 Patrycja A. Krawczyk
Patrycja A. Krawczyk Marco Laub
Marco Laub Patrycja Kozik
Patrycja Kozik- MRC Laboratory of Molecular Biology, Protein and Nucleic Acid Chemistry Division, Cambridge Biomedical Campus, Cambridge, United Kingdom
Pore-forming proteins (PFPs) are present in all domains of life, and play an important role in host-pathogen warfare and in the elimination of cancers. They can be employed to deliver specific effectors across membranes, to disrupt membrane integrity interfering with cell homeostasis, and to lyse membranes either destroying intracellular organelles or entire cells. Considering the destructive potential of PFPs, it is perhaps not surprising that mechanisms controlling their activity are remarkably complex, especially in multicellular organisms. Mammalian PFPs discovered to date include the complement membrane attack complex (MAC), perforins, as well as gasdermins. While the primary function of perforin-1 and gasdermins is to eliminate infected or cancerous host cells, perforin-2 and MAC can target pathogens directly. Yet, all mammalian PFPs are in principle capable of generating pores in membranes of healthy host cells which—if uncontrolled—could have dire, and potentially lethal consequences. In this review, we will highlight the strategies employed to protect the host from destruction by endogenous PFPs, while enabling timely and efficient elimination of target cells.
Introduction and Overview
The emergence of cell membranes was critical for the evolution of all modern organisms. They provide a physical barrier to separate an organism from its environment and enable compartmentalization of biochemical processes inside cells. In modern multicellular organisms, interfering with membrane integrity is one the most effective strategies employed in immune defense, and membrane disrupting pore-forming proteins (PFPs) have evolved as key effectors in both innate and adaptive immune responses.
PFPs can be found in all kingdoms of life. Bacteria use them to facilitate their entry into cells (e.g., listeriolysin), to aid in the delivery of effector molecules across membranes (e.g., streptolysin O) or as toxic agents (e.g., diphtheria or anthrax toxins) (1). Eukaryotic multicellular organisms, including mammals, use PFPs as either membranolytic pores assembled directly on the surface of invading pathogens or as effectors to selectively eliminate infected or cancerous host cells (2, 3). While bacteria can specifically target eukaryotic membranes through recognition of host-specific molecules, mammals are faced with the more challenging task of eliminating unwanted cells without accidentally damaging surrounding healthy tissues. In fact, mammalian PFPs evolved to show limited target membrane specificity in isolation and therefore depend on other proteins of the immune system to safely guide their activity.
In this review, we discuss how mammalian PFPs are controlled by both the innate and adaptive arms of the immune system. Our goal is to provide a comprehensive overview of the variety of mechanisms, ranging from inducible expression and regulated trafficking to post-translational modifications and proteolytic processing, that collectively ensure tight spatial and temporal regulation of pore formation during immune responses.
Mammalian PFPs in Innate and Adaptive Immunity
Toward the end of the 19th century, George Nuttall and Hans Ernst August Buchner noted that blood contains a heat-sensitive component with killing activity against bacteria (4, 5). The proteins responsible were later named membrane attack complex (MAC) or terminal complement complex. Today we know, that the soluble MAC components C5, C6, C7, C8 (comprising C8α, C8β, and C8γ), and C9 (Figure 1) present in the serum assemble to form membranolytic pores on the surface of Gram-negative bacteria, enveloped viruses, parasites, and host cells (6–10). The mediators that initiate the assembly of MAC pores include components of both the innate and adaptive arms of the immune system. Specific receptors of the complement system are able to recognize a wide range of structures including unique pathogen-associated molecular patterns (PAMPs) and microbes opsonized with antibodies (key components of the humoral adaptive immune system) such that MAC can in principle assemble in response to any antigen. Both, the important contribution of MAC to anti-bacterial immunity as well as its potential toxicity are reflected in population genetics. On the one hand, genetic deficiencies in MAC components are associated with susceptibility to neisserial disease including endemic meningococcal infections (11, 12), on the other hand, these deficiencies can also confer a selective advantage (13).
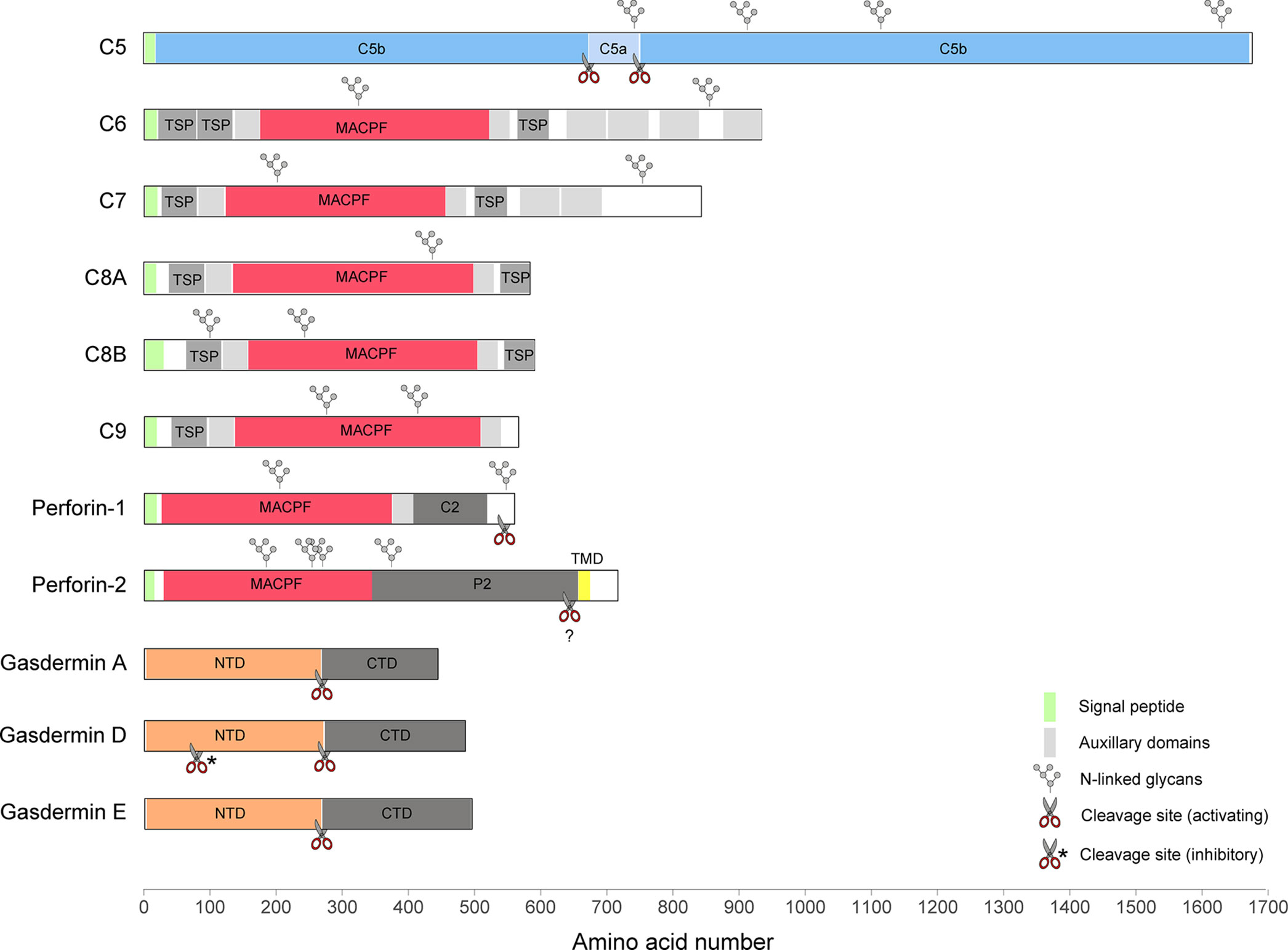
Figure 1 A diagram illustrating the domain structures and selected regulatory features of mammalian PFPs.
Five of the seven MAC subunits (the exceptions being C5 and C8γ) are evolutionarily related and, together with two perforins, they belong to the membrane attack complex/perforin (MACPF) superfamily of PFPs. Perforin-1 (Figure 1) is stored together with pro-apoptotic effectors, granzyme A and B, in specialized secretory granules of natural killer (NK) cells and cytotoxic T cells (CTLs). When released into the immune synapse formed between the cytotoxic lymphocyte and an infected or cancerous cell, it rapidly assembles into homo-oligomeric pores on the target membrane (14). The pores can be lytic at high concentration (15–17), but their primary function is to facilitate entry of granzymes into target cells inducing apoptosis (18). NK cells belong to the innate arm of the immune response and recognize a variety of stress signals presented on the cell surface. In CTLs, key effectors of adaptive immunity, the specificity during immune synapse formation relies on the interaction between the T cell receptor (TCR) and MHC class I complexed with a foreign or a mutated peptide. In line with the fundamental function of perforin-1 in immunity, deleterious variants in the PRF1 gene in humans lead to an aggressive immunoregulatory disorder called familial hemophagocytic lymphohistiocytosis (FHL, which can be fatal without bone-marrow transplantation) (19, 20), as well as hematological malignancies (21–23). Perforin-1−/− mice exhibit an increased mortality in response to viral infections (24), fail to control tumor growth (25), and are highly prone to the development of spontaneous lymphomas (26).
Perforin-2 (also known as macrophage-expressed gene 1, MPEG1, Figure 1) has been discovered only recently (27), but appears to be the most ancient member of the MACPF family. It is encoded by the intronless MPEG1 gene which is found already in some of the earliest multicellular organisms, such as sponges (28). In mammals, perforin-2 is predominantly expressed in cells of monocytic origin, such as macrophages. During bacterial infection, it is recruited to pathogen-containing vacuoles where it damages membranes of diverse bacterial species limiting their proliferation (29–34). Consistent with its proposed function in antimicrobial defense, four deleterious MPEG1 variants have recently been identified through whole exome sequencing of patients with pulmonary non-tuberculous mycobacterial infection (35).
The second family of mammalian PFPs, gasdermins (Figure 1), has been discovered while searching for molecular mechanisms involved in an inflammatory cell death pathway termed pyroptosis (36, 37). Gasdermins are employed during both innate and adaptive immune responses, for example, in response to inflammasome assembly or following CTL-mediated delivery of granzymes into the cytosol. The best characterized member of this family, gasdermin D (GSDMD), is stored in the cytosol, and when activated, assembles into pores on the plasma membrane. The pores initially facilitate the secretion of cytosolic pro-inflammatory cytokines, such as IL-1β and IL-18, but if they persist unrepaired, the cell undergoes pyroptosis (38–40). In neutrophils, GSDMD pores contribute to the generation of neutrophil extracellular traps (NETs), secreted chromatin structures which capture extracellular pathogens (41). In vitro, GSDMD has also been shown to target cytosolic bacteria directly (38, 42).
In total there are six gasdermin paralogues in humans (GSDMA, GSDMB, GSDMC, GSDMD, GSDME/DFNA5, and PJVK/DFNB59) and 10 in mice (Gsdma1-3, Gsdmc1-4, Gsdmd, Gsdme, and Pjvk). Notably, gasdermin orthologues are also found in lower vertebrates, such as zebrafish (43), and more distant homologues are present in fungi (44). Mutations in gasdermin genes have been associated with a variety of disease phenotypes including skin and developmental defects (GSDMA3), susceptibility to asthma (GSDMA3 and GSDMB) (45–47), and autosomal dominant and recessive hearing loss (GSDME and DFNB59) (48, 49). The precise functions, mechanisms of activation, and physiological relevance for the majority of the gasdermins remain to be uncovered.
