 Caroline Dudreuilh1,2,3*
Caroline Dudreuilh1,2,3* Sumoyee Basu1,2,3
Sumoyee Basu1,2,3 Cristiano Scottà2,3,4
Cristiano Scottà2,3,4 Anthony Dorling1,2,3†
Anthony Dorling1,2,3† Giovanna Lombardi2,3,4†
Giovanna Lombardi2,3,4†- 1Department of Inflammation Biology, King’s College London (KCL), Guy’s Hospital, London, United Kingdom
- 2Centre for Nephrology, Urology and Transplantation, School of Immunology and Microbial Sciences, King’s College London, London, United Kingdom
- 3NIHR Biomedical Research Centre-Transplant Theme, Guy’s Hospital, London, United Kingdom
- 4Peter Gorer Department of Immunobiology, School of Immunology and Microbial Science, King’s College London (KCL), Guy’s Hospital, London, United Kingdom
Regulatory T cells (Tregs) constitute a small proportion of circulating CD4+ T cells that function to maintain homeostasis and prevent autoimmunity. In light of their powerful immunosuppressive and tolerance-promoting properties, Tregs have become an interesting potential candidate for therapeutic use in conditions such as solid organ transplant or to treat autoimmune and inflammatory conditions. Clinical studies have demonstrated the safety of polyclonally expanded Tregs in graft-versus-host disease, type 1 diabetes, and more recently in renal and liver transplantation. However, Tregs are heterogenous. Recent insights indicate that only a small proportion of Tregs, called T follicular regulatory cells (Tfr) regulate interactions between B cells and T follicular helper (Tfh) cells within the germinal center. Tfr have been mainly described in mouse models due to the challenges of sampling secondary lymphoid organs in humans. However, emerging human studies, characterize Tfr as being CD4+CD25+FOXP3+CXCR5+ cells with different levels of PD-1 and ICOS expression depending on their localization, in the blood or the germinal center. The exact role they play in transplantation remains to be elucidated. However, given the potential ability of these cells to modulate antibody responses to allo-antigens, there is great interest in exploring translational applications in situations where B cell responses need to be regulated. Here, we review the current knowledge of Tfr and the role they play focusing on human diseases and transplantation. We also discuss the potential future applications of Tfr therapy in transplantation and examine the evidence for a role of Tfr in antibody production, acute and chronic rejection and tertiary lymphoid organs. Furthermore, the potential impact of immunosuppression on Tfr will be explored. Based on preclinical research, we will analyse the rationale of Tfr therapy in solid organ transplantation and summarize the different challenges to be overcome before Tfr therapy can be implemented into clinical practice.
Introduction
Over the last three decades, despite an improvement in short-term outcomes after solid organ transplantation, long-term outcomes have not drastically improved (1). There has been a massive leap in understanding the mechanisms that cause chronic allograft dysfunction (CAD) leading to graft loss, particularly in kidney transplantation. Immune mediated injury is the predominant cause of CAD, many cases of which are associated with the presence of donor specific antibodies (DSA), directed predominantly against donor human leukocyte antigens (HLA). These DSA are produced by B cells in response to alloantigen stimulus through a process that is T-cell regulated (2). The majority of traditional therapeutic strategies attempted have focussed on either targeting B cells or whole T cell population or on removing the DSA themselves, without more precise targeting. Targeting the immune allo-responses to regulate DSA responses might be a way to improve patient outcomes.
CD4+ T cells play an important role in both activating other cells (including B cells) and in regulating the immune response (regulatory T cells-Tregs) (3). The old concept of T cell ability to help B cells has been further clarified recently with the discovery of a new small subpopulation of CD4+ T cells, called T follicular helper cells (Tfh) (4–6). Tfh are responsible for the interactions with B cells in the germinal centers (GC) within the secondary lymphoid organs (SLO) (spleen or lymph nodes) (7). They support B cells in the process of antibody production (4). In addition, Tfh help in promoting the differentiation of B cells into memory B cells and long-lived plasma cells (8). Human and murine Tfh display similarities in phenotype (expressing CD4, CXCR5, PD1, and Bcl6; lacking expression of CCR7 and IL-7Rα and secreting IL-21) (9–11) and function, responsible of interaction and activation of GC B cells leading to antibody production.
Tfh display a Treg counterpart population called T follicular regulatory cells (Tfr), which have the role of regulating specific interactions between B cells and Tfh (12–14). Tfr have been extensively studied in mouse models, with fewer studies in humans, due to the challenges of sampling human SLO. In human, they have been characterized as being Tfh-like [expressing CD4, the C-X-C chemokine receptor type 5 (CXCR5) and Bcl-6 in association with CD25 and FOXP3] and express varying levels of PD-1 and ICOS depending on their localization. They can be found in SLO like tonsil, lymph nodes, spleen, and ectopic lymphoid structures and even in blood (bTfr) (15). The bTfr remain to be further characterized in humans and their function remains poorly understood. In general, while the role of Tfr in modulating autoimmune responses seems to be crucial, the exact role of these cells in the transplantation settings remains to be elucidated. Here, we review the current evidence around the origin of Tfr, the different subtypes and associated functions. We will discuss the role of Tfr in human diseases with a focus on transplantation and explore the potential of cell therapy using Tfr.
Definition and Origin of Tfr
Definition of Tfr and Tfh With Focus on the Differences Between Mouse and Human Tf Cells
The discovery that only a small fraction of CD4+ T cells was involved in antibody production (Tfh) and modulation (Tfr) within the GC is recent. Interestingly, although the role of Tregs in regulation of the GC response was first described more than fifteen years ago in human (16, 17), the formal discovery of the Tfr subtype occurred later in murine models (12–14). Tfr are proposed to form when FOXP3+ precursors acquire a Tfh-like phenotype that includes expression of Bcl-6, CXCR5, PD-1, and ICOS. Although Tfr share some Tfh features, they do not express/produce the same cytokines which characterize Tfh, such as IL-21, IL-4. In addition to FOXP3, Tfr cells express the typical markers expressed by Tregs such as GITR, Blimp-1, and CTLA-4 (12–14).
Precise definition of Tfr as a sole entity is complicated by the dynamic expression of some chemokine receptors, particularly as the cells navigate between compartments within the SLO, and within the body (see section on Maturation of Tfr), as they can be found in blood and SLO like tonsil, lymph nodes, spleen and ectopic lymphoid structures. However, distinguishing between Tfh and/or Tfr with only one specific marker is almost impossible. Most of the work on these two subtypes of T cells has been done in mouse models with this work inevitably influencing study of human counterparts. CXCR5 is probably the most accepted and used marker for Tfh and Tfr (12–14). CXCR5 is a G protein receptor for the chemokine CXCL13. It enables T cells to migrate to the B cell zones of the lymph nodes. This is supported by the evidence that CXCR5 knock out mice present a complex pattern of lymph node developmental defects and a completely disorganized splenic microarchitecture, lacking segregated T- and B-cell areas (18). However, it has been demonstrated recently that some Tfr can access the GC independently of CXCR5 (19), and that after interaction with B cells they proliferate less than their CXCR5+ counterparts. Interestingly CXCR5 expression seems to be regulated by nuclear factor of activated T cells 2 (NFAT2) in Tfr (20) but by achaete-scute homologue-2 (ASCL2) in Tfh (21). The involvement of NFAT2 regulation in Tfr was confirmed in Nfat2fl/fl x Cd4cre mice by the demonstration that NFAT2 knock out mice displayed reduced numbers of Tfr (20).
Until recently, studies describing CD4+CXCR5+ cells did not differentiate between Tfh and Tfr (4, 7, 11). It is not clear what proportion of the CD4+ CXCR5+ population are Tfr cells. A recent report suggested that CD4+CXCR5+FOXP3+ cells accounted for only 12.8% of circulating CD4+ CXCR5+cells (22), implying that the majority of circulating CD4+CXCR5+cells were Tfh, although without confirmatory evidence of a more detailed phenotypic characterization (15).
Current evidence suggest that Tfr derive from Tregs, at least in mouse (see below), and represent 18.57±6.55% of the total CD25+FOXP3+ T cells (23). Therefore, FOXP3 is expressed in Tfr at different levels through their differentiation process (see Figure 1). Another way to identify Tfr would be the use of the combination of markers CD4+CD25high CXCR5+CD127low, as circulating Tfr are CD25highCD127low as they originate from Tregs, while blood Tfh express heterogenous amount of CD25 (negative to low) and a low level of CD127 (24).
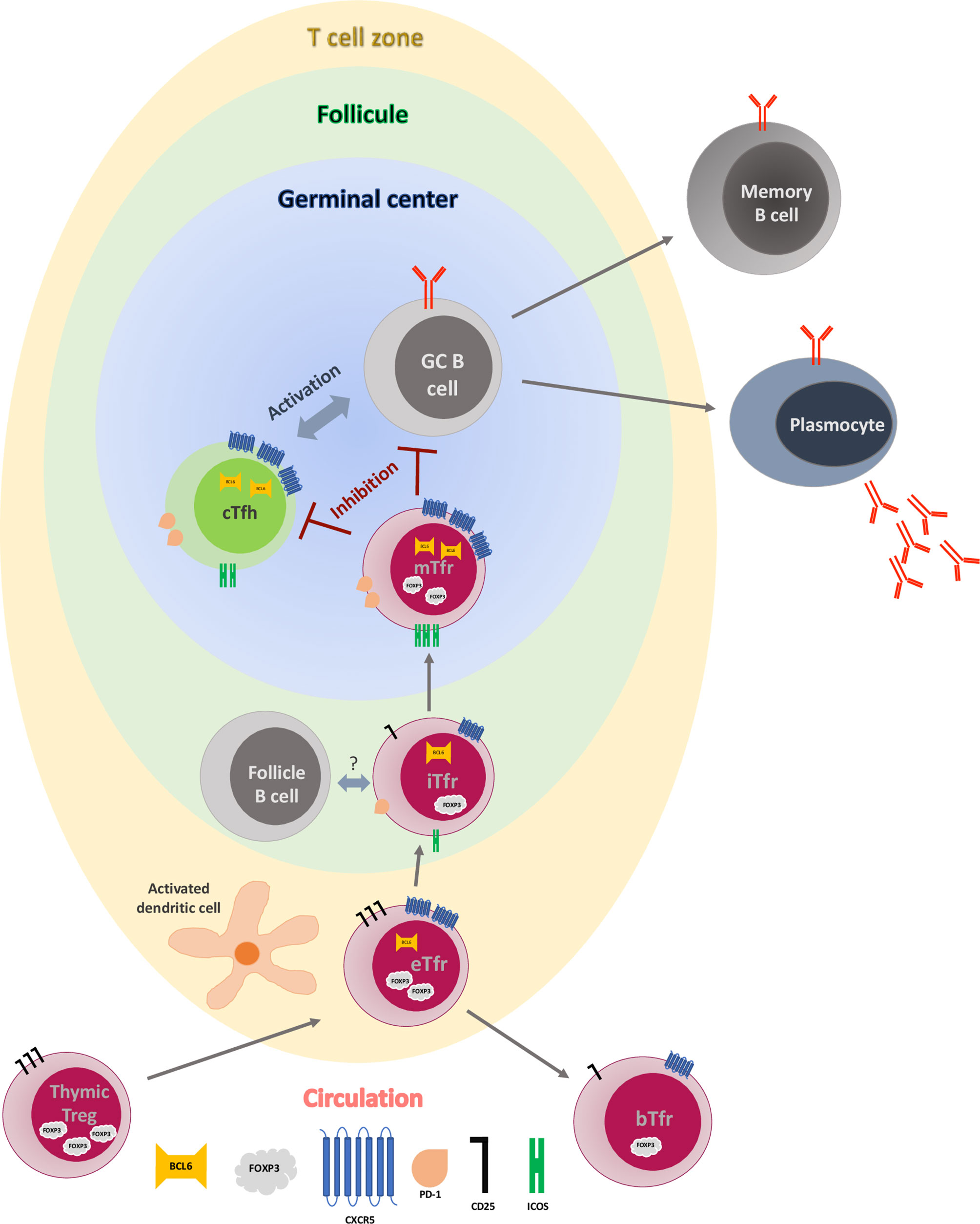
Figure 1 Early germinal center reaction and T follicular regulatory cell maturation process. Early Tfr (eTfr) derive from natural regulatory T cells (Tregs) after expression of CXCR5 and Bcl-6 and down regulation of FOXP3. After interaction with activated dendritic cells (DC) in the T cell zone, some eTfr lose expression of Bcl-6 and enter the circulation (blood Tfr-bTfr), while some migrate to the follicle (intermediate Tfr-iTfr), where they interact with the follicular B cells and start expressing PD-1 and ICOS. Eventually, they move to the germinal center (GC), becoming mature Tfr (mTfr), where they inhibit both the central T follicular helper cell (cTfh) and the GC B cells, leading to regulation of antibody production and B cell differentiation.
The phenotype and function of Tfr and Tfh both depend on the expression of Bcl-6 and STAT3 (5, 12, 14, 25, 26). However, only Tfr express Bcl-6 alongside the Bcl-6 antagonist Blimp-1, although this has only been demonstrated in mouse and not yet in human Tfr (23). While Bcl-6 is important for the Tfh-like properties of Tfr, Blimp-1 is associated with the Treg-like phenotype and function of Tfr (12, 27–29). Blimp1 is a transcriptional repressor protein that suppresses Bcl6 expression. Tfr numbers are regulated through a balance between Bcl-6 and Blimp-1 (12). IL-2 is a key factor regulating Tfr differentiation, promoting Blimp1 expression while repressing Bcl6 in Tregs to preclude Tfr cell development (30).
In humans, they have been characterized as being CD4+CD25+FOXP3+CXCR5+ cells with different levels of PD-1 and ICOS expression depending on their localization (31, 32) (and on the transcription factor Bcl-6 for differentiation and localization into the B cell follicle (4–6, 14, 33). ICOS expression in human does not discriminate Tfr from other Tregs (34).
As none of the individual markers described above seems to be specific for Tfh/Tfr and blood, a combination of surface markers is necessary to be able to characterize these two subsets properly, from their origin to the fully matured T cells (see Table 1 and Figure 1). The function of Tfr in a normal immunological response is described below.
