 Walter L. Miller
Walter L. Miller- Department of Pediatrics, Center for Reproductive Sciences, and Institute of Human Genetics, University of California, San Francisco, CA, United States
Introduction
Tenascin-X (TNX) is a large extracellular matrix protein discovered because its TNXB gene overlaps the CYP21A2 gene encoding steroid 21-hydroxylase (P450c21), whose mutations cause congenital adrenal hyperplasia (CAH). In the 1980s, several laboratories worked to clone the “CAH gene”. We sought a P450c21 clone in a cDNA library prepared from a CAH adrenal and identified a transcript larger than P450c21 cDNA; sequencing indicated it was encoded by an unknown (”X“) gene overlapping CYP21A2 on the opposite DNA strand. Extensive genomic sequencing revealed the structure of a tenascin: N-terminal EGF-like repeats, multiple fibronectin-III repeats, and a C-terminal fibrinogen-like domain; we named this “Tenascin-X” (TNX). To study TNX function, we postulated a “contiguous gene syndrome”—a single mutation affecting both CYP21A2 and TNXB, causing CAH plus another disorder that might suggest the role of TNX. A patient with CAH and Ehlers-Danlos syndrome (EDS) had partial deletions encompassing both genes. With collaborators, we described patients with recessive TNX-deficient EDS (now termed “classic-like EDS”), which was clinically distinct from dominant EDS caused by collagen mutations. TNX haploinsufficiency causes the mild “hypermobility form” of EDS, often associated with CAH, comprising the unique CAH-X syndrome. The discovery of TNX illustrates scientific serendipity and the value of pursuing unexpected results.
Stumbling Onto “Gene X”—An Unexpected Finding in an Endocrine Study
The discovery of TNX and its associated deficiency disease (a form of Ehlers-Danlos Syndrome) was done in studies of human steroidogenesis and its disorders rather than in studies of connective tissues. In the 1980s, application of then-new molecular biologic techniques revolutionized understanding of steroidogenesis (1). A driving force behind this early work was steroid 21-hydroxylase deficiency (21OHD), a form of congenital adrenal hyperplasia (CAH) causing cortisol deficiency, potentially-lethal aldosterone deficiency, and androgen excess with prenatal virilization of affected females. There are many forms of CAH, but 21OHD, with an incidence of ~1:15,000 (2) was responsible for >90% of cases; 21OHD is now well understood, but requires intensive management (3). Adrenal 21-hydroxylation is catalyzed by P450c21 (CYP21), a microsomal cytochrome P450 enzyme. In 1986, we reported the bovine Cyp21 gene sequence (4) and others (5, 6) reported the human gene. The gene lay in the human leukocyte antigen (HLA) locus (major histocompatibility locus, MHC) on chromosome 6p21.33, the most gene-dense and highly recombinogenic region of the human genome. Duplicated 30-kb units contained the functional CYP21A2 gene and a non-functional CYP21A1P pseudogene duplicated in tandem with the C4A and C4B genes encoding the fourth component of serum complement (7–9) (Figure 1). CYP21A1P is transcribed (10, 11), but is considered a pseudogene because its RNAs do not encode protein. Human CYP21A2 encodes P450c21, in mice the cyp21a1 gene corresponding to CYP21A1P is active (12, 13), in cattle both genes function (4, 14), and some other mammals have single copies of this locus (15); thus the gene duplication post-dates mammalian speciation (16).
