 Llorenç Rovirosa1
Llorenç Rovirosa1 Biola M. Javierre
Biola M. Javierre- 13D Chromatin Organization Group, Josep Carreras Leukaemia Research Institute (IJC), Germans Trias i Pujol, Badalona, Spain
- 2Institute for Health Science Research Germans Trias i Pujol (IGTP), Badalona, Spain
Associations between blood cancer and genetic predisposition, including both inherited variants and acquired mutations and epimutations, have been well characterized. However, the majority of these variants affect noncoding regions, making their mechanisms difficult to hypothesize and hindering the translation of these insights into patient benefits. Fueled by unprecedented progress in next-generation sequencing and computational integrative analysis, studies have started applying combinations of epigenetic, genome architecture, and functional assays to bridge the gap between noncoding variants and blood cancer. These complementary tools have not only allowed us to understand the potential malignant role of these variants but also to differentiate key variants, cell-types, and conditions from misleading ones. Here, we briefly review recent studies that have provided fundamental insights into our understanding of how noncoding mutations at enhancers predispose and promote blood malignancies in the context of spatial genome architecture.
Introduction: Why We Need to Study Enhancers and Spatial Genome Organization to Understand Blood Cancer
Noncoding regions of the genome, which comprise more than 98% of the genome, have historically been overlooked. However, more than 95% of the risk variants associated with genetically complex diseases, such as blood cancer, map at noncoding regions and remain unexplored due to the lack of an obvious disease mechanism (1). In addition, the oncogenic potential of somatically acquired noncoding mutations has become increasingly evident (2). The mutational rate of the noncoding genome is significantly higher than its coding counterpart, although the selective pressure is weaker unless the mutations have a functional effect that confers an advantage to cell survival (3). For this reason, in the search for noncoding mutations it is important to find and focus on functional regions that impact gene regulation such as enhancers.
Enhancers are frequent targets of genetic and epigenetic alterations in many diseases including blood cancer (1, 4–6). The human genome contains around one million enhancers, many of which are cell-type or stimulus-specific (7). They modulate the activation of promoters and fine-tune transcription over large genomic distances, independent of sequence orientation and position (8). It is now generally accepted that long-range enhancers are brought into close spatial proximity with the promoters they regulate. This proximity is determined by the DNA folding into loops, which are cell-type and stimulus-specific (6, 9–11).
Though enhancers have been relatively easy to identify—since they harbor a high density of DNA motifs recognized by transcription factors (12–14), have specific histone modification and co-factor binding profiles (13, 15–17), and produce enhancer RNAs (18, 19) —their target genes are harder to pinpoint (7, 20). Enhancers can be up to a few megabases away from their targets, often jumping over several intervening genes or even being located in the intron of a non-target gene (21). In addition, a given gene is frequently controlled by more than one enhancer, and an enhancer can control more than one gene (22, 23) (Figure 1). Enhancers cannot simply be assigned to the nearest gene, and most of their target genes in each cell-type and stimulus condition remain unknown. For this reason, during the last years chromatin conformation capture (3C) techniques, such as Hi-C (24), a technique designed to identify the entire ensemble of chromosomal interactions within a cell population, have emerged to support that spatio-temporal chromatin organization not only plays a key role in the function of enhancers, but will also help scientists identify target genes, thus helping us determine the disease mechanism of enhancer mutations.
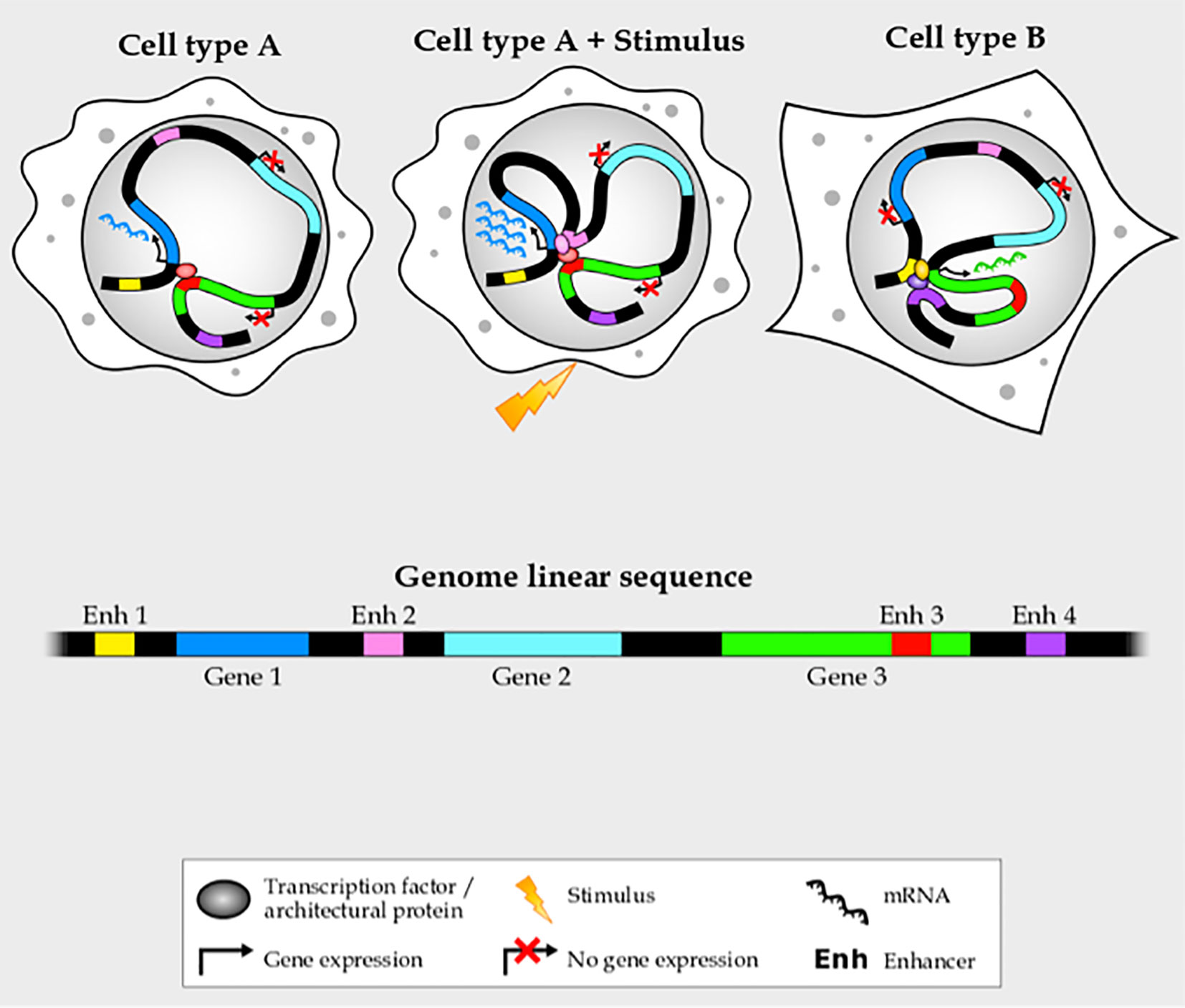
Figure 1 Enhancer–promoter interactions are complex and cell- and stimulus-specific.
