 K. Michael Pollard
K. Michael Pollard- Department of Molecular Medicine, The Scripps Research Institute, La Jolla, CA, United States
Studies have shown that a wide range of factors including drugs, chemicals, microbes, and other environmental agents can induce pre-clinical autoimmunity. However, only a few have been confidently linked to autoimmune diseases. Among these are exposures to inhaled particulates that are known to be associated with autoimmune diseases such as lupus and rheumatoid arthritis. In this article, the potential of particle, fiber, and nanomaterial exposures to induce autoimmunity is discussed. It is hypothesized that inhalation of particulate material known to be associated with human autoimmune diseases, such as cigarette smoke and crystalline silica, results in a complex interplay of a number of pathological processes, including, toxicity, oxidative stress, cell and tissue damage, chronic inflammation, post-translational modification of self-antigens, and the formation of lymphoid follicles that provide a milieu for the accumulation of autoreactive B and T cells necessary for the development and persistence of autoimmune responses, leading to disease. Although experimental studies show nanomaterials are capable of inducing several of the above features, there is no evidence that this matures to autoimmune disease. The procession of events hypothesized here provides a foundation from which to pursue experimental studies to determine the potential of other environmental exposures to induce autoimmunity and autoimmune disease.
Introduction
Numerous drugs, chemicals, microbes, and other environmental factors have been identified as possible causes of pre-clinical autoimmunity (1). However, very few have been confidently associated with human autoimmune diseases (2). Among these are respirable particulates such as cigarette smoke and silica dust (2) which have been linked to systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and systemic sclerosis (SSc) (2, 3). Asbestos, a fibrous silicate mineral, has also been associated with autoimmunity, although linkage to specific autoimmune diseases is less well established (2, 4). In contrast, a role for nanomaterial [materials with at least one dimension between 1 and 100 nanometers (5, 6)] exposure as a causative agent in human autoimmunity remains to be established.
In reviewing the etiopathogenesis of environment-induced autoimmunity, we (7) proposed that the toxic insult of an environmental exposure initiates a multi-step process characterized by tissue damage, and the release of nucleic acids and other damage associated molecular patterns (DAMPs) including self- and modified self-antigens, which induces an innate inflammatory response, leading to adaptive immunity involving the presentation of self- and modified self-antigens to non-tolerant lymphocytes. Included within this process is the engagement of Toll-like receptors (TLRs) and other innate sensors, the production of inflammatory mediators, the expansion of autoreactive B and T effector cell populations, and the production of autoantibodies. These events often occur at the site of exposure, where they can lead to the development of tertiary lymphoid structures (TLSs), and/or germinal centers in secondary lymphoid organs draining the site of exposure. In either case the expansion of autoreactive lymphocytes and their migration to target tissues, such as the kidney in SLE or the joints in RA, results in autoimmune disease (7).
To include an additional perspective to the above hypothesis, this article compares the current state of knowledge regarding the response of the immune system to selected particle, fiber, and nanomaterial exposures. Specific particulate and fibrous materials are discussed because of their known role in pre-clinical autoimmunity and autoimmune diseases (2). This is contrasted with the immunological responses to nanomaterials, particularly engineered nanoparticles (ENPs), where evidence for induction of autoimmunity is less convincing.
Particulate Material
Cigarette Smoke
Cigarette smoke is a complex mixture of particulate-phase and chemical compounds (8, 9), and is a significant risk factor for chronic obstructive pulmonary disease (COPD) (10). Smoking is also a significant risk factor for RA (11), and has been linked to other autoimmune diseases including SLE, multiple sclerosis, and Crohn’s disease (2). The linkage of smoking and RA is strongest for seropositive disease particularly patients with anti-citrullinated protein antibody (ACPA) (2) which is itself strongly linked to the HLA shared epitope (12). The association between smoking, RA, and the HLA-linked ACPA response identifies an important gene-environment interaction for autoimmune diseases (13).
The particulate material of cigarette smoke consists of partially combusted plant material between >0.25 to <1 um in size (8, 9, 14). The chronic inflammation resulting from cigarette smoke exposure is thought to be due to this particulate material and gas-phase chemicals but may also involve bacterial and fungal contaminants of tobacco (15). Cigarette smoke also contains reactive oxygen species (ROS), that may contribute to post-translational modification of proteins and other markers of oxidative stress (16). Tissue damage from inhaled smoke leads to activation of lung epithelial cells and alveolar macrophages (AM) via interaction between DAMPs and pattern recognition receptors (PRRs) such as TLRs (16). This initiates an innate immune response, characterized by inflammasome formation and the production of IL-1β, IL-18, and other proinflammatory mediators including IL-6 and TNF-α (16). The resulting infiltration of neutrophils and monocytes further exacerbates oxidative stress and inflammation. Additional processes, such as neutrophil death, mediated by NETosis, also leads to cell and tissue damage (17).
The innate inflammatory response progresses to adaptive immunity consisting of T helper cell responses, including Th1 and Th17 (18, 19), and B cells that can form lymphoid aggregates in the lung (10). These structures are similar to TLSs found in target organs of autoimmune diseases where they are argued to provide a microenvironment for the survival of autoreactive lymphocytes (20, 21). The presence of plasma cells in lung TLSs (21) suggests that the lungs are a source of autoantibodies, and this is supported by the presence of autoantibodies in bronchoalveolar lavage (BAL) fluid (21, 22). Moreover, autoantibodies to modified self-antigens have been described in COPD (21, 23, 24) including autoantibodies known to be important in seropositive RA [rheumatoid factor (RF) and ACPA] (11, 25). These events may occur years before clinical diagnosis of RA (26). It has been hypothesized that localized chronic inflammatory processes in the lungs following smoke exposure are important in the pre-clinical phase of not only diseases such as COPD but also autoimmune diseases such as RA (25, 27). Importantly, experimental studies have shown that TLSs and autoantibodies persist after ceasing smoking and are dependent upon IL-1 receptor 1 (28).
While COPD and RA are comorbidities, their pathological relationship remains under investigation (26, 29). Development of COPD in RA patients (29) is consistent with the known pulmonary involvement in RA (25, 30). Conversely, a recent study identified COPD as a risk factor for RA with the strongest association between COPD and seropositive RA in older smokers (31). Interestingly, individuals who were ACPA positive before RA diagnosis were at increased risk of developing COPD (32). This suggests a linkage between appearance of ACPA and susceptibility to respiratory disease. ACPA may also be an outcome of inhalation of other forms of particulate matter, and may identify inhalant exposures that increase the risk of RA or other autoimmune diseases (33).
