 Jill E. Weatherhead1,2,3*
Jill E. Weatherhead1,2,3* Pedro Gazzinelli-Guimaraes4
Pedro Gazzinelli-Guimaraes4 John M. Knight5
John M. Knight5 Ricardo Fujiwara6Peter J. Hotez2,3,7,8,9,10Maria Elena Bottazzi2,3,7,8
Ricardo Fujiwara6Peter J. Hotez2,3,7,8,9,10Maria Elena Bottazzi2,3,7,8 David B. Corry5,11,12
David B. Corry5,11,12- 1Department of Medicine, Infectious Diseases, Baylor College of Medicine, Houston, TX, United States
- 2Department of Pediatrics, Pediatric Tropical Medicine, Baylor College of Medicine, Houston, TX, United States
- 3National School of Tropical Medicine, Baylor College of Medicine, Houston, TX, United States
- 4Laboratory of Parasitic Diseases, NIAID, National Institutes of Health, Bethesda, MD, United States
- 5Department of Medicine, Pathology and Immunology, and the Biology of Inflammation Center, Baylor College of Medicine, Houston, TX, United States
- 6Departamento de Parasitologia, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
- 7Texas Children’s Center for Vaccine Development, Houston, TX, United States
- 8Department of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, TX, United States
- 9Department of Biology, Baylor University, Waco, TX, United States
- 10Hagler Institute for Advanced Study at Texas A&M University, College State, TX, United States
- 11Department of Medicine, Immunology, Allergy, Rheumatology, Baylor College of Medicine, Houston, TX, United States
- 12Michael E. DeBakey VA Center for Translational Research in Inflammatory Diseases, Houston, TX, United States
Helminths, including nematodes, cestodes and trematodes, are complex parasitic organisms that infect at least one billion people globally living in extreme poverty. Helminthic infections are associated with severe morbidity particularly in young children who often harbor the highest burden of disease. While each helminth species completes a distinct life cycle within the host, several helminths incite significant lung disease. This impact on the lungs occurs either directly from larval migration and host immune activation or indirectly from a systemic inflammatory immune response. The impact of helminths on the pulmonary immune response involves a sophisticated orchestration and activation of the host innate and adaptive immune cells. The consequences of activating pulmonary host immune responses are variable with several helminthic infections leading to severe, pulmonary compromise while others providing immune tolerance and protection against the development of pulmonary diseases. Further delineation of the convoluted interface between helminth infection and the pulmonary host immune responses is critical to the development of novel therapeutics that are critically needed to prevent the significant global morbidity caused by these parasites.
Introduction
Helminths are multicellular parasitic organisms belonging to a diverse taxonomic group of metazoans that compromise the phylum Platyhelminths, known as flatworms, including cestodes and trematodes, and Nematoda, known as roundworms, including Ascaris, hookworm, whipworms, filarial parasites, and others. Helminths overwhelmingly infect people living in extreme poverty in tropical and sub-tropical regions. Helminths cause significant disease burden globally, particularly in young children, infecting 1.5 billion people worldwide, nearly 20% of the world’s population (1). Together, the impact of helminth infections contributes to more than 12 million disability adjusted life years (DALYS), a measure of significant global mortality and morbidity (2). Helminth induced morbidity is largely due to the direct impact of the helminth on host tissues and indirectly from the host inflammatory response reflecting the complex helminth-host interface.
Host organs such as the lungs are a frequent target of helminth infection. For some human helminth infections, such as paragonomiasis, the adult stage of the parasite takes up its final residence in the lung (3). More commonly, the lung hosts the migratory pathways of the helminth larval stages either through the lung parenchyma or vasculature. As a general principle, helminth tissue invasion causes profound mechanical and chemical damage to the human lung and is linked to vigorous host inflammatory responses. The consequences are huge but the global burden of pulmonary disease as a result of helminths remains unknown.
A coordinated type-2 innate and adaptive immune response aimed at pathogen containment and management of tissue restoration occurs during helminth larval migration. This highly regulated type-2 immune response is communicated through the production of the cytokines interleukin (IL)-4, IL-5, IL-9, IL-10, and IL-13 and chemokines targeting the recruitment and activation of immune cells (4). In addition to the direct tissue damage incurred from the worm itself, helminths also release inflammatory mediators, typically through shedding of the outer chitin layer during molting and through release of soluble excretory-secretory (ES) product or extracellular vesicles (EVs) that contain immunomodulatory proteins including proteases and protease inhibitors, glycolytic enzymes, allergens, and lectins (5, 6). ES product and EVs are released by helminths at all stages of development, and can have an impact on the local environment in a paracrine-like mechanism, but also can influence the immunologic milieu in distant tissues (7, 8). The antigen profile of ES product is diverse amongst helminthic species but remains essential for the maturation and migration of these organisms (9–11). Because parasites are macropathogens, unable to be phagocytized by classic antigen presenting cells (APC), antigens within ES product or EVs are a major mode of communication with the host immune system leading in some cases to immune activation and in others to immunmodulation (12). The variation in antigenic components and concentration within ES product and EVs in different helminths and at different developmental stages contributes to the challenges in understanding the direct relationship between helminth derived factors and host immune responses. Given the link between helminth products, helminth survival, and modification of the host immune response, understanding the communication mechanism between the helminths and host may provide targets for future therapeutic interventions and highlights the need for further investigation.
Helminth Life Cycles
The impact of helminths on the host lungs is varied and largely depends on the burden of disease and the life cycle of the helminth species. Large burden of helminth disease contributes to a greater degree of tissue damage and a more profound immunologic response aimed at larval control and tissue repair (13). Additionally, helminths have distinct, complex life cycles, which support maturation of larvae into adult worms, as they migrate through varying host tissue compartments including the lungs (14). Several helminths including Ascaris, Strongyloides, and hookworms (Necator americanus, Ancylostoma duodenale) have mandatory but transient life cycles that involve larval migration through human lung tissue as an essential larval developmental step. In contrast, Toxocara, Dirofilaria, and Echinococcus larvae, all of which are zoonotic infections, travel to the human lungs and are unable to complete their life cycle; while filaria (Wuchereria bancrofiti and Brugia malayi) and Schistosoma larvae travel through the pulmonary vasculature (15, 16). Anisakis and Trichinella larvae do not typically migrate to the lungs but do induce a profound systemic inflammatory response during their migration cycle that impacts lung function. The purpose of the species-specific life cycle, particularly one that involves the lungs, remains unclear. Despite the extreme energy expenditure endured during migration, larval migration likely provides a survival advantage allowing for more rapid maturation and larger body size for some species and may also serve as an immune-evasion mechanism during larval development (13, 17). However, the transient migration process of helminths can have a significant impact on the host lungs through activation of the pulmonary and systemic immune responses.
Helminth Associated-Lung Diseases
Lung pathology induced by helminths occurs in three general categories: 1) diffuse lung disease, 2) focal lung lesions 3) and systemic inflammatory responses and hypersensitivity (Table 1). Helminths that cause lung disease typically have either a larval migratory phase directly through the lung parenchyma or through the lung vasculature, form cyst, and nodules in the lung tissue or have indirect systemic effects on the lungs (Figure 1).
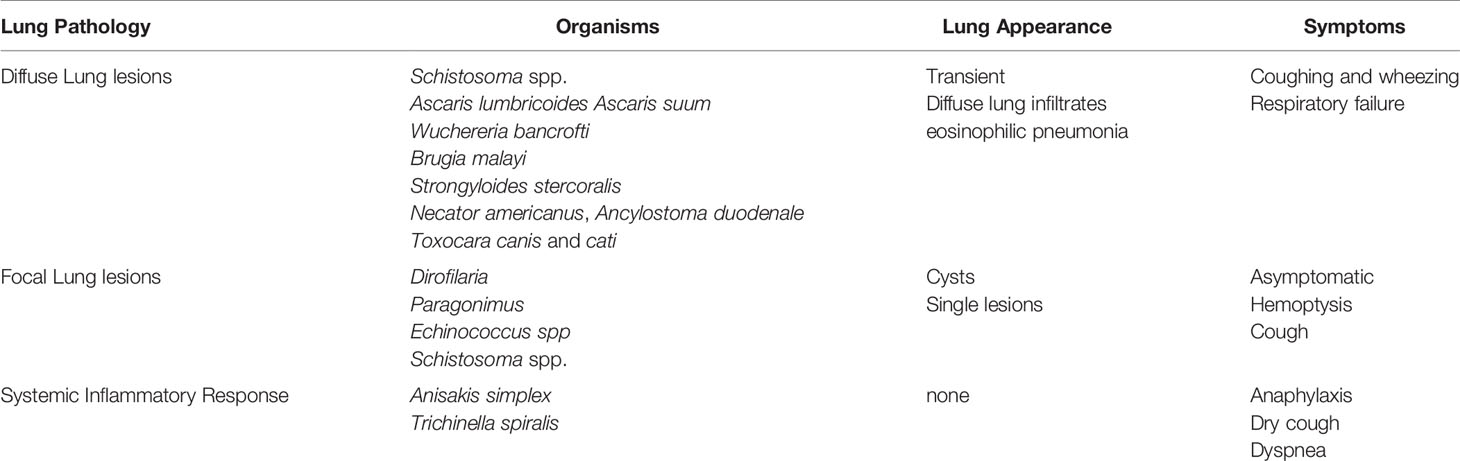
Table 1 Lung pathology induced by helminth infection.