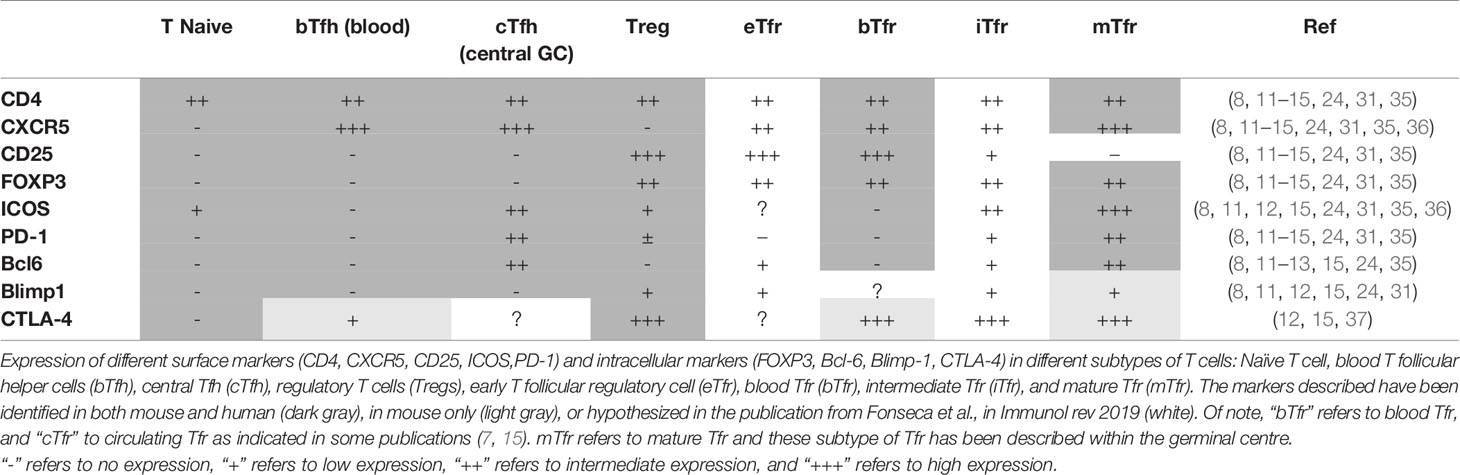
Table 1 Different expression of markers in different types of Tfr and Tfh compared to Tregs and T naive.
Germinal Center Reaction
GC are defined structures that develop within the SLO during ongoing immune responses; they have been extensively studied and described in mouse models. Through a process called GC reaction, a naïve mature B cell first undergoes clonal expansion and somatic hypermutation within the dark zone of the GC before moving to the light zone (38). There, B cells demonstrating a relevant affinity toward the antigen of interest form cognate interactions with Tfh cells primed by the same antigen. These Tfh help promote B cell responses by providing cytokines (such as IL-21, IL-4) and co-stimulation (through the inducible costimulatory molecules ICOS and CD40L) (7, 35, 39–41). Continued cognate interaction between Tfh and B cells drives immunoglobulin class switching, somatic hypermutation, and B cell differentiation (42) leading to the production of long-lived plasma cells and memory B cells.
Mechanisms of Regulation by Tfr and Antibody Production
In adoptive transfer experiments of Tfr and Tfh in mice lacking these population of cells, the group of Sage et al. have demonstrated that Tfr have a direct impact on B cell effector function by decreasing antibody secretion, and inhibiting somatic hypermutation and class-switch recombination (23, 31, 36). However, Tfr can also act indirectly to inhibit antibody responses by specifically suppressing production of IL-4 and IL-21 by Tfh, leaving other functions intact (36).
Different approaches have been used to address the specific role of Tfr in regulation of GC responses. The first series of reports used Bcl-6 as a surrogate marker of Tfr in genetic and/or bone marrow chimera models. In these models, Bcl-6 was deleted in FOXP3+ cells. The results were contradictory, with some studies indicating that Bcl-6 was essential for Tfr function (12, 43). Fu et al., for example, using Bcl-6fl/flFOXP3Cre (KO) mice, which have reduced numbers of CXCR5+PD1+CD4+FOXP3+ Tfr cells, demonstrated enhanced protection against influenza virus associated with an increase in humoral autoimmunity (43). Others, however have demonstrated that the lack of Bcl-6+FOXP3+ cells did not impact on the development of the Tfh-cell population and numbers of GC B cells, but did alter the levels and avidity of the antigen-specific IgG response (44).
As described earlier, Bcl-6 is not an absolute marker of Tfr and could be expressed by other Tregs. Therefore, Clement et al. (45), designed an inducible Tfr cell-deletion model aiming to study the role of Tfr in an intact host. They generated a strain of mice called TFR-DTR (for Diphtheria Toxin receptor) where DTR expression is under the control of a recombinant Cxcr5 gene in cells expressing FOXP3. Thus, only Tfr cells expressing both FOXP3 and CXCR5 expressed DTR on their surface so were susceptible to deletion by DT. After immunization with (4-hydroxy-3-niotrophneyl)acetyl-ovalbumin (NP-OVA) Tfr in these mice regulated only early GC responses to antigen-specific antibody and B cell memory. Lack of Tfr was associated with a surge of self-reactive IgG and IgE, demonstrating a key role of Tfr in preventing these potentially deleterious responses post-vaccination. Both blood Tfh (bTfh) (46) in HIV+ patients, and bTfr seem to have a memory function and able to be redirected toward antigen re-exposition in other diseases (8, 36).
Although the mechanisms of regulation by Tfr in vivo are still under investigation and what is known has been reviewed recently (23, 47, 48), a summary of some of the most important functions of Tfr is set out below. CTLA-4 expression by Tfr has been shown to be key for their function, as conditional deletion of CTLA-4 on Tfr inhibits their function, GC B cells are not inhibited and this leads to increased amounts of antibody produced (47). Furthermore, Tfr cells inhibit antigen-specific IgG levels when adoptively transferred into CTLA-4 inducible knockout (KO) mice (47) or mice immunised with NP-OVA. MOG-CFA and NP-HEL (31, 36). This will be discussed more when we describe the effect of immunosuppressive drugs on Tfr later on in this review. The modulation of the metabolism of GC B cells and Tfh could be another regulatory mechanism used by Tfr, leading to inhibition of production of IL-21 and IL-4 by Tfh and inhibition of class-switch recombination and antibody production by B cells (23). Tfr have been shown to produce TGF-β and IL-10 which is turn could lead to inhibition of B cells responses (47). It could be possible that Tfr produce granzyme B leading to B-cell and/or Tfh cytolysis (23, 47). Eventually, a direct mechanical disruption of Tfh and GC B cell has been hypothesised, but this has not been proven yet (47). Finally, Tfr regulates the interaction between Tfh and B cell during the GC reaction and limits the size of the GC reaction. They inhibit the production of high-affinity antibodies specific for self-antigens (48) and limits both self-reactive and non-specific responses (see Figure 1).
A distinct population of helper cells involved in B cell responses has been recently described by Rao et al. (49), as PD-1hi CXCR5− Bcl6loand called T peripheral helper (Tph) cells. The original description of these cells was in a model of Rheumatoid Arthritis, but they have also been recently described as important in type 1 diabetes (50) and in the pathogenesis of lupus (51). It is currently not known whether Tfr can regulate this population, nor whether Tph are relevant to transplantation.
Origin of Follicular Regulatory T Cells
Tfr have been found in spleen, lymph nodes and lymphoid tissues as well as in the lymphatic and blood circulations. Tfr cells were initially thought to arise from natural (thymus-derived) Tregs (12), that become induced upon TGFβ signaling in the periphery (14). Linterman et al. (12) found Tfr resembled Treg more closely than Tfh due to the elevated expression of many Treg associated genes; FOXP3, Ctla4, Gitr, Klrg1, and Prdm1 as detailed above. However, Tfr also expressed high amounts of the CXCR5, Pdcd1, Bcl6, CXCL13 (9) and ICOS, the typical Tfh genes. Tfr did not express receptors for the helper cytokines IL-21 or IL-4 or the costimulatory ligand CD40L. Furthermore Linterman et al., reported that 97% of Tfr cells express Helios, a transcription factor expressed by thymus-derived Treg cells. Thus, the origin of Tfr cannot be determined by genetic analysis alone.
To shed further light on the origin of Tfr, Linterman et al., transferred naïve cells (CD4+CD44loCD25-) from mice expressing the 3A9 TCR transgene recognising a hen egg lysozyme peptide (HEL) into congenic mice. After being challenged with HEL, no donor originating Tfr could be identified with all Tfr deriving from recipient cells. Furthermore 6 days after selective ablation of all FOXP3+ Tregs using a diptheria toxin receptor inserted in the FOXP3 locus, Tfr were absent in diptheria treated mice, indicating that Tfr cannot form if FOXP3+ cells are absent, suggesting that Tfr development requires the presence of FOXP3+ Tregs.
Chung et al. (14), similarly sought to trace the origin of CXCR5+ Treg in mice. They found that CXCR5+FOXP3+ Tregs were essentially absent in the thymus compared to the spleen. To determine whether Bcl6+CXCR5+ Treg cells were generated from naïve CD4+ or natural Treg precursors in the periphery, they mixed CD45.1+ naïve CD4+ T cells (CD25-GITRCD44loCD62Lhi) and CD45.2+CXCR5- Treg from FOXP3gfp mice, The T cells were injected into Tcrb-/- mice, which were deficient in alpha beta T-cell receptor and consequently had ~ 6% CD4+CD8+ of wt (52). This was followed by immunization with keyhole limpet hemocyanin (KLH) in complete Freund’s adjuvant (CFA). 98.2% of Bcl6+FOXP3+ cells in the recipient mice were CD45.2+, indicating that the origin of Bcl6+CXCR5+ Treg is from CXCR5- Treg. Furthermore, they found that the majority of CXCR5+ FOXP3+ cells expressed Helios. While Chung et al. and Linterman et al., showed Tfr differentiate primarily from FOXP3+ Treg precursors, the former concluded that Bcl6+CXCR5+ Treg cells are absent in the thymus but induced in the periphery from CXCR5-FOXP3+ natural Tregs.
It may be that the differentiation of Tfr requires numerous stimulations. Thus, the thymus provides the microenvironment for Treg precursors to acquire CD31 (53) and Helios but then the subsequent differentiation of Tfr occurs by further stimulation in peripheral lymphoid tissue (15).
As a counter to Tfr deriving solely from FOXP3+ natural Tregs, Aloulou et al. (54), proposed they may also derive from FOXP3 negative precursors such as naive CD4+ T cells. They demonstrated that naive CD4+ T cells can become Tfr cells in murine models using an adjuvant that promotes peripheral Treg cell formation. This may occur in the context of a stimulus that promotes the conversion of CD4+ FOXP3− cells into FOXP3+ Treg cells, specifically, one that enhances PD-L1 expression on antigen presenting cells. Whether these ‘induced’ Tfr cells emerge from Tfh cells that acquire FOXP3 expression, or from peripheral Treg cells that acquire the follicular fate through CXCR5 expression awaits further investigation. However, Tfh cells cannot be induced to switch on FOXP3 in vitro (13), once again suggesting that it is more likely that it is peripheral Treg cells that give rise to Tfr cells.
Again, the concept that Tfr can arise from FOXP3- T cells has been countered by Maceiras et al. (55). They used congenic mice to investigate the precursors of Tfr cells following immunization in two distinct genetic backgrounds without the confounding issue of lymphopenia. They found that the adoptive transfer of thymic-derived FOXP3+ Treg in mice can differentiate into Tfr but FOXP3– T cells only differentiate into Tfh. Additionally, they demonstrated that Tfh and Tfr pools are generated from distinct TCR repertoires, with Tfh cells expressing antigen-responsive TCRs to promote antibody responses, and Tfr cells expressing potentially autoreactive TCRs to suppress autoimmunity, strengthening the idea that Tfr and Tfh are derived from distinct populations. The proposed origin of Tfr in murine models is summarized in Table 2.
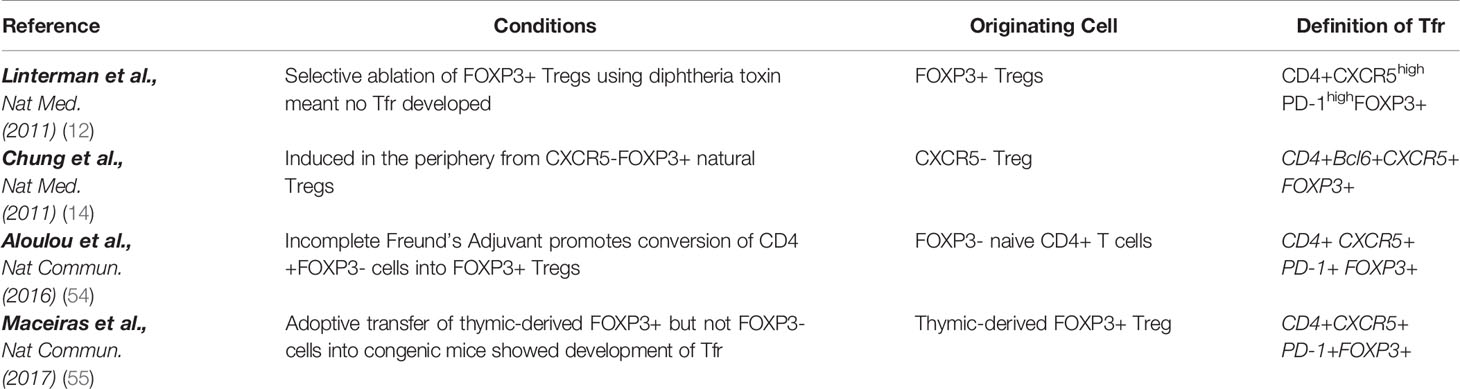
Table 2 Proposed cells of origin of Tfr in mouse models.
