (2014) 159:1312–26. doi: 10.1016/j.cell.2014.11.018
41. Muller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. (2017) 18:234. doi: 10.1186/s13059-017-1362-4
42. Hutter G, Theruvath J, Graef CM, Zhang M, Schoen MK, Manz EM, et al. Microglia are effector cells of CD47-SIRPalpha antiphagocytic axis disruption against glioblastoma. Proc Natl Acad Sci USA. (2019) 116:997–1006. doi: 10.1073/pnas.1721434116
43. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. (2017) 77:2266–78. doi: 10.1158/0008-5472.CAN-16-2310
44. Chen Z, Ross JL, Hambardzumyan D. Intravital 2-photon imaging reveals distinct morphology and infiltrative properties of glioblastoma-associated macrophages. Proc Natl Acad Sci USA. (2019) 116:14254–9. doi: 10.1073/pnas.1902366116
45. Couto M, Coelho-Santos V, Santos L, Fontes-Ribeiro C, Silva AP, Gomes CMF. The interplay between glioblastoma and microglia cells leads to endothelial cell monolayer dysfunction via the interleukin-6-induced JAK2/STAT3 pathway. J Cell Physiol. (2019) 234:19750–60. doi: 10.1002/jcp.28575
46. Wang Q, He Z, Huang M, Liu T, Wang Y, Xu H, et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2alpha. Nat Commun. (2018) 9:559. doi: 10.1038/s41467-018-03050-0
47. Zhang Y, Yu G, Chu H, Wang X, Xiong L, Cai G, et al. Macrophage-associated PGK1 phosphorylation promotes aerobic glycolysis and tumorigenesis. Mol Cell. (2018) 71:201–15 e207. doi: 10.1016/j.molcel.2018.06.023
48. Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC, et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci. (2019) 22:729–40. doi: 10.1038/s41593-019-0370-y
49. Chen P, Zhao D, Li J, Liang X, Li J, Chang A, et al. Symbiotic macrophage-glioma cell interactions reveal synthetic lethality in PTEN-Null glioma. Cancer Cell. (2019) 35:868–84 e866. doi: 10.1016/j.ccell.2019.05.003
50. Wei J, Marisetty A, Schrand B, Gabrusiewicz K, Hashimoto Y, Ott M, et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J Clin Invest. (2019) 129:137–49. doi: 10.1172/JCI121266
51. Kuppner MC, Hamou MF, Sawamura Y, Bodmer S, De Tribolet N. Inhibition of lymphocyte function by glioblastoma-derived transforming growth factor beta 2. J Neurosurg. (1989) 71:211–7. doi: 10.3171/jns.1989.71.2.0211
52. Jackson CM, Kochel CM, Nirschl CJ, Durham NM, Ruzevick J, Alme A, et al. Systemic tolerance mediated by melanoma brain tumors is reversible by radiotherapy and vaccination. Clin Cancer Res. (2016) 22:1161–72. doi: 10.1158/1078-0432.CCR-15-1516
53. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res. (2013) 19:3165–75. doi: 10.1158/1078-0432.CCR-12-3314
54. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. (2007) 13:84–8. doi: 10.1038/nm1517
55. Wischhusen J, Friese MA, Mittelbronn M, Meyermann R, Weller M. HLA-E protects glioma cells from NKG2D-mediated immune responses in vitro: implications for immune escape in vivo. J Neuropathol Exp Neurol. (2005) 64:523–8. doi: 10.1093/jnen/64.6.523
56. Didenko VV, Ngo HN, Minchew C, Baskin DS. Apoptosis of T lymphocytes invading glioblastomas multiforme: a possible tumor defense mechanism. J Neurosurg. (2002) 96:580–4. doi: 10.3171/jns.2002.96.3.0580
57. Dusoswa SA, Verhoeff J, Abels E, Mendez-Huergo SP, Croci DO, Kuijper LH, et al. Glioblastomas exploit truncated O-linked glycans for local and distant immune modulation via the macrophage galactose-type lectin. Proc Natl Acad Sci USA. (2020) 117:3693–703. doi: 10.1073/pnas.1907921117
58. Lai SW, Lin HJ, Liu YS, Yang LY, Lu DY. Monocarboxylate transporter 4 regulates glioblastoma motility and monocyte binding ability. Cancers. (2020) 12:380. doi: 10.3390/cancers12020380
59. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
60. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
61. Silva LS, Poschet G, Nonnenmacher Y, Becker HM, Sapcariu S, Gaupel AC, et al. Branched-chain ketoacids secreted by glioblastoma cells via MCT1 modulate macrophage phenotype. EMBO Rep. (2017) 18:2172–85. doi: 10.15252/embr.201744154
62. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol. (2020) 6:1003–10. doi: 10.1001/jamaoncol.2020.1024
63. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, Lopez-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. (2019) 25:470–6. doi: 10.1038/s41591-018-0339-5
64. Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. (2019) 25:477–86. doi: 10.1038/s41591-018-0337-7
65. Maxwell JA, Johnson SP, Mclendon RE, Lister DW, Horne KS, Rasheed A, et al. Mismatch repair deficiency does not mediate clinical resistance to temozolomide in malignant glioma. Clin Cancer Res. (2008) 14:4859–68. doi: 10.1158/1078-0432.CCR-07-4807
66. Bouffet E, Larouche V, Campbell BB, Merico D, De Borja R, Aronson M, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. (2016) 34:2206–11. doi: 10.1200/JCO.2016.66.6552
67. Johanns TM, Miller CA, Dorward IG, Tsien C, Chang E, Perry A, et al. Immunogenomics of hypermutated glioblastoma: a patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov. (2016) 6:1230–6. doi: 10.1158/2159-8290.CD-16-0575
68. Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature. (2020) 580:517–23. doi: 10.1038/s41586-020-2209-9
69. Yang C, Austin F, Richard H, Idowu M, Williamson V, Sabato F, et al. Lynch syndrome-associated ultra-hypermutated pediatric glioblastoma mimicking a constitutional mismatch repair deficiency syndrome. Cold Spring Harb Mol Case Stud. (2019) 5:a003863. doi: 10.1101/mcs.a003863
70. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. (2017) 357:409–13. doi: 10.1126/science.aan6733
71. Galstyan A, Markman JL, Shatalova ES, Chiechi A, Korman AJ, Patil R, et al. Blood-brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat Commun. (2019) 10:3850. doi: 10.1038/s41467-019-11719-3
72. Bateman ME, Strong AL, Gimble JM, Bunnell BA. Concise review: using fat to fight disease: a systematic review of nonhomologous adipose-derived stromal/stem cell therapies. Stem Cells. (2018) 36:1311–28. doi: 10.1002/stem.2847
73. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. (2017) 18:1373–85. doi: 10.1016/S1470-2045(17)30517-X
74. Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. (2018) 16:142. doi: 10.1186/s12967-018-1552-1
75. Narita Y, Arakawa Y, Yamasaki F, Nishikawa R, Aoki T, Kanamori M, et al. A randomized, double-blind, phase III trial of personalized peptide vaccination for recurrent glioblastoma. Neuro Oncol. (2019) 21:348–59. doi: 10.1093/neuonc/noy200
76. Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. (2019) 565:240–5. doi: 10.1038/s41586-018-0810-y
77. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. (2019) 565:234–9. doi: 10.1038/s41586-018-0792-9
78. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
79. O'rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:984. doi: 10.1126/scitranslmed.aaa0984
80. Pellegatta S, Savoldo B, Di Ianni N, Corbetta C, Chen Y, Patane M, et al. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: implications for CAR-T cell therapy. Sci Transl Med. (2018) 10:eaao2731. doi: 10.1126/scitranslmed.aao2731
81. Krenciute G, Prinzing BL, Yi Z, Wu MF, Liu H, Dotti G, et al. Transgenic expression of IL15 improves antiglioma activity of IL13Ralpha2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. (2017) 5:571–81. doi: 10.1158/2326-6066.CIR-16-0376
82. Wang D, Aguilar B, Starr R, Alizadeh D, Brito A, Sarkissian A, et al. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight. (2018) 3. doi: 10.1172/jci.insight.99048
83. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. (2019) 10:4016. doi: 10.1038/s41467-019-11869-4
84. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. (2019) 37:1049–58. doi: 10.1038/s41587-019-0192-1
85. Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep. (2015) 5:11483. doi: 10.1038/srep11483
86. Nehama D, Di Ianni N, Musio S, Du H, Patane M, Pollo B, et al. B7-H3-redirected chimeric antigen receptor T cells target glioblastoma and neurospheres. EBioMedicine. (2019) 47:33–43. doi: 10.1016/j.ebiom.2019.08.030