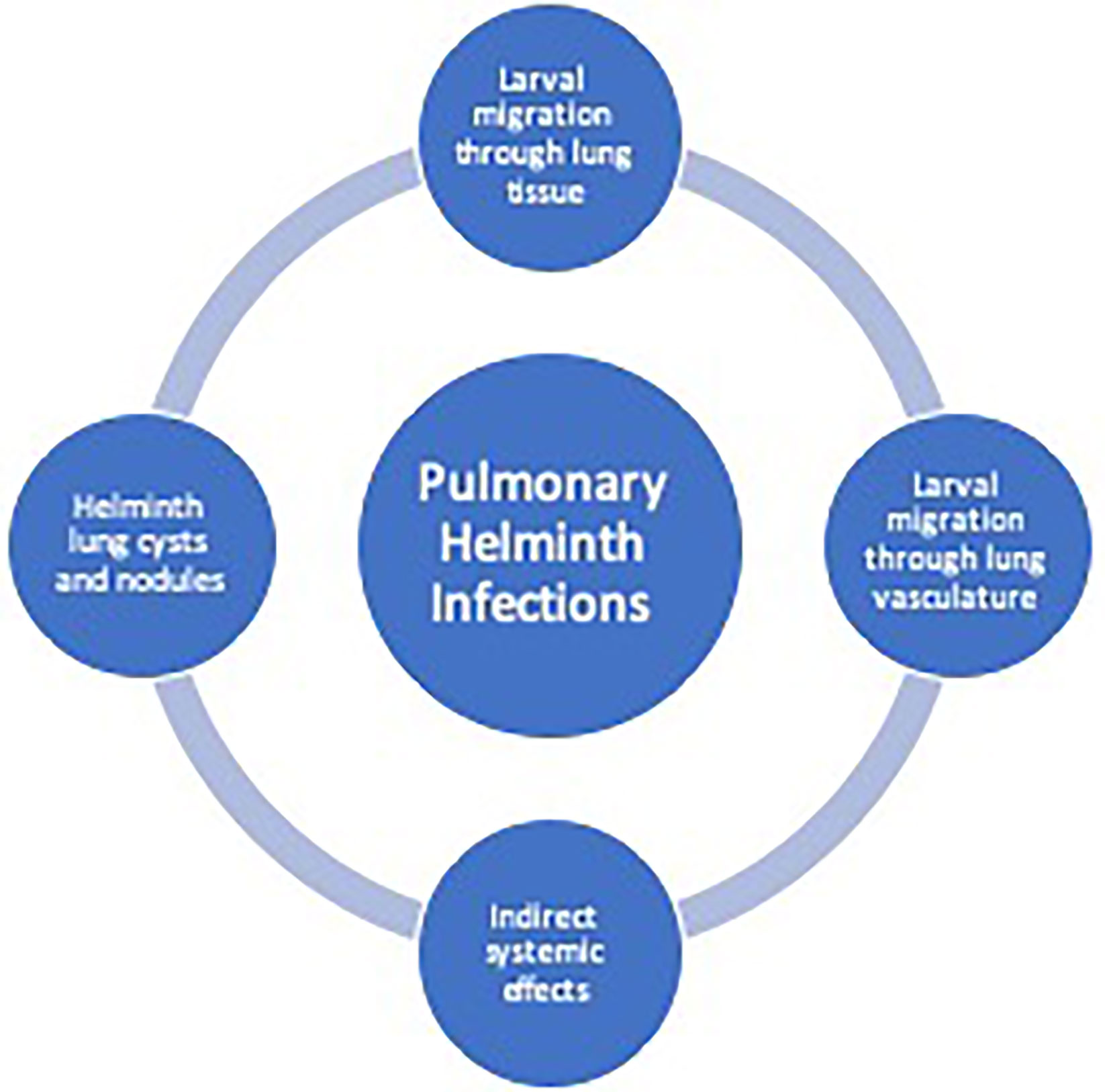
Figure 1 Helminth-induced pathogenesis of human pulmonary disease. Helminth infections can cause pulmonary pathology due to larval migration through the lung vasculature or through the lung tissue causing a diffuse lung disease. Additionally, trapped larvae or eggs can cause focal lesions within the lungs. Indirect systemic effects from helminths may cause pulmonary disease from release of parasite-derived or host-derived factors leading to system inflammation.
Diffuse Lung Disease
Organisms that have a larval migration stage through the lung parenchyma include the major soil-transmitted helminths, Ascaris lumbricoides and Ascaris suum, Strongyloides stercoralis, hookworm (Necator americanus, Ancylostoma duodenale), and the zoonotic infection Toxocara canis and cati (14). These interactions can lead to transient, diffuse lung infiltrates (14), and eosinophilic pneumonia termed Löeffler’s Syndrome (18, 19). Similar to allergic diseases such as asthma, this syndrome is characterized by a severe type-2 immune response with eosinophilia, goblet cell hyperplasia, increased mucus production and manifests clinically as coughing and wheezing and, in severe cases, respiratory failure (13). Given the similar clinical features, helminths such as ascarids may be a major cause of allergic airway disease globally (20). In addition, there are clinical consequences due to mechanical and chemical tissue destruction from these pulmonary helminth invasions, which can produce manifestations that resemble chronic obstructive pulmonary disease (COPD) (21).
Moreover, pulmonary vascular helminths, including filaria (Wuchereria bancrofti or Brugia malayi) and schistosomes also induce diffuse lung disease. The filarial parasites, for instance, cause a severe pulmonary syndrome called Tropical Pulmonary Eosinophilia (TPE) as a result of marked immunologic hyperresponsiveness to microfilariae trapped in the pulmonary vasculature. Within the pulmonary vasculature, microfilariae degenerate and release antigenic components eliciting a robust local inflammatory processes (22). Clinical manifestations include nocturnal cough, dyspnea, and wheezing in addition to interstitial infiltrates on chest radiograph, predominantly restrictive but also obstructive lung function abnormalities, peripheral blood eosinophilia, and markedly elevated levels of anti-filarial antibodies (23, 24). Other examples of vascular helminths that may trigger diffuse lung diseases are Schistosoma spp., primarily associated with Schistosoma mansoni, which can lead to different pathways of disease from either the larvae or the eggs. The larval migratory stage known as lung-stage schistosomula sometimes leads to an acute febrile illness known as Katayama fever which can include fever, rash, cough and associated diffuse, interstitial pulmonary infiltrates secondary to release of larvae derived products (25, 26). Additionally, chronic pulmonary schistosomiasis can cause nodular lesions composed of granulomatous inflammation from egg deposition in lung tissue appearing as “ground-glass” on pulmonary imaging (27). Late stage, chronic schistosomiasis can lead to pulmonary arterial hypertension (Sch-PAH). The etiology of Sch-PAH is unknown but may be due to egg deposition in pulmonary vessels causing vascular inflammation or due to portal hypertension. PAH most commonly presents clinically as dyspnea and reduced exercise tolerance. Given the widespread distribution, schistosomiasis may be a leading cause of PAH globally (27–29).
Focal Lung Lesions and Cysts
Other organisms such as Dirofilaria and Echinococcus, in which humans are accidental hosts and do not permit completion of the life cycle, worms are sequestered in the lungs and induce an inflammatory response manifesting as focal lung lesions (13). Dirofilaria immitis, the dog heartworm, cause formation of granulomas in the pulmonary vasculature seen on pulmonary imaging as “coin lesions”. Likewise, Echinococcus causes fluid-filled hydatid cysts in the lungs (13). Paragonimus also forms focal lung lesions. Larvae migrate to the lungs and develop into adult worms in pulmonary cysts manifesting clinically as cough, fever, chest pain and hemoptysis (16). Because paragonimiasis geographically overlaps with tuberculosis in East Asia, it is not uncommon to confuse these two causes of hemoptysis (30).
Indirect Effects of Systemic Inflammation
Several helminths, which do not directly infect host lungs, cause significant systemic inflammation through parasite-derived immunomodulatory molecules (13). Heavy infection with Anisakis simplex, thorough ingestion of raw or uncooked fish or handling fresh fish, can cause allergic airway disease or life-threatening anaphylaxis despite no direct contact with the host lungs. Fish processing workers specifically are at increased risk of developing Anisakis-induced bronchial hyperresponsiveness from consuming, touching or even inhaling Anisakis proteins (31). Trichinella spiralis larvae obtained from consuming undercooked pork or wild game can migrate through the lung vasculature on the way to striated muscle causing a dry cough (32). However, more commonly lung pathology occurs from either the systemic release of larval products during acute trichinellosis causing cough and dyspnea or from larvae developing into cysts in the chest wall accessory muscles and the diaphragm impacting lung dynamics (33).
These distinct clinical manifestations occur regularly in helminth endemic areas or can occur seasonally based on region specific transmission patterns (34). However, understanding the role of the host immune response to helminths and the resulting clinical impact on human lungs can be challenging and has lead to contradictory outcomes in clinical studies (20).
Helminths and the Host Immune Response
The challenge in gaining an in-depth understanding of the host innate and adaptive pulmonary immune response to helminths is multifactorial. Human studies have led to varying outcomes based on inconsistent diagnostic capabilities, variation in study populations, seasonality of infection, genetic predisposition, population age, and disease burden. As a result, animal models have been used to gain a greater understanding of the host pulmonary immune response to helminths. Nippostrongylus brasiliensiss, Strongyloides venezuelensis, Schistosoma mansoni, Brugia malayi, and Ascaris suum have all been used to evaluate the impact of helminths on host lung function in animal models (35, 36). These animal models are critical to elucidate the complex balance between helminth induced innate and adaptive lung immunity. Mouse models of Nippostrongylus, Schistosoma, and Ascaris have been especially helpful in elucidating immunological mechanisms. It is critical to dissect the complex lung innate and adaptive immune response as a result of helminth infection in order to inform on-going therapeutic and vaccine development that is needed to reduce global morbidity.
Innate Immune Response in the Lungs to Helminthic Infections
Early recognition of helminths by the host pulmonary innate immune response is critical for disease control. Helminth molecules, glycans and lipids, serve as pathogen-associated molecular patterns (PAMPs). Recognition and processing of helminth PAMPS by APCs through pattern recognition receptors (PRR) such as toll-like receptors (TLR), C-type lectins and intracellular nucleotide-binding oligomerization domain-containing protein (NOD)-like receptors are initial steps in the host innate immune response engagement (17, 37, 38). Intracellular signaling as a result of PRR activation, leads to release of cytokines, particularly IL-4, IL-5, IL-9, IL-13, IL-25, IL-33, and IL-17 as well as chemokines that propagate the host immune response. Cytokines and chemokines activate and recruit additional innate inflammatory cells including neutrophils, macrophages, basophils, eosinophils, dendritic cells, mast cells, natural killer (NK) cells, and type 2 innate lymphoid cells (ILC2) to the lungs, further propagating the innate immune response (13, 39). Helminths also expel cytokine- and chemokine-like natural products and molecules displaying immunologic mimicry, further aiding in cellular recruitment and activation. The recruitment and activation of innate cells in coordination with epithelial cell release of alarmins (e.g., IL-33; HMGB-1) and cellular hyperplasia, goblet cell production and release of intraluminal mucous and smooth muscle contraction are all necessary for the eradication of parasites from host lungs. A variety of specialized innate cells direct type-2 immune polarization and subsequent synchronization with adaptive immune pulmonary responses against helminth infections. The major innate immunity effector mechanisms to helminth infections include the following major elements.
Complement System
The classical, lectin and alternative complement pathways are prominently activated in early helminth pulmonary infection (40). Mice deficient in C3 infected with Strongyloides have increased larval survival (41). Despite activation of complement during helminth infection, helminths are able to evade complement mediated immunity potentially through binding complement inhibitory protein factor H, during molting, a process allowing for shedding of the larval outer layer (40), and by expressing surface proteinases that continuously degrade complement proteins binding to the helminth cuticle. While not necessarily playing a large role in opsonization and pathogenic killing, complement remains an important regulator of leukocyte-mediated immunity during helminth infection. Specifically, C3a and C5a act as chemotactic factors aiding eosinophil recruitment (40). Despite all three pathways being engaged, the independent role of the three individual pathways may differ depending on the helminth species, helminth developmental stage and the location of infection (40). In N. brasilliensis infection, the alternative pathway, mediated by hydrolysis of C3, plays a major role in eosinophil adherence in the infective larval stage (L3) occurring in the host lungs. In contrast, during the adult stage of N. brasilliensis, which resides within the gastrointestinal tract, lectin pathway plays a larger role in helminthic control (40).