As discussed above, early Tfr (eTfr), derive most probably from natural Tregs in the periphery, and, after expression of Bcl-6 and CXCR5, are attracted to the T cell zone of SLO, where they interact with activated dendritic cells (Figure 1). This priming step is required by the Tfr as the number of these cells were reduced in a model of immunised mice with 4-hydroxy-3-nitrophenylacetyl hapten–conjugated OVA (NP-OVA), where dendritic cells have been ablated (36). Within the T-zone eTfr can have two different fates. Following the interaction with follicular B cells, they can lose their Blimp-1 expression, upregulate Bcl-6, ICOS, and PD-1 and transfer to the T-B border in a CXCR5 dependent manner, becoming “intermediate Tfr” (iTfr) (15). Again, this step is crucial for full differentiation of Tfr, as Tfr were almost absent in draining lymph nodes of immunized mice that lack B cells (36). The molecular mechanisms associated with the interactions between follicular B cells and iTfr remains to be determined, however, there is some evidence that this step could be antigen-independent (36, 55–57). Conversely, eTfr can retain an immature phenotype, fail to express Bcl6, and access the circulation, becoming CD25+CXCR5+FOXP3+Bcl6- blood Tfr (bTfr) (34, 36). Therefore, the presence of bTfr could be the footprint of a truncated GC formation regulation and the consequence of this could be an increase proliferation of Tfh and antibody production. In some auto-immune diseases the presence of bTfr in the blood correlated with the severity of disease (53, 58, 59). Although bTfr have been described in human (15), they have not been fully characterized and their function remain poorly understood. In particular, their precise role in transplantation remains to be elucidated.
The iTfr migrate then to the GC where they can become fully mature Tfr (mTfr) with very efficient suppressive capacities (30). It is at this stage that they lose the CD25 expression and upregulate Bcl-6, ICOS and PD-1 (60). In human, these cells are able, not only to inhibit Tfh activation (therefore decreasing their production of IL-4, IL-21) and suppress Tfh cell-GC B cell interactions leading to antibody production (61) but also decrease the activation of B cells through PD-1/PD-L1 interactions and the inhibitory function of CTLA4 molecules. Moreover, it has been demonstrated that they inhibit the capacity of class switching from IgM to IgG in mouse (23) and decrease IgA production by B cells in human (62), therefore inhibiting the selection of non-antigen-specific B cells (including those with self-reacting BCR) and limiting the number of B cells (indirect regulation). The regulation of B cells by Tfr is mechanistically complex and context-dependant as demonstrated very recently by Lopez-Ocasio Maria et al. (63). They showed with experiments in vitro that when the BCR was engaged, B cells were more resistant to suppression by Tfr, and this was dependent on a CD40-CD40L-associated mechanism.
The localization of these potential Tfr subtypes are not exclusive, and it has been demonstrated histologically that both iTfr and mTfr can be present in the Follicle and in the GC (60). It remains unknown if mTfr and iTfr could recirculate in the blood. However, it seems to be possible for bTfr to migrate back to Follicle and GCs (36). The Tfr regulation within GC seems to be happening in early GC formation stages (45), as Tfr seem to be less frequent in fully developed GCs (64) and might be inhibited by cytokines produced in GCs (65).
In summary, even if further work is needed in human, Tfr seem to derive from thymic Trefs. bTfr seems to be CD4+CD25+FOXP3+CXCR5+PD-1lowICOSlow, whereas mTfr could be described as CD4+CD25+FOXP3+CXCR5+PD-1+ICOS+Bcl-6+. The exact role of these different subpopulations still needs to be investigated, particularly in the context of transplantation.
Role of Tfr in Human Diseases
Tfr have been described in responses to influenzae vaccination and chronic infections associated with hepatitis C virus (HCV), human immunodeficiency virus (HIV) and hepatitis B virus (HBV) (46, 66–68) and they are particularly relevant in the settings of autoimmune (AI) diseases. Tregs are at the core of the physiopathology of autoimmune diseases as their role is to regulate the responses to self-antigen as demonstrated by an association between autoimmune conditions and defects in Treg function (37). Tfr have been described and characterized in several autoimmune diseases (53, 58, 59, 69). They represent a critical peripheral tolerance mechanism, to prevent GC derived auto-immunity. Patients suffering from auto-immune diseases may have an unbalance of Tfr between blood and LN in favour of blood naïve Tfr, leading to a non-specialized response in the LN (59). In this review, we will focus on the role of Tfr and their impact on the immune responses in the setting of bone marrow and solid-organ transplantation.
Relevance of Tfr in Bone Marrow Transplantation
Graft versus host disease (GvHD) is a significant complication of allogeneic hematopoietic stem cell transplantation, whereby transplanted donor cells recognise recipient antigens as foreign. This may be acute (aGvHD) or chronic (cGvHD). In cGvHD, alloreactive Tfh cells and germinal center (GC) B cells have a crucial role in GC reactions to produce pathogenic antibodies, as evidenced by the reduction in severity of cGvHD in mouse models when these antibodies are inhibited (70). Although Tfr can inhibit GC reactions by acting as negative regulators of B cell function (71), Treg numbers are reduced in patient samples of cGvHD (72) likely contributing to cGvHD pathogenesis.
McDonald-Hyman et al. (73) found mice with cGvHD had significantly fewer Tfr in line with patient data, suggesting that a loss of regulation by Tfr associates with cGvHD. However daily therapeutic interleukin-2 complexed with the JES6-1 clone of anti-IL-2 antibody (IL-2/mAb) increased Tfr numbers, due to the fact they preferentially bind to CD25hi cells, while Tfh numbers were consequently reduced. Markers of cGvHD were also reduced as assessed by tissue pathology scores and pulmonary function tests. This effect was not seen in aGvHD since treatment with IL-2/mAb complexes led to an expansion of total Tregs and CD8+ Tconv likely counterbalancing Treg expansion. They did not examine Tfh or Tfr subsets in this context.
The same authors also tested wt Treg infusions in cGvHD, which increased Tfr, while Tfh and GC B-cell frequency, GC size, and tissue pathology scores were significantly reduced in these mice. Thus this implies boosting Tfr can ameliorate cGvHD. They found that these events were CXCR5 dependent since CXCR5KO Tregs given during cGvHD once GC had formed had no effect on lung function, Tfh or GC B cells but did have improve lung function if given prophylactically. The lack of any effect correlated with the Tfr numbers that was not increased but was to a similar level to the one observed in mice with cGvHD. This suggests the importance of targeting Tregs homing to formed GC. Furthermore, the miR‐17–92 cluster has been found to facilitate Tfh‐cell differentiation and impair Tfr/Tfh balance, thus accelerating the development of cGvHD in mice (74).
Comparatively, Kamihara et al. (75) performed functional assays and flow cytometry on cryopreserved human PBMC of healthy donors and those undergoing allogeneic stem cell transplantation and those with active GvHD on IL2 therapy. Numbers of Tfr were significantly reduced in those who had undergone allo-SCT compared to healthy donors (median 0.08 vs 0.34% of CD4+ T cells respectively). Patients with active cGvHD also had significantly lower Tfr cell frequency compared to matched patients with none or resolved cGvHD. In vivo administration of low dose IL-2 therapy for one week led a selective expansion of Tfr which remained stable during the 12 weeks of therapy. The Tfr had increased expression of CD25, FOXP3, CTLA-4, ICOS, Helios, Ki67 Bcl6, and p-STAT5. In contrast, activated ICOS+PD-1+ circulating Tfh were suppressed during IL-2 therapy. The selective activation of circulating Tfr and suppression of circulating Tfh provide a mechanism whereby low dose IL-2 therapy can promote both B and T cell tolerance in patients with cGvHD. In summary, these studies suggest a reduction of Tfr correlates with active or chronic GvHD and by boosting numbers of Tfr, either with cell infusion or IL-2, this may prove an effective therapeutic strategy.
Relevance of Tfr in Solid Organ Transplantation
Tfr in Transplantation and Alloantibody Responses
The role of Tfh in transplantation and alloantibody formation has been extensively studied over the last decade (76–78). Some human observational studies have described an increase in bTfh cell numbers in patients with transplant rejection (78, 79), and a reduced proportion of bTfh cells in patients with operational tolerance (80). Tfh cells have been shown in biopsies of patients with acute kidney rejection (77) and in ectopic lymphoid structures in kidney biopsies of acute T cell rejection (81). However, the role of Tfr in the alloimmune context needs to be explored. Some have postulated that Tfr prevent antibody responses in the context of low levels of antigen (36) and/or when low-affinity BCR are produced after somatic hypermutation (12). These situations do not fit with the transplant setting where the antigens are persistent.
Tfr in Acute and Chronic Rejection in Solid Organ Transplantation
Although the exact role of Tfr in transplantation and in antibody production needs further investigation, analysis of phenotype and/or frequency and/or function of these cells might be helpful for diagnostic purpose. Extensive work has been done to link Tfh with rejection (82, 83), or exploring Tfh/Tfr ratio in the context of autoimmune diseases (59). The published data evaluating Tfr in solid organ transplantation other than kidney is scarce. One study in a mouse model of lung transplantation (84) has demonstrated that bronchus-associated lymphoid tissue-resident FOXP3+ T lymphocytes expressing CXCR5 were responsible for the prevention of antibody-mediated rejection. Table 3 summarize the studies of Tfr in kidney transplant patients. The definition of Tfr varies and the markers selected to characterize the Tfr do not allow to distinguish between the different subtypes of Tfr (bTfr vs iTfr or eTfr). However, there seems to be a trend toward lower levels of bTfr and chronic rejection (79, 86) and a potential correlation between low levels (79, 86) of bTfr and reduced kidney function as demonstrated by low estimated glomerular filtration rate (eGFR) (79). However, the number of bTfr in the transplanted patients was not compared to a group of patients with kidney dysfunction. Moreover, the definition of CAD used in these studies was not uniform or not clearly stated. Therefore, it is difficult to know if the decrease in bTfr numbers was secondary to the degree of uraemia (87). As yet Tfr in patients with end-stage renal disease (ESRD) or on haemodialysis has not been assessed. These results could seem inconstant with the findings of high bTfr in the auto-immune settings described above, however there are no definite explanation for these apparent inconstancies. Only two studies managed to collect some lymphoid structures from transplanted patients: Tfr were present in LN from a patient who had a kidney transplantation (62) but rare in kidney tertiary lymphoid organs in patients with either chronic or acute rejection (85). In conclusion, the current evidence is trending toward a decrease of bTfr in patients with CAD, however this needs to be confirmed with bigger numbers of patients and a more consensual definition of CAD.
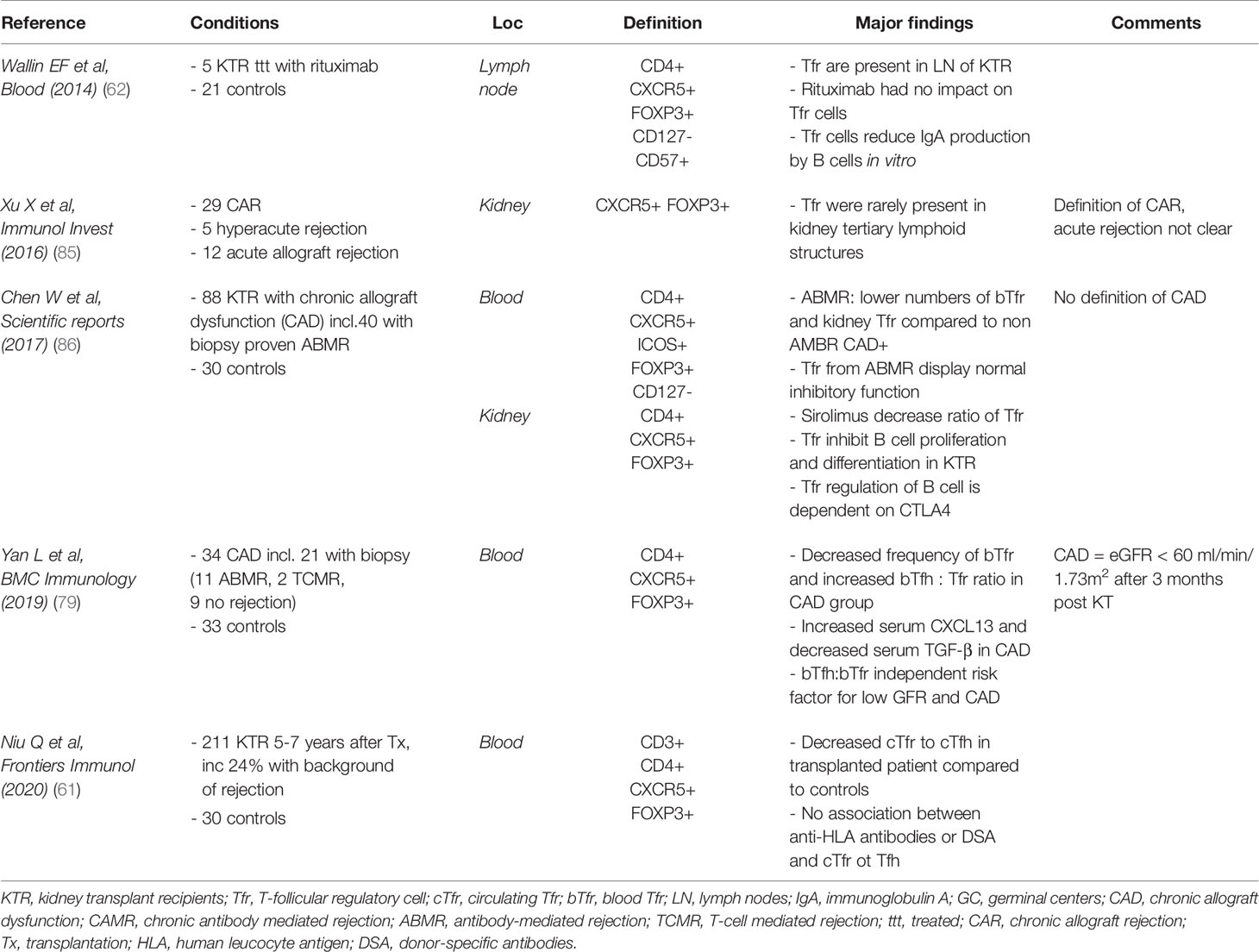
Table 3 Studies focusing on T-follicular regulatory cells in human and solid-organ transplantation.
Tertiary Lymphoid Organs
Tertiary lymphoid organs (TLOs) are ectopic lymphoid aggregates frequently observed in tissues affected by non-resolving inflammation as a result of infection, autoimmunity, cancer, and allograft rejection (88–91). They vary from tight clusters of T and B cells to highly ordered structures resembling the cellular composition of lymphoid follicles typically associated with secondary lymphoid organs (SLOs), such spleen and LN (90). The process whereby inflammatory cells infiltrate chronically rejected grafts and are progressively organised into structures has been termed lymphoid neogenesis (92). Although TLOs within tissues show varying degrees of organization, they frequently demonstrate segregated T and B cell zones, follicular dendritic cell networks, a supporting stromal reticulum, and high endothelial venules. In this respect, they mimic the activities of germinal centers and contribute to the local control of adaptive immune responses. However, unlike SLOs but akin to mucosa-associated lymphoid tissue, TLOs do not have afferent lymph vessels and are not encapsulated, suggesting they are directly exposed to local antigens or cytokines (93). Studies in various disease settings have described how these structures can contribute to either beneficial or harmful outcomes. In the context of transplantation and DSA production whether lymphoid neogenesis is harmful, beneficial, or simply a bystander occurrence remains to be fully elucidated.
