 Santosh V. Suryavanshi
Santosh V. Suryavanshi Igor Kovalchuk
Igor Kovalchuk Olga Kovalchuk
Olga Kovalchuk- Department of Biological Sciences, University of Lethbridge, Lethbridge, AB, Canada
Inflammasomes are cytoplasmic inflammatory signaling protein complexes that detect microbial materials, sterile inflammatory insults, and certain host-derived elements. Inflammasomes, once activated, promote caspase-1–mediated maturation and secretion of pro-inflammatory cytokines, interleukin (IL)-1β and IL-18, leading to pyroptosis. Current advances in inflammasome research support their involvement in the development of chronic inflammatory disorders in contrast to their role in regulating innate immunity. Cannabis (marijuana) is a natural product obtained from the Cannabis sativa plant, and pharmacologically active ingredients of the plant are referred to as cannabinoids. Cannabinoids and cannabis extracts have recently emerged as promising novel drugs for chronic medical conditions. Growing evidence indicates the potent anti-inflammatory potential of cannabinoids, especially Δ9-tetrahydrocannabinol (Δ9-THC), cannabidiol (CBD), and synthetic cannabinoids; however, the mechanisms remain unclear. Several attempts have been made to decipher the role of cannabinoids in modulating inflammasome signaling in the etiology of chronic inflammatory diseases. In this review, we discuss recently published evidence on the effect of cannabinoids on inflammasome signaling. We also discuss the contribution of various cannabinoids in human diseases concerning inflammasome regulation. Lastly, in the milieu of coronavirus disease-2019 (COVID-19) pandemic, we confer available evidence linking inflammasome activation to the pathophysiology of COVID-19 suggesting overall, the importance of cannabinoids as possible drugs to target inflammasome activation in or to support the treatment of a variety of human disorders including COVID-19.
Introduction
Animals, from lower vertebrates, such as hagfish, to higher ones, such as mammals, use innate, adaptive immune responses to protect themselves from external pathogens and injuries (1, 2). An innate immune response is the first event upon any hazard where external microbes are sensed by a group of diverse germline-encoded receptors. These receptors-pattern recognition receptors (PRRs)-recognize conserved microbial structures called pathogen-associated molecular patterns (PAMPs) (3). Innate immune cells can also mount an infection-independent immune response by sensing endogenous substances released from host tissue damage: damage (or danger)-associated molecular patterns (DAMPs) (4). This type of DAMP-mediated inflammatory response is often called “sterile inflammation” due to no pathogen involvement (5). Overall, both PAMPs and DAMPs can stimulate an initial immune response by activating different types of PRRs, including toll-like receptors (TLRs), nucleotide-binding domain (NBD) and leucine-rich-repeat-(LRR)-containing or simply nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), C-type lectin receptors (CLRs), and several cytosolic DNA sensor-like receptors absent in melanoma 2 (AIM2)-like receptors (ALRs) (4, 6). These PRRs are expressed by immune cells (macrophages, dendritic cells, etc.) and non-immune cells (endothelial cells, fibroblasts, etc.) and are present in various subcellular compartments (3, 4). Among all PRRs, the NLR family is the most extensively described in the literature due to numerous known sterile and pathogenic activators (7). NLRs and ALRs form a multimeric complex known as the “inflammasome” following the detection of respective PAMPs and/or DAMPs.
TLR-induced priming is often required to activate and assemble the inflammasome (8). Once formed and activated at the cellular level, canonical inflammasomes activate caspase-1 by autoproteolysis, resulting in proteolytic maturation of IL-1β and IL-18, which in turn leads to respective cell death by pyroptosis. Non-canonical inflammasome activation by bacterial pathogens leads to caspase-11 activation and the subsequent hyper-activation of innate immunity in mice (9). Nevertheless, IL-1β and IL-18 are crucial pro-inflammatory cytokines implicated in a variety of human disorders, such as aging, lung cancer, cardiovascular diseases, gout, etc (10, 11). Besides pyroptosis, inflammasomes may be involved in eicosanoid synthesis, phagosome maturation, glycolysis, lipid metabolism, and autophagy in a cytokine- and pyroptosis-independent manner (12).
Cannabis sativa (C. sativa) has been cultivated for centuries around the world for many purposes, and it is the most frequently used illegal plant. “Marijuana” is the term used to describe cannabis varieties that contain more than 0.3% Δ9-THC by dry weight, while “hemp” is used for varieties with lower than 0.3% Δ9-THC. C. sativa is a versatile plant that provides food, feed, shelter, and medicine. Since ancient times, various cannabis preparations have been used in both traditional and professional medicine. Although cannabis can be beneficial in treating various human diseases (13), evidence-based medical conditions for which cannabis can be usefully prescribed are chronic pain, nausea and vomiting after chemotherapy, seizures in Lennox-Gastaut and Dravet syndrome, and spasticity (14, 15). On the other hand, a recent study suggests that newly prescribed cannabinoid use (either nabilone or dronabinol) among older adults with the chronic obstructive pulmonary disorder (COPD) was associated with higher rates of adverse events. Although further research is needed to confirm the same, the physicians should weigh benefits against risks while prescribing new cannabinoids to older COPD patients (16). At least 554 compounds, including 113 phytocannabinoids and 120 terpenes, have been identified in C. sativa (13). Terpenes in cannabis give the plant a characteristic odor based on percentages of various volatile aromatic compounds. Terpenes are believed to be partially responsible for the “entourage effect,” with minor cannabinoids and other molecules such as phenolic compounds having additional effects. To date, no clear evidence has emerged for the role of any molecule or their combination in the entourage effect. In fact, recent experiments have demonstrated that terpenes do not add to the activation of cannabinoid receptors triggered by cannabinoids (17, 18). It remains to be shown, however, whether they can contribute to the entourage effect through interaction with other receptors.
Several publications have demonstrated the potent anti-inflammatory effect of cannabinoids (19). Their mechanisms of action include activating cannabinoid and other receptors, inhibiting cytokines and cell proliferation, inducing apoptosis, and so on (19–21). Inflammation occurs when innate immune cells detect pathogens, injury, or danger signals via PRRs on cell membranes and in cytosols. Activated PRRs then form inflammasomes, triggering signaling cascades leading to the recruitment of leukocytes to the injury site (22). Under normal conditions, acute inflammatory events characterized by the influx of neutrophils at the injured tissue are crucial parts of innate immunity. However, dysregulated acute inflammation, sterile inflammation, and recurrent acute inflammatory insults result in chronic inflammation. Chronic inflammation has been implicated in the pathophysiology of a variety of diseases. Inflammasomes are activated during microbial invasion, tissue injury, and sterile inflammation, which all lead to cell death. Cell death can also result in the secretion of another round of inflammasome activators, such as uric acid and ATP, which both activate inflammasomes in a paracrine manner. These signaling cascades eventually give rise to chronic inflammatory disorders, such as cardiovascular disease, cancer, metabolic disorders, autoimmune disorders, and neurodegenerative disorders (23). Besides, recent developments in inflammasome research suggest that the anti-inflammatory action of cannabinoids is mediated in part by modulating inflammasome assembly and function. Hence, our goals in this review are to cover all published research on the action of cannabinoids on the inflammasome to propose the future therapeutic potential of cannabis in chronic inflammatory disorders.
CANNABINOIDS Signaling
The first documented evidence of the medicinal use of C. sativa showed that its extracts were already in use around 5,000 years ago in ancient China to alleviate pain (24). Three types of cannabinoids exist: endocannabinoids produced by the human body; phytocannabinoids produced naturally by C. sativa; and synthetic cannabinoids synthesized under laboratory conditions. After the discovery of Δ9-THC (25), extensive research efforts were carried out to understand the pharmacological effects of cannabis. Eventually, two members of the G-protein coupled receptor (GPCR) family, the cannabinoid receptors CB1R and CB2R, were successfully cloned from rat cerebral cortex and rat spleen, respectively (26, 27). Many cannabinoids were demonstrated to bind these receptors, albeit with different efficiencies. Cannabinoids were also shown to bind to receptors other than CB1R and CB2R, as reviewed by our group elsewhere (28). All three types of cannabinoids exert their biological actions by binding to these receptors, and each cannabinoid may bind different combinations of receptors at a given time.
The endocannabinoid system (ECS) comprises of two endocannabinoids (anandamide and 2-arachidonoylglycerol), cannabinoid receptors (CB1 and CB2), and enzymes that metabolize endocannabinoids (29). Anandamide (AEA) (30) and 2-arachidonylglycerol (2-AG) (31) are the two most important endocannabinoids, although other arachidonic acid derivatives may produce similar effects (28). Both AEA and 2-AG are produced from postsynaptic terminals owing to increased intracellular Ca2+ influx (24, 32). Once produced, AEA and majorly 2-AG travel in a retrograde fashion due to their high lipophilicity to activate CB1 receptors in presynaptic terminals. The activation of CB1R inhibits neurotransmitter release via the reduction of Ca2+ inflow and inhibition of cyclic adenosine monophosphate (cAMP). However, AEA activates the intracellular transient receptor potential cation channel subfamily V member 1 (TRPV1) receptor, inhibits L-type Ca2+ channels, and inhibits 2-AG biosynthesis (24). The major role of ECS in the body is to maintain homeostasis, and it is involved in the regulation of a variety of processes, including immune, digestive, neurological, metabolic, and reproductive functions (29, 33). Variations in the ECS are pathophysiological and depend on cell and tissue type, age, and sex, and fluctuations in the function of ECS components are associated with the onset of many disorders, such as neurodegenerative, gastrointestinal, chronic inflammatory, cardiovascular, reproductive, circulatory, and metabolic disorders, including cancers (24, 34, 35).
The human CB1R and CB2R crystal structures reveal that both the intramembrane and the extracellular surface of the receptor play significant role in the ligand binding, depending on the type of ligand, unlike other lipid-stimulated GPCRs (36, 37). The extracellular-facing lid over intracellular binding pocket with its acidic residues facing outside disfavors interactions with lipophilic ligands in the extracellular space, which gain entrance to the binding pocket via an intramembrane “tunnel” (37). CB1 and CB2 receptors share only 44% of the homology of the protein sequence in humans, and CB1 receptors are highly conserved among mammals in contrast to CB2 (38). Overall, CB1R is the most extensively expressed GPCR in the central nervous system (CNS) and the peripheral nervous system (PNS), whereas CB2R is highly expressed in peripheral immune cells. However, the functional CB1Rs are also expressed in several non-neuronal peripheral tissues including heart, intestines, and liver (39). Additionally, the functional CB2 receptors are also expressed in microglia and resident macrophages in the CNS under neuroinflammatory conditions (40, 41). CB2R expression was confirmed in the neurons, as well, governing synaptic plasticity (42). Both receptors are coupled to the Gi/o family of G-proteins to inhibit cAMP production by reducing adenylyl cyclase activity, leading to lower protein kinase A (PKA) activity and the phosphorylation of mitogen-activated protein kinase (MAPK) activity (p38, c-Jun N-terminal kinase (JNK), and p42/44). They also can activate phosphatidylinositol-3-kinase-protein kinase B (PI3K-AKT), ceramide production, and the expression of various genes (24, 41, 43, 44). Interestingly, CB1 and CB2 receptors interact with Gs proteins as well to induce cAMP production under specific circumstances (45, 46). CB1 receptor activation specifically stimulates G-protein-gated inwardly rectifying potassium (GIRK) channels and inhibits voltage-gated (N-type) Ca2+ channels. Both receptors exhibit constitutive activity suggestive of G-protein activation in the absence of agonists (42). Lastly, CB1 and CB2 receptors also signal via β-arrestin and a few other biased, cell-, and ligand-specific cannabinoid receptor–mediated signal transduction mechanisms (47).
AEA is a highly specific partial agonist of CB1R with negligible or weak partial agonist activity at CB2R, whereas 2-AG is a full agonist at both CB1 and CB2 receptors. Δ9-THC has the highest affinity toward CB1R and CB2R among all phytocannabinoids (48). CBD is a weak antagonist at CB1 and an inverse agonist at CB2 receptors (49), although a meta-analysis study revealed that CBD mostly acts indirectly through other signaling pathways (50). CBD acts as an agonist at adenosine receptors and 5-HT1A receptors, and it increases AEA levels to elicit TRPV1 channel activation (50). Other minor cannabinoids also bind to cannabinoid and other receptors, as reviewed by our group elsewhere (28). By modulating intracellular cAMP levels and thereby PKA activity, cannabinoids regulate the phosphorylation of a plethora of downstream proteins, resulting in major changes in cellular activities. MAPK activation is an important pathway by which cannabinoids regulate the expression of various genes. Changes in the extracellular and intracellular ions and activation of ion channels contribute to crucial downstream cellular effects (44). Due to the possibility of binding to different receptors at a given time, including CB1R and CB2R, along with different respective agonist/antagonist/inverse agonist potentials at those receptors, cannabinoids exhibit complexity in their mechanisms of action and downstream signaling transduction pathways.
Cannabinoids in Inflammation
Cannabinoids and cannabinoid-like compounds have proven potent anti-inflammatory and immunomodulatory properties (19, 21, 35, 51, 52). In general, cannabinoids work by inducing apoptosis, preventing cell proliferation, reducing cytokine production, and enhancing T-regulatory cells (Tregs) to produce anti-inflammatory effects (19). Interestingly, cannabinoids may change the balance between the response involving T-helper 1 (Th-1) and Th-2 cells, inhibiting the expression of Th-1–induced cytokines and stimulating the expression of Th-2–induced cytokines (53). More than 350 patents have been filed on cannabinoids in the treatment of inflammation (54). Ajulemic acid (anabasum), a novel selective CB2R agonist, is currently undergoing phase II and phase III clinical trials owing to its potent anti-inflammatory effect on neutrophil migration in response to ultraviolet (UV)‐killed E. coli‐triggered dermal inflammation in humans. Notably, ajulemic acid removed the pathogenic bacteria that caused the inflammation and promoted the biosynthesis of special pro‐resolution lipid mediators to boost the body’s innate immunity (55, 56). The data from recently concluded RESOLVE-1 phase III trial of anabasum for the treatment of systemic sclerosis failed to provide any efficacy in the primary or secondary end points (Corbus Pharmaceuticals Press Release Sept 8, 2020). However, the additional post-hoc analyses released by Corbus recently showed that anabasum treatment was associated with a benefit in the lung function (forced vital capacity) in subjects on established background of immunosuppressant therapies (Corbus Pharmaceuticals Press Release Nov 9, 2020). Overall, anabasum was able to improve the lung function in patients with systemic sclerosis although was not effective in improving the actual end points of the clinical trial (NCT03398837).