Mucosal Barrier (Epithelium, Smooth Muscle, Mucus Production)
Direct helminth-induced damage to and death of lung epithelial cells can be one of the initial triggering events in helminth infection, leading to release of epithelial derived alarmins IL-25, IL-33, thymic stromal lymphopoietin (TSLP), and chemokines CXCL1, CXCL2, CXCL8, and eotaxins (42, 43). These alarmins signal to innate cell populations such as Innate lymphoid cells type 2 (ILC2), basophils, eosinophils, neutrophils, macrophages, and dendritic cells during the early phases of infection (43). The influx of type-2 cytokines into the lung compartment as a result of innate immune cell activation, particularly through the action of IL-4, IL-9, and IL-13, aids in goblet cell hyperplasia andmucin production needed for helminth expulsion and epithelial turn-over and repair (37, 43). In Schistosoma mansoni infection in the lung, rapid influx of IL-9 leads to robust generation of goblet cell hyperplasia (44). IL-4 receptor alpha (IL-4Rα) signaling via IL-4 and IL-13 cytokines also plays a role in mucin production and smooth muscle responsiveness during Nippostrongylus infection (45). Smooth muscle cells responding to IL-4Rα activation on smooth muscle leads to airway hyperresponsiveness, the exaggerated tendency of the airway to constrict, during Nippostrongylus infection in addition to T cell recruitment to the lungs (45). Mice deficient in smooth muscle IL-4Rα lack coordination of acetylcholine responsiveness with reduced M3 muscarinic receptor expression, delayed goblet cell hyperplasia, reduced type-2 cytokine production, and have a delayed ability to expel Nippostrongylus (46). Independent of IL-4Rα signaling, secreted proteins Ym1 and Restin-link molecule alpha (RELMα/Fizz1) from epithelial cells in the lungs contribute to lung repair through inducing IL-17A and neutrophilic infiltration promoting type-2 immunity and remodeling (47). Pulmonary tuft cells, termed brush cells, along the airway epithlium contain microvilli and potentially function as chemosensory cells playing an additional role in innate epithelial immunity (48). Tuft cells in the intestines have been shown to be involved in the generation of type-2 immunity to helminths (49). However, more research is needed to evaluate the interaction between helminths and pulmonary tuft cells.
Neurons and Neurotransmitter Signaling
The role of the neuroimmune network in helminth infection has more recently been highlighted as a critical modulator in helminth control and tissue repair particularly at heavily innervated mucosal sites (50). Pulmonary neuroendocrine cells, located at airway branching points release neuropeptides and neurotransmitters that influence smooth muscle tone (51). Bovine lungworm infection, Dictyocaulus vivparus, leads to upregulation of nictotinic acetylcholine receptors and increased cholinergic signaling on immune cells including epithelium, leukocytes, and macrophages (52). Additionally, helminth-derived acetylcholinesterase (AChE) has been identified in ES product of lung stage helminth larvae in different species. AChE secreted by Nippostrongylus regulates hydrolysis of endogenous Ach in mucosal tissue and co-localizes with cholinergic mucosal neurons that express the neuropeptide neuromedin U (NMU) suggesting that helminth derived products are involved in neurotransmitter modulation in mucosal tissue (50, 52, 53). NMU is particularly involved in coordination of the host innate response in the lungs during helminth infections. Neuron stimulation by either host alarmin IL-33 or IL-25 or Nippostrongylus ES product leads to direct neuronal release of NMU showing that neurons are sensing host damage and larval products (54). High concentrations of IL-25 in combination with NMU are associated with enhanced expression of type-2 cytokines IL-5 and IL-33 (55). Furthermore, intranasal administration of NMU has been linked to initiation of type-2 immune responses such as increased eosinophils, decreased tissue hemorrhage, enhanced mucus production and reduced infectious burden (50, 54). This may in part be an indirect effect of NMU given its role in enhancing maturation, proliferation and cytokine expression of lung ILC2s. ILC2 and NMU seem to have an inter-dependent relationship during helminth infection in the lungs. ILC2 selectively express neuromedin U receptor 1 (Nmur1), deletion of which impairs type-2 immune response and parasite control (54). NMU neuroimmune effects during helminth infection is counter regulated by the neuropeptide CGRP. The cognate receptor of CGRP is highly expressed on IL-5 secreting ILC2 populations, and when activated, decreases ILC2 populations (56).
ILC2s
ILC2s are innate tissue-resident cells particularly abundant at mucosal barriers including the lungs. They are found in high concentrations around the adventitia of lung bronchi and large vessels and co-localize with subsets of dendritic cells, T regulatory (Treg) cells, adventitial stromal cells, and mesenchymal fibroblasts (57). ILC2 activation and proliferation in the lungs is dependent on the release of alarmins IL-25, IL-33, and TSLP, in addition to the neuropeptide NMU. ILC2s also express IL-4Rα enabling the cells to respond to the local type-2 cytokine milieu during helminth infection. Responding to IL-4 and IL-13 signals leads to further cellular expansion of ILC2s and a robust release of type-2 cytokines particularly IL-5 and IL-13 in a STAT6-independent mechanism (58, 59). The significant influx of IL-5 and IL-13 supports eosinophil recruitment, epithelial cell hyperplasia, and goblet cell hyperplasia (60). STAT6 is also not required for IL-13 production in ILC2s response to Nippostrongylus in the lungs (59). Once thought to be a homogenous population, single cell analysis during Nippostrongylus infection demonstrates significant heterogeneity within ILC2 populations in the lungs (56). Transient IL-25 responsive ILC2 (iILC2) are circulating ILC2s that mobilize to lung tissue in response to chemokines and cytokines (58, 61). Basic Leucine Zipper ATF-Like Transcription Factor (BATF), an AP-1 superfamily transcription factor, is critical for activation of iILC2 during Nippostrongylus infection (58). BATF-deficient Nippostrongylus infected mice lack iILC2 in lungs, have reduced early influx of type-2 cytokines and impaired mucosal barrier integrity. Conversely, tissue resident ILC2 (nILC2) are IL-33 responsive and not BATF- dependent (58, 61). Migration of ILC2 to the lungs may be secondary to increased expression of prostaglandin D2 (PGD2) receptor CRTH2 (chemoattractant receptor-homologous molecule) on the cell service (62). IL-33 coordinates the PGD2–CRTH2 pathway further regulating ILC2 migration patterns to the lungs (63).
Granulocytes (Basophils, Mast Cells, Eosinophils)
In addition to ILC2, basophils, mast cells, and eosinophilia constitute IL-4– and IL-13–producing cells of the innate immune system during lung helminth infection. The role of basophil and eosinophil production of IL-4 and IL-13 is sufficient enough to induce allergic airway disease in defense against migrating worms (64).
Basophils are granulocytes that produce type-2 cytokines including IL-4, IL-5, and IL-13 in addition to histamine, leukotrienes, and prostaglandins during degranulation as a result of helminth infection (65). Production of IL-4 by basophils is STAT-6 independent. Nippostrongylus infection as well as Schistosoma egg antigen (SEA) increase the number of basophils in the lungs as a result of FcR cross-linkage by IgE or IgG, complement activation of C5a or cytokine stimulation through T cell derived IL-3 or IL-18 (66–69). As a result, basophils can potentially serve as an early source of IL-4 during helminth lung infection prior to T cell activation. Schistosoma ES product IPSE/alpha-1 can trigger basophil production of IL-4 and IL-13 and influence cellular commitment toward type-2 immunity such as alternatively activated macrophage (AAM) formation, smooth muscle activity, goblet cell hyperplasia, and eosinophilic infiltration into the lungs (65, 70). Basophils are also capable of processing antigen to naïve CD4 T cells and promoting Th2 differentiation via MHC Class II expression (65, 71). While basophils are not essential for Th2 differentiation they are likely a critical initial trigger in the type-2 immune cascade (65). Recently, up-regulation of Notch2 receptor in basophils was shown to cause increase cytokine production, including IL-4 and IL-6, in intestinal helminth infections. Moreover, basophil-intrinsic Notch signaling promoted worm clearance and type 2 inflammation in the cecum (72). Whether basophil Notch signaling occurs in the lungs remains unclear.
Mast cells located along the epithelial lining of the host lungs additionally play a role in helminth infection. At baseline, mast cells are rare resident cells in host lung tissue but can rise significantly during helminth infection. However, the inciting chemoattractants that induce mastocystosis in the lungs during helminth infection remains largely unknown (73). IL-9 signaling through IL-9R on mast cells is an important factor for maturation and activation of mast cells. Depletion of IL-9 during Schistosoma mansoni infection demonstrates 8-fold fewer mast cells in the lungs (44). Additionally IL-3, derived from varying cell sources including T cells, may contribute to further influx of mast cells. In IL-3 deficient, Strongyloides infected mice, mast-cell development is stalled and parasite killing is impaired (67). Degranulation and rapid release of inflammatory mediators (histamine, leukotrienes, and prostaglandins), cytokines (IL-4, IL-6, and TNFα), and proteases (mcpt1 and mcpt2) occurs secondary to high affinity mast cell FcϵR1 bound to IgE, which plays an important role in helminth killing (11, 43). However, release of these mediators including IL-4 from mast cells is not required to generate a robust type-2 immune response. Mast-cell deficient mice infected with helminths can still generate normal type-2 immune response in the lungs (65).
Eosinophils are critical effector cells during helminth infection through direct killing of parasites and, potentially acting as APCs directing T cell differentiation to type-2 immune cells, although this function remains controversial (39, 74, 75). The early presence of helminth larvae in the lungs triggers rapid eosinophilic activation and recruitment through the release of chemoattractants eotaxins and MIP-1a as well as cytokine IL-5 production (76). Eosinophilic degranulation occurs with engagement of the FcϵR1 on eosinophils surface to Fc of IgE bound parasites (11). Eosinophilic cellular cytotoxicity of helminthic larvae in the lungs occurs through release of major basic protein (MBP) and eosinophil peroxidase (EPO)-dependent mechanisms particularly during secondary infection and through induction of histamine release from mast cells (75, 77). The release of eosinophil DNA-based extracellular traps (EETs) may be an additional mechanism of helminth larvae-killing or immune response against extracellular pathogens (78). The eosinophilic immune response restricts larval development and reduces parasite burden in the lungs particularly in secondary exposure to helminths or in hosts with allergen presensitization (79). Presensitized lung tissue and airways in HDM-allergic lungs or after multiple helminth exposures, contain IL-4 and IL-13-rich AAMs and eosinophil-dominated type 2 cellular infiltration, which dramatically reduces the burden of disease, including lower hemorrhage and mechanical damage in the tissue as a result of parasite migration through tissue (76, 79). Likewise, deletion of eosinophils in animal models has been shown to increase parasite survival (38, 39). Reduction of eosinophils through anti-IL-5 or anti-CCR3 monoclonal antibody blocks innate protective immunity to Strongyloides and leads to reduced control of larvae (39). However, eosinophils may not be essential for larval killing as neutrophils provide redundency (77). In the complete absence of eosinophils during Strongyloides infection, neutrophils were capable of partial control of infection (39). While eosinophils release IL-4 and aid in type 2 T cell polarization, eosinophil deficient mice still mount a normal T helper type 2 (Th2) response to helminth infection (65).