In the following sections, we will review the current knowledge on the effect of noncoding mutations on enhancers and genomic 3D structure in blood malignancies. We will highlight the cognate clinical implications, not only in better understanding oncogenesis, but also in the identification of potential new biomarkers and therapeutic strategies to improve disease diagnosis, monitoring and treatment.
The Three-Dimensional Context of the Genome Plays a Major Role in the Development and Progression of Blood Cancer
Chromatin interactions are crucial for cellular health, and errors in these interactions give rise to a broad range of diseases, including blood cancers (25–27). Cancer-associated alterations in chromatin architecture increase proliferation and decrease differentiation capacity by blocking cell differentiation, altering the expression of oncogenic or tumor suppressor genes, and/or creating unnatural cell states with deregulated expression of key developmental genes. Genomic regions brought into close proximity by the chromatin architecture have a higher frequency of producing translocations, thus explaining the frequency of recurrent pathological translocations such as BCR-ABL and MYC-IGH (26, 27). In addition to chromosome-scale alterations, three-dimensional chromatin organization works on a smaller scale to impact gene regulation by rewiring physical interactions between gene promoters and regulatory elements. This important spatial organization is often altered in blood cancer through the genetic alteration or altered binding of the proteins involved in establishing and maintaining chromatin loops.
Architectural proteins, including CTCF and cohesins, play an important role in establishing and maintaining chromatin loops (28, 29), facilitating communication of gene promoters with some regulatory elements while reducing contacts with others (30–32). Cohesin complex mutations, which impair hematopoietic progenitor differentiation (33–35), occur in ~13% of acute myeloid leukemia (AML) patients (36–38). CTCF generally only binds non-methylated DNA sequences (39–42), so cancer-related methylation changes can impact genomic architecture broadly or at specific loci, although in some sites CTCF binding remains independent of DNA methylation suggesting that methylation may be one of many synergistic factors impacting transcription factor binding (43). A recent study by Kloetgen and colleagues characterized global alterations in genome architecture in T-cell precursor acute lymphoblastic leukemia (T-ALL) (44). The authors found that the lack of CTCF-mediated insulation at a specific locus allowed the cancer-driver gene MYC to come into direct contact with a distal super-enhancer, thus increasing this transcription factor’s expression, allowing MYC to turn on signal transduction pathways leading to cell growth and proliferation.
Interestingly, altered DNA methylation at enhancers in cancer is more closely related to changes in gene expression than at promoters (45). Hypomethylated enhancers bind transcription factors better than methylated ones (46–49), thus promoting gene expression and potentially affecting genome architecture. Transcription factors can also influence spatial chromatin organization, from ubiquitously expressed factors such as YY1 (50) to cell-type specific factors including GATA1, LBD1, HSPs, KLF4, LacI, MYOD, and OCT4 (51–56). In addition to mutating these proteins, genetic or epigenetic alterations can have profound effects on chromatin architecture by affecting the binding of these structural proteins (48, 57, 58).
Acquired mutations, regardless of whether they directly affect transcription, can alter spatial chromatin organization and chromatin states to ultimately promote cancer-specific transcriptional programs. This complex interplay is nicely deciphered by Yun and colleagues who used an allelic series of mutant mice to model normal, pre-malignant and AML states using the commonly co-occurring FLT3 and NPM1 mutations. Each mutation in isolation altered chromatin state, but together, they synergized to cause global alterations in spatial chromatin organization that produced a leukemia-associated transcriptional program (59), despite these mutations lacking direct influence on transcription or epigenetics. For this reason, the development of therapeutic approaches against mutations also needs to take into account cancer-associated genome-wide epigenetic and conformational landscapes. Interestingly, some anti-leukemic drugs partially reverse the altered topology of some genomic regions. For example THZ1, a covalent CDK7 inhibitor, has been shown to abrogate MYC-enhancer aberrant contacts (44) and deter growth in T-ALL cell lines (60), potentially accounting for the anti-tumoral effect of these small-molecule inhibitors.
Although we have just started understanding the role of spatial-temporal genome architecture in blood cancer, there is no doubt about the potential therapeutic opportunities it encompasses. In order to understand changes in genomic spatial architecture, we must first understand the causal changes in the underlying DNA, resulting from both inherited variants and somatic mutations.
Inherited Genetic Susceptibility to Blood Malignancy From the Three-Dimensional Perspective
Blood cancer is not entirely explained by acquired genomic rearrangements, amplifications, deletions or mutations. In fact, inherited genetic predisposition plays a key role (61–63). In fact, survival rates and phenotype differ between racial and ethnic groups (64–66). The effect of genetic background in blood malignancies has been elegantly demonstrated by a recent work from Young and colleagues in which they cross the Mll-AF9 knockin mouse with differing inbred strains (67). The authors show that genetic background not only impacts blood composition and survival rates, but also the type of malignancy, from ALL to AML or mixed phenotype acute leukemia.
