 Ziwei Xu1†
Ziwei Xu1† Pingzheng Zhou
Pingzheng Zhou- 1Guangdong Provincial Key Laboratory of New Drug Screening, School of Pharmaceutical Sciences, Southern Medical University, Guangzhou, China
- 219th grade, Pharmacy Major, School of Pharmaceutical Sciences, Southern Medical University, Guangzhou, China
- 3Department of Neurology, Nanfang Hospital, Southern Medical University, Guangzhou, China
The NLRP3 inflammasome is a core component of innate immunity, and dysregulation of NLRP3 inflammasome involves developing autoimmune, metabolic, and neurodegenerative diseases. Potassium efflux has been reported to be essential for NLRP3 inflammasome activation by structurally diverse pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs). Thus, the molecular mechanisms underlying potassium efflux to activate NLRP3 inflammasome are under extensive investigation. Here, we review current knowledge about the distinction channels or pore-forming proteins underlying potassium efflux for NLRP3 inflammasome activation with canonical/non-canonical signaling or following caspase-8 induced pyroptosis. Ion channels and pore-forming proteins, including P2X7 receptor, Gasdermin D, pannexin-1, and K2P channels involved present viable therapeutic targets for NLRP3 inflammasome related diseases.
NLRP3 Inflammasome
Inflammasomes are intracellular multiprotein complexes and core components of innate immunity (1–3). To date, the NOD-like receptor (NLR) family and the PYHIN family have been reported to form inflammasomes (4). These are composed of six NLR family proteins, including NLRP1, NLRP2, NLRP3, NLRP6, NLRC4, NLRP12, and two members of the PYHIN family, including AIM2 and IFI16 (5, 6).
Among various inflammasomes, NLRP3 inflammasome has been widely under investigation because of its most significant clinical relevance (7, 8). NLRP3 inflammasome consists of sensory protein NLRP3, adaptor protein ASC (the adaptor molecule apoptosis-associated speck-like protein containing a CARD), and effector protein caspase-1 (7, 8). Canonical NLRP3 inflammasome activation requires two steps: priming and activation. The priming process leads to the expression of NLRP3, pro-IL-1β, and pro-IL-18, which could be initiated by Toll-like receptors (TLR) ligands (9). The activation process promotes the assembly of inflammasome complexes, cleaving pro-caspase-1 to form active caspase-1, thereby cleaving pro-IL-1β and pro-IL-18 to release mature IL-1β and IL-18 (Figure 1).
