Abstract
Staphylococcus aureus is a leading cause of bacterial infections globally in both healthcare and community settings. The success of this bacterium is the product of an expansive repertoire of virulence factors in combination with acquired antibiotic resistance and propensity for biofilm formation. S. aureus leverages these factors to adapt to and subvert the host immune response. With the burgeoning field of immunometabolism, it has become clear that the metabolic program of leukocytes dictates their inflammatory status and overall effectiveness in clearing an infection. The metabolic flexibility of S. aureus offers an inherent means by which the pathogen could manipulate the infection milieu to promote its survival. The exact metabolic pathways that S. aureus influences in leukocytes are not entirely understood, and more work is needed to understand how S. aureus co-opts leukocyte metabolism to gain an advantage. In this review, we discuss the current knowledge concerning how metabolic biases dictate the pro- vs. anti-inflammatory attributes of various innate immune populations, how S. aureus metabolism influences leukocyte activation, and compare this with other bacterial pathogens. A better understanding of the metabolic crosstalk between S. aureus and leukocytes may unveil novel therapeutic strategies to combat these devastating infections.
Introduction
Staphylococcus aureus (S. aureus) is an opportunistic pathogen that colonizes approximately one-third of the human population and can cause invasive disease at an array of different sites throughout the body, including endocarditis, skin and soft tissue infection, bacteremia, pneumonia, osteomyelitis, and medical implant-associated infection (1, 2). The ability to successfully infect and persist in such a wide range of tissue niches is due to a number of characteristics that allow the bacterium to evade immune-mediated clearance. Such attributes include the production of various toxins, the acquisition of antibiotic resistance or tolerance, and the ability to form biofilm (3–6). Each of these factors, often in combination with one another, contribute to the ability of S. aureus to counteract immune effector mechanisms.
Throughout the course of an infection, both host and pathogen undergo substantial changes in their metabolic programs to facilitate the production of different effector molecules that aid in their respective goals (7, 8). In the case of leukocytes, this takes the form of regulating the production of cytokines, such as IL-1β. For S. aureus, metabolic reprogramming allows for the production of various leukocidins and lactate, among other virulence factors, that combat the immune system (9). As an infection progresses, nutrient concentrations within the tissue milieu can rapidly fluctuate as host and pathogen compete for the same extracellular energy sources (10, 11). The intrinsic metabolic flexibility of S. aureus allows it to quickly adapt to these evolving conditions to promote its survival (12, 13). While S. aureus may be able to overcome the depletion of specific nutrient sources, the leukocyte population may not be as flexible. As metabolism is inextricably linked to immune cell function, the intentional depletion and/or release of specific metabolites that can impair leukocyte microbicidal activity could represent a system by which the bacteria influences host cell metabolism to its benefit. Here we provide an overview of how S. aureus interacts with leukocytes at the metabolic level. We will focus on immunomodulatory metabolites and how they contribute to the crosstalk between host and pathogen during an infection, with a particular emphasis on S. aureus biofilm formation. We also provide examples of metabolic crosstalk between leukocytes and other bacterial species as further mechanisms to consider in the context of S. aureus infection.
Immunometabolism
The study of immunometabolism is focused on linking changes in metabolic programs to effector functions. Over the years, it has become apparent that activated leukocytes experience a shift in metabolism that regulates inflammatory mediator production (14–16). Activation usually coincides with a metabolic shift from the energy (ATP)-rich resting state to an increase in the production of effector metabolites necessary for biosynthetic processes for inflammatory effector function, such as fatty acid biosynthesis for prostaglandin production (Figure 1) (17–20). Since this observation was made, there has been considerable interest in defining metabolic programs that are characteristic of either the pro- or anti-inflammatory status of various leukocyte populations. Much of this fervor originated from the idea that immune cell function could potentially be orchestrated via the manipulation of the nutrient milieu or by therapeutically targeting specific metabolic pathways.
Figure 1

Metabolic shifts in leukocytes define resting from inflammatory states. Immune cells normally respire under resting conditions to provide sufficient energy (ATP) for survival. Upon activation, although energy production is somewhat increased, a significant portion of the metabolic flow is dedicated to producing metabolic intermediates (Effector Metabolites) that are used to generate inflammatory mediators (i.e. cytokines, inflammatory lipids, etc.) to promote leukocyte effector functions (Immune Response). S. aureus, either through competition for metabolic resources or by releasing immunomodulatory metabolites, can interfere with this process to disrupt a productive immune response. Monocyte (MO), macrophage (Mφ), neutrophil (PMN), and granulocytic myeloid-derived suppressor cell (G-MDSC). Figure created with BioRender.
In the context of an infection, immunometabolism is only half of the story, since pathogens must also acquire essential nutrient sources for their survival (21). This leads to direct competition between the host and pathogen on a metabolic scale. Much like responding leukocytes, S. aureus must also undergo some degree of metabolic adaptation to counteract the effects of the immune response. Switching between alternative forms of metabolism is common in S. aureus due to its significant degree of metabolic plasticity (22–24). This flexibility greatly increases the chances that the bacteria will win the battle of attrition between host and pathogen for specific nutrient sources. If this ability to shift between different metabolic modes is compromised, it could drastically alter the landscape of an infection. For instance, work from our laboratory has established that S. aureus biofilm skews leukocytes towards an anti-inflammatory phenotype in a murine model of prosthetic joint infection (PJI) that is mediated, in part, by IL-10 production (25–28). Recent work demonstrated that the loss of an essential metabolic process in S. aureus, such as ATP synthesis (ΔatpA), significantly decreased the chronicity of biofilm infection by eliciting a heightened pro-inflammatory response (29). This is likely attributable to the respiratory defect in S. aureus ΔatpA that limits the energy required for virulence factor production (30), which subsequently led to increased leukocyte viability and pro-inflammatory activity (29). Another group has shown that metabolic adaptation of Pseudomonas aeruginosa is essential for establishing chronic biofilm infection in patients with debilitating diseases such as cystic fibrosis (CF). Host-adapted P. aeruginosa strains preferentially utilized the tricarboxylic acid (TCA) cycle to limit stress from reactive oxygen species (ROS) produced by leukocytes in the airway. This TCA metabolic bias in P. aeruginosa enhanced biofilm formation, which served to increase the chronicity of infection (31). How metabolic changes can influence the function of various leukocyte populations and an overview of critical S. aureus metabolic pathways in the host will be discussed in the following sections.
Macrophage Metabolism
The macrophage is currently the prototypical cell studied in the immunometabolism field. Much progress has been made in characterizing how macrophages shift their metabolism after exposure to various stimuli in vitro and how this influences their effector functions. These experiments led to the discovery that pro- and anti-inflammatory polarized macrophages have distinct metabolic programs (32–34). For example, exposure to Toll-like receptor (TLR) agonists such as bacterial lipoproteins, peptidoglycan, DNA, or lipopolysaccharide (LPS) biases macrophages towards glycolysis. In contrast, anti-inflammatory cytokines (i.e. IL-4 or IL-10) or growth factors promote oxidative phosphorylation (35, 36). In pro-inflammatory macrophages, the increase in glycolytic metabolism following TLR activation is due, in part, to two TCA cycle blocks (Figure 2) (15, 35). These break points create an anaplerotic cycle that leads to the progressive accumulation of metabolites such as succinate and aconitate, which are immunomodulatory in nature. Initial experiments showed that the intracellular concentration of succinate dramatically increases following TLR stimulation (37–39). Early work into understanding metabolic reprogramming found that this succinate pool was diverted from normal TCA cycle metabolism and oxidized by succinate dehydrogenase (SDH) to produce high amounts of ROS, which stabilized the transcription factor hypoxia-inducible factor-1α (HIF-1α) by preventing its ubiquitination and degradation (40–44). HIF-1α stabilization leads to the production of pro-IL-1β, which is cleaved into its mature form by the NOD-like receptor pyrin domain containing 3 (NLRP3) inflammasome (45–47). A secondary effect of this process is the HIF-1α-dependent induction of glycolytic enzymes by that feed back to promote biosynthetic pathways to augment pro-inflammatory activity (40, 48, 49). Increased glycolysis serves to produce ATP and sustain the mitochondrial membrane potential that is necessary for succinate oxidation.