CBD is the most abundant non-psychoactive cannabinoid of C. sativa and hence has been extensively studied for its anti-inflammatory properties. CBD is currently undergoing clinical trials for its effectiveness in schizophrenia (57), refractory epileptic encephalopathy (58), and tuberous sclerosis (59, 60). In addition to CB1, CB2, TRPV1, and adenosine receptors, the activation of GPR55, inhibition of fatty acid amide hydrolase (FAAH), stimulation of peroxisome proliferator-activated receptor-gamma (PPAR-γ), and heterodimerization of CB2/5HT1A are also involved in mediating the anti-inflammatory effects of CBD (20, 61, 62). Subsequently, CBD was also found to extensively inhibit the production of pro-inflammatory cytokines, such as IL-1α, IL-1β, IL-6, and tissue necrosis factor α (TNF-α), etc., in pre-clinical in vitro and in vivo models of inflammation and cancer (20). The anti-inflammatory activity of CBD was paralleled by the modulation of downstream gene expression, reduction in lipid peroxidation, Ca2+ homeostasis, and reduction of oxidative stress (61, 63, 64). However, Δ9-THC mediates its anti-inflammatory actions mainly via CB2 receptor activation, decreased production of cytokines, inhibition of Th-1, promotion of Th-2 cells, induction of apoptosis, and downregulation of cell proliferation (35, 52). Cannabichromene (CBC) has been reported to inhibit the expression and activity of TRPV1-4 channels (65). Cannabigerol (CBG) has exhibited protective properties in a murine model of inflammatory bowel disease (IBD) by regulating cytokine (IL-1β, IL-10, and interferon-γ) levels and inhibiting inducible nitric oxide synthase (iNOS) expression (66). Cannabinol (CBN), like CBD and THC, is shown to inhibit pro-inflammatory cytokine production (67). Cannabidiolic acid (CBDA) has been demonstrated to be a selective inhibitor of cyclooxygenase-2 (COX-2), and it likely plays an essential role in the reduction of inflammation (68). Data on other minor cannabinoids are limited at this point.
Inflammasome Signaling
The “inflammasome” is the name given to the high molecular weight scaffold formed by an assembly of different proteins. This scaffold mostly consists of three parts (1): a sensor protein, (2) typically the adaptor protein ASC (an apoptosis-associated, speck-like protein containing a C-terminal caspase recruitment domain [CARD]), and (3) the effector protein caspase-1 (cysteine protease) (12, 69). Sensor proteins are six NLRs (NLRP1, NLRP3, NLRP6, NLRP7, NLRP12, and NLRC4) or two ALRs (AIM2 and interferon-gamma inducible protein 16 (IFI-16) or pyrin (PYD). The NLR family contains the central nucleotide-binding and oligomerization (NACHT) domain flanked by C-terminal leucine-rich repeats (LRRs) and N-terminal CARD or PYD domains. LRRs specifically govern the ligand sensing for each NLR and autoregulation, whereas CARD or PYD domains regulate the protein-protein interactions required for downstream signaling cascades. The NACHT domain is shared by all NLRs and regulates NLR activation via adenosine 5′-triphosphate (ATP)-induced oligomerization (69). ASC is required for certain PRRs, NLRP3, AIM2, and PYD to recruit caspase-1, and NLRs such as NLRP1 and NLRC4 contain CARD and hence directly recruit caspase-1. Although other PRRs might not need ASC for caspase-1 recruitment, downstream cytokine processing depends on ASC in the complex (70) (Figure 1).
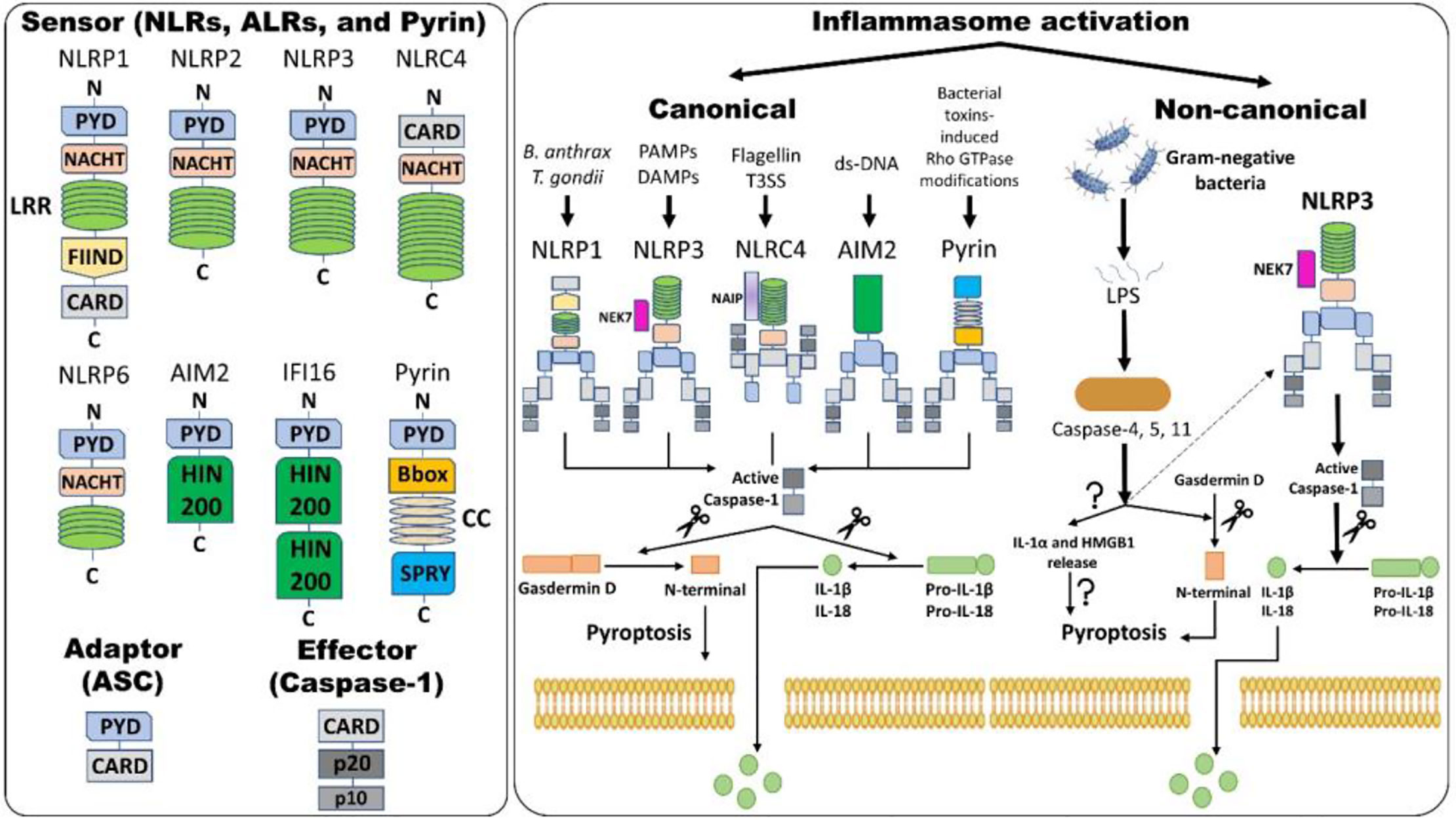
Figure 1 Inflammasome assembly and activation pathways. An inflammasome consists of three proteins: sensor, adaptor, and effector. Please note that NLRP3 and NLRC4 require NIMA (Never in Mitosis Gene A)-related Kinase 7 (NEK7) and NLR family apoptosis inhibitory protein (NAIP), respectively, for activation. Canonical inflammasome activation results in the formation of active caspase-1 (activation) by removing caspase activation and recruitment domain (CARD; processing). Active caspase-1 cleaves gasdermin D (GSDMD) and pro-interleukins into the N-terminal of GSDMD and mature interleukins, respectively. The N-terminal of GSDMD forms pores within the cell membrane, allowing mature IL-1β and IL-18 release along with changes in ion fluxes. Such caspase-1-dependent formation of plasma membrane pores releasing inflammatory intracellular materials resulting in cell lysis is termed pyroptosis. Alternatively, in non-canonical inflammasome activation, gram-negative bacteria release lipopolysaccharides (LPS) that activate caspase-4 and -5 in humans and caspase-11 in mice, which results in pyroptosis via several mechanisms. Firstly, GSDMD-mediated pyroptosis occurs as explained above and, secondly, activation of caspases leads to the release of IL-1α and high-mobility group box protein 1 (HMGB1) via unknown mechanisms, which results in pyroptosis. Caspase-4, -5, and -11 activation by LPS also indirectly activates the NLRP3 inflammasome, culminating in the maturation of IL-1β and IL-18 via activating caspase-1. Abbreviations: Nucleotide-binding domain (NBD) and leucine-rich-repeat-(LRR)-containing receptors (NLRs); Absent in melanoma 2 (AIM2)-like receptors (ALRs); pyrin domain (PYD); Caspase activation and recruitment domain (CARD); Nucleotide-binding and oligomerization or NAIP, CIITA, HET-E and TP1 (NACHT) domain; Apoptosis-associated speck-like protein containing CARD (ASC); Interferon-gamma inducible protein 16 (IFI-16); Function-to-find domain (FIIND); Coiled-coil (CC); Bacterial type III secretion system (T3SS); Hematopoietic interferon-inducible nuclear protein with 200 amino acids (HIN-200).
Inflammasome amplification is regulated by ASC via three mechanisms: first, sensors nucleate ASC, forming oligomers and ASC nucleates caspase-1 such that the sensor, adaptor, and enzyme are always present at cumulative concentrations; second, cytokines formed by caspase-1 infiltrate immune cells, lowering their activation; third, “ASC specks” released after pyroptosis can be engulfed by neighboring cells, forming an inflammasome in recipient cells (70, 71). These ASC specks are formed by phosphorylated-ASC and are crucial to inflammasome activity (72) (Figure 2). After the oligomerization of sensor proteins, inactive zymogen caspase-1 attaches itself to the scaffold and is activated by self-proteolysis into an active enzyme. Mouse caspase-1, -11, and -12 and human caspase-1, -4, and -5 are pro-inflammatory (73). Once activated, caspase-1 cleaves pro-IL-1β and pro-IL-18 and initiates their secretion, leading to an inflammatory form of cell death, pyroptosis. Pyroptosis consists of a formation of cell-membrane pores followed by cell swelling, osmotic lysis, and the release of intracellular debris (70, 74). Inflammatory caspase–dependent pyroptosis is carried out by the protein gasdermin D (GSDMD). Caspase-1, -4, -5, and -11 recognize and cleave to the same site in GSDMD, releasing its N-terminus, which signifies the autoinhibitory function of the C-terminus (75, 76). The N-terminus is the active form of GSDMD that forms pores, causing pyroptosis. Inflammasomes with caspase-1 as an effector are termed “canonical inflammasomes,” and caspase-11-mediated inflammasome activation is termed “non-canonical” (77). Nonetheless, activating the inflammasome works on an all-or-nothing principle (78), and multiple inflammasome sensors can orchestrate an inflammation response against a single pathogen inside a single host cell (79). In this review, we briefly discuss the most widely described inflammasomes in the literature (Figure 1).
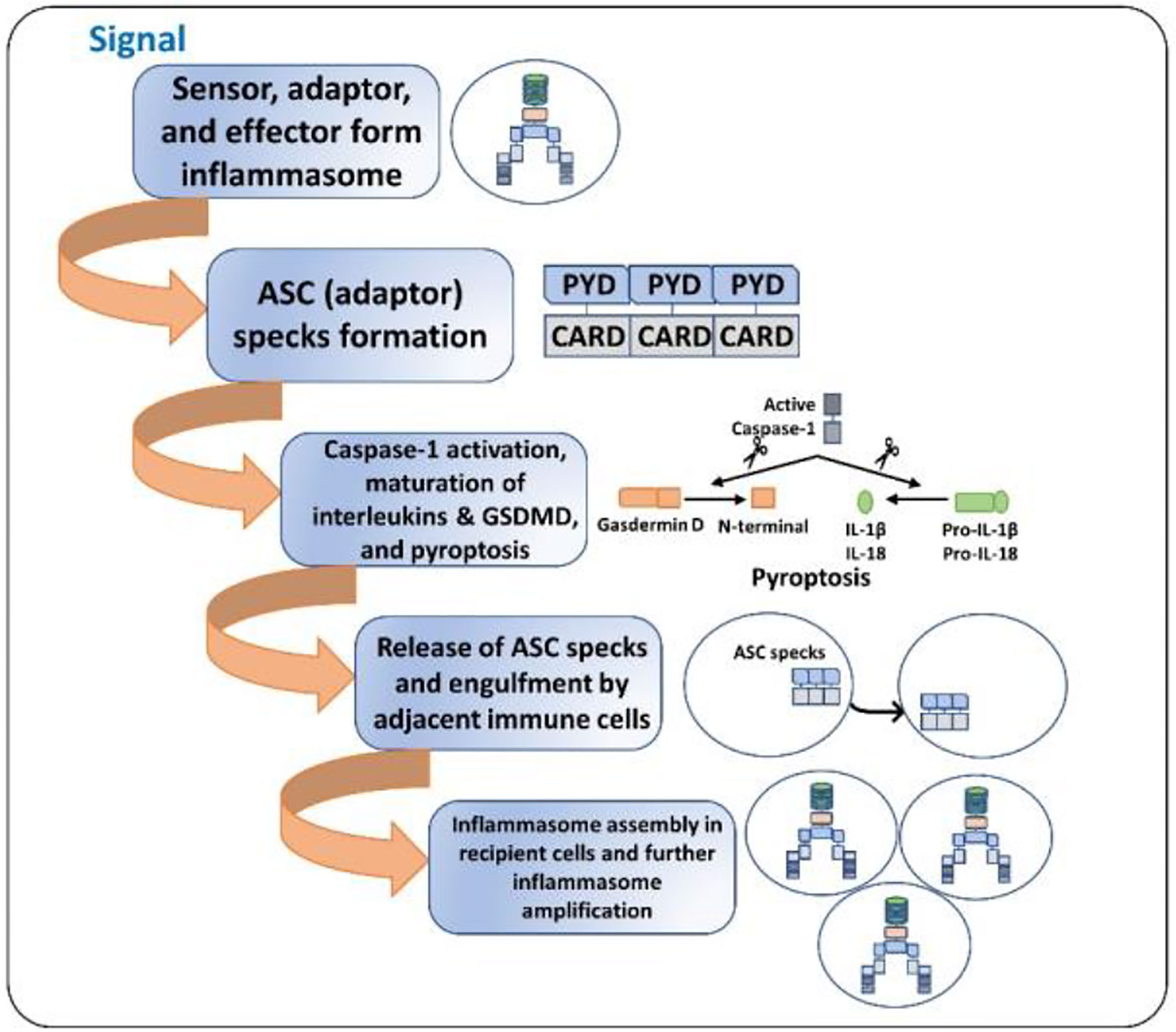
Figure 2 ASC specks and inflammasome activation amplification. 1. Initial signal (bacterial toxins, PAMPs, DAMPs, ds-DNA, etc.) leads to the assembly of the inflammasome. 2. ASC specks (oligomers) are formed such that the sensor, the adaptor, and the caspase-1 are always present at increasing concentrations. 3. The cell undergoes pyroptosis, resulting in cell lysis and the release of inflammatory cytokines and ASC specks. 4. The released ASC specks can be engulfed by adjacent immune cells, leading to the transduction of the upstream signal from one cell to the other. 5. The process of inflammasome assembly, activation, and pyroptosis repeats in the recipient immune cells, leading to amplification of inflammasome activation.
The NLRP1
It is the first PRR to be discovered that forms an inflammasome scaffold to mediate caspase-1–induced pyroptosis (80). Mice have three orthologs of NLRP1 (NLRP1a–c), whereas humans have a single gene coding for NLRP1. Murine orthologs have the CARD domain and lack the PYD domain, thus recruiting caspase-1 without ASC. In contrast, human NLRP1 has the N-terminal PYD domain, and NLRP1 is vital during host defense against anthrax (81). Mice lacking NLRP1 activation were more prone to the toxin due to aberrant host defense (82). In humans, macrophages induce apoptosis, not pyroptosis, in response to anthrax due to the absence of the Nlrp1b gene. Hence, NLRP1 is activated via binding to muramyl dipeptide (MDP) in the presence of the MDP sensor in humans (83). Single-nucleotide polymorphisms in the Nlrp1 gene in humans are also associated with congenital toxoplasmosis, proving their importance in Toxoplasma infection as well (70).