Silica Dust
Exposure to crystalline silica as a result of the breakdown of quartz (e.g. mining, sandblasting, quarrying, ceramics), or during the fabrication of artificial stone (34, 35), can lead to silicosis (36), COPD (36), lung cancer (37), and autoimmune diseases including SLE, RA, SSc, and antineutrophil cytoplasmic antibody (ANCA)-related vasculitis (2, 35, 38). The occurrence of COPD in silica-exposed individuals may also reflect coincident cigarette smoke exposure (39). Significantly, when silica exposure and smoking occur together it results in a synergistic interaction which, for example, greatly increases the risk of ACPA-positive RA in Asian (40) and Caucasian (41) populations. Silica dust exposure is most often via occupational inhalation with deposition of respirable particles [<10μm (42)] in the alveoli of the lungs leading to chronic inflammation and development of fibrotic nodules (36). Although the mechanistic aspects of the pathogenic process of silicosis have not been completely defined, a number of important checkpoints have been identified (36, 43, 44). The cumulative dose and physicochemical properties of silica dust are important in inflammatory and fibrogenic responses (43, 45). Inhaled respirable silica particles can be deposited throughout the lung including the distal airways and alveoli leading to direct cytotoxicity (46). Silica particles also interact with endogenous molecules leading to the formation of a surface corona (43). Subsequent phagocytosis by AM leads to lysosomal damage resulting in activation of inflammatory and cell death pathways. A significant contributor to these processes is oxidative stress due to ROS originating from both silica particles and lung cells (47). The chronic persistence of silica particles in the lung results in fibrosis, silicosis (43, 48), and development of silica-induced autoimmunity (49).
An important observation in the initiation of pulmonary inflammation following crystalline silica inhalation is the release of IL-1α from AM following silica induced cellular damage (50). Release of IL-1α from dying cells is known as an important mediator of sterile inflammation (51) following interaction between IL-1α released from crystal damaged cells and IL-1 receptor on surrounding macrophages (48, 50). Silica-induced cell death also results in the release of numerous types of DAMPs that are recognized by TLRs and other PRR on the cell surface as well as within cytoplasmic components such as endosomes. These interactions lead to intracellular signaling, activation of transcription factors such as NF-κB and AP-1, and the expression of inflammatory cytokines such as IL-1β, IL-6, TNF-α, and interferons (IFNs) (48). Phagocytosis of silica particles via scavenger receptors such as MARCO (SR-A6) and SR-A1 leads to lysosomal destabilization and release of proteases including cathepsins that contribute to the activation of the NLRP3 inflammasome and cleavage of IL-1β and IL-18 into their bioactive forms (52–54). Generation of ROS, either from the surface of silica crystals or phagocytosis and lysosomal/mitochondrial damage, also contributes to these events (45, 47). The end result is an inflammatory response consisting of neutrophils, monocytes and lymphocytes that exhibits acute and chronic phases which have differing molecular and cellular features (35). The failure to clear silica particles from the lungs exacerbates this chronic inflammatory process leading to fibroblast proliferation and collagen production resulting in fibrosis and silicosis (35, 36, 44).
Epidemiological studies have found associations between occupational silica exposure and SLE, SSc, and ANCA-related vasculitis (2, 55, 56). More recently, exposure to silica-containing dust from artificial stone during fabrication of countertops has been linked to both silicosis and autoimmune disease (34, 57). Pre-clinical features of autoimmunity, such as autoantibodies, as well as autoimmune diseases can occur in the absence of silicosis (35, 55) suggesting that events prior to fibrosis are important for silica-induced autoimmunity. The fibrosis following particulate exposure, including silica, may be the result of exaggerated and persistent immunosuppression due to TGF-β and IL-10, rather than chronic inflammation (58).
A significant outcome of the chronic pulmonary inflammation induced by crystalline silica, in animal models, is the formation of TLS (59). These structures comprise accumulations of B and T cells as well as follicular dendritic cells (FDC) and plasma cells (59–61). The presence of immunoglobulin including autoantibodies in the BALF of silica-exposed mice (60, 61) suggests that the lung is a site of autoantibody production. A variety of autoantibody specificities are found in both humans (62) and experimental models (59, 60, 63) after silica exposure. These include anti-DNA, -SS-A/Ro, -SS-B/La, -Scl70, -Sm, and -RNP. ACPA have also been described in RA patients exposed to silica (40). A possible mechanism for citrullination may involve silica-induced increases in peptidylargininedeiminase (PAD) activity (64). This is consistent with the formation of neutrophil extracellular traps (NETs) during NETosis induced by silica (65) which requires PAD mediated protein citrullination (66). Although autoantibody responses in silica exposed patients with autoimmune diseases are often consistent with those of idiopathic autoimmune diseases, it is unclear if they play a pathogenic role. As with smoking, it is also unclear how expression of silica-induced autoreactivity in the lung results in pathogenesis in organs such as the kidney in SLE or the joints in RA.
Fibrous Material
Asbestos
Asbestos describes six naturally occurring fibrous silicate minerals categorized into two groups, serpentine (chrysotile) and amphibole (crocidolite, amosite, tremolite, anthophyllite, actinolite) (67). While there are significant differences in chemical composition and crystalline structure, serpentine fibers are more curvilinear and softer than the needle-like, brittle amphibole fibers (67). Occupational exposures, primarily from mining, construction, and automotive industries, can result in asbestosis and malignancy (68). The properties of asbestos fibers that cause toxicity and pathology, especially inflammation, are uncertain (69), consequently very little is known about the fiber properties necessary for autoimmunity. Short fibers (<5 um), although abundant in the lungs (70), are considered unlikely to cause malignancy (71), but it is unclear if this applies to autoimmunity.
Evidence from both human and experimental animal studies suggest that amphibole asbestos exposure can lead to autoimmunity (4, 72). Several different cohorts including Libby, Montana (73), Wittenoom, Western Australia (74), and Biancavilla, Sicily (75), have linked autoantibodies, including ANA, to asbestos exposure. Studies of the communities of Libby and Troy in Montana have found that almost 14% have been diagnosed with an autoimmune disease other than diabetes (76, 77) and mortality to autoimmune diseases is higher than expected (78). However, a causal role for asbestos exposure in autoimmune disease has not been established due to study limitations (4), and other confounders, including the possibility of concurrent silica and asbestos exposures (2), and smoking history (79).