Risk loci frequently span noncoding regions (6, 68) and, in fact, cluster at enhancers (1). The single-nucleotide polymorphisms (SNPs) or structural variants (SVs) can alter the binding of transcription factors or structural proteins to regulatory elements by modifying recognition motifs or modulating accessibility, thus altering the expression of their target genes (Figure 2). These inherited variants may either disrupt enhancer function, leading to reduced or lost target gene expression, or increase enhancer function, leading to increased target gene expression or even ectopic expression (5). In both cases, if the binding of structural proteins is affected, the enhancer–promoter interaction frequency shifts. However, if transcription factor binding is altered, chromatin looping can either be compromised or not.
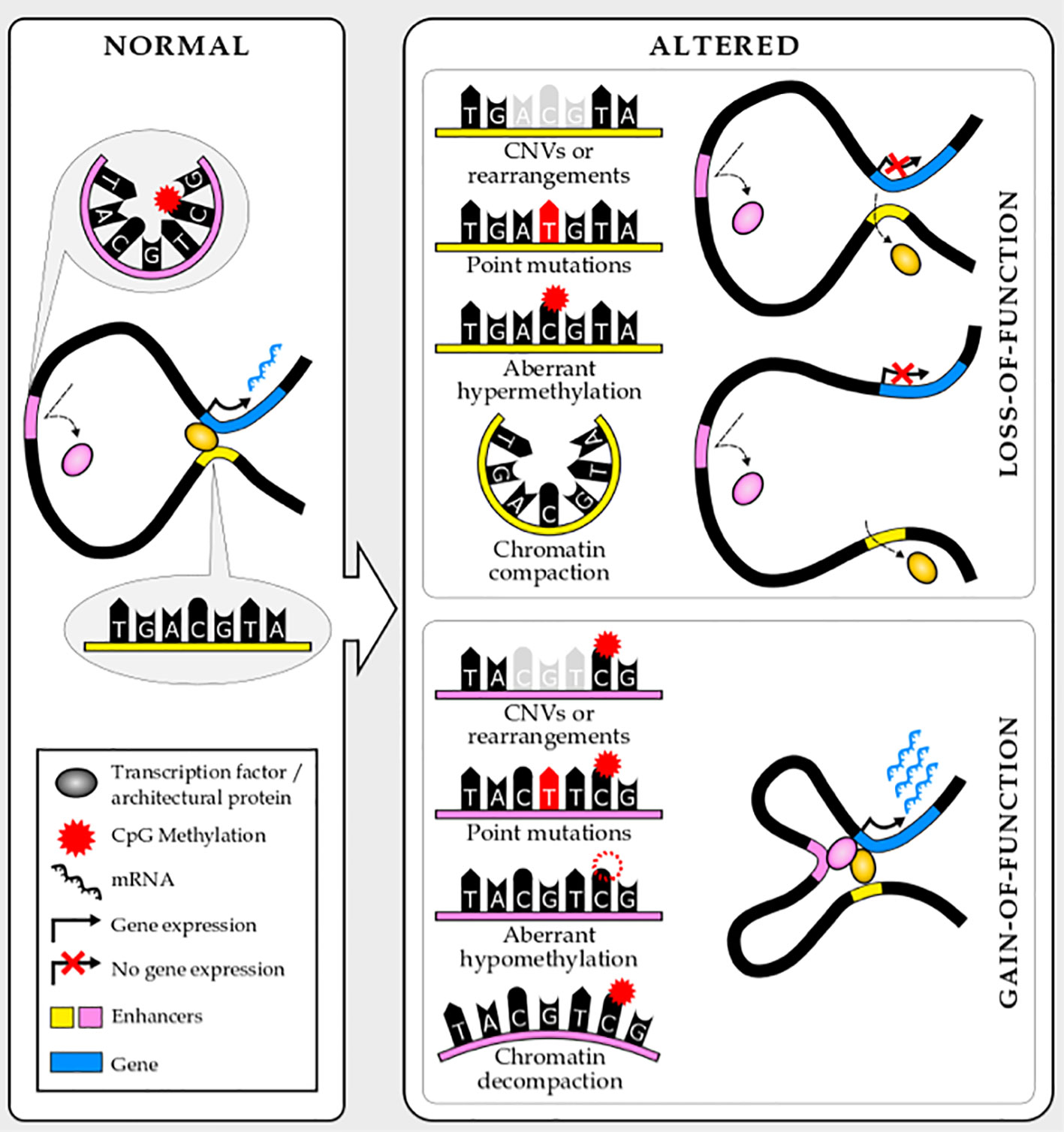
Figure 2 Noncoding mutations and epimutations lead to enhancer loss- or gain-of-function.
Demonstrating this, Speedy and colleagues found that noncoding genetic variants associated with chronic lymphocytic leukemia (CLL) predisposition map to CLL active chromatin, with evidence of that chromatin state being CLL-specific or differentially regulated in normal B-cell differentiation (69). The risk alleles disrupt SPI1, NFKB, and PAX5 binding motifs, or affect accessibility, suggesting a potential impact on enhancer activity. Using chromatin conformation data in parallel with expression data, they connected variants at enhancers with target gene promoters and observed that candidate genes are involved in B-cell biology including immune response, Wnt signaling and apoptosis. Although experimental validation is needed to completely understand the molecular mechanism by which CLL germline variants contribute to malignant transformation, this study underlines the value of epigenetics and genome architecture in interpreting genetic susceptibility.