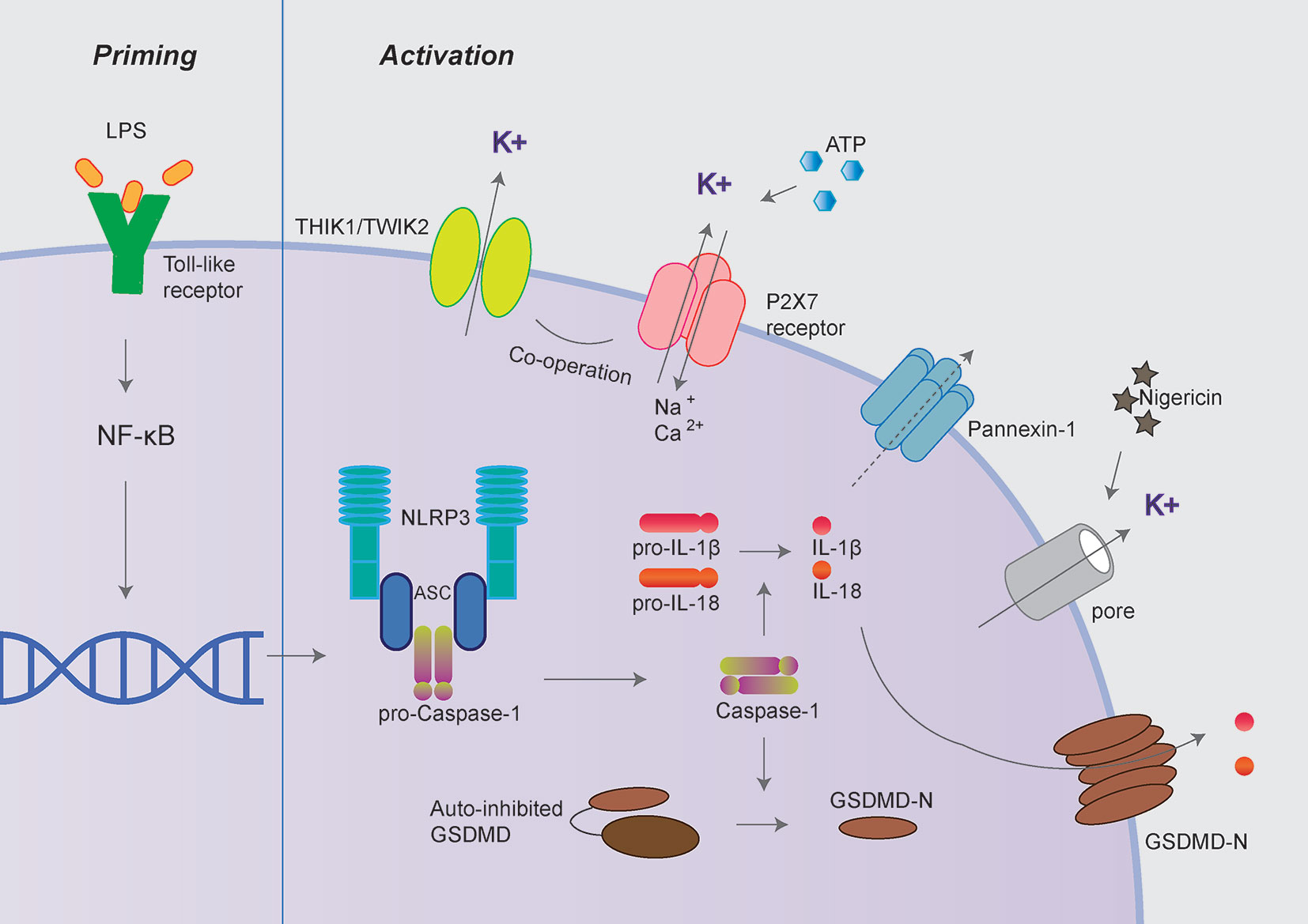
Figure 1 Canonical NLRP3 inflammasome activation. Canonical NLRP3 inflammasome activation includes two signals: priming and activation. The priming process leads to the expression of NLRP3, pro-IL-1β and pro-IL-18 could be provided by Toll-like receptors activation. The activation process promotes the assembly of inflammasome complexes through various PAMPs or DAMPs, including extracellular ATP, nigericin, and particulate matters. The activation of NLRP3 inflammasome cleaves pro-caspase-1 to active caspase-1, thereby cleaves pro-IL-1β and pro-IL-18 to produce mature IL-1β and IL-18. Besides, activated caspase-1 also cleaves GSDMD to release its N-terminal domain, which forms pores at the plasma membrane and mediates the release of mature IL-1β and IL-18. The P2X7 receptor, Pannexin-1, TWIK2, and THIK1 have been proposed to mediate potassium efflux during NLRP3 inflammasome activation under different circumstances.
Besides, activated caspase-1 also cleaves Gasdermin D (GSDMD) to release its N-terminal domain, which forms pores at the plasma membrane and induces a rapid, pro-inflammatory form of cell death termed “pyroptosis” (10–12). Intriguingly, the activation process of NLRP3 inflammasome could be provided by surprisingly various types of PAMPs (pathogen-associated molecular pattern) or DAMPs (danger-associated molecular pattern). These include extracellular ATP, pore-forming toxins (nigericin and maitotoxin, etc.), particulate matter (urate crystalline MSU, aluminum adjuvant, silica, and asbestos), and misfolded proteins related to neurodegenerative diseases (fibrillar Aβ protein; α-synuclein) (10–12). The dysregulated activation of NLRP3 inflammasome is closely related to various auto-inflammatory or chronic inflammations, such as gout, atherosclerosis, obesity, Alzheimer’s disease, Parkinson’s disease, and type 2 diabetes (13–15). Besides the canonical activation process, the non-canonical inflammasome pathway is mediated by caspase-11 in mouse cell or caspase-4/caspase-5 in a human cell in response to cytoplasmic bacterial lipopolysaccharide (LPS) (Figure 2) (16, 17).