The NLRP3
The most widely studied inflammasome with respect to several human disorders is formed by NLRP3. It contains three domains—LRR, NACHT, and PYD—to bind ASC. The formation of the NLRP3 inflammasome is extensively studied in macrophages, where it is a two-step process: priming and activation (23, 84). PAMPs/DAMPs and cytokines induce non-transcriptional priming via post-translational changes (e.g., deubiquitination) and nuclear factor kappa B (NF-κB) activation via TLR4-induced transcriptional priming, leading to higher NLRP3 expression. The phosphorylation of ASC is also mandatory for NLRP3 scaffold formation. Non-transcriptional priming lasts a short (10–30 min) to intermediate time (30 min–1 h), while transcriptional priming lasts longer (>3 h) (23). TLR-mediated MyD88/IL-1 receptor-associated kinase 1 (IRAK1) regulates non-transcriptional NLRP3 priming, where NLRP3 expression does not change but priming is enough to secrete cleaved caspase-1 (85). A second signal is required for the activation step of NLRP3 and the formation of the NLRP3 signaling complex. A variety of “second signals” activate NLRP3, including potassium (K+) efflux, cathepsin release from lysosomal rupture, mitochondrial ROS and DNA, cardiolipin, calcium signaling, Na+, and Cl- efflux, among others (23, 84). A newly identified NLRP3 binding partner, NEK7 (NIMA-related kinase 7), forms a NLRP3 inflammasome after K+ efflux (86). Many particulate substances, such as amyloid β (Aβ) (87), silica, alum, monosodium urate, etc., can cause lysosomal destabilization to activate NLRP3 inflammasome (88). Autophagy and related proteins inhibit the mitochondrial DNA release into cytosol, regulating NLRP3 activation (89). The dysregulation of the NLRP3 inflammasome is involved in the pathogenesis of a variety of human disorders, and a better understanding of NLRP3 activation would help identify drug targets for NLRP3-related diseases.
The NLRC4
NLRC4 [also called ice protease-activating factor (IPAF)] contains the CARD domain to directly recruit caspase-1 to the inflammasome. The ASC is not needed for NLRC4-mediated pyroptosis but essential for amplifying the response and IL-1β release (10, 90). NLRC4 requires NLR-family apoptosis inhibitory proteins (NAIPs) to interact with bacterial pathogens upstream for its activation. NAIPs and their binding to NLRC4 have been reviewed in-depth elsewhere (91). After NAIPs bind to bacteria, NLRC4-NAIP-complexes are formed, leading to their activation. Besides pyroptosis, NLRC4 causes an actin polymerization response against Salmonella, highlighting the non-conventional role of NLRC4 inflammasome activation (92).
The AIM2
It is a cytosolic DNA sensor from the ALR family characterized by a hematopoietic interferon-inducible nuclear protein with a 200-amino-acid (HIN-200) domain. AIM2 is activated by binding to double-stranded DNA (dsDNA) of a minimum 250–300 bp in a non-sequence-specific manner via the HIN200 domain (93). In the absence of cytosolic dsDNA, HIN200 binds to AIM2 PYD as an autoinhibition. The AIM2 PYD domain is displaced after dsDNA binding, forming the PYD-PYD interaction of ASC (84). The AIM2 inflammasome plays an incomparable role in host defense against bacteria, Listeria, and DNA viruses, as the impaired secretion of cytokines has been observed in macrophages lacking AIM2 (94). Interestingly, a recent report documented the requirement of AIM2 inflammasome surveillance of DNA damage for normal brain maturation and function. AIM2 contributes to the cell death of genetically compromised CNS cells and shapes overall behavior in mice (95). The dysfunction of AIM2 is linked to various human conditions.
The Non-Canonical Inflammasome
Recent advances have identified a complex innate immune response phenomenon in which inflammasomes cleave caspase-11 in mice, termed “non-canonical inflammasome activation” (9). However, caspase-11 activation itself may stimulate the caspase-1–mediated release of canonical interleukins, as shown by the inhibition of the secretion of IL-1β and IL-18 under lipopolysaccharide (LPS) treatment in caspase-11-knockout mice (9). Additionally, caspase-11 is required for host defense against gram-negative bacterial infections since it detects specific acylated lipid A present in the LPS of this bacterial population (96). Caspase-11 is not activated in gram-positive bacterial infections, and caspase-11 activation results in the secretion of IL-1α and specific high-mobility group box-1 (HMGB1), triggering direct pyroptosis (77). TLR4 activation by LPS as a priming step is also dispensable in non-canonical signaling transduction, as HMGB1 promotes TLR4 signaling (97). It has been predicted that a caspase-11-bound pannexin-1–dependent decrease of intracellular K+ might activate the NLRP3 inflammasome (70), but further research is needed to understand whether the drop in intracellular K+ is sufficient to activate inflammasomes and/or whether other possible mechanisms are involved.
Inflammasomes in Chronic Inflammatory Disorders
Inflammasomes are critical regulators of theoretically all chronic inflammatory disorders. Irregular signaling by inflammasomes inflicts perturbations on innate immune cells, eventually affecting adaptive immunity and being involved in the pathogenesis of acute and chronic inflammatory disorders. We discuss a few representative data that link various inflammasomes and the development of inflammatory diseases (Table 1).
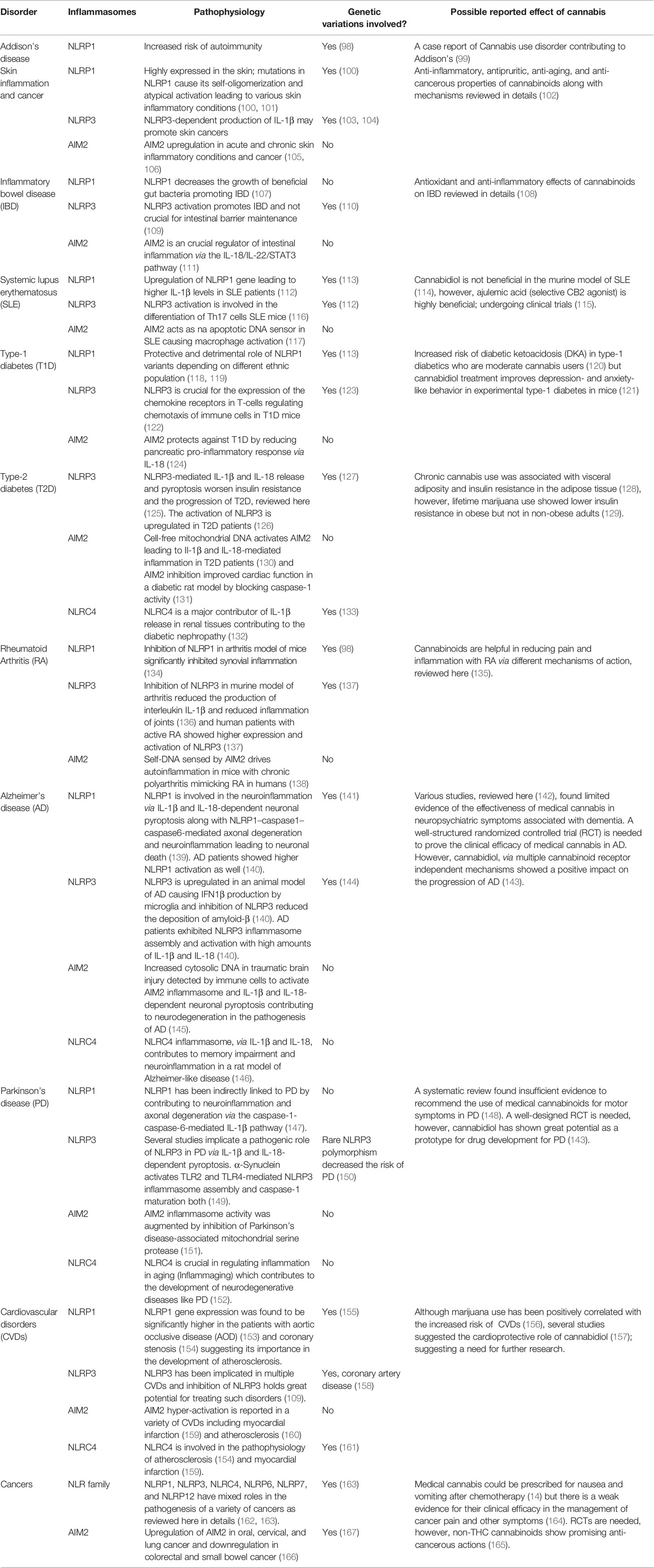
Table 1 Role of major Inflammasomes in the representative chronic inflammatory disorders along with known cannabinoid effect.
Role of NLRP1
Genomic studies of NLRP1 have identified mutations associated with autoinflammatory diseases in humans, including systemic sclerosis, Crohn’s disease, Addison’s disease, rheumatoid arthritis, type-1 diabetes, and vitiligo (70, 98). NLRP1 is the most highly expressed inflammasome in human skin, and gain-of-function NLRP1 mutations cause chronic skin inflammation and skin cancer. These mutations result in the higher self-oligomerization of NLRP1, disrupting the PYD-LRR interaction crucial in keeping NLRP1 dormant under physiological situations (100). Genetic variations in NLRP1 are also positively associated with a higher susceptibility to psoriasis (101). Interestingly, higher NLRP1 expression is correlated to dry skin–induced chronic itch in a sex- and age-dependent manner in mice (168). NLRP1 has recently been implicated in the pathophysiology of IBD by restricting the beneficial butyrate-producing Clostridiales in the gut of the dextran sulphate sodium (DSS)-induced colitis murine model of IBD (107). Single-nucleotide polymorphisms in NLRP1 are associated with an increased risk of developing type-1 diabetes and systemic lupus erythematosus (SLE) in Brazilian population cohorts (113). Patients with aortic occlusive disease (AOD) have exhibited higher mRNA expression of NLRP1 than healthy individuals (153).
Role of NLRP3
NLRP3 has been considered the gold standard of inflammasome signaling, as many NLRP3 inhibitors are under investigation in clinical trials for coronary artery disease (169) and gout (170); more than 50 clinical studies are currently underway to elucidate the role of NLRP3 in various diseases. Cryopyrin‐associated periodic syndrome (CAPS) is a well-documented autosomal‐dominant autoinflammatory disorder caused by gain-of-function mutations in NLRP3 in pediatric patients that lead to increased plasma IL-1β levels (171, 172). An enormous amount of data shows that NLRP3 is involved in the pathophysiology of Alzheimer’s, stroke and cardiovascular diseases, asthma, gout, IBD, non-alcoholic fatty liver disease, non-alcoholic steatohepatitis, multiple sclerosis, rheumatoid arthritis, myelodysplastic syndrome, obesity-induced inflammation or insulin resistance, type-1 diabetes, oxalate-induced nephropathy, graft-versus-host disease, and silicosis (109). Inflammaging is a condition marked by higher-than-normal levels of inflammatory markers in the blood, indicating a high risk of frailty. A major mechanism of inflammaging is abnormal NLRP3 inflammasome activation (173). In type-2 diabetes patients, NLRP3 inflammasome activation is higher in myeloid cells, and NLRP3 inflammasome inhibitors might be clinically useful in treating ischemic stroke concomitant with diabetes (126, 174). The activation of NLRP3 is involved in renal disorders, such as chronic kidney disease (CKD), diabetic nephropathy (DN), and acute kidney injury (AKI), by both canonical and non-canonical pathways (175). The NLR family (NLRP1, NLRP3, NLRC4, NLRP6, and NLRP12) and their binding partners have mixed roles in the pathogenesis of a variety of cancers (162). The serum levels of alpha-synuclein (α-synuclein) and caspase-1 are lower in Parkinson’s disease (PD) patients than in healthy individuals. Cleaved α-synuclein from caspase-1 enzymatic activity can form aggregates (176). Lower levels of caspase-1 and α-synuclein in PD patients’ serum are indicative of cell aggregate formation by both. Additionally, α-synuclein itself can activate the NLRP3 inflammasome, raising cytokine levels in PD patients (177). The field of targeting NLRP3 is continuously evolving and holds immense potential for the future of anti-inflammatory drug therapy.
Role of NLRC4
Mutations in NLRC4 are associated with various autoimmune disorders. Three gain-of-function mutations (V341A, T337S, and H443P) in humans are linked to constitutive NLRC4 activation with recurrent macrophage activation syndrome (178, 179). NLRC4 activation primarily in neutrophils is enough to induce severe systemic autoinflammatory disease (180). Significant upregulation in mRNA and the protein levels of NLRC4 and NLRP3 has been found in urinary tract–infected female patients (181). Lastly, aberrant activation of NLRC4 is evident in non-alcoholic fatty liver disease (182), memory impairment in Alzheimer-like disease (146), myocardial infarction (159), and coronary stenosis (154). A genome-wide association study discovered that genetic variations in NLRC4 play vital roles in determining IL-18 levels in acute coronary syndrome patients (161).
Role of AIM2
Circulating cell-free mitochondrial DNA (ccf-mtDNA) has been detected in the serum and plasma samples of type-2 diabetes patients. Research has shown that ccf-mtDNA-mediated AIM2 inflammation activation might be one of the contributing mechanisms of chronic inflammation in diabetic patients (130). AIM2 inflammasome is hyper-activated in type-2 diabetic mice with myocardial infarction (159). Blocking AIM2 expression improves cardiac function in a streptozotocin-induced diabetic rat model by preventing caspase-1–mediated cell signaling (131). Interestingly, AIM2 expression is higher in the kidney sections of patients with DN or hypertensive sclerosis than in healthy volunteers (183). The kidneys of mice with CKD have exhibited a higher expression of AIM2 mRNA, whereas AIM2-knockout mice kidneys have shown decreased maturation of IL-1β and IL-18 (184). AIM2 expression is positively associated with the severity of SLE in human patients and mice. Inhibition of the apoptotic DNA-induced macrophagic AIM2 activation is a key in AIM2 gene silencing–ameliorated SLE symptoms in mice (117). Furthermore, AIM2 expression is upregulated in oral, cervical, and lung cancer and downregulated in colorectal and small bowel cancer (166). AIM2 expression is protective in rheumatoid arthritis patients, leading to higher IL-1β release in the absence of AIM2 (185).
Cannabinoids, Inflammasomes, and Human Diseases
So far, we have discussed the anti-inflammatory potential of various cannabinoids and the close association of inflammasomes with chronic inflammatory disorders. Several studies in the last few decades have suggested the potential of cannabinoids to modulate the inflammasome pathway. Below, we discuss research publications that established a mechanistic link between cannabinoids and inflammasome-related human diseases (Tables 2 and 3).