The mechanistic insights acquired from inherited variants can be extrapolated to acquired mutations and epimutations. Indeed, germline risk variants co-segregate with acquired genomic abnormalities, although the interplay between these remains poorly understood (70, 71). A possible mechanism is that inherited variants alter the expression of pioneer transcription factors, which could synergize with the action of driver mutations to promote genome-wide gene deregulation and oncogenesis. Supporting this hypothesis, Yang and colleagues identified a germline variant at the GATA3 intron that is strongly associated with Philadelphia-like ALL (72). This enhancer gain-of-function mutation upregulates GATA3 expression, which in turn, reprograms spatial genome architecture and chromatin accessibility genome-wide. This reprogramming puts the oncogene CRLF2 in proximity to a distal enhancer, promoting CRLF2-mediated constitutive activation of the JAK-STAT pathway, which has been implicated in leukemogenesis (73). This variant is not enough to cause cancer on its own, but it sets the stage for a driver mutation to tip the scales. Not only does GATA3 overexpression facilitate enhancer hijacking by oncogenes, but the authors propose that it also causes chromosomal instability and translocations, due to GATA3 binding sites clustering near Philadelphia-like ALL translocation breakpoints.
Acquired Mutations During Blood Malignancy Transformation Frequently Target Noncoding Regulatory Elements
Similar to inherited genetic variants, acquired mutations commonly target enhancers, altering gene expression, in blood cancer (74). Mutations preferentially cluster at regulatory elements characteristic to the cell-type from which the tumor originates (75). These mutations may be enhancer gain-of-function, enhancer loss-of-function or enhancer hijacking. Of note, the last category can also be considered a gain-of-function and is frequently linked with major chromosomal rearrangements or insulator alteration (76, 77). Similar to mutations, epimutations such as DNA methylation alteration, also target enhancers (45), and in principle, can affect enhancers in similar ways (Figure 2). Silencers and other distal cis-regulatory elements can also be targeted by mutations. However, the narrow characterization of these elements and the incomplete knowledge of their mechanism of action limit their study. Nevertheless, breakthroughs in genomics and molecular biology will put into play these elements in cancer genomics very soon.
Many noncoding gain-of-function mutations, including copy number variation (duplications and deletions) and point mutations, have been associated with activating proto-oncogene transcription in blood cancer. For instance, Herranz and colleagues identified a long-range enhancer controlled by NOTCH1 that has recurrent chromosomal duplications in human T-ALL (78). This region is activated through a mechanism of nucleosome eviction mediated by GATA3 (79), it physically interacts with the MYC promoter 1.4 megabases away to induce MYC transcription, and it has a fundamental role in the homeostasis of immature T cells. This gain-of-function amplification drives MYC expression downstream of NOTCH1 in T-ALL and helps to connect MYC and Notch signaling in driving oncogenesis; a different mechanism but with the same effect as the previous mentioned study in which an impaired CTCF binding (due to reduced chromatin accessibility) leads to MYC enhancer hijacking (44). Regarding deletions, Liu and colleagues identified a recurrent deletion of a noncoding region in T-ALL using cis-X, a computational method for identifying regulatory noncoding alterations (80). This deletion destroys the nodal CTCF binding site that forms the boundary of two independent, insulated genomic neighborhoods. As a consequence, this structural variation allows the hijacking of an active enhancer from one neighborhood by the PRLR promoter located in the other neighborhood, leading to PRLR upregulation and JAK2 signaling activation. In the same paper, the authors also report a recurrent intronic point mutation that activates TAL1 oncogene transcription, a frequently mutated oncogene in T-ALL (81, 82). This point mutation creates a new active enhancer characterized by a de novo recognition motif for YY1. Interestingly, this transcription factor has structural properties similar to CTCF in bridging promoter–enhancer chromatin interactions (50), in addition to previous studies that show that ectopic expression of TAL1 can be also due to CTCF binding alterations that cause loss of TAD boundary insulation and subsequently enhancer hijacking (83, 84). Functional validation will be needed to better understand the complex interplay between noncoding genetic mutations, transactivation and three-dimensional chromatin organization to promote malignant transformation.
Noncoding copy number variation and point mutations are not exclusive to T-ALL. For instance, Cornish and colleagues identified recurrent mutations in B-cell lymphoma at cis-regulatory elements of naive B lymphocytes (85). Using structural data to connect cis-regulatory elements harboring structural and point mutations to promoters, they identified putative noncoding driver mutations. These mutations alter distal regulatory elements, leading to deregulation of target gene transcription. Intriguingly, they also observed that coding and noncoding mutations often converged on the same genes. For instance, MMP14 is a negative regulator of Notch signaling. It plays a key role in normal B cell differentiation, the development of diffuse large B-cell lymphoma and patient survival (86, 87). Distal cis-regulatory elements of MMP14 are frequently deleted in B-cell lymphoma, leading to MMP14 downregulation. On top of that, coding mutations frequently affect the MMP14 gene in several malignancies, often resulting in loss-of-function.
This convergence needs to be investigated in other contexts, for example, whether recurrent coding mutations in blood malignancies, such as in histone modifiers and chromatin remodeling factors, operate in parallel with noncoding mutations. Is there co-occurrence between the coding mutations of oncogenes and noncoding mutations at their regulatory elements in the same malignant cell or tumor type? If so, is this a synergy, an exclusion, or a redundant event? Could a cancer-related reduction of a given transcription factor level be accompanied by a reduction of its binding at regulatory elements due to mutations or epimutations at its recognition motifs? Or do tumoral cells use one or the other mechanism exclusively to silence a given transcription factor? Many questions remain open that the scientific community will have to address using the fast-growing number of methodological breakthroughs.
Noncoding loss-of-function mutations at enhancers also contribute to cancer pathology by silencing tumor-suppressor genes, although these are less well characterized. These noncoding mutations, similar to gain-of-function ones, involve different types of structural, copy number, and point mutations (Figure 2). A recent paper by Li and colleagues identified and validated more than two hundred tumor suppressive or oncogenic enhancers recurrently mutated in hematopoietic malignancies using enhancer CRISPR/dCas9 perturbation (activation or repression) (88). Interestingly, some of these reside in proximity to nuclear receptor–binding genomic regions, contributing to aberrant nuclear receptor signaling in blood malignancies. One example is two noncoding variants within the PER2 enhancer that have tumor suppressive properties in AML and are potentially controlled by the nuclear receptor program. PER2 controls circadian rhythm and it has been suggested as a tumor suppressor gene (89–91). This work elegantly addresses the major challenge of prioritizing noncoding mutations and assigning functional relevance to them. Approaches like this will be needed to translate the knowledge about the noncoding genome in blood cancer into patient benefits.