Neutrophils
Neutrophils also perform as effector cells during helminthic infection in the lungs. Facilitated by the release of myeloperoxidase, neutrophils can independently kill Strongyloides larvae in the lungs (39). Interestingly, Strongyloides larvae alone can directly induce the activation and recruitment of neutrophils to infected tissues (80). Strongyloides-specific molecules released during the larval stage promote neutrophil release of chemokines MIP-2 and KC, further enhancing recruitment of neutrophils (39). Additionally, chitinase-like proteins Ym1 and Ym2 lead to expansion of IL-17A–producing γδT cells with increased IL-17A production and subsequent neutrophil recruitment. The enhanced neutrophil recruitment prevents parasite survival but at the expense of enhanced lung injury (81). Likewise Anisaki release of ES products elicits neutrophil recruitment to the lungs via production of IL-6, IL-8, and CXCL1 which has been shown to further enhance lung inflammation and contribute to lung disease during anisakiasis despite no lung larval migratory phase (82). Such tissue damage is not seen to the same degree with type 2 immunity-dependent, eosinophil predominant inflammation, suggesting that type 2 immunity may have evolved to provide control over chronic, inescapable parasitic infections while minimizing bystander tissue injury.
Natural Killer Cells
NK cells contribute to helminthic control in the lungs through direct cytotoxicity and cytokine production. NK cells expand early in helminth lung infection potentially secondary to recognition of helminth ES product (83, 84). Filarial infective-stage larvae and microfilariae modulate NK cell activation and release of cytokines including gamma interferon (IFN-γ) and tumor necrosis factor-α (TNF-α) (85). In filarial disease animal models, in vivo depletion of NK cells increased worm burden in the pleural cavity and influenced IL-4 and IL-5 plasma levels (84). Likewise, pulmonary schistosomiasis increases NK cells within the lungs and NK cells localize near Schistosoma pulmonary granulomas (86). NK cells additionally promote dendritic cell maturation through IL-12 production and directly interact with T cells providing a link between innate and adaptive immunity (86).
Professional APCs (Macrophages and Dendritic Cells)
Dendritic cells (DCs) serve as mediators in the innate and adaptive orchestration of type-2 immunity during lung helminth infection (87). DCs recognize helminth ES-derived antigens or parasite surface molecules. Proteins and lipids that are highly glycosylated are recognized by different innate PRR including TLR, c-type lectin receptors, and NOD-like receptors (11, 87, 88). DCs capture helminthic antigen and present to T cells propagating the host type-2 immune response (11). Schistosoma and filaria both activate TLR4 on dendritic cells to engage type-2 immunity in the lungs potentially through reduction in dendritic cells ability to produce IL-12. However, evaluation of th immunologic pathways involving TLR4’s role in the development of type-2 immunity is on-going (11).
Activated macrophages in the lungs serve as APCs that aid in transitioning to a type-2 adaptive immune response during helminthic infection and selectively polarize toward type-2 mediated AAMs. In the context of a predominant type-2 immune response environment, IL-4 and IL-13 signaling via the STAT6 pathway induce expression of mediators that promote AAM. The IL-4/IL-13 and STAT6 signaling pathway allow for upregulation of genes associated with AAM (YM1, YM2, RELMα, ARG1) and upregulation of class II MHC on macrophages to further promote type-2 immune responses (89, 90). Amplified concentrations of AAM during helminth infection in the lungs impedes parasite migration and are sufficient to kill Nippostrongylus in vitro (80). Additionally, AAM associated RELMα specifically supports tissue repair, extracellular matrix turnover and homeostasis and is essential to prevent fatal lung damage as a result of helminth infection in the lungs (89, 90).
Additional cells that play a role in innate immunity including γδT cells (81) and coagulation factors (platelets, thrombin, fibrinogen) (91, 92) may also contribute to helminth disease control and tissue damage in the lungs. However, additional studies are required. Overall, the coordinated work of innate cells play a critical role in early recognition and termination of helminth infections as well as initiation of tissue repair in the lungs. However, perhaps the most important role of the innate cells during pulmonary helminthic infections is engagement and activation of the adaptive immune response (Figure 2).
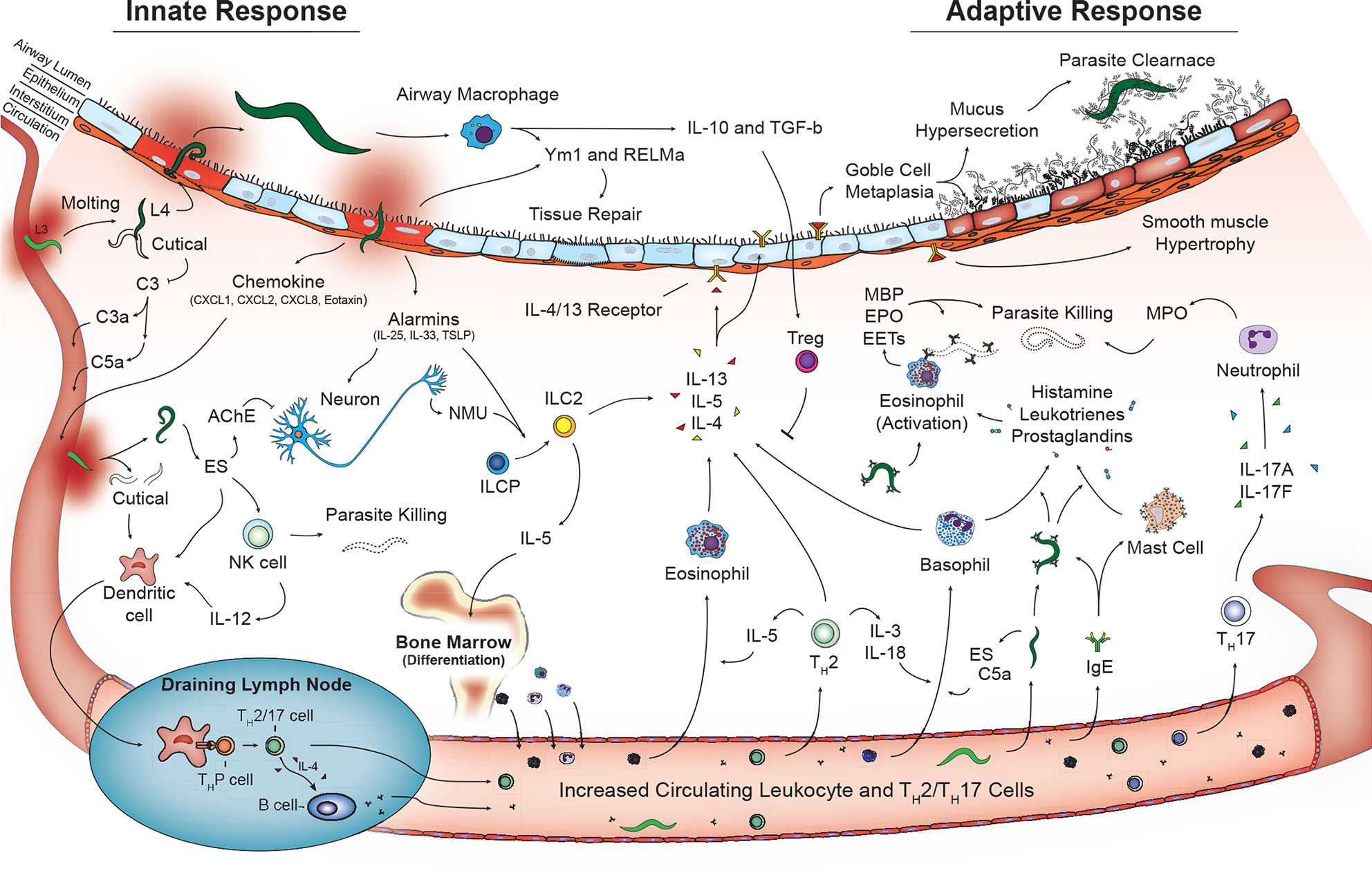
Figure 2 Activated innate and adaptive immune pathways in the lungs during helminth infection. Innate phase of immune activation to helminths in the lungs is mediated by antigen presenting cells including dendritic cells and macrophages in addition to release of alarmins from damaged epithelium that drive a type-2 immune response. Release of type-2 cytokines IL-4, IL-5, and IL-13 contributed from granulocytes and type 2 innate lymphocytes promote a lung phenotype with goblet cell metaplasia and mucus hypersecretion and smooth muscle hypertrophy. T lymphocytes including Th2, Th17 in combination with B lymphocyte aiding immunoglobulin class switching further contribute to type-2 immune response promoting parasite killing and clearance as well as tissue repair and recovery.
Adaptive Immune Response in the Lungs to Helminthic Infections
Innate immunity is not just important as the first line of defense but also influences the nature of the adaptive response (93), by creating environments with signature cytokines that have the ability to drive the proliferation and differentiation of the effector T helper (Th) cells. In response to helminth infection, naive CD4+ T‐cells differentiate into several possible effector subsets, including Th2, Th17, T regulatory cells (Treg), and T follicular helper cells (Tfh) as well as influence the maturation of B cells and isotype switching of immunoglobulins favoring a type-2 immune response (94). Conversely, Th1 cells that secrete principally IFN-γ are activated most commonly in intracellular parasitic infections such as protozoa (e.g., Leishmania and Plasmodium) and are not predominately linked to helminth infections with some exceptions (95). Several cytokines, mostly originated from innate cells, and transcription factors tune the differentiation and expansion of these cell populations. Overall, helminth-induced immunoregulation occurs through the induction T cell subsets and B cell maturation influencing the immunologic landscape during helminth infection, providing effector function, tissue repair, and memory in the lungs. Of note, very few studies have analyzed CD8+ T cell responses to helminth infection in the lung tissue. While, CD4+ T cells classically play a major effector role in pulmonary helminthic disease, CD8+ cells can enhance granulomatous inflammation in the lungs to schistosomiasis (96). More detailed analysis of the role of CD8+ cells during pulmonary helminth infection are needed. While helminths classically create a type-2 immune response, the specific balance of adaptive immune cells is intrinsically associated with the helminth species, the stage of infection (acuteness or chronicity) and what organ/tissue has been affected. In the lung tissue, as most of the pulmonary helminthic infections are transient, the role of the adaptive response can be divided in 2 phases: helminth control and tissue repair.
Helminth Control in the Lungs
During parasitic invasion of the lungs, Th2 effector cells are essential to rapidly polarize and amplify a multifactorial type-2 inflammatory response to control parasite burdens. Although the mechanisms are not fully elucidated, the circumstances that promote the acute Th2 cell protective immunity in the lungs occurs by cross-talk with innate immunity through cytokines, chemokines and growth factors that result in the establishment of a robust type-2 inflammatory environment rich in AAM and dominated by eosinophils. Initial Th2-cell differentiation largely involves the cytokines IL-2, IL-4, and collectively IL-25, TSLP, and IL-33 as well as the transcription factor GATA3 directing effector function in the lungs (95, 97, 98). The Th2 effector response to helminth infections in the lungs is thus typified by the differentiation of antigen-specific CD4+ T cells to Th2, high levels of IL-4, IL-5, and IL-13, accompanied by eosinophilia, mucus production, and abundant IgE production, phenotypically similar to allergic airway disease (99).
IL-4–, IL-13–, and IL-5–producing CD4+ Th2 cells have been implicated as one of the most important effector mechanism in adaptive pulmonary immunity against helminths. IL-5–producing Th2 cells provide support for IL-5–mediated eosinophil-dependent larval killing. IL-5 enhances the differentiation, maturation, and survival of eosinophils derived from bone marrow precursors (100, 101). In animal models of Strongyloides stercoralis, Th2 production of IL-5 is not only required to elicit additional effector function but also to induce protective antibodies (102). IL-4– and IL-13–producing Th2 cells induce STAT6-dependent changes in epithelial-cell function resulting in goblet cell hyperplasia and increased mucus production via IL-4Rα and IL-13Rα1 signaling. Additionally, IL-4Rα expression on lung-resident CD4+ T cells is required to generate a protective recall immunity to N. brasiliensis re-infection (103). In subsequent infection, the presences of primed memory Th2 cells aid in the accumulation of eosinophils expressing high levels of MBP. The recruitment of eosinophils by memory Th2 cells facilitates more rapid reduction of helminth burden in the lungs compared to primary infection (104).