General Mechanism of Pore Formation
Mammalian PFPs are synthesized and stored in an inactive conformation as monomeric, usually soluble proteins. Structural studies revealed that the pore-forming fold of the MACPF proteins is highly similar to the pore-forming domain of bacterial cholesterol-dependent cytolysins (CDC) (50–52). In contrast, the pore-forming N-terminal domain of gasdermins is structurally distinct from the MACPF/CDC fold and is thought to have evolved independently (53, 54). Nevertheless, the MACPF/CDC and gasdermin family members follow a broadly similar mechanism of pore formation which can be roughly divided into three stages: membrane binding, oligomerization, and membrane insertion (55, 56). Membrane insertion is accompanied by dramatic structural rearrangements that include refolding of α-helical regions into transmembrane β-hairpins, termed TMH1 and TMH2 in MACPF/CDC proteins, and HP1 and HP2 in gasdermins. In the resulting β-barrel pores, each monomer typically contributes one four-stranded lipid-embedded β-sheet.
Challenges in Storing and Targeting of Mammalian PFPs
Despite considerable similarity in the pore assembly process, the mechanisms involved in selecting target membranes differ strikingly between bacterial CDCs and mammalian MACPF proteins and gasdermins. For bacterial PFPs, the transition between soluble monomer and membrane pore is initiated by binding of the PFP to proteins, sugars, or lipids unique to the host. Thus, CDCs form pores preferentially on membranes with 25–35% cholesterol content, a lipid present only in eukaryotic cells (57). As mammalian PFPs themselves show limited target membrane selectivity, additional mechanisms need to be in place to enable spatiotemporal control of pore formation and to limit damage to both PFP-producing cells and surrounding tissues.
Transcriptional Control of PFP Expression
While some PFPs can be safely expressed in the majority of cells (e.g., gasdermins), others (e.g., perforins) can be toxic soon after translation and are only produced by specialized cells of the immune system (Figure 2A).
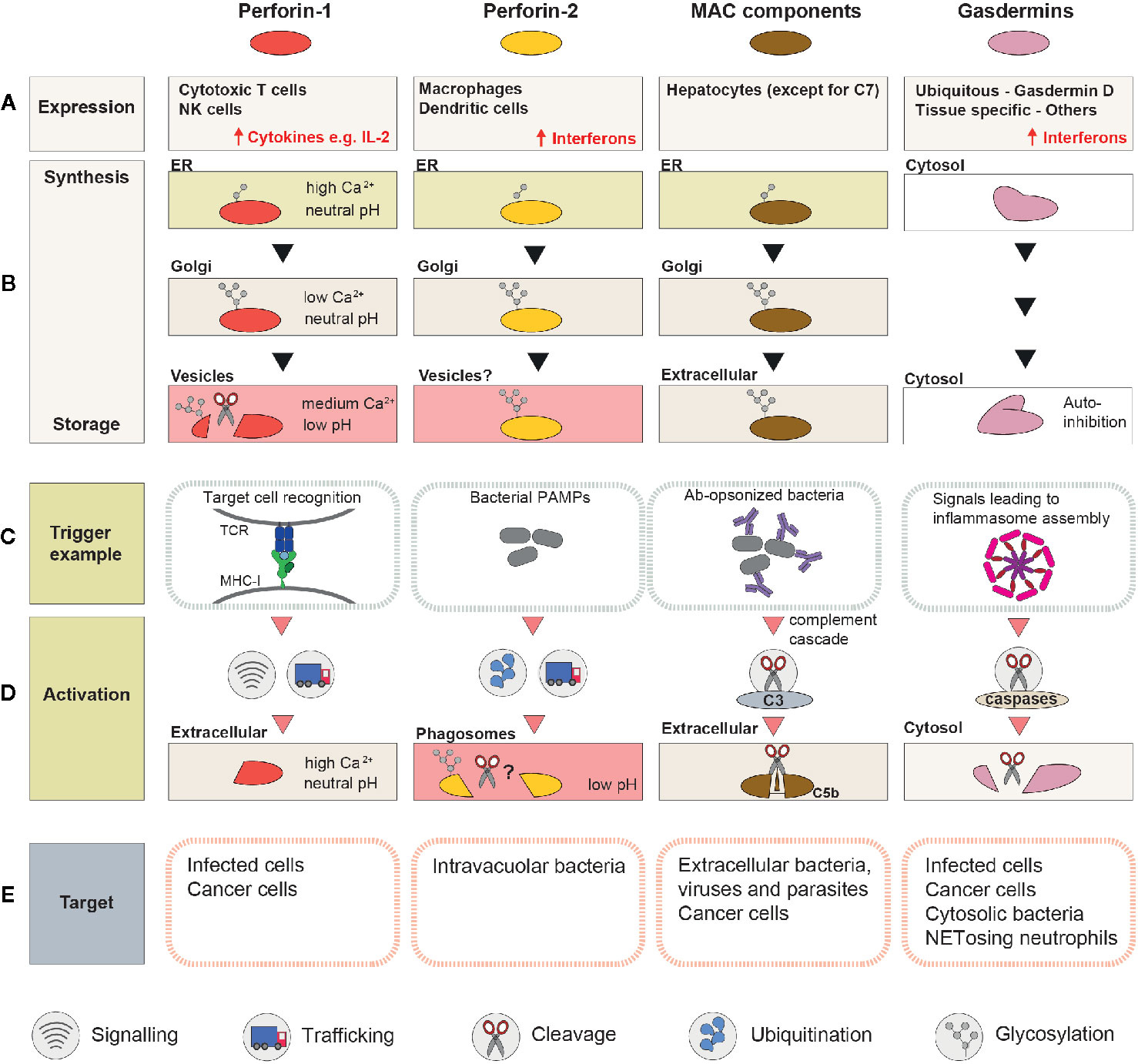
Figure 2 A schematic summary of the mechanisms involved in regulation and activation of perforin-1, perforin-2, MAC, and gasdermins. (A) Cell types or tissues in which the PFP is constitutively expressed (inducible expression is highlighted with ↑). (B) Processing and trafficking steps involved in synthesis of the “stored” form of the PFP. The last row corresponds to the storage compartment. (C) Immune system components which initiate pore formation, and ligands they recognize. (D) A schematic representation of the events that precede pore assembly. (E) Targeted membranes.
MAC Components Are Produced in the Liver
The majority of the MAC components, similar to other serum proteins, are produced by hepatocytes in the liver (58). The exception is C7, which is expressed primarily extrahepatically as shown by the fact that, in liver transplant patients, as little as 10% of plasma C7 originated from the donor cells (59), compared to nearly 100% for C6 and C9 (60, 61). It has been proposed that local synthesis of C7 might be important for modulation of MAC activity (62), but this hypothesis has not yet been verified with tissue- or cell type-specific C7 knockouts. Intriguingly, liver-derived C7 is actually not produced by hepatocytes, but by endothelial and stellate cells (63, 64), raising the question whether co-expression of C7 with the remaining MAC components might be toxic for the cells.
Gasdermins Can Be Expressed Ubiquitously or in a Tissue-Specific Manner
Members of the gasdermin family employ a wide range of different transcription factors to regulate their constitutive and inducible expression, and as a result, they all display different expression patterns [summarized in a recent review by Broz et al. (65)]. GSDMD and GMDMB are the most abundant and ubiquitous family members, while expression of the remaining gasdermins is more restricted. GSDMA, for example, is predominantly expressed in the skin, while GSDME is mainly present in the blood, spinal cord and uterus (58). Expression of gasdermins can be further regulated during a variety of pathological conditions, such as infection (66), cancer (67, 68), or in response to DNA damage (69). For example, GSDMD expression is strongly upregulated during bacterial, viral, and parasitic infections by interferon-γ-dependent signaling pathways (66).
Perforin-1—an Effector Restricted to Killer Lymphocytes
In contrast to MAC components and gasdermins, perforins are primarily expressed in immune cells. PRF1 promoter activity is restricted to the T and NK cell lineages during development (70). In T cells, PRF1 expression is induced upon maturation of naïve T cells into CTLs during an active immune response [reviewed in (71)]. T cell activation requires three distinct signals delivered by dendritic cells: signal 1—MHC class I-bound peptides, identical to those present in target cells (recognized by TCR); signal 2—cell surface molecules that act as stimulatory or inhibitory co-receptors, and signal 3—chemokines and cytokines that modulate T cell proliferation and differentiation. These requirements ensure that only T cells with relevant TCRs are activated, expanded, and upregulate perforin-1 (72). Mechanistically, regulation of perforin-1 transcription is a complex process controlled by a 150 kb territory surrounding the PRF1 gene (70) and several transcription factors (Sp-1, AP-1, MEF, MITF, T-bet, and EOMES) [reviewed in (73, 74)].
NK cells upregulate Prf1 expression early during their development (75, 76). Yet, resting NK cells isolated from mice bred under pathogen-free conditions are only minimally cytotoxic and they contain only a small amount of perforin-1 protein (77). Instead, they store large quantities of Prf1 mRNA, which is only translated upon NK cell activation (e.g., by cytokines IL-15 or IL-2). How mRNA translation is regulated in NK cells is not fully understood. The lymphocyte specific miRNA, miR-150, has been shown to target Prf1 mRNA and downregulate its translation in resting NK cells (78), but additional mechanisms involving cytoplasmic mRNA-binding proteins and mRNA modifications are likely to be involved (79, 80).
MPEG1 Is Expressed Predominantly in Antigen-Presenting Cells
MPEG1 is constitutively expressed in macrophages, dendritic cells, monocytes, and granulocytes (81), but interferon-inducible expression has been observed in other cells types, e.g., fibroblasts and neurons (32, 82). Thus, MPEG1 is strongly upregulated during bacterial, viral, and parasitic infections. For example, in the lungs of mice infected with influenza virus, it is among the top 30 genes with highest expression (66). Interestingly, pathogens that disrupt interferon signaling during infection have been reported to escape perforin-2-mediated killing. This has been demonstrated for Chlamydia trachomatis, which prevents interferon-dependent translocation of the STAT1 transcription factor into the nucleus (83). While ectopic expression of MPEG1 in HeLa cells suppresses chlamydial growth, MPEG1 is upregulated only in cells exposed to heat-killed Chlamydiae, but not live bacteria (29).
Maturation and Storage
Newly synthesized PFPs are rarely immediately activated. Instead, they are stored as inactive monomers that can be rapidly deployed in response to the appropriate signals (Figure 2B). These nascent PFP are unable to bind membranes as monomers, are synthesized as inactive immature propeptides, and/or are stored in a compartment where the ionic environment is incompatible with pore formation.
MAC Components Are Abundant in the Plasma
All seven MAC subunits are secreted into the plasma and circulate the body through the blood vessels. C8 isolated from plasma is a stable heterotrimer of C8α, C8β, and C8γ (with C8α covalently bound to C8γ via a disulfide bridge) (84), but the remaining subunits are monomeric. In vitro, C9 can be forced to form homooligomeric pores (85, 86) but the poly-C9 complex is unable to insert into cell membranes (85), and under physiological conditions, the TMH1 domain of C9 precludes polymerization in the absence of other MAC components (87). The secreted MAC components do not bind membranes efficiently on their own and as a consequence they are non-lytic at steady-state. C5 is the only MAC subunit which is proteolytically processed prior to pore formation, and this cleavage event is what initiates a cascade of conformational changes that drives MAC assembly.
Gasdermins Are Stored as Immature Propeptides in the Cytosol
In contrast to MAC components, gasdermins are synthesized and stored as immature cytosolic propeptides, which adopt an auto-inhibited state immediately after folding (38, 54, 88). Gasdermins contain an N-terminal pore-forming domain (NTD) connected to an inhibitory C-terminal domain (CTD) by a flexible linker (with the exception of PJVK/DFNB59 which has a truncated C-terminus). Proteolytic cleavage at the linker region destabilizes the NTD-CTD intramolecular interaction releasing the NTD to assemble into pores (36, 37). Indeed, ectopic expression of the NTD alone is sufficient to trigger pyroptotic cell death (42). It has been concluded that the CTD masks the lipid-binding motif of the NTD preventing membrane association of full-length gasdermins (54), but it does not sterically hinder pore formation. Mutations, including known disease variants, that weaken the NTD-CTD interdomain interactions are sufficient to expose the lipid binding motif and trigger constitutive gasdermin activity without cleavage (38) raising a possibility of alternative activation mechanisms.