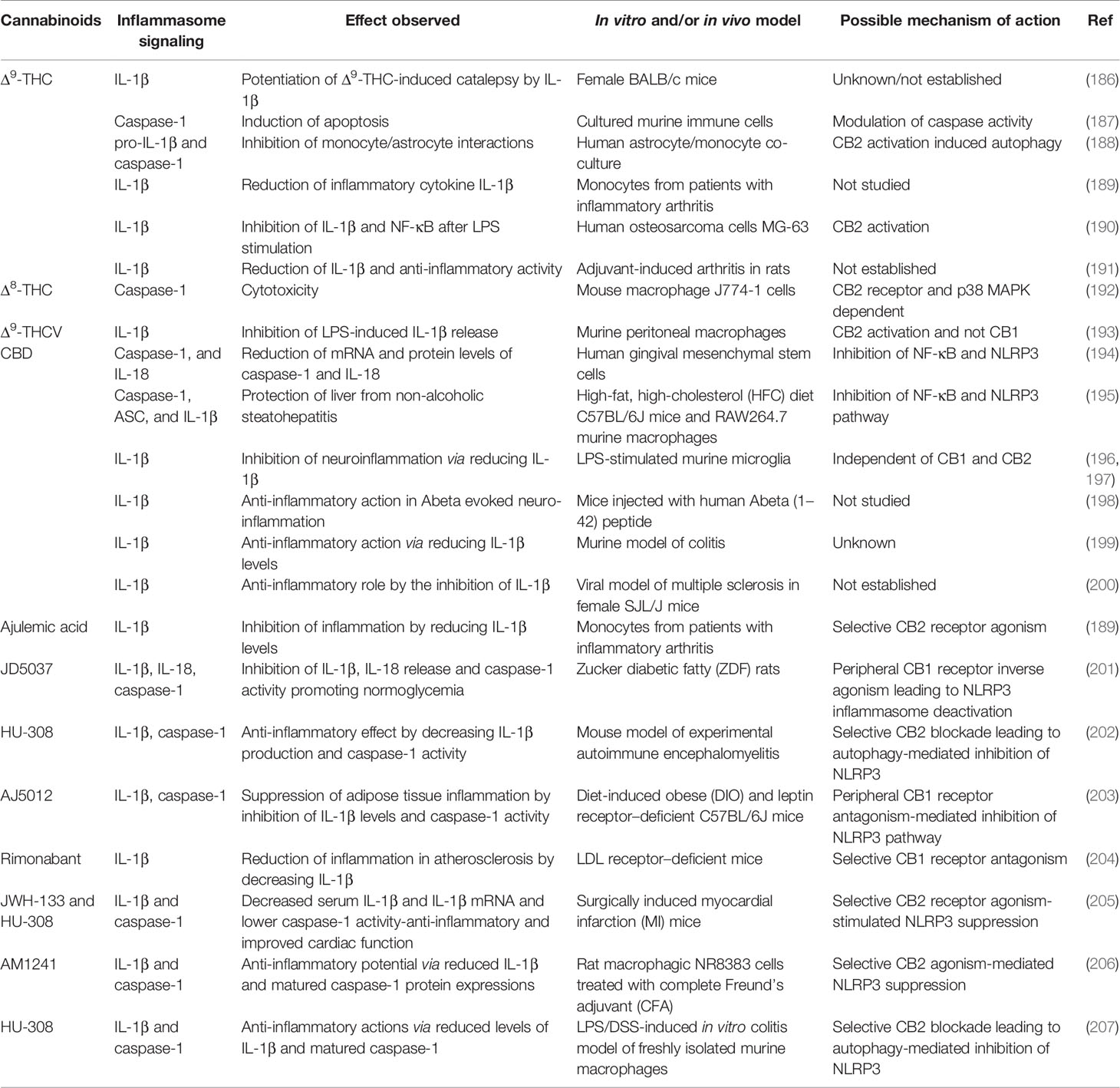
Table 2 Effects of cannabinoids on inflammasome proteins.
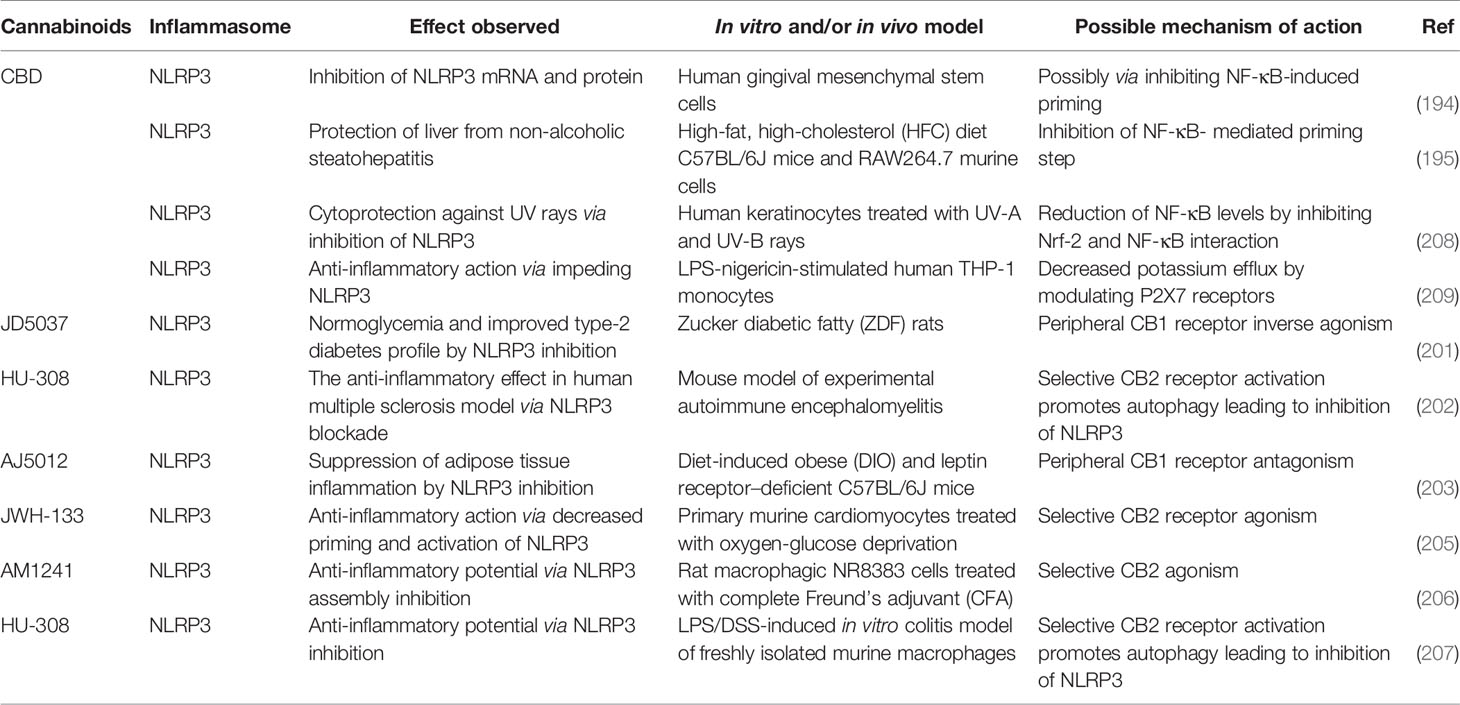
Table 3 Effects of cannabinoids on inflammasomes.
Tetrahydrocannabinol (THC) and Analogs
The first reports on the effect of Δ9 THC on IL-1β (186) and caspase-1 (187) were published in the 1990s. Δ9 THC has been found to reduce the levels of pro-IL-1β and inflammasome-induced caspase-1 activation in human astrocyte-monocyte co-culture in vitro (188). In these cells, Δ9 THC inhibited caspase-1 activity, as shown by a reduction in IL-1β levels at a concentration as low as 0.5 μM. The authors confirmed that the CB2R activation-mediated induction of autophagy was the best possible mechanism by which Δ9 THC was inhibiting inflammasome activation, as both Δ9 THC and JWH-015 (selective CB2 agonists) showed similar results (188). Δ8 THC, an isomer of Δ9 THC, was reported to induce cell death via a caspase-1–dependent pathway in mouse macrophages, activating CB2R followed by the activation of p38 MAPK (192). The effect of Δ9 THC on reducing IL-1β mRNA and protein levels was comparable to ajulemic acid, a novel CB2 agonist, in monocytes isolated from patients with inflammatory arthritis (189). In another study, Δ9 THC was able to reduce IL-1β and NF-κB levels via CB2R activation in a human osteosarcoma cell line after LPS stimulation (190). Δ9-tetrahydrocannabivarin (THCV) was also shown to inhibit IL-1β levels in LPS-challenged murine macrophages (193). Feeding Δ9 THC mixed with sesame oil orally to rats with chemically induced rheumatoid arthritis (RA) for 21 days significantly reduced IL-1β concentration to baseline, suggesting the possible inhibition of inflammasomes as a promising target in RA (191). Although Δ9 THC displayed a significant effect on the inflammasome pathway, direct action on inflammasome sensor proteins has not yet been reported. It has been pointed out that a Ca2+ channel, TRPV2, is activated by Δ9 THC in myeloid cells and that TRPV2 stimulation leads to NLRP3 inflammasome activation, but no studies have confirmed this (210).
Cannabidiol (CBD)
The first report on the direct effect of CBD on the inflammasome came in 2016 from a group of Italian researchers (194). They treated human gingival mesenchymal stem cells (hGMSCs) for 24 h with CBD (5 μM) and performed gene expression analysis and immunocytochemistry. They discovered that CBD-treated hGMSCs suppressed NLRP3, caspase-1, and IL-18 at the gene and protein levels and inhibited NF-κB. As NF-κB is involved in the priming of the NLRP3 inflammasome, the CBD treatment–induced inactive state of the NLRP3 inflammasome in hGMSCs (194) suggested that CBD-treated gingival stem cells were more immunocompetent, avoiding the risk of inflammatory reactions and promoting survival. Mice fed with a high-fat, high-cholesterol diet (HFC) for 8 weeks showed significantly higher expressions of NLRP3 inflammasome pathway proteins (NLRP3, ASC, IL-1β, and caspase-1) in the liver; these proteins were significantly attenuated by simultaneous treatment with CBD (5 mg/kg/day for 8 weeks). Similarly, the phosphorylation of NF-κB was significantly reduced in the liver of CBD-treated HFC mice compared to the non-treated group, corroborating the role of NF-κB in priming the NLRP3 inflammasome. To further confirm the role of the inflammasome in liver inflammation, the authors studied the effect of CBD on an LPS + ATP treated mouse macrophage cell line, confirming with in vivo data that the expressions of NLRP3, ASC, IL-1β, NF-κB, and caspase-1 were lower in CBD-treated cells (195). Mouse microglial cells treated with LPS to simulate neuroinflammatory conditions exhibited a robust activation of pro-inflammatory cytokine repertoire, and CBD (1–10 μM) was able to suppress the secretion of IL-1β and inhibit the NF-κB signaling pathway (196). A similar reduction in the secretion of IL-1β by CBD (10 μM) was also reported by another independent report (197). In the in vitro skin inflammation model, human keratinocytes were treated with ultraviolet (UV) rays A and B (UVA and UVB) and treated with CBD (1 μM) for 24 h. CBD inhibited protein-protein interaction between nuclear factor erythroid 2-related factor 2 (Nrf2) and NF-κB in UVA- and UVB-treated skin cells. CBD increased NRF2 expression, leading to decreased ROS, which in turn may have partially suppressed NLRP3 inflammasome activation by reducing NF-κB levels (208). Aβ-induced neurotoxicity (198) and the severity of inflammatory colitis (199) were significantly suppressed by CBD treatment in mice partly due to inhibiting the expression and release of IL-1β. A significant reduction of IL-1β by CBD treatment in a murine viral model of multiple sclerosis was also shown, suggesting its role in combating inflammation in multiple sclerosis (200). Recently, CBD was shown to inhibit NLRP3 inflammasome by reducing K+ efflux by binding to the P2X7 receptor in human monocytes (209). Remarkably, the fact that only CBD (non-psychoactive), not THC (psychoactive), was found to inhibit NF-κB signaling (197), coupled with the direct proven inhibitory action of CBD on inflammasomes (195) and downstream proteins, indicates the incomparable potential of CBD as an inflammasome-inhibitory drug target.
Synthetic Cannabinoids
Synthetic cannabinoids often act by modulating the action of endocannabinoids on CB1 and CB2 receptors to exert their anti-inflammatory effects. Endocannabinoids mediate insulin resistance by activating peripheral CB1R. Beta-cell failure in Zucker diabetic fatty (ZDF) rats was not associated with CB1R in beta cells; but rather, macrophages infiltrating the pancreas activated the NLRP3 inflammasome machinery. Macrophage-specific knockdown and/or peripheral blocking of CB1R via the non-CNS-penetrant CB1R inverse agonist JD5037 improved the pathology of type-2 diabetes and re-established normoglycemia by reducing the expression of cardinal NLRP3 inflammasome proteins (201). However, CB1R has detrimental effects on beta-cell function, and its activation promotes islet inflammation under pathological insults. Beta-cell-specific CB1R gene knockout increased insulin secretion and cAMP levels in islets. High-fat/high-sugar-induced inflammation was attenuated significantly by the absence of CB1R in beta cells via the reduction of ROS and suppression of NLRP3 inflammasome activation (211). Contrary to the protective role of CB2R in diabetic inflammation, in a mouse model of experimental autoimmune encephalomyelitis (EAE) (an experimental model for human multiple sclerosis), CB2R mRNA and NLRP3 protein expressions were significantly higher with unchanged CB1R mRNA expression (212). The authors did not study the protein expression of CB1R and CB2R in this report, but using the same multiple sclerosis model, another group of researchers showed an exacerbated NLRP3 inflammasome response in CB2R knockout mice and amelioration of the response in wild-type mice by selective CB2R activation by HU‐308 (202). It was discovered that CB2R activation by HU‐308 induces autophagy in mouse microglial cells, inhibiting NLRP3 activation. The discrepancy in the results may be due to the use of only female mice and not analyzing cannabinoid receptors at the protein level (212). An antagonist of peripheral CB1R, AJ5012, prevents adipose tissue inflammation in leptin receptor–deficient and diet-induced obese (DIO) mice by inhibiting NLRP3 inflammasome signaling. Reductions in the protein expressions of caspase-1, IL-1β, and NLRP3, along with caspase-1 activity, were noticed in the presence of AJ5012 in DIO mice. AEA-induced increases in the expressions of NLRP3, CB1R, and IL-1β in murine macrophage cells were also significantly attenuated by AJ5012 (203, 213). These data indicate the critical role of the NLRP3 inflammasome in adipose tissue inflammation via peripheral CB1R signaling. Rimonabant, a selective CB1R antagonist, inhibits the development of atherosclerosis in low-density lipoprotein (LDL) receptor–deficient mice, partially by reducing IL-1β–mediated pro-inflammatory gene expression (204). Myocardial infarction (MI) is a major cardiovascular event with a high mortality rate in post-MI heart failure. A selective CB2R agonist, JWH-133, improved heart function in a surgically induced MI mice model. JWH-133 treatment reduced serum IL-1β and IL-1β mRNA expressions even at a 1 mg/kg concentration. The administration of JWH-133 at a 10 mg/kg dose significantly diminished the priming and activation of the NLRP3 inflammasome, as shown by levels of inflammasome protein expression (205). Electroacupuncture (EA) attenuated inflammatory pain induced by complete Freund’s adjuvant (CFA) in rats by blocking NLRP3 activation in skin macrophages. This effect was largely attributed to CB2R, as CB2R-knockout mice exhibited a loss of EA effects on pain. In rat macrophagic cell lines, CFA- and LPS+ATP–induced NLRP3 activation was significantly inhibited by the selective CB2R agonist AM1241 (206). In freshly isolated mice macrophages, the CB2R selective agonist HU-308 significantly blocked the expression of NLRP3 inflammasome assembly proteins in an LPS/DSS-induced in vitro colitis model (207). Selective inhibitors of CB1R and activators of CB2R have shown tremendous potential in alleviating inflammatory disorders partly by affecting NLRP3 inflammasome assembly and activation.