Asbestos inhalation results in chronic pulmonary inflammation (48), mediated in part by NLRP3 inflammasome activation, and expression of IL-1β and other inflammatory cytokines (80, 81). This is facilitated by asbestos fiber-induced damage to lung epithelial cells, phagocytosis of asbestos particles by macrophages, lysosomal destabilization, subsequent cellular stress and damage leading to production of ROS and oxidative injury, culminating in a fibrogenic response including release of TGF-β, TNF-α, and IL-1β, that promotes collagen deposition and fibrosis (82, 83). Pulmonary inflammation can include increases in AMs and neutrophils, as well as CD4+ T cells that spontaneously release IFN-γ, identified as diffuse lymphocytic-macrophage alveolitis (84).
Autoimmune diseases associated with Libby Asbestiform Amphibole (LAA) exposure include SLE, SSc, and RA (76, 78). ANA are common and include autoantibodies against dsDNA, SS-A/Ro52, Scl70, Sm, and RNP (76). Anti-mesothelial cell antibodies, and ANA, have been linked to pleural abnormalities (85). Autoantibodies against cyclic citrullinated peptide (CCP) and RF are not elevated (86) even though there is evidence of increased PAD2/4 and citrullination in the lung following asbestos exposure (87). ANA have also been found in subjects exposed to asbestos without systemic autoimmune diseases (75). This may be an indicator of pre-clinical autoimmunity, similar to the appearance of ACPA prior to COPD or RA (11, 25), as appearance of autoantibodies can precede diagnosis of autoimmune disease (88, 89). Whether autoantibodies can be linked to TLS in the lung in asbestosis, as has been observed for cigarette smoke and crystalline silica exposures, remains to be investigated.
Nanomaterials
Nanomaterials constitute two major groups, ambient ultrafine particles (UFPs) and ENPs (5, 6, 90). Numerous nanomaterial approaches are being developed to modulate immune responses (91, 92). This includes nanocarriers for drugs, vaccines, antigens and adjuvants (91–93) as therapies for chronic inflammation (91), infection (92), and autoimmunity (91, 92, 94) including specific tolerance approaches for treating autoimmunity (94, 95). The safety of these nanomedicines is dependent upon their ability to avoid toxicity and immune recognition (96, 97) which may result in adverse toxicological (98) and immunological outcomes (99). Although UPFs and ENPs display a range of physicochemical properties (6), inhalation is a common route of exposure (90, 100) and can lead to tissue damage, protein corona, oxidative stress, inflammasome activation, proinflammatory mediators, and inflammation (5, 6, 99, 101), as well as pathological outcomes including fibrosis (58, 102). However, a role for NPs, particularly ENPs, in the causation of pre-clinical human autoimmunity or autoimmune diseases has not been established.
Nanodiamond NPs induce lysosomal damage and NETosis (103) but this leads to resolution of inflammation (103, 104) presumably by sequestration of the offending particles (105). There is evidence for this as NET-related proteins were found in BAL fluid in the acute phase response to TiO2 NPs but disappeared over time, and were not associated with histopathological changes (106). Alternatively, solubility or clearance by phagocytes may lead to resolution of NP induced inflammation (103, 107). Nonetheless, nanoparticles appear capable of inducing post-translational protein modification and autoantibodies. Several nanoparticles including, SiO2 NPs, cadmium NPs, ultrafine carbon black, and single-wall carbon nanotubes (SWCNT), elicited in vitro and/or in vivo increases in PAD activity and/or protein citrullination (64, 87, 108), and nickel nanowires stimulated anti-cyclic citrullinated (anti-CCP3) autoantibodies in female C57BL/6 mice (87). However, histological images do not show evidence of pulmonary TLSs following SWCNT exposure (109) or at sites of protein citrullination (64). Thus, although ENPs appear capable of inducing features of experimental pre-clinical autoimmunity (Table 1), studies have yet to show that this can mature to autoimmune disease.
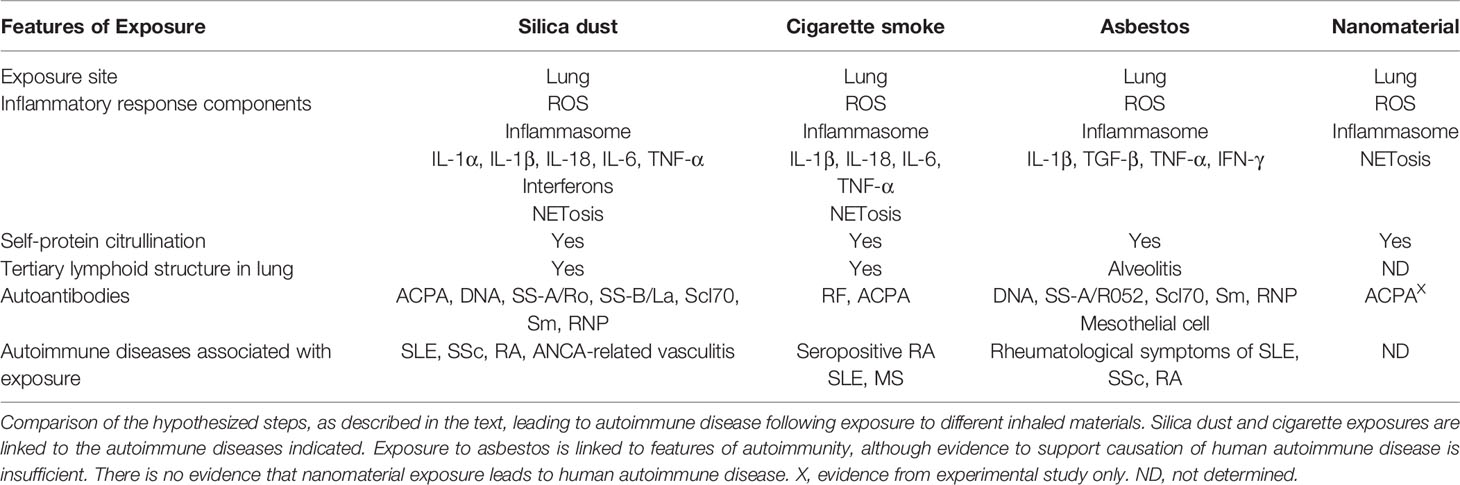
Table 1 Comparison of hypothesized steps leading to autoimmune disease following exposure to inhaled materials.