Fully Processed Perforin-1 Is Stored in Cytotoxic Granules
Perforin-1 is also initially synthesized as an inactive propeptide but, in contrast to gasdermins, it is stored in a mature form. Besides the MACPF domain, immature perforin-1 consists of two additional domains: an epidermal growth factor (EGF) domain, and a C-terminal, Ca2+-binding domain (C2) required for association with the membrane (89, 90). Perforin-1 maturation involves cleavage of a short (~2 kDa) fragment of the C-terminal domain which contains a bulky Asn549-glycan (91). The Asn549-glycan inhibits pore formation by steric hindrance interfering with oligomerization (92). Indeed, full length (uncleaved) perforin-1 purified from human NK KHYG1 cells does not form pores (92), while the full-length Asn549-glycosylation deficient mutant is lytic when purified and toxic when overexpressed (93). Intriguingly, full-length perforin-1 purified from insect cells is fully functional (52, 93), suggesting that the size of the glycan moiety is critical for inhibition of perforin-1 activity [N-glycoproteins produced in standard insect cell expression systems acquire simple side-chains instead of complex N-glycans found in mammalian proteins (94)]. Similarly, the unbranched N-glycans acquired initially in the ER offer only partial protection from unwanted lysis, and N-glycosylated perforin-1 is still toxic when ER exit is slow (e.g., in BFA-treated cells or upon fusion of an ER retention signal to perforin-1 C-terminus) (93).
Following further branching of glycans in the Golgi, perforin-1 is transported into lysosome-related organelles, called cytotoxic granules. Once in the granules, lysosomal proteases cleave the C-terminus, along with the inhibitory Asn549-glycan, to prime perforin-1 for activation upon secretion. In vitro, perforin-1 can be cleaved by cathepsins L or B (95, 96), but Ctsl−/− or Ctsb−/− mice do not show a defect in perforin-1-mediated killing (96, 97). In contrast, mice lacking asparaginyl endopeptidase (AEP) show degranulation defects, but the potential contribution of AEP to perforin-1 cleavage has not been addressed directly (98). Consistent with the potential involvement of multiple (or redundant) proteases, the cleavage site itself does not appear to be precise and mass spectrometry analysis of perforin-1 immunoprecipitated from KHYG1 cells identified fragments with multiple C-termini (92). In line with this observation, site-directed mutagenesis of the C-terminal region did not reveal any critical positions and cleavage occurred even when every residue from Gln540 through to Gly548 was replaced with a serine. Thus, perforin-1 proteolytic processing may be mediated by non-specific lysosomal proteases and the susceptibility of the region to cleavage is likely to be due to its disordered character.
Regardless of the exact nature of the protease, the cleavage takes place only after perforin-1 reaches a low pH compartment (91). This is critical, as acidic pH (~5.5 in the granules) prevents the fully processed perforin-1 from pore-formation (89). At pH < 6.2 the key acidic residues involved in ionic interactions between monomers are protonated preventing oligomerization (14). Furthermore, protonation of Ca2+-binding Asp residues is expected to reduce Ca2+ binding to the C2 domain, which is critical for the association of perforin-1 with the target membrane (89, 90, 99). Ca2+ binding might be additionally prevented by granule-resident calreticulin, which sequesters free Ca2+ ions to further inhibit perforin-1 activity (100). Thus, cleavage only occurs when perforin-1 reaches a compartment with an environment incompatible with pore formation, providing an important safeguarding mechanism.
Where Is Perforin-2 Stored at Steady State?
Perforin-2 is a type I transmembrane protein comprising the MACPF domain, a unique membrane-binding P2 domain, a transmembrane domain, and a short cytosolic tail. The protective mechanisms involved in its biosynthesis and storage are not well characterized. In membrane-anchored perforin-2, the pore-forming TMH regions would face away from the membrane after unfurling (101, 102), suggesting that the transmembrane domain might be important for preventing perforin-2 assembly on host cell membranes. This unusual topological feature might therefore offer an elegant safeguard against accidental autolysis during biosynthesis and storage.
Overexpressed GFP- or RFP-tagged perforin-2 shows diffuse staining, which could correspond to small post-Golgi vesicles (29, 32). Hence, it appears that similar to perforin-1, perforin-2 might also be sorted into a specialized storage compartment. However, in contrast to perforin-1, the ectodomain of perforin-2 forms pores at low pH (101, 102), suggesting that the hypothetical storage compartment would have to have elevated rather than low luminal pH.
Intriguingly, overexpression of perforin-2 in HEK-293Ts is toxic (28, 103), yet in professional phagocytes it is one of the most abundant proteins. For example, in mouse dendritic cells, perforin-2 is ranked as the top 55th protein by abundance (104) with an estimated 3x106 copies safely stored within each cell (105). It appears likely, therefore, that antigen-presenting cells (APCs) employ specialized mechanisms to protect their intracellular membranes from perforin-2 mediated damage, but this has not been addressed to date. Considering that in non-immune cells, perforin-2 is expressed during infection or upon stimulation with interferons (30, 32), it is possible that interferons also facilitate expression of additional factors that control perforin-2 activity.
Initiation of Pore Formation
Despite broadly similar mechanisms of pore formation, the signals that trigger PFP activation and the events that precede pore formation are strikingly different between MAC, perforins, and gasdermins (Figures 2C, D).
MAC: Cleavage of C5 Triggers a Cascade of Conformational Changes
MAC assembly can be initiated by multiple enzymatic chain reactions known as the classical, lectin and alternative pathways [reviewed in (106)]. All three pathways converge on the formation of a C5 convertase, which catalyzes the cleavage of C5, the key event in initiation of MAC assembly. The activation of the classical pathway starts when the C1q component of the complement system binds to antigen-antibody complexes, e.g., IgM- or IgG-opsonized bacteria. The lectin pathway is initiated following the recognition of pathogen-specific carbohydrates on the bacterial surface (e.g., by mannose-binding lectins, collectins, or ficolins). Ligand-bound C1q or lectin receptors initiate distinct proteolytic cascades, but both lead to cleavage of C4 and C2. The resulting cleavage products, C4b and C2a, assemble into the C3 convertase, which then cleaves C3 to generate C3b. The C4bC2aC3b complex forms the classical C5 convertase. The alternative pathway starts with spontaneous C3 hydrolysis or by deposition of C3b directly on bacterial surface during ongoing complement activation. These events, in the presence of factors B and D, lead to formation of the alternative C5 convertase, C3bBbC3b.
The cleaved C5 initiates a cascade of conformational changes that lead to MAC assembly and unfurling of the TMH domains. Cleavage generates a small C5a fragment (74-77 residues in length) and a large 170 kDa C5b fragment formed by two peptide chains, β (residues 19–673) and α (residues 752–1676), linked by a disulfide bond. C5b is very labile and it decays (aggregates) within 2 min unless stabilized by binding of C6 (107). The C5bC6 associates weakly with the membrane but remains soluble as the TMHs of C6 are not yet deployed in the dimer (108). Membrane binding interfaces may instead be provided by auxiliary domains such as C6 thrombospondin-like domain which has amphipathic properties at its base (109). The C5bC6 complex recruits C7 driving a cascade of conformational changes within auxiliary domains of C6 and C7 that ultimately trigger unfurling of TMHs in both C6 and C7 and membrane anchoring (110–113). The C6 and C7 β-hairpins, however, do not fully penetrate the membrane. Instead, recruitment and irreversible binding of the C8 trimer and unfurling of the four additional β-hairpins (in C8α/C8β) leads to formation of a stable C5bC6C7C8 complex that can no longer be removed from the membrane by washing (114, 115). The C5b-C8 complex initiates polymerization and membrane insertion of up to 18 copies of C9, recruited directly from solution to the growing pore (111). Notably, MAC pores are the only known PFPs that form hetero-oligomeric, asymmetric pores.
Gasdermins Are Cleaved to Release a Pore-Forming Domain
Gasdermin pores are formed following a regulated cleavage event of the immature propeptide. This cleavage is primarily executed by caspases, which themselves are stored as inactive propeptides that require proteolytic processing for activation.
GSDMD is primarily cleaved by caspases-1 and -4/5/11 (36, 37). Caspase-1 is activated by so-called canonical inflammasomes, a group of large multiprotein complexes composed of distinct pattern recognition receptors (LRP1, NLRP3, NLRC4, AIM2, and pyrin). Canonical inflammasomes assemble in response to a wide range of PAMPs and damage-associated molecular patterns (DAMPs), including bacterial flagellin, cytosolic dsDNA, ROS, ionic imbalances, and many others [reviewed in (116)]. By contrast, caspases-4 and -5 (and caspase-11 in mice) are activated within non-canonical inflammasomes by directly binding to bacterial lipopolysaccharides (LPS) (117). Activated, proteolytically processed caspases-1/4/5/11 bind to the GSDMD C-terminal domain executing cleavage within the GSDMD linker region at position Asp276, in the Phe-Leu-Thr-Asp (humans) or Leu-Leu-Ser-Asp (mice) tetrapeptide (118, 119). In Yersinia-infected cells, a caspase-8-dependent processing of GSDMD has also been reported (120, 121). However, considering the low efficiency with which caspase-8 cleaves GSDMD in vitro, it is unclear whether the observed effect was specifically due to cleavage of GSDMD or other co-factors (121, 122). Other proteases implicated in GSDMD activation include neutrophil elastase (released from cytoplasmic granules) (123, 124) and cathepsin G (released from lysosomes) (125), but the physiological relevance of these pathways remains to be addressed.
Non-inflammatory signals might also regulate the activity of gasdermins. For example, GSDME can be cleaved and activated by caspase-3, an executioner of canonical apoptosis. While this cleavage results in conversion of apoptotic cell death into pyroptosis (39, 40), intriguingly, caspase-3-mediated cleavage of GSDMD at position Asp87 inhibits pore formation, and as a result inhibits pyroptosis (40, 122, 126). Furthermore, two recent studies suggested that granzymes B and A, delivered by CTLs/NKs through perforin-1 pores, can cleave gasdermins E and B, respectively, and promote pyroptotic rather than apoptotic death of cancer cells (127–129). Hence, unexpectedly, gasdermin-mediated pyroptosis might also contribute to CTL-mediated killing during adaptive immune responses. These findings reveal an intriguing cooperation between mammalian PFPs and further strengthen the notion of plasticity between cell death pathways.
Perforin-1 Is Released From Cytotoxic Granules Into a Neutral pH Environment
The key event that triggers the assembly of perforin-1 pores involves the formation of an immune synapse between a CTL/NK cell and the target cell, followed by fusion of the lytic granules with the plasma membrane and secretion of their contents into the synaptic space.
For CTLs, secretion of the cytotoxic granules is triggered by the association of antigen-specific TCR with MHC I loaded with a foreign or a mutated peptide. Exocytosis of NK granules is controlled by integration of signals delivered from activating and inhibitory cell surface receptors [reviewed in (130)]. The best characterized inhibitory receptors, killer immunoglobulin-like receptors (KIRs) in humans (Ly49 in mice), also bind MHC I and are maximally engaged at the MHC I density found on healthy host cells (131). This, therefore, provides a mechanism to specifically target cells that escape CTL killing by downregulating MHC I—an evasion mechanisms employed by both viruses (132) and cancers [reviewed in (133)]. The NK activating receptors recognize either host proteins upregulated in response to cellular stress (134, 135) or viral proteins expressed on the cell (e.g., viral hemagglutinins bound by receptors NKp46 and NKp44).