Figure 2
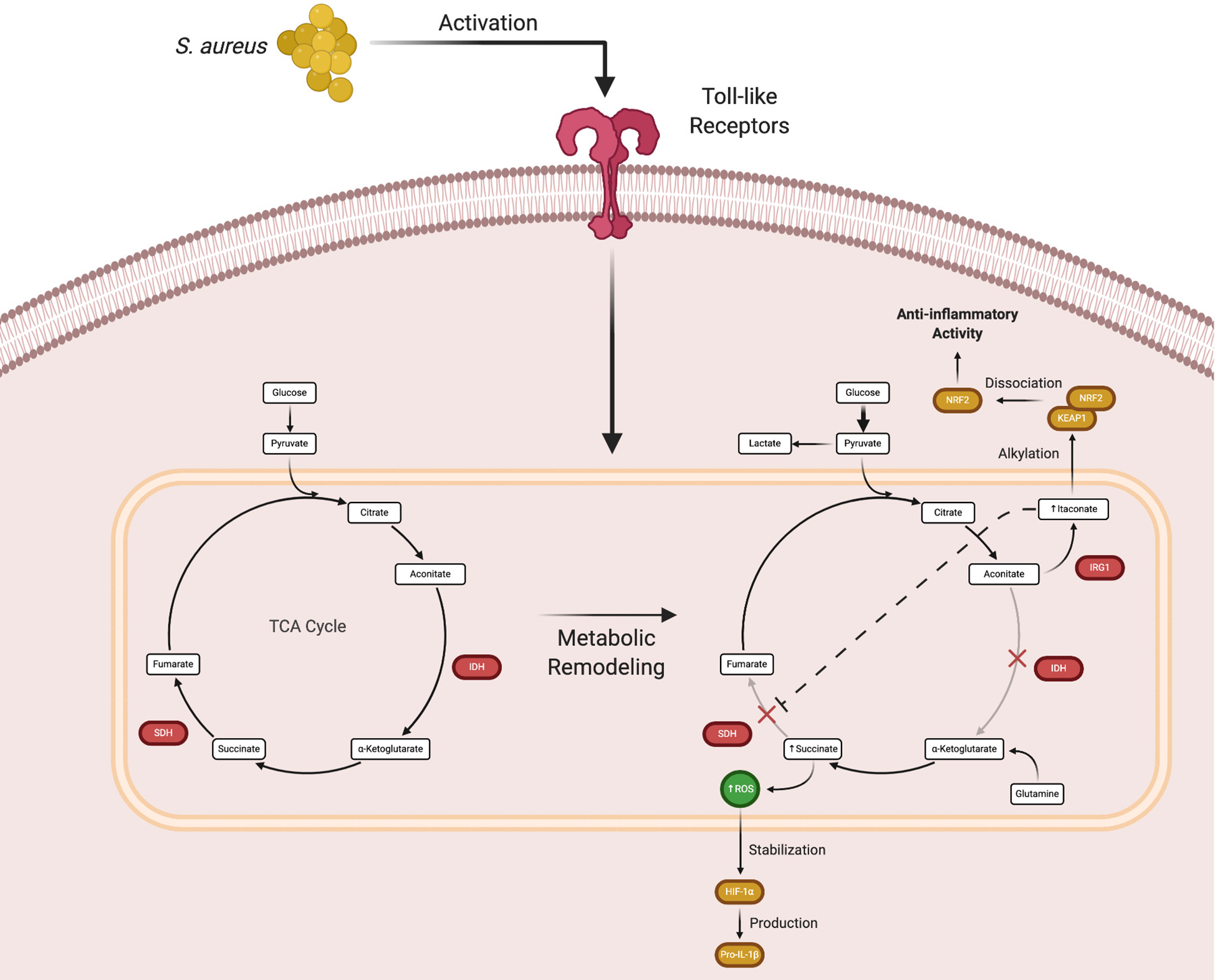
Macrophage metabolic remodeling during an immune response. Following Toll-like receptor (TLR) activation, macrophages undergo metabolic rewiring to promote inflammatory mediator production. The TCA cycle breaks at two points, isocitrate dehydrogenase (IDH) that causes the accumulation of aconitate, which is converted by immune responsive gene 1 (IRG1) to form itaconate, and succinate dehydrogenase (SDH) that leads to succinate accumulation. While this process normally augments proinflammatory activity, excess itaconate production eventually causes a shift to promote the expression of anti-inflammatory genes. Figure created with BioRender.
In macrophages, NLRP3 inflammasome activation generally occurs via a two signal model (45). Signal 1 is mediated by TLR, IL-1R, or TNFR activation that elicits maximal expression of pro-IL-1β and inflammasome components. Numerous stimuli have been shown to provide signal 2 and the diverse structure of these molecules has led to the concept that cellular stress is a unifying factor responsible for NLRP3 inflammasome activation (45). With regard to S. aureus, α-toxin acts as signal 2 to activate a primed NLRP3 inflammasome (50). This has been attributed to K+ efflux from cells as a consequence of toxin-mediated membrane disruption, which can be potentiated by gasdermin D cleavage by the NLRP3 inflammasome that forms another transmembrane pore leading to pyroptosis (51). However, in vivo studies have shown that α-toxin alone is not sufficient for NLRP3 activation. Another requisite is S. aureus lipoproteins that provide signal 1 via TLR2 activation to elicit maximal expression of pro-IL-1β and inflammasome components (52). Recently, it was shown that S. aureus packages its pore-forming toxins into extracellular vesicles that are then internalized by immune cells. Upon uptake, S. aureus-derived vesicles contain all of the requisite factors to induce inflammasome activation, thus providing the bacteria with another mechanism to modulate the immune response (53). A paradox is why S. aureus augments NLRP3 inflammasome activation given its ability to produce the pro-inflammatory cytokines IL-1β and IL-18. However, a recent study has demonstrated that S. aureus α-toxin exploits NLRP3 inflammasome activation in macrophages by recruiting mitochondria away from the phagosome, which inhibits mitochondrial ROS production via complex II (SDH) of the electron transport chain (ETC), phagosomal acidification, and bacterial killing (54). This effect was independent of NLRP3-mediated IL-1β and IL-18 production. Intriguingly, this represents another mechanism that S. aureus exploits to prevent immune-mediated clearance.
Subsequent in vitro studies examining the mechanisms of macrophage metabolic remodeling revealed that the increase in succinate following TLR activation was due to the action of itaconate, a derivative of the TCA intermediate aconitate. Itaconate is produced by immune-responsive gene 1 (IRG1) and as the concentration of itaconate increases, SDH is progressively inhibited. This leads to succinate accumulation and decreased oxygen consumption via the inhibition of SDH, which is also complex II of the mitochondrial ETC (Figure 2) (55–59). Although the initial production of itaconate augments macrophage pro-inflammatory activity, its accumulation begins to exert anti-inflammatory effects (60). As itaconate accumulates, it is transported out of mitochondria where it can interact with cytoplasmic protein targets, namely Kelch-like ECH-associated protein 1 (KEAP1) (61–63). Under homeostatic conditions, KEAP1 is bound to nuclear factor erythroid 2-related factor 2 (Nrf2), which targets the complex for proteasomal degradation. Under stress conditions, such as S. aureus-induced toxin action, the complex dissociates and Nrf2 translocates to the nucleus where it acts as a transcription factor for numerous anti-inflammatory genes (64). Itaconate is capable of disrupting the KEAP1-Nrf2 association via alkylation of cysteine residues in KEAP1 to promote Nrf2 nuclear translocation and the transcriptional activation of anti-inflammatory genes. Via this mechanism, itaconate represents a way to counterbalance the proinflammatory activity of succinate accumulation (65, 66).