Future Perspectives
Even though compelling evidence for the contribution of noncoding mutations and epimutations to oncogenesis exists, there is still a long way to go before we can translate them into new strategies to characterize, treat and monitor blood cancer. To achieve this translational challenge, many considerations should be taken into account.
First, reliably identifying noncoding driver mutations from passenger ones remains a great endeavor due to sequencing and mapping artifacts, poorly understood mutational processes, and inaccurate estimation of the mutational background. To face these impediments, adequate statistical methods, larger datasets, higher sequence coverage, and longer and more accurate sequencing reads will be fundamental. In addition, the sequencing of normal tissues alongside malignant ones can help separate acquired from germline mutations and shed light on differentiating driver from passenger mutations.
Second, given the vastness of the noncoding genome, we need to restrict the search to relevant noncoding driver mutations. To this end, comparative genomic analysis, high-throughput in vitro reporter assays, and genome-wide histone modification profiling, coupled with chromatin accessibility and expression analysis, are indispensable. However, due to the highly dynamic, cell- and stimulus-specific nature of regulatory regions, it is critical to identify the right cell-type and to extend this descriptive interrogation not only to the cells of origin but also to the transformed cells.
Third, the unknown functional role of noncoding mutations imposes important limitations. To ascertain biological and mechanistic relevance, it is essential to integrate genetic and epigenetic profiling with genome conformation data and CRISPR-based functional validation. As previously described, many noncoding mutations map to enhancers, and these can exert their pathological function by altering the expression level of their target genes. However, connecting enhancers and target genes is not trivial, and due to cell-specificity and complexity, most of the associations remain unknown. Quantitative associations between noncoding variants and gene expression, and regulatory biochemical properties combined with sequencing-based chromatin conformation capture methods, such as Hi-C (24), have started providing some insights. However, reliable and reproducible identification of significant interactions between individual restriction fragments is not feasible unless Hi-C libraries are subjected to ultra-deep sequencing, which is not an economically viable solution for analyzing a comprehensive collection of cell-types or tumoral samples. To overcome it, the development of sequence-specific capturing approaches to enrich for promoter interactions and mutations in Hi-C libraries (6, 11, 69, 85), or other methods such as ChIA-PET (92) or HiChIP (93), is crucial. Nonetheless, all these methods need millions of cells, which hinders the analysis of rare cell populations such as hematopoietic stem cells or hematopoietic precursors, which are the origin cells of the majority of leukemias. Methodological breakthroughs allowing lower inputs will be fundamental in the incipient era of noncoding driver mutations.
In conclusion, the current methodological breakthroughs have positioned the scientific community in a perfect situation to explore the noncoding genome in the context of cancer. Cancer genomics is rapidly moving from a static, one-dimensional picture, to a time-dependent three-dimensional scenario to provide biological relevance of noncoding mutations at regulatory elements. We anticipate a very exciting time ahead, in which we will be fascinated by the power of noncoding mutations and epimutations in malignant transformation and the new clinical opportunities these genetic alterations will involve.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
LR is funded by AGAUR project number 2019FI-B00017 of the Catalan Government (Generalitat de Catalunya). AR-M is funded by the José Carreras Leukämie-Stiftung (08R/2019). BJ is funded by FEDER/Spanish Ministry of Science and Innovation (project number RTI2018-094788-A-I00), by La Caixa Banking Foundation Junior Leader project (LCF/BQ/PI19/11690001), by the José Carreras Leukämie-Stiftung (08R/2019), and by the European Hematology Association Advance Research Grant. The funder bodies were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to the authors of many relevant studies for not citing their work due to space limitations. We thank the members of the Javierre laboratory for helpful discussion and Christina Usher for her editing suggestions.
References
1. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Sci (80- ) (2012) 337(6099):1190–5. doi: 10.1126/science.1222794
2. Khurana E, Fu Y, Chakravarty D, Demichelis F, Rubin MA, Gerstein M. Role of non-coding sequence variants in cancer. Nat Rev Genet (2016) 17:93–108. doi: 10.1038/nrg.2015.17
3. Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat Genet (2014) 46(11):1160–5. doi: 10.1038/ng.3101
4. Farh KKH, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature (2015) 518(7539):337–43. doi: 10.1038/nature13835
5. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell (2013) 155(4):934. doi: 10.1016/j.cell.2013.09.053
6. Javierre BM, Sewitz S, Cairns J, Wingett SW, Várnai C, Thiecke MJ, et al. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell (2016) 167(5):1369–1384.e19. doi: 10.1016/j.cell.2016.09.037
7. Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al. An integrated encyclopedia of DNA elements in the human genome. Nature (2012) 489(7414):57–74. doi: 10.1038/nature11247
8. Banerji J, Rusconi S, Schaffner W. Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell (1981) 27(2 PART 1):299–308. doi: 10.1016/0092-8674(81)90413-X
9. Carter D, Chakalova L, Osborne CS, Dai YF, Fraser P. Long-range chromatin regulatory interactions in vivo. Nat Genet (2002) 32(4):623–6. doi: 10.1038/ng1051
10. Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell (2014) 159(7):1665–80. doi: 10.1016/j.cell.2014.11.021
11. Burren OS, Rubio García A, Javierre BM, Rainbow DB, Cairns J, Cooper NJ, et al. Chromosome contacts in activated T cells identify autoimmune disease candidate genes. Genome Biol (2017) 18(1):165. doi: 10.1186/s13059-017-1285-0
12. Zaret KS, Carroll JS. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev Genes Dev (2011) 25:2227–41. doi: 10.1101/gad.176826.111
13. Long HK, Prescott SL, Wysocka J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell Cell Press (2016) 167:1170–87. doi: 10.1016/j.cell.2016.09.018
14. Spitz F, Furlong EEM. Transcription factors: From enhancer binding to developmental control. Nat Rev Genet (2012) 13:613–26. doi: 10.1038/nrg3207
15. Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res (2011) 21(8):1273–83. doi: 10.1101/gr.122382.111
16. Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature (2011) 470(7333):279–85. doi: 10.1038/nature09692
17. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A (2010) 107(50):21931–6. doi: 10.1073/pnas.1016071107
18. Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature (2010) 465(7295):182–7. doi: 10.1038/nature09033
19. de Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, et al. A large fraction of extragenic RNA Pol II transcription sites overlap enhancers. PloS Biol (2010) 8(5), e1000384. doi: 10.1371/journal.pbio.1000384
20. Shen Y, Yue F, Mc Cleary DF, Ye Z, Edsall L, Kuan S, et al. A map of the cis-regulatory sequences in the mouse genome. Nature (2012) 488(7409):116–20. doi: 10.1038/nature11243
21. Amano T, Sagai T, Tanabe H, Mizushina Y, Nakazawa H, Shiroishi T. Chromosomal Dynamics at the Shh Locus: Limb Bud-Specific Differential Regulation of Competence and Active Transcription. Dev Cell (2009) 16(1):47–57. doi: 10.1016/j.devcel.2008.11.011
22. Perry MW, Boettiger AN, Levine M. Multiple enhancers ensure precision of gap gene-expression patterns in the Drosophila embryo. Proc Natl Acad Sci U S A (2011) 108(33):13570–5. doi: 10.1073/pnas.1109873108
23. Tasic B, Nabholz CE, Baldwin KK, Kim Y, Rueckert EH, Ribich SA, et al. Promoter choice determines splice site selection in protocadherin α and γ pre-mRNA splicing. Mol Cell (2002) 10(1):21–33. doi: 10.1016/S1097-2765(02)00578-6
24. Lieberman-Aiden E, Van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Sci (80- ) (2009) 326(5950):289–93. doi: 10.1126/science.1181369
25. Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet (2013) 45(10):1232–7. doi: 10.1038/ng.2731
26. Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell (2011) 147(1):95–106. doi: 10.1016/j.cell.2011.07.048
27. Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell (2011) 147(1):107–19. doi: 10.1016/j.cell.2011.07.049
28. Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep (2016) 15(9):2038–49. doi: 10.1016/j.celrep.2016.04.085
29. Sanborn AL, Rao SSP, Huang SC, Durand NC, Huntley MH, Jewett AI, et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A (2015) 112(47):E6456–65. doi: 10.1073/pnas.1518552112
30. Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, et al. Cohesin Loss Eliminates All Loop Domains. Cell (2017) 171(2):305–320.e24. doi: 10.1016/j.cell.2017.09.026
31. Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, et al. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell (2017) 169(5):930–944.e22. doi: 10.1016/j.cell.2017.05.004
32. Wutz G, Várnai C, Nagasaka K, Cisneros DA, Stocsits RR, Tang W, et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J (2017) Dec 1536(24):3573–99. doi: 10.15252/embj.201798004
33. Mazumdar C, Shen Y, Xavy S, Zhao F, Reinisch A, Li R, et al. Leukemia-Associated Cohesin Mutants Dominantly Enforce Stem Cell Programs and Impair Human Hematopoietic Progenitor Differentiation. Cell Stem Cell (2015) 17(6):675–88. doi: 10.1016/j.stem.2015.09.017
34. Viny AD, Ott CJ, Spitzer B, Rivas M, Meydan C, Papalexi E, et al. Dose-dependent role of the cohesin complex in normal and malignant hematopoiesis. J Exp Med (2015) 212(11):1819–32. doi: 10.1084/jem.20151317
35. Mullenders J, Aranda-Orgilles B, Lhoumaud P, Keller M, Pae J, Wang K, et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J Exp Med (2015) 212(11):1833–50. doi: 10.1084/jem.20151323
36. Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature (2012) 481(7382):506–10. doi: 10.1038/nature10738
37. Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell (2012) 150(2):264–78. doi: 10.1016/j.cell.2012.06.023
38. Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med (2012) 4(149):149ra118. doi: 10.1126/scitranslmed.3004315
39. Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature (2000) 405(6785):486–9. doi: 10.1038/35013106
40. Ong C-T, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet (2014) 15(4):234–46. doi: 10.1038/nrg3663
41. Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nat (2011) 480(7378):490–5. doi: 10.1038/nature10716
42. Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res (2012) 22(9):1680–8. doi: 10.1101/gr.136101.111
43. Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K, et al. Role of DNA Methylation in Modulating Transcription Factor Occupancy. Cell Rep (2015) 12(7):1184–95. doi: 10.1016/j.celrep.2015.07.024
44. Kloetgen A, Thandapani P, Ntziachristos P, Ghebrechristos Y, Nomikou S, Lazaris C, et al. Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat Genet (2020) 52(4):388–400. doi: 10.1038/s41588-020-0602-9
45. Aran D, Sabato S, Hellman A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol (2013) 14(3):R21. doi: 10.1186/gb-2013-14-3-r21
46. Mann IK, Chatterjee R, Zhao J, He X, Weirauch MT, Hughes TR, et al. CG methylated microarrays identify a novel methylated sequence bound by the CEBPB|ATF4 heterodimer that is active in vivo. Genome Res (2013) 23(6):988–97. doi: 10.1101/gr.146654.112
47. Gaston K, Fried M. CpG methylation has differential effects on the binding of YY1 and ETS proteins to the bi-directional promoter of the Surf-1 and Surf-2 genes. Nucleic Acids Res (1995) 23(6):901–9. doi: 10.1093/nar/23.6.901
48. Iguchi-Ariga SM, Schaffner W. CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev (1989) 3(5):612–9. doi: 10.1101/gad.3.5.612
49. Watt F, Molloy PL. Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev (1988) 2(9):1136–43. doi: 10.1101/gad.2.9.1136
50. Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell (2017) 171(7):1573–1588.e28. doi: 10.1016/j.cell.2017.11.008
51. Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, et al. Controlling Long-Range Genomic Interactions at a Native Locus by Targeted Tethering of a Looping Factor. Cell (2012) 149(6):1233–44. doi: 10.1016/j.cell.2012.03.051
52. Chowdhary S, Kainth AS, Pincus D, Gross DS. Heat Shock Factor 1 Drives Intergenic Association of Its Target Gene Loci upon Heat Shock. Cell Rep (2019) 26(1):18–28.e5. doi: 10.1016/j.celrep.2018.12.034
53. Wei Z, Gao F, Kim S, Yang H, Lyu J, An W, et al. Klf4 Organizes Long-Range Chromosomal Interactions with the Oct4 Locus in Reprogramming and Pluripotency. Cell Stem Cell (2013) 13(1):36–47. doi: 10.1016/j.stem.2013.05.010
54. Hao N, Shearwin KE, Dodd IB. Positive and Negative Control of Enhancer-Promoter Interactions by Other DNA Loops Generates Specificity and Tunability. Cell Rep (2019) 26(9):2419–2433.e3. doi: 10.1016/j.celrep.2019.02.002
55. Dall’Agnese A, Caputo L, Nicoletti C, di Iulio J, Schmitt A, Gatto S, et al. Transcription Factor-Directed Re-wiring of Chromatin Architecture for Somatic Cell Nuclear Reprogramming toward trans-Differentiation. Mol Cell (2019) 76(3):453–472.e8. doi: 10.1016/j.molcel.2019.07.036
56. Levasseur DN, Wang J, Dorschner MO, Stamatoyannopoulos JA, Orkin SH. Oct4 dependence of chromatin structure within the extended Nanog locus in ES cells. Genes Dev (2008) 22(5):575–80. doi: 10.1101/gad.1606308
57. Zhao J, Li D, Seo J, Allen AS, Gordân R. Quantifying the Impact of Non-coding Variants on Transcription Factor-DNA Binding. Res Comput Mol Biol (2017) 10229:336–52. doi: 10.1007/978-3-319-56970-3_21
58. Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Sci (80- ) (2014) 346(6215):1373–7. doi: 10.1126/science.1259037
59. Yun H, Vohra S, Mupo A, Giotopoulos G, Sasca D, Horton SJ, et al. Mutational Synergy Coordinately Remodels Chromatin Accessibility, Enhancer Landscape and 3-Dimensional DNA Topology to Alter Gene Expression during Leukemia Induction. Blood (2019) 134(Supplement_1):278–8. doi: 10.1182/blood-2019-122413
60. Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nat (2014) 511(7511):616–20. doi: 10.1038/nature13393
61. Speedy HE, Di Bernardo MC, Sava GP, Dyer MJS, Holroyd A, Wang Y, et al. A genome-wide association study identifies multiple susceptibility loci for chronic lymphocytic leukemia. Nat Genet (2014) 46(1):56–60. doi: 10.1038/ng.2843
62. Vijayakrishnan J, Studd J, Broderick P, Kinnersley B, Holroyd A, Law PJ, et al. Genome-wide association study identifies susceptibility loci for B-cell childhood acute lymphoblastic leukemia. Nat Commun (2018) 9(1):1–9. doi: 10.1038/s41467-018-03178-z
63. Perez-Andreu V, Roberts KG, Xu H, Smith C, Zhang H, Yang W, et al. Lymphoid neoplasia: A genome-wide association study of susceptibility to acute lymphoblastic leukemia in adolescents and young adults. Blood (2015) 125(4):680–6. doi: 10.1182/blood-2014-09-595744
64. Pollock BH, DeBaun MR, Camitta BM, Shuster JJ, Ravindranath Y, Pullen DJ, et al. Racial Differences in the Survival of Childhood B-Precursor Acute Lymphoblastic Leukemia: A Pediatric Oncology Group Study. J Clin Oncol (2000) 18(4):813–3. doi: 10.1200/JCO.2000.18.4.813
65. Kadan-Lottick NS, Ness KK, Bhatia S, Gurney JG. Survival variability by race and ethnicity in childhood acute lymphoblastic leukemia. JAMA (2003) 290(15):2008–14. doi: 10.1001/jama.290.15.2008
66. Bhatia S, Sather HN, Heerema NA, Trigg ME, Gaynon PS, Robison LL. Racial and ethnic differences in survival of children with acute lymphoblastic leukemia. Blood (2002) 100(6):1957–64. doi: 10.1182/blood-2002-02-0395
67. Young K, Loberg MA, Eudy E, Schwartz LS, Mujica KD, Trowbridge JJ. Heritable Genetic Background Alters Survival and Phenotype of Mll-AF9-Induced Leukemias. Exp Hematol (2020) 89:61–67. e3. doi: 10.1016/j.exphem.2020.07.012
68. Qian M, Zhao X, Devidas M, Yang W, Gocho Y, Smith C, et al. Genome-Wide Association Study of Susceptibility Loci for T-Cell Acute Lymphoblastic Leukemia in Children. JNCI J Natl Cancer Inst (2019) 111(12):1350–7. doi: 10.1093/jnci/djz043