The precision in killing is ensured by polarized secretion of the cytotoxic granules toward the immune synapse [reviewed in (136–139)]. The polarization depends on the phospholipase C-γ and Ca2+-dependent signaling of the TCR or NK activating receptors and is followed by dynein-dependent movement and docking of the microtubule organizing center at the synapse. Subsequent microtubule-dependent transport and exocytosis of the granules release perforin-1 from its inhibitory storage compartment into an environment highly favorable for pore formation. The high extracellular Ca2+ concentration (~1–1.3 mM) stabilizes the perforin-1 C2 domain and induces a conformational change that permits four key hydrophobic residues to anchor perforin-1 to the plasma membrane of the target cell (99). The neutral pH further facilitates ionic intermolecular interactions of perforin-1 monomers driving their oligomerization into ring- and arc-shaped pores (14, 140, 141).
Perforin-2 Pores Are Assembled at Low pH
Perforin-2 activity has been observed only at low pH in vitro and is likely controlled by regulated trafficking to an acidic compartment. Indeed, RFP-tagged perforin-2 redistributes to bacteria-containing phagosomes during infection with Escherichia coli or Salmonella typhimurium (32). In cells infected with enteropathogenic E. coli strain E2348/69 or treated with LPS, the C-terminal cytosolic tail of perforin-2 is monoubiquitinated by a cullin-RING E3 ubiquitin ligase (CRL) complex and mutation of the lysines in the cytosolic tails of perforin-2 prevents its recruitment to phagosomes and bactericidal activity (31). Yet, the signaling pathways that promote ubiquitination, the mechanisms involved in trafficking of ubiquitinated perforin-2 or whether ubiquitination is indeed required for perforin-2 recruitment to pathogen-containing vacuoles remain to be carefully addressed.
Several lines of evidence suggest that the activation of perforin-2 in acidic compartments might involve cleavage of its ectodomain from the transmembrane domain. Firstly, the ectodomain alone assembles into pre-pores and pores on liposomes in vitro (101, 102). Secondly, in HEK-293 cells perforin-2 was able to form ring-like structures only following trypsin treatment (32). Finally, perforin-2 that was present on bacteria isolated from MEFs expressing perforin-2-GFP was recognized by antibodies specific to MACPF and P2 domains but not to the cytosolic tail (32). Nevertheless, neither the cleavage site nor the relevant proteases have been identified to date and future work will need to address whether ectodomain release is indeed physiologically relevant.
Intriguingly, perforin-2 does not restrict bacterial growth without pre-stimulation of cells with IFN or LPS (32). It is not known, however, whether pre-stimulation is required for processing/trafficking of perforin-2 itself, whether it stimulates expression of co-factors required to trigger pore formation, or whether the pore forming ability of perforin-2 is insufficient to restrict bacterial growth in the absence of additional interferon-stimulated genes that facilitate killing of pathogens.
Safety Mechanisms for Protection of Bystander Membranes
After the appropriate trigger signals have been received, the newly unleashed lytic activity of PFPs requires continuous control as unrestrained pore formation would not only be highly damaging to bystander membranes but would also reduce the availability of monomers for a productive lytic response at the target membrane (Figure 2E).
Lipid-Binding Selectivity of Gasdermins Prevents Bystander Cell Lysis
Gasdermins preferentially bind to negatively charged lipid species [cardiolipin, phosphatidylinositol phosphates (PIPs), phosphatidic acid (PA), and phosphatidylserine (PS)] which are found on the inner leaflet of the plasma membrane but are absent from its extracellular leaflet (38, 39, 42). This lipid-binding preference therefore appears to be sufficient to protect bystander cells from activated gasdermins released during pyroptosis (142). Given that the cytosolic leaflets of endosomes and phagosomes contain the same lipid species as the inner leaflet of the plasma membrane, it is likely that gasdermins pores are not restricted to the plasma membrane. Whether intracellular membranes [other than the mitochondrial membrane (143)] are indeed disrupted by gasdermins and, if not, how they are protected remains to be investigated.
Inactivation of MAC Assembly
The soluble C5bC6 complex can in principle diffuse away from the target membrane and initiate pore assembly on bystander cells. MAC formation, however, can be inhibited at multiple stages during pore formation, even after proteolysis of C5. The key factors that disarm MAC pores include CD59, clusterin, and vitronectin.
CD59 is a small GPI-anchored glycoprotein widely expressed on the surface of mammalian cells (58). CD59 inhibits MAC formation by binding to C8 in the C5b-8 complex thus preventing C9 incorporation, as well as by binding to C9 in the preformed C5b-9 complex suppressing further polymerization (144). Specifically, CD59 interacts with C8α and C9b in regions exposed during MAC formation (114, 145). The protective role of CD59 is most evident when its levels are pathologically low. Deficiencies in CD59 or in proteins required for biosynthesis of its GPI anchor result in inflammatory neuropathy, recurrent strokes, and chronic hemolysis (146–148).
In contrast to CD59, clusterin and vitronectin are soluble glycoproteins found in plasma (149). Clusterin can inhibit the lytic activity of C5b-7, C5b-8, and C5b-9 subcomplexes by interacting with C7, C8β, and C9 through binding sites exposed during pore formation (150). Vitronectin, also known as S-protein, has been reported to inhibit MAC insertion at two stages: either by binding to the nascent C5b-7, rendering the complex soluble (151) or by inhibiting polymerization of the C9 subunits (152). Interestingly, some Gram-negative bacteria including Moraxella catarrhalis, Haemophilus influenzae, and Neisseria gonorrhoeae recruit vitronectin to prevent MAC deposition on their surface and escape MAC-mediated killing [reviewed in (153)].
How Are Cytotoxic Lymphocytes Protected From Perforin-1 at the Immune Synapse?
Both CTL and NK cells can sequentially kill several target cells suggesting that the killing cell itself does not undergo a bystander death (154–157). Indeed, several studies have demonstrated that various T cell lines as well as primary T cells are more resistant to killing by other CTLs compared to, for example, cancer cell lines (158–160). A similar increased resistance was demonstrated to granule extracts and purified perforin-1 alone (158, 161, 162). Nevertheless, lymphocytes are not invulnerable to CTLs, especially when the attack is directed against them (160, 163, 164). This so-called fratricide (i.e., killing of CTLs by CTLs) might not only be crucial to eliminate CTLs that have been infected or accumulated mutations, but could also help to dampen an excessive immune response (165). Importantly, when an immune synapse between two CTLs is formed, only one cell gets polarized to inflict cell death and the killer always survives (166). Moreover, while a CTL engaged with the target cell avoids destruction by their own lytic mediators, it is not refractory to bystander lysis when induced by neighboring CTLs (167). This apparent paradox suggests that cytotoxic lymphocytes acquire additional resistance to perforin-1 primarily within the immune synapse following degranulation.
Several models were proposed to explain why the degranulating lymphocyte might be resistant to perforin-1, but none has been widely accepted to date. Earlier studies suggested that other proteins contained within the granules might have protective functions during degranulation. For example, Balaji et al. (95) observed that CTLs are more prone to death in the presence of membrane impermeable cathepsin B inhibitors and proposed that secreted cathepsin B cleaves perforin-1 on the surface of degranulating CTLs to protect them. More recent work, however, revealed that CTLs of cathepsin B-null mice survive their encounter with target cells normally (97). Jiang et al. (168) suggested in turn that glycosylation and sialylation of membrane protein(s) on the CTL surface might provide negative charges that repel perforin-1 and in later work by Cohnen et al. (169), LAMP1/CD107a was implicated as a key O-glycosylated and sialylated protein involved. In line with this model, LAMP1 deficient NK cells were more susceptible to apoptosis after an encounter with the target and overexpression of truncated LAMP1 (targeted directly to the cell surface) reduced apoptosis caused by cytotoxic granules. A separate study, however, did not report a reduction in perforin-1 binding to the surface of primary mouse T cells that overexpress LAMP1 (162). Considering the putative role of LAMP1 in trafficking perforin-1 toward cytotoxic granules (170), the exact contribution of LAMP1 to preventing perforin-1 mediated damage might be difficult to decipher.
Alternatively, perforin-1 resistance could be conferred by local changes in lipid composition that follow degranulation. This model is supported by the observations that perforin-1 preferentially forms pores on phosphatidylcholine-rich, disordered lipid phases, avoiding sphingomyelin/cholesterol-rich ordered domains abundant within the immune synapse (162, 171–173). Furthermore, degranulation is associated with a transient increase of surface exposed PS which is also believed to provide a membrane composition unfavorable for pore assembly (174, 175). On the one hand, the presence of PS could simply interfere with perforin-1 membrane binding (42). On the other hand, PS might act as a negatively charged sink that binds perforin-1 in a conformation incompatible with pore assembly (162).
Finally, unidirectional killing might be facilitated by mechanopotentiation, the process of increasing membrane tension on the target cell via the exertion of synaptic forces (176). The forces at the immune synapse are generated by the concerted action of cytosolic proteins that regulate actin dynamics, myosin II, and integrins (177–179). A resulting increase in membrane tension on the target cell was proposed to lower the necessary concentration of perforin-1 required for pore assembly (176). This discovery implies an additional function of the immune synapse in protecting from perforin-1-mediated damage: not only does it protect bystander cells by limiting perforin-1 diffusion, but also the cytotoxic cells themselves, by lowering the effective concentration of perforin-1 required for pore assembly on the target membrane.
(How) Are Phagosomal Membranes Protected From Perforin-2 Activity?
Little is known about the mechanisms involved in the protection of host cells from perforin-2 pores formed in phagosomes. In vitro, perforin-2 displays preference for negatively charged lipids including PS, PIPs as well as cardiolipin, which is found in the membranes of most bacteria (101, 102). However, considering that overexpressed perforin-2 can be toxic (103) and that perforin-2 pores have been observed also on mammalian membranes (32), it is unclear whether in infected cells the pores are solely formed on the pathogen surface.
Pore Insertion and Membrane Repair Pathways
Even a small injury to the plasma membrane can lead to local spikes in cytosolic Ca2+ and trigger membrane repair pathways in the affected cell. In general, these repair pathways involve endocytosis to internalize damaged membranes, exocytosis to shed damaged membranes, and membrane patching to reseal any damage using internal endolysosome-derived donor membranes (180).
Perforin-1 insertion primarily triggers membrane patching using lysosomal and endosomal donor membranes (140, 181–183). It has also been observed that, in addition to patching, perforin-1-mediated membrane destabilization promotes clathrin- and dynamin-dependent endocytosis resulting in the internalization of both perforin-1 and granzymes (183–185). These data led to the hypothesis that endolysosomal compartments (gigantosomes) rather than the plasma membrane are the primary site of perforin-1 pore assembly and granzyme entry into the cytosol. Later studies, however, did not support this model. Firstly, it remains controversial whether the luminal pH and Ca2+ concentration in gigantosomes are permissive for assembly of perforin-1 pores (140, 186). Secondly, the relatively slow kinetics of granzyme endocytosis and release (~ 15 min) are inconsistent with the rapid (~ 2 min) induction of Bid cleavage reported in cells exposed to granzyme B and sublytic amounts of perforin-1 (140). Hence, Lopez et al. proposed that, while membrane repair pathways do indeed negatively regulate perforin-1 activity at the plasma membrane, they allow the formation of transient pores that persist for 20–80 s providing sufficient permeability to deliver granzymes into the cytosol of the target cell and to initiate apoptosis (140). Future work involving high-resolution electron tomography might be necessary to resolve the controversy surrounding the primary location of functional perforin-1 pores, but what remains clear is that membrane repair pathways are important to prevent uncontrolled perforin-1 mediated lysis of the target cells.