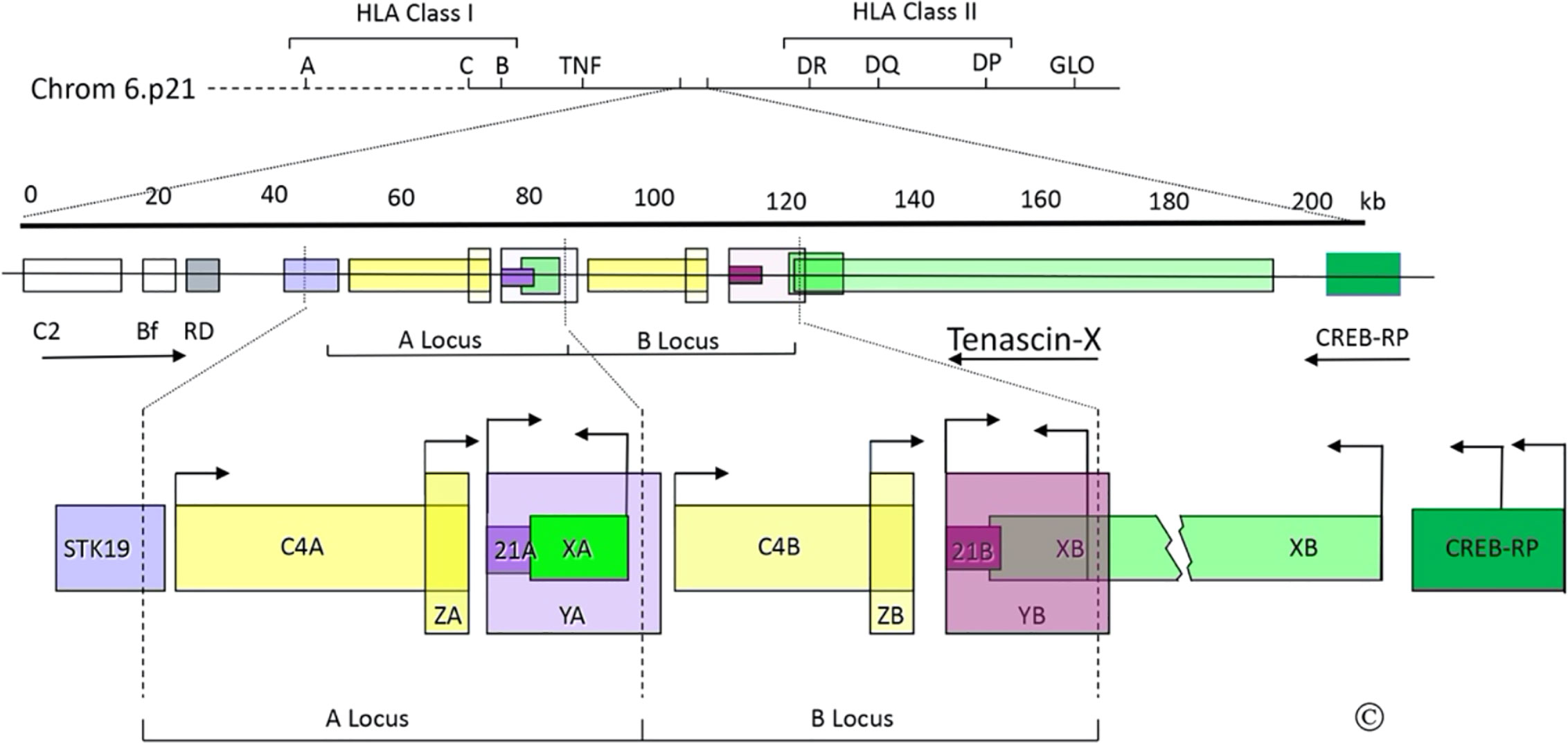
Figure 1 The C4/CYP21/TNX gene locus. Top: Diagram of the short arm of chromosome 6; the telomere is to the left and the centromere is to the right. The MHC Class I and Class II regions are indicated with their principal human leukocyte antigen (HLA) genes; the ~1 megabase region between these is the “Class III region”, which includes the gene for tumor necrosis factor (TNF). Middle: Scale bar in kilobases (kb) and enlarged view of a portion of the Class III region (chrom 6p21.33); the arrows indicate transcriptional orientations. C2, complement factor C2; Bf, properedin factor Bf; RD is now known as NEFLE, negative elongation factor subunit E; CREB-RP, CREB-related protein. Bottom: The duplicated 30 kb C4/CYP21/TNX units and adjacent regions: STK19, serine/threonine kinase 19; C4A and C4B, genes for complement component 4; 21A, CYP21A1P pseudogene; 21B CYP21A2 gene; XA, YA, and YB, adrenal transcripts that lack open reading frames; XB, the TNXB gene; XB-S the short, adrenal-specific form of TNX, arises from the leftward transcription arrow within the XB gene, analogously to XA; ZA and ZB, adrenal-specific transcripts with open reading frames arising from promoters within the C4 genes; the ZA and ZB promoters are enhancer elements of the CYP21A1P and CYP21A2 promoters. Most TNX transcription arises from the untranslated exon at the 5’end of TNXB, but some also arises from two sites within CREB-RP. The vertical dotted lines designate the boundaries of the gene duplication event. © WL Miller.
There was great interest in studying CYP21 genes in patients with 21OHD. We obtained adrenal mRNA from an abortus with 21OHD, prepared a cDNA library, and screened it with radiolabeled double-stranded P450c21 cDNA to obtain the cDNA for the responsible mutant gene (17). Both the known cDNA sequence and RNA blotting showed that the full-length cDNA would be 2.0 kb, but several clones were longer than that, suggesting that the 21OHD might have arisen by an RNA splicing error. Restriction endonuclease mapping of the longest (2.7 kb) clone indicated that it contained only the 3’ end of CYP21. A 2.7 kb clone might have been a cloning artifact, a recombination between CYP21 and something else (as sometimes happened with the reagents then available), but because we had screened the cDNA library with a double-stranded probe, we considered that the 2.7 kb clone could have arisen from a transcript on the opposite strand of DNA from the CYP21 genes. DNA sequencing showed that the 2.7 kb clone had a 3’ poly(A) tail with upstream DNA that matched the predicted opposite-strand sequence of final exon of CYP21. The complete 2.7 kb sequence showed an open reading frame with repeating units whose structural significance was not clear (17). Because the CYP21/C4 locus is duplicated, we knew that this new “gene X” must also be duplicated as “XA” and “XB” genes with the arrangement 5’-C4A-21A-XA-C4B-21B-XB-3’ (Figure 1). Deletions in the gene causing 21OHD did not appear to extend into the XB gene, but deletions of the XA region were found in 14% of human chromosomes (18, 19), suggesting that the 2.7-kb cDNA arose from the XB gene (17). But the nature of the gene encoding the 2.7-kb cDNA was unknown—the operational name “gene X” thus became the source of the name “Tenascin-X”.