Several groups have reported that TLOs can amplify anti-graft immunity and accelerate tissue destruction. In murine studies of heart and skin transplantation, TLO formation has been associated with rejection (89). Infiltrating lymphocytes were found to be composed of both B cells and follicular-helper like CD4+ T cells in rat aortic allografts, and to be associated with antibody production independent of SLOs, suggesting local antibody production (85). This group went on to find ectopic GCs in all explanted human cardiac (n=5) and renal allografts (n=24) undergoing chronic rejection but not control organs (88). Histological examination revealed B cells near CD23+ follicular DC surrounded by CD3+ T cells, although further phenotyping to identify Tfr was not done.
Other studies suggest that the presence of Tregs in TLO might promote graft tolerance, thereby slowing down the kinetics of chronic rejection (92, 94–98). Xu et al. (85) measured the distribution of TLOs and the expression of FOXP3 and CXCR5 in explanted human renal allografts with chronic rejection. FOXP3+ Tregs were detected in 10/29 chronically rejected grafts and 1/12 acutely rejected grafts and this did not correlate with lymphoid neogenesis or prolonged graft functioning. CXCR5+ FOXP3+ Tfr cells were rare in both chronically and acutely rejected grafts with TLO vs. those without. CXCR5+ FOXP3+ cells were present in 7/29 chronically rejected grafts but none of the acutely rejected grafts, implying Th17 but not Tfh could be involved in lymphoid neogenesis.
In summary, the possibilities remain that 1) lymphoid neogenesis is simply an epiphenomenon related to graft duration as proposed by Thaunat (92), 2) that Treg numbers are diminished in rejected organs in TLO, or 3) that there is another regulatory cell that aids TLO-mediated tolerance.
Effect of Immunosuppression
Regardless of the type of organ transplanted, recipients require some degree of immunosuppression to prevent allograft rejection; this is usually in the form of induction and then subsequent lifelong maintenance immunosuppression. Many of the immunosuppressive agents are T cell targeted and result in disruption of T cell homeostasis. A summary of studies focusing on the effect of immunosuppressive agents on human Tfr is presented in Table 4.
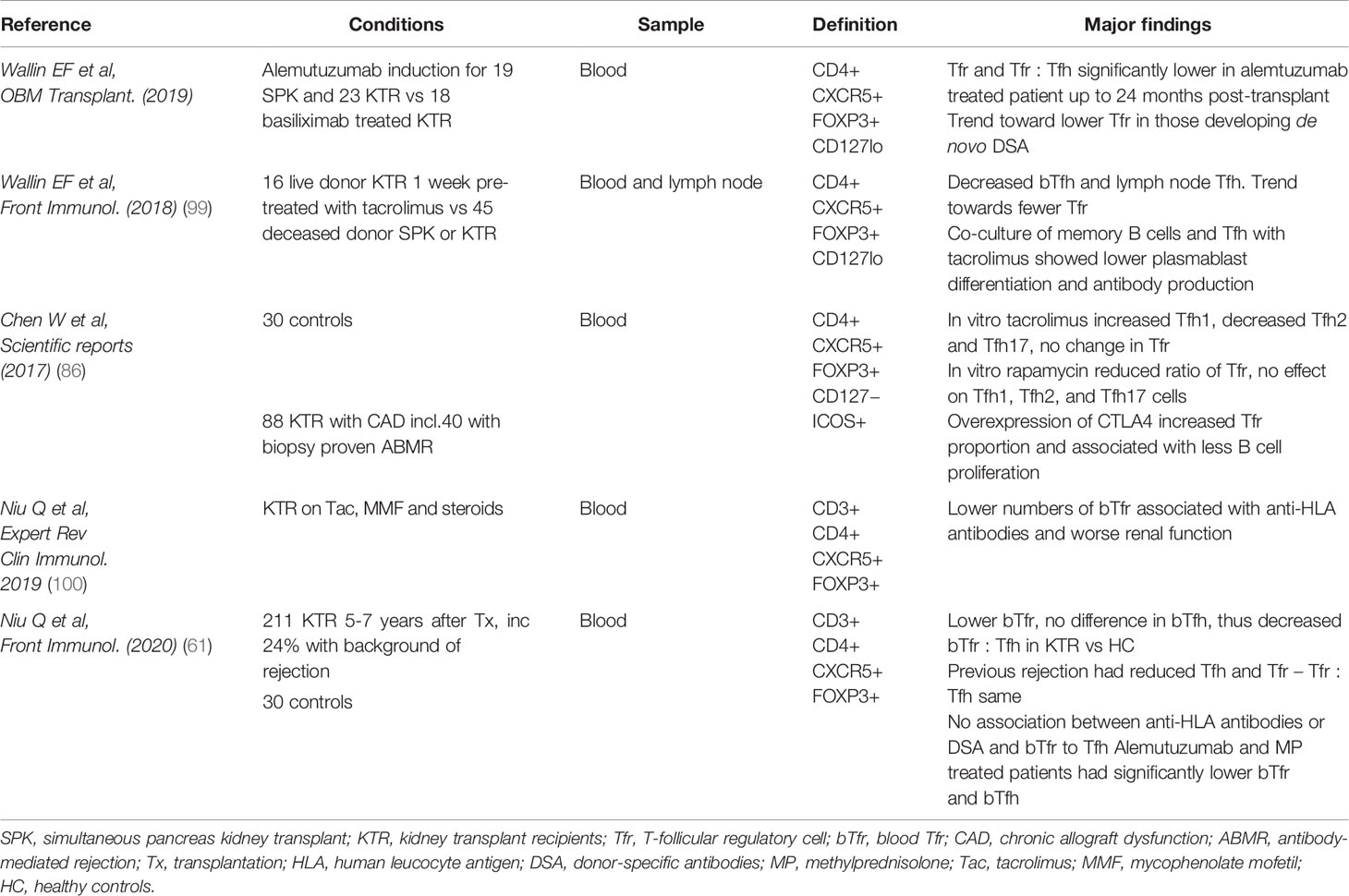
Table 4 Summary of studies focusing on the effect of immunosuppressive agents on human Tfr.
Early Transplant Immunosuppression
Induction T cell depleting agents include anti-thymocyte globulin (ATG) and alemtuzumab which is a monoclonal antibody against CD52 expressed by most lymphocytes. Comparatively, another induction agent basiliximab, is a non–T cell depleting monoclonal antibody specific for the IL-2Rα receptor (CD25). Blockade of CD25 is designed to prevent T cells activation, in part by blocking the effect of autocrine IL-2 production and also to reduce T cell activation of B cells (101). Whether it affects Tfh and Tfr equally is unclear.
Lymphopenia-induced proliferation after depletion initiates repopulation of CD4+ T cells with a phenotype skewed toward effector memory pool and a significant decrease in the naïve pool (102). These cells have a lower threshold for activation, can circulate to the graft and are less dependent on costimulation for activation (56). Thus, a predominance of memory T cells could contribute to graft injury and rejection. Conversely, the frequency and memory differentiation of CD4+ T cells post-basiliximab induction remains unchanged (103).
In an ATG treated group of renal transplant patients, Macedo et al. (104) found that the absolute numbers of circulating Tfh (CD45RO+CXCR5+CD4+CD3+) were significantly decreased at all time points up to 360 days post induction. The percentage of Tregs was overall decreased in the ATG group, although they did not evaluate the Tfr numbers. Furthermore, the Tfh repopulation following ATG was found to be skewed toward Th1 polarization and effector memory. There was also a correlation of higher Th1 polarized cTfh cells relative to Tregs numbers in those that developed DSA. This suggests that rising Tfh numbers post-transplant may associate with DSA occurrence in the context of ATG induction.
Wallin et al. (105) compared induction with alemtuzumab in simultaneous pancreas-kidney (SPK) vs. renal transplant patients receiving basiliximab. CXCR5+IL-7RloFOXP3+CD4+ bTfr cells remained significantly lower in alemtuzumab treated patients, both in proportion of total CD4 population and absolute cell count (compared to basiliximab patients at almost all time points up to 24 months post-transplant) despite being significantly higher in this group prior to transplant. There was also a trend toward a lower proportion of CXCR5+IL-7RloFOXP3+CD4+ Tfr cells in patients developing de novo DSA compared to those who did not, but this was not statistically significant owing to low patient numbers developing de novo DSA.
Overall, the ratio of circulating Tfr : Tfh between treatment groups was significantly lower in all alemtuzumab patients compared to basiliximab treated patients up to 24 months post-transplant, reflecting the persistent low levels of Tfr cells in alemtuzumab treated patients despite a recovering bTfh population. The fact that both alemtuzumab use and de novo DSAs associates with a pattern of low bTfr:cTfh is one putative explanation of why alemtuzumab patients develop higher rates of de novo DSA post-transplant (106), but necessitates further elucidation to establish causality.
Glucocorticoids are frequently used at least in the early stages of post-transplant immunosuppression or during rejection. They act via inhibition of cytokines such as IL-1, TNF alpha, IFNγ and IL-6. A study by Wen et al. (107) found a correlation between 3 months of glucocorticoid use in thirteen previously untreated myasthenic patients and an increase in circulating Tregs and Tfr cells with a reduction in circulating Tfh. They inferred that treatment with steroids can attenuate the symptoms of myasthenia gravis by restoration of the imbalances between circulating Treg, Tfr, and Tfh and maintaining immune homeostasis. This suggests that steroids may exert a similar effect in transplant recipients by promoting a positive ratio of Tfr : Tfh. This was also seen in autoimmune treatment with 5mg prednisolone causing an increase in Tfr compared to pre-treatment.
In the context of renal transplant rejection Seissler et al. (108), found methylprednisolone (125-250mg) for 3 days did not affect the percentage of Treg numbers (CD4+FOXP3+CD127lo+/−) within the total CD4+ T cell population. However, the ratio of different Treg subsets changed such that DR+CD45RA− Tregs increased significantly, while the naïve DR−CD45RA+ Tregs decreased significantly. Moreover, they observed a disproportionally strong expansion of the DRhigh+CD45RA−Tregs which have been shown to have maximal suppressive properties (108). However, this proportional increase in DRhigh+CD45RA−Tregs was not sustained beyond 3 days and may be confounded by the effects of increasing other immunosuppressive doses.
Longer-Term Immunosuppression
For maintenance immunosuppression, post-transplant patients are commonly on calcineurin inhibitors which remain a cornerstone of immunosuppression regimes. Tacrolimus (Tac) and cyclosporin (Csa) are often used and exhibit their action via blockade of the dephosphorylation of the nuclear factor in activated T cells (NFAT) (109). This prevents translocation into the DNA promoter region in the nucleus, thereby selectively suppressing the cytokine gene transcription for IL-2 (110), TNF-a, IL-3 and IL-4 (111). This affects T cell proliferation and activation (112). Vaeth et al. (20), found that Tfr are highly dependent on NFAT signaling indicating that CNI could plausibly impair the function of these subsets (113).
Wallin et al. (99), compared the effect of tacrolimus on paired blood and lymph node samples from transplant recipients. Living-donor kidney transplant recipients were treated with tacrolimus for a week prior to transplantation while the deceased-donor recipients received no pre-transplantation tacrolimus. One week of treatment reduced the frequency of both circulating and lymph node Tfh cells in the transplant recipients. At the same time, Treg remained the same in both tacrolimus treated and untreated recipients. There was a trend to toward fewer Tfr (CXCR5+IL-7RloFOXP3+CD4+ cells) numbers and as a proportion of total CD4+. Comparatively, 11 days of tacrolimus administered to Tfh in co-culture with memory B cells lead to lower PD-1 expression, plasmablast differentiation and antibody production compared to control treated cells.
Further in vitro evidence indicates Tac administration to Tfh-B co-culture prevented plasmablasts and IgG formation in cells from renal transplant patients, suggesting it is Tac targeting of Tfh that prevents DSA formation (114). Similarly, in healthy volunteers, Chen et al. found that Tfr cell proportions were unaffected by treatment with CNI but the Tfh1 percentage increased while the IL-21 producing Tfh2 and Tfh17 cells decreased (86). The results from these studies may be influenced by the use of healthy volunteers in Chen’s study vs. transplant recipients. Thus, CNIs are likely to have a dominant effect on Tfh rather than Tfr. This may be explained by higher expression of NFAT in Tfh cells than in other CD4 subsets (20). Alternatively, suppression of Tfr cells, like Tregs, may require higher doses or longer duration of CNI treatment than required for suppression of Tfh cells.
Antiproliferative agents, such as mycophenolate mofetil (MMF), are another commonly used immunosuppressive drug. This is a pro-drug which undergoes hydrolyzation by gut esterases to give the active mycophenolic acid. This acts by inhibiting inosine monophosphate dehydrogenase, which is crucial for purine synthesis in T and B cells. However, there are conflicting reports of MMF promoting induction of Tregs from Tconvs (115), while others suggest a dose dependent reduction in Treg viability and proliferative capacity (116). The specific effect on Tfh and Tfr has not been examined in the literature.
A less commonly used agent is sirolimus, a mammalian target of rapamycin inhibitor (mTOR). The mTOR signaling pathway plays a crucial role in dictating T cell fate through the interaction and balance of two mTOR containing complexes, mTORC1 and mTORC2. Xu et al. (117) found mTORC1 was expressed at high levels in mouse Tfr cells. By deleting the essential components of mTORC1 and mTORC2 they demonstrated that mTORC 1 but not mTORC2 was essential for Tfr differentiation, which was via the p-STAT3-TCF-1-Bcl-6 pathway. Essig et al. also found mTOR inhibitors suppressing PI3K-mTOR signaling inhibits the conversion of Treg to Tfr cells (118). Xu et al. showed that Tfr differentiated from mouse Tregs in the presence of rapamycin had lower expression of CXCR5, GITR and CTLA-4 compared to vehicle treated precursors. Tfr derived in the presence of rapamycin also had reduced suppressive function as indicated by an increased proportion and total number of GC B cells in spleens compared with the spleens of mice that had received wt Tfh and vehicle-treated Tfr cells. Essig et al. (118) used Roquin, an RNA-binding protein, to inhibit the PI3K-mTOR pathway at several levels, noting that differentiation toward Th17 and Tfh (PD1intCXCR5intCD4+) was inhibited as well as Treg to Tfr (CXCR5hi PD-1hi FOXP3 CD4+).