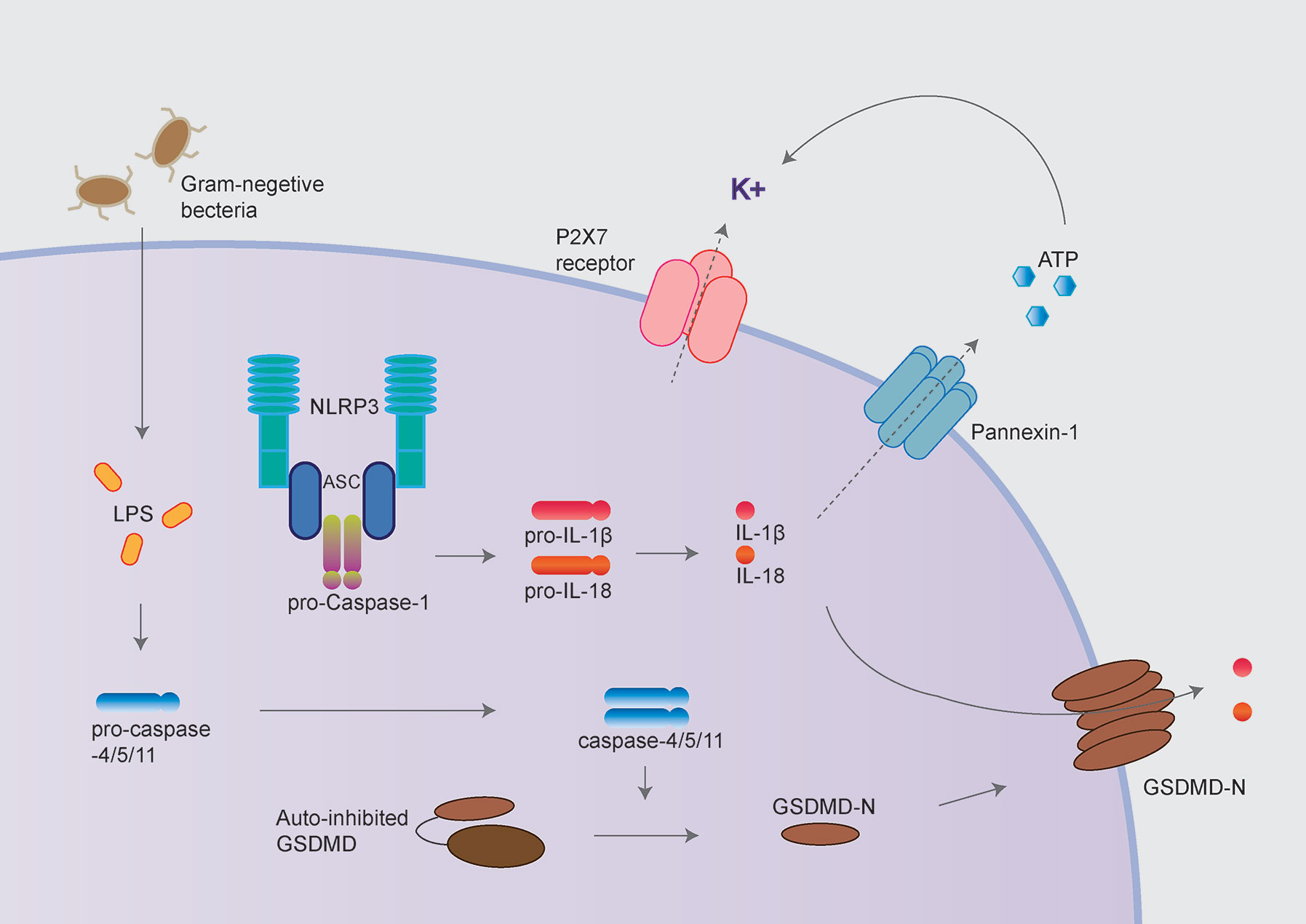
Figure 2 NLRP3 inflammasome activation following non-canonical inflammasome activation. The non-canonical inflammasome pathway is mediated by caspase-11 in mouse cell and caspase-4/caspase-5 in a human cell in response to cytoplasmic bacterial lipopolysaccharide (LPS). Caspase-4/5/11 cleaves GSDMD to initiate pyroptosis, thus leads to NLRP3 inflammasome activation. GSDMD and P2X7 receptor/Pannexin-1 have been proposed to mediate potassium efflux, which underlies the mechanism of NLRP3 inflammasome activation following non-canonical inflammasome.
Cytoplasmic LPS directly binds the caspase recruitment domain (CARD) of caspase-4/5/11, triggering caspase-4/5/11 cleaves GSDMD to initiate pyroptosis (18, 19). Caspase-11 mediated pyroptosis in response to cytosolic LPS is critical for antibacterial defense and septic shock in mice as demonstrated that GSDMD–/– and caspase11–/– mice could be protected against LPS-induced lethality (20, 21). Besides directly causing pyroptosis, the non-canonical inflammasome also promotes the canonical NLRP3 inflammasome to cause the maturation and release of IL-1β and IL-18 (19).
Ion Channels and Pore-Forming Proteins Mediating Potassium Efflux During NLRP3 Inflammasome Activation
It has been well accepted that potassium (K+) efflux is both necessary and sufficient for NLRP3 inflammasome activation in most cases (22–25). First, a large reduction of intracellular potassium concentration was observed to activate the NLRP3 inflammasome by ATP, nigericin, and crystal molecules (23). Furthermore, incubation of primed macrophages in a K+-free medium was sufficient to trigger NLRP3 inflammasome activation (26). In contrast, NLRP3 inflammasome activation could be blocked by high concentrations of extracellular potassium (30-45 mM) (23, 26). Besides, AIM2 and NLRC4 inflammasomes activation was not affected by high concentrations of extracellular K+, indicating potassium’s specific role in modulating NLRP3 inflammasome (23–25).