Amino acid metabolism is also important for influencing macrophage polarization, where arginine is differentially utilized by macrophages to exert distinct effector functions (67). For example, in response to planktonic S. aureus and other pro-inflammatory stimuli, macrophages utilize arginine to drive inducible nitric oxide synthase (iNOS) activity and nitric oxide (NO) production (68). Nitric oxide is a highly reactive free radical that exerts potential bactericidal activity by inducing DNA and membrane damage as well as targeting oxidative metabolism (69–71). However, S. aureus is capable of evading host NO production, which differentiates it from other Staphylococcal species. One example is flavohemoprotein (hmp) expression that allows S. aureus to detoxify its environment by converting NO into nitrate, making it an iNOS-dependent virulence determinant (72). Another component of the nitrosative stress response in S. aureus is L-lactate dehydrogenase (ldh1), which is a NO-inducible gene. While hmp serves to detoxify the environment, Ldh allows S. aureus to maintain redox homeostasis by promoting the conversion of pyruvate to lactate (73). Although this metabolic program generates less ATP compared to oxidative metabolism, it provides a mechanism by which S. aureus can maintain its reducing equivalents until Hmp can decrease NO levels. In contrast to iNOS, arginine is used by arginase-1 (Arg-1) in anti-inflammatory polarized macrophages to produce ornithine. Ornithine is further metabolized into polyamines and proline for wound repair and cell growth processes (74, 75). Increased Arg1 expression has been linked to macrophage anti-inflammatory activity during S. aureus biofilm infection (76–78). To determine if Arg1 expression was required for the immune suppression associated with S. aureus biofilm infection, our laboratory utilized Arg-1fl/fl; Tie-2Cre conditional knockout mice where myeloid cells lacked Arg-1. Arg1 was dispensable for myeloid immunosuppression during biofilm formation but was critical for S. aureus containment during abscess formation (79). This was in agreement with a prior study showing that host polyamine production was important for controlling S. aureus growth in a mouse SSTI model (80). Taken together, these results indicate that the effects of Arg1 expression are context-dependent in terms of myeloid cell function during S. aureus biofilm vs. planktonic infection.
Monocyte/macrophage metabolism was recently shown by our laboratory to be important for influencing the outcome of S. aureus biofilm infection in a mouse model of PJI (81). S. aureus biofilm biased monocytes towards oxidative metabolism which, much like macrophages exposed to biofilm in vitro, were largely anti-inflammatory in nature (82). Therefore, a nanoparticle approach was used to deliver oligomycin, an inhibitor of complex V of the ETC, to redirect monocyte metabolism towards glycolysis. Oligomycin nanoparticles augmented monocyte pro-inflammatory activity, which coincided with reduced biofilm burden in vivo, indicating that metabolic remodeling could prove to be an effective therapeutic approach for chronic biofilm infection (81). Importantly, monocyte metabolic reprogramming was capable of attenuating an established biofilm infection, whose efficacy was heightened by concominant antibiotic treatment.
Granulocyte Metabolism
Although much progress has been made in understanding the role of metabolism in macrophages, comparatively less is known about how metabolic changes affect granulocyte function. Neutrophils have been shown to rely almost entirely on glycolytic modes of metabolism to fuel their effector functions, which agrees with a reduced mitochondrial abundance (83, 84). Similar to pro-inflammatory macrophages, activated neutrophils adopt a metabolic program that is similar to the aerobic glycolysis that was first described by Otto Warburg in the 1920’s (85), which has since become known as “Warburg metabolism” (86, 87). This heavy reliance on glycolysis, even in oxygen replete conditions, is necessary to increase carbon flux through the pentose phosphate pathway (PPP) to increase the NADPH pool that is required for NADPH oxidase activity and ROS production (16, 88). Like pro-inflammatory macrophages, metabolic intermediates from the TCA cycle that is fueled by glycolysis, are used for anabolic processes to promote granulocyte functional activity. For example, citrate from the TCA cycle can be diverted for fatty acid synthesis to drive the production of pro-inflammatory mediators such as prostaglandins and leukotrienes (89–92). Although granulocytes are typically considered as purely glycolytic, some studies have also identified granulocytes that instead utilize mitochondrial oxidative metabolism (93). In the context of cancer, glucose within the tumor microenvironment can quickly become a limiting factor. Under these conditions, a population of immature, c-Kit+ neutrophils has been shown to utilize fatty acid oxidation to maintain ROS production by NADPH oxidase (93). These immature neutrophils in tumor-bearing mice were regulated through aberrant SCF/c-Kit signaling and metabolically adapted for the lack of glucose within the tumor microenvironment. Functionally, this meant the adapted neutrophils retained the ability to produce ROS, which can interfere with CD4+ T cell anti-tumor activities (94–96). Although not definitively established, these c-Kit+ neutrophils possess many characteristics of granulocytic MDSCs (G-MDSCs, see below). A similar reduction in glucose availability occurs during S. aureus biofilm formation, as reflected by a shift towards fermentative metabolism and lactate production (23), which shapes the metabolic attributes of infiltrating leukocytes, which is discussed below.
MDSCs are a heterogenous population of immature myeloid cells that are grouped into two categories based on their shared characteristics with mature monocytes or neutrophils, namely M-MDSCs and G-/PMN-MDSCs (97–99). Both subsets are thought to exert their suppressive activity through increased ROS production, although M-MDSCs can also utilize Arg-1 to deplete arginine that is required for TCR expression to inhibit T cell activation (94, 100, 101). Although MDSCs have been best characterized in cancer, they have also been implicated in promoting chronic infection, including S. aureus biofilm (25–28), bone, and skin infection (102, 103). Due to the extensive heterogeneity of MDSCs and context-dependent modes of action, describing a singular metabolic program that is characteristic of these cells has proved challenging. Nevertheless, a few reports have examined MDSC metabolism and how this affects their suppressive activity (104–106). One study found that M-MDSCs suppress T cell activation by inhibiting glycolysis through direct physical contact. Interestingly, M-MDSCs were metabolically dormant, characterized by an accumulation of the α-dicarbonyl methylglyoxal. Methylglyoxal was found in T cells following co-culture with M-MDSCs and treatment with dimethylbiguanide (DMBG), which neutralizes dicarbonyls, restored T cell activation, supporting the importance of methylglyoxal in MDSC-mediated T cell suppression (107, 108). Another study has pointed to fatty acid accumulation and subsequent prostaglandin production as a metabolic mechanism for MDSC suppression. Specifically, PMN-MDSCs overexpress a fatty acid transporter (FATP2), which led to fatty acid accumulation and prostaglandin E2 (PGE2) production that promoted their immunosuppressive effects (109). These studies highlight the metabolic diversity that MDSCs can adopt to influence their inhibitory activity. S. aureus biofilm elicits G-MDSCs that may utilize a distinct metabolic program to exert their suppressive effects than those described here, which remains to be determined. Further investigation into how MDSC metabolism evolves throughout the course of infection is required to appreciate the role that these cells play in shaping the host immune response.
Host–Pathogen Metabolic Crosstalk
In an infectious milieu, not only will leukocytes and the pathogen have to compete for the same nutrient sources, but this also creates a new environment in which the two could interact. For instance, a recent study demonstrated how host adapted strains of P. aeruginosa responded to the secretion of itaconate by selecting for variants that were able to utilize the host-derived metabolite as a nutrient source (110). This selection process coincided with modifications of membrane structural components in P. aeruginosa to augment host-derived itaconate release, thereby establishing a positive feedback loop to promote chronic infection. This illustrates the importance of not only considering the metabolism of either the host or pathogen in isolation, but also the byproducts that they excrete and exchange. Such molecules constitute a new avenue for cellular signaling that the host and/or pathogen could leverage to their benefit during infection. These systems become even more complex in the context of polymicrobial infections, a common occurrence with P. aeruginosa and S. aureus in the lungs of CF patients (111–113). With the additional layering of another organism, the potential number of molecules and interactions increases exponentially.
S. aureus Metabolism and Competition for Nutrients
Competition for the various nutrient sources in an infectious milieu is probably the most intuitive level by which metabolic crosstalk occurs. In an infection, there are at least two entities (i.e. host and pathogen) racing to consume the same resources, which becomes more complicated in the context of polymicrobial infections. Glucose and oxygen are among the first resources that become restricted in these settings and their deprivation can be enough to bias the actions of responding immune cells or pathogens alike. These events can also dictate the nature of immune cell death, which has been reported to occur via two types of programmed necrosis, namely necroptosis or pyroptosis. Necroptosis is induced by the interaction of TNF with receptor-interacting protein kinase 1 (RIPK1), eliciting a cascade that culminates in the phosphorylation of mixed lineage kinase domain-like protein (MLKL) that damages the plasma membrane leading to cell death. Pyroptosis occurs in response to inflammasome activation that cleaves gasdermin D, which oligomerizes to form pores in the cell membrane. While both forms of cell death induce inflammation, the heightened production of inflammatory mediators associated with inflammasome activation makes pyroptosis more inflammatory in nature than necroptosis (114). The roles of S. aureus metabolism in dictating different modes of immune cell death will be discussed below.