Ca2+ influx and membrane repair have also been reported upon membrane insertion of gasdermins (187). Gasdermin pores trigger recruitment of endosomal sorting complexes required for transport (ESCRTs) which mediate repair of damaged membranes through exocytosis (188). ESCRT-mediated membrane repair negatively regulates GSDMD-induced pyroptotic death as well as the release of IL-1β and IL-18 from infected cells (187). Considering ESCRTs are also recruited to membranes exposed to CDCs such as streptolysin O and listeriolysin O (188), it appears likely that they may contribute to the removal of perforin-1 pores as well. Interestingly, ESCRTs are also involved in repair of small perforations in endolysosomes to facilitate recovery of damaged intracellular membranes (189). This pathway might provide a safety mechanism against possible accidental damage of PFP storage compartments and an additional layer of protection for PFP-producing cells.
Concluding Remarks
It is striking how both innate and adaptive immune systems employ PFPs as their key effectors. In this review, we aimed to provide an overview of the pathways and immune system components involved in controlling the activity of these membrane-disrupting molecules. However, it is important to recognize that PFP biology is tightly linked to fundamental processes that go well beyond what we discussed here including positive and negative T cell selection, antibody affinity maturation as well as signaling pathways associated with different types of cell death, all of which ultimately contribute to the regulation of PFP activity.
Despite the century of research since the discovery of MAC, many questions regarding PFPs remain unanswered. Is extrahepatic production of MAC components physiologically relevant during infections? What are the precise conformational changes that govern the initial membrane insertion of MAC and the final pore closure? Is mechanopotentiation involved in protecting degranulating CTLs and NKs cells from perforin-1 activity in vivo? How is translation of perforin-1 and other NK effectors suppressed in resting cells?
The recent discovery of perforin-2 and gasdermins has also opened new avenues to explore. What are the physiological functions of all gasdermins? Are they able to assemble on endolysosomal membranes and if so, what are the consequences of the potential damage? Is perforin-2 released from the phagosomal membrane to form pores on intravacuolar pathogens and if so, by which proteases? Does perforin-2 damage phagosomes or does it assemble exclusively on bacterial membranes?
The latest advances in the PFP field have uncovered an unexpected link between perforin-1-mediated granzyme delivery and gasdermin activation, and future work is likely to reveal other examples of such cooperativity. We now also only begin to appreciate the different mechanisms involved in the sensing and repair of damaged membranes and how they can affect the consequences of pore formation. Finally, many of the regulatory pathways discussed in this review can be disrupted by pathogens, and the full picture of the mechanisms involved in evasion of PFP-mediated immunity is yet to emerge.
There is no doubt that the PFP field continues to rapidly expand. Following the recent advances in cryo-electron microscopy, the structures of MAC, perforin-1/2, and gasdermin pores are now available shedding some light on the conformational changes involved in pore assembly. Whole exome sequencing data from immunodeficient patients is helping to uncover novel disease-associated variants both in PFPs themselves and in their regulators. Finally, CRISPR-Cas9 technology is facilitating the generation of cell-type specific knockouts to address the contribution of candidate proteins in PFP biology using primary cells and animal models of disease. Novel scalable assays to study PFPs in vitro, identification of co-regulators through genetic screens in the relevant primary cell types, and structural insights into pre-pore and pore intermediates should provide us with a more complete picture of the mechanisms involved in the regulation of these powerful effectors and facilitate development of targeted immunomodulatory therapeutics.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
We would like to thank Dr. Greg Slodkowicz for comments on the manuscript. The authors were supported by the MRC grant MC_UP_1201/26. PAK was also supported by a Boehringer Ingelheim Fonds PhD Fellowship and the Cambridge Trust.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Peraro MD, van der Goot FG. Pore-forming toxins: ancient, but never really out of fashion. Nat Rev Microbiol (2016) 14(2):77–92. doi: 10.1038/nrmicro.2015.3
2. Anderluh G, Kisovec M, Kraševec N, Gilbert RJC. Distribution of MACPF/CDC Proteins. In: Anderluh G, Gilbert R, editors. MACPF/CDC Proteins - Agents of Defence, Attack and Invasion. Dordrecht: Springer Netherlands (2014). doi: 10.1007/978-94-017-8881-6_2
3. Liu X, Lieberman J. Knocking ‘em dead: pore-forming proteins in immune defense. Annu Rev Immunol (2020) 38(1):455–85. doi: 10.1146/annurev-immunol-111319-023800
4. Nuttall G. Experimente über die bacterienfeindlichen Einflüsse des thierischen Körpers. Z Hyg Infektionskr (1888) 4(1):353–94. doi: 10.1007/BF02188097
5. Buchner HEA. Über die bakterientötende Wirkung des Zellfreien Blutserums. I and II. Zentralbl Bakteriol (1889) 5:817–23.
6. Tomlinson S, Taylor PW, Morgan BP, Luzio JP. Killing of gram-negative bacteria by complement. Fractionation of cell membranes after complement C5b-9 deposition on to the surface of Salmonella minnesota Re595. Biochem J (1989) 263(2):505–11. doi: 10.1042/bj2630505
7. Nakamura M, Okada H, Sasaki H, Yoshida K, Kamada M, Okada N, et al. Quantification of the CD55 and CD59, membrane inhibitors of complement on HIV-1 particles as a function of complement-mediated virolysis. Microbiol Immunol (1996) 40(8):561–7. doi: 10.1111/j.1348-0421.1996.tb01109.x
8. Hoover DL, Berger M, Nacy CA, Hockmeyer WT, Meltzer MS. Killing of Leishmania tropica amastigotes by factors in normal human serum. J Immunol (1984) 132(2):893.
9. Koski CL, Ramm LE, Hammer CH, Mayer MM, Shin ML. Cytolysis of nucleated cells by complement: cell death displays multi-hit characteristics. Proc Natl Acad Sci U S A (1983) 80(12):3816–20. doi: 10.1073/pnas.80.12.3816
10. Morgan BP. The membrane attack complex as an inflammatory trigger. Immunobiology (2016) 221(6):747–51. doi: 10.1016/j.imbio.2015.04.006
11. Ross SC, Densen P. Complement deficiency states and infection: epidemiology, pathogenesis and consequences of neisserial and other infections in an immune deficiency. Medicine (Baltimore) (1984) 63(5):243–73. doi: 10.1097/00005792-198409000-00001
12. Orren A, Potter PC, Cooper RC, du Toit E. Deficiency of the sixth component of complement and susceptibility to Neisseria meningitidis infections: studies in 10 families and five isolated cases. Immunology (1987) 62(2):249–53.
13. Khajoee V, Ihara K, Kira R, Takemoto M, Torisu H, Sakai Y, et al. Founder effect of the C9 R95X mutation in Orientals. Hum Genet (2003) 112(3):244–8. doi: 10.1007/s00439-002-0870-8
14. Baran K, Dunstone M, Chia J, Ciccone A, Browne KA, Clarke CJP, et al. The molecular basis for perforin oligomerization and transmembrane pore assembly. Immunity (2009) 30(5):684–95. doi: 10.1016/j.immuni.2009.03.016
15. Zychlinsky A, Zheng LM, Liu CC, Young JD. Cytolytic lymphocytes induce both apoptosis and necrosis in target cells. J Immunol (1991) 146(1):393.
16. Waterhouse NJ, Sutton VR, Sedelies KA, Ciccone A, Jenkins M, Turner SJ, et al. Cytotoxic T lymphocyte-induced killing in the absence of granzymes A and B is unique and distinct from both apoptosis and perforin-dependent lysis. J Cell Biol (2006) 173(1):133–44. doi: 10.1083/jcb.200510072
17. Oshimi Y, Oshimi K, Miyazaki S. Necrosis and apoptosis associated with distinct Ca2+ response patterns in target cells attacked by human natural killer cells. J Physiol (1996) 495(Pt 2):319–29. doi: 10.1113/jphysiol.1996.sp021596
18. Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol (2006) 6(12):940–52. doi: 10.1038/nri1983
19. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science (1999) 286(5446):1957–9. doi: 10.1126/science.286.5446.1957
20. Baker KS, DeLaat CA, Steinbuch M, Gross TG, Shapiro RS, Loechelt B, et al. Successful correction of hemophagocytic lymphohistiocytosis with related or unrelated bone marrow transplantation. Blood (1997) 89(10):3857–63. doi: 10.1182/blood.V89.10.3857.3857_3857_3863
21. Brennan AJ, Chia J, Trapani JA, Voskoboinik I. Perforin deficiency and susceptibility to cancer. Cell Death Differ (2010) 17(4):607–15. doi: 10.1038/cdd.2009.212
22. Clementi R, Locatelli F, Dupre L, Garaventa A, Emmi L, Bregni M, et al. A proportion of patients with lymphoma may harbor mutations of the perforin gene. Blood (2005) 105(11):4424–8. doi: 10.1182/blood-2004-04-1477
23. Chia J, Yeo KP, Whisstock JC, Dunstone MA, Trapani JA, Voskoboinik I. Temperature sensitivity of human perforin mutants unmasks subtotal loss of cytotoxicity, delayed FHL, and a predisposition to cancer. Proc Natl Acad Sci U S A (2009) 106(24):9809–14. doi: 10.1073/pnas.0903815106
24. Badovinac VP, Hamilton SE, Harty JT. Viral infection results in massive CD8+ T cell expansion and mortality in vaccinated perforin-deficient mice. Immunity (2003) 18(4):463–74. doi: 10.1016/s1074-7613(03)00079-7
25. van den Broek ME, Kägi D, Ossendorp F, Toes R, Vamvakas S, Lutz WK, et al. Decreased tumor surveillance in perforin-deficient mice. J Exp Med (1996) 184(5):1781–90. doi: 10.1084/jem.184.5.1781
26. Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey D II, Trapani JA. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med (2000) 192(5):755–60. doi: 10.1084/jem.192.5.755
27. Spilsbury K, O’Mara MA, Wu W, Rowe PB, Symonds G, Takayama Y. Isolation of a novel macrophage-specific gene by differential cDNA analysis. Blood (1995) 85(6):1620–9. doi: 10.1182/blood.V85.6.1620.bloodjournal8561620
28. McCormack R, Podack ER. Perforin-2/Mpeg1 and other pore-forming proteins throughout evolution. J Leukoc Biol (2015) 98(5):761–8. doi: 10.1189/jlb.4MR1114-523RR
29. Fields KA, McCormack R, de Armas LR, Podack ER. Perforin-2 restricts growth of Chlamydia trachomatis in macrophages. Infect Immun (2013) 81(8):3045–54. doi: 10.1128/IAI.00497-13
30. McCormack R, de Armas LR, Shiratsuchi M, Ramos JE, Podack ER. Inhibition of intracellular bacterial replication in fibroblasts is dependent on the perforin-like protein (perforin-2) encoded by macrophage-expressed gene 1. J innate Immun (2013) 5(2):185–94. doi: 10.1159/000345249
31. McCormack RM, Lyapichev K, Olsson ML, Podack ER, Munson GP. Enteric pathogens deploy cell cycle inhibiting factors to block the bactericidal activity of Perforin-2. Elife (2015) 4:e06505. doi: 10.7554/eLife.06505