Discussion
Particulate exposures are among the initiating events most closely linked to human autoimmune diseases (2, 3), with the lungs as a major site of pathological and immunological events leading to clinical disease (26, 27, 33, 110). Such exposures can be protracted and involve particulates resistant to degradation, both of which contributes to the chronic pulmonary inflammation associated with these exposures (48, 79). A complex interplay between pathological processes, including, cellular toxicity, oxidative stress, tissue damage, persistent inflammation, post-translational modification of self-antigens, and the formation of TLSs, promote the generation of autoantibodies that contribute to development of autoimmunity. An unresolved issue is how expression of particulate-induced autoreactivity in the lung results in disease specific pathogenesis in distant organs (e.g. kidney in SLE or joints in RA). However, a recent hypothesis suggests that activated B cells act on preinflamamtory mesenchymal (PRIME) cells which then migrate to the joint in RA (111). The role of pulmonary mesenchymal cells in inflammation (112) supports the possibility of their interaction with B cells in lung TLSs.
Fibrous (asbestos) and non-fibrous (cigarette smoke, crystalline silica) fine (PM2.5) particulates have been linked to pre-clinical autoimmunity and autoimmune diseases (2, 76). However, there is little information to support a role for ultrafine (PM0.1) particulate matter in human autoimmune disease. This may reflect a difference in size which allows NPs to be cleared from the lung more readily. However, other aspects of pulmonary inflammation including the chronicity and/or resolution of the response, and the apparent absence of TLS, are likely to limit the severity of the adaptive autoimmune response and subsequent development of pathology in target organs other than the lung.
The pathological processes discussed above provide a foundation from which to determine the potential of other particles and fibers to induce autoimmunity. Such studies will provide a better understanding of the physical and chemical properties of particulate matter that lead to the induction and propagation of autoimmune diseases.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
Acknowledgments
This is manuscript MS#30012 from The Scripps Research Institute. The author acknowledges that only some of the important contributions of many investigators in the field are mentioned here because of space constraints. Where possible recent reviews, containing a more complete listing of applicable literature, have been cited. The cited publications from the author’s laboratory were made possible by National Institutes of Health grants ES029581, ES024485, ES022625, ES021464, and ES007511.
References
1. Germolec D, Kono DH, Pfau JC, Pollard KM. Animal models used to examine the role of the environment in the development of autoimmune disease: Findings from an NIEHS Expert Panel Workshop. J Autoimmun (2012) 39(4):285–93. doi: 10.1016/j.jaut.2012.05.020
2. Miller FW, Alfredsson L, Costenbader KH, Kamen DL, Nelson LM, Norris JM, et al. Epidemiology of environmental exposures and human autoimmune diseases: Findings from a National Institute of Environmental Health Sciences Expert Panel Workshop. J Autoimmun (2012) 39(4):259–71. doi: 10.1016/j.jaut.2012.05.002
3. Parks CG, Miller FW, Pollard KM, Selmi C, Germolec D, Joyce K, et al. Expert panel workshop consensus statement on the role of the environment in the development of autoimmune disease. Int J Mol Sci (2014) 15(8):14269–97. doi: 10.3390/ijms150814269
4. Pfau JC. Immunotoxicity of asbestos. Curr Opin Toxicol (2018) 10:1–7. doi: 10.1016/j.cotox.2017.11.005
5. Madl AK, Plummer LE, Carosino C, Pinkerton KE. Nanoparticles, lung injury, and the role of oxidant stress. Annu Rev Physiol (2014) 76:447–65. doi: 10.1146/annurev-physiol-030212-183735
6. Yang W, Wang L, Mettenbrink EM, DeAngelis PL, Wilhelm S. Nanoparticle Toxicology. Annu Rev Pharmacol Toxicol (2020). doi: 10.1146/annurev-pharmtox-032320-110338
7. Pollard KM, Christy JM, Cauvi DM, Kono DH. Environmental Xenobiotic Exposure and Autoimmunity. Curr Opin Toxicol (2018) 10:15–22. doi: 10.1016/j.cotox.2017.11.009
8. John G, Kohse K, Orasche J, Reda A, Schnelle-Kreis J, Zimmermann R, et al. The composition of cigarette smoke determines inflammatory cell recruitment to the lung in COPD mouse models. Clin Sci (Lond) (2014) 126(3):207–21. doi: 10.1042/CS20130117
9. Braun M, Koger F, Klingelhofer D, Muller R, Groneberg DA. Particulate Matter Emissions of Four Different Cigarette Types of One Popular Brand: Influence of Tobacco Strength and Additives. Int J Environ Res Public Health (2019) 16(2):263. doi: 10.3390/ijerph16020263
10. Brandsma CA, Van den Berge M, Hackett TL, Brusselle G, Timens W. Recent advances in chronic obstructive pulmonary disease pathogenesis: from disease mechanisms to precision medicine. J Pathol (2020) 250(5):624–35. doi: 10.1002/path.5364
11. Ishikawa Y, Terao C. The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review. Cells (2020) 9(2)475. doi: 10.3390/cells9020475