Structurally diverse DAMPs/PAMPs employ distinct mechanisms to cause potassium efflux to activate the NLRP3 inflammasome. Firstly, the existing research mainly focuses on the molecular mechanism of potassium efflux during ATP-induced NLRP3 inflammasome activation. The P2X7 receptor, pannexin-1, and K2P channels have been reported to participate in the process above (24, 25, 27). Secondly, toxins such as nigericin directly promote potassium efflux by forming pores on the plasma membrane. NLRP3 inflammasome could also be activated following the non-canonical inflammasome or caspase-8 mediated pyroptosis, which also depends on potassium efflux (28–31). Controversially, GSDMD, and pannexin-1 have been proposed to mediate potassium efflux in the above process to activate NLRP3 inflammasome (19, 29, 30, 32, 33). This section will review the current knowledge about ion channels’ roles and pore-forming proteins mediating potassium efflux during NLRP3 inflammasome activation under different circumstances.
P2X7 Receptor
The P2X7 receptor is an ATP-gated cation-selective channel widely expressed in various immune cells (34). At resting conditions, extracellular ATP concentration is at low levels (<10 nM/L), which will be massively increased to several tens or hundreds of μmoles/l within stressed or dying cells (35). The elevated extracellular ATP activates the P2X7 receptor, which then mediates potassium efflux and thus leads to NLRP3 inflammasome activation (34, 36–38).
Besides, the canonical NLRP3 inflammasome, P2X7 receptor, and pannexin-1 (see Pannexin-1) also have been reported to participate in non-canonical inflammasome (39) coordinately. It was reported that the activated caspase-11 cleaves pannexin-1 followed up by ATP release, which in turn activates the P2X7 receptor to mediate potassium efflux and NLRP3 inflammasome activation (39). Correspondingly, the P2X7 receptor ablation significantly reduced the mortality of mice and IL-1β secretion in peritoneal fluid in a sepsis mice model (39). However, this study is contradicted with studies by several other groups that we will discuss in the next section.
Gasdermin D
Gasdermin D (GSDMD) has been identified as the executor of pyroptosis activated by caspase-1/4/5/11 in 2015 (19, 31, 40). Full-length GSDMD includes the N-terminal (GSDMD-N) and C-terminal repressor domain (GSDMD-C) interacting with each other in the absence of stimulation. This auto-inhibitory conformation is released upon efficient cleavage at a conserved glutamic acid residue (D276 in mouse and D275 in human GSDMD) caspase-1/4/5/11, dividing GSDMD into GSDMD-N and GSDMD-C. The generation of GSDMD-N allows it to insert into the plasma membrane and form large oligomeric pores, leading to IL-1β and IL-18 secretion and pyroptosis. Kayagaki et al. and Shi et al. reported that potassium pass through the pore-forming GSDMD, which further leads to NLRP3 inflammasome activation during non-canonical inflammasome activation (18, 19). Besides mediating pyroptosis and NLRP3 inflammasome activation, GSDMD was recently reported to restrain type I interferon response to cytosolic DNA by driving potassium efflux (41).
Caspase-8 has long been considered to play key roles in extrinsic apoptosis and suppress necroptosis by inhibiting RIPK1/RIPK3 and MLKL. More recently, three independent studies have demonstrated the “apoptotic” caspase-8 also could cleave GSDMD leading to pyroptosis-like cell death, further triggering NLRP3 inflammasome activation in murine macrophages (Figure 3) (29, 30, 32). It has been proposed that potassium efflux underlies NLRP3 inflammasome activation, followed by caspase-8 mediated pyroptosis. However, three groups disagree with the molecular mechanism underlying potassium efflux in the process above. Orning et al. and Sarhan et al. suggest that NLRP3 inflammasome activation is dependent on GSDMD-mediated potassium efflux based on delays in ASC oligomerization in GSDMD-/- cells (29, 30). However, Chen et al. observed normal caspase-1 processing in GSDMD-/- and/or GSDME-/- (Gasdermin E; another member of Gasdermin protein) cells, which suggests NLRP3 inflammasome activation is not dependent on GSDMD or GSDME (32).