32. McCormack RM, de Armas LR, Shiratsuchi M, Fiorentino DG, Olsson ML, Lichtenheld MG, et al. Perforin-2 is essential for intracellular defense of parenchymal cells and phagocytes against pathogenic bacteria. Elife (2015) 4:e06508. doi: 10.7554/eLife.06508
33. McCormack R, Bahnan W, Shrestha N, Boucher J, Barreto M, Barrera CM, et al. Perforin-2 protects host cells and mice by restricting the vacuole to cytosol transitioning of a bacterial pathogen. Infect Immun (2016) 84(4):1083–91. doi: 10.1128/IAI.01434-15
34. Bai F, McCormack RM, Hower S, Plano GV, Lichtenheld MG, Munson GP. Perforin-2 breaches the envelope of phagocytosed bacteria allowing antimicrobial effectors access to intracellular targets. J Immunol (2018) 201(9):2710–20. doi: 10.4049/jimmunol.1800365
35. McCormack RM, Szymanski EP, Hsu AP, Perez E, Olivier KN, Fisher E, et al. MPEG1/perforin-2 mutations in human pulmonary nontuberculous mycobacterial infections. JCI Insight (2017) 2(8):e89635. doi: 10.1172/jci.insight.89635
36. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
37. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
38. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535(7610):111–6. doi: 10.1038/nature18590
39. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature (2017) 547(7661):99–103. doi: 10.1038/nature22393
40. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun (2017) 8(1):14128. doi: 10.1038/ncomms14128
41. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol (2018) 3(26):eaar6676. doi: 10.1126/sciimmunol.aar6676
42. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
43. Li JY, Wang YY, Shao T, Fan DD, Lin AF, Xiang LX, et al. The zebrafish NLRP3 inflammasome has functional roles in ASC-dependent interleukin-1beta maturation and gasdermin E-mediated pyroptosis. J Biol Chem (2020) 295(4):1120–41. doi: 10.1074/jbc.RA119.011751
44. Daskalov A, Mitchell PS, Sandstrom A, Vance RE, Glass NL. Molecular characterization of a fungal gasdermin-like protein. Proc Natl Acad Sci U S A (2020) 117(31):18600–7. doi: 10.1073/pnas.2004876117
45. Runkel F, Marquardt A, Stoeger C, Kochmann E, Simon D, Kohnke B, et al. The dominant alopecia phenotypes Bareskin, Rex-denuded, and Reduced Coat 2 are caused by mutations in gasdermin 3. Genomics (2004) 84(5):824–35. doi: 10.1016/j.ygeno.2004.07.003
46. Lunny DP, Weed E, Nolan PM, Marquardt A, Augustin M, Porter RM. Mutations in gasdermin 3 cause aberrant differentiation of the hair follicle and sebaceous gland. J Invest Dermatol (2005) 124(3):615–21. doi: 10.1111/j.0022-202X.2005.23623.x
47. Yu J, Kang M-J, Kim B-J, Kwon J-W, Song Y-H, Choi W-A, et al. Polymorphisms in GSDMA and GSDMB are associated with asthma susceptibility, atopy and BHR. Pediatr Pulmonol (2011) 46(7):701–8. doi: 10.1002/ppul.21424
48. Laer LV, Huizing EH, Verstreken M, v. Zuijlen D, Wauters JG, Bossuyt PJ, et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet (1998) 20(2):194–7. doi: 10.1038/2503
49. Delmaghani S, del Castillo FJ, Michel V, Leibovici M, Aghaie A, Ron U, et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat Genet (2006) 38(7):770–8. doi: 10.1038/ng1829
50. Hadders MA, Beringer DX, Gros P. Structure of C8 -MACPF Reveals Mechanism of membrane attack in complement immune defense. Science (2007) 317(5844):1552–4. doi: 10.1126/science.1147103
51. Rosado CJ, Buckle AM, Law RHP, Butcher RE, Kan W-T, Bird CH, et al. A Common fold mediates vertebrate defense and bacterial attack. Science (2007) 317(5844):1548. doi: 10.1126/science.1144706
52. Law RHP, Lukoyanova N, Voskoboinik I, Caradoc-Davies TT, Baran K, Dunstone MA, et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature (2010) 468(7322):447–51. doi: 10.1038/nature09518PMID-21037563
53. Ruan J, Xia S, Liu X, Lieberman J, Wu H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature (2018) 557(7703):62–7. doi: 10.1038/s41586-018-0058-6
54. Liu Z, Wang C, Yang J, Zhou B, Yang R, Ramachandran R, et al. Crystal structures of the full-length murine and human gasdermin D reveal mechanisms of autoinhibition, lipid binding, and oligomerization. Immunity (2019) 51(1):43–49.e4. doi: 10.1016/j.immuni.2019.04.017
55. Dunstone MA, Tweten RK. Packing a punch: the mechanism of pore formation by cholesterol dependent cytolysins and membrane attack complex/perforin-like proteins. Curr Opin Struct Biol (2012) 22(3):342–9. doi: 10.1016/j.sbi.2012.04.008
56. Mulvihill E, Sborgi L, Mari SA, Pfreundschuh M, Hiller S, Müller DJ. Mechanism of membrane pore formation by human gasdermin-D. EMBO J (2018) 37(14):e98321. doi: 10.15252/embj.201798321
57. Johnson BB, Moe PC, Wang D, Rossi K, Trigatti BL, Heuck AP. Modifications in perfringolysin O domain 4 alter the cholesterol concentration threshold required for binding. Biochemistry (2012) 51(16):3373–82. doi: 10.1021/bi3003132
58. G. T. Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science (2020) 369(6509):1318–30. doi: 10.1126/science.aaz1776
59. Würzner R, Joysey VC, Lachmann PJ. Complement component C7. Assessment of in vivo synthesis after liver transplantation reveals that hepatocytes do not synthesize the majority of human C7. J Immunol (1994) 152(9):4624–9.
60. Hobart MJ, Lachmann PJ, Calne RY. C6: synthesis by the liver in vivo. J Exp Med (1977) 146(2):629–30. doi: 10.1084/jem.146.2.629
61. Alper CA, Raum D, Awdeh ZL, Petersen BH, Taylor PD, Starzl TE. Studies of hepatic synthesis in vivo of plasma proteins, including orosomucoid, transferrin, alpha 1-antitrypsin, C8, and factor B. Clin Immunol Immunopathol (1980) 16(1):84–9. doi: 10.1016/0090-1229(80)90169-5
62. Würzner R. Modulation of complement membrane attack by local C7 synthesis. Clin Exp Immunol (2000) 121(1):8–10. doi: 10.1046/j.1365-2249.2000.01263.x
63. Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature (2019) 572(7768):199–204. doi: 10.1038/s41586-019-1373-2
64. MacParland SA, Liu JC, Ma X-Z, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun (2018) 9(1):4383–3. doi: 10.1038/s41467-018-06318-7
65. Broz P, Pelegrin P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol (2020) 20(3):143–57. doi: 10.1038/s41577-019-0228-2
66. Singhania A, Graham CM, Gabryšová L, Moreira-Teixeira L, Stavropoulos E, Pitt JM, et al. Transcriptional profiling unveils type I and II interferon networks in blood and tissues across diseases. Nat Commun (2019) 10(1):2887. doi: 10.1038/s41467-019-10601-6
67. Saeki N, Usui T, Aoyagi K, Kim DH, Sato M, Mabuchi T, et al. Distinctive expression and function of four GSDM family genes (GSDMA-D) in normal and malignant upper gastrointestinal epithelium. Genes Chromosomes Cancer (2009) 48(3):261–71. doi: 10.1002/gcc.20636
68. Akino K, Toyota M, Suzuki H, Imai T, Maruyama R, Kusano M, et al. Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci (2007) 98(1):88–95. doi: 10.1111/j.1349-7006.2006.00351.x
69. Masuda Y, Futamura M, Kamino H, Nakamura Y, Kitamura N, Ohnishi S, et al. The potential role of DFNA5, a hearing impairment gene, in p53-mediated cellular response to DNA damage. J Hum Genet (2006) 51(8):652–64. doi: 10.1007/s10038-006-0004-6
70. Pipkin ME, Ljutic B, Cruz-Guilloty F, Nouzova M, Rao A, Zúñiga-Pflücker JC, et al. Chromosome transfer activates and delineates a locus control region for perforin. Immunity (2007) 26(1):29–41. doi: 10.1016/j.immuni.2006.11.009
71. Podack ER, Hengartner H, Lichtenheld MG. A Central Role of Perforin in Cytolysis? Annu Rev Immunol (1991) 9(1):129–57. doi: 10.1146/annurev.iy.09.040191.001021PMID-1910674
72. Liu CC, Rafii S, Granelli-Piperno A, Trapani JA, Young JD. Perforin and serine esterase gene expression in stimulated human T cells. Kinetics, mitogen requirements, and effects of cyclosporin A. J Exp Med (1989) 170(6):2105–18. doi: 10.1084/jem.170.6.2105
73. Glimcher LH, Townsend MJ, Sullivan BM, Lord GM. Recent developments in the transcriptional regulation of cytolytic effector cells. Nat Rev Immunol (2004) 4(11):900–11. doi: 10.1038/nri1490
74. Pipkin ME, Rao A, Lichtenheld MG. The transcriptional control of the perforin locus. Immunol Rev (2010) 235(1):55–72. doi: 10.1111/j.0105-2896.2010.00905.xPMID-20536555
75. Jacobs R, Hintzen G, Kemper A, Beul K, Kempf S, Behrens G, et al. CD56bright cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells. Eur J Immunol (2001) 31(10):3121–7. doi: 10.1002/1521-4141(2001010)31:10<3121::aid-immu3121>3.0.co;2-4
76. Lacorazza HD, Miyazaki Y, Di Cristofano A, Deblasio A, Hedvat C, Zhang J, et al. The ETS protein MEF plays a critical role in perforin gene expression and the development of natural killer and NK-T cells. Immunity (2002) 17(4):437–49. doi: 10.1016/s1074-7613(02)00422-3
77. Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, et al. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity (2007) 26(6):798–811. doi: 10.1016/j.immuni.2007.04.010
78. Kim N, Kim M, Yun S, Doh J, Greenberg PD, Kim TD, et al. MicroRNA-150 regulates the cytotoxicity of natural killers by targeting perforin-1. J Allergy Clin Immunol (2014) 134(1):195–203. doi: 10.1016/j.jaci.2014.02.018
79. Anderson P. Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat Rev Immunol (2010) 10(1):24–35. doi: 10.1038/nri2685
80. Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol (2017) 18(1):31–42. doi: 10.1038/nrm.2016.132
81. Heng TS, Painter MW, Consortium IGP. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol (2008) 9(10):1091–4. doi: 10.1038/ni1008-1091
82. Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, et al. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res (2013) 41(Database issue):D1040–6. doi: 10.1093/nar/gks1215
83. Ibana JA, Sherchand SP, Fontanilla FL, Nagamatsu T, Schust DJ, Quayle AJ, et al. Chlamydia trachomatis-infected cells and uninfected-bystander cells exhibit diametrically opposed responses to interferon gamma. Sci Rep (2018) 8(1):8476. doi: 10.1038/s41598-018-26765-y
84. Bubeck D, Roversi P, Donev R, Morgan BP, Llorca O, Lea SM. Structure of human complement C8, a precursor to membrane attack. J Mol Biol (2011) 405(2):325–30. doi: 10.1016/j.jmb.2010.10.031
85. Tschopp J, Müller-Eberhard HJ, Podack ER. Formation of transmembrane tubules by spontaneous polymerization of the hydrophilic complement protein C9. Nature (1982) 298(5874):534–8. doi: 10.1038/298534a0
86. Dudkina NV, Spicer BA, Reboul CF, Conroy PJ, Lukoyanova N, Elmlund H, et al. Structure of the poly-C9 component of the complement membrane attack complex. Nat Commun (2016) 7(1):10588. doi: 10.1038/ncomms10588PMID-26841934
87. Spicer BA, Law RHP, Caradoc-Davies TT, Ekkel SM, Bayly-Jones C, Pang S-S, et al. The first transmembrane region of complement component-9 acts as a brake on its self-assembly. Nat Commun (2018) 9(1):3266. doi: 10.1038/s41467-018-05717-0