Gene X Becomes Tenascin-X
The publication of the 2.7 kb cDNA led us and others to study its gene structure. In 1989, Dr. Russell Doolittle (UC-San Diego), told us that the sequence of our 2.7 kb cDNA resembled chicken tenascin, and published a paper describing a fibrinogen-like sequence in an invertebrate, saying “The sea cucumber protein also corresponds exactly with a segment found as part of the previously unidentified gene product found in human adrenals” (20). Our 2.7 kb cDNA sequence had domains for fibrinogen and fibronectin type III (Fn-III) repeats, thus resembling tenascin (16). Matsumoto et al. confirmed the identification of fibrinogen and Fn-III domains, identified tenascin-like EGF-like domains and provided additional information about exonic organization (21, 22). To determine how this locus was duplicated, we sequenced genomic DNA at the predicted duplication boundaries and through the entire 7 kb between 21A and C4B that had to comprise the XA locus, providing the entire XA gene sequence and the precise boundaries of the human gene duplication (16). These boundaries were substantially different from the corresponding duplication loci in the mouse genome, as expected for independent duplication events that post-dated mammalian speciation. Although XA was abundantly expressed in the adrenal, its gene was truncated at its 5’ end (compared to XB) by the gene duplication (16), suggesting that it is a pseudogene and that XB would be the more important locus.
Manual sequencing of overlapping genomic clones revealed the nearly complete structure of the XB gene: 39 exons spanning 65 kb encoding a protein of >400-kDa (23). Some Fn-III repeats underwent alternative splicing; current data show 44 exons spanning 68 kb encoding 4,244 amino acids totaling 458,220 Da (24). The structure contained the five domains expected of a tenascin. First, the N-terminus comprised a 22AA signal peptide that directs the protein to the secretory pathway, used by extracellular matrix proteins. Second is a hydrophobic domain containing three heptad repeats that encode the tenascin “head piece”, which permits polymerization of tenascin monomers into multi-armed “brachion” structures. The three heptad repeats suggested that TNX should form a “tri-brachion”, similar to the hexabrachion structure of chicken (25) and human (26) tenascin. TNX lacked the additional cysteine residues in this domain of tenascin and restrictin, which permit two tri-brachions to pair into a hexabrachion. The tri-brachion structure of TNX was subsequently confirmed (27); with glycosylation, a TNX tri-brachion is ~1.5 million Da. Third, a single exon encoded a series of 18.5 EGF-like repeats having 55% similarity to the 13.5 EGF-like repeats of human tenascin/cytotactin. Fourth, a series of evolutionarily duplicated exons encoded 32 Fn-III repeats, including the cell-binding domain identified in chicken tenascin (25). Finally, the last five exons encode the carboxy-terminal fibrinogen-like domain and the 3’-untranslated region, including the domains that overlap CYP21A2 (17). The carboxy-terminal fibrinogen-like domain was widely conserved in evolution (20, 28), and the sequence and intron/exon arrangement of the 3’ end of XB were very similar to the β- and γ- chains of fibrinogen (29). Thus, the product of the XB gene was a member of the family of tenascins. We said “We suggest that this category of proteins be termed ‘brachions’ or ‘tenascins’. We favor the latter. Tenascin, the first-described member, which is also widely termed ‘cytotactin’, would be termed tenascin-C or TN-C to designate tenascin-cytotactin; restrictin would be termed tenascin-R or TN-R; and the product of the XB gene described in this paper would be tenascin-X or TN-X. This system would emphasize the relatedness among the monomeric units of these proteins and would, to the extent possible, incorporate terminologies and letterings favored by various groups. It seems unlikely that the number of tenascins will exceed the confines of the alphabet.” (23). Thus, the currently used nomenclature for the tenascins, TNC, TNR, and TNX, was established, soon to be followed by TNW (30, 31).
Additional Studies and Genes in the TNX Gene Locus
The structure of TNX is conserved in mice, with subunits of ~500 kD expressed in a pattern distinct from TNC (32). Expression of TNXB is tissue-specific and developmentally regulated (33, 34). In fetal adrenal, fetal muscle, and skin HT1080 cells, TNXB transcription begins with an untranslated exon ~10 kb upstream from the first coding exon (35). The CREB-RP gene encoding the transcription factor CREB-related protein lies immediately upstream from TNXB (36). TNXB transcripts arise from multiple Sp1/Sp3 sites near to and within CREB-BP (35, 37); thus, both ends of TNXB overlap other genes (Figure 1). Because XA is transcribed despite lacking promoter sequences comparable to those of TNXB, we characterized the 128 bp XA promoter lying between XA and C4B (38). This sequence is identical in TNXB and drives the adrenal-specific expression of a truncated 74 kDa form of TNX, called XB-Short (XB-S), which is identical to the carboxy-terminal 673 amino acids of TNX (38) (Figure 1). Expression of XB-S is induced by hypoxia (39), and XB-S associates with mitotic motor kinesin Eg5 (40), but its precise function remains unclear. Additional transcripts termed YA and YB arise from the CYP21A1P and CYP21A2 promoters, but do not encode protein (10), and transcripts termed ZA and ZB arise from a promoter element within intron 35 of the C4 genes, but it is not clear whether these open reading frames encode protein (41). The ZB promoter is an upstream adrenal enhancer element for CYP21A2 (42). The location of this essential CYP21A2 element within C4B (also seen in the mouse) (43), explains why the C4, CYP21, and TNX genes remain intimately linked in mammalian genomes.
Tenascin-X Deficiency Causes an Autosomal Recessive Form of Ehlers-Danlos Syndrome
Developmental expression of TNXB showed a recurring pattern, appearing first in connective tissue surrounding muscle and then in a subset of intramuscular cells, suggesting roles in muscle morphogenesis (33). To find a biological role for TNX, we hypothesized that an HLA-linked deficiency disease for TNX might exist, but no clinical candidates emerged. Another approach was to postulate existence of a “contiguous gene syndrome” comprising a partial deletion of both the CYP21A2 and TNXB genes, so we sought a patient with 21OHD “and something else”. Serendipitously, Dr. Cynthia Curry (Fresno CA), asked us about a patient with 21OHD and a connective tissue disorder that resembled Ehlers-Danlos Syndrome (EDS). EDS was then known as an autosomal dominant disorder of collagen deposition, with rare recessive forms in collagen-modifying enzymes, (lysyl hydroxylase or pro-collagen N-proteinase) (44), hence TNX was not an obvious candidate. The patient’s skin had ultrastructural findings atypical for known forms of EDS. An antiserum that recognized multiple TNX epitopes detected TNX in cultured dermal fibroblasts from controls and from the obligately-heterozygous parents, but not in the patient’s fibroblasts; similarly, control, but not patient fibroblasts contained TNX mRNA, confirming TNX-deficiency. Because the protein-coding regions of CYP21A2 and TNXB do not overlap, we sought gene deletions rather than point mutations. Genomic PCR and Southern blotting identified a deletion extending from XA through CYP21A2 to the corresponding point in TNXB, demonstrating that TNX deficiency causes EDS (45). In collaboration with Prof. Joost Schalkwijk (U. Nijmegen, Netherlands), we found an immunoassayable TNX fragment in the sera of 146 of 151 patients with EDS; the five patients lacking serum TNX had TNXB mutations, none of which encompassed CYP21A2 (46). Subsequent work has confirmed that TNX deficiency causes a clinically distinct, severe form of EDS (47, 48). Similarly, Tnxb-knockout mice had skin hyperextensibility, reduced skin tensile strength and reduced skin collagen content (49); whereas mouse knockouts of TNC and TNR lacked abnormal phenotypes (50–52). TNX appears to associate with and stabilize newly produced collagen fibrils (27, 53, 54), thus all recessive forms of EDS concern post-translational modification of collagens.
Congenital Adrenal Hyperplasia and Tenascin-X—The CAH-X Syndrome
TNX has functions beyond EDS (55); it promotes epithelial-mysenchymal transitions in development (56), and may be associated with tumor invasion (57–59). TNX-deficiency has been associated with primary myopathy (60, 61), recurrent gastrointestinal perforation (62), and vesicoureteral reflux (63, 64). TNX is expressed in leptomeninges and choroid plexus (34, 65), suggesting neurologic roles: TNXB single nucleotide polymorphisms are associated with schizophrenia (66, 67), and Tnxb-knockout mice have increased anxiety, improved memory, and higher sensorimotor coordination than controls (68).
While TNX-deficient EDS is autosomal recessive, heterozygous TNXB mutations cause TNX haploinsufficiency, with joint hypermobility, recurring joint dislocations and joint pain—the “hypermobility type EDS”. Among 20 obligate heterozygotes for a severely defective TNXB allele, 9 of 14 females but no males had hypermobility EDS (69). Dr. Deborah Merke (NIH, Bethesda MD) found that 7% of patients with 21OHD had symptomatic TNX haploinsufficiency (70, 71), and a recent study reported 14% (72); this association is now regarded as a sub-type of 21OHD termed CAH-X (73). Thus, studies of 21OHD and TNX, like their genes, have been linked from the beginning and continue together.
Author Contributions
WLM assembled the literature, wrote and edited the manuscript, drew the figure, and approved it for publication.
Funding
WLM’s research was supported in the past by grants from the NIH and the March of Dimes.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank James Bristow for his indispensable role in the discovery and functional elucidation of Tenascin-X, first in my laboratory and then independently.
References
1. Miller WL. Molecular biology of steroid hormone synthesis. Endocr Rev (1988) 9:295–318. doi: 10.1210/edrv-9-3-295
2. Miller WL, Levine LS. Molecular and clinical advances in congenital adrenal hyperplasia. J Pediatr (1987) 111:1–17. doi: 10.1016/S0022-3476(87)80334-7
3. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An Endocrine Society clinical practice guideline. J Clin Endocrinol Metab (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
4. Chung B, Matteson KJ, Miller WL. Structure of the bovine gene for P450c21 (steroid 21-hydroxylase) defines a novel cytochrome P450 gene family. Proc Natl Acad Sci USA (1986) 83:4243–7. doi: 10.1073/pnas.83.12.4243
5. Higashi Y, Yoshioka H, Yamane M, Gotoh O, Fujii-Kuriyama Y. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: A pseudogene and genuine gene. Proc Natl Acad Sci USA (1986) 83:2841–5. doi: 10.1073/pnas.83.9.2841
6. White PC, New MI, Dupont B. Structure of the human steroid 21-hydroxylase genes. Proc Natl Acad Sci USA (1986) 83:5111–5. doi: 10.1073/pnas.83.14.5111
7. Amor M, Tosi M, Duponchel C, Steinmetz M, Meo T. Liver cDNA probes disclose two cytochrome P450 genes duplicated in tandem with the complement C4 loci of the mouse H-2S region. Proc Natl Acad Sci USA (1985) 82:4453–7. doi: 10.1073/pnas.82.13.4453
8. Carroll MC, Campbell RD, Porter RR. Mapping of steroid 21-hydroxylase genes to complement component C4 genes in HLA, the major histocompatibility locus in man. Proc Natl Acad Sci USA (1985) 82:521–5. doi: 10.1073/pnas.82.2.521
9. White PC, Grossberger D, Onufer BJ, Chaplin DD, New MI, Dupont B, et al. Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proc Natl Acad Sci USA (1985) 82:1089–93. doi: 10.1073/pnas.82.4.1089
10. Bristow J, Gitelman SE, Tee MK, Staels B, Miller WL. Abundant adrenal-specific transcription of the human P450c21A “pseudogene”. J Biol Chem (1993) 268:12919–24.
11. Chang SF, Chung BC. Difference in transcriptional activity of two homologous CYP21A genes. Mol Endocrinol (1995) 9:1330–6. doi: 10.1210/me.9.10.1330
12. Parker KL, Chaplin DD, Wong M, Seidman JG, Smith JA, Schimmer BP. Expression of murine 21-hydroxylase in mouse adrenal glands and in transfected Y1 adrenocortical tumor cells. Proc Natl Acad Sci USA (1985) 82:7860–4. doi: 10.1073/pnas.82.23.7860
13. Chaplin DD, Galbraith LJ, Seidman JG, White PC, Parker KL. Nucleotide sequence analysis of murine 21-hydroxylase genes: mutations affecting gene expression. Proc Natl Acad Sci USA (1986) 83:9601–5. doi: 10.1073/pnas.83.24.9601
14. John ME, Okamura T, Dee A, Adler B, John MC, White PC, et al. Bovine steroid 21-hydroxylase: Regulation of biosynthesis. Biochemistry (1986) 25:2846–53. doi: 10.1021/bi00358a016
15. Geffrotin C, Chardon P, DeAndres-Cara DR, Feil R, Renard C, Vaiman M. The swine steroid 21-hydroxylase gene (CYP21): Cloning and mapping within the swine leukocyte antigen locus. Anim Genet (1990) 21:1–13. doi: 10.1111/j.1365-2052.1990.tb03202.x
16. Gitelman SE, Bristow J, Miller WL. Mechanism and consequences of the duplication of the human C4/P450c21/gene X locus. Mol Cell Biol (1992) 12:2124–34. doi: 10.1128/MCB.12.5.2124. Correction: Mol Cell Biol (1992) 12:3313-3314.
17. Morel Y, Bristow J, Gitelman SE, Miller WL. Transcript encoded on the opposite strand of the human steroid 2l-hydroxylase/complement component C4 gene locus. Proc Natl Acad Sci USA (1989) 86:6582–6. doi: 10.1073/pnas.86.17.6582
18. Miller WL, Morel Y. The molecular genetics of 21-hydroxylase deficiency. Annu Rev Genet (1989) 23:371–93. doi: 10.1146/annurev.genet.23.1.371
19. Morel Y, Miller WL. Clinical and molecular genetics of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Adv Hum Genet (1991) 20:1–68. doi: 10.1007/978-1-4684-5958-6_1
20. Xu X, Doolittle RF. Presence of a vertebrate fibrinogen-like sequence in an echinoderm. Proc Natl Acad Sci USA (1990) 87:2097–101. doi: 10.1073/pnas.87.6.2097
21. Matsumoto K, Arai M, Ishihara N, Ando A, Inoko H, Ikemura T. Cluster of fibronectin type III repeats found in the human major histocompatibility complex class III region shows the highest homology with the repeats in an extracellular matrix protein, tenascin. Genomics (1992) 12:485–91. doi: 10.1016/0888-7543(92)90438-X
22. Matsumoto K, Ishihara N, Ando A, Inoko H, Ikemura T. Extracellular matrix protein tenascin-like gene found in human MHC class III region. Immunogenetics (1992) 36:400–3. doi: 10.1007/BF00218048
23. Bristow J, Tee MK, Gitelman SE, Mellon SH, Miller WL. Tenascin-X: A novel extracellular matrix protein encoded by the human XB gene overlapping P450c21B. J Cell Biol (1993) 122:265–78. doi: 10.1083/jcb.122.1.265
25. Spring J, Beck K, Chiquet-Ehrismann R. Two contrary functions of tenascin: dissection of the active sites by recombinant tenascin fragments. Cell (1989) 59:325–34. doi: 10.1016/0092-8674(89)90294-8
26. Gulcher JR, Nies DE, Alexakos MJ, Ravikant NA, Sturgill ME, Marten LS, et al. Structure of the human hexabrechion (tenascin) gene. Proc Natl Acad Sci USA (1991) 88:9438–42. doi: 10.1073/pnas.86.5.1588
27. Lethias C, Carisey A, Comte J, Cluzel C, Exposito J-Y. A model of tenascin-X integration within the collagenous network. FEBS Lett (2006) 80:6281–5. doi: 10.1016/j.febslet.2006.10.037
28. Baker NE, Mlodzik M, Rubin GM. Spacing differentiation in the developing Drosophila eye: a fibrinogen-related lateral inhibitor encoded by scabrous. Science (1990) 250:1370–7. doi: 10.1126/science.2175046
29. Pan Y, Doolittle RF. cDNA sequence of a second fibrinogen α chain in lamprey: an archetypal version aliguable with full-length β- and γ- chains. Proc Natl Acad Sci USA (1992) 89:2066–70. doi: 10.1073/pnas.89.6.2066
30. Weber P, Montag D, Schachner M, Bernhardt RR, Zebrafish tenascin- W. a new member of the tenascin family. J Neurobiol (1998) 35:1–16. doi: 10.1002/(SICI)1097-4695(199804)35:1<1::AID-NEU1>3.0.CO;2-9
31. Scherberich A, Tucker RP, Samandari E, Brown-Luedi M, Martin D, Chiquet-Ehrismann R. Murine tenascin-W: a novel mammalian tenascin expressed in kidney and at sites of bone and smooth muscle development. J Cell Sci (2004) 17:571–81. doi: 10.1242/jcs.00867
32. Matsumoto K, Saga Y, Ikemura T, Sakakura T, Chiquet-Ehrismann R. The distribution of tenascin-X is distinct and often reciprocal to that of tenascin-C. J Cell Biol (1994) 125:483–93. doi: 10.1083/jcb.125.2.483
33. Burch GH, Bedolli MA, McDonough S, Rosenthal SM, Bristow J. Embryonic expression of tenascin-X suggests a role in limb, muscle, and heart development. Dev Dynamics (1995) 203:491–504. doi: 10.1038/ng0997-104
34. Geffrotin C, Garrido JJ, Tremet L, Vaiman M. Distinct tissue distribution in pigs of Tenascin-X and Tenascin-C transcripts. Eur J Biochem (1995) 231:83–92. doi: 10.1111/j.1432-1033.1995.0083f.x
35. Speek M, Barry F, Miller WL. Alternate promoters and alternate splicing of human Tenascin-X, a gene with 5’ and 3’ ends buried in other genes. Hum Mol Genet (1996) 5:1749–58. doi: 10.1093/hmg/5.11.1749
36. Min J, Shukla H, Kozono H, Bronson SK, Weissman SM, Chaplin DD. A novel Creb family gene telomeric of HLA-DRA in the HLAcomplex. Genomics (1995) 30:149–56. doi: 10.1006/geno.1995.9891
37. Wijesuriya SD, Bristow J, Miller WL. Localization and analysis of the principal promoter for human Tenascin-X. Genomics (2002) 80:443–52. doi: 10.1006/geno.2002.6852
38. Tee MK, Thomson AA, Bristow J, Miller WL. Sequences promoting the transcription of the human XA gene overlapping P450c21A correctly predict the presence of a novel, adrenal-specific, truncated form of Tenascin-X. Genomics (1995) 28:171–8. doi: 10.1006/geno.1995.1128
39. Kato A, Endo T, Abiko S, Ariga H, Matsumoto K. Induction of truncated form of Tenascin-X (XB-S) through dissociation of HDAC1 from SP-1/HDAC1 complex in response to hypoxic conditions. Exp Cell Res (2008) 314:2661–73. doi: 10.1016/j.yexcr.2008.05.019
40. Endo T, Ariga H, Matsumoto K. Truncated form of Tenascin-X, XB-S, interacts with mitotic motor kinesin Eg5. Mol Cell Biochem (2009) 320:53–66. doi: 10.1007/s11010-008-9898-y
41. Tee MK, Babalola GO, Aza-Blanc P, Speek M, Gitelman SE, Miller WL. A promoter within intron 35 of the human C4A gene initiates adrenal-specific transcription of a 1kb RNA: location of a cryptic CYP21 promoter element? Hum Mol Genet (1995) 4:2109–16. doi: 10.1093/hmg/4.11.2109
42. Wijesuriya SD, Zhang G, Dardis A, Miller WL. Transcriptional regulatory elements of the human gene for cytochrome P450c21 (steroid 21-hydroxylase) lie within intron 35 of the linked C4B gene. J Biol Chem (1999) 274:38097–106. doi: 10.1074/jbc.274.53.38097
43. Milstone DS, Shaw SK, Parker KL, Szyf M, Seidman JG. An element regulating adrenal-specific steroid 21-hydroxylase expression is located within the slp gene. J Biol Chem (1992) 267:21924–7.
44. Byers PH. Ehlers-Danlos syndrome: recent advances and current understanding of the clinical and genetic heterogeneity. J Invest Dermatol (1994) 103:475–525. doi: 10.1038/jid.1994.9
45. Burch GH, Gong Y, Liu W, Dettman RW, Curry CJ, Smith L, et al. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat Genet (1997) 17:104–8. doi: 10.1038/ng0997-104
46. Schalkwijk J, Zweers MC, Steijlen PM, Dean WB, Taylor G, von Vlijmen IM, et al. A recessive form of Ehlers-Danlos syndrome caused by Tenascin-X deficiency. New Engl J Med (2001) 345:1167–75. doi: 10.1056/NEJMoa002939
47. Lindor NM, Bristow J. Tenascin-X deficiency in autosomal recessive Ehlers-Danlos syndrome. Am J Med Genet (2005) 135A:75–80. doi: 10.1002/ajmg.a.30671
48. Demirdas S, Dulfer E, Robert L, Kempers M, van Beek D, Micha D, et al. Recognizing the tenascin-X deficient type of Ehlers–Danlos syndrome: a cross-sectional study in 17 patients. Clin Genet (2017) 91:411–25. doi: 10.1111/cge.12853
49. Mao JR, Taylor G, Dean WB, Wagner DR, Afzal V, Lotz JC, et al. Tenascin-X deficiency mimics Ehlers–Danlos syndrome in mice through alteration of collagen deposition. Nat Genet (2002) 30:421–5. doi: 10.1038/ng850
50. Saga Y, Yagi T, Ikawa Y, Sakakura T, Aizawa S. Mice develop normally without tenascin. Genes Dev (1992) 6:1821–31. doi: 10.1101/gad.6.10.1821
51. Forsberg E, Hirsch E, Frohlich L, Meyer M, Ekblom P, Aszodi A, et al. Skin wounds and severed nerves heal normally in mice lacking tenascin-C. Proc Natl Acad Sci USA (1996) 93:6594–9. doi: 10.1073/pnas.93.13.6594
52. Weber P, Bartsch U, Rasband MN, Czaniera R, Lang Y, Bluethmann H, et al. Mice deficient for tenascin-R display alterations of the extracellular matrix and decreased axonal conduction velocities in the CNS. J Neurosci (1999) 19:4245–62. doi: 10.1523/jneurosci.19-11-04245.1999
53. Elefteriou F, Exposito JY, Garrone R, Lethias C. Characterization of the bovine Tenascin-X. J Biol Chem (1997) 272:22866–74. doi: 10.1074/jbc.272.36.22866
54. Mao JR, Bristow J. The Ehlers-Danlos syndrome: on beyond collagens. J Clin Invest (2001) 107:1063–9. doi: 10.1172/JCI12881
55. Valcourt U, Alcaraz LB, Esposito JY, Lethias C, Bartholin L. Tenascin-X: beyond the architectural function. Cell Adhesion Migration (2015) 9:154–65. doi: 10.4161/19336918.2014.994893
56. Alcaraz LB, Exposito JY, Chuvin N, Pommier RM, Cluzel C, Martel S, et al. Tenascin-X promotes epithelial-to-mesenchymal transition by activating latent TGF-β. J Cell Biol (2014) 205:409–28. doi: 10.1083/jcb.201308031
57. Geffrotin C, Horak V, Crechet F, Tricaud Y, Lethias C, Vincent-Naulleau S, et al. Opposite regulation of tenascin-C and tenascin-X in MeLiM swine heritable cutaneous malignant melanoma. Biochim Biophys Acta (2000) 1524:196–202. doi: 10.1016/S0304-4165(00)00158-6
58. Matsumoto K, Takayama N, Ohnishi J, Ohnishi E, Shirayoshi Y, Nakatsuji N, et al. Tumour invasion and metastasis are promoted in mice deficient in tenascin-X. Genes Cells (2001) 6:1101–11. doi: 10.1046/j.1365-2443.2001.00482.x
59. Levy P, Ripoche H, Laurendeau I, Lazar V, Ortonne N, Parfait B, et al. Microarray-based identification of Tenascin C and Tenascin XB, genes possibly involved in tumorigenesis associated with neurofibromatosis type 1. Clin Cancer Res (2007) 13:398–407. doi: 10.1158/1078-0432.CCR-06-0182
60. Voermans NC, van Alfen N, Pillen S, Lammens M, Schalkwijk J, Zwarts MJ, et al. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol (2009) 65:687–97. doi: 10.1002/ana.21643
61. Penisson-Besnier I, Allamand V, Beurrier P, Martin L, Schalkwijk J, van Vlijmen-Willems I, et al. Compound heterozygous mutations of the TNXB gene cause primary myopathy. Neuromuscular Disord (2013) 23:664–9. doi: 10.1016/j.nmd.2013.04.009
62. Sakiyama T, Kubo A, Sasaki T, Yamada T, Yabe N, Matsumoto K, et al. Recurrent gastrointestinal perforation in a patient with Ehlers–Danlos syndrome due to tenascin-X deficiency. J Dermatol (2015) 42:511–4. doi: 10.1111/1346-8138.12829
63. Gbadegesin RA, Brophy PD, Adeyemo A, Hall G, Gupta IR, Hains D, et al. TNXB mutations can cause vesicoureteral reflux. J Am Soc Nephrol (2013) 24:1313–22. doi: 10.1681/ASN.2012121148
64. Tokhmafshan F, Brophy PD, Gbadegesin RA, Gupta IR. Vesicoureteral reflux and the extracellular matrix connection. Pediatr Nephrol (2017) 32:565–76. doi: 10.1007/s00467-016-3386-5
65. Imura K, Sato I. Identification of the novel localization of tenascin X in the monkey choroid plexus and comparison with the mouse. Euro J Histochem (2009) 53:225–31. doi: 10.4081/ejh.2009.225
66. Wei J, Hemmings GP. TNXB locus may be a candidate gene predisposing to schizophrenia. Am J Med Genet B Neuropsychiatr Genet (2004) 125:43–9. doi: 10.1002/ajmg.b.20093
67. Tochigi M, Zhang X, Ohashi J, Hibino H, Otowa T, Rogers M, et al. Association study between the TNXB locus and schizophrenia in a Japanese population. Am J Med Genet B Neuropsychiatr Genet (2007) 144B:305–9. doi: 10.1002/ajmg.b.30441
68. Kawakami K, Matsumoto K. Behavioral alterations in mice lacking the gene for Tenascin-X. Biol Pharm Bull (2011) 34:590–3. doi: 10.1248/bpb.34.590
69. Zweers MC, Bristow J, Steijlen PM, Dean WB, Hamel BC, Otero M, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos Syndrome. Am J Hum Genet (2003) 73:214–7. doi: 10.1086/376564
70. Merke DP, Chen W, Morissette R, Xu Z, Van Ryzin C, Sachdev V, et al. Tenascin-X haploinsufficiency associated with Ehlers-Danlos syndrome in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab (2013) 98:E379–87. doi: 10.1210/jc.2012-3148
71. Morissette R, Chen W, Perritt AF, Dreiling JL, Arai AE, Sachdev V, et al. Broadening the spectrum of Ehlers Danlos syndrome in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab (2015) 100:E1143–52. doi: 10.1210/jc.2015-2232
72. Gao Y, Lu L, Yu B, Mao J, Wang X, Ni M, et al. The prevalence of chimeric TNXA/TNXB gene and clinical symptoms of Ehlers-Danlos syndrome with 21-hydroxylase deficiency. J Clin Endocrinol Metab (2020) 105:2288–99. doi: 10.1210/clinem/dgaa199
Keywords: adrenal, 21-hydroxylase, human leukocyte antigen locus, extracellular matrix, Ehlers-Danlos syndrome, major histocompatibility locus, CYP21, cytochrome P450
Citation: Miller WL (2021) Tenascin-X—Discovery and Early Research. Front. Immunol. 11:612497. doi: 10.3389/fimmu.2020.612497
Received: 30 September 2020; Accepted: 04 December 2020;
Published: 11 January 2021.
Edited by:
Sermin Genc, Dokuz Eylul University, TurkeyReviewed by:
Deborah Merke, National Institutes of Health Clinical Center (NIH), United StatesDelfien Syx, Ghent University, Belgium
Copyright © 2021 Miller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Walter L. Miller, d2xtbGFiQHVjc2YuZWR1