12. Bowes J, Barton A. Recent advances in the genetics of RA susceptibility. Rheumatol (Oxford) (2008) 47(4):399–402. doi: 10.1093/rheumatology/ken005
13. Lee YH, Bae SC, Song GG. Gene-environmental interaction between smoking and shared epitope on the development of anti-cyclic citrullinated peptide antibodies in rheumatoid arthritis: a meta-analysis. Int J Rheum Dis (2014) 17(5):528–35. doi: 10.1111/1756-185X.12307
14. Kant N, Muller R, Braun M, Gerber A, Groneberg D. Particulate Matter in Second-Hand Smoke Emitted from Different Cigarette Sizes and Types of the Brand Vogue Mainly Smoked by Women. Int J Environ Res Public Health (2016) 13(8):479. doi: 10.3390/ijerph13080799
15. Pauly JL, Smith LA, Rickert MH, Hutson A, Paszkiewicz GM. Review: Is lung inflammation associated with microbes and microbial toxins in cigarette tobacco smoke? Immunol Res (2010) 46(1-3):127–36. doi: 10.1007/s12026-009-8117-6
16. Hikichi M, Mizumura K, Maruoka S, Gon Y. Pathogenesis of chronic obstructive pulmonary disease (COPD) induced by cigarette smoke. J Thorac Dis (2019) 11(Suppl 17):S2129–S40. doi: 10.21037/jtd.2019.10.43
17. Grabcanovic-Musija F, Obermayer A, Stoiber W, Krautgartner WD, Steinbacher P, Winterberg N, et al. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir Res (2015) 16:59. doi: 10.1186/s12931-015-0221-7
18. Shan M, Cheng HF, Song LZ, Roberts L, Green L, Hacken-Bitar J, et al. Lung myeloid dendritic cells coordinately induce TH1 and TH17 responses in human emphysema. Sci Trans Med (2009) 1(4):4ra10. doi: 10.1126/scitranlsmed.3000154
19. Lee SH, Goswami S, Grudo A, Song LZ, Bandi V, Goodnight-White S, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med (2007) 13(5):567–9. doi: 10.1038/nm1583
20. Pipi E, Nayar S, Gardner DH, Colafrancesco S, Smith C, Barone F. Tertiary Lymphoid Structures: Autoimmunity Goes Local. Front Immunol (2018) 9:1952. doi: 10.3389/fimmu.2018.01952
21. Polverino F, Seys LJ, Bracke KR, Owen CA. B cells in chronic obstructive pulmonary disease: moving to center stage. Am J Physiol Lung Cell Mol Physiol (2016) 311(4):L687–L95. doi: 10.1152/ajplung.00304.2016
22. Reynisdottir G, Karimi R, Joshua V, Olsen H, Hensvold AH, Harju A, et al. Structural changes and antibody enrichment in the lungs are early features of anti-citrullinated protein antibody-positive rheumatoid arthritis. Arthritis Rheumatol (2014) 66(1):31–9. doi: 10.1002/art.38201
23. Wen L, Krauss-Etschmann S, Petersen F, Yu X. Autoantibodies in Chronic Obstructive Pulmonary Disease. Front Immunol (2018) 9:66. doi: 10.3389/fimmu.2018.00066
24. Byrne R, Todd I, Tighe PJ, Fairclough LC. Autoantibodies in chronic obstructive pulmonary disease: A systematic review. Immunol Lett (2019) 214:8–15. doi: 10.1016/j.imlet.2019.08.007
25. Sparks JA, Karlson EW. The Roles of Cigarette Smoking and the Lung in the Transitions Between Phases of Preclinical Rheumatoid Arthritis. Curr Rheumatol Rep (2016) 18(3):15. doi: 10.1007/s11926-016-0563-2
26. Friedlander HM, Ford JA, Zaccardelli A, Terrio AV, Cho MH, Sparks JA. Obstructive lung diseases and risk of rheumatoid arthritis. Expert Rev Clin Immunol (2020) 16(1):37–50. doi: 10.1080/1744666X.2019.1698293
27. Pentony P, Duquenne L, Dutton K, Mankia K, Gul H, Vital E, et al. The initiation of autoimmunity at epithelial surfaces: a focus on rheumatoid arthritis and systemic lupus erythematosus. Discov Med (2017) 24(133):191–200.
28. Morissette MC, Jobse BN, Thayaparan D, Nikota JK, Shen P, Labiris NR, et al. Persistence of pulmonary tertiary lymphoid tissues and anti-nuclear antibodies following cessation of cigarette smoke exposure. Respir Res (2014) 15:49. doi: 10.1186/1465-9921-15-49
29. Gergianaki I, Tsiligianni I. Chronic obstructive pulmonary disease and rheumatic diseases: A systematic review on a neglected comorbidity. J Comorb (2019) 9:1–01 doi: 10.1177/2235042X18820209
30. Spagnolo P, Lee JS, Sverzellati N, Rossi G, Cottin V. The Lung in Rheumatoid Arthritis: Focus on Interstitial Lung Disease. Arthritis Rheumatol (2018) 70(10):1544–54. doi: 10.1002/art.40574
31. Ford JA, Liu X, Chu SH, Lu B, Cho MH, Silverman EK, et al. Asthma, Chronic Obstructive Pulmonary Disease, and Subsequent Risk for Incident Rheumatoid Arthritis Among Women: A Prospective Cohort Study. Arthritis Rheumatol (2020) 72(5):704–13. doi: 10.1002/art.41194
32. Zaccardelli A, Liu X, Ford JA, Cui J, Lu B, Chu SH, et al. Elevated anti-citrullinated protein antibodies prior to rheumatoid arthritis diagnosis and risks for chronic obstructive pulmonary disease or asthma. Arthritis Care Res (Hoboken) (2020). doi: 10.1002/acr.24140
33. Prisco LC, Martin LW, Sparks JA. Inhalants other than personal cigarette smoking and risk for developing rheumatoid arthritis. Curr Opin Rheumatol (2020) 32(3):279–88. doi: 10.1097/BOR.0000000000000705
34. Barnes H, Goh NSL, Leong TL, Hoy R. Silica-associated lung disease: An old-world exposure in modern industries. Respirology (2019) 24(12):1165–75. doi: 10.1111/resp.13695
35. Pollard KM. Silica, Silicosis, and Autoimmunity. Front Immunol (2016) 7:97. doi: 10.3389/fimmu.2016.00097
36. Leung CC, Yu IT, Chen W. Silicosis. Lancet (2012) 379(9830):2008–18. doi: 10.1016/S0140-6736(12)60235-9
37. Steenland K, Ward E. Silica: a lung carcinogen. CA Cancer J Clin (2014) 64(1):63–9. doi: 10.3322/caac.21214