88. Liu Z, Wang C, Rathkey JK, Yang J, Dubyak GR, Abbott DW, et al. Structures of the gasdermin D C-terminal domains reveal mechanisms of autoinhibition. Structure (London Engl 1993) (2018) 26(5):778–784.e3. doi: 10.1016/j.str.2018.03.002
89. Voskoboinik I, Thia M-C, Fletcher J, Ciccone A, Browne K, Smyth MJ, et al. Calcium-dependent plasma membrane binding and Cell Lysis by Perforin Are Mediated through Its C2 Domain A CRITICAL ROLE FOR ASPARTATE RESIDUES 429, 435, 483, AND 485 BUT NOT 491. J Biol Chem (2005) 280(9):8426–34. doi: 10.1074/jbc.m413303200PMID-15576364
90. Tschopp J, Schäfer S, Masson D, Peitsch MC, Heusser C. Phosphorylcholine acts as a Ca2+-dependent receptor molecule for lymphocyte perforin. Nature (1989) 337(6204):272–4. doi: 10.1038/337272a0
91. Uellner R, Zvelebil MJ, Hopkins J, Jones J, MacDougall LK, Morgan BP, et al. Perforin is activated by a proteolytic cleavage during biosynthesis which reveals a phospholipid-binding C2 domain. EMBO J (1997) 16(24):7287–96. doi: 10.1093/emboj/16.24.7287
92. House IG, House CM, Brennan AJ, Gilan O, Dawson MA, Whisstock JC, et al. Regulation of perforin activation and pre-synaptic toxicity through C-terminal glycosylation. EMBO Rep (2017) 18(10):1775–85. doi: 10.15252/embr.201744351
93. Brennan AJ, Chia J, Browne KA, Ciccone A, Ellis S, Lopez JA, et al. Protection from endogenous perforin: glycans and the C terminus regulate exocytic trafficking in cytotoxic lymphocytes. Immunity (2011) 34(6):879–92. doi: 10.1016/j.immuni.2011.04.007
94. Shi X, Jarvis D. Protein N-glycosylation in the baculovirus-insect cell system. Curr Drug Targets (2007) 8(10):1116–25. doi: 10.2174/138945007782151360
95. Balaji KN, Schaschke N, Machleidt W, Catalfamo M, Henkart PA. Surface cathepsin b protects cytotoxic lymphocytes from self-destruction after degranulation. J Exp Med (2002) 196(4):493–503. doi: 10.1084/jem.20011836
96. Konjar Š., Sutton VR, Hoves S, Repnik U, Yagita H, Reinheckel T, et al. Human and mouse perforin are processed in part through cleavage by the lysosomal cysteine proteinase cathepsin L. Immunology (2010) 131(2):257–67. doi: 10.1111/j.1365-2567.2010.03299.x
97. Baran K, Ciccone A, Peters C, Yagita H, Bird P II, Villadangos JA, et al. Cytotoxic T lymphocytes from cathepsin B-deficient mice survive normally in vitro and in vivo after encountering and killing target cells. J Biol Chem (2006) 281(41):30485–91. doi: 10.1074/jbc.M602007200
98. Chan C-B, Abe M, Hashimoto N, Hao C, Williams IR, Liu X, et al. Mice lacking asparaginyl endopeptidase develop disorders resembling hemophagocytic syndrome. Proc Natl Acad Sci U S A (2009) 106(2):468–73. doi: 10.1073/pnas.0809824105
99. Traore DAK, Brennan AJ, Law RHP, Dogovski C, Perugini MA, Lukoyanova N, et al. Defining the interaction of perforin with calcium and the phospholipid membrane. Biochem J (2013) 456(3):323–35. doi: 10.1042/bj20130999
100. Dupuis M, Schaerer E, Krause KH, Tschopp J. The calcium-binding protein calreticulin is a major constituent of lytic granules in cytolytic T lymphocytes. J Exp Med (1993) 177(1):1–7. doi: 10.1084/jem.177.1.1
101. Pang SS, Bayly-Jones C, Radjainia M, Spicer BA, Law RHP, Hodel AW, et al. The cryo-EM structure of the acid activatable pore-forming immune effector Macrophage-expressed gene 1. Nat Commun (2019) 10(1):4288. doi: 10.1038/s41467-019-12279-2
102. Ni T, Jiao F, Yu X, Aden S, Ginger L, Williams S II, et al. Structure and mechanism of bactericidal mammalian perforin-2, an ancient agent of innate immunity. Sci Adv (2020) 6(5):eaax8286. doi: 10.1126/sciadv.aax8286
103. de Armas LR. Characterization of Perforin-2, a Novel Anti-bacterial, Pore-forming Protein of the Innate Immune System. (2013) University of Miami.
104. Luber CA, Cox J, Lauterbach H, Fancke B, Selbach M, Tschopp J, et al. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity (2010) 32(2):279–89. doi: 10.1016/j.immuni.2010.01.013
105. Kozik P, Gros M, Itzhak DN, Joannas L, Heurtebise-Chrétien S, Krawczyk PA, et al. Small Molecule Enhancers of Endosome-to-Cytosol Import Augment Anti-tumor Immunity. Cell Rep (2020) 32(2):107905. doi: 10.1016/j.celrep.2020.107905
106. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11(9):785–97. doi: 10.1038/ni.1923
107. DiScipio RG, Smith CA, Muller-Eberhard HJ, Hugli TE. The activation of human complement component C5 by a fluid phase C5 convertase. J Biol Chem (1983) 258(17):10629–36.
108. Hadders MA, Bubeck D, Roversi P, Hakobyan S, Forneris F, Morgan BP, et al. Assembly and Regulation of the Membrane Attack Complex Based on Structures of C5b6 and sC5b9. Cell Rep (2012) 1(3):200–7. doi: 10.1016/j.celrep.2012.02.003
109. Aleshin AE, Schraufstatter IU, Stec B, Bankston LA, Liddington RC, DiScipio RG. Structure of Complement C6 Suggests a Mechanism for Initiation and Unidirectional, Sequential Assembly of Membrane Attack Complex (MAC). J Biol Chem (2012) 287(13):10210–22. doi: 10.1074/jbc.m111.327809
110. Aleshin AE, DiScipio RG, Stec B, Liddington RC. Crystal Structure of C5b-6 Suggests Structural Basis for Priming Assembly of the Membrane Attack Complex. J Biol Chem (2012) 287(23):19642–52. doi: 10.1074/jbc.m112.361121
111. Parsons ES, Stanley GJ, Pyne ALB, Hodel AW, Nievergelt AP, Menny A, et al. Single-molecule kinetics of pore assembly by the membrane attack complex. Nat Commun (2019) 10(1):2066. doi: 10.1038/s41467-019-10058-7
112. Preissner KT, Podack ER, Müller-Eberhard HJ. The membrane attack complex of complement: relation of C7 to the metastable membrane binding site of the intermediate complex C5b-7. J Immunol (1985) 135(1):445–51.
113. DiScipio RG, Chakravarti DN, Muller-Eberhard HJ, Fey GH. The structure of human complement component C7 and the C5b-7 complex. J Biol Chem (1988) 263(1):549–60.
114. Menny A, Serna M, Boyd CM, Gardner S, Joseph AP, Morgan BP, et al. CryoEM reveals how the complement membrane attack complex ruptures lipid bilayers. Nat Commun (2018) 9(1):5316. doi: 10.1038/s41467-018-07653-5
115. Steckel EW, Welbaum BE, Sodetz JM. Evidence of direct insertion of terminal complement proteins into cell membrane bilayers during cytolysis. Labeling by a photosensitive membrane probe reveals a major role for the eighth and ninth components. J Biol Chem (1983) 258(7):4318–24.
116. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16(7):407–20. doi: 10.1038/nri.2016.58
117. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514(7521):187–92. doi: 10.1038/nature13683
118. Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, et al. Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell (2020) 180(5):941–955 e20. doi: 10.1016/j.cell.2020.02.002
119. Liu Z, Wang C, Yang J, Chen Y, Zhou B, Abbott DW, et al. Caspase-1 engages full-length gasdermin D through two distinct interfaces that mediate caspase recruitment and substrate cleavage. Immunity (2020) 53(1):106–114 e5. doi: 10.1016/j.immuni.2020.06.007
120. Sarhan J, Liu BC, Muendlein H II, Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during. Proc Natl Acad Sci U S A (2018) 115(46):E10888–97. doi: 10.1073/pnas.1809548115
121. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science (2018) 362(6418):1064–9. doi: 10.1126/science.aau2818
122. Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J (2019) 38(10):e101638. doi: 10.15252/embj.2019101638
123. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol (2018) 3(26):eaar6689. doi: 10.1126/sciimmunol.aar6689
124. Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep (2018) 22(11):2924–36. doi: 10.1016/j.celrep.2018.02.067
125. Burgener SS, Leborgne NGF, Snipas SJ, Salvesen GS, Bird P II, Benarafa C. Cathepsin G inhibition by Serpinb1 and Serpinb6 prevents programmed necrosis in neutrophils and monocytes and reduces GSDMD-driven inflammation. Cell Rep (2019) 27(12):3646–3656.e5. doi: 10.1016/j.celrep.2019.05.065
126. Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem Biol (2017) 24(4):507–514.e4. doi: 10.1016/j.chembiol.2017.03.009
127. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science (2020) 368(6494):eaaz7548. doi: 10.1126/science.aaz7548
128. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature (2020) 579(7799):415–20. doi: 10.1038/s41586-020-2071-9
129. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol (2020) 5(43):eaax7969. doi: 10.1126/sciimmunol.aax7969
130. Lanier LL. NK CELL RECOGNITION. Annu Rev Immunol (2004) 23(1):225–74. doi: 10.1146/annurev.immunol.23.021704.115526
131. Almeida CR, Davis DM. Segregation of HLA-C from ICAM-1 at NK cell immune synapses is controlled by its cell surface density. J Immunol (2006) 177(10):6904. doi: 10.4049/jimmunol.177.10.6904
132. Coscoy L, Ganem D. Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc Natl Acad Sci U S A (2000) 97(14):8051–6. doi: 10.1073/pnas.140129797
133. Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol (2016) 39:44–51. doi: 10.1016/j.coi.2015.12.007
134. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999) 285(5428):727. doi: 10.1126/science.285.5428.727
135. Cosman D, Müllberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, et al. ULBPs, Novel MHC class I–related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity (2001) 14(2):123–33. doi: 10.1016/S1074-7613(01)00095-4
136. Dieckmann NMG, Frazer GL, Asano Y, Stinchcombe JC, Griffiths GM. The cytotoxic T lymphocyte immune synapse at a glance. J Cell Sci (2016) 129(15):2881. doi: 10.1242/jcs.186205
137. Kabanova A, Zurli V, Baldari CT. Signals controlling lytic granule polarization at the cytotoxic immune synapse. Front Immunol (2018) 9:307. doi: 10.3389/fimmu.2018.00307
138. Krzewski K, Coligan JE. Human NK cell lytic granules and regulation of their exocytosis. Front Immunol (2012) 3:335. doi: 10.3389/fimmu.2012.00335
139. Luzio JP, Hackmann Y, Dieckmann NMG, Griffiths GM. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harbor Perspect Biol (2014) 6(9):a016840. doi: 10.1101/cshperspect.a016840PMID-25183830
140. Lopez JA, Susanto O, Jenkins MR, Lukoyanova N, Sutton VR, Law RH, et al. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood (2013) 121(14):2659–68. doi: 10.1182/blood-2012-07-446146
141. Leung C, Hodel AW, Brennan AJ, Lukoyanova N, Tran S, House CM, et al. Real-time visualization of perforin nanopore assembly. Nat Nanotechnol (2017) 12(5):467–73. doi: 10.1038/nnano.2016.303
142. Liu X, Lieberman J. A Mechanistic understanding of pyroptosis: the fiery death triggered by invasive infection. Adv Immunol (2017) 135:81–117. doi: 10.1016/bs.ai.2017.02.002
143. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun (2019) 10(1):1689. doi: 10.1038/s41467-019-09397-2
144. Meri S, Morgan BP, Davies A, Daniels RH, Olavesen MG, Waldmann H, et al. Human protectin (CD59), an 18,000-20,000 MW complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipid bilayers. Immunology (1990) 71(1):1–9.