69. Speedy HE, Beekman R, Chapaprieta V, Orlando G, Law PJ, Martín-García D, et al. Insight into genetic predisposition to chronic lymphocytic leukemia from integrative epigenomics. Nat Commun (2019) 10(1):1–9. doi: 10.1038/s41467-019-11582-2
70. Walsh KM, de Smith AJ, Chokkalingam AP, Metayer C, Dahl GV, Hsu L, et al. Novel childhood ALL susceptibility locus BMI1-PIP4K2A is specifically associated with the hyperdiploid subtype. Blood (2013) 121(23):4808–9. doi: 10.1182/blood-2013-04-495390
71. Perez-Andreu V, Roberts KG, Harvey RC, Yang W, Cheng C, Pei D, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet (2013) 45(12):1494–8. doi: 10.1038/ng.2803
72. Yang H, Zhang H, Luan Y, Liu T, Roberts KG, Qian M, et al. Non-coding germline GATA3 variants alter chromatin topology and contribute to pathogenesis of acute lymphoblastic leukemia. bioRxiv (2020) 2020.02.23.961672. doi: 10.1101/2020.02.23.961672
73. Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia (2004) 18:189–218. doi: 10.1038/sj.leu.2403241
74. Sur I, Taipale J. The role of enhancers in cancer. Nat Rev Cancer (2016) 16:483–93. doi: 10.1038/nrc.2016.62
75. Polak P, Karlic R, Koren A, Thurman R, Sandstrom R, Lawrence MS, et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature (2015) 518(7539):360–4. doi: 10.1038/nature14221
76. Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature (2016) 529(7584):110–4. doi: 10.1038/nature16490
77. Hnisz D, Weintrau AS, Day DS, Valton AL, Bak RO, Li CH, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Sci (80- ) (2016) 351(6280):1454–8. doi: 10.1126/science.aad9024
78. Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med (2014) 20(10):1130–7. doi: 10.1038/nm.3665
79. Belver L, Yang AY, Albero R, Herranz D, Brundu FG, Quinn SA, et al. GATA3-controlled nucleosome eviction drives MYC enhancer activity in T-cell development and leukemia. Cancer Discovery (2019) 9(12):1774–91. doi: 10.1158/2159-8290.CD-19-0471
80. Liu Y, Li C, Shen S, Chen X, Szlachta K, Edmonson MN, et al. Discovery of regulatory noncoding variants in individual cancer genomes by using cis-X. Nat Genet (2020) 52(8):811–18. doi: 10.1038/s41588-020-0659-5
81. Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell (2002) 1(1):75–87. doi: 10.1016/S1535-6108(02)00018-1
82. Ferrando AA, Neuberg DS, Dodge RK, Paietta E, Larson RA, Wiernik PH, et al. Prognostic importance of TLX1 (HOX11) oncogene expression in adults with T-cell acute lymphoblastic leukaemia. Lancet (2004) 363(9408):535–6. doi: 10.1016/S0140-6736(04)15542-6
83. Zhou Y, Kurukuti S, Saffrey P, Vukovic M, Michie AM, Strogantsev R, et al. Chromatin looping defines expression of TAL1, its flanking genes, and regulation in T-ALL. Blood (2013) 122(26):4199–209. doi: 10.1182/blood-2013-02-483875
84. Li Y, Liao Z, Luo H, Benyoucef A, Kang Y, Lai Q, et al. Alteration of CTCF-associated chromatin neighborhood inhibits TAL1-driven oncogenic transcription program and leukemogenesis. Nucleic Acids Res (2020) 48(6):3119–33. doi: 10.1093/nar/gkaa098
85. Cornish AJ, Hoang PH, Dobbins SE, Law PJ, Chubb D, Orlando G, et al. Identification of recurrent noncoding mutations in B-cell lymphoma using capture Hi-C. Blood Adv (2019) 3(1):21–32. doi: 10.1182/bloodadvances.2018026419
86. Jin G, Zhang F, Chan KM, Xavier Wong HL, Liu B, Cheah KSE, et al. MT1-MMP cleaves Dll1 to negatively regulate Notch signalling to maintain normal B-cell development. EMBO J (2011) 30(11):2281–93. doi: 10.1038/emboj.2011.136
87. Lee SY, Kumano K, Nakazaki K, Sanada M, Matsumoto A, Yamamoto G, et al. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer Sci (2009) 100(5):920–6. doi: 10.1111/j.1349-7006.2009.01130.x
88. Li K, Zhang Y, Liu X, Liu Y, Gu Z, Cao H, et al. Noncoding Variants Connect Enhancer Dysregulation with Nuclear Receptor Signaling in Hematopoietic Malignancies. Cancer Discovery (2020) May10(5):724–45. doi: 10.1158/2159-8290.CD-19-1128
89. Albrecht U, Sun ZS, Eichele G, Lee CC. A Differential Response of Two Putative Mammalian Circadian Regulators, mper1and mper2, to Light. Cell (1997) 91(7):1055–64. doi: 10.1016/S0092-8674(00)80495-X
90. Gery S, Gombart AF, Yi WS, Koeffler C, Hofmann W-K, Koeffler HP. Transcription profiling of C/EBP targets identifies Per2 as a gene implicated in myeloid leukemia. Blood (2005) 106(8):2827–36. doi: 10.1182/blood-2005-01-0358
91. Fu L, Pelicano H, Liu J, Huang P, Lee CC. The Circadian Gene Period2 Plays an Important Role in Tumor Suppression and DNA Damage Response In Vivo. Cell (2002) 111(1):41–50. doi: 10.1016/S0092-8674(02)00961-3
92. Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Bin MY, et al. An oestrogen-receptor-α-bound human chromatin interactome. Nature (2009) 462(7269):58–64. doi: 10.1038/nature08497
Keywords: spatial genome architecture, 3D chromatin organization, DNA loops, noncoding mutations and epimutations, enhancers, blood cancer, hematopoietic malignancies
Citation: Rovirosa L, Ramos-Morales A and Javierre BM (2020) The Genome in a Three-Dimensional Context: Deciphering the Contribution of Noncoding Mutations at Enhancers to Blood Cancer. Front. Immunol. 11:592087. doi: 10.3389/fimmu.2020.592087
Received: 06 August 2020; Accepted: 21 September 2020;
Published: 07 October 2020.
Edited by:
Ageliki Tsagaratou, The University of North Carolina at Chapel Hill, United StatesReviewed by:
Panagiotis Ntziachristos, Northwestern Medicine, United StatesJuan Ignacio Fuxman Bass, Boston University, United States
Copyright © 2020 Rovirosa, Ramos-Morales and Javierre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Biola M. Javierre, Ym1qYXZpZXJyZUBjYXJyZXJhc3Jlc2VhcmNoLm9yZw==