Terpenoids and flavonoids are abundantly present in cannabis extracts, and both molecule groups exhibit anti-inflammatory activities. Many terpenoids and flavonoids are inflammasome inhibitors in general and are reviewed extensively elsewhere (214–217), a subject beyond the scope of this review.
Relevance to The Coronavirus Disease 2019 (COVID-19) PANDEMIC
The ongoing COVID-19 outbreak is a global pandemic caused by a novel coronavirus named “severe acute respiratory syndrome coronavirus 2” (SARS-CoV-2). The angiotensin-converting enzyme 2 (ACE2), a metallopeptidase, is a known functional receptor for coronaviruses, and their surface spike glycoproteins (S) bind physically to ACE2 (218–220). A high expression of ACE2 is correlated to innate and acquired immune response, cytokine secretion, and enhanced inflammatory response in COVID-19 patients. A clinical study in Wuhan pointed out that the levels of IL-1β, IL-10, and IL-8 were significantly higher in critically ill patients with SARS-CoV-2 infection, indicating a cytokine-mediated inflammatory response (221). Recently, it was established that the SARS-CoV open reading frame (ORF)-8b activates the NLRP3 inflammasome affecting innate immunity (222) and SARS-CoV viroporin 3a protein independently activates NLRP3 inflammasome in macrophages isolated from adult mice (223). Very recently published reviews strongly suggest that SARS-CoV-2 could directly activate NLRP3 inflammasome and NLRP3 activation could be a potential drug target in the treatment of COVID-19 (224, 225). In our laboratory, we established several novel high-CBD C. sativa extracts that significantly inhibited the expression of ACE2, entry point of the SARS-CoV-2 (226). The reason for choosing high-CBD cannabis extracts was to avoid psychoactive side-effects of Δ9-THC and to avoid CB1 agonism-mediated pathologic changes observed in the pulmonary tissues (227). Therefore, with the established evidence suggesting the role of cannabinoids as key regulators of inflammasome signaling, the vital cannabinoid moieties (CBD and THC) might be beneficial in alleviating the inflammatory aspects of COVID-19 by blocking inflammasome signaling.
Conclusions and Clinical Relevance
Inflammation is a crucial phenomenon in understanding the pathophysiology of a variety of inflammatory disorders, and many anti-inflammatory antibodies are important treatment options for moderate-severe inflammatory diseases. The contribution of inflammasomes in the regulation of human disorders has been emphasized in research within the last few decades. However, no inflammasome-targeted therapy is currently approved for human use.
Cannabis has been shown to possess anti-inflammatory effects owing to its constituents, cannabinoids and terpenoids. New evidence is accumulating on the potential inhibitory action of cannabinoids on NLRP3 and other inflammasomes leading to their potent anti-inflammatory effects. On the other hand, cannabinoids with CB1 receptor agonist activity exhibit pro-inflammatory effects by inflammasome activation via CB1 agonism. The summarized reports here showed in vitro and in vivo data on cannabinoids modulating inflammasome activity and proving beneficial in reducing the pathogenicity of chronic inflammatory diseases. Cannabinoids also target crucial proteins involved in the inflammasome signaling, including NF-κB, IL-1β, etc.
The exact molecular mechanisms by which cannabinoids modulate inflammasome signaling have not been investigated completely, nevertheless, the current evidence supports their importance as promising therapeutic targets to regulate inflammasome signaling (Figure 3). Targeted inhibition of inflammasome by cannabinoids may prove beneficial over the global inhibition of cytokines, which increases the chances of infection as a side effect. Overall, cannabinoids hold a great promise as additional therapeutics to support the current treatment of chronic inflammatory diseases, along with COVID-19, however it should be weighed against pro-inflammatory actions mediated by CB1-agonism. Hence, cannabinoids with CB1 receptor antagonist and CB2 receptor agonist activity, for instance cannabidiol, should be considered for future research.
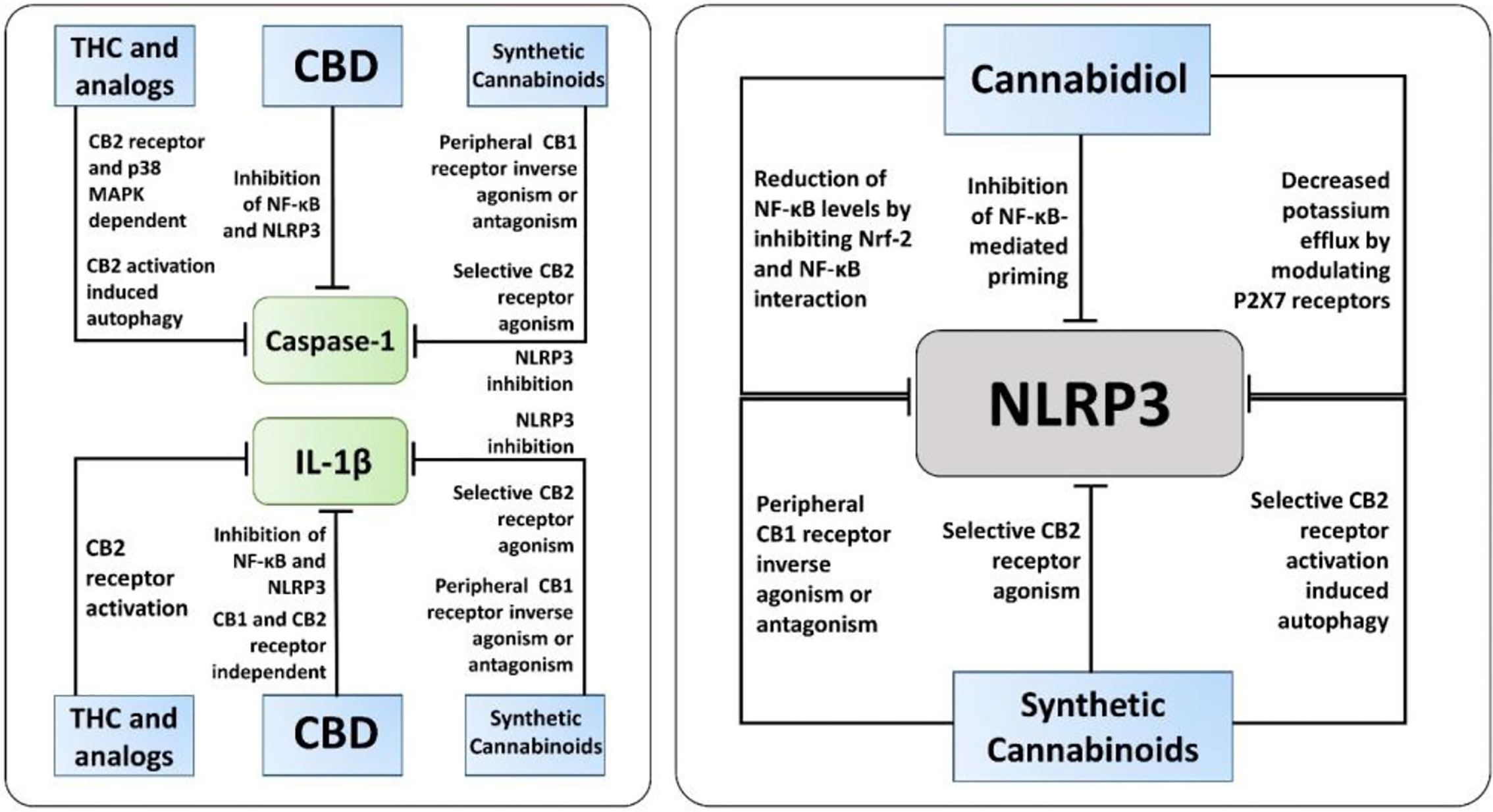
Figure 3 The known molecular pathways of inhibition of Caspase-1, IL-1β, and NLRP3 by cannabinoids. Along with NLRP3 activation, cannabidiol (CBD), synthetic cannabinoids, and Δ9-tetrahydrocannabinol (THC) and its analogs inhibit caspase-1, IL-1β, and IL-18 via different mechanisms independent of inflammasome activation. Notably, only CBD and synthetic cannabinoids, not THC and its analogs, have been reported to have a direct inhibitory action on NLRP3 activation.
Author Contributions
Conceptualization: SS. Writing, reviewing, and editing: SS, IK, and OK. All authors contributed to the article and approved the submitted version.
Funding
Research was supported by the CIHR and MITACs grants to IK and OK.
Conflict of Interest
IK and OK are involved with Pathway RX, a biotech start-up company focused on medical cannabis, albeit this start-up has no bearing on the materials summarized in the current review.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Buchmann K. Evolution of Innate Immunity: Clues from Invertebrates via Fish to Mammals. Front Immunol (2014) 5:459. doi: 10.3389/fimmu.2014.00459
2. Cooper MD, Alder MN. The evolution of adaptive immune systems. Cell (2006) 124(4):815–22. doi: 10.1016/j.cell.2006.02.001
3. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
4. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol (2020) 20(2):95–112. doi: 10.1038/s41577-019-0215-7
5. Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol (2010) 28:321–42. doi: 10.1146/annurev-immunol-030409-101311
6. Gray EE, Winship D, Snyder JM, Child SJ, Geballe AP, Stetson DB. The AIM2-like Receptors Are Dispensable for the Interferon Response to Intracellular DNA. Immunity (2016) 45(2):255–66. doi: 10.1016/j.immuni.2016.06.015
7. Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol (2011) 29:707–35. doi: 10.1146/annurev-immunol-031210-101405
8. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res (2015) 25(12):1285–98. doi: 10.1038/cr.2015.139
9. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
10. Howrylak JA, Nakahira K. Inflammasomes: Key Mediators of Lung Immunity. Annu Rev Physiol (2017) 79:471–94. doi: 10.1146/annurev-physiol-021115-105229
11. Mantovani A, Dinarello CA, Molgora M, Garlanda C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity (2019) 50(4):778–95. doi: 10.1016/j.immuni.2019.03.012
12. Rathinam VA, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell (2016) 165(4):792–800. doi: 10.1016/j.cell.2016.03.046
13. Aizpurua-Olaizola O, Soydaner U, Ozturk E, Schibano D, Simsir Y, Navarro P, et al. Evolution of the Cannabinoid and Terpene Content during the Growth of Cannabis sativa Plants from Different Chemotypes. J Nat Prod (2016) 79(2):324–31. doi: 10.1021/acs.jnatprod.5b00949
14. Allan GM, Finley CR, Ton J, Perry D, Ramji J, Crawford K, et al. Systematic review of systematic reviews for medical cannabinoids: Pain, nausea and vomiting, spasticity, and harms. Can Fam Physician (2018) 64(2):e78–94.
15. Allan GM, Ramji J, Perry D, Ton J, Beahm NP, Crisp N, et al. Simplified guideline for prescribing medical cannabinoids in primary care. Can Fam Physician (2018) 64(2):111–20.
16. Vozoris NT, Pequeno P, Li P, Austin PC, Stephenson AL, O’Donnell DE, et al. Morbidity and mortality associated with prescription cannabinoid drug use in COPD. Thorax (2020) 76(1):29–36. doi: 10.1136/thoraxjnl-2020-215346
17. Finlay DB, Sircombe KJ, Nimick M, Jones C, Glass M. Terpenoids From Cannabis Do Not Mediate an Entourage Effect by Acting at Cannabinoid Receptors. Front Pharmacol (2020) 11:359. doi: 10.3389/fphar.2020.00359
18. Santiago M, Sachdev S, Arnold JC, McGregor IS, Connor M. Absence of Entourage: Terpenoids Commonly Found in Cannabis sativa Do Not Modulate the Functional Activity of Delta(9)-THC at Human CB1 and CB2 Receptors. Cannabis Cannabinoid Res (2019) 4(3):165–76. doi: 10.1089/can.2019.0016
19. Nagarkatti P, Pandey R, Rieder SA, Hegde VL, Nagarkatti M. Cannabinoids as novel anti-inflammatory drugs. Future Med Chem (2009) 1(7):1333–49. doi: 10.4155/fmc.09.93
20. Nichols JM, Kaplan BLF. Immune Responses Regulated by Cannabidiol. Cannabis Cannabinoid Res (2020) 5(1):12–31. doi: 10.1089/can.2018.0073
21. Zurier RB, Burstein SH. Cannabinoids, inflammation, and fibrosis. FASEB J (2016) 30(11):3682–9. doi: 10.1096/fj.201600646R
22. Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol (2012) 4(3):a006049. doi: 10.1101/cshperspect.a006049
23. Groslambert M, Py BF. Spotlight on the NLRP3 inflammasome pathway. J Inflammation Res (2018) 11:359–74. doi: 10.2147/JIR.S141220
24. Zou S, Kumar U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int J Mol Sci (2018) 19(3):833. doi: 10.3390/ijms19030833
25. Gaoni Y, Mechoulam R. Isolation, Structure, and Partial Synthesis of an Active Constituent of Hashish. J Am Chem Soc (1964) 86(8):1646–7. doi: 10.1021/ja01062a046
26. Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature (1990) 346(6284):561–4. doi: 10.1038/346561a0
27. Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature (1993) 365(6441):61–5. doi: 10.1038/365061a0
28. Kovalchuk O, Kovalchuk I. Cannabinoids as anticancer therapeutic agents. Cell Cycle (2020) 19(9):961–89. doi: 10.1080/15384101.2020.1742952
29. Cristino L, Bisogno T, Di Marzo V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat Rev Neurol (2020) 16(1):9–29. doi: 10.1038/s41582-019-0284-z
30. Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science (1992) 258(5090):1946–9. doi: 10.1126/science.1470919
31. Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol (1995) 50(1):83–90. doi: 10.1016/0006-2952(95)00109-D
32. Castillo PE, Younts TJ, Chavez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron (2012) 76(1):70–81. doi: 10.1016/j.neuron.2012.09.020
33. Khan MI, Sobocinska AA, Czarnecka AM, Krol M, Botta B, Szczylik C. The Therapeutic Aspects of the Endocannabinoid System (ECS) for Cancer and their Development: From Nature to Laboratory. Curr Pharm Des (2016) 22(12):1756–66. doi: 10.2174/1381612822666151211094901
34. Pacher P, Kunos G. Modulating the endocannabinoid system in human health and disease–successes and failures. FEBS J (2013) 280(9):1918–43. doi: 10.1111/febs.12260
35. Katchan V, David P, Shoenfeld Y. Cannabinoids and autoimmune diseases: A systematic review. Autoimmun Rev (2016) 15(6):513–28. doi: 10.1016/j.autrev.2016.02.008
36. Shao Z, Yin J, Chapman K, Grzemska M, Clark L, Wang J, et al. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature (2016) 540(7634):602–6. doi: 10.1038/nature20613
37. Li X, Hua T, Vemuri K, Ho JH, Wu Y, Wu L, et al. Crystal Structure of the Human Cannabinoid Receptor CB2. Cell (2019) 176(3):459–67 e13. doi: 10.1016/j.cell.2018.12.011
38. Hourani W, Alexander SPH. Cannabinoid ligands, receptors and enzymes: Pharmacological tools and therapeutic potential. Brain Neurosci Adv (2018) 2:2398212818783908. doi: 10.1177/2398212818783908
39. Maccarrone M, Bab I, Biro T, Cabral GA, Dey SK, Di Marzo V, et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol Sci (2015) 36(5):277–96. doi: 10.1016/j.tips.2015.02.008
40. Bie B, Wu J, Foss JF, Naguib M. An overview of the cannabinoid type 2 receptor system and its therapeutic potential. Curr Opin Anaesthesiol (2018) 31(4):407–14. doi: 10.1097/ACO.0000000000000616
41. Atwood BK, Mackie K. CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol (2010) 160(3):467–79. doi: 10.1111/j.1476-5381.2010.00729.x
42. Kendall DA, Yudowski GA. Cannabinoid Receptors in the Central Nervous System: Their Signaling and Roles in Disease. Front Cell Neurosci (2016) 10:294. doi: 10.3389/fncel.2016.00294
43. Viscomi MT, Oddi S, Latini L, Pasquariello N, Florenzano F, Bernardi G, et al. Selective CB2 receptor agonism protects central neurons from remote axotomy-induced apoptosis through the PI3K/Akt pathway. J Neurosci (2009) 29(14):4564–70. doi: 10.1523/JNEUROSCI.0786-09.2009
44. Demuth DG, Molleman A. Cannabinoid signalling. Life Sci (2006) 78(6):549–63. doi: 10.1016/j.lfs.2005.05.055
45. Jarrahian A, Watts VJ, Barker EL. D2 dopamine receptors modulate Galpha-subunit coupling of the CB1 cannabinoid receptor. J Pharmacol Exp Ther (2004) 308(3):880–6. doi: 10.1124/jpet.103.057620