Chen et al. (86) found 48-h in vitro culture with rapamycin could reduce the ratio of CD4+CXCR5+ICOS+FOXP3+CD127− Tfr cells in healthy volunteers but had no effect on Tfh1, Tfh2 and Tfh17 cells. Thus, mTOR inhibitors may adversely affect Tfr cell function, skewing the balance toward Tfh cells. This is supported by mouse models suggesting that the use of mTOR inhibitor rapamycin after alemtuzumab induction increased the proportion of Tfh cells while significantly reduced the number of Tregs 2 weeks post cardiac transplantation and was associated with an increase in DSAs (119).
Another key mediator of Treg function is CTLA-4, which contributes to the suppressive function of Tfr by downregulating the expression of CD80/86 on antigen presenting cells and consequently reducing CD28 engagement (120). Treg-specific deletion of CTLA-4 results in a massive increase in antibody production, pointing to a crucial role for CTLA-4 on Treg cells in limiting B cell responses (120). Wing et al. subsequently (71) showed that murine CTLA-4 deficient Tfr (CXCR5+ Bcl6+ FOXP3+) either from CTLA-4 KO mice or using anti-CTLA Fab were less able to reduce the expression of CD80 and CD86 on B cells. They also were less able to prevent effector T cell proliferation when purified B cells were used as stimulators. Thus, CTLA-4-deficient Tregs and Tfr exhibited significantly reduced, but not a total loss of, suppressive function in vitro.
Sage et al. (47) demonstrated murine Tfr (CD4+CXCR5+ICOS+FOXP3+CD19−) had very high expression of CTLA-4 compared to Tfh CD4+CXCR5+ICOS+FOXP3−CD19−. In their mouse model where CTLA-4 is conditionally deleted on FOXP3+Tregs upon tamoxifen administration, they found substantial increases in total Treg and Tfr numbers, ICOS expression and an increase in Tfr : Tfh after tamoxifen. However, these Tfr showed diminished suppressive capacity of B cell function in suppression assays both in vitro and in vivo, as indicated by a substantial increase in antigen specific IgG. Chen et al. (86) used a lentivirus to overexpress CTLA-4 in Tregs from renal patients with CAD finding an increased proportion of human Tfr (CD4+CXCR5+ICOS+FOXP3+CD127−) and significantly less B cell proliferation. ELISA results showed Tfr inhibited IgG and IgA production from plasma cells. Conversely, there was increased proliferation and differentiation to plasma cells in the Tfr deleted group. This suggests that selectively increasing Tfr via CTLA-4 may be a good strategy to treat AMR by preventing B cell proliferation and differentiation.
Belatacept is a CTLA-IgG fusion protein which binds to the CD80 and CD86 molecules on antigen presenting cells preventing T cell co-stimulation and promoting expression of indoleamine 2,3-dioxygenase. Oh et al. (114) showed CTLA-Ig in addition to rapamycin increased Tfh (ICOS+ PD-1+ CD4+) cells but not Tregs (CD4+ CD25+ FOXP3+) in a murine cardiac transplant model with alemtuzumab induction. In clinical transplantation, the impact of belatacept on Tregs has been difficult to assess because it is administered in combination with blocking antibodies targeting IL-2Rα and cyclosporin (121). Despite this, it is currently widely accepted that the use of high doses of CTLA4 Ig is detrimental to Treg survival, whereas low doses of CTLA4 Ig, unable to saturate CD80 and CD86, may favour Treg expansion to some extent in the long term (122). Kim et al. (123) in a primate renal transplant model found CXCR5+Bcl6+PD-1hiCD4+ T cells were greatly reduced in lymph nodes of costimulatory blockade treated (belatacept or anti-CD40 mAb) animals with AMR that had lower levels of DSA compared to primates with AMR receiving the control immunosuppression regimen.
While these studies involving immunosuppression are confounded by small patient numbers, differences in HLA typing and variation in immunosuppression regimes, there is a consistent finding that a positive ratio of Tfr : Tfh correlates with immune regulation and a trend toward less DSA formation. Niu et al. (100) observed that kidney transplant recipients on immunosuppressive therapy with tacrolimus (Tac), mycophenolate mofetil (MMF), and steroids, with anti-HLA antibodies including DSA had lower numbers of circulating Tfr (CD3+ CD4+ CXCR5+ FOXP3+) cells than patients without anti-HLA antibodies. Inverse correlations between the kidney function parameters (serum creatinine level), and number of circulating Tfr cells and number of Helios+ circulating Tfr cells were found, indicating that the reduction of number of bTfr cells might reflect the lack of regulation of active B cell immunit directed against the allograft in kidney transplant recipients.
Niu et al. (61), evaluated Tfr in the clinical context of long term multiple agent immunosuppression in 211 patients with a functioning renal transplant over 5 years post transplantation. They found absolute numbers of Tfr (total CD3+CD4+CXCR5+FOXP3−) including subsets that were PD1+ and Helios+, were lower in transplant recipients compared to healthy controls. There was no difference in total Tfh (CD3+CD4+CXCR5+FOXP3−), meaning consequently there was also a decrease in the Tfr : Tfh. Although different induction agents were used according to clinical indication, the majority being anti-CD25 mAb, most patients had similar maintenance regimes; tacrolimus, MMF and tapering prednisolone to 0 at 4–5 months. They compared 162 patients with no history of rejection vs 49 with rejection (5 presumed and 44 with biopsy proven), noting that those with previous rejection had lower numbers of Tfr and Tfh such that the Tfr : Tfh remained the same as those without rejection. However, blood samples were taken at a median of 4.9 years post rejection episode thus cell numbers may be as a consequence of specific anti-rejection treatment, enhanced overall immunosuppression or reflecting a pattern that predated rejection. Of the anti-rejection treatments, Alemtuzumab in combination with methylprednisolone was the only one associated with significantly lower numbers of both bTfr and bTfh cells, including their subsets.
One of the limitations of this study was the use of healthy controls as a comparison without CKD. Although the transplant recipients had functioning grafts, their eGFR was <40ml/min and they demonstrated that lower eGFR correlated with lower Tfr. Thus, a comparison to CKD patients with a GFR may have controlled for the confounding effect of uraemia rather than contrasting with healthy controls.
Overall, there appears to be a reduction of Tfr associated with most immunosuppression regimens. However, elucidating each agent’s effect is substantially confounded by the use of other drugs and frequent clinician adjustment in the context of rejection, infection etc. There has also been a trend in some studies (100) of a positive Tfr : Tfh associating with less DSA formation. However, Niu et al. (61), found patients with anti-HLA antibodies or DSA at 5–7 years post-transplant or any form of rejection had similar bTfr and bTfh cell numbers as those without antibodies or rejection. This is in opposition to Macedo et al. (104), who demonstrated lower Tfh. Similarly, a longitudinal study by Cano-Romero et al. (124), indicated that cTfh (CD4+CXCR5+PD1hiCCR7lo) expanded significantly more after transplantation in patients who developed de novo anti‐HLA antibodies than in patients who remained unsensitised. Thus, a contemporaneous longitudinal study of both Tfr and Tfh numbers and their functional assessment may indicate which patterns associate with immunosuppression changes, rejection and de novo DSA development.
Cell Therapy Using Tfr
Tregs Therapy
Over the last decade, clinical trials using autologous Tregs have grown exponentially, with not less than 52 current Treg clinical trials registered in January 2020 on ClinicalTrials.gov, two of which have involved our group. In solid organ transplantation, two trials in liver transplantation have been published (125, 126). One was suspended because of rejection in three patients even if seven were successfully weaned of immunosuppression (125), though there were some concerns about the potential presence of antigen-specific effector cells in the cell product used in this study. The ThRIL study (126) demonstrated that Treg infusion in liver transplant recipients was safe. It was easier to achieve the aim of delivering 4.5 millions of Tregs per kilogram after expansion if the Tregs were isolated 6–12 months after liver transplantation, in comparison to trying to identify patients to treat while they were awaiting their transplant. Treg infusion resulted in a transient increase of the pool of circulating Tregs. Patients who received the full 4.5 million/kg Tregs product displayed a decrease in T cell responses against donor cells (assessed by the upregulation of CD154 on memory CD8+ Tcells).
The One study (127) presented the results of seven trials using different types of regulatory cell products, used in place of induction treatment for kidney transplantation. The results were not individualized for each type of cell products but demonstrated that the use of regulatory cell products is safe and could even lead to a decrease to post-transplant viral infections. Patients receiving Tregs products displayed more stable Treg-specific demethylated region compared to those receiving standard of care. However, those studies have used autologous polyclonally expanded Tregs and the proportion of Tfr in these products was not investigated.
These have led to the development of an ongoing Phase IIb study in kidney recipients (TWO Study, https://doi.org/10.1186/ISRCTN11038572) and to a Phase IV study in liver recipients, with the association of low dose IL-2 infusions (LITE study: NCT02949492). The latter, which tested the efficacy of low dose IL-2 infusions in liver transplant recipient as potential therapy to increase autologous Tregs in vivo was stopped prematurely, as some patients developed rejection on protocol biopsies.
Access to Tfr and Cell-Engineering
The biology and role of different subsets of Tfr remains to be further elucidated in humans. However, bTfr seem to be good candidates for autologous therapy. First, they are accessible by leukapheresis (in contrast to their lymph nodes counterparts) and display a memory-type phenotype (36). Moreover, they are easily identifiable and could potentially be sorted from fresh blood as they could be defined as CD4+CD25+ CXCR5+ cells. However, their suppressive capacity, in comparison to mTfr has not been tested and it remains unknown whether they will be recruited into GC and become fully mature. Moreover, the frequency of bTfr in human blood and whether they can be expanded in vitro are both unclear.
As Tfr are derived from Tregs, it could be possible to use general Tregs, and modify them so that they become Tfr. Kim et al. (128), used retroviral transduction of the CXCR5 gene in FOXP3+ Tregs and demonstrated stable expression of functional CXCR5 on transduced-Tregs. CXCR5-transduced Tregs maintained a Treg signature and suppressive activity in vivo after adoptive transfer in mice. Moreover, using a transwell culture system, Kim et al. demonstrated that they migrated efficiently down a CXCL13 gradient and suppressed antibody production by B cells.
The field of tailored cell therapy has advanced enormously in recent years thanks to new engineering techniques including CRISPR-Cas9 (129) and chimeric antigen receptor (CAR) technologies (130, 131). The latter involves synthetic fusion proteins which typically combine an extracellular antibody-derived antigen targeting moiety and an intracellular TCR complex-derived signaling domain (or domains). The resultant protein is consequently able to bind designated target antigens, in an MHC-independent manner, and translate this engagement into activation of customized T cell signaling cascades [reviewed by us (132)]. The first Phase 1/2 human clinical trial using CAR-Tregs (in a setting other than cancer) will launch in 2020 (STEADFAST study, Sangamo Therapeutics) and will test CAR-Tregs in prevention of immune-mediated rejection following HLA-A2 mismatched kidney transplantation. Using a CXCR5-CAR on Tregs could help them to migrate to the lymph node and potentially inhibit antibody responses. Fine tuning the expression of CXCR5 seems to be more appropriate and as the Bcl-6 dependant CXCR5 expression is a dynamic process, using CRISPR-Cas9 technology to incorporate Bcl-6 to Tregs genome might be another track to explore.
Although promising and full of potential, cell-engineering might not be appropriate for Tfr production, as these cells cannot be defined by the presence of one parameter. More than producing Tfr through an engineering process, expanding Tfr from isolated Tregs might be another tempting option.
Applicability of Treg Expansions Protocols to Tfr: IL-2/Rapamycin
The production and expansion of polyclonal autologous Tregs for cell therapy has been extensively described over the last few years, with a few protocol variations between centers (126, 127, 133–137). Others groups have been focusing on delivering autologous donor-antigen reactive Tregs to prevent transplant rejection (138, 139). In order to obtain a pure cell product which complies with Good Manufacturing Product (GMP) regulations before being reinfused to patients, experts have made a step forward standardization of required tests in a recent publication (140). The different options from isolation to culturing these cells are presented in Figure 2.