Th2 effector cells and Tfh cells in germinal centers coordinate the humoral response in type-2 inflammation by promoting a helminth-specific B cell response and stimulating B cell class switching to high-affinity IgE, IgG1, and IgG4 (38). Tfh is reliant on Notch signaling in an IL-4 dependent pathway and the transcription factor BCL6. However, deficiency of Tfh does no impact worm clearance, overall IL-4 production or Th2 mediated immunity (105, 106). Antibodies are the quintessential adaptive immune effector molecules and are a central feature of Th2 immunity (97). Moreover, B cell activation has been investigated over the years as the target for effective vaccine development against helminth parasites and antigen-specific IgG antibodies generate a protective adaptive immune response against Ascaris parasites specifically (107).
Lung Tissue Repair and Restoration
After parasites have migrated through the lungs, Th2 effector cells are critical to activating efficient tissue repair responses. IL-4– and IL-13–producing Th2 cells are important for differentiation and maintenance of AAM in the tissue, encoding genes such as YM1, RELMα, and Arg1, key players in tissue repair and remodeling (108, 109). The crosstalk between Th2 effectors cells and macrophages has been demonstrated in experimental models of the filarial nematode Brugia malayi. Filarial infection in recombinase activating gene (RAG) or MHC class II-deficient mice (mice lacking T cells) demonstrate a striking lack of AAM, suggesting that Th2 cells are required for maintenance and full activation of AAM directed repair immunity to helminth parasites (110).
Treg populations, “natural” Tregs and “induced” Tregs, are activated in an effort to dampen host immune response through production of IL-10 and TGF-β during helminthic infection (111). The role of Treg varies depending on the developmental stage and the parasite species. Lung damage, blood clots, damage-associated molecular patterns (DAMPS) as a result of helminth infection in the lungs, promotes differentiation of macrophages and release of macrophage-derived growth factors and anti-inflammatory IL-10 and TGF-β that activate Treg (38). Depletion of Treg heightens Th2 cell proliferation and clearance of infection; however, this is associated with increased lung damage due to prolonged effector activation (112). Conversely, Treg production early in infection can promote worm survival by dampening the acute Th2 effector cell response but promoting tissue integrity (95). In helminth re-infection models, Tregs suppress memory Th2 cells in order to reduce the recruitment of eosinophils into the lungs and prevent excessive lung damage and further pathology (104, 111). Additionally, Treg activation during helminth infection can generate non-specific host immune suppression across tissue compartments and thus can be important in parasitic co-infection models particularly regarding progression of malaria and tuberculosis (112).
Additional T helper subtypes can play important roles during helminthic infection. Driven largely by pro-inflammatory cytokines, IL-6 and IL-17A and the transcription factor RORγt and maintained by IL-23, Th17 cells at mucosal barrier sites aid in mucosal defense and tissue restoration (95). Lung tissue expression of receptors for the Th17 cytokines IL-17A, IL-21, GM-CSF, and IL-22 allows for both protective and pathogenic responses in the lungs to helminth challenge (113). During Nippostrongylus infection in the lungs, Th17 activation is associated with early release of IL-17A which aids in IFNγ suppression and type-2 immunity, recruitment of neutrophils and subsequent pulmonary damage and hemorrhage (38, 114). Prolonged Th17 activation and excessive early IL-17A can be associated with immune-mediated disease in the lungs including asthma (114). It is also linked with the extravasation of eosinophils from the bone marrow and eosinophilic infiltrations in host lung tissues. However, once established, IL-17A can also act as a negative regulator of the type-2 response in the lungs (114). On that basis Th17 responses, mediated through IL-17A, may be as important as Th2 responses in the pathogenesis of helminth-inducted pulmonary disease (115). However, Th17-driven IL-22 aids in epithelial cell repair and regeneration of mucosal barrier after helminth infection in the lungs (116). The IL-17/IL-22 axis is critical to the generation of epithelial homeostasis post-infection. IL-6 promotes differentiation of Th22 cells and release of IL-22, which also likely aids in tissue repair although the role of IL-22 in pulmonary helminth infections remains unknown. Additionally, Th9 in gastrointestinal helminth infection has been shown to cause rapid worm expulsion and basophil activation however further investigation is needed in the lungs (117).
Systemic Impact of the Adaptive Immune Response
The complexity of the adaptive immune response to helminths is varied and can be influential in co-infection and co-morbid models of disease. Helminth co-infection models with different infected host compartments impacts not only the local adaptive immune response but also the host immune response to helminth infection in distant tissues. H. polygyrus intestinal infection induces a IL-33–mediated activation of IL-5 secreting Th2 cells and causes upregulation of IL-5–mediated eosinophils, leading to a significant increase in immune-mediated killing of N. brasiliensis larvae in the lungs of the co-infected mice (118). Additionally, co-morbidities may also influence the adaptive immune response to helminth infections. In an animal model of house dust mite (HDM) allergic sensitization, subsequent Ascaris infection is mitigated by a primed memory Th2 response in the lungs. Allergen-driven inflammation, increases IL-5– and IL-13–producing Th2 cells in the lungs leading to an IL-4– and IL-13–rich environment that drives the differentiation of lung macrophages toward an AAM phenotype expressing arginase-1, as well as, an eosinophil infiltration. This strict type-2 immune milieu leads to a marked reduction in the number of lung-stage Ascaris larvae, reducing the intensity of infection, inducing lower pulmonary hemorrhage and mechanical damage in the lung tissue during larval migration (79). These results suggest a sophisticated and efficient feedback loop among Th2 cells, eosinophils and AAM in coordinating innate and adaptive immunity against lung tissue helminths (95).
Implications for Vaccines
Several human helminth vaccines are in phase 1 or phase 2 stages of clinical development, including hookworm infection and schistosomiasis (119). For human hookworm infection, vaccine immunity appears to operate via directing anti-enzyme antibody responses to parasite gut digestive enzymes, including a glutathione S-transferase and a hemoglobinase, while schistosome vaccines focus on larval and adult surface antigens (120). Extensive studies in mice show how vaccine immunity against schistosomes and possibly other helminths operate through adaptive immune responses in the lungs. In some cases, these responses actually trap larval helminths in the lung and block further migration (121). Furthermore, Ascaris vaccines targeting larval-stage immunodominant proteins that are highly conserved between helminths suggest the possibility of developing a pan-helminth vaccine that may prevent larval migration (122). Therefore, helminth vaccine development targeting effector immunity in the lung may prevent larvae from completing their life cycle while preventing host lung damage.
Helminths and Immune Modulation
Helminths can directly cause lung pathology as a result of direct larval migration or indirectly through systemic activation of a type-2 immune response. Many of the molecules released from helminths have been associated with development of lung disease, including allergic airway disease. Ascaris ABA-1 fatty acid binding protein, a glutathione transferase (GST) involved in distribution of lipids, is released at different stages of Ascaris development including the lung stage. ABA-1 has significant homology to mite and cockroach GST and has been linked to the development of asthma (123). Conversely, Anisakis, which is the most allergenic helminth and does not typically have a lung larval stage, contains the allergens molecules Ani s 1, Ani s 4, and Ani s 9, in ES product, all of which are proteins central to the development of systemic anaphylaxis and lung pathology (123). Similar proteins are found in the ES product of hookworm and Schistosoma spp. that have been linked to lung disease (123). This acute lung pathology can lead to chronic lung abnormalities. Nippostrongylus larval migration causes destruction of alveoli, long-term airway hyperresponsiveness and chronic low level hemorrhaging in the lungs through chronically activated AAM and release of matrix metalloproteinase 12 (MMP-12; macrophage metalloelastase) (21). The release of high levels of IL-13 causes persistent type-2 immune polarization of macrophages leading to on-going, chronic lung damage over months (14). However, these acute and chronic changes in the lungs are likely developmental stage- and helminth-specific.
While some larval stages induce end organ disease within the lungs, others, particularly those without larval migration through the lungs or once the larvae develop into adult worms, induce immunomodulation and may prevent underlying inflammatory disease states. Components of ES product as well as secreted EVs can be associated with immune evasion and immunomodulatory mechanisms (5). Helminths can produce mediators within the ES product that influence regulatory immunity using immune mimicry through production of TGFβ cyotkine-like molecules and altering the gut microbiome to promote Treg differentiation (38, 124). Additionally, helminth derived chemokine-like molecules such as IPSE/alpha-1 have also been linked to immune modulation (70). Ascaris ES product at larval and adult stages contains protein-1 of Ascaris suum (PAS-1) which contributes to increased concentrations of IL-10, a cytokine that is both made by and influences the generation of Treg and thus an immunomodulatory environment (125). Likewise, anti-inflammatory protein-2 (AIP-2) secreted from hookworms, has also been noted to enhance Treg and suppress airway inflammation. This influence over the host immune response balance can lead to long-term protection of lung structure and function (14). Animal models of Heligmosomoides polygyrus, a non-pathogenic helminthic infection in the gastrointestinal tract, has been shown to attenuate airway inflammation (38, 126, 127). Helminth-derived immunomodulatory molecules can influence all facets of the host immune response including cytokine and chemokine signaling and gene expression (126). This immunomodulation and immune-mimicry allows parasites to live within the host for extended time periods. Further characterization of helminthic molecules and their cognate host receptors may contribute knowledge in the development of future therapeutic approaches in the treatment of diverse inflammatory conditions.
Conclusions
Helminth infections remain a significant global issue impacting the lives of billions of people. Several human helminth infections lead to profound morbidity including pulmonary disease. Conversely, some helminths may prevent the development of pulmonary disease. This dichotomy in outcomes in the lungs and the global impact on pulmonary disease is likely helminth-species and helminth-stage specific. Dissecting the intricacies of the parasite-host pulmonary immune response is critical knowledge in order to develop therapeutic strategies to reduce helminth burden worldwide.
Author Contributions
JW developed, researched, wrote, edited, and reviewed the manuscript. PG-G wrote, edited, and reviewed the manuscript. JK wrote, edited, and reviewed the manuscript. RF reviewed the manuscript. PH edited and reviewed the manuscript. MB edited and reviewed the manuscript. DC edited and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Work for this manuscript was supported by NIH NIAID K08 AI143968 01A1 supported by the Division of Intramural Research, NIAID, NIH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with several of the authors PH, MB.