38. Turner MT, Samuel SR, Silverstone EJ, Yates DH. Silica Exposure and Connective Tissue Disease: An Under-Recognised Association in Three Australian Artificial Stone Workers. Am J Respir Crit Care Med (2020) 201(3):378–80 doi: 10.1164/rccm.201905-1057LE
39. Fan Y, Xu W, Wang Y, Wang Y, Yu S, Ye Q. Association of occupational dust exposure with combined chronic obstructive pulmonary disease and pneumoconiosis: a cross-sectional study in China. BMJ Open (2020) 10(9):e038874. doi: 10.1136/bmjopen-2020-038874
40. Yahya A, Bengtsson C, Larsson P, Too CL, Mustafa AN, Abdullah NA, et al. Silica exposure is associated with an increased risk of developing ACPA-positive rheumatoid arthritis in an Asian population: evidence from the Malaysian MyEIRA case-control study. Mod Rheumatol (2013). doi: 10.1007/s10165-013-0890-3
41. Stolt P, Yahya A, Bengtsson C, Kallberg H, Ronnelid J, Lundberg I, et al. Silica exposure among male current smokers is associated with a high risk of developing ACPA-positive rheumatoid arthritis. Ann Rheum Dis (2010) 69(6):1072–6. doi: 10.1136/ard.2009.114694
42. Chubb LG, Cauda EG. Characterizing Particle Size Distributions of Crystalline Silica in Gold Mine Dust. Aerosol Air Qual Res (2017) 17(1):24–33. doi: 10.4209/aaqr.2016.05.0179
43. Pavan C, Fubini B. Unveiling the Variability of “Quartz Hazard” in Light of Recent Toxicological Findings. Chem Res Toxicol (2017) 30(1):469–85. doi: 10.1021/acs.chemrestox.6b00409
44. Kawasaki H. A mechanistic review of silica-induced inhalation toxicity. Inhal Toxicol (2015) 27(8):363–77. doi: 10.3109/08958378.2015.1066905
45. Pavan C, Delle Piane M, Gullo M, Filippi F, Fubini B, Hoet P, et al. The puzzling issue of silica toxicity: are silanols bridging the gaps between surface states and pathogenicity? Part Fibre Toxicol (2019) 16(1):32. doi: 10.1186/s12989-019-0315-3
46. Lopes-Pacheco M, Bandeira E, Morales MM. Cell-Based Therapy for Silicosis. Stem Cells Int (2016) 2016:5091838. doi: 10.1155/2016/5091838
47. Fubini B, Hubbard A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic Biol Med (2003) 34(12):1507–16. doi: 10.1016/S0891-5849(03)00149-7
48. Franklin BS, Mangan MS, Latz E. Crystal Formation in Inflammation. Annu Rev Immunol (2016) 34:173–202. doi: 10.1146/annurev-immunol-041015-055539
49. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med (2019) 25(12):1822–32. doi: 10.1038/s41591-019-0675-0
50. Rabolli V, Badissi AA, Devosse R, Uwambayinema F, Yakoub Y, Palmai-Pallag M, et al. The alarmin IL-1alpha is a master cytokine in acute lung inflammation induced by silica micro- and nanoparticles. Part Fibre Toxicol (2014) 11:69. doi: 10.1186/s12989-014-0069-x
51. Rider P, Voronov E, Dinarello CA, Apte RN, Cohen I. Alarmins: Feel the Stress. J Immunol (2017) 198(4):1395–402. doi: 10.4049/jimmunol.1601342
52. Borthwick LA. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin Immunopathol (2016) 38(4):517–34. doi: 10.1007/s00281-016-0559-z
53. Sayan M, Mossman BT. The NLRP3 inflammasome in pathogenic particle and fibre-associated lung inflammation and diseases. Part Fibre Toxicol (2016) 13(1):51. doi: 10.1186/s12989-016-0162-4
54. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol (2008) 9(8):847–56. doi: 10.1038/ni.1631
55. Hoy RF, Chambers DC. Silica-related diseases in the modern world. Allergy (2020) 75(11):2805–17 doi: 10.1111/all.14202
56. Caplan A. Certain unusual radiological appearances in the chest of coal-miners suffering from rheumatoid arthritis. Thorax (1953) 8(1):29–37. doi: 10.1136/thx.8.1.29
57. Shtraichman O, Blanc PD, Ollech JE, Fridel L, Fuks L, Fireman E, et al. Outbreak of autoimmune disease in silicosis linked to artificial stone. Occup Med (Lond) (2015) 65(6):444–50. doi: 10.1093/occmed/kqv073
58. Huaux F. Emerging Role of Immunosuppression in Diseases Induced by Micro- and Nano-Particles: Time to Revisit the Exclusive Inflammatory Scenario. Front Immunol (2018) 9:2364. doi: 10.3389/fimmu.2018.02364
59. Bates MA, Brandenberger C, Langohr I, Kumagai K, Harkema JR, Holian A, et al. Silica Triggers Inflammation and Ectopic Lymphoid Neogenesis in the Lungs in Parallel with Accelerated Onset of Systemic Autoimmunity and Glomerulonephritis in the Lupus-Prone NZBWF1 Mouse. PloS One (2015) 10(5):e0125481. doi: 10.1371/journal.pone.0125481
60. Foster MH, Ord JR, Zhao EJ, Birukova A, Fee L, Korte FM, et al. Silica Exposure Differentially Modulates Autoimmunity in Lupus Strains and Autoantibody Transgenic Mice. Front Immunol (2019) 10:2336. doi: 10.3389/fimmu.2019.02336
61. Bates MA, Akbari P, Gilley KN, Wagner JG, Li N, Kopec AK, et al. Dietary Docosahexaenoic Acid Prevents Silica-Induced Development of Pulmonary Ectopic Germinal Centers and Glomerulonephritis in the Lupus-Prone NZBWF1 Mouse. Front Immunol (2018) 9:2002. doi: 10.3389/fimmu.2018.02002
62. Conrad K, Mehlhorn J. Diagnostic and prognostic relevance of autoantibodies in uranium miners. Int Arch Allergy Immunol (2000) 123(1):77–91. doi: 10.1159/000024426
63. Mayeux JM, Escalante GM, Christy JM, Pawar RD, Kono DH, Pollard KM. Silicosis and Silica-Induced Autoimmunity in the Diversity Outbred Mouse. Front Immunol (2018) 9:874. doi: 10.3389/fimmu.2018.00874