46. Saroz Y, Kho DT, Glass M, Graham ES, Grimsey NL. Cannabinoid Receptor 2 (CB2) Signals via G-alpha-s and Induces IL-6 and IL-10 Cytokine Secretion in Human Primary Leukocytes. ACS Pharmacol Transl Sci (2019) 2(6):414–28. doi: 10.1021/acsptsci.9b00049
47. Ibsen MS, Connor M, Glass M. Cannabinoid CB1 and CB2 Receptor Signaling and Bias. Cannabis Cannabinoid Res (2017) 2(1):48–60. doi: 10.1089/can.2016.0037
48. Reekie TA, Scott MP, Kassiou M. The evolving science of phytocannabinoids. Nat Rev Chem (2017) 2(1):0101. doi: 10.1038/s41570-017-0101
49. Thomas A, Baillie GL, Phillips AM, Razdan RK, Ross RA, Pertwee RG. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol (2007) 150(5):613–23. doi: 10.1038/sj.bjp.0707133
50. McPartland JM, Duncan M, Di Marzo V, Pertwee RG. Are cannabidiol and Delta(9) -tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br J Pharmacol (2015) 172(3):737–53. doi: 10.1111/bph.12944
51. Donvito G, Nass SR, Wilkerson JL, Curry ZA, Schurman LD, Kinsey SG, et al. The Endogenous Cannabinoid System: A Budding Source of Targets for Treating Inflammatory and Neuropathic Pain. Neuropsychopharmacology (2018) 43(1):52–79. doi: 10.1038/npp.2017.204
52. Klein TW, Cabral GA. Cannabinoid-induced immune suppression and modulation of antigen-presenting cells. J Neuroimmune Pharmacol (2006) 1(1):50–64. doi: 10.1007/s11481-005-9007-x
53. Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol (2005) 5(5):400–11. doi: 10.1038/nri1602
54. Nascimento Menezes PM, Valenca Pereira EC, Gomes da Cruz Silva ME, da Silva BAO, de Souza Duarte Filho LAM, de Lima Araujo TC, et al. Cannabis and Cannabinoids on Treatment of Inflammation: A Patient Review. Recent Pat Biotechnol (2019) 13(4):256–67. doi: 10.2174/1872208313666190618124345
55. Drew L. Cannabis research round-up. Nature (2019) 572(7771):S20–S1. doi: 10.1038/d41586-019-02532-5
56. Motwani MP, Bennett F, Norris PC, Maini AA, George MJ, Newson J, et al. Potent Anti-Inflammatory and Pro-Resolving Effects of Anabasum in a Human Model of Self-Resolving Acute Inflammation. Clin Pharmacol Ther (2018) 104(4):675–86. doi: 10.1002/cpt.980
57. McGuire P, Robson P, Cubala WJ, Vasile D, Morrison PD, Barron R, et al. Cannabidiol (CBD) as an Adjunctive Therapy in Schizophrenia: A Multicenter Randomized Controlled Trial. Am J Psychiatry (2018) 175(3):225–31. doi: 10.1176/appi.ajp.2017.17030325
58. Reithmeier D, Tang-Wai R, Seifert B, Lyon AW, Alcorn J, Acton B, et al. The protocol for the Cannabidiol in children with refractory epileptic encephalopathy (CARE-E) study: a phase 1 dosage escalation study. BMC Pediatr (2018) 18(1):221. doi: 10.1186/s12887-018-1191-y
59. Herlopian A, Hess EJ, Barnett J, Geffrey AL, Pollack SF, Skirvin L, et al. Cannabidiol in treatment of refractory epileptic spasms: An open-label study. Epilepsy Behav (2020) 106:106988. doi: 10.1016/j.yebeh.2020.106988
60. Hess EJ, Moody KA, Geffrey AL, Pollack SF, Skirvin LA, Bruno PL, et al. Cannabidiol as a new treatment for drug-resistant epilepsy in tuberous sclerosis complex. Epilepsia (2016) 57(10):1617–24. doi: 10.1111/epi.13499
61. Burstein S. Cannabidiol (CBD) and its analogs: a review of their effects on inflammation. Bioorg Med Chem (2015) 23(7):1377–85. doi: 10.1016/j.bmc.2015.01.059
62. Hegde VL, Singh UP, Nagarkatti PS, Nagarkatti M. Critical Role of Mast Cells and Peroxisome Proliferator-Activated Receptor gamma in the Induction of Myeloid-Derived Suppressor Cells by Marijuana Cannabidiol In Vivo. J Immunol (2015) 194(11):5211–22. doi: 10.4049/jimmunol.1401844
63. Atalay S, Jarocka-Karpowicz I, Skrzydlewska E. Antioxidative and Anti-Inflammatory Properties of Cannabidiol. Antioxidants (Basel) (2019) 9(1):21. doi: 10.3390/antiox9010021
64. Pellati F, Borgonetti V, Brighenti V, Biagi M, Benvenuti S, Corsi L. Cannabis sativa L. and Nonpsychoactive Cannabinoids: Their Chemistry and Role against Oxidative Stress, Inflammation, and Cancer. BioMed Res Int (2018) 2018:1691428. doi: 10.1155/2018/1691428
65. De Petrocellis L, Orlando P, Moriello AS, Aviello G, Stott C, Izzo AA, et al. Cannabinoid actions at TRPV channels: effects on TRPV3 and TRPV4 and their potential relevance to gastrointestinal inflammation. Acta Physiol (Oxf) (2012) 204(2):255–66. doi: 10.1111/j.1748-1716.2011.02338.x
66. Borrelli F, Fasolino I, Romano B, Capasso R, Maiello F, Coppola D, et al. Beneficial effect of the non-psychotropic plant cannabinoid cannabigerol on experimental inflammatory bowel disease. Biochem Pharmacol (2013) 85(9):1306–16. doi: 10.1016/j.bcp.2013.01.017
67. Gu Z, Singh S, Niyogi RG, Lamont GJ, Wang H, Lamont RJ, et al. Marijuana-Derived Cannabinoids Trigger a CB2/PI3K Axis of Suppression of the Innate Response to Oral Pathogens. Front Immunol (2019) 10:2288. doi: 10.3389/fimmu.2019.02288
68. Takeda S, Misawa K, Yamamoto I, Watanabe K. Cannabidiolic acid as a selective cyclooxygenase-2 inhibitory component in cannabis. Drug Metab Dispos (2008) 36(9):1917–21. doi: 10.1124/dmd.108.020909
69. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140(6):821–32. doi: 10.1016/j.cell.2010.01.040
70. Sharma D, Kanneganti TD. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol (2016) 213(6):617–29. doi: 10.1083/jcb.201602089
71. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol (2014) 15(8):727–37. doi: 10.1038/ni.2913
72. Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez-Cuellar E, Shen Y, et al. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol (2013) 14(12):1247–55. doi: 10.1038/ni.2749
73. Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ (2007) 14(1):10–22. doi: 10.1038/sj.cdd.4402038
74. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol (2006) 8(11):1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
75. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
76. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
77. Diamond CE, Khameneh HJ, Brough D, Mortellaro A. Novel perspectives on non-canonical inflammasome activation. Immunotargets Ther (2015) 4:131–41. doi: 10.2147/ITT.S57976
78. Liu T, Yamaguchi Y, Shirasaki Y, Shikada K, Yamagishi M, Hoshino K, et al. Single-cell imaging of caspase-1 dynamics reveals an all-or-none inflammasome signaling response. Cell Rep (2014) 8(4):974–82. doi: 10.1016/j.celrep.2014.07.012
79. Karki R, Man SM, Malireddi RKS, Gurung P, Vogel P, Lamkanfi M, et al. Concerted activation of the AIM2 and NLRP3 inflammasomes orchestrates host protection against Aspergillus infection. Cell Host Microbe (2015) 17(3):357–68. doi: 10.1016/j.chom.2015.01.006
80. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10(2):417–26. doi: 10.1016/S1097-2765(02)00599-3
81. Muehlbauer SM, Evering TH, Bonuccelli G, Squires RC, Ashton AW, Porcelli SA, et al. Anthrax lethal toxin kills macrophages in a strain-specific manner by apoptosis or caspase-1-mediated necrosis. Cell Cycle (2007) 6(6):758–66. doi: 10.4161/cc.6.6.3991
82. Terra JK, Cote CK, France B, Jenkins AL, Bozue JA, Welkos SL, et al. Cutting edge: resistance to Bacillus anthracis infection mediated by a lethal toxin sensitive allele of Nalp1b/Nlrp1b. J Immunol (2010) 184(1):17–20. doi: 10.4049/jimmunol.0903114
83. Hsu LC, Ali SR, McGillivray S, Tseng PH, Mariathasan S, Humke EW, et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci U S A (2008) 105(22):7803–8. doi: 10.1073/pnas.0802726105
84. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21(7):677–87. doi: 10.1038/nm.3893
85. Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem (2012) 287(43):36617–22. doi: 10.1074/jbc.M112.407130
86. He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature (2016) 530(7590):354–7. doi: 10.1038/nature16959
87. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol (2008) 9(8):857–65. doi: 10.1038/ni.1636
88. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis (2019) 10(2):128. doi: 10.1038/s41419-019-1413-8
89. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12(3):222–30. doi: 10.1038/ni.1980
90. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature (2004) 430(6996):213–8. doi: 10.1038/nature02664
91. von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol (2013) 31:73–106. doi: 10.1146/annurev-immunol-032712-095944
92. Man SM, Ekpenyong A, Tourlomousis P, Achouri S, Cammarota E, Hughes K, et al. Actin polymerization as a key innate immune effector mechanism to control Salmonella infection. Proc Natl Acad Sci U S A (2014) 111(49):17588–93. doi: 10.1073/pnas.1419925111
93. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458(7237):514–8. doi: 10.1038/nature07725
94. Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol (2010) 11(5):395–402. doi: 10.1038/ni.1864
95. Lammert CR, Frost EL, Bellinger CE, Bolte AC, McKee CA, Hurt ME, et al. AIM2 inflammasome surveillance of DNA damage shapes neurodevelopment. Nature (2020) 580(7805):647–52. doi: 10.1038/s41586-020-2174-3
96. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science (2013) 341(6151):1250–3. doi: 10.1126/science.1240988
97. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (2013) 341(6151):1246–9. doi: 10.1126/science.1240248
98. Levandowski CB, Mailloux CM, Ferrara TM, Gowan K, Ben S, Jin Y, et al. NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1beta processing via the NLRP1 inflammasome. Proc Natl Acad Sci U S A (2013) 110(8):2952–6. doi: 10.1073/pnas.1222808110
99. Lazare K. Addison’s Disease and Possible Cannabis Withdrawal Syndrome Presenting as an Eating Disorder in a Thirty-Year-Old Female. Case Rep Endocrinol (2017) 2017:4096021. doi: 10.1155/2017/4096021
100. Zhong FL, Mamai O, Sborgi L, Boussofara L, Hopkins R, Robinson K, et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell (2016) 167(1):187–202 e17. doi: 10.1016/j.cell.2016.09.001
101. Ekman AK, Verma D, Fredrikson M, Bivik C, Enerback C. Genetic variations of NLRP1: susceptibility in psoriasis. Br J Dermatol (2014) 171(6):1517–20. doi: 10.1111/bjd.13178
102. Sheriff T, Lin MJ, Dubin D, Khorasani H. The potential role of cannabinoids in dermatology. J Dermatolog Treat (2019) 31(8):839–45. doi: 10.1080/09546634.2019.1675854
103. Verma D, Bivik C, Farahani E, Synnerstad I, Fredrikson M, Enerback C, et al. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment Cell Melanoma Res (2012) 25(4):506–13. doi: 10.1111/j.1755-148X.2012.01008.x
104. Lamkanfi M, Kanneganti TD. Nlrp3: an immune sensor of cellular stress and infection. Int J Biochem Cell Biol (2010) 42(6):792–5. doi: 10.1016/j.biocel.2010.01.008
105. de Koning HD, Bergboer JG, van den Bogaard EH, van Vlijmen-Willems IM, Rodijk-Olthuis D, Simon A, et al. Strong induction of AIM2 expression in human epidermis in acute and chronic inflammatory skin conditions. Exp Dermatol (2012) 21(12):961–4. doi: 10.1111/exd.12037
106. Farshchian M, Nissinen L, Siljamaki E, Riihila P, Piipponen M, Kivisaari A, et al. Tumor cell-specific AIM2 regulates growth and invasion of cutaneous squamous cell carcinoma. Oncotarget (2017) 8(28):45825–36. doi: 10.18632/oncotarget.17573
107. Tye H, Yu CH, Simms LA, de Zoete MR, Kim ML, Zakrzewski M, et al. NLRP1 restricts butyrate producing commensals to exacerbate inflammatory bowel disease. Nat Commun (2018) 9(1):3728. doi: 10.1038/s41467-018-06125-0
108. Carvalho ACA, Souza GA, Marqui SV, Guiguer EL, Araujo AC, Rubira CJ, et al. Cannabis and Canabidinoids on the Inflammatory Bowel Diseases: Going Beyond Misuse. Int J Mol Sci (2020) 21(8):2940. doi: 10.3390/ijms21082940
109. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov (2018) 17(8):588–606. doi: 10.1038/nrd.2018.97
110. Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet (2009) 41(1):71–6. doi: 10.1038/ng.285
111. Ratsimandresy RA, Indramohan M, Dorfleutner A, Stehlik C. The AIM2 inflammasome is a central regulator of intestinal homeostasis through the IL-18/IL-22/STAT3 pathway. Cell Mol Immunol (2017) 14(1):127–42. doi: 10.1038/cmi.2016.35
112. da Cruz HLA, Cavalcanti CAJ, de Azevedo Silva J, de Lima CAD, Fragoso TS, Barbosa AD, et al. Differential expression of the inflammasome complex genes in systemic lupus erythematosus. Immunogenetics (2020) 72(4):217–24. doi: 10.1007/s00251-020-01158-6
113. Pontillo A, Girardelli M, Kamada AJ, Pancotto JA, Donadi EA, Crovella S, et al. Polimorphisms in inflammasome genes are involved in the predisposition to systemic lupus erythematosus. Autoimmunity (2012) 45(4):271–8. doi: 10.3109/08916934.2011.637532
114. Katz-Talmor D, Kivity S, Blank M, Katz I, Perry O, Volkov A, et al. Cannabidiol Treatment in a Murine Model of Systemic Lupus Erythematosus Accelerates Proteinuria Development. Isr Med Assoc J (2018) 20(12):741–5.