87. Tang X, Zhao S, Zhang Y, Wang Y, Zhang Z, Yang M, et al. B7-H3 as a novel CAR-T therapeutic target for glioblastoma. Mol Ther Oncolytics. (2019) 14:279–87. doi: 10.1016/j.omto.2019.07.002
88. Burger MC, Zhang C, Harter PN, Romanski A, Strassheimer F, Senft C, et al. CAR-engineered NK cells for the treatment of glioblastoma: turning innate effectors into precision tools for cancer immunotherapy. Front Immunol. (2019) 10:2683. doi: 10.3389/fimmu.2019.02683
89. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest. (2019) 129:3464. doi: 10.1172/JCI131246
90. Shen L, Li H, Bin S, Li P, Chen J, Gu H, et al. The efficacy of third generation antiHER2 chimeric antigen receptor T cells in combination with PD1 blockade against malignant glioblastoma cells. Oncol Rep. (2019) 42:1549–57. doi: 10.3892/or.2019.7263
91. Yi Z, Prinzing BL, Cao F, Gottschalk S, Krenciute G. Optimizing EphA2-CAR T cells for the adoptive immunotherapy of glioma. Mol Ther Methods Clin Dev. (2018) 9:70–80. doi: 10.1016/j.omtm.2018.01.009
92. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
93. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. (2018) 20:506–18. doi: 10.1093/neuonc/nox182
94. Wang D, Starr R, Chang WC, Aguilar B, Alizadeh D, Wright SL, et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci Transl Med. (2020) 12:eaaw2672. doi: 10.1126/scitranslmed.aaw2672
95. Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. (2018) 379:150–61. doi: 10.1056/NEJMoa1716435
96. Ji N, Weng D, Liu C, Gu Z, Chen S, Guo Y, et al. Adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of recurrent high-grade glioma. Oncotarget. (2016) 7:4369–78. doi: 10.18632/oncotarget.6737
97. Wheeler LA, Manzanera AG, Bell SD, Cavaliere R, Mcgregor JM, Grecula JC, et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro Oncol. (2016) 18:1137–45. doi: 10.1093/neuonc/now002
98. Xu B, Ma R, Russell L, Yoo JY, Han J, Cui H, et al. An oncolytic herpesvirus expressing E-cadherin improves survival in mouse models of glioblastoma. Nat Biotechnol. (2018) 37:45–54. doi: 10.1038/nbt.4302
99. Zhu Z, Mesci P, Bernatchez JA, Gimple RC, Wang X, Schafer ST, et al. Zika virus targets glioblastoma stem cells through a SOX2-Integrin alphavbeta5 Axis. Cell Stem Cell. (2020) 26:187–204 e110. doi: 10.1016/j.stem.2019.11.016
100. Saha D, Martuza RL, Rabkin SD. Curing glioblastoma: oncolytic HSV-IL12 and checkpoint blockade. Oncoscience. (2017) 4:67–9. doi: 10.18632/oncoscience.359
101. Gholamin S, Mitra SS, Feroze AH, Liu J, Kahn SA, Zhang M, et al. Disrupting the CD47-SIRPalpha anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci Transl Med. (2017) 9:eaaf2968. doi: 10.1126/scitranslmed.aaf2968
102. Von Roemeling CA, Wang Y, Qie Y, Yuan H, Zhao H, Liu X, et al. Therapeutic modulation of phagocytosis in glioblastoma can activate both innate and adaptive antitumour immunity. Nat Commun. (2020) 11:1508. doi: 10.1038/s41467-020-15129-8
103. Buonfiglioli A, Efe IE, Guneykaya D, Ivanov A, Huang Y, Orlowski E, et al. let-7 MicroRNAs regulate microglial function and suppress glioma growth through toll-like receptor 7. Cell Rep. (2019) 29:3460–71. e3467. doi: 10.1016/j.celrep.2019.11.029
Keywords: glioblastoma, immune response, macrophage, immunotherapy, CART-T cell
Citation: Daubon T, Hemadou A, Romero Garmendia I and Saleh M (2020) Glioblastoma Immune Landscape and the Potential of New Immunotherapies. Front. Immunol. 11:585616. doi: 10.3389/fimmu.2020.585616
Received: 21 July 2020; Accepted: 07 September 2020;
Published: 14 October 2020.
Edited by:
Xiaoxing Xiong, Renmin Hospital of Wuhan University, ChinaReviewed by:
Stephen Gottschalk, St. Jude Children's Research Hospital, United StatesDipongkor Saha, Texas Tech University Health Sciences Center, Abilene, United States
Copyright © 2020 Daubon, Hemadou, Romero Garmendia and Saleh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Daubon, dGhvbWFzLmRhdWJvbkB1LWJvcmRlYXV4LmZy; Maya Saleh, bWF5YS5zYWxlaEB1LWJvcmRlYXV4LmZy
†These authors share first authorship