References
1. GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional and national incidence, prevalence, and years lived with disability fo 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet (2018) 392:1789–858. doi: 10.1016/S0140-6736(18)32279-7
2. GBD 2017 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 359 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet (2018) 392:1859–922. doi: 10.1016/S0140-6736(18)32335-3
3. Blair D. Paragonimiasis. in Advances in Experimental Medicine and Biology Vol. 1154. New York LLC: Springer (2019) p. 105–38. doi: 10.1007/978-3-030-18616-6_5
4. Van Dyken SJ, Mohapatra A, Nussbaum JC, Molofsky AB, Thornton EE, Ziegler Steven F, et al. Chitin activates parallel immune modules that direct distinct inflammatory responses via innate lymphoid type 2 and γδ T cells. Immunity (2014) 40:414–24. doi: 10.1016/j.immuni.2014.02.003
5. Zakeri A, Hansen EP, Andersen SD, Williams AR, Nejsum P. Immunomodulation by helminths: Intracellular pathways and extracellular vesicles. Front Immunol (2018) 9:2349. doi: 10.3389/fimmu.2018.02349
6. Lightowlers MW, Rickard MD. Excretory-secretory products of helminth parasites: Effects on host immune responses. Parasitology (1988) 96:S123–66. doi: 10.1017/S0031182000086017
7. Coakley G, McCaskill JL, Borger JG, Simbari F, Robertson E, Millar M, et al. Extracellular Vesicles from a Helminth Parasite Suppress Macrophage Activation and Constitute an Effective Vaccine for Protective Immunity. Cell Rep (2017) 19:1545–57. doi: 10.1016/j.celrep.2017.05.001
8. Wangchuk P, Kouremenos K, Eichenberger RM, Pearson M, Susianto A, Wishart DS, et al. Metabolomic profiling of the excretory-secretory products of hookworm and whipworm. Metabolomics (2019) 15:101. doi: 10.1007/s11306-019-1561-y
9. Sotillo J, Sanchez-Flores A, Cantacessi C, Harcus Y, Pickering J, Bouchery T, et al. Secreted proteomes of different developmental stages of the gastrointestinal nematode Nippostrongylus brasiliensis. Mol Cell Proteomics (2014) 13:2736–51. doi: 10.1074/mcp.M114.038950
10. Wang T, Van Steendam K, Dhaenens M, Vlaminck J, Deforce D, Jex AR, et al. Proteomic analysis of the excretory-secretory products from larval stages of Ascaris suum reveals high abundance of glycosyl hydrolases. PloS Negl Trop Dis (2013) 7:e2467. doi: 10.1371/journal.pntd.0002467
11. Motran CC, Silvane L, Chiapello LS, Theumer MG, Ambrosio LF, Volpini X, et al. Helminth infections: Recognition and modulation of the immune response by innate immune cells. Front Immunol (2018) 9:664. doi: 10.3389/fimmu.2018.00664
12. Mekonnen GG, Pearson M, Loukas A, Sotillo J. Extracellular vesicles from parasitic helminths and their potential utility as vaccines. Expert Rev Vaccines (2018) 17:197–205. doi: 10.1080/14760584.2018.1431125
13. Craig JM, Scott AL. Helminths in the lungs. Parasite Immunol (2014) 36:463–74. doi: 10.1111/pim.12102
14. Schwartz C, Hams E, Fallon PG. Helminth Modulation of Lung Inflammation. Trends Parasitol (2018) 34(5):388–403. doi: 10.1016/j.pt.2017.12.007
15. Ferraz Frezza T. Helminthiasis and its Relationship with Lung Symptoms in Humans. EC Pulm Respir Med (2019) 8.3:217–26.
16. Kunst H, Mack D, Kon OM, Banerjee AK, Chiodini P, Grant A. Parasitic infections of the lung: a guide for the respiratory physician. Thorax (2011) 66:528–36. doi: 10.1136/thx.2009.132217
17. Mulcahy G, O’Neill S, Fanning J, McCarthy E, Sekiya M. Tissue migration by parasitic helminths - An immunoevasive strategy? Trends Parasitol (2005) 21:273–7. doi: 10.1016/j.pt.2005.04.003
18. Oliveira FMS, Matias PHdP, Kraemer L, Gazzinelli-Guimarães AC, Santos FV, Amorim CCO, et al. Comorbidity associated to ascaris suum infection during pulmonary fibrosis exacerbates chronic lung and liver inflammation and dysfunction but not affect the parasite cycle in mice. PloS Negl Trop Dis (2019) 13:1–30. doi: 10.1371/journal.pntd.0007896
19. Chitkara R, Krishna G. Parasitic Pulmonary Eosinophilia. Semin Respir Crit Care Med (2006) 27:171–84. doi: 10.1055/s-2006-939520
20. Briggs N, Weatherhead J, Sastry KJ, Hotez PJ. The Hygiene Hypothesis and Its Inconvenient Truths about Helminth Infections. PloS Negl Trop Dis (2016) 10:e0004944. doi: 10.1371/journal.pntd.0004944
21. Marsland BJ, Kurrer M, Reissmann R, Harris NL, Kopf M. Nippostrongylus brasiliensis infection leads to the development of emphysema associated with the induction of alternatively activated macrophages. Eur J Immunol (2008) 38:479–88. doi: 10.1002/eji.200737827
22. Pinkston P, Vijayan VK, Nutman TB, Rom WN, O'Donnell KM, Cornelius MJ, et al. Acute tropical pulmonary eosinophilia. Characterization of the lower respiratory tract inflammation and its response to therapy. J Clin Invest (1987) 80:216–25. doi: 10.1172/JCI113050
23. Ottesen E, Nutman T. Tropical Pulmonary Eosinophilia. Annu Rev Med (1992) 43:417–24. doi: 10.1146/annurev.me.43.020192.002221
24. Mullerpattan JB, Udwadia ZF, Udwadia FE. Tropical pulmonary eosinophilia - A review. Indian J Med Res (2013) 138:295–302.
25. Gobert GN, Chai M, McManus DP. Biology of the schistosome lung-stage schistosomulum. Parasitology (2007) 134:453–60. doi: 10.1017/S0031182006001648
26. Ross AG, Vickers D, Olds GR, Shah SM, McManus DP. Katayama syndrome. Lancet Infect Dis (2007) 7:218–24. doi: 10.1016/S1473-3099(07)70053-1
27. Gobbi F, Tamarozzi F, Buonfrate D, Van Lieshout L, Bisoffi Z, Bottieau E. New Insights on Acute and Chronic Schistosomiasis: Do We Need a Redefinition? on behalf of TropNet Schisto Task Force The Pathophysiology and Duration of the Early Phase of Schistosomiasis Are Still Poorly Defined. Trends Parasitol (2020) 36:660–7. doi: 10.1016/j.pt.2020.05.009
28. Knafl D, Gerges C, King CH, Humbert M, Bustinduy AL. Schistosomiasis-associated pulmonary arterial hypertension: A systematic review. Eur Respir Rev (2020) 29:190089. doi: 10.1183/16000617.0089-2019
29. Fernandes CJCDS, Jardim CVP, Hovnanian A, Hoette S, Morinaga LK, Souza R. Schistosomiasis and pulmonary hypertension. Expert Rev Respir Med (2011) 5:675–81. doi: 10.1586/ers.11.58
30. Lall M, Sahni AK, Rajput AK. Pleuropulmonary paragonimiasis: Mimicker of tuberculosis. Pathog Glob Health (2013) 107:40–2. doi: 10.1179/2047773212Y.0000000067
31. Nieuwenhuizen N, Lopata AL, Jeebhay MF, Herbert BR, Robins TG, Brombacher F, et al. Food allergy, dermatologic diseases, and anaphylaxis Exposure to the fish parasite Anisakis causes allergic airway hyperreactivity and dermatitis. J Allergy Clin Immunol (2006) 117:1098–105. doi: 10.1016/j.jaci.2005.12.1357
32. Diaz JH, Warren RJ, Oster MJ. The Disease Ecology, Epidemiology, Clinical Manifestations, and Management of Trichinellosis Linked to Consumption of Wild Animal Meat. Wilderness Environ Med (2020) 31:235–44. doi: 10.1016/j.wem.2019.12.003
33. Gottstein B, Pozio E, Nöckler K. Epidemiology, Diagnosis, Treatment, and Control of Trichinellosis. Clin Microbiol Rev (2009) 22:127–45. doi: 10.1128/CMR.00026-08
34. Gelpi AP, Mustafa A. Seasonal pneumonitis with eosinophilia. A study of larval ascariasis in Saudi Arabs. Am J Trop Med Hyg (1967) 16:646–57. doi: 10.4269/ajtmh.1967.16.646
35. Negrão-Corrêa D, Silveira MR, Borges CM, Souza DG, Teixeira MM. Changes in pulmonary function and parasite burden in rats infected with Strongyloides venezuelensis concomitant with induction of allergic airway inflammation. Infect Immun (2003) 71:2607–14. doi: 10.1128/IAI.71.5.2607-2614.2003
36. Weatherhead JE, Porter P, Coffey A, Haydel D, Versteeg L, Zhan B, et al. Ascaris Larval Infection and Lung Invasion Directly Induces Severe Allergic Airway Disease in Mice. Infect Immun (2018) IAI.00533-18:e00533-18. doi: 10.1128/IAI.00533-18
37. Perrigoue JG, Marshall FA, Artis D. On the hunt for helminths: Innate immune cells in the recognition and response to helminth parasites. Cell Microbiol (2008) 10:1757–64. doi: 10.1111/j.1462-5822.2008.01174.x
38. Schwartz C, Hams E, Fallon PG. Helminth Modulation of Lung Inflammation. Trends Parasitol (2018) 34:388–403. doi: 10.1016/j.pt.2017.12.007
39. Bonne-Anneé S, Hess JA, Abraham D. Innate and adaptive immunity to the nematode Strongyloides stercoralis in a mouse model. Immunol Res (2011) 51:205–14. doi: 10.1007/s12026-011-8258-2
40. Giacomin PR, Gordon DL, Botto M, Daha MR, Sanderson SD, Taylor SM, et al. The role of complement in innate, adaptive and eosinophil-dependent immunity to the nematode Nippostrongylus brasiliensis. Mol Immunol (2008) 45:446–55. doi: 10.1016/j.molimm.2007.05.029
41. Kerepesi LA, Hess JA, Nolan TJ, Schad GA, Abraham D. Complement Component C3 Is Required for Protective Innate and Adaptive Immunity to Larval Strongyloides stercoralis in Mice. J Immunol (2006) 176:4315–22. doi: 10.4049/jimmunol.176.7.4315
42. Maya A, Jayaraman K, Balakrishnan A. Necrosis of lung epithelial cells by filarial parasitic protein via an early induction of c-H-ras and TNFα expression. Cell Biol Int (1997) 21:273–80. doi: 10.1006/cbir.1997.0139
43. Inclan-Rico JM, Siracusa MC. First Responders: Innate Immunity to Helminths. Trends Parasitol (2018) 34:861–80. doi: 10.1016/j.pt.2018.08.007
44. Townsend MJ, Fallon PG, Matthews DJ, Smith P, Jolin HE, McKenzie ANJ. IL-9-deficient mice establish fundamental roles for IL-9 in pulmonary mastocytosis and goblet cell hyperplasia but not T cell development. Immunity (2000) 13:573–83. doi: 10.1016/S1074-7613(00)00056-X
45. Horsnell WGC, Vira A, Kirstein F, Mearns H, Hoving JC, Cutler AJ, et al. IL-4Rα-responsive smooth muscle cells contribute to initiation of TH2 immunity and pulmonary pathology in Nippostrongylus brasiliensis infections. Mucosal Immunol (2011) 4:83–92. doi: 10.1038/mi.2010.46
46. Horsnell WGC, Cutler AJ, Hoving CJ, Mearns H, Myburgh E, Arendse B, et al. Delayed Goblet Cell Hyperplasia, Acetylcholine Receptor Expression, and Worm Expulsion in SMC-Specific IL-4Rα-Deficient Mice. PloS Pathog (2007) 3:e1. doi: 10.1371/journal.ppat.0030001