Carbohydrate Availability and S. aureus Metabolism
Much work has been done to elucidate S. aureus metabolic adaptations under a variety of in vitro and in vivo conditions and the reader is directed to several excellent and comprehensive reviews on the topic (9, 23, 24), since only a brief overview is provided here. S. aureus utilizes a number of two-component regulatory systems to sense changes in its environment (115, 116). These systems interface with complex transcriptional networks to tightly control nutrient use throughout different phases of growth and infection. This is referred to as carbon catabolite repression and ensures that bacteria optimally utilize available nutrient resources in a hierarchical manner (117–119). S. aureus uses two catabolite control proteins (CcpA & CcpE) to modulate glucose utilization through central carbon metabolism. In glucose replete conditions, oxidative metabolism is repressed by CcpA, which inhibits the expression of critical TCA cycle enzymes (120–122). As glucose becomes depleted, CcpA activity is reduced which induces the TCA cycle and citrate production. Citrate is sensed by CcpE, which bolsters TCA cycle activity and increases the expression of S. aureus virulence factors (123, 124). It has been shown that S. aureus can quickly outcompete host keratinocytes for available glucose, which leads to keratinocyte death by pyroptosis (125, 126). Pyroptosis fails to clear S. aureus infection because NLPR3 inflammasome activation by α-hemolysin redirects mitochondria away from bacteria-containing phagosomes, thereby preventing acidification and killing (54). Thus, pyroptosis is beneficial for the bacteria because it promotes its intracellular escape and dissemination, while simultaneously introducing any host-derived metabolic intermediates into the infection milieu to be utilized for pathogen survival and replication.
The importance of this battle for glucose is particularly relevant in the setting of diabetes. Diabetic patients are characterized by hyperglycemia and often present with persistent and invasive S. aureus infections (127, 128). A recent study examining S. aureus SSTI in a diabetic mouse model has shown that although there is more bioavailable glucose in diabetic tissues, phagocytes failed to take up the carbohydrate with either GLUT-1 or GLUT-3 transporters. This resulted in impaired oxidative burst activity and increased bacterial burden (129). S. aureus infection in diabetic patients has been linked to an increase in the recruitment of low-density neutrophils (LDNs) (130). LDNs are associated with increased rates of NETosis, or the production of neutrophil extracellular traps (NETs). Although, NETs are typically considered to exert anti-microbial effects, elevated levels of NETosis have been linked to impaired wound healing in patients with diabetes, whose neutrophils are more prone to NET formation (131). Increased NETosis in diabetic patients could result from the elevated levels of glucose within diabetic tissues, as glucose is a metabolic requirement for the process. Of note, MDSCs that are expanded in the blood of tumor patients have been characterized as LDNs (132, 133). By extension, it is intruging to speculate that the LDNs described in diabetic patients are actually MDSCs, which might explain why these individuals are more prone to chronic and recurrent S. aureus infections, although this remains to be determined.
Hypoxia and S. aureus Metabolism
Hypoxia is particularly relevant in the context of biofilm infections, since bacteria in the innermost regions of the biofilm experience low oxygen bioavailability. Anaerobic conditions have been shown to induce the expression of biofilm-associated genes such as icaADBC that encode polysaccharide intercellular adhesins that promote bacterial aggregation and adherence to host surfaces (134–136). Bone, a niche that is often targeted by S. aureus biofilm, has a low oxygen tension since it is less vascularized compared to other tissues (137, 138). The immune response elicited during S. aureus bone infection exacerbates this hypoxic environment as infiltrating immune cells quickly increase their oxygen demand upon activation (139, 140). The biofilm responds to this progressive decline in oxygen tension by switching from respiration to fermentation concomitant with an increase in virulence factor production to attack immune cells to promote infection persistence (141). The transition from respiration to fermentation in S. aureus is regulated by several factors including SrrAB and Rex. The SrrAB two-component system was initially identified through homology comparisons with the ResDE two-component system of Bacillus subtilis, which controls the switch between aerobic and anaerobic respiration (142). While its ligand remains unknown, SrrAB increases the expression of fermentative genes such as pflAB, adhE, and nrdDG that allow it to thrive in an anaerobic environment (141, 143, 144). S. aureus can also respond to changes in oxygen availability indirectly through the transcriptional repressor Rex, which inhibits genes that are important for anaerobic respiration when oxygen is present. Rex senses redox conditions within the cell through changes in the NADH/NAD+ pools. As NADH levels rise, Rex becomes derepressed and is released from the DNA, thereby allowing for the transcription of fermentative genes in an effort to maintain reducing equivalents within the cell (9, 145). Therefore, oxygen depletion could either be a consequence of bacterial growth, or a strategy enacted by the biofilm to ensure persistent infection.
Epigenetic Changes Induced by Immunomodulatory Metabolites
Bacterial-derived metabolites are also capable of affecting leukocyte activation and function. Recent work has shown that lactate production by S. aureus biofilm induces epigenetic changes in leukocytes at the level of histone acetylation (146). Utilizing a number of S. aureus lactate dehydrogenase (ldh) mutants, our group demonstrated that biofilm-derived lactate was imported into MDSCs and macrophages where it inhibited histone deacetylase 11 (HDAC11; Figure 3). HDAC11 inhibition prevented its normal function of counterbalancing HDAC6 activity that is a positive regulator of IL-10 transcription, resulting in enhanced IL-10 production and biofilm persistence. Synovial fluid from patients with PJI contained elevated amounts of both D-lactate and IL-10 compared with control subjects and IL-10 production by human monocyte-derived macrophages was induced by biofilm-derived lactate, supporting the translational relevance of these findings (146). This demonstrates how a bacterial-derived metabolite can significantly rewire the host-pathogen dynamic to favor persistent infection. Recently, lactate has also been shown to directly modify histones with functional genomic implications (147–149). This lactate modification of histone lysine residues (termed lactylation) was shown to operate as a sort of “clock”. As inflammation progressed, the lactate produced by increased glycolysis lactylated histones. As these lactate marks accumulated, homeostatic genes were induced that led to a resolution of inflammation. Thus, histone lactylation functions as an endogenous timer for inflammatory events.