115. Burstein SH. Ajulemic acid: potential treatment for chronic inflammation. Pharmacol Res Perspect (2018) 6(2):e00394. doi: 10.1002/prp2.394
116. Yu Y, Fu S, Zhang X, Wang L, Zhao L, Wan W, et al. Leptin facilitates the differentiation of Th17 cells from MRL/Mp-Fas lpr lupus mice by activating NLRP3 inflammasome. Innate Immun (2020) 26(4):294–300. doi: 10.1177/1753425919886643
117. Zhang W, Cai Y, Xu W, Yin Z, Gao X, Xiong S. AIM2 facilitates the apoptotic DNA-induced systemic lupus erythematosus via arbitrating macrophage functional maturation. J Clin Immunol (2013) 33(5):925–37. doi: 10.1007/s10875-013-9881-6
118. Sun X, Xia Y, Liu Y, Wang Y, Luo S, Lin J, et al. Polymorphisms in NLRP1 Gene Are Associated with Type 1 Diabetes. J Diabetes Res (2019) 2019:7405120. doi: 10.1155/2019/7405120
119. Soares JLS, Fernandes FP, Patente TA, Monteiro MB, Parisi MC, Giannella-Neto D, et al. Gain-of-function variants in NLRP1 protect against the development of diabetic kidney disease: NLRP1 inflammasome role in metabolic stress sensing? Clin Immunol (2018) 187:46–9. doi: 10.1016/j.clim.2017.10.003
120. Kinney GL, Akturk HK, Taylor DD, Foster NC, Shah VN. Cannabis Use Is Associated With Increased Risk for Diabetic Ketoacidosis in Adults With Type 1 Diabetes: Findings From the T1D Exchange Clinic Registry. Diabetes Care (2020) 43(1):247–9. doi: 10.2337/dc19-0365
121. Chaves YC, Genaro K, Stern CA, de Oliveira Guaita G, de Souza Crippa JA, da Cunha JM, et al. Two-weeks treatment with cannabidiol improves biophysical and behavioral deficits associated with experimental type-1 diabetes. Neurosci Lett (2020) 729:135020. doi: 10.1016/j.neulet.2020.135020
122. Hu C, Ding H, Li Y, Pearson JA, Zhang X, Flavell RA, et al. NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets. Proc Natl Acad Sci U S A (2015) 112(36):11318–23. doi: 10.1073/pnas.1513509112
123. Pontillo A, Brandao L, Guimaraes R, Segat L, Araujo J, Crovella S. Two SNPs in NLRP3 gene are involved in the predisposition to type-1 diabetes and celiac disease in a pediatric population from northeast Brazil. Autoimmunity (2010) 43(8):583–9. doi: 10.3109/08916930903540432
124. Leite JA, Pessenda G, Guerra-Gomes IC, de Santana AKM, Andre Pereira C, Ribeiro Campos Costa F, et al. The DNA Sensor AIM2 Protects against Streptozotocin-Induced Type 1 Diabetes by Regulating Intestinal Homeostasis via the IL-18 Pathway. Cells (2020) 9(4):959. doi: 10.3390/cells9040959
125. Ding S, Xu S, Ma Y, Liu G, Jang H, Fang J. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules (2019) 9(12):850. doi: 10.3390/biom9120850
126. Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes (2013) 62(1):194–204. doi: 10.2337/db12-0420
127. Zheng Y, Zhang D, Zhang L, Fu M, Zeng Y, Russell R. Variants of NLRP3 gene are associated with insulin resistance in Chinese Han population with type-2 diabetes. Gene (2013) 530(1):151–4. doi: 10.1016/j.gene.2013.07.082
128. Muniyappa R, Sable S, Ouwerkerk R, Mari A, Gharib AM, Walter M, et al. Metabolic effects of chronic cannabis smoking. Diabetes Care (2013) 36(8):2415–22. doi: 10.2337/dc12-2303
129. Ngueta G, Ndjaboue R. Lifetime marijuana use in relation to insulin resistance in lean, overweight, and obese US adults. J Diabetes (2020) 12(1):38–47. doi: 10.1111/1753-0407.12958
130. Bae JH, Jo SI, Kim SJ, Lee JM, Jeong JH, Kang JS, et al. Circulating Cell-Free mtDNA Contributes to AIM2 Inflammasome-Mediated Chronic Inflammation in Patients with Type 2 Diabetes. Cells (2019) 8(4):328. doi: 10.3390/cells8040328
131. Wang X, Pan J, Liu H, Zhang M, Liu D, Lu L, et al. AIM2 gene silencing attenuates diabetic cardiomyopathy in type 2 diabetic rat model. Life Sci (2019) 221:249–58. doi: 10.1016/j.lfs.2019.02.035
132. Yuan F, Kolb R, Pandey G, Li W, Sun L, Liu F, et al. Involvement of the NLRC4-Inflammasome in Diabetic Nephropathy. PloS One (2016) 11(10):e0164135. doi: 10.1371/journal.pone.0164135
133. Zeller T, Haase T, Muller C, Riess H, Lau D, Zeller S, et al. Molecular Characterization of the NLRC4 Expression in Relation to Interleukin-18 Levels. Circ Cardiovasc Genet (2015) 8(5):717–26. doi: 10.1161/CIRCGENETICS.115.001079
134. Li F, Guo N, Ma Y, Ning B, Wang Y, Kou L. Inhibition of P2X4 suppresses joint inflammation and damage in collagen-induced arthritis. Inflammation (2014) 37(1):146–53. doi: 10.1007/s10753-013-9723-y
135. Lowin T, Schneider M, Pongratz G. Joints for joints: cannabinoids in the treatment of rheumatoid arthritis. Curr Opin Rheumatol (2019) 31(3):271–8. doi: 10.1097/BOR.0000000000000590
136. Guo C, Fu R, Wang S, Huang Y, Li X, Zhou M, et al. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin Exp Immunol (2018) 194(2):231–43. doi: 10.1111/cei.13167
137. Choulaki C, Papadaki G, Repa A, Kampouraki E, Kambas K, Ritis K, et al. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res Ther (2015) 17:257. doi: 10.1186/s13075-015-0775-2
138. Jakobs C, Perner S, Hornung V. AIM2 Drives Joint Inflammation in a Self-DNA Triggered Model of Chronic Polyarthritis. PloS One (2015) 10(6):e0131702. doi: 10.1371/journal.pone.0131702
139. Yap JKY, Pickard BS, Chan EWL, Gan SY. The Role of Neuronal NLRP1 Inflammasome in Alzheimer’s Disease: Bringing Neurons into the Neuroinflammation Game. Mol Neurobiol (2019) 56(11):7741–53. doi: 10.1007/s12035-019-1638-7
140. Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener (2016) 11:23. doi: 10.1186/s13024-016-0088-1
141. Pontillo A, Catamo E, Arosio B, Mari D, Crovella S. NALP1/NLRP1 genetic variants are associated with Alzheimer disease. Alzheimer Dis Assoc Disord (2012) 26(3):277–81. doi: 10.1097/WAD.0b013e318231a8ac
142. Peprah K, McCormack S. Medical Cannabis for the Treatment of Dementia: A Review of Clinical Effectiveness and Guidelines. In: CADTH Rapid Response Reports. Ottawa, ON: CADTH (2019).
143. Cassano T, Villani R, Pace L, Carbone A, Bukke VN, Orkisz S, et al. From Cannabis sativa to Cannabidiol: Promising Therapeutic Candidate for the Treatment of Neurodegenerative Diseases. Front Pharmacol (2020) 11:124. doi: 10.3389/fphar.2020.00124
144. Tan MS, Yu JT, Jiang T, Zhu XC, Wang HF, Zhang W, et al. NLRP3 polymorphisms are associated with late-onset Alzheimer’s disease in Han Chinese. J Neuroimmunol (2013) 265(1-2):91–5. doi: 10.1016/j.jneuroim.2013.10.002
145. Choubey D. Type I interferon (IFN)-inducible Absent in Melanoma 2 proteins in neuroinflammation: implications for Alzheimer’s disease. J Neuroinflammation (2019) 16(1):236. doi: 10.1186/s12974-019-1639-5
146. Saadi M, Karkhah A, Pourabdolhossein F, Ataie A, Monif M, Nouri HR. Involvement of NLRC4 inflammasome through caspase-1 and IL-1beta augments neuroinflammation and contributes to memory impairment in an experimental model of Alzheimer’s like disease. Brain Res Bull (2020) 154:81–90. doi: 10.1016/j.brainresbull.2019.10.010
147. Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, et al. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ (2015) 22(10):1676–86. doi: 10.1038/cdd.2015.16
148. Bougea A, Koros C, Simitsi AM, Chrysovitsanou C, Leonardos A, Stefanis L. Medical cannabis as an alternative therapeutics for Parkinsons’ disease: Systematic review. Complement Ther Clin Pract (2020) 39:101154. doi: 10.1016/j.ctcp.2020.101154
149. Lang Y, Chu F, Shen D, Zhang W, Zheng C, Zhu J, et al. Role of Inflammasomes in Neuroimmune and Neurodegenerative Diseases: A Systematic Review. Mediators Inflammation (2018) 2018:1549549. doi: 10.1155/2018/1549549
150. von Herrmann KM, Salas LA, Martinez EM, Young AL, Howard JM, Feldman MS, et al. NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson’s disease. NPJ Parkinsons Dis (2018) 4:24. doi: 10.1038/s41531-018-0061-5
151. Rodrigue-Gervais IG, Doiron K, Champagne C, Mayes L, Leiva-Torres GA, Vanie P Jr., et al. The mitochondrial protease HtrA2 restricts the NLRP3 and AIM2 inflammasomes. Sci Rep (2018) 8(1):8446. doi: 10.1038/s41598-018-26603-1
152. Mejias NH, Martinez CC, Stephens ME, de Rivero Vaccari JP. Contribution of the inflammasome to inflammaging. J Inflammation (Lond) (2018) 15:23. doi: 10.1186/s12950-018-0198-3
153. Gonzalez-Hidalgo C, De Haro J, Bleda S, Canibano C, Michel I, Acin F. Differential mRNA expression of inflammasome genes NLRP1 and NLRP3 in abdominal aneurysmal and occlusive aortic disease. Ther Adv Cardiovasc Dis (2018) 12(4):123–9. doi: 10.1177/1753944717750338
154. Borborema MEA, Crovella S, Oliveira D, de Azevedo Silva J. Inflammasome activation by NLRP1 and NLRC4 in patients with coronary stenosis. Immunobiology (2020) 225(3):151940. doi: 10.1016/j.imbio.2020.151940
155. Clipman SJ, Henderson-Frost J, Fu KY, Bern C, Flores J, Gilman RH. Genetic association study of NLRP1, CARD, and CASP1 inflammasome genes with chronic Chagas cardiomyopathy among Trypanosoma cruzi seropositive patients in Bolivia. PloS One (2018) 13(2):e0192378. doi: 10.1371/journal.pone.0192378
156. DeFilippis EM, Bajaj NS, Singh A, Malloy R, Givertz MM, Blankstein R, et al. Marijuana Use in Patients With Cardiovascular Disease: JACC Review Topic of the Week. J Am Coll Cardiol (2020) 75(3):320–32. doi: 10.1016/j.jacc.2019.11.025
157. Stanley CP, Hind WH, O’Sullivan SE. Is the cardiovascular system a therapeutic target for cannabidiol? Br J Clin Pharmacol (2013) 75(2):313–22. doi: 10.1111/j.1365-2125.2012.04351.x
158. Zhou D, Wang X, Chen T, Wen W, Liu Y, Wu Y, et al. The NLRP3 rs10754558 Polymorphism Is Associated with the Occurrence and Prognosis of Coronary Artery Disease in the Chinese Han Population. BioMed Res Int (2016) 2016:3185397. doi: 10.1155/2016/3185397
159. Durga Devi T, Babu M, Makinen P, Kaikkonen MU, Heinaniemi M, Laakso H, et al. Aggravated Postinfarct Heart Failure in Type 2 Diabetes Is Associated with Impaired Mitophagy and Exaggerated Inflammasome Activation. Am J Pathol (2017) 187(12):2659–73. doi: 10.1016/j.ajpath.2017.08.023
160. Pan J, Han L, Guo J, Wang X, Liu D, Tian J, et al. AIM2 accelerates the atherosclerotic plaque progressions in ApoE-/- mice. Biochem Biophys Res Commun (2018) 498(3):487–94. doi: 10.1016/j.bbrc.2018.03.005
161. Johansson A, Eriksson N, Becker RC, Storey RF, Himmelmann A, Hagstrom E, et al. NLRC4 Inflammasome Is an Important Regulator of Interleukin-18 Levels in Patients With Acute Coronary Syndromes: Genome-Wide Association Study in the PLATelet inhibition and patient Outcomes Trial (PLATO). Circ Cardiovasc Genet (2015) 8(3):498–506. doi: 10.1161/CIRCGENETICS.114.000724
162. Sharma N, Jha S. NLR-regulated pathways in cancer: opportunities and obstacles for therapeutic interventions. Cell Mol Life Sci (2016) 73(9):1741–64. doi: 10.1007/s00018-015-2123-8
163. Velloso FJ, Trombetta-Lima M, Anschau V, Sogayar MC, Correa RG. NOD-like receptors: major players (and targets) in the interface between innate immunity and cancer. Biosci Rep (2019) 39(4):BSR20181709. doi: 10.1042/BSR20181709
164. Meng H, Dai T, Hanlon JG, Downar J, Alibhai SMH, Clarke H. Cannabis and cannabinoids in cancer pain management. Curr Opin Support Palliat Care (2020) 14(2):87–93. doi: 10.1097/SPC.0000000000000493
165. Afrin F, Chi M, Eamens AL, Duchatel RJ, Douglas AM, Schneider J, et al. Can Hemp Help? Low-THC Cannabis and Non-THC Cannabinoids for the Treatment of Cancer. Cancers (Basel) (2020) 12(4):1633. doi: 10.3390/cancers12041033
166. Choubey D. Absent in melanoma 2 proteins in the development of cancer. Cell Mol Life Sci (2016) 73(23):4383–95. doi: 10.1007/s00018-016-2296-9
167. Woerner SM, Kloor M, Schwitalle Y, Youmans H, Doeberitz M, Gebert J, et al. The putative tumor suppressor AIM2 is frequently affected by different genetic alterations in microsatellite unstable colon cancers. Genes Chromosomes Cancer (2007) 46(12):1080–9. doi: 10.1002/gcc.20493
168. Fan JJ, Gao B, Song AQ, Zhu YJ, Zhou J, Li WZ, et al. Spinal cord NLRP1 inflammasome contributes to dry skin induced chronic itch in mice. J Neuroinflammation (2020) 17(1):122. doi: 10.1186/s12974-020-01807-3
169. Zhou W, Chen C, Chen Z, Liu L, Jiang J, Wu Z, et al. NLRP3: A Novel Mediator in Cardiovascular Disease. J Immunol Res (2018) 2018:5702103. doi: 10.1155/2018/5702103
170. Jansen T, Klück V, Janssen M, Comarniceanu A, Efdé M, Scribner C, et al. P160 The first phase 2A proof-of-concept study of a selective NLRP3 inflammasome inhibitor, dapansutrile™ (OLT1177™), in acute gout. Ann Rheum Dis (2019) 78(Suppl 1):A70–A1. doi: 10.1136/annrheumdis-2018-EWRR2019.142
171. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet (2001) 29(3):301–5. doi: 10.1038/ng756
172. Brogan PA, Hofer M, Kuemmerle-Deschner JB, Kone-Paut I, Roesler J, Kallinich T, et al. Rapid and Sustained Long-Term Efficacy and Safety of Canakinumab in Patients With Cryopyrin-Associated Periodic Syndrome Ages Five Years and Younger. Arthritis Rheumatol (2019) 71(11):1955–63. doi: 10.1002/art.41004
173. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol (2018) 15(9):505–22. doi: 10.1038/s41569-018-0064-2
174. Hong P, Gu RN, Li FX, Xiong XX, Liang WB, You ZJ, et al. NLRP3 inflammasome as a potential treatment in ischemic stroke concomitant with diabetes. J Neuroinflammation (2019) 16(1):121. doi: 10.1186/s12974-019-1498-0
175. Xiang H, Zhu F, Xu Z, Xiong J. Role of Inflammasomes in Kidney Diseases via Both Canonical and Non-canonical Pathways. Front Cell Dev Biol (2020) 8:106. doi: 10.3389/fcell.2020.00106
176. Wang W, Nguyen LT, Burlak C, Chegini F, Guo F, Chataway T, et al. Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein alpha-synuclein. Proc Natl Acad Sci U S A (2016) 113(34):9587–92. doi: 10.1073/pnas.1610099113
177. Wijeyekoon RS, Kronenberg-Versteeg D, Scott KM, Hayat S, Kuan WL, Evans JR, et al. Peripheral innate immune and bacterial signals relate to clinical heterogeneity in Parkinson’s disease. Brain Behav Immun (2020) 87:473–88. doi: 10.1016/j.bbi.2020.01.018
178. Vance RE. The NAIP/NLRC4 inflammasomes. Curr Opin Immunol (2015) 32:84–9. doi: 10.1016/j.coi.2015.01.010
179. Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet (2014) 46(10):1140–6. doi: 10.1038/ng.3089
180. Nichols RD, von Moltke J, Vance RE. NAIP/NLRC4 inflammasome activation in MRP8(+) cells is sufficient to cause systemic inflammatory disease. Nat Commun (2017) 8(1):2209. doi: 10.1038/s41467-017-02266-w
181. Verma V, Gupta S, Kumar P, Yadav S, Dhanda RS, Gaind R, et al. Involvement of NLRP3 and NLRC4 Inflammasome in Uropathogenic E. coli Mediated Urinary Tract Infections. Front Microbiol (2019) 10:2020. doi: 10.3389/fmicb.2019.02020
182. Chen Y, Ma K. NLRC4 inflammasome activation regulated by TNF-alpha promotes inflammatory responses in nonalcoholic fatty liver disease. Biochem Biophys Res Commun (2019) 511(3):524–30. doi: 10.1016/j.bbrc.2019.02.099
183. Linkermann A, Parmentier SP, Hugo C. We AIM2 Inflame. J Am Soc Nephrol (2018) 29(4):1077–9. doi: 10.1681/ASN.2018020116
184. Komada T, Chung H, Lau A, Platnich JM, Beck PL, Benediktsson H, et al. Macrophage Uptake of Necrotic Cell DNA Activates the AIM2 Inflammasome to Regulate a Proinflammatory Phenotype in CKD. J Am Soc Nephrol (2018) 29(4):1165–81. doi: 10.1681/ASN.2017080863
185. Mendez-Frausto G, Medina-Rosales MN, Uresti-Rivera EE, Baranda-Candido L, Zapata-Zuniga M, Bastian Y, et al. Expression and activity of AIM2-inflammasome in rheumatoid arthritis patients. Immunobiology (2019) 225(2):151880. doi: 10.1016/j.imbio.2019.11.015
186. Gibertini M, Newton C, Friedman H, Klein TW. IL-1 beta and TNF alpha modulate delta 9-tetrahydrocannabinol-induced catalepsy in mice. Pharmacol Biochem Behav (1995) 50(2):141–6. doi: 10.1016/0091-3057(94)00270-S
187. Zhu W, Friedman H, Klein TW. Delta9-tetrahydrocannabinol induces apoptosis in macrophages and lymphocytes: involvement of Bcl-2 and caspase-1. J Pharmacol Exp Ther (1998) 286(2):1103–9.