47. Sutherland TE, Rückerl D, Logan N, Duncan S, Wynn TA, Allen JE. Ym1 induces RELMα and rescues IL-4Rα deficiency in lung repair during nematode infection. PloS Pathog (2018) 14:1–27. doi: 10.1371/journal.ppat.1007423
48. Reid L, Meyrick B, Antony VB, Chang LY, Crapo JD, Reynolds HY. The mysterious pulmonary brush cell: A cell in search of a function. Am J Respir Crit Care Med (2005) 172:136–9. doi: 10.1164/rccm.200502-203WS
49. Grencis RK, Worthington JJ. Tuft Cells: A New Flavor in Innate Epithelial Immunity. Trends Parasitol (2016) 32:583–5. doi: 10.1016/j.pt.2016.04.016
50. Klose CSN, Mahlakõiv T, Moeller JB, Rankin LC, Flamar A-L, Kabata H, et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nat Publ Gr (2017) 549:282–6. doi: 10.1038/nature23676
51. Mindt BC, Fritz JH, Duerr CU. Group 2 innate lymphoid cells in pulmonary immunity and tissue homeostasis. Front Immunol (2018) 9:840. doi: 10.3389/fimmu.2018.00840
52. Lazari O, Kipar A, Johnson DR, Selkirk ME, Matthews JB. Up-regulated expression of the α 7 nicotinic acetylcholine receptor subunit on inflammatory infiltrates during Dictyocaulus viviparus infection. Parasite Immunol (2006) 28:429–37. doi: 10.1111/j.1365-3024.2006.00873.x
53. Hussein AS, Harel M, Selkirk ME. A distinct family of acetylcholinesterases is secreted by Nippostrongylus brasiliensis. Mol Biochem Parasitol (2002) 123:125–34. doi: 10.1016/S0166-6851(02)00141-X
54. Cardoso V, Chesné J, Ribeiro H, Garcia-Cassani B, Carvalho T, Bouchery T, et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature (2017) 549:277–81. doi: 10.1038/nature23469
55. Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour REE, Nyman J, Dionne D, et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature (2017) 549:351–6. doi: 10.1038/nature24029
56. Nagashima H, Mahlakõ T, Shih H-Y, Kanno Y, Artis D, O' JJ. Neuropeptide CGRP Limits Group 2 Innate Lymphoid Cell Responses and Constrains Type 2 Inflammation. Immunity (2009) 51:682–95. doi: 10.1016/j.immuni.2019.06.009
57. Dahlgren MW, Jones SW, Cautivo KM, Dubinin A, Ortiz-Carpena JF, Farhat S, et al. Adventitial Stromal Cells Define Group 2 Innate Lymphoid Cell Tissue Niches. Immunity (2019) 50:707–722.e6. doi: 10.1016/j.immuni.2019.02.002
58. Miller MM, Patel PS, Bao K, Danhorn T, O'Connor BP, Reinhardt RL, et al. BATF acts as an essential regulator of IL-25-responsive migratory ILC2 cell fate and function. Sci Immunol (2020) 5(43):eaay3994. doi: 10.1126/sciimmunol.aay3994
59. Symowski C, Voehringer D. Th2 cell-derived IL-4/IL-13 promote ILC2 accumulation in the lung by ILC2-intrinsic STAT6 signaling in mice. Eur J Immunol (2019) 49:1421–32. doi: 10.1002/eji.201948161
60. Webb LM, Wojno EDT. The role of rare innate immune cells in Type 2 immune activation against parasitic helminths. Parasitology (2017) 144:1288–301. doi: 10.1017/S0031182017000488
61. Huang Y, Mao K, Chen X, Sun MA, Kawabe T, Li W, et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Sci (80- ) (2018) 359:114–9. doi: 10.1126/science.aam5809
62. Tait Wojno E, Monticelli LA, Tran SV, Alenghat T, Osborne LC, Thome JJ, et al. The prostaglandin D2 receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol (2015) 8:1313–23. doi: 10.1038/mi.2015.21
63. Oyesola OO, Duque C, Huang LC, Larson EM, Früh SP, Webb LM, et al. The Prostaglandin D 2 Receptor CRTH2 Promotes IL-33-Induced ILC2 Accumulation in the Lung. J Immunol (2020) 204:1001–11. doi: 10.4049/jimmunol.1900745
64. Voehringer D, Reese TA, Huang X, Shinkai K, Locksley RM. Type 2 immunity is controlled by IL-4/IL-13 expression in hematopoietic non-eosinophil cells of the innate immune system. J Exp Med (2006) 203:1435–46. doi: 10.1084/jem.20052448
65. Voehringer D. The role of basophils in helminth infection. Trends Parasitol (2009) 25:551–6. doi: 10.1016/j.pt.2009.09.004
66. Min B, Prout M, Hu-Li J, Zhu J, Jankovic D, Morgan ES, et al. Basophils produce IL-4 and accumulate in tissues after infection with a Th2-inducing parasite. J Exp Med (2004) 200:507–17. doi: 10.1084/jem.20040590
67. Lantz CS, Boeslger J, Song CH, Mach N, Kobayashi T, Mulligan RC, et al. Role for interleukin-3 in mast cell and basophil development and in immunity to parasites. Nature (1998) 392:90–3. doi: 10.1038/32190
68. Falcone FH, Dahinden CA, Gibbs BF, Noll T, Amon U, Hebestreit H, et al. Human basophils release interleukin-4 after stimulation with Schistosoma mansoni egg antigen. Eur J Immunol (1996) 26:1147–55. doi: 10.1002/eji.1830260528
69. Sullivan BM, Liang H-E, Bando JK, Wu D, Cheng LE, Mckerrow JK, et al. Genetic analysis of basophil function in vivo. Nature (2011) 12:527–35. doi: 10.1038/ni.2036
70. Knuhr K, Langhans K, Nyenhuis S, Viertmann K, Overgaard Kildemoes AM, Doenhoff MJ, et al. Schistosoma mansoni egg-released IPSE/alpha-1 dampens inflammatory cytokine responses viabasophil interleukin (IL)-4 and IL-13. Front Immunol (2018) 9:2293. doi: 10.3389/fimmu.2018.02293
71. Perrigoue JG, Saenz SA, Siracusa MC, Allenspach EJ, Taylor BC, Giacomin PR, et al. MHC class II-dependent basophil-CD4+ T cell interactions promote TH2 cytokine-dependent immunity. Nat Immunol (2009) 10:697–705. doi: 10.1038/ni.1740
72. Webb LM, Oyesola OO, Früh SP, Kamynina E, Still KM, Patel RK, et al. The Notch signaling pathway promotes basophil responses during helminth-induced type 2 inflammation. J Exp Med (2019) 216:1268–79. doi: 10.1084/jem.20180131
73. Gurish MF, Boyce JA. Mast cells: Ontogeny, homing, and recruitment of a unique innate effector cell. J Allergy Clin Immunol (2006) 117:1285–91. doi: 10.1016/j.jaci.2006.04.017
74. Padigel UM, Hess JA, Lee JJ, Lok JB, Nolan TJ, Schad GA, et al. Eosinophils Act as Antigen-Presenting Cells to Induce Immunity to Strongyloides stercoralis in Mice. J Infect Dis (2007) 196:1844–51. doi: 10.1086/522968
75. Cadman ET, Lawrence RA. Granulocytes: Effector cells or immunomodulators in the immune response to helminth infection? Parasite Immunol (2010) 32:1–19. doi: 10.1111/j.1365-3024.2009.01147.x
76. Culley FJ, Brown A, Girod N, Pritchard D II, Williams TJ. Innate and cognate mechanisms of pulmonary eosinophilia in helminth infection. Eur J Immunol (2002) 32:1376–85. doi: 10.1002/1521-4141(200205)32:5<1376::AID-IMMU1376>3.0.CO;2-8
77. O’Connell AE, Hess JA, Santiago GA, Nolan TJ, Lok JB, Lee JJ, et al. Major basic protein from eosinophils and myeloperoxidase from neutrophils are required for protective immunity to Strongyloides stercoralis in mice. Infect Immun (2011) 79:2770–8. doi: 10.1128/IAI.00931-10
78. Ueki S, Konno Y, Takeda M, Moritoki Y, Hirokawa M, Matsuwaki Y, et al. Eosinophil extracellular trap cell death-derived DNA traps: Their presence in secretions and functional attributes. J Allergy Clin Immunol (2016) 137:258–67. doi: 10.1016/j.jaci.2015.04.041
79. Gazzinelli-Guimaraes PH, De Queiroz Prado R, Ricciardi A, Bonne-Année S, Sciurba J, Karmele EP, et al. Allergen presensitization drives an eosinophil-dependent arrest in lung-specific helminth development. J Clin Invest (2019) 129:3686–701. doi: 10.1172/JCI127963
80. Chen F, Wu W, Millman A, Craft JF, Chen E, Patel N, et al. Neutrophils prime a long-lived effector macrophage phenotype that that Mediates accelerated helminth exxpulsion. Nat Immunol (2014) 15:938–46. doi: 10.1038/ni.2984
81. Sutherland TE, et al. Chitinase-like proteins promote IL-17-mediated neutrophilia in a tradeoff between nematode killing and host damage. Nat Immunol (2014) 15:1116–25. doi: 10.1038/ni.3023
82. Cho MK, Ahn SC, Kim DH, Yu HS. Parasite excretory-secretory proteins elicit TRIF dependent CXCL1 and IL-6 mediated allergic inflammation. Parasite Immunol (2010) 32:354–60. doi: 10.1111/j.1365-3024.2009.01195.x
83. Hsieh GC-F, Loukas A, Wahl AM, Bhatia M, Wang Y, Williamson AL, et al. A Secreted Protein from the Human Hookworm Necator americanus Binds Selectively to NK Cells and Induces IFN-γ Production. J Immunol (2004) 173:2699–704. doi: 10.4049/jimmunol.173.4.2699
84. Korten S, Volkmann L, Saeftel M, Fischer K, Taniguchi M, Fleischer B, et al. Expansion of NK Cells with Reduction of Their Inhibitory Ly-49A, Ly-49C, and Ly-49G2 Receptor-Expressing Subsets in a Murine Helminth Infection: Contribution to Parasite Control. J Immunol (2002) 168:5199–206. doi: 10.4049/jimmunol.168.10.5199
85. Babu S, Blauvelt CP, Nutman TB. Filarial Parasites Induce NK Cell Activation, Type 1 and Type 2 Cytokine Secretion, and Subsequent Apoptotic Cell Death. J Immunol (2007) 179:2445–56. doi: 10.4049/jimmunol.179.4.2445
86. Cha H, Qin W, Yang Q, Xie H, Qu J, Wang M, et al. Differential pulmonic NK and NKT cell responses in Schistosoma japonicum-infected mice. Parasitol Res (2017) 116:559–67. doi: 10.1007/s00436-016-5320-y
87. White RR, Artavanis-Tsakonas K. How helminths use excretory secretory fractions to modulate dendritic cells. Virulence (2012) 3:668–77. doi: 10.4161/viru.22832
88. Cardenas PA, Cooper PJ, Cox MJ, Chico M, Arias C, Moffatt MF, et al. Upper Airways Microbiota in Antibiotic-Naïve Wheezing and Healthy Infants from the Tropics of Rural Ecuador. PloS One (2012) 7:e46803. doi: 10.1371/journal.pone.0046803
89. Krljanac B, Schubart C, Naumann R, Wirtz S, Culemann S, Krönke G, et al. RELM-expressing macrophages protect against fatal lung damage and reduce parasite burden during helminth infection. Sci Immunol (2019) 4:1–12. doi: 10.1126/sciimmunol.aau3814
90. Reece JJ, Siracusa MC, Scott AL. Innate immune responses to lung-stage helminth infection induce alternatively activated alveolar macrophages. Infect Immun (2006) 74:4970–81. doi: 10.1128/IAI.00687-06
91. Joseph M, Auriault C, Capron M, Ameisen J-C, Pancré V, Torpier G, et al. IgE-Dependent Platelet cytotoxicity against helminths. In: Henkart P, Martz E, editors. Advances in Experimental MEdicine and Biology. New York, NY: Springer (1985). p. 184.