Figure 3
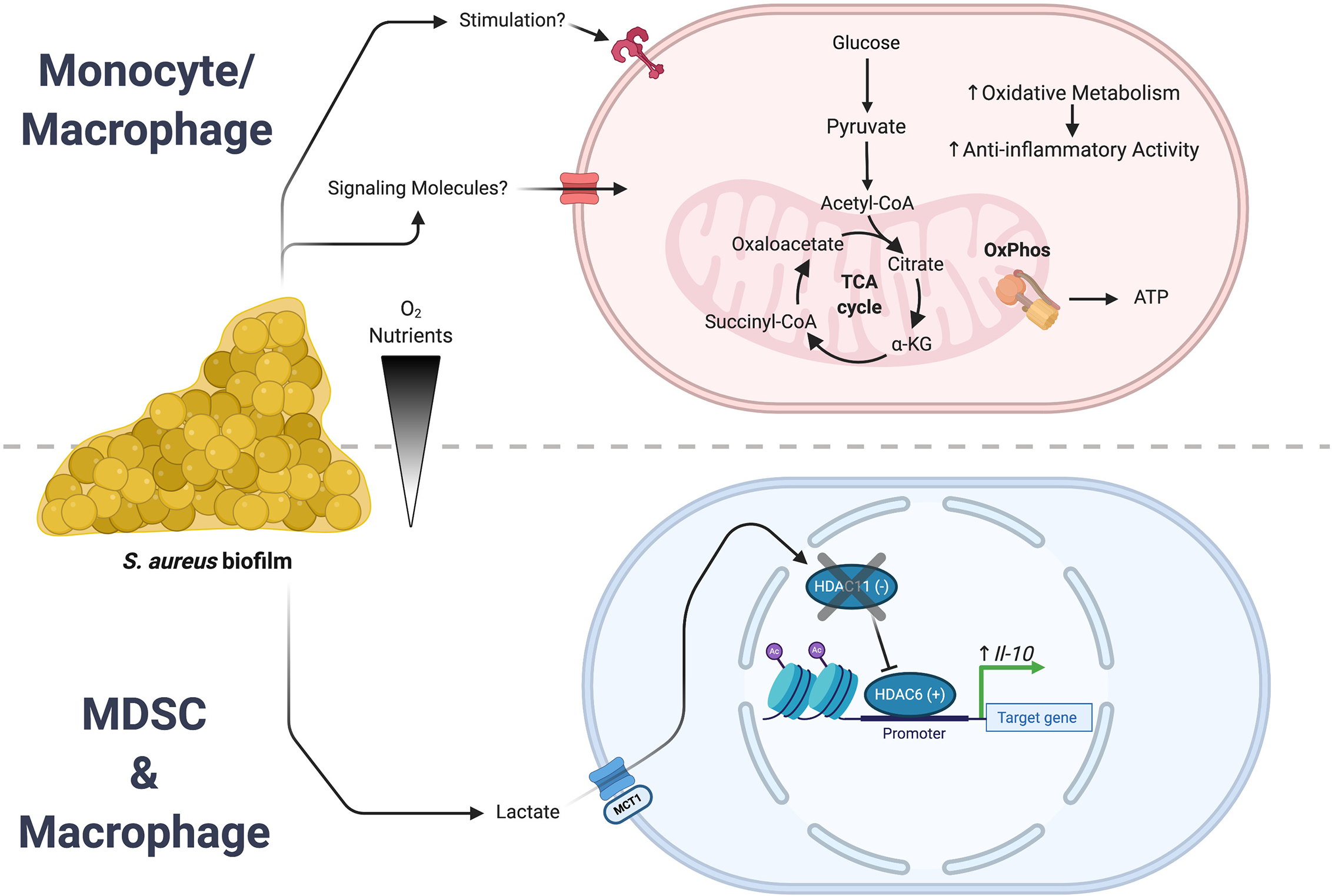
S. aureus biofilm regulates leukocyte inflammatory activity. S. aureus biofilms employ a number of strategies to create an infection milieu to ensure persistence. Two of these approaches involve the action of either bacterial-derived metabolites or intrinsic reprogramming of monocyte/macrophage metabolism that culminate in the expression of anti-inflammatory genes. (Top) Biofilm augments oxidative metabolism in infiltrating monocytes and macrophages that biases them towards an anti-inflammatory phenotype. (Bottom) In MDSCs and macrophages, biofilm-derived lactate causes epigenomic remodeling that leads to an increase in IL-10 expression. Figure created with BioRender.
While innate immune cells do not possess the characteristic long-lasting immunity of lymphocytes, they are capable of trained immune memory (150, 151). Trained immunity arises from epigenetic modifications that prime leukocytes for a subsequent encounter with another stimulus (152–154). Interestingly, trained immunity is not stimulus-specific and can induce widespread changes in how a leukocyte responds to a second insult. Several studies have shown that prior infection with S. aureus establishes trained immunity in macrophages that provides temporary protection from a second bacterial challenge (155–157). Trained immunity in response to S. aureus has been shown to occur via epigenetic changes induced by the metabolite fumarate. Fumarate, like lactate, interferes with the epigenomic remodeling of histone marks by acting as an antagonist for lysine demethylases (KDMs) (158–162). A recent study demonstrated the importance of fumarate during S. aureus infection by contrasting wild type infection with ΔhemB small colony variants (SCVs) that were able to deplete local fumarate stores via enhanced fumarate hydratase (fumC) expression (163, 164). Trained immunity was assessed by comparing bacterial burden after a secondary challenge with S. aureus 28 days following the primary infection. The reduction in fumarate by SCVs increased host cell glycolysis, which inhibited trained immunity by necroptosis (Figure 4) rather than inflammasome-dependent pyroptosis (165). This is beneficial for bacterial persistence since necroptosis elicits less inflammation compared to pyroptosis (166). These effects were not observed with wild type S. aureus, which was less effective at utilizing fumarate. This difference may represent one explanation for why S. aureus SCVs are typically associated with chronic infections.
Figure 4
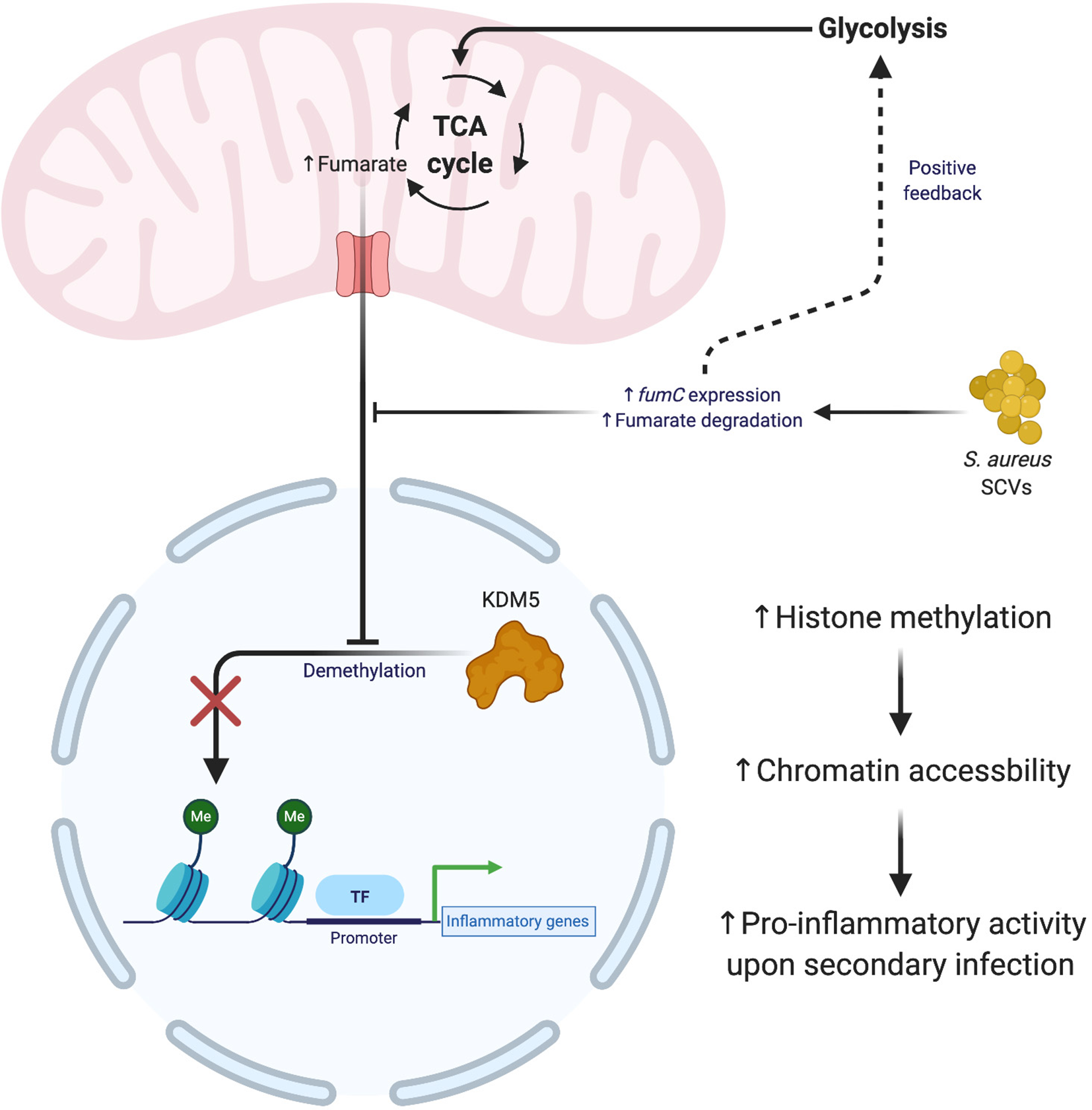
S. aureus small colony variants (SCVs) interfere with the establishment of trained immunity. The generation of SCVs with mutations in metabolic pathways exert influence over the formation of trained immunity. S. aureus SCVs with hyperactive fumarate hydratase (fumC) quickly degrade mitochondrial pools of fumarate, which leads to an upregulation of glycolysis and impaired formation of trained immunity. The absence of fumuarate causes KDM5 to become active, which removes the methylation marks around the promoters of pro-inflammatory genes, thereby decreasing the accessilibity of the chromatin in these regions. Figure created with BioRender.