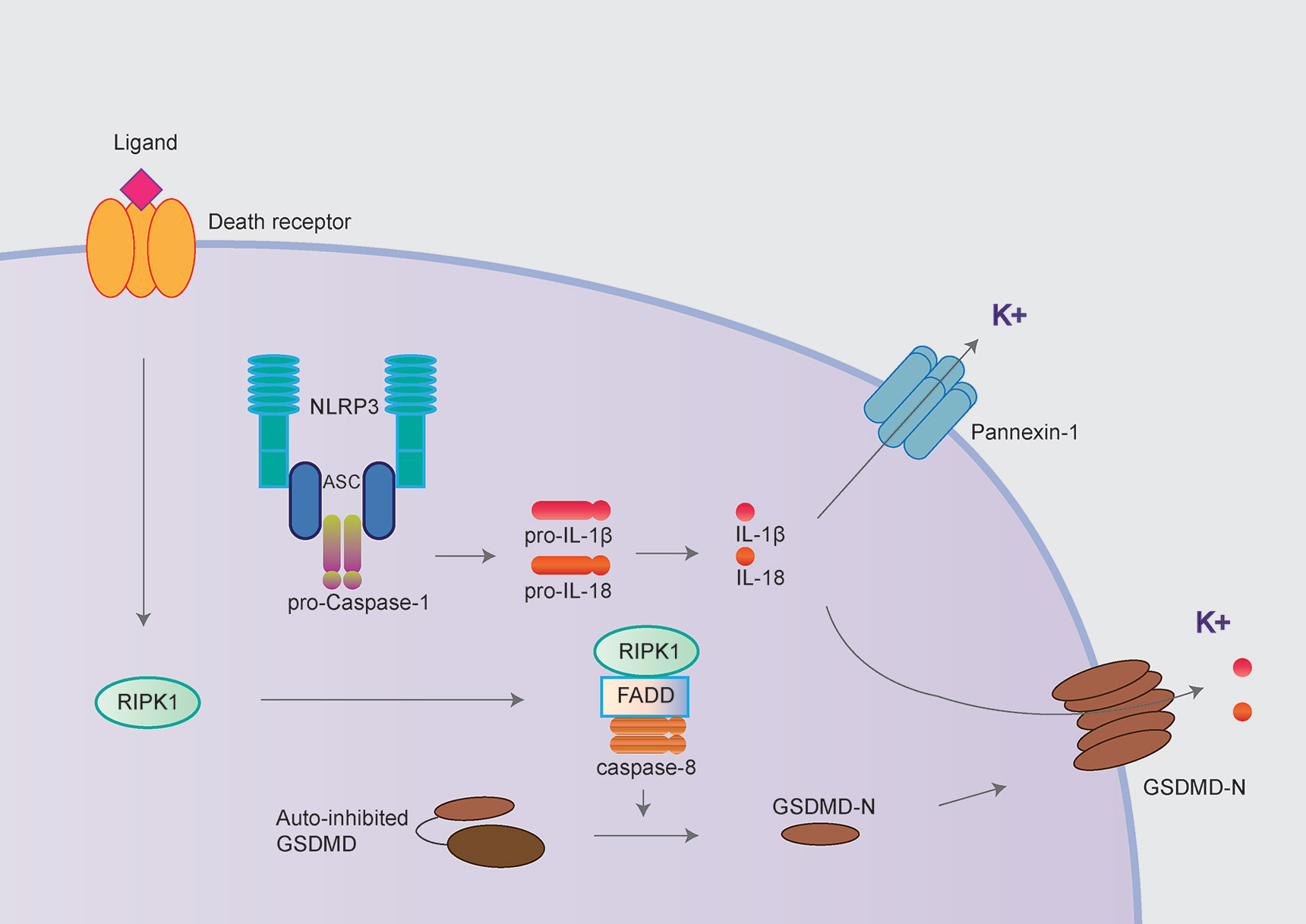
Figure 3 NLRP3 inflammasome activation following caspase-8 mediated pyroptosis. The NLRP3 inflammasome could also be activated following by caspase-8 mediated pyroptosis. The “apoptotic” caspase-8 cleaves GSDMD and further mediated potassium efflux leading to NLRP3 inflammasome activation in murine macrophages. In contrast, Chen et al. demonstrated the pannexin-1 but not GSDMD mediating potassium efflux contributed to NLRP3 inflammasome activation following caspase-8 mediated pyroptosis.
Pannexin-1
The pannexin-1 is a non-selective, large-pore channel that releases potassium and nucleotides, including ATP (42, 43). Pannexin-1 is expressed in most cell types and functionally auto-inhibited by its cytoplasmic C-terminal domain. In response to apoptosis, the pannexin-1 channel can be functionally activated by caspase-3 mediated cleavage of the inhibitory C-terminal domain (44, 45).
The relationship between pannexin-1 and NLRP3 inflammasome is still controversial. By using pannexin-1 inhibitors or siRNA, Pelegrin et al. reported that pannexin-1 is responsible for IL-1β release upon NLRP3 inflammasome agonists ATP or nigericin (46–48). However, this channel was lately reported to be dispensable for canonical NLRP3 inflammasome activation using pannexin-1 knockout mice (49).