145. Ninomiya H, Sims PJ. The human complement regulatory protein CD59 binds to the alpha-chain of C8 and to the “b”domain of C9. J Biol Chem (1992) 267(19):13675–80.
146. Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell (1993) 73(4):703–11. doi: 10.1016/0092-8674(93)90250-t
147. Nevo Y, Ben-Zeev B, Tabib A, Straussberg R, Anikster Y, Shorer Z, et al. CD59 deficiency is associated with chronic hemolysis and childhood relapsing immune-mediated polyneuropathy. Blood (2013) 121(1):129–35. doi: 10.1182/blood-2012-07-441857
148. Haliloglu G, Maluenda J, Sayinbatur B, Aumont C, Temucin C, Tavil B, et al. Early-onset chronic axonal neuropathy, strokes, and hemolysis: inherited CD59 deficiency. Neurology (2015) 84(12):1220–4. doi: 10.1212/WNL.0000000000001391
149. Chauhan AK, Moore TL. Presence of plasma complement regulatory proteins clusterin (Apo J) and vitronectin (S40) on circulating immune complexes (CIC). Clin Exp Immunol (2006) 145(3):398–406. doi: 10.1111/j.1365-2249.2006.03135.x
150. Tschopp J, Chonn A, Hertig S, French LE. Clusterin, the human apolipoprotein and complement inhibitor, binds to complement C7, C8 beta, and the b domain of C9. J Immunol (1993) 151(4):2159–65.
151. Podack ER, Kolb WP, Müller-Eberhard HJ. The SC5b-7 complex: formation, isolation, properties, and subunit composition. J Immunol (1977) 119(6):2024–9.
152. Podack ER, Preissner KT, Müller-Eberhard HJ. Inhibition of C9 polymerization within the SC5b-9 complex of complement by S-protein. Acta Pathol Microbiol Immunol Scand Suppl (1984) 284:89–96.
153. Singh B, Su YC, Riesbeck K. Vitronectin in bacterial pathogenesis: a host protein used in complement escape and cellular invasion. Mol Microbiol (2010) 78(3):545–60. doi: 10.1111/j.1365-2958.2010.07373.x
154. Rothstein TL, Mage M, Jones G, McHugh LL. Cytotoxic T lymphocyte sequential killing of immobilized allogeneic tumor target cells measured by time-lapse microcinematography. J Immunol (Baltimore Md. 1950) (1978) 121(5):1652–6.
155. Bhat R, Watzl C. Serial killing of tumor cells by human natural killer cells–enhancement by therapeutic antibodies. PloS One (2007) 2(3):e326. doi: 10.1371/journal.pone.0000326
156. Choi PJ, Mitchison TJ. Imaging burst kinetics and spatial coordination during serial killing by single natural killer cells. Proc Natl Acad Sci U S A (2013) 110(16):6488–93. doi: 10.1073/pnas.1221312110
157. Vanherberghen B, Olofsson PE, Forslund E, Sternberg-Simon M, Khorshidi MA, Pacouret S, et al. Classification of human natural killer cells based on migration behavior and cytotoxic response. Blood (2013) 121(8):1326–34. doi: 10.1182/blood-2012-06-439851
158. Blakely A, Gorman K, Ostergaard H, Svoboda K, Liu CC, Young JD, et al. Resistance of cloned cytotoxic T lymphocytes to cell-mediated cytotoxicity. J Exp Med (1987) 166(4):1070–83. doi: 10.1084/jem.166.4.1070
159. Kranz DM, Eisen HN. Resistance of cytotoxic T lymphocytes to lysis by a clone of cytotoxic T lymphocytes. Proc Natl Acad Sci U S A (1987) 84(10):3375–9. doi: 10.1073/pnas.84.10.3375
160. Lopez JA, Jenkins MR, Rudd-Schmidt JA, Brennan AJ, Danne JC, Mannering S II, et al. Rapid and unidirectional perforin pore delivery at the cytotoxic immune synapse. J Immunol (2013) 191(5):2328–34. doi: 10.4049/jimmunol.1301205
161. Verret CR, Firmenich AA, Kranz DM, Eisen HN. Resistance of cytotoxic T lymphocytes to the lytic effects of their toxic granules. J Exp Med (1987) 166(5):1536–47. doi: 10.1084/jem.166.5.1536
162. Rudd-Schmidt JA, Hodel AW, Noori T, Lopez JA, Cho HJ, Verschoor S, et al. Lipid order and charge protect killer T cells from accidental death. Nat Commun (2019) 10(1):5396. doi: 10.1038/s41467-019-13385-x
163. Golstein P. Sensitivity of cytotoxic T cells to T-cell mediated cytotoxicity. Nature (1974) 252(5478):81–3. doi: 10.1038/252081a0
164. Kuppers RC, Henney CS. Studies on the mechanism of lymphocyte-mediated cytolysis. IX. Relationships between antigen recognition and lytic expression in killer T cells. J Immunol (1977) 118(1):71–6.
165. Huang JF, Yang Y, Sepulveda H, Shi W, Hwang I, Peterson PA, et al. TCR-mediated internalization of peptide-MHC complexes acquired by T cells. Science (1999) 286(5441):952–4. doi: 10.1126/science.286.5441.952
166. Kupfer A, Singer SJ, Dennert G. On the mechanism of unidirectional killing in mixtures of two cytotoxic T lymphocytes. Unidirectional polarization of cytoplasmic organelles and the membrane-associated cytoskeleton in the effector cell. J Exp Med (1986) 163(3):489–98. doi: 10.1084/jem.163.3.489
167. Burrows SR, Fernan A, Argaet V, Suhrbier A. Bystander apoptosis induced by CD8+ cytotoxic T cell (CTL) clones: implications for CTL lytic mechanisms. Int Immunol (1993) 5(9):1049–58. doi: 10.1093/intimm/5.9.1049
168. Jiang SB, Ojcius DM, Persechini PM, Young JD. Resistance of cytolytic lymphocytes to perforin-mediated killing. Inhibition of perforin binding activity by surface membrane proteins. J Immunol (1990) 144(3):998–1003.
169. Cohnen A, Chiang SC, Stojanovic A, Schmidt H, Claus M, Saftig P, et al. Surface CD107a/LAMP-1 protects natural killer cells from degranulation-associated damage. Blood (2013) 122(8):1411–8. doi: 10.1182/blood-2012-07-441832
170. Krzewski K, Gil-Krzewska A, Nguyen V, Peruzzi G, Coligan JE. LAMP1/CD107a is required for efficient perforin delivery to lytic granules and NK-cell cytotoxicity. Blood (2013) 121(23):4672–83. doi: 10.1182/blood-2012-08-453738
171. Antia R, Schlegel RA, Williamson P. Binding of perforin to membranes is sensitive to lipid spacing and not headgroup. Immunol Lett (1992) 32(2):153–7. doi: 10.1016/0165-2478(92)90108-z
172. Burack WR, Lee KH, Holdorf AD, Dustin ML, Shaw AS. Cutting edge: quantitative imaging of raft accumulation in the immunological synapse. J Immunol (2002) 169(6):2837–41. doi: 10.4049/jimmunol.169.6.2837
173. Gaus K, Chklovskaia E, Fazekas de St Groth B, Jessup W, Harder T. Condensation of the plasma membrane at the site of T lymphocyte activation. J Cell Biol (2005) 171(1):121–31. doi: 10.1083/jcb.200505047
174. Fischer K, Voelkl S, Berger J, Andreesen R, Pomorski T, Mackensen A. Antigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cells. Blood (2006) 108(13):4094–101. doi: 10.1182/blood-2006-03-011742
175. Zech T, Ejsing CS, Gaus K, de Wet B, Shevchenko A, Simons K, et al. Accumulation of raft lipids in T-cell plasma membrane domains engaged in TCR signalling. EMBO J (2009) 28(5):466–76. doi: 10.1038/emboj.2009.6
176. Basu R, Whitlock BM, Husson J, Le Floc’h A, Jin W, Oyler-Yaniv A, et al. Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell (2016) 165(1):100–10. doi: 10.1016/j.cell.2016.01.021
177. Tamzalit F, Wang MS, Jin W, Tello-Lafoz M, Boyko V, Heddleston JM, et al. Interfacial actin protrusions mechanically enhance killing by cytotoxic T cells. Sci Immunol (2019) 4(33):eaav5445. doi: 10.1126/sciimmunol.aav5445
178. Tamzalit F, Tran D, Jin W, Boyko V, Bazzi H, Kepecs A, et al. Centrioles control the capacity, but not the specificity, of cytotoxic T cell killing. Proc Natl Acad Sci U S A (2020) 117(8):4310–9. doi: 10.1073/pnas.1913220117
179. Gawden-Bone CM, Griffiths GM. Phospholipids: pulling back the actin curtain for granule delivery to the immune synapse. Front Immunol (2019) 10:700. doi: 10.3389/fimmu.2019.00700
180. Andrews NW, Corrotte M. Plasma membrane repair. Curr Biol (2018) 28(8):R392–7. doi: 10.1016/j.cub.2017.12.034
181. Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell (2001) 106(2):157–69. doi: 10.1016/s0092-8674(01)00421-4
182. McNeil PL, Steinhardt RA. Plasma membrane disruption: repair, prevention, adaptation. Annu Rev Cell Dev Biol (2003) 19:697–731. doi: 10.1146/annurev.cellbio.19.111301.140101
183. Keefe D, Shi L, Feske S, Massol R, Navarro F, Kirchhausen T, et al. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity (2005) 23(3):249–62. doi: 10.1016/j.immuni.2005.08.001
184. Thiery J, Keefe D, Saffarian S, Martinvalet D, Walch M, Boucrot E, et al. Perforin activates clathrin- and dynamin-dependent endocytosis, which is required for plasma membrane repair and delivery of granzyme B for granzyme-mediated apoptosis. Blood (2010) 115(8):1582–93. doi: 10.1182/blood-2009-10-246116
185. Thiery J, Keefe D, Boulant S, Boucrot E, Walch M, Martinvalet D, et al. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat Immunol (2011) 12(8):770–7. doi: 10.1038/ni.2050
186. Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol (2015) 15(6):388–400. doi: 10.1038/nri3839
187. Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science (2018) 362(6417):956. doi: 10.1126/science.aar7607
188. Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F. ESCRT machinery is required for plasma membrane repair. Science (2014) 343(6174):1247136. doi: 10.1126/science.1247136
Keywords: immunity, pore-forming proteins, membrane attack complex, perforins, gasdermins, membrane integrity
Citation: Krawczyk PA, Laub M and Kozik P (2020) To Kill But Not Be Killed: Controlling the Activity of Mammalian Pore-Forming Proteins. Front. Immunol. 11:601405. doi: 10.3389/fimmu.2020.601405
Received: 31 August 2020; Accepted: 20 October 2020;
Published: 13 November 2020.
Edited by:
George P. Munson, University of Miami, United StatesReviewed by:
Ilia Voskoboinik, Peter MacCallum Cancer Centre, AustraliaTsan Sam Xiao, Case Western Reserve University, United States
Copyright © 2020 Krawczyk, Laub and Kozik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrycja Kozik, cGtvemlrQG1yYy1sbWIuY2FtLmFjLnVr
†These authors have contributed equally to this work