64. Mohamed BM, Verma NK, Davies AM, McGowan A, Crosbie-Staunton K, Prina-Mello A, et al. Citrullination of proteins: a common post-translational modification pathway induced by different nanoparticles in vitro and in vivo. Nanomedicine (Lond) (2012) 7(8):1181–95. doi: 10.2217/nnm.11.177
65. Li Y, Cao X, Liu Y, Zhao Y, Herrmann M. Neutrophil Extracellular Traps Formation and Aggregation Orchestrate Induction and Resolution of Sterile Crystal-Mediated Inflammation. Front Immunol (2018) 9:1559. doi: 10.3389/fimmu.2018.01559
66. Wong SL, Wagner DD. Peptidylarginine deiminase 4: a nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J (2018) 32:6358–70 doi: 10.1096/fj.201800691R
67. Sporn TA. Mineralogy of asbestos. Recent Results Cancer Res (2011) 189:1–11. doi: 10.1007/978-3-642-10862-4_1
68. Solbes E, Harper RW. Biological responses to asbestos inhalation and pathogenesis of asbestos-related benign and malignant disease. J Invest Med (2018) 66(4):721–7. doi: 10.1136/jim-2017-000628
69. Aust AE, Cook PM, Dodson RF. Morphological and chemical mechanisms of elongated mineral particle toxicities. J Toxicol Environ Health B Crit Rev (2011) 14(1-4):40–75. doi: 10.1080/10937404.2011.556046
70. Dodson RF, Atkinson MA, Levin JL. Asbestos fiber length as related to potential pathogenicity: a critical review. Am J Ind Med (2003) 44(3):291–7. doi: 10.1002/ajim.10263
71. Barlow CA, Grespin M, Best EA. Asbestos fiber length and its relation to disease risk. Inhal Toxicol (2017) 29(12-14):541–54. doi: 10.1080/08958378.2018.1435756
72. Ferro A, Zebedeo CN, Davis C, Ng KW, Pfau JC. Amphibole, but not chrysotile, asbestos induces anti-nuclear autoantibodies and IL-17 in C57BL/6 mice. J Immunotoxicol (2014) 11(3):283–90. doi: 10.3109/1547691X.2013.847510
73. Marchand LS, St-Hilaire S, Putnam EA, Serve KM, Pfau JC. Mesothelial cell and anti-nuclear autoantibodies associated with pleural abnormalities in an asbestos exposed population of Libby MT. Toxicol Lett (2012) 208(2):168–73. doi: 10.1016/j.toxlet.2011.10.024
74. Reid A, Franklin P, de Klerk N, Creaney J, Brims F, Musk B, et al. Autoimmune antibodies and asbestos exposure: Evidence from Wittenoom, Western Australia. Am J Ind Med (2018) 61(7):615–20. doi: 10.1002/ajim.22863
75. Ledda C, Caltabiano R, Loreto C, Cina D, Senia P, Musumeci A, et al. Prevalence of anti-nuclear autoantibodies in subjects exposed to natural asbestiform fibers: a cross-sectional study. J Immunotoxicol (2018) 15(1):24–8. doi: 10.1080/1547691X.2017.1415398
76. Diegel R, Black B, Pfau JC, McNew T, Noonan C, Flores R. Case series: rheumatological manifestations attributed to exposure to Libby Asbestiform Amphiboles. J Toxicol Environ Health A (2018) 81(15):734–47. doi: 10.1080/15287394.2018.1485124
77. Noonan CW, Pfau JC, Larson TC, Spence MR. Nested case-control study of autoimmune disease in an asbestos-exposed population. Environ Health Perspect (2006) 114(8):1243–7. doi: 10.1289/ehp.9203
78. Larson TC, Williamson L, Antao VC. Follow-Up of the Libby, Montana Screening Cohort: A 17-Year Mortality Study. J Occup Environ Med (2020) 62(1):e1–6. doi: 10.1097/JOM.0000000000001760
79. Wong J, Magun BE, Wood LJ. Lung inflammation caused by inhaled toxicants: a review. Int J Chron Obstruct Pulmon Dis (2016) 11:1391–401. doi: 10.2147/COPD.S106009
80. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science (2008) 320(5876):674–7. doi: 10.1126/science.1156995
81. Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A (2008) 105(26):9035–40. doi: 10.1073/pnas.0803933105
82. Liu G, Cheresh P, Kamp DW. Molecular basis of asbestos-induced lung disease. Annu Rev Pathol (2013) 8:161–87. doi: 10.1146/annurev-pathol-020712-163942
83. Pailes WH, Judy DJ, Resnick H, Castranova V. Relative effects of asbestos and wollastonite on alveolar macrophages. J Toxicol Environ Health (1984) 14(4):497–510. doi: 10.1080/15287398409530601
84. Rom WN, Travis WD. Lymphocyte-macrophage alveolitis in nonsmoking individuals occupationally exposed to asbestos. Chest (1992) 101(3):779–86. doi: 10.1378/chest.101.3.779
85. Pfau JC, McNew T, Hanley K, Swan L, Black B. Autoimmune markers for progression of Libby amphibole lamellar pleural thickening. Inhal Toxicol (2019) 31(11-12):409–19. doi: 10.1080/08958378.2019.1699616
86. Pfau JC, Barbour C, Black B, Serve KM, Fritzler MJ. Analysis of autoantibody profiles in two asbestiform fiber exposure cohorts. J Toxicol Environ Health A (2018) 81(19):1015–27. doi: 10.1080/15287394.2018.1512432
87. Mohamed BM, Boyle NT, Schinwald A, Murer B, Ward R, Mahfoud OK, et al. Induction of protein citrullination and auto-antibodies production in murine exposed to nickel nanomaterials. Sci Rep (2018) 8(1):679. doi: 10.1038/s41598-017-19068-1
88. Aberle T, Bourn RL, Munroe ME, Chen H, Roberts VC, Guthridge JM, et al. Clinical and serological features distinguish patients with incomplete lupus classification from systemic lupus erythematosus patients and controls. Arthritis Care Res (Hoboken) (2017) 69(12):1780–8 doi: 10.1002/acr.23201
89. Wither J, Johnson SR, Liu T, Noamani B, Bonilla D, Lisnevskaia L, et al. Presence of an interferon signature in individuals who are anti-nuclear antibody positive lacking a systemic autoimmune rheumatic disease diagnosis. Arthritis Res Ther (2017) 19(1):41. doi: 10.1186/s13075-017-1243-y