Together with the P2X7 receptor, pannexin-1 was also implicated in promoting pyroptosis and NLRP3 activation during non-canonical inflammasome activation (discussed in P2X7 Receptor) (39). In LPS-induced sepsis mouse models, the ablation of pannexin-1 significantly reduced mice mortality, which indicates the role of pannexin-1 in non-canonical inflammasome activation (39). This finding is at odds with the observation that caspase-11 drives NLRP3 inflammasome activation through GSDMD pores (18, 19, 40). A recent study further pointed out that pannexin-1 is dispensable for canonical or non-canonical inflammasome activation within pharmacological inhibition and two other macrophages strain with pannexin-1 ablation (33).
Interestingly, during the NLRP3 inflammasome activation following caspase-8 activated pyroptosis, Chen et al. observed that potassium efflux mediated by pannexin-1 but not GSDMD is critical for NLRP3 inflammasome activation following caspase-8 mediated pyroptosis (32, 33).
K2P Channels
Two-pore domain potassium (K2P) channels comprise a major and structurally distinct subset of mammalian K+ channel superfamily, including fifteen K2P subtypes that form six subfamilies (TWIK, TASK, TRESK, TALK, THIK, and TRESK) (50, 51). K2P channels contribute to the background leak currents, responsible for maintaining the resting membrane potential in nearly all cells. They are regulated by various physical, chemical, and biological stimuli and implicated in multiple physiological processes. In recent years, significant roles of K2P channels for the activation of NLRP3 inflammasome and innate immunity have been gradually revealed (27, 52).
TWIK2 is a member of K2P channels, highly expressed in the gastrointestinal tract, blood vessels, and immune system (53). Given that TWIK2 showed no or little conductance in heterologous expression systems, the physiological functions of TWIK2 is poorly understood (54). Interestingly, a recent study demonstrated that pharmacological inhibition or genetic deletion of the TWIK2 channel blocked the activation of NLRP3 inflammasome induced by ATP and thus reduced the release of caspase-1 and IL-1β (27). In contrast, the TWIK2 channel had no effect on the activation of NLRP3 inflammasome activated by imiquimod or nigericin. The TWIK2 channel was mechanistically suggested to cooperate with the P2X7 receptor activated by extracellular ATP, thus mediated potassium efflux required for NLRP3 inflammasome activation.
Furthermore, TWIK2 deletion prevents inflammatory lung injury in sepsis mice (27). Besides TWIK2, THIK1 channel, another member of K2P channels, was recently discovered to play key roles in microglia (52). THIK1 channel was reported to be the main potassium channel expressed in microglia. Pharmacological inhibition or gene knockout of THIK1 depolarizes microglia, decreasing microglial ramification, reducing surveillance function, and IL-1β secretion. This study indicates that THIK1 is necessary for NLRP3 inflammasome activation and immune surveillance in microglia.
The Mechanisms of Potassium Efflux During NLRP3 Inflammasome Activation
K+ efflux is proposed as an important event upstream of NLRP3 inflammasome activation, and the decrease in intracellular K+ can activate the NLRP3 inflammasome; however, the mechanisms of potassium efflux during NLRP3 inflammasome activation is not understood. Macrophages expressing a constitutively active mutant NLRP3 R258W, which could not be suppressed by high extracellular concentrations of potassium, suggests that potassium efflux may be related to NLRP3 protein conformational change (23). Two individual studies show that potassium efflux is essential for NLRP3 and NEK7 interaction, which is an important part of the assembly of the NLRP3 inflammasome, given that the interaction disappears with high extracellular concentrations of potassium (55, 56). These studies suggest that potassium efflux may be closely related to the conformational change of NLRP3 protein and NLRP3-NEK7 interaction during NLRP3 activation, and the underlying mechanism ought to be further investigated. Moreover, K+ efflux might promote NLRP3 activation by mitochondrial dysfunction and mtROS production (57).
Summary and Outlook
Given the critical role of NLRP3 inflammasome in autoimmune, metabolic, and neurodegenerative diseases and the essential role of potassium efflux in NLRP3 inflammasome activation, it is of great significance to explore the molecular mechanisms underlying potassium efflux during NLRP3 inflammasome activation under different circumstances.
The important role of the P2X7 receptor and GSDMD in immune responses has gained a lot of attention, both academically and industrially (31, 34). An inhibitor JNJ-55308942 targeting the P2X7 receptor is now in phase I clinical study to treat neuroinflammation (58). The role of pannexin-1 in NLRP3 inflammasome activation following caspase-11 or caspase-8 induced pyroptosis is still under debate. Furthermore, although crystalline substances also depend on potassium efflux to activate NLRP3 inflammasome, this process’s mechanism is not clear and needed to be resolved in the future.