Modulating Metabolism as a Therapeutic Target
The selective targeting of either host or pathogen metabolism represents an exciting therapeutic prospect that would bolster traditional antibiotic therapies that are commonly used for infections. Metabolic interventions have the potential to be highly efficacious as was shown in our work where metabolically reprogramming monocytes synergized with antibiotics to reduce established S. aureus biofilm infection to below the limit of detection (81). Of particular interest are therapies targeting immunometabolism rather than bacterial metabolism, since pathogen resistance would be less prevalent with a host-targeted approach. If we can gain a better understanding of the underlying metabolic modules that dictate beneficial vs. detrimental immune responses, then it should be feasible to tailor leukocyte metabolism to enhance pathogen neutralization. The main caveat of this approach is that the metabolic pathways utilized by the host and pathogens share many attributes. Therefore, potential metabolic therapies would need to be targeted to avoid non-specific effects.
Conclusion and Future Directions
In a time of increasing interest in leukocyte metabolism, it is important to think globally in terms of the interplay between host and pathogen metabolic states. Bacterial pathogens, such as S. aureus, undergo their own metabolic programming (10, 11), often in direct competition with responding immune cells. In addition, the metabolic attributes of tissue resident parenchymal cells have the potential to shape the metabolic responses of both infiltrating leukocytes and bacteria, revealing another level of complexity. Considerable care must be taken when designing experiments to deconstruct these complex systems in vitro, since the composition of mammalian cell culture media can have drastic deviations from metabolite concentrations found in human plasma (167). Therefore, findings must be validated in leukocytes and bacteria immediately ex vivo, as the selection of in vitro culture conditions could result in metabolic changes that are not reflective of in vivo infection (167).
As discussed in this review, S. aureus as well as other bacterial pathogens possess the ability to not only modify their metabolism, but also that of the host. Further work is needed to understand the numerous intricacies that underpin metabolism, especially in the context of an infection where multiple players are involved. Fortunately, the field is advancing rapidly with the development of novel tools and methods to dissect metabolism with single cell resolution (168, 169). The continued development of these technologies will be integral for future studies. Should these efforts be successful, metabolic modulation could advance therapeutic approaches for S. aureus infection in combination with conventional antibiotic treatment regimens.
Funding
The Kielian laboratory is supported by NIH grants R01 NS107369 and 3P01AI083211 (Project 4 to TK).
Statements
Author contributions
CH wrote the manuscript draft that was edited by TK and CH. All authors contributed to the article and approved the submitted version.
Acknowledgments
We apologize to authors whose work could not be cited due to space constraints.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Lee AS de Lencastre H Garau J Kluytmans J Malhotra-Kumar S Peschel A et al . Methicillin-resistant Staphylococcus aureus. Nat Rev Dis Primers (2018) 4:18033.
2
Turner NA Sharma-Kuinkel BK Maskarinec SA Eichenberger EM Shah PP Carugati M et al . Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat Rev Microbiol (2019) 17(4):203–18.
3
Schilcher K Horswill AR . Staphylococcal Biofilm Development: Structure, Regulation, and Treatment Strategies. Microbiol Mol Biol Rev (2020) 84(3):e00026–19.
4
Tam K Torres VJ . Staphylococcus aureus Secreted Toxins and Extracellular Enzymes. Microbiol Spectr (2019) 7(2). doi: 10.1128/microbiolspec.GPP3-0039-2018
5
Foster TJ . Immune evasion by staphylococci. Nat Rev Microbiol (2005) 3(12):948–58.
6
Askarian F Wagner T Johannessen M Nizet V . Staphylococcus aureus modulation of innate immune responses through Toll-like (TLR), (NOD)-like (NLR) and C-type lectin (CLR) receptors. FEMS Microbiol Rev (2018) 42(5):656–71.
7
Olive AJ Sassetti CM . Metabolic crosstalk between host and pathogen: sensing, adapting and competing. Nat Rev Microbiol (2016) 14(4):221–34.
8
Ayres JS . Immunometabolism of infections. Nat Rev Immunol (2020) 20(2):79–80.
9
Somerville GA Proctor RA . At the crossroads of bacterial metabolism and virulence factor synthesis in Staphylococci. Microbiol Mol Biol Rev (2009) 73(2):233–48.
10
Potter AD Butrico CE Ford CA Curry JM Trenary IA Tummarakota SS et al . Host nutrient milieu drives an essential role for aspartate biosynthesis during invasive Staphylococcus aureus infection. Proc Natl Acad Sci USA (2020) 117(22):12394–401.
11
Lehman MK Nuxoll AS Yamada KJ Kielian T Carson SD Fey PD . Protease-Mediated Growth of Staphylococcus aureus on Host Proteins Is opp3 Dependent. mBio (2019) 10(2):e02553–18.
12
Riquelme SA Wong Fok Lung T Prince A . Pulmonary Pathogens Adapt to Immune Signaling Metabolites in the Airway. Front Immunol (2020) 11:385.
13
Mazharul Islam M Thomas VC Van Beek M Ahn JS Alqarzaee AA Zhou C et al . An integrated computational and experimental study to investigate Staphylococcus aureus metabolism. NPJ Syst Biol Appl (2020) 6(1):3.
14
Mathis D Shoelson SE . Immunometabolism: an emerging frontier. Nat Rev Immunol (2011) 11(2):81.
15
O’Neill LA Kishton RJ Rathmell J . A guide to immunometabolism for immunologists. Nat Rev Immunol (2016) 16(9):553–65.
16
Pearce EL Pearce EJ . Metabolic pathways in immune cell activation and quiescence. Immunity (2013) 38(4):633–43.
17
Galvan-Pena S O’Neill LA . Metabolic reprograming in macrophage polarization. Front Immunol (2014) 5:420.
18
Infantino V Convertini P Cucci L Panaro MA Di Noia MA Calvello R et al . The mitochondrial citrate carrier: a new player in inflammation. Biochem J (2011) 438(3):433–6.
19
Ricciotti E FitzGerald GA . Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol (2011) 31(5):986–1000.
20
O’Neill LA . A critical role for citrate metabolism in LPS signalling. Biochem J (2011) 438(3):e5–6.
21
Traven A Naderer T . Central metabolic interactions of immune cells and microbes: prospects for defeating infections. EMBO Rep (2019) 20(7):e47995.
22
Tuchscherr L Loffler B Proctor RA . Persistence of Staphylococcus aureus: Multiple Metabolic Pathways Impact the Expression of Virulence Factors in Small-Colony Variants (SCVs). Front Microbiol (2020) 11:1028.
23
Richardson AR . Virulence and Metabolism. Microbiol Spectr (2019) 7(2) doi: 10.1128/microbiolspec.GPP3-0011-2018
24
Balasubramanian D Harper L Shopsin B Torres VJ . Staphylococcus aureus pathogenesis in diverse host environments. Pathog Dis (2017) 75(1). doi: 10.1093/femspd/ftx005
25
Heim CE Vidlak D Kielian T . Interleukin-10 production by myeloid-derived suppressor cells contributes to bacterial persistence during Staphylococcus aureus orthopedic biofilm infection. J Leukoc Biol (2015) 98(6):1003–13.
26
Heim CE Vidlak D Scherr TD Hartman CW Garvin KL Kielian T . IL-12 promotes myeloid-derived suppressor cell recruitment and bacterial persistence during Staphylococcus aureus orthopedic implant infection. J Immunol (2015) 194(8):3861–72.
27
Heim CE Vidlak D Scherr TD Kozel JA Holzapfel M Muirhead DE et al . Myeloid-derived suppressor cells contribute to Staphylococcus aureus orthopedic biofilm infection. J Immunol (2014) 192(8):3778–92.
28
Heim CE West SC Ali H Kielian T . Heterogeneity of Ly6G(+) Ly6C(+) Myeloid-Derived Suppressor Cell Infiltrates during Staphylococcus aureus Biofilm Infection. Infect Immun (2018) 86(12):e00684–18.
29
Bosch ME Bertrand BP Heim CE Alqarzaee AA Chaudhari SS Aldrich AL et al . Staphylococcus aureus ATP Synthase Promotes Biofilm Persistence by Influencing Innate Immunity. mBio (2020) 11(5):e01581–20.
30
Grosser MR Paluscio E Thurlow LR Dillon MM Cooper VS Kawula TH et al . Genetic requirements for Staphylococcus aureus nitric oxide resistance and virulence. PloS Pathog (2018) 14(3):e1006907.
31
Gabryszewski SJ Wong Fok Lung T Annavajhala MK Tomlinson KL Riquelme SA Khan IN et al . Metabolic Adaptation in Methicillin-Resistant Staphylococcus aureus Pneumonia. Am J Respir Cell Mol Biol (2019) 61(2):185–97.
32
Newsholme P Gordon S Newsholme EA . Rates of utilization and fates of glucose, glutamine, pyruvate, fatty acids and ketone bodies by mouse macrophages. Biochem J (1987) 242(3):631–6.
33
Freemerman AJ Johnson AR Sacks GN Milner JJ Kirk EL Troester MA et al . Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem (2014) 289(11):7884–96.
34
Fukuzumi M Shinomiya H Shimizu Y Ohishi K Utsumi S . Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT1. Infect Immun (1996) 64(1):108–12.
35
Jha AK Huang SC Sergushichev A Lampropoulou V Ivanova Y Loginicheva E et al . Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity (2015) 42(3):419–30.
36
Rodriguez-Prados JC Traves PG Cuenca J Rico D Aragones J Martin-Sanz P et al . Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol (2010) 185(1):605–14.
37
Kelly B O’Neill LA . Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res (2015) 25(7):771–84.
38
Ryan DG Murphy MP Frezza C Prag HA Chouchani ET O’Neill LA et al . Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat Metab (2019) 1:16–33.
39
Lachmandas E Boutens L Ratter JM Hijmans A Hooiveld GJ Joosten LA et al . Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat Microbiol (2016) 2:16246.
40
Tannahill GM Curtis AM Adamik J Palsson-McDermott EM McGettrick AF Goel G et al . Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496(7444):238–42.
41
Mills E O’Neill LA . Succinate: a metabolic signal in inflammation. Trends Cell Biol (2014) 24(5):313–20.
42
Mills EL Kelly B Logan A Costa ASH Varma M Bryant CE et al . Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell (2016) 167(2):457–70.
43
Wang D Malo D Hekimi S . Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1alpha in long-lived Mclk1+/- mouse mutants. J Immunol (2010) 184(2):582–90.
44
Benit P Letouze E Rak M Aubry L Burnichon N Favier J et al . Unsuspected task for an old team: succinate, fumarate and other Krebs cycle acids in metabolic remodeling. Biochim Biophys Acta (2014) 1837(8):1330–7.
45
Swanson KV Deng M Ting JP . The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89.
46
Hughes MM O’Neill LAJ . Metabolic regulation of NLRP3. Immunol Rev (2018) 281(1):88–98.
47
Haneklaus M O’Neill LA . NLRP3 at the interface of metabolism and inflammation. Immunol Rev (2015) 265(1):53–62.
48
McGettrick AF O’Neill LAJ . The Role of HIF in Immunity and Inflammation. Cell Metab (2020) 32(4):524–36.
49
Cramer T Yamanishi Y Clausen BE Forster I Pawlinski R Mackman N et al . HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell (2003) 112(5):645–57.
50
Craven RR Gao X Allen IC Gris D Bubeck Wardenburg J McElvania-Tekippe E et al . Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PloS One (2009) 4(10):e7446.
51
Liu X Zhang Z Ruan J Pan Y Magupalli VG Wu H et al . Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535(7610):153–8.
52
Munoz-Planillo R Franchi L Miller LS Nunez G . A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol (2009) 183(6):3942–8.
53
Wang X Eagen WJ Lee JC . Orchestration of human macrophage NLRP3 inflammasome activation by Staphylococcus aureus extracellular vesicles. Proc Natl Acad Sci U S A (2020) 117(6):3174–84.
54
Cohen TS Boland ML Boland BB Takahashi V Tovchigrechko A Lee Y et al . S. aureus Evades Macrophage Killing through NLRP3-Dependent Effects on Mitochondrial Trafficking. Cell Rep (2018) 22(9):2431–41.
55
Michelucci A Cordes T Ghelfi J Pailot A Reiling N Goldmann O et al . Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci USA (2013) 110(19):7820–5.
56
Degrandi D Hoffmann R Beuter-Gunia C Pfeffer K . The proinflammatory cytokine-induced IRG1 protein associates with mitochondria. J Interferon Cytokine Res (2009) 29(1):55–67.
57
Ackermann WW Potter VR . Enzyme inhibition in relation to chemotherapy. Proc Soc Exp Biol Med (1949) 72(1):1–9.
58
Nemeth B Doczi J Csete D Kacso G Ravasz D Adams D et al . Abolition of mitochondrial substrate-level phosphorylation by itaconic acid produced by LPS-induced Irg1 expression in cells of murine macrophage lineage. FASEB J (2016) 30(1):286–300.
59
Tomlinson K Dach F Riquelme SA Wong T Moustafa AM Planet PJ et al . In Vivo Adaptation of S. Aureus to the Lung Is Driven by Itaconate, A Mitochondrial Immune Metabolite. Am J Respiratory Crit Care Med (2020) doi: 10.1164/ajrccm-conference.2020.201.1_MeetingAbstracts.A7715
60
O’Neill LAJ Artyomov MN . Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol (2019) 19(5):273–81.
61
Mills EL Ryan DG Prag HA Dikovskaya D Menon D Zaslona Z et al . Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature (2018) 556(7699):113–7.
62
Bambouskova M Gorvel L Lampropoulou V Sergushichev A Loginicheva E Johnson K et al . Electrophilic properties of itaconate and derivatives regulate the IkappaBzeta-ATF3 inflammatory axis. Nature (2018) 556(7702):501–4.
63
Saito R Suzuki T Hiramoto K Asami S Naganuma E Suda H et al . Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol Cell Biol (2016) 36(2):271–84.
64
Kobayashi EH Suzuki T Funayama R Nagashima T Hayashi M Sekine H et al . Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun (2016) 7:11624.
65
Luan HH Medzhitov R . Food Fight: Role of Itaconate and Other Metabolites in Antimicrobial Defense. Cell Metab (2016) 24(3):379–87.
66
Cordes T Wallace M Michelucci A Divakaruni AS Sapcariu SC Sousa C et al . Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J Biol Chem (2016) 291(27):14274–84.
67
Rath M Muller I Kropf P Closs EI Munder M . Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front Immunol (2014) 5:532.
68
MacMicking J Xie QW Nathan C . Nitric oxide and macrophage function. Annu Rev Immunol (1997) 15:323–50.
69
Fang FC . Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol (2004) 2(10):820–32.
70
Nathan CF Hibbs JB Jr. Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr Opin Immunol (1991) 3(1):65–70.
71
Sasaki S Miura T Nishikawa S Yamada K Hirasue M Nakane A . Protective role of nitric oxide in Staphylococcus aureus infection in mice. Infect Immun (1998) 66(3):1017–22.
72
Richardson AR Dunman PM Fang FC . The nitrosative stress response of Staphylococcus aureus is required for resistance to innate immunity. Mol Microbiol (2006) 61(4):927–39.
73
Richardson AR Libby SJ Fang FC . A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science (2008) 319(5870):1672–6.
74
Albina JE Mills CD Barbul A Thirkill CE Henry WL Jr. Mastrofrancesco B et al . Arginine metabolism in wounds. Am J Physiol (1988) 254(4 Pt 1):E459–67.
75
Sica A Mantovani A . Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122(3):787–95.
76
Hanke ML Heim CE Angle A Sanderson SD Kielian T . Targeting macrophage activation for the prevention and treatment of Staphylococcus aureus biofilm infections. J Immunol (2013) 190(5):2159–68.
77
Hanke ML Angle A Kielian T . MyD88-dependent signaling influences fibrosis and alternative macrophage activation during Staphylococcus aureus biofilm infection. PloS One (2012) 7(8):e42476.
78
Heim CE Hanke ML Kielian T . A mouse model of Staphylococcus catheter-associated biofilm infection. Methods Mol Biol (2014) 1106:183–91.
79
Yamada KJ Heim CE Aldrich AL Gries CM Staudacher AG Kielian T . Arginase-1 Expression in Myeloid Cells Regulates Staphylococcus aureus Planktonic but Not Biofilm Infection. Infect Immun (2018) 86(7):e00206–18.
80
Thurlow LR Joshi GS Clark JR Spontak JS Neely CJ Maile R et al . Functional modularity of the arginine catabolic mobile element contributes to the success of USA300 methicillin-resistant Staphylococcus aureus. Cell Host Microbe (2013) 13(1):100–7.
81
Yamada KJ Heim CE Xi X Attri KS Wang D Zhang W et al . Monocyte metabolic reprogramming promotes pro-inflammatory activity and Staphylococcus aureus biofilm clearance. PloS Pathog (2020) 16(3):e1008354.
82
Thurlow LR Hanke ML Fritz T Angle A Aldrich A Williams SH et al . Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J Immunol (2011) 186(11):6585–96.
83
Kominsky DJ Campbell EL Colgan SP . Metabolic shifts in immunity and inflammation. J Immunol (2010) 184(8):4062–8.
84
Sbarra AJ Karnovsky ML . The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J Biol Chem (1959) 234(6):1355–62.
85
Warburg O Wind F Negelein E . The Metabolism of Tumors in the Body. J Gen Physiol (1927) 8(6):519–30.
86
DeBerardinis RJ Chandel NS . We need to talk about the Warburg effect. Nat Metab (2020) 2(2):127–9.
87
Borregaard N Herlin T . Energy metabolism of human neutrophils during phagocytosis. J Clin Invest (1982) 70(3):550–7.
88
Dale DC Boxer L Liles WC . The phagocytes: neutrophils and monocytes. Blood (2008) 112(4):935–45.
89
Palsson-McDermott EM O’Neill LAJ . Targeting immunometabolism as an anti-inflammatory strategy. Cell Res (2020) 30(4):300–14.
90
Howard NC Khader SA . Immunometabolism during Mycobacterium tuberculosis Infection. Trends Microbiol (2020) 28(10):832–50.
91
O’Neill LA Pearce EJ . Immunometabolism governs dendritic cell and macrophage function. J Exp Med (2016) 213(1):15–23.
92
Kumar R Singh P Kolloli A Shi L Bushkin Y Tyagi S et al . Immunometabolism of Phagocytes During Mycobacterium tuberculosis Infection. Front Mol Biosci (2019) 6:105.
93
Rice CM Davies LC Subleski JJ Maio N Gonzalez-Cotto M Andrews C et al . Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat Commun (2018) 9(1):5099.
94
Corzo CA Cotter MJ Cheng P Cheng F Kusmartsev S Sotomayor E et al . Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol (2009) 182(9):5693–701.
95
Chen X Song M Zhang B Zhang Y . Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid Med Cell Longev (2016) 2016:1580967.
96
Zhang H Li ZL Ye SB Ouyang LY Chen YS He J et al . Myeloid-derived suppressor cells inhibit T cell proliferation in human extranodal NK/T cell lymphoma: a novel prognostic indicator. Cancer Immunol Immunother (2015) 64(12):1587–99.
97
Gabrilovich DI Nagaraj S . Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74.
98
Ostrand-Rosenberg S Fenselau C . Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J Immunol (2018) 200(2):422–31.
99
Mastio J Condamine T Dominguez G Kossenkov AV Donthireddy L Veglia F et al . Identification of monocyte-like precursors of granulocytes in cancer as a mechanism for accumulation of PMN-MDSCs. J Exp Med (2019) 216(9):2150–69.
100
Ohl K Tenbrock K . Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front Immunol (2018) 9:2499.
101
Aarts CEM Hiemstra IH Beguin EP Hoogendijk AJ Bouchmal S van Houdt M et al . Activated neutrophils exert myeloid-derived suppressor cell activity damaging T cells beyond repair. Blood Adv (2019) 3(22):3562–74.
102
Tebartz C Horst SA Sparwasser T Huehn J Beineke A Peters G et al . A major role for myeloid-derived suppressor cells and a minor role for regulatory T cells in immunosuppression during Staphylococcus aureus infection. J Immunol (2015) 194(3):1100–11.
103
Skabytska Y Wolbing F Gunther C Koberle M Kaesler S Chen KM et al . Cutaneous innate immune sensing of Toll-like receptor 2-6 ligands suppresses T cell immunity by inducing myeloid-derived suppressor cells. Immunity (2014) 41(5):762–75.
104
Wang Y Jia A Bi Y Wang Y Liu G . Metabolic Regulation of Myeloid-Derived Suppressor Cell Function in Cancer. Cells (2020) 9(4). doi: 10.3390/cells9041011
105
Sica A Strauss L . Energy metabolism drives myeloid-derived suppressor cell differentiation and functions in pathology. J Leukoc Biol (2017) 102(2):325–34.
106
Yan D Adeshakin AO Xu M Afolabi LO Zhang G Chen YH et al . Lipid Metabolic Pathways Confer the Immunosuppressive Function of Myeloid-Derived Suppressor Cells in Tumor. Front Immunol (2019) 10:1399.
107
Baumann T Dunkel A Schmid C Schmitt S Hiltensperger M Lohr K et al . Regulatory myeloid cells paralyze T cells through cell-cell transfer of the metabolite methylglyoxal. Nat Immunol (2020) 21(5):555–66.
108
Ramesh V Locasale JW . A reactive metabolite as an immune suppressant. Nat Immunol (2020) 21(5):497–8.
109
Veglia F Tyurin VA Blasi M De Leo A Kossenkov AV Donthireddy L et al . Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature (2019) 569(7754):73–8.
110
Riquelme SA Liimatta K Wong Fok Lung T Fields B Ahn D Chen D et al . Pseudomonas aeruginosa Utilizes Host-Derived Itaconate to Redirect Its Metabolism to Promote Biofilm Formation. Cell Metab (2020) 31(6):1091–106 e6.
111
Fischer AJ Singh SB LaMarche MM Maakestad LJ Kienenberger ZE Pena TA et al . Sustained Coinfections with Staphylococcus aureus and Pseudomonas aeruginosa in Cystic Fibrosis. Am J Respir Crit Care Med (2020). doi: 10.1164/rccm.202004-1322OC
112
Bernardy EE Petit R Raghuram V Alexander AM Read TD Goldberg JB . Genotypic and Phenotypic Diversity of Staphylococcus aureus Isolates from Cystic Fibrosis Patient Lung Infections and Their Interactions with Pseudomonas aeruginosa. mBio (2020) 11(3):e00735–20.
113
Limoli DH Hoffman LR . Help, hinder, hide and harm: what can we learn from the interactions between Pseudomonas aeruginosa and Staphylococcus aureus during respiratory infections? Thorax (2019) 74(7):684–92.
114
Nagata S Tanaka M . Programmed cell death and the immune system. Nat Rev Immunol (2017) 17(5):333–40.
115
Rapun-Araiz B Haag AF De Cesare V Gil C Dorado-Morales P Penades JR et al . Systematic Reconstruction of the Complete Two-Component Sensorial Network in Staphylococcus aureus. mSystems (2020) 5(4):e00511–20.
116
Bronner S Monteil H Prevost G . Regulation of virulence determinants in Staphylococcus aureus: complexity and applications. FEMS Microbiol Rev (2004) 28(2):183–200.
117
Titgemeyer F Hillen W . Global control of sugar metabolism: a gram-positive solution. Antonie Van Leeuwenhoek (2002) 82(1-4):59–71.
118
Gorke B Stulke J . Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol (2008) 6(8):613–24.
119
Warner JB Lolkema JS . CcpA-dependent carbon catabolite repression in bacteria. Microbiol Mol Biol Rev (2003) 67(4):475–90.
120
Strasters KC Winkler KC . Carbohydrate Metabolism of Staphylococcus Aureus. J Gen Microbiol (1963) 33:213–29.
121
Seidl K Goerke C Wolz C Mack D Berger-Bachi B Bischoff M