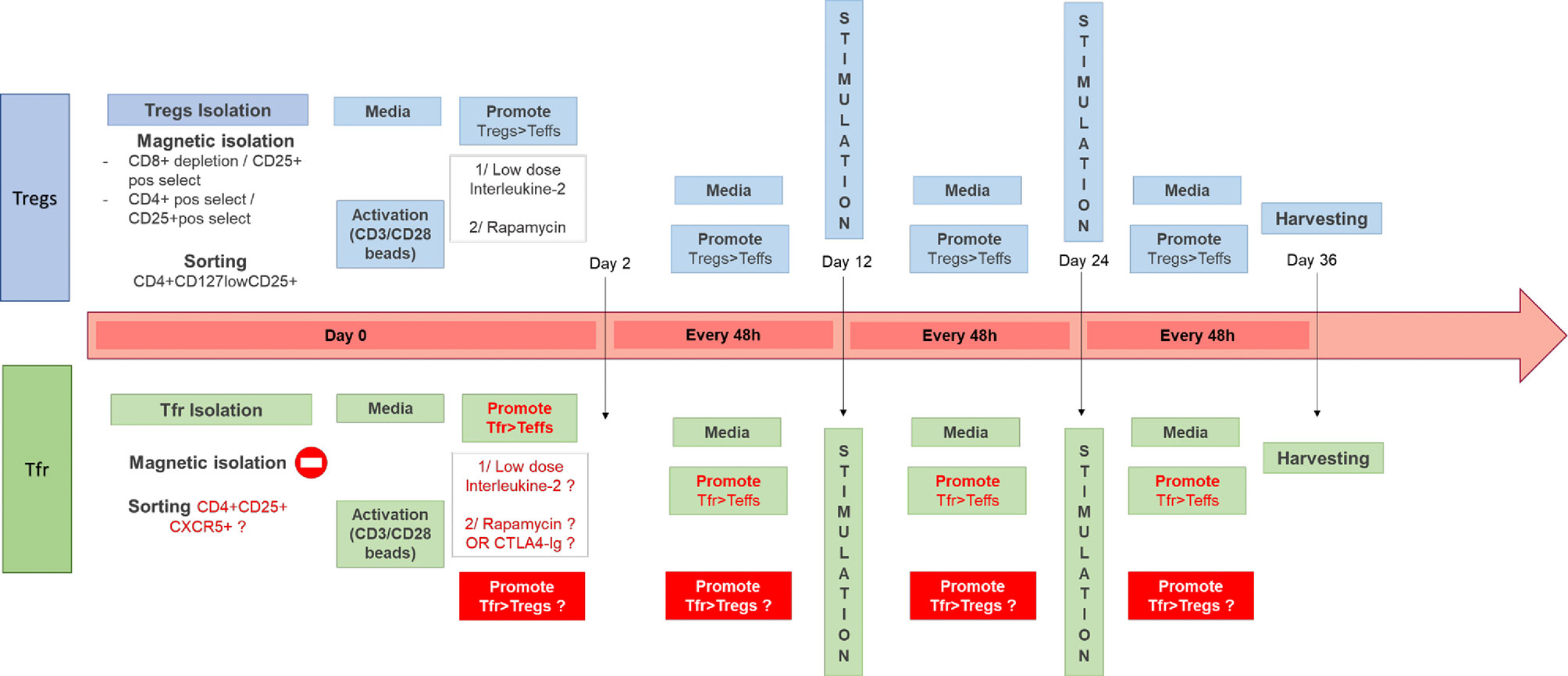
Figure 2 Different Tregs Good Manufacturing Product (GMP)-compatible expansion protocols and potential areas to improve for Tfr cell therapy. In the top diagram are described some of the protocols in use in our GMP facility (126, 127, 135) for the expansion of Tregs from the isolation step (using either magnetic beads or cell sorting) to the other steps such as stimulation every 12 days (with anti-CD3/CD28 beads) and feeding every 48 h. The use of IL-2 and rapamycin has been shown necessary to expand functional and stable Tregs in the absence of T effectors. In the bottom diagram is described a similar process for the generation of a Tfr product and in red are all the different steps which remains to be optimized.
The communally accepted GMP-compliant process to expand polyclonal autologous Tregs developed in our laboratory which has been in use and demonstrated to be safe during the Phase I ONE Study (127) and ThRIL (126) includes harvesting Tregs from the (potential) recipient using CD8+ depletion and CD25+ positive selection (Miltenyi), followed by polyclonal expansion using CD3/CD28 beads (Dynabeads, Thermofisher) in the presence of Rapamycin (116, 135) and Interleukin(IL)-2 (141). This protocol allows the preferential expansion of Tregs compared to other T cell population. The aim of the expansion is to increase the numbers of cell present while maintaining phenotype, purity, and suppressive abilities. Other centers have used sorted cells as the initial Treg product (142), followed by expansion with or without Rapamycin present (138, 142, 143). As discussed previously the mTOR pathway seems to be essential for Tfr functions, therefore using Rapamycin to expand Tfr ex vivo might not be appropriate (86). Li et al. (144) demonstrated that patients on Sirolimus had lower numbers of CD4+CXCR5+ cells, but in this study Tfr and Tfh were not differentiated. Therefore, further exploratory work is needed before using Rapamycin in Tfr culture. Guinan et al. (139) have developed a technique using ex vivo costimulatory blockage with Belatacept to support the expansion of potent allo-specific Tregs. They perform a mixed lymphocyte reaction using T cells and allogenic PBMC stimulators in the presence of co-stimulatory blockade for 72 h. The cell product is then washed and the Treg fraction is isolated using CD8 and CD19 depletion, followed by CD25+ isolation prior to administration. These Tregs may represent induced Tregs, by co-stimulatory-deficient activation trough the TCR. This approach has been demonstrated to be safe, as part of the recently published ONE Study and is currently under investigation in a Phase I/II liver transplant study (NCT03577431). Using Belatacept to promote Tfr might be an attractive alternative to Rapamycin.
IL-2 is a crucial cytokine for T cell expansion. However, as Tregs constitutively express CD25, which is the α subunit of the IL-2 receptor, Treg proliferation requires less IL-2. Using low-dose IL-2 is therefore a way to foster Treg expansion in preference to Teffs, which require higher doses of IL-2 to expand. Low dose IL-2 has been used in all GMP-compatible Treg expansion protocols (126, 127, 134, 142) except in the antigen-specific Treg expansion protocol published by Guinan et al. mentioned above (where there is no expansion per se). The sensitivity of Tfr to IL-2 remains to be determined. As described earlier, low dose IL-2 in the context of GvHD seems to positively influence Tfr. On the other hand, some mouse and human (30, 56) bTfr may express lower levels of CD25 than other Tregs, and thus may not respond as well to lower doses of IL-2 compared to other Tregs. Moreover, high levels of IL-2 may be detrimental for Tfr expansion (30), as described by Botta et al. using a mouse model of influenza, although Tfr in this model seem to be differentially regulated by IL-2 during the early and later stages of infection. Therefore, the relevance and importance of low-dose IL-2 in protocols to isolate and expand autologous Tfr remains to be determined.
It remains to be explored if current protocols in place for Treg expansion could be extrapolated to Tfr expansion.
Potential Applications in Transplantation
Despite having a non-clear potential diagnostic and prognostic applications in the transplant settings, cell therapy using Tfr could have a high potential both in prophylactic and therapeutic applications. Assuming the challenges associated with isolation and expansion of Tfr are solved, there are still multiple uncertainties regarding optimal indication (tolerance induction, treatment of CAD secondary to DSA), dose (in relation to number of Tfh), timing (in relation to the transplant and the need to re-dosing), and antigen specificity (to promote specific suppression of DSA production).
In summary, improving the understanding of Tfr-Tfh interactions might be the relevant to enhance long-term outcomes after transplantation. While Tfr have just started to be explored in the transplantation setting, their role remains to be further defined in human. Cell therapy using autologous Tfr has never been done, but should be technically feasible, with some refinement of existing protocols. Work to define the exact potential of such therapy is required.
Author Contributions
CD participated in manuscript writing, editing, and coordination of its submission. SB contributed to manuscript writing. AD, GL, and CS contributed to manuscript writing and editing. AD and GL are co-last author of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The authors have received funding from the Medical Research Council (MRC): award number MR/S000852/1 for CD and MR/T006560/1 for SB. This work was further supported by the Department of Health (DoH) via the National Institute for Health Research (NIHR) Comprehensive Biomedical Research Centre award to King’s Health Partners. Institutional Open Access funds to support article publication were also received. Views expressed are those of the authors and not necessarily those of the NHS, NIHR, or DoH.
Conflict of Interest
GL is co-Founder of Quell Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Manook M, Koeser L, Ahmed Z, Robb M, Johnson R, Shaw O, et al. Post-listing survival for highly sensitised patients on the UK kidney transplant waiting list: a matched cohort analysis. Lancet (2017) 389(10070):727–34. doi: 10.1016/S0140-6736(16)31595-1
2. Benichou G, Gonzalez B, Marino J, Ayasoufi K, Valujskikh A. Role of Memory T Cells in Allograft Rejection and Tolerance. Front Immunol (2017) 8:170. doi: 10.3389/fimmu.2017.00170
3. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155(3):1151–64.
4. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 Are Reciprocal and Antagonistic Regulators of T Follicular Helper Cell Differentiation. Science (2009) 325(5943):1006–10. doi: 10.1126/science.1175870
5. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 Mediates the Development of T Follicular Helper Cells. Science (2009) 325(5943):1001–5. doi: 10.1126/science.1176676
6. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity (2009) 31(3):457–68. doi: 10.1016/j.immuni.2009.07.002
7. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41(4):529–42. doi: 10.1016/j.immuni.2014.10.004
8. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5+ CD4+ T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity (2013) 39(4):770–81. doi: 10.1016/j.immuni.2013.09.007
9. Chtanova T, Tangye SG, Newton R, Frank N, Hodge MR, Rolph MS, et al. T Follicular Helper Cells Express a Distinctive Transcriptional Profile, Reflecting Their Role as Non-Th1/Th2 Effector Cells That Provide Help for B Cells. J Immunol (2004) 173(1):68–78. doi: 10.4049/jimmunol.173.1.68
10. Rasheed A-U, Rahn H-P, Sallusto F, Lipp M, Müller G. Follicular B helper T cell activity is confined to CXCR5hiICOShi CD4 T cells and is independent of CD57 expression. Eur J Immunol (2006) 36(7):1892–903. doi: 10.1002/eji.200636136
11. Tangye SG, Ma CS, Brink R, Deenick EK. The good, the bad and the ugly — TFH cells in human health and disease. Nat Rev Immunol (2013) 13(6):412–26. doi: 10.1038/nri3447
12. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control T follicular helper cells and the germinal center response. Nat Med (2011) 17(8):975–82. doi: 10.1038/nm.2425
13. Wollenberg I, Agua-Doce A, Hernández A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol (2011) 187(9):4553–60. doi: 10.4049/jimmunol.1101328
14. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med (2011) 17(8):983–8. doi: 10.1038/nm.2426
15. Fonseca VR, Ribeiro F, Graca L. T follicular regulatory (Tfr) cells: Dissecting the complexity of Tfr-cell compartments. Immunol Rev (2019) 288(1):112–27. doi: 10.1111/imr.12739
16. Lim HW, Hillsamer P, Kim CH. Regulatory T cells can migrate to follicles upon T cell activation and suppress GC-Th cells and GC-Th cell–driven B cell responses. J Clin Invest (2004) 114(11):1640–9. doi: 10.1172/JCI200422325
17. Lim HW, Hillsamer P, Banham AH, Kim CH. Cutting Edge: Direct Suppression of B Cells by CD4+CD25+ Regulatory T Cells. J Immunol (2005) 175(7):4180–3. doi: 10.4049/jimmunol.175.7.4180
18. Müller G, Höpken UE, Lipp M. The impact of CCR7 and CXCR5 on lymphoid organ development and systemic immunity. Immunol Rev (2003) 195:117–35. doi: 10.1034/j.1600-065X.2003.00073.x
19. Vanderleyden I, Fra-Bido SC, Innocentin S, Stebegg M, Okkenhaug H, Evans-Bailey N, et al. Follicular Regulatory T Cells Can Access the Germinal Center Independently of CXCR5. Cell Rep (2020) 30(3):611–9. doi: 10.1016/j.celrep.2019.12.076
20. Vaeth M, Müller G, Stauss D, Dietz L, Klein-Hessling S, Serfling E, et al. Follicular regulatory T cells control humoral autoimmunity via NFAT2-regulated CXCR5 expression. J Exp Med (2014) 211(3):545–61. doi: 10.1084/jem.20130604
21. Liu X, Chen X, Zhong B, Wang A, Wang X, Chu F, et al. Transcription factor achaete-scute homologue 2 initiates follicular T-helper-cell development. Nature (2014) 507(7493):513–8. doi: 10.1038/nature12910
22. Fonseca VR, Graca L. Contribution of FoxP3+ Tfr cells to overall human blood CXCR5+ T cells. Clin Exp Immunol (2019) 195(3):302–4. doi: 10.1111/cei.13245
23. Maceiras AR, Fonseca VR, Agua-Doce A, Graca L. T follicular regulatory cells in mice and men. Immunology (2017) 152(1):25–35. doi: 10.1111/imm.12774
24. Wei Y, Feng J, Hou Z, Wang XM, Yu D. Flow cytometric analysis of circulating follicular helper T (Tfh) and follicular regulatory T (Tfr) populations in human blood. Methods Mol Biol (2015) 1291:199–207. doi: 10.1007/978-1-4939-2498-1_17
25. Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity (2014) 40(3):367–77. doi: 10.1016/j.immuni.2014.02.005
26. Wu H, Xie MM, Liu H, Dent AL. Stat3 Is Important for Follicular Regulatory T Cell Differentiation. PloS One (2016) 11(5):e0155040. doi: 10.1371/journal.pone.0155040
27. Xie MM, Koh B-H, Hollister K, Wu H, Sun J, Kaplan MH, et al. Bcl6 promotes follicular helper T-cell differentiation and PD-1 expression in a Blimp1-independent manner in mice. Eur J Immunol (2017) 47(7):1136–41. doi: 10.1002/eji.201747034
28. Wing JB, Tekgüç M, Sakaguchi S. Control of Germinal Center Responses by T-Follicular Regulatory Cells. Front Immunol (2018) 9:1910. doi: 10.3389/fimmu.2018.01910
29. Xie MM, Dent AL. Unexpected Help: Follicular Regulatory T Cells in the Germinal Center. Front Immunol (2018) 9:1536. doi: 10.3389/fimmu.2018.01536
30. Botta D, Fuller MJ, Marquez-Lago TT, Bachus H, Bradley JE, Weinmann AS, et al. Dynamic regulation of T follicular regulatory cell responses by interleukin 2 during influenza infection. Nat Immunol (2017) 18(11):1249–60. doi: 10.1038/ni.3837
31. Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat Immunol (2013) 14(2):152–61. doi: 10.1038/ni.2496
32. Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, et al. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med (2008) 205(12):2873–86. doi: 10.1084/jem.20080840
33. Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, et al. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J Exp Med (2011) 208(7):1377–88. doi: 10.1084/jem.20102065