188. Rizzo MD, Crawford RB, Bach A, Sermet S, Amalfitano A, Kaminski NE. Delta(9)-Tetrahydrocannabinol Suppresses Monocyte-Mediated Astrocyte Production of Monocyte Chemoattractant Protein 1 and Interleukin-6 in a Toll-Like Receptor 7-Stimulated Human Coculture. J Pharmacol Exp Ther (2019) 371(1):191–201. doi: 10.1124/jpet.119.260661
189. Zurier RB, Rossetti RG, Burstein SH, Bidinger B. Suppression of human monocyte interleukin-1beta production by ajulemic acid, a nonpsychoactive cannabinoid. Biochem Pharmacol (2003) 65(4):649–55. doi: 10.1016/S0006-2952(02)01604-0
190. Yang L, Li FF, Han YC, Jia B, Ding Y. Cannabinoid receptor CB2 is involved in tetrahydrocannabinol-induced anti-inflammation against lipopolysaccharide in MG-63 cells. Mediators Inflamm (2015) 2015:362126. doi: 10.1155/2015/362126
191. Ismail M, Hasan H, El-Orfali Y, Ismail H, Khawaja G. Anti-Inflammatory, Antioxidative, and Hepatoprotective Effects of Trans Delta9-Tetrahydrocannabinol/Sesame Oil on Adjuvant-Induced Arthritis in Rats. Evid Based Complement Alternat Med (2018) 2018:9365464. doi: 10.1155/2018/9365464
192. Yamaori S, Ishii H, Chiba K, Yamamoto I, Watanabe K. Delta-Tetrahydrocannabinol induces cytotoxicity in macrophage J774-1 cells: involvement of cannabinoid receptor 2 and p38 MAPK. Toxicology (2013) 314(2-3):254–61. doi: 10.1016/j.tox.2013.10.007
193. Romano B, Pagano E, Orlando P, Capasso R, Cascio MG, Pertwee R, et al. Pure Delta(9)-tetrahydrocannabivarin and a Cannabis sativa extract with high content in Delta(9)-tetrahydrocannabivarin inhibit nitrite production in murine peritoneal macrophages. Pharmacol Res (2016) 113(Pt A):199–208. doi: 10.1016/j.phrs.2016.07.045
194. Libro R, Diomede F, Scionti D, Piattelli A, Grassi G, Pollastro F, et al. Cannabidiol Modulates the Expression of Alzheimer’s Disease-Related Genes in Mesenchymal Stem Cells. Int J Mol Sci (2016) 18(1):26. doi: 10.3390/ijms18010026
195. Huang Y, Wan T, Pang N, Zhou Y, Jiang X, Li B, et al. Cannabidiol protects livers against nonalcoholic steatohepatitis induced by high-fat high cholesterol diet via regulating NF-kappaB and NLRP3 inflammasome pathway. J Cell Physiol (2019) 234(11):21224–34. doi: 10.1002/jcp.28728
196. Dos-Santos-Pereira M, Guimaraes FS, Del-Bel E, Raisman-Vozari R, Michel PP. Cannabidiol prevents LPS-induced microglial inflammation by inhibiting ROS/NF-kappaB-dependent signaling and glucose consumption. Glia (2020) 68(3):561–73. doi: 10.1002/glia.23738
197. Rimmerman N, Juknat A, Kozela E, Levy R, Bradshaw HB, Vogel Z. The non-psychoactive plant cannabinoid, cannabidiol affects cholesterol metabolism-related genes in microglial cells. Cell Mol Neurobiol (2011) 31(6):921–30. doi: 10.1007/s10571-011-9692-3
198. Esposito G, Scuderi C, Savani C, Steardo L Jr., De Filippis D, Cottone P, et al. Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1beta and iNOS expression. Br J Pharmacol (2007) 151(8):1272–9. doi: 10.1038/sj.bjp.0707337
199. Borrelli F, Aviello G, Romano B, Orlando P, Capasso R, Maiello F, et al. Cannabidiol, a safe and non-psychotropic ingredient of the marijuana plant Cannabis sativa, is protective in a murine model of colitis. J Mol Med (Berl) (2009) 87(11):1111–21. doi: 10.1007/s00109-009-0512-x
200. Mecha M, Feliu A, Inigo PM, Mestre L, Carrillo-Salinas FJ, Guaza C. Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: a role for A2A receptors. Neurobiol Dis (2013) 59:141–50. doi: 10.1016/j.nbd.2013.06.016
201. Jourdan T, Godlewski G, Cinar R, Bertola A, Szanda G, Liu J, et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat Med (2013) 19(9):1132–40. doi: 10.1038/nm.3265
202. Shao BZ, Wei W, Ke P, Xu ZQ, Zhou JX, Liu C. Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP3 inflammasome. CNS Neurosci Ther (2014) 20(12):1021–8. doi: 10.1111/cns.12349
203. Han JH, Shin H, Park JY, Rho JG, Son DH, Kim KW, et al. A novel peripheral cannabinoid 1 receptor antagonist, AJ5012, improves metabolic outcomes and suppresses adipose tissue inflammation in obese mice. FASEB J (2019) 33(3):4314–26. doi: 10.1096/fj.201801152RR
204. Dol-Gleizes F, Paumelle R, Visentin V, Mares AM, Desitter P, Hennuyer N, et al. Rimonabant, a selective cannabinoid CB1 receptor antagonist, inhibits atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol (2009) 29(1):12–8. doi: 10.1161/ATVBAHA.108.168757
205. Yu W, Jin G, Zhang J, Wei W. Selective Activation of Cannabinoid Receptor 2 Attenuates Myocardial Infarction via Suppressing NLRP3 Inflammasome. Inflammation (2019) 42(3):904–14. doi: 10.1007/s10753-018-0945-x
206. Gao F, Xiang HC, Li HP, Jia M, Pan XL, Pan HL, et al. Electroacupuncture inhibits NLRP3 inflammasome activation through CB2 receptors in inflammatory pain. Brain Behav Immun (2018) 67:91–100. doi: 10.1016/j.bbi.2017.08.004
207. Ke P, Shao BZ, Xu ZQ, Wei W, Han BZ, Chen XW, et al. Activation of Cannabinoid Receptor 2 Ameliorates DSS-Induced Colitis through Inhibiting NLRP3 Inflammasome in Macrophages. PloS One (2016) 11(9):e0155076. doi: 10.1371/journal.pone.0155076
208. Jastrzab A, Gegotek A, Skrzydlewska E. Cannabidiol Regulates the Expression of Keratinocyte Proteins Involved in the Inflammation Process through Transcriptional Regulation. Cells (2019) 8(8):827. doi: 10.3390/cells8080827
209. Liu C, Ma H, Slitt AL, Seeram NP. Inhibitory Effect of Cannabidiol on the Activation of NLRP3 Inflammasome Is Associated with Its Modulation of the P2X7 Receptor in Human Monocytes. J Nat Prod (2020) 83(6):2025–9. doi: 10.1021/acs.jnatprod.0c00138
210. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev (2015) 265(1):35–52. doi: 10.1111/imr.12286
211. Gonzalez-Mariscal I, Montoro RA, Doyle ME, Liu QR, Rouse M, O’Connell JF, et al. Absence of cannabinoid 1 receptor in beta cells protects against high-fat/high-sugar diet-induced beta cell dysfunction and inflammation in murine islets. Diabetologia (2018) 61(6):1470–83. doi: 10.1007/s00125-018-4576-4
212. Contarini G, Franceschini D, Facci L, Barbierato M, Giusti P, Zusso M. A co-ultramicronized palmitoylethanolamide/luteolin composite mitigates clinical score and disease-relevant molecular markers in a mouse model of experimental autoimmune encephalomyelitis. J Neuroinflammation (2019) 16(1):126. doi: 10.1186/s12974-019-1514-4
213. Han JH, Shin H, Rho JG, Kim JE, Son DH, Yoon J, et al. Peripheral cannabinoid 1 receptor blockade mitigates adipose tissue inflammation via NLRP3 inflammasome in mouse models of obesity. Diabetes Obes Metab (2018) 20(9):2179–89. doi: 10.1111/dom.13350
214. Hortelano S, Gonzalez-Cofrade L, Cuadrado I, de Las Heras B. Current status of terpenoids as inflammasome inhibitors. Biochem Pharmacol (2020) 172:113739. doi: 10.1016/j.bcp.2019.113739
215. Yi YS. Regulatory Roles of Flavonoids on Inflammasome Activation during Inflammatory Responses. Mol Nutr Food Res (2018) 62(13):e1800147. doi: 10.1002/mnfr.201800147
216. Lim H, Min DS, Park H, Kim HP. Flavonoids interfere with NLRP3 inflammasome activation. Toxicol Appl Pharmacol (2018) 355:93–102. doi: 10.1016/j.taap.2018.06.022
217. Gallily R, Yekhtin Z, Hanus LO. The Anti-Inflammatory Properties of Terpenoids from Cannabis. Cannabis Cannabinoid Res (2018) 3(1):282–90. doi: 10.1089/can.2018.0014
218. Wong SK, Li W, Moore MJ, Choe H, Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J Biol Chem (2004) 279(5):3197–201. doi: 10.1074/jbc.C300520200
219. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature (2003) 426(6965):450–4. doi: 10.1038/nature02145
220. Yuan M, Wu NC, Zhu X, Lee CD, So RTY, Lv H, et al. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science (2020) 368(6491):630–3. doi: 10.1126/science.abb7269
221. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
222. Shi CS, Nabar NR, Huang NN, Kehrl JH. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov (2019) 5:101. doi: 10.1038/s41420-019-0181-7
223. Chen IY, Moriyama M, Chang MF, Ichinohe T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front Microbiol (2019) 10:50. doi: 10.3389/fmicb.2019.00050
224. Shah A. Novel Coronavirus-Induced NLRP3 Inflammasome Activation: A Potential Drug Target in the Treatment of COVID-19. Front Immunol (2020) 11:1021. doi: 10.3389/fimmu.2020.01021
225. Freeman TL, Swartz TH. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front Immunol (2020) 11:1518. doi: 10.3389/fimmu.2020.01518
226. Wang B, Kovalchuk A, Li D, Rodriguez-Juarez R, Ilnytskyy Y, Kovalchuk I, et al. In search of preventive strategies: novel high-CBD Cannabis sativa extracts modulate ACE2 expression in COVID-19 gateway tissues. Aging (Albany NY) (2020) 12(22):22425–44. doi: 10.20944/preprints202004.0315.v1
Keywords: inflammasome, pro-inflammatory cytokines, inflammasome signaling, cannabidiol, Delta-9 tetrahydrocannabinol, cannabinoids
Citation: Suryavanshi SV, Kovalchuk I and Kovalchuk O (2021) Cannabinoids as Key Regulators of Inflammasome Signaling: A Current Perspective. Front. Immunol. 11:613613. doi: 10.3389/fimmu.2020.613613
Received: 02 October 2020; Accepted: 10 December 2020;
Published: 28 January 2021.
Edited by:
Kuo-Feng Hua, National Ilan University, TaiwanReviewed by:
George Kunos, National Institutes of Health (NIH), United StatesAntonella Naldini, University of Siena, Italy
Liying Li, Capital Medical University, China
Copyright © 2021 Suryavanshi, Kovalchuk and Kovalchuk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olga Kovalchuk, b2xnYS5rb3ZhbGNodWtAdWxldGguY2E=