92. Yang Y, Wen YJ, Cai YN, Vallée I, Boireau P, Liu MY, et al. Serine proteases of parasitic helminths. Korean J Parasitol (2015) 53:1–11. doi: 10.3347/kjp.2015.53.1.1
93. Palucka K, Banchereau J. Linking innate and adaptive immunity. Nat Med (1999) 5:868–70. doi: 10.1038/11303
94. Nuyen Q, Deng T, Witherden D, Goldrath A. Origins of CD4 circulating and tissue-resident memory T-cells. Immunology (2019) 157:3–12. doi: 10.1111/imm.13059
95. Anthony RM, Rutitzky L II, Urban JF Jr., Standecker MJ, Gause WC. Protective immune mechanisms in helminth infection. Nat Rev Immunol (2007) 7:975–87. doi: 10.1038/nri2199
96. Zhao Y, Yang Q, Jin C, Feng Y, Xie S, Xie H, et al. Changes of CD103-expressing pulmonary CD4 + and CD8 + T cells in S. japonicum infected C57BL/6 mice. BMC Infect Dis (2019) 19:999. doi: 10.1186/s12879-019-4633-8
97. Allen JE, Sutherland TE. Host protective roles of type 2 immunity: Parasite killing and tissue repair, flip sides of the same coin. Semin Immunol (2014) 26:329–40. doi: 10.1016/j.smim.2014.06.003
98. Vannella KM, Ramalingam TR, Borthwick LA, Barron L, Hart KM, Thompson RW, et al. Combinatorial targeting of TSLP, IL-25, and IL-33 in type 2 cytokine-driven inflammation and fibrosis. Sci Transl Med (2016) 8:337ra65–337ra65. doi: 10.1126/scitranslmed.aaf1938
99. Yazdanbakhsh M, van den Biggelaar A, Maizels R. Th2 responses without atopy: immunoregulation in chronic helminth infections and reduced allergic disease. Trends Immunol (2001) 22:P372–377. doi: 10.1016/S1471-4906(01)01958-5
100. Roboz G, Rafii S. Interleukin-5 and the Regulation of Eosinophil Production. Curr Opin Hematol (1999) 5:164–8. doi: 10.1097/00062752-199905000-00007
101. Lalani T, Simmons R, Ahmed A. biology of IL-5 in health and disease. Ann Allergy Asthma Immunol (1999) 82:P331–333. doi: 10.1016/S1081-1206(10)63281-4
102. Herbert DR, Lee JJ, Lee NA, Nolan TJ, Schad GA, Abraham D. Role of IL-5 in Innate and Adaptive Immunity to Larval Strongyloides stercoralis in Mice. J Immunol (2000) 165:4544–51. doi: 10.4049/jimmunol.165.8.4544
103. Thawer SG, Horsnell WGC, Darby M, Hoving JC, Dewals B, Cutler AJ, et al. Lung-resident CD4+ T cells are sufficient for IL-4R-dependent recall immunity to Nippostrongylus brasiliensis infection. Mucosal Immunol (2014) 7:239–48. doi: 10.1038/mi.2013.40
104. Obata-Ninomiya K, Ishiwata K, Nakano H, Endo Y, Ichikawa T, Onodera A, et al. CXCR6+ST2+ memory T helper 2 cells induced the expression of major basic protein in eosinophils to reduce the fecundity of helminth. Proc Natl Acad Sci U S A (2018) 115:E9849–58. doi: 10.1073/pnas.1714731115
105. Dell’aringa M, Lee Reinhardt R. Notch signaling represents an important checkpoint between follicular T-helper and canonical T-helper 2 cell fate article. Mucosal Immunol (2018) 11:1079–91. doi: 10.1038/s41385-018-0012-9
106. Meli AP, Fontés G, Leung Soo C, King IL. T Follicular Helper Cell-Derived IL-4 Is Required for IgE Production during Intestinal Helminth Infection. J Immunol (2017) 199:244–52. doi: 10.4049/jimmunol.1700141
107. Gazzinelli-Guimarães AC, Gazzinelli-Guimarães PH, Nogueira DS, Oliveira FMS, Barbosa FS, Amorim CCO, et al. IgG induced by vaccination with ascaris suumExtracts is protective against infection. Front Immunol (2018) 9:2535. doi: 10.3389/fimmu.2018.02535
108. Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Sci (80- ) (2017) 356:1072–6. doi: 10.1126/science.aai8132
109. Thawer S, Auret J, Schnoeller C, Chetty A, Smith K, Darby M, et al. Surfactant Protein-D Is Essential for Immunity to Helminth Infection. PloS Pathog (2016) 12:1–18. doi: 10.1371/journal.ppat.1005461
110. Loke P, Gallagher I, Nair MG, Zang X, Brombacher F, Mohrs M, et al. Alternative Activation Is an Innate Response to Injury That Requires CD4 + T Cells to be Sustained during Chronic Infection. J Immunol (2007) 179:3926–36. doi: 10.4049/jimmunol.179.6.3926
111. McSorley HJ, Maizels RM. Helminth infections and host immune regulation. Clin Microbiol Rev (2012) 25:585–608. doi: 10.1128/CMR.05040-11
112. Boer MC, Joosten SA, Ottenhoff THM. Regulatory T-cells at the interface between human host and pathogens in infectious diseases and vaccination. Front Immunol (2015) 6:217. doi: 10.3389/fimmu.2015.00217
113. Weaver CT, Elson CO, Fouser LA, Kolls JK. The Th17 Pathway and Inflammatory Diseases of the Intestines, Lungs, and Skin. Annu Rev Pathol Mech Dis (2013) 8:477–512. doi: 10.1146/annurev-pathol-011110-130318
114. Ajendra J, Chenery AL, Parkinson JE, Chan BHK, Pearson S, Colombo SAP, et al. IL-17A both initiates, via IFNγ suppression, and limits the pulmonary type-2 immune response to nematode infection. Mucosal Immunol (2020). doi: 10.1038/s41385-020-0318-2
115. Hotez PJ, Bottazzi ME, Corry DB. The potential role of Th17 immune responses in coronavirus immunopathology and vaccine-induced immune enhancement. Microbes Infect (2020) 22:165. doi: 10.1016/j.micinf.2020.04.005
116. Gurczynski SJ, Moore BB. IL-17 in the lung: The good, the bad, and the ugly. Am J Physiol - Lung Cell Mol Physiol (2018) 314:L6–L16. doi: 10.1152/ajplung.00344.2017
117. Bouchery T, Kyle R, Ronchese F, Le Gros G. The differentiation of CD4+ T-helper cell subsets in the context of helminth parasite infection. Front Immunol (2014) 5:487. doi: 10.3389/fimmu.2014.00487
118. Filbey KJ, Camberis M, Chandler J, Turner R, Kettle AJ, Eichenberger RM, et al. Intestinal helminth infection promotes IL-5- and CD4 + T cell-dependent immunity in the lung against migrating parasites. Mucosal Immunol (2019) 12:352–62. doi: 10.1038/s41385-018-0102-8
119. Diemert DJ, Bottazzi ME, Plieskatt J, Hotez PJ, Bethony JM. Lessons along the Critical Path: Developing Vaccines against Human Helminths. Trends Parasitol (2018) 34:747–58. doi: 10.1016/j.pt.2018.07.005
120. Hotez PJ, Strych U, Lustigman S, Bottazzi ME. Human anthelminthic vaccines: Rationale and challenges. Vaccine (2016) 34:3549–55. doi: 10.1016/j.vaccine.2016.03.112
121. Coulson PS, Wilson RA. Recruitment of lymphocytes to the lung through vaccination enhances the immunity of mice exposed to irradiated schistosomes. Infect Immun (1997) 65:42–8. doi: 10.1128/IAI.65.1.42-48.1997
122. Versteeg L, Wei J, Liu Z, Keegan B, Fujiwara RT, Jones KM, et al. Protective immunity elicited by the nematode-conserved As37 recombinant protein against Ascaris suum infection. PloS Negl Trop Dis (2020) 14:e0008057. doi: 10.1371/journal.pntd.0008057
123. Caraballo L, Coronado S. Parasite allergens. Mol Immunol (2018) 100:113–9. doi: 10.1016/j.molimm.2018.03.014
124. Weinstock JV. Helminths and mucosal immune modulation. Ann N Y Acad Sci (2006) 1072:356–64. doi: 10.1196/annals.1326.033
125. Antunes MFP, Titz TO, Batista IFC, Marques-Porto R, Oliveira CF, Alves De Araujo CA, et al. Immunosuppressive PAS-1 is an excretory/secretory protein released by larval and adult worms of the ascarid nematode Ascaris suum. J Helminthol (2015) 89:367–74. doi: 10.1017/S0022149X14000200
126. Maizels RM, Smits HH, McSorley HJ. Modulation of Host Immunity by Helminths: The Expanding Repertoire of Parasite Effector Molecules. Immunity (2018) 49:801–18. doi: 10.1016/j.immuni.2018.10.016
Keywords: hookworm, Ascaris, Schistosoma, lung pathologies, inflammation, host immune response
Citation: Weatherhead JE, Gazzinelli-Guimaraes P, Knight JM, Fujiwara R, Hotez PJ, Bottazzi ME and Corry DB (2020) Host Immunity and Inflammation to Pulmonary Helminth Infections. Front. Immunol. 11:594520. doi: 10.3389/fimmu.2020.594520
Received: 13 August 2020; Accepted: 30 September 2020;
Published: 20 October 2020.
Edited by:
Paul Giacomin, James Cook University, AustraliaReviewed by:
James Hewitson, University of York, United KingdomTomabu Adjobimey, University Hospital Bonn, Germany
Roland Ruscher, James Cook University, Australia
Copyright © 2020 Weatherhead, Gazzinelli-Guimaraes, Knight, Fujiwara, Hotez, Bottazzi and Corry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jill E. Weatherhead, d2VhdGhlcmhAYmNtLmVkdQ==