34. Fonseca VR, Agua-Doce A, Maceiras AR, Pierson W, Ribeiro F, Romão VC, et al. Human blood Tfr cells are indicators of ongoing humoral activity not fully licensed with suppressive function. Sci Immunol (2017) 2(14). doi: 10.1126/sciimmunol.aan1487
35. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
36. Sage PT, Alvarez D, Godec J, von Andrian UH, Sharpe AH. Circulating T follicular regulatory and helper cells have memory-like properties. J Clin Invest (2014) 124(12):5191–204. doi: 10.1172/JCI76861
37. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713
38. Stoler-Barak L, Biram A, Davidzohn N, Addadi Y, Golani O, Shulman Z. B cell dissemination patterns during the germinal center reaction revealed by whole-organ imaging. J Exp Med (2019) 216(11):2515–30. doi: 10.1084/jem.20190789
39. Reinhardt RL, Liang H-E, Locksley RM. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol (2009) 10(4):385–93. doi: 10.1038/ni.1715
40. Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med (2010) 207(2):365–78. doi: 10.1084/jem.20091777
41. Lüthje K, Kallies A, Shimohakamada Y, Belz GT, Light A, Tarlinton DM, et al. The development and fate of follicular helper T cells defined by an IL-21 reporter mouse. Nat Immunol (2012) 13(5):491–8. doi: 10.1038/ni.2261
42. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
43. Fu W, Liu X, Lin X, Feng H, Sun L, Li S, et al. Deficiency in T follicular regulatory cells promotes autoimmunity. J Exp Med (2018) 215(3):815–25. doi: 10.1084/jem.20170901
44. Wu H, Chen Y, Liu H, Xu L-L, Teuscher P, Wang S, et al. Follicular regulatory T cells repress cytokine production by follicular helper T cells and optimize IgG responses in mice. Eur J Immunol (2016) 46(5):1152–61. doi: 10.1002/eji.201546094
45. Clement RL, Daccache J, Mohammed MT, Diallo A, Blazar BR, Kuchroo VK, et al. Follicular regulatory T cells control humoral and allergic immunity by restraining early B cell responses. Nat Immunol (2019) 20(10):1360–71. doi: 10.1038/s41590-019-0472-4
46. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity (2013) 39(4):758–69. doi: 10.1016/j.immuni.2013.08.031
47. Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity (2014) 41(6):1026–39. doi: 10.1016/j.immuni.2014.12.005
48. Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol (2009) 9(12):845–57. doi: 10.1038/nri2637
49. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature (2017) 542(7639):110–4. doi: 10.1038/nature20810
50. Ekman I, Ihantola E-L, Viisanen T, Rao DA, Näntö-Salonen K, Knip M, et al. Circulating CXCR5-PD-1hi peripheral T helper cells are associated with progression to type 1 diabetes. Diabetologia (2019) 62(9):1681–8. doi: 10.1007/s00125-019-4936-8
51. Bocharnikov AV, Keegan J, Wacleche VS, Cao Y, Fonseka CY, Wang G, et al. PD-1hiCXCR5- T peripheral helper cells promote B cell responses in lupus via MAF and IL-21. JCI Insight (2019) 4(20). doi: 10.1172/jci.insight.130062
52. Mombaerts P, Clarke AR, Rudnicki MA, Iacomini J, Itohara S, Lafaille JJ, et al. Mutations in T-cell antigen receptor genes alpha and beta block thymocyte development at different stages. Nature (1992) 360(6401):225–31. doi: 10.1038/360225a0
53. Dhaeze T, Peelen E, Hombrouck A, Peeters L, Van Wijmeersch B, Lemkens N, et al. Circulating Follicular Regulatory T Cells Are Defective in Multiple Sclerosis. JI (2015) 195(3):832–40. doi: 10.4049/jimmunol.1500759
54. Aloulou M, Carr EJ, Gador M, Bignon A, Liblau RS, Fazilleau N, et al. Follicular regulatory T cells can be specific for the immunizing antigen and derive from naive T cells. Nat Commun (2016) 7(1):10579. doi: 10.1038/ncomms10579
55. Maceiras AR, Almeida SCP, Mariotti-Ferrandiz E, Chaara W, Jebbawi F, Six A, et al. T follicular helper and T follicular regulatory cells have different TCR specificity. Nat Commun (2017) 8. doi: 10.1038/ncomms15067
56. Ritvo P-GG, Churlaud G, Quiniou V, Florez L, Brimaud F, Fourcade G, et al. Tfr cells lack IL-2Rα but express decoy IL-1R2 and IL-1Ra and suppress the IL-1-dependent activation of Tfh cells. Sci Immunol (2017) 2(15):eaan0368. doi: 10.1126/sciimmunol.aan0368
57. Ritvo P-G, Saadawi A, Barennes P, Quiniou V, Chaara W, El Soufi K, et al. High-resolution repertoire analysis reveals a major bystander activation of Tfh and Tfr cells. Proc Natl Acad Sci USA (2018) 115(38):9604–9. doi: 10.1073/pnas.1808594115
58. Shan Y, Qi C, Zhao J, Liu Y, Gao H, Zhao D, et al. Higher frequency of peripheral blood follicular regulatory T cells in patients with new onset ankylosing spondylitis. Clin Exp Pharmacol Physiol (2015) 42(2):154–61. doi: 10.1111/1440-1681.12330
59. Fonseca VR, Romão VC, Agua-Doce A, Santos M, López-Presa D, Ferreira AC, et al. The Ratio of Blood T Follicular Regulatory Cells to T Follicular Helper Cells Marks Ectopic Lymphoid Structure Formation While Activated Follicular Helper T Cells Indicate Disease Activity in Primary Sjögren’s Syndrome. Arthritis Rheumatol (Hoboken NJ) (2018) 70(5):774–84. doi: 10.1002/art.40424
60. Wing JB, Kitagawa Y, Locci M, Hume H, Tay C, Morita T, et al. A distinct subpopulation of CD25- T-follicular regulatory cells localizes in the germinal centers. Proc Natl Acad Sci USA (2017) 114(31):E6400–9. doi: 10.1073/pnas.1705551114
61. Niu Q, Mendoza Rojas A, Dieterich M, Roelen DL, Clahsen-van Groningen MC, Wang L, et al. Immunosuppression Has Long-Lasting Effects on Circulating Follicular Regulatory T Cells in Kidney Transplant Recipients. Front Immunol (2020) 11:1972. doi: 10.3389/fimmu.2020.01972
62. Wallin EF, Jolly EC, Suchánek O, Bradley JA, Espéli M, Jayne DRW, et al. Human T-follicular helper and T-follicular regulatory cell maintenance is independent of germinal centers. Blood (2014) 124(17):2666–74. doi: 10.1182/blood-2014-07-585976
63. Lopez-Ocasio M, Buszko M, Blain M, Wang K, Shevach EM. T Follicular Regulatory Cell Suppression of T Follicular Helper Cell Function Is Context-Dependent in vitro. Front Immunol (2020) 11:637. doi: 10.3389/fimmu.2020.00637
64. Sayin I, Radtke AJ, Vella LA, Jin W, Wherry EJ, Buggert M, et al. Spatial distribution and function of T follicular regulatory cells in human lymph nodes. J Exp Med (2018) 215(6):1531–42. doi: 10.1084/jem.20171940
65. Sage PT, Ron-Harel N, Juneja VR, Sen DR, Maleri S, Sungnak W, et al. Suppression by TFR cells leads to durable and selective inhibition of B cell effector function. Nat Immunol (2016) 17(12):1436–46. doi: 10.1038/ni.3578
66. Colineau L, Rouers A, Yamamoto T, Xu Y, Urrutia A, Pham H-P, et al. HIV-Infected Spleens Present Altered Follicular Helper T Cell (Tfh) Subsets and Skewed B Cell Maturation. PloS One (2015) 10(10):e0140978. doi: 10.1371/journal.pone.0140978
67. Wang L, Qiu J, Yu L, Hu X, Zhao P, Jiang Y. Increased numbers of CD5+CD19+CD1dhighIL-10+ Bregs, CD4+Foxp3+ Tregs, CD4+CXCR5+Foxp3+ follicular regulatory T (TFR) cells in CHB or CHC patients. J Transl Med (2014) 12:251. doi: 10.1186/s12967-014-0251-9
68. Wu X, Su Z, Cai B, Yan L, Li Y, Feng W, et al. Increased Circulating Follicular Regulatory T-Like Cells May Play a Critical Role in Chronic Hepatitis B Virus Infection and Disease Progression. Viral Immunol (2018) 31(5):379–88. doi: 10.1089/vim.2017.0171
69. Ding T, Niu H, Zhao X, Gao C, Li X, Wang C. T-Follicular Regulatory Cells: Potential Therapeutic Targets in Rheumatoid Arthritis. Front Immunol (2019) 10:2709. doi: 10.3389/fimmu.2019.02709
70. Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol (2012) 12(6):443–58. doi: 10.1038/nri3212
71. Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity (2014) 41(6):1013–25. doi: 10.1016/j.immuni.2014.12.006
72. Zorn E, Kim HT, Lee SJ, Floyd BH, Litsa D, Arumugarajah S, et al. Reduced frequency of FOXP3+ CD4+CD25+ regulatory T cells in patients with chronic graft-versus-host disease. Blood (2005) 106(8):2903–11. doi: 10.1182/blood-2005-03-1257
73. McDonald-Hyman C, Flynn R, Panoskaltsis-Mortari A, Peterson N, MacDonald KPA, Hill GR, et al. Therapeutic regulatory T-cell adoptive transfer ameliorates established murine chronic GVHD in a CXCR5-dependent manner. Blood (2016) 128(7):1013–7. doi: 10.1182/blood-2016-05-715896
74. Wu Y, Schutt S, Paz K, Zhang M, Flynn RP, Bastian D, et al. MicroRNA-17-92 is required for T-cell and B-cell pathogenicity in chronic graft-versus-host disease in mice. Blood (2018) 131(17):1974–86. doi: 10.1182/blood-2017-06-789321
75. Kamihara Y, Forcade E, Koreth J, Kim HT, Liu H, Douchet I, et al. Low-Dose Interleukin-2 Therapy Activates Circulating T Follicular Regulatory Cells (cTFR) and Suppresses Circulating T Follicular Helper Cells (cTFH) in Patients with Chronic Gvhd. Biol Blood Marrow Transplant (2018) 24(3):S72. doi: 10.1016/j.bbmt.2017.12.617
76. Conlon TM, Cole JL, Motallebzadeh R, Harper I, Callaghan CJ, Bolton EM, et al. Unlinked memory helper responses promote long-lasting humoral alloimmunity. J Immunol (2012) 189(12):5703–12. doi: 10.4049/jimmunol.1202257
77. de Graav GN, Dieterich M, Hesselink DA, Boer K, Clahsen-van Groningen MC, Kraaijeveld R, et al. Follicular T helper cells and humoral reactivity in kidney transplant patients. Clin Exp Immunol (2015) 180(2):329–40. doi: 10.1111/cei.12576
78. Shi J, Luo F, Shi Q, Xu X, He X, Xia Y. Increased circulating follicular helper T cells with decreased programmed death-1 in chronic renal allograft rejection. BMC Nephrol (2015) 16:182. doi: 10.1186/s12882-015-0172-8
79. Yan L, Li Y, Li Y, Wu X, Wang X, Wang L, et al. Increased circulating Tfh to Tfr ratio in chronic renal allograft dysfunction: a pilot study. BMC Immunol (2019) 20(1):26. doi: 10.1186/s12865-019-0308-x
80. Chenouard A, Chesneau M, Bui Nguyen L, Le Bot S, Cadoux M, Dugast E, et al. Renal Operational Tolerance Is Associated With a Defect of Blood Tfh Cells That Exhibit Impaired B Cell Help. Am J Transpl (2017) 17(6):1490–501. doi: 10.1111/ajt.14142
81. de Leur K, Clahsen-van Groningen MC, van den Bosch TPP, de Graav GN, Hesselink DA, Samsom JN, et al. Characterization of ectopic lymphoid structures in different types of acute renal allograft rejection. Clin Exp Immunol (2018) 192(2):224–32. doi: 10.1111/cei.13099
82. Kwun J, Manook M, Page E, Burghuber C, Hong J, Knechtle SJ, et al. and B Cells in the Germinal Center After Transplantation. Transplantation (2017) 101(4):704–12. doi: 10.1097/TP.0000000000001588
83. Walters GD, Vinuesa CG. T Follicular Helper Cells in Transplantation. Transplantation (2016) 100(8):1650–5. doi: 10.1097/TP.0000000000001217
84. Li W, Gauthier JM, Higashikubo R, Hsiao H-M, Tanaka S, Vuong L, et al. Bronchus-associated lymphoid tissue-resident Foxp3+ T lymphocytes prevent antibody-mediated lung rejection. J Clin Invest (2019) 129(2):556–68. doi: 10.1172/JCI122083
85. Xu X, Han Y, Wang Q, Cai M, Qian Y, Wang X, et al. Characterisation of Tertiary Lymphoid Organs in Explanted Rejected Donor Kidneys. Immunol Invest (2016) 45(1):38–51. doi: 10.3109/08820139.2015.1085394
86. Chen W, Bai J, Huang H, Bi L, Kong X, Gao Y, et al. Low proportion of follicular regulatory T cell in renal transplant patients with chronic antibody-mediated rejection. Sci Rep (2017) 7(1):1322. doi: 10.1038/s41598-017-01625-3
88. Motallebzadeh R, Bolton EM, Pettigrew GJ. Lymphoid tissue formation in allografts: innocent until proven guilty. Transplantation (2008) 85(3):309–11. doi: 10.1097/TP.0b013e318162d2d0
89. Nasr IW, Reel M, Oberbarnscheidt MH, Mounzer RH, Baddoura FK, Ruddle NH, et al. Tertiary Lymphoid Tissues Generate Effector and Memory T Cells That Lead to Allograft Rejection. Am J Transpl (2007) 7(5):1071–9. doi: 10.1111/j.1600-6143.2007.01756.x
90. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol (2014) 14(7):447–62. doi: 10.1038/nri3700
91. Thaunat O, Field A-C, Dai J, Louedec L, Patey N, Bloch M-F, et al. Lymphoid neogenesis in chronic rejection: evidence for a local humoral alloimmune response. Proc Natl Acad Sci USA (2005) 102(41):14723–8. doi: 10.1073/pnas.0507223102
92. Thaunat O. Pathophysiologic Significance of B-Cell Clusters in Chronically Rejected Grafts: Transplantation. (2011) 92(2):121–6. doi: 10.1097/TP.0b013e31821f74fe
93. Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol (2006) 6(3):205–17. doi: 10.1038/nri1786
94. Waldmann H, Adams E, Fairchild P, Cobbold S. Infectious tolerance and the long-term acceptance of transplanted tissue. Immunol Rev (2006) 212:301–13. doi: 10.1111/j.0105-2896.2006.00406.x