Last but not least, the lately identified TWIK2 and THIK1 channels were the only “specific” potassium channels involved in NLRP3 inflammasome activation (27, 52). Both TWIK2 and THIK1 channels could be attractive therapeutic targets for the treatment of NLRP3 inflammasome related autoimmune diseases in the future.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
This study was supported by research grants from the National Natural Science Foundation of China (81973333), the Young Scholars of Pearl River in Guangdong Province, and the Natural Science Foundation of Guangdong Province (2019A1515010964) to PZ.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
2. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi: 10.1038/nri.2016.58
3. Martinon F, Burns K, Tschopp J. The Inflammasome. Mol Cell (2002) 10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3
4. He Y, Hara H, Núñez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
5. Swanson KV, Deng M, Ting JP. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
6. Compan V, Baroja-Mazo A, López-Castejón G, Gomez AI, Martínez CM, Angosto D, et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity (2012) 37:487–500. doi: 10.1016/j.immuni.2012.06.013
7. Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature (2006) 440:228–32. doi: 10.1038/nature04515
8. de Zoete MR, Palm NW, Zhu S, Flavell RA. Inflammasomes. Cold Spring Harb Perspect Biol (2014) 6:a016287. doi: 10.1101/cshperspect.a016287
9. He Y, Franchi L, Núñez G. TLR agonists stimulate Nlrp3-dependent IL-1β production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J Immunol (2013) 190:334–9. doi: 10.4049/jimmunol.1202737
10. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol (2008) 9:857–65. doi: 10.1038/ni.1636
11. Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med (2018) 10(465):eaah4066. doi: 10.1126/scitranslmed.aah4066
12. Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol (2012) 28:137–61. doi: 10.1146/annurev-cellbio-101011-155745
13. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature (2012) 481:278–86. doi: 10.1038/nature10759
14. Lee H-M, Kim J-J, Kim HJ, Shong M, Ku BJ, Jo E-K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes (2013) 62:194–204. doi: 10.2337/db12-0420
15. Vandanmagsar B, Youm Y-H, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med (2011) 17:179–88. doi: 10.1038/nm.2279
16. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479:117–21. doi: 10.1038/nature10558
17. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514:187–92. doi: 10.1038/nature13683
18. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526:666–71. doi: 10.1038/nature15541
19. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
20. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science (2013) 341:1250–3. doi: 10.1126/science.1240988
21. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Non-canonical inflammasome activation by intracellular LPS independent of TLR4. Science (2013) 341:1246–9. doi: 10.1126/science.1240248
22. Walev I, Reske K, Palmer M, Valeva A, Bhakdi S. Potassium-inhibited processing of IL-1 beta in human monocytes. EMBO J (1995) 14:1607–14. doi: 10.1002/j.1460-2075.1995.tb07149.x
23. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
24. Hafner-Bratkovič I, Pelegrín P. Ion homeostasis and ion channels in NLRP3 inflammasome activation and regulation. Curr Opin Immunol (2018) 52:8–17. doi: 10.1016/j.coi.2018.03.010
25. Gong T, Yang Y, Jin T, Jiang W, Zhou R. Orchestration of NLRP3 Inflammasome Activation by Ion Fluxes. Trends Immunol (2018) 39:393–406. doi: 10.1016/j.it.2018.01.009
26. Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ (2007) 14:1583–9. doi: 10.1038/sj.cdd.4402195
27. Di A, Xiong S, Ye Z, Malireddi RK, Kometani S, Zhong M, et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity (2018) 49:56–65.e4. doi: 10.1016/j.immuni.2018.04.032
28. Rivers-Auty J, Brough D. Potassium efflux fires the canon: Potassium efflux as a common trigger for canonical and non-canonical NLRP3 pathways. Eur J Immunol (2015) 45:2758–61. doi: 10.1002/eji.201545958
29. Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci USA (2018) 115:E10888–97. doi: 10.1073/pnas.1809548115
30. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science (2018) 362:1064–9. doi: 10.1126/science.aau2818
31. Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol (2020) 20:143–57. doi: 10.1038/s41577-019-0228-2
32. Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J (2019) 38(10):e101638. doi: 10.15252/embj.2019101638
33. Chen KW, Demarco B, Broz P. Pannexin-1 promotes NLRP3 activation during apoptosis but is dispensable for canonical or non-canonical inflammasome activation. Eur J Immunol (2020) 50:170–7. doi: 10.1002/eji.201948254
34. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in Infection and Inflammation. Immunity (2017) 47:15–31. doi: 10.1016/j.immuni.2017.06.020
35. Gordon JL. Extracellular ATP: effects, sources and fate. Biochem J (1986) 233:309–19. doi: 10.1042/bj2330309
36. Gombault A, Baron L, Couillin I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol (2013) 3:414. doi: 10.3389/fimmu.2012.00414
37. Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E. Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATP. Nat Commun (2016) 7:10555. doi: 10.1038/ncomms10555
38. Perregaux D, Gabel CA. Interleukin-1 Beta Maturation and Release in Response to ATP and Nigericin. Evidence That Potassium Depletion Mediated by These Agents Is a Necessary and Common Feature of Their Activity. J Biol Chem (1994) 269.
39. Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity (2015) 43:923–32. doi: 10.1016/j.immuni.2015.10.009
40. Aglietti RA, Dueber EC. Recent Insights into the Molecular Mechanisms Underlying Pyroptosis and Gasdermin Family Functions. Trends Immunol (2017) 38:261–71. doi: 10.1016/j.it.2017.01.003
41. Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, et al. Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity (2018) 49:413–26.e5. doi: 10.1016/j.immuni.2018.07.006
42. Sandilos JK, Bayliss DA. Physiological mechanisms for the modulation of pannexin 1 channel activity. J Physiol (Lond ) (2012) 590:6257–66. doi: 10.1113/jphysiol.2012.240911
43. MacVicar BA, Thompson RJ. Non-junction functions of pannexin-1 channels. Trends Neurosci (2010) 33:93–102. doi: 10.1016/j.tins.2009.11.007
44. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature (2010) 467:863–7. doi: 10.1038/nature09413
45. Sandilos JK, Chiu Y-H, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS, et al. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J Biol Chem (2012) 287:11303–11. doi: 10.1074/jbc.M111.323378
46. Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J (2006) 25:5071–82. doi: 10.1038/sj.emboj.7601378
47. Kanneganti T-D, Lamkanfi M, Kim Y-G, Chen G, Park J-H, Franchi L, et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity (2007) 26:433–43. doi: 10.1016/j.immuni.2007.03.008
48. Pelegrin P, Surprenant A. Pannexin-1 couples to maitotoxin- and nigericin-induced interleukin-1beta release through a dye uptake-independent pathway. J Biol Chem (2007) 282:2386–94. doi: 10.1074/jbc.M610351200
49. Qu Y, Misaghi S, Newton K, Gilmour LL, Louie S, Cupp JE, et al. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J Immunol (2011) 186:6553–61. doi: 10.4049/jimmunol.1100478
50. Enyedi P, Czirják G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev (2010) 90:559–605. doi: 10.1152/physrev.00029.2009
51. Feliciangeli S, Chatelain FC, Bichet D, Lesage F. The family of K2P channels: salient structural and functional properties. J Physiol (Lond ) (2015) 593:2587–603. doi: 10.1113/jphysiol.2014.287268
52. Madry C, Kyrargyri V, Arancibia-Cárcamo IL, Jolivet R, Kohsaka S, Bryan RM, et al. Microglial Ramification, Surveillance, and Interleukin-1β Release Are Regulated by the Two-Pore Domain K+ Channel THIK-1. Neuron (2018) 97:299–312.e6. doi: 10.1016/j.neuron.2017.12.002
53. Patel AJ, Maingret F, Magnone V, Fosset M, Lazdunski M, Honoré E. TWIK-2, an inactivating 2P domain K+ channel. J Biol Chem (2000) 275:28722–30. doi: 10.1074/jbc.M003755200
54. Lloyd EE, Crossland RF, Phillips SC, Marrelli SP, Reddy AK, Taffet GE, et al. Disruption of K(2P)6.1 produces vascular dysfunction and hypertension in mice. Hypertension (2011) 58:672–8. doi: 10.1161/HYPERTENSIONAHA.111.175349
55. Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol (2016) 17:250–8. doi: 10.1038/ni.3333
56. He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature (2016) 530:354–7. doi: 10.1038/nature16959
57. Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun (2017) 8:202. doi: 10.1038/s41467-017-00227-x
Keywords: P2X7 receptor, pannexin-1, Gasdermin D, K2P channels, TWIK protein-related acid-sensitive potassium channel 2, THIK-1, inflammasome
Citation: Xu Z, Chen Z-m, Wu X, Zhang L, Cao Y and Zhou P (2020) Distinct Molecular Mechanisms Underlying Potassium Efflux for NLRP3 Inflammasome Activation. Front. Immunol. 11:609441. doi: 10.3389/fimmu.2020.609441
Received: 23 September 2020; Accepted: 09 November 2020;
Published: 07 December 2020.
Edited by:
Walter Gottlieb Land, Université de Strasbourg, FranceReviewed by:
Marco Di Gioia, Boston Children’s Hospital and Harvard Medical School, United StatesChunfu Zheng, Fujian Medical University, China
Copyright © 2020 Xu, Chen, Wu, Zhang, Cao and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pingzheng Zhou, cHp6aG91QHNtdS5lZHUuY24=
†These authors have contributed equally to this work