90. Xia T, Li N, Nel AE. Potential health impact of nanoparticles. Annu Rev Public Health (2009) 30:137–50. doi: 10.1146/annurev.publhealth.031308.100155
91. Prosperi D, Colombo M, Zanoni I, Granucci F. Drug nanocarriers to treat autoimmunity and chronic inflammatory diseases. Semin Immunol (2017) 34:61–7. doi: 10.1016/j.smim.2017.08.010
92. Singha S, Shao K, Ellestad KK, Yang Y, Santamaria P. Nanoparticles for Immune Stimulation Against Infection, Cancer, and Autoimmunity. ACS Nano (2018) 12(11):10621–35. doi: 10.1021/acsnano.8b05950
93. Kelly HG, Kent SJ, Wheatley AK. Immunological basis for enhanced immunity of nanoparticle vaccines. Expert Rev Vaccines (2019) 18(3):269–80. doi: 10.1080/14760584.2019.1578216
94. Serra P, Santamaria P. Nanoparticle-based approaches to immune tolerance for the treatment of autoimmune diseases. Eur J Immunol (2018) 48(5):751–6. doi: 10.1002/eji.201747059
95. Pearson RM, Podojil JR, Shea LD, King NJC, Miller SD, Getts DR. Overcoming challenges in treating autoimmuntity: Development of tolerogenic immune-modifying nanoparticles. Nanomedicine (2019) 18:282–91. doi: 10.1016/j.nano.2018.10.001
96. Boraschi D, Castellano LRC, Italiani P. Editorial: Interaction of Nanomaterials with the Immune System: Role in Nanosafety and Nanomedicine. Front Immunol (2017) 8:1688. doi: 10.3389/fimmu.2017.01688
97. Leso V, Fontana L, Iavicoli I. Nanomaterial exposure and sterile inflammatory reactions. Toxicol Appl Pharmacol (2018) 355:80–92. doi: 10.1016/j.taap.2018.06.021
98. Wu T, Tang M. Review of the effects of manufactured nanoparticles on mammalian target organs. J Appl Toxicol (2018) 38(1):25–40. doi: 10.1002/jat.3499
99. Pallardy MJ, Turbica I, Biola-Vidamment A. Why the Immune System Should Be Concerned by Nanomaterials? Front Immunol (2017) 8:544. doi: 10.3389/fimmu.2017.00544
100. Leikauf GD, Kim SH, Jang AS. Mechanisms of ultrafine particle-induced respiratory health effects. Exp Mol Med (2020) 52(3):329–37. doi: 10.1038/s12276-020-0394-0
101. You R, Lu W, Shan M, Berlin JM, Samuel EL, Marcano DC, et al. Nanoparticulate carbon black in cigarette smoke induces DNA cleavage and Th17-mediated emphysema. Elife (2015) 4:e09623. doi: 10.7554/eLife.09623
102. Ma Q. Polarization of Immune Cells in the Pathologic Response to Inhaled Particulates. Front Immunol (2020) 11:1060. doi: 10.3389/fimmu.2020.01060
103. Munoz LE, Bilyy R, Biermann MH, Kienhofer D, Maueroder C, Hahn J, et al. Nanoparticles size-dependently initiate self-limiting NETosis-driven inflammation. Proc Natl Acad Sci USA (2016) 113(40):E5856–E65. doi: 10.1073/pnas.1602230113
104. Biermann MH, Podolska MJ, Knopf J, Reinwald C, Weidner D, Maueroder C, et al. Oxidative Burst-Dependent NETosis Is Implicated in the Resolution of Necrosis-Associated Sterile Inflammation. Front Immunol (2016) 7:557. doi: 10.3389/fimmu.2016.00557
105. Yang H, Marion TN, Liu Y, Zhang L, Cao X, Hu H, et al. Nanomaterial Exposure Induced Neutrophil Extracellular Traps: A New Target in Inflammation and Innate Immunity. J Immunol Res (2019) 2019:3560180. doi: 10.1155/2019/3560180
106. Chezeau L, Kohlstaedt LA, Le Faou A, Cosnier F, Rihn B, Gate L. Proteomic analysis of bronchoalveolar lavage fluid in rat exposed to TiO2 nanostructured aerosol by inhalation. J Proteomics (2019) 207:103451. doi: 10.1016/j.jprot.2019.103451
107. Napierska D, Thomassen LC, Lison D, Martens JA, Hoet PH. The nanosilica hazard: another variable entity. Part Fibre Toxicol (2010) 7(1):39. doi: 10.1186/1743-8977-7-39
108. Hutchinson D, Muller J, McCarthy JE, Gun’ko YK, Verma NK, Bi X, et al. Cadmium nanoparticles citrullinate cytokeratins within lung epithelial cells: cadmium as a potential cause of citrullination in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis (2018) 13:441–9. doi: 10.2147/COPD.S152028
109. Lam CW, James JT, McCluskey R, Hunter RL. Pulmonary toxicity of single-wall carbon nanotubes in mice 7 and 90 days after intratracheal instillation. Toxicol Sci (2004) 77(1):126–34. doi: 10.1093/toxsci/kfg243
110. Holers VM, Demoruelle MK, Kuhn KA, Buckner JH, Robinson WH, Okamoto Y, et al. Rheumatoid arthritis and the mucosal origins hypothesis: protection turns to destruction. Nat Rev Rheumatol (2018) 14(9):542–57. doi: 10.1038/s41584-018-0070-0
111. Orange DE, Yao V, Sawicka K, Fak J, Frank MO, Parveen S, et al. RNA Identification of PRIME Cells Predicting Rheumatoid Arthritis Flares. N Engl J Med (2020) 383(3):218–28. doi: 10.1056/NEJMoa2004114
Keywords: lung, autoimmunity, particle, nanoparticle, fiber
Citation: Pollard KM (2020) Perspective: The Lung, Particles, Fibers, Nanomaterials, and Autoimmunity. Front. Immunol. 11:587136. doi: 10.3389/fimmu.2020.587136
Received: 24 July 2020; Accepted: 17 November 2020;
Published: 18 December 2020.
Edited by:
Heiko Mühl, Goethe University Frankfurt, GermanyReviewed by:
Farrah Kheradmand, Baylor College of Medicine, United StatesMiquéias Lopes-Pacheco, University of Lisbon, Portugal
Sabina Halappanavar, Health Canada, Canada
Copyright © 2020 Pollard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: K. Michael Pollard, bXBvbGxhcmRAc2NyaXBwcy5lZHU=