95. Katakai T, Nomura T, Gonda H, Sugai M, Agata Y, Nishio A, et al. Spontaneous Large-Scale Lymphoid Neogenesis and Balanced Autoimmunity versus Tolerance in the Stomach of H + /K + -ATPase-Reactive TCR Transgenic Mouse. J Immunol (2006) 177(11):7858–67. doi: 10.4049/jimmunol.177.11.7858
96. Xu Q, Lee J, Jankowska-Gan E, Schultz J, Roenneburg DA, Roennburg DA, et al. Human CD4+CD25low adaptive T regulatory cells suppress delayed-type hypersensitivity during transplant tolerance. J Immunol (2007) 178(6):3983–95. doi: 10.4049/jimmunol.178.6.3983
97. Brown K, Sacks SH, Wong W. Tertiary lymphoid organs in renal allografts can be associated with donor-specific tolerance rather than rejection. Eur J Immunol (2011) 41(1):89–96. doi: 10.1002/eji.201040759
98. Cowan ML, Sciammas R, Chong AS. Experimental models of B cell tolerance in transplantation. Semin Immunol (2012) 24(2):77–85. doi: 10.1016/j.smim.2011.08.018
99. Wallin EF, Hill DL, Linterman MA, Wood KJ. The Calcineurin Inhibitor Tacrolimus Specifically Suppresses Human T Follicular Helper Cells. Front Immunol (2018) 9:1184. doi: 10.3389/fimmu.2018.01184
100. Niu Q, Kraaijeveld R, Li Y, Mendoza Rojas A, Shi Y, Wang L, et al. An overview of T follicular cells in transplantation: spotlight on their clinical significance. Expert Rev Clin Immunol (2019) 15(12):1249–62. doi: 10.1080/1744666X.2020.1693262
101. Iwasaki K, Kitahata N, Hiramitsu T, Yamamoto T, Noda T, Okada M, et al. Increased CD40L+PD-1+ follicular helper T cells (Tfh) as a biomarker for predicting calcineurin inhibitor sensitivity against Tfh-mediated B-cell activation/antibody production after kidney transplantation. Int Immunol (2018) 30(8):345–55. doi: 10.1093/intimm/dxy039
102. Gurkan S, Luan Y, Dhillon N, Allam SR, Montague T, Bromberg JS, et al. Immune reconstitution following rabbit antithymocyte globulin. Am J Transpl (2010) 10(9):2132–41. doi: 10.1111/j.1600-6143.2010.03210.x
103. Bouvy AP, Kho MML, Klepper M, Litjens NHR, Betjes MGH, Weimar W, et al. Kinetics of homeostatic proliferation and thymopoiesis after rATG induction therapy in kidney transplant patients. Transplantation (2013) 96(10):904–13. doi: 10.1097/TP.0b013e3182a203e4
104. Macedo C, Hadi K, Walters J, Elinoff B, Marrari M, Zeevi A, et al. Impact of Induction Therapy on Circulating T Follicular Helper Cells and Subsequent Donor-Specific Antibody Formation After Kidney Transplant. Kidney Int Rep (2019) 4(3):455–69. doi: 10.1016/j.ekir.2018.11.020
105. Transplant Research Immunology Group, Nuffield Dept Surgical Sciences, University of Oxford, Oxford, UK, Wallin EF, Linterman MA, Lymphocyte Signalling and Development ISP, Babraham Institute, Cambridge, UK, Wood KJ, Transplant Research Immunology Group, Nuffield Dept Surgical Sciences, University of Oxford, Oxford, UK. Alemtuzumab Treatment Leads to Delayed Recovery of T Follicular Regulatory Cells, and May Therefore Predispose Patients to de novo Donor-Specific Antibody Formation. obm Transpl (2019) 3(3):1–1. doi: 10.21926/obm.transplant.1903079
106. 3C Study Collaborative Group, Haynes R, Harden P, Judge P, Blackwell L, Emberson J, et al. Alemtuzumab-based induction treatment versus basiliximab-based induction treatment in kidney transplantation (the 3C Study): a randomised trial. Lancet (2014) 384(9955):1684–90. doi: 10.1016/S0140-6736(14)61095-3
107. Wen Y, Yang B, Lu J, Zhang J, Yang H, Li J. Imbalance of circulating CD4(+)CXCR5(+)FOXP3(+) Tfr-like cells and CD4(+)CXCR5(+)FOXP3(-) Tfh-like cells in myasthenia gravis. Neurosci Lett (2016) 630:176–82. doi: 10.1016/j.neulet.2016.07.049
108. Seissler N, Schmitt E, Hug F, Sommerer C, Zeier M, Schaier M, et al. Methylprednisolone treatment increases the proportion of the highly suppressive HLA-DR(+)-Treg-cells in transplanted patients. Transpl Immunol (2012) 27(4):157–61. doi: 10.1016/j.trim.2012.09.003
109. Shrestha BM. Two Decades of Tacrolimus in Renal Transplant: Basic Science and Clinical Evidences. Exp Clin Transplant (2017) 15(1):1–9. doi: 10.6002/ect.2016.0157
110. Vafadari R, Kraaijeveld R, Weimar W, Baan CC. Tacrolimus inhibits NF-κB activation in peripheral human T cells. PloS One (2013) 8(4):e60784. doi: 10.1371/journal.pone.0060784
111. Hošková L, Málek I, Kopkan L, Kautzner J. Pathophysiological mechanisms of calcineurin inhibitor-induced nephrotoxicity and arterial hypertension. Physiol Res (2017) 66(2):167–80. doi: 10.33549/physiolres.933332
112. Laskin BL, Jiao J, Baluarte HJ, Amaral S, Furth SL, Akimova T, et al. The Effects of Tacrolimus on T-Cell Proliferation Are Short-Lived: A Pilot Analysis of Immune Function Testing. Transplant Direct (2017) 3(8):e199. doi: 10.1097/TXD.0000000000000715
113. Martinez GJ, Hu JK, Pereira RM, Crampton JS, Togher S, Bild N, et al. Cutting Edge: NFAT Transcription Factors Promote the Generation of Follicular Helper T Cells in Response to Acute Viral Infection. J Immunol (2016) 196(5):2015–9. doi: 10.4049/jimmunol.1501841
114. de Graav GN, Hesselink DA, Dieterich M, Kraaijeveld R, Verschoor W, Roelen DL, et al. Belatacept Does Not Inhibit Follicular T Cell-Dependent B-Cell Differentiation in Kidney Transplantation. Front Immunol (2017) 8:641. doi: 10.3389/fimmu.2017.00641
115. Lagaraine C, Lemoine R, Baron C, Nivet H, Velge-Roussel F, Lebranchu Y. Induction of human CD4+ regulatory T cells by mycophenolic acid-treated dendritic cells. J Leukoc Biol (2008) 84(4):1057–64. doi: 10.1189/jlb.1007716
116. Scotta C, Fanelli G, Hoong SJ, Romano M, Lamperti EN, Sukthankar M, et al. Impact of immunosuppressive drugs on the therapeutic efficacy of ex vivo expanded human regulatory T cells. Haematologica (2016) 101(1):91–100. doi: 10.3324/haematol.2015.128934
117. Xu L, Huang Q, Wang H, Hao Y, Bai Q, Hu J, et al. The Kinase mTORC1 Promotes the Generation and Suppressive Function of Follicular Regulatory T Cells. Immunity (2017) 47(3):538–51. doi: 10.1016/j.immuni.2017.08.011
118. Essig K, Hu D, Guimaraes JC, Alterauge D, Edelmann S, Raj T, et al. Roquin Suppresses the PI3K-mTOR Signaling Pathway to Inhibit T Helper Cell Differentiation and Conversion of Treg to Tfr Cells. Immunity (2017) 47(6):1067–1082.e12. doi: 10.1016/j.immuni.2017.11.008
119. Oh B, Yoon J, Farris A, Kirk A, Knechtle S, Kwun J. Rapamycin Interferes With Postdepletion Regulatory T Cell Homeostasis and Enhances DSA Formation Corrected by CTLA4-Ig. Am J Transpl (2016) 16(9):2612–23. doi: 10.1111/ajt.13789
120. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi: 10.1126/science.1160062
121. Vincenti F, Rostaing L, Grinyo J, Rice K, Steinberg S, Gaite L, et al. Belatacept and Long-Term Outcomes in Kidney Transplantation. N Engl J Med (2016) 374(4):333–43. doi: 10.1056/NEJMoa1506027
122. Schwarz C, Unger L, Mahr B, Aumayr K, Regele H, Farkas AM, et al. The Immunosuppressive Effect of CTLA4 Immunoglobulin Is Dependent on Regulatory T Cells at Low But Not High Doses. Am J Transplant (2016) 16(12):3404–15. doi: 10.1111/ajt.13872
123. Kim EJ, Kwun J, Gibby AC, Hong JJ, Farris AB, Iwakoshi NN, et al. Costimulation blockade alters germinal center responses and prevents antibody-mediated rejection. Am J Transpl (2014) 14(1):59–69. doi: 10.1111/ajt.12526
124. Cano-Romero FL, Laguna Goya R, Utrero-Rico A, Gómez-Massa E, Arroyo-Sánchez D, Suárez-Fernández P, et al. Longitudinal profile of circulating T follicular helper lymphocytes parallels anti-HLA sensitization in renal transplant recipients. Am J Transpl (2019) 19(1):89–97. doi: 10.1111/ajt.14987
125. Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology (2016) 64(2):632–43. doi: 10.1002/hep.28459
126. Sánchez-Fueyo A, Whitehouse G, Grageda N, Cramp ME, Lim TY, Romano M, et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am J Transplant (2020) 20(4):1125–36. doi: 10.1111/ajt.15700
127. Sawitzki B, Harden PN, Reinke P, Moreau A, Hutchinson JA, Game DS, et al. Regulatory cell therapy in kidney transplantation (The ONE Study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet (2020) 395(10237):1627–39. doi: 10.1016/S0140-6736(20)30167-7
128. Kim YU, Kim B-S, Lim H, Wetsel RA, Chung Y. Enforced Expression of CXCR5 Drives T Follicular Regulatory-Like Features in Foxp3 + T Cells. Biomolecules Ther (2017) 25(2):130–9. doi: 10.4062/biomolther.2016.075
129. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science (2020) 367(6481):eaba7365. doi: 10.1126/science.aba7365
130. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood (2015) 125(26):4017–23. doi: 10.1182/blood-2014-12-580068
131. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377(26):2531–44.
132. Mohseni YR, Tung SL, Dudreuilh C, Lechler RI, Fruhwirth GO, Lombardi G. The Future of Regulatory T Cell Therapy: Promises and Challenges of Implementing CAR Technology. Front Immunol (2020) 11:1608. doi: 10.3389/fimmu.2020.01608
133. Hoffmann P, Eder R, Kunz-Schughart LA, Andreesen R, Edinger M. Large-scale in vitro expansion of polyclonal human CD4(+)CD25high regulatory T cells. Blood (2004) 104(3):895–903. doi: 10.1182/blood-2004-01-0086
134. Landwehr-Kenzel S, Zobel A, Hoffmann H, Landwehr N, Schmueck-Henneresse M, Schachtner T, et al. Ex vivo expanded natural regulatory T cells from patients with end-stage renal disease or kidney transplantation are useful for autologous cell therapy. Kidney Int (2018) 93(6):1452–64. doi: 10.1016/j.kint.2018.01.021
135. Fraser H, Safinia N, Grageda N, Thirkell S, Lowe K, Fry LJ, et al. A Rapamycin-Based GMP-Compatible Process for the Isolation and Expansion of Regulatory T Cells for Clinical Trials. Mol Ther Methods Clin Dev (2018) 8:198–209. doi: 10.1016/j.omtm.2018.01.006
136. Safinia N, Vaikunthanathan T, Fraser H, Thirkell S, Lowe K, Blackmore L, et al. Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget (2016) 7(7):7563–77. doi: 10.18632/oncotarget.6927
137. Safinia N, Grageda N, Scottà C, Thirkell S, Fry LJ, Vaikunthanathan T, et al. Cell Therapy in Organ Transplantation: Our Experience on the Clinical Translation of Regulatory T Cells. Front Immunol (2018) 9:354. doi: 10.3389/fimmu.2018.00354
138. Putnam AL, Safinia N, Medvec A, Laszkowska M, Wray M, Mintz MA, et al. Clinical Grade Manufacturing of Human Alloantigen-Reactive Regulatory T Cells for Use in Transplantation. Am J Transpl (2013) 13(11):3010–20. doi: 10.1111/ajt.12433
139. Guinan EC, Cole GA, Wylie WH, Kelner RH, Janec KJ, Yuan H, et al. Ex Vivo Costimulatory Blockade to Generate Regulatory T Cells From Patients Awaiting Kidney Transplantation. Am J Transplant (2016) 16(7):2187–95. doi: 10.1111/ajt.13725
140. Fuchs A, Gliwiński M, Grageda N, Spiering R, Abbas AK, Appel S, et al. Minimum Information about T Regulatory Cells: A Step toward Reproducibility and Standardization. Front Immunol (2017) 8:1844. doi: 10.3389/fimmu.2017.01844
141. Whitehouse G, Gray E, Mastoridis S, Merritt E, Kodela E, Yang JHM, et al. IL-2 therapy restores regulatory T-cell dysfunction induced by calcineurin inhibitors. Proc Natl Acad Sci U S A (2017) 114(27):7083–8. doi: 10.1073/pnas.1620835114
142. Chandran S, Tang Q, Sarwal M, Laszik ZG, Putnam AL, Lee K, et al. Polyclonal Regulatory T Cell Therapy for Control of Inflammation in Kidney Transplants. Am J Transpl (2017) 17(11):2945–54. doi: 10.1111/ajt.14415
143. Berglund D, Korsgren O, Lorant T, Schneider K, Tufveson G, Carlsson B. Isolation, expansion and functional assessment of CD4+CD25+FoxP3+ regulatory T cells and Tr1 cells from uremic patients awaiting kidney transplantation. Transpl Immunol (2012) 26(1):27–33. doi: 10.1016/j.trim.2011.09.003
Keywords: regulatory T cell, T-follicular regulatory cell, transplantation, cell therapy, immunosuppression
Citation: Dudreuilh C, Basu S, Scottà C, Dorling A and Lombardi G (2021) Potential Application of T-Follicular Regulatory Cell Therapy in Transplantation. Front. Immunol. 11:612848. doi: 10.3389/fimmu.2020.612848
Received: 30 September 2020; Accepted: 14 December 2020;
Published: 02 February 2021.
Edited by:
Michaela Lucas, University of Western Australia, AustraliaReviewed by:
Federica Casiraghi, Mario Negri Institute of Pharmacological Research, ItalyJianing Fu, Columbia University, United States
Copyright © 2021 Dudreuilh, Basu, Scottà, Dorling and Lombardi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline Dudreuilh, Y2Fyb2xpbmUuZHVkcmV1aWxoQGtjbC5hYy51aw==
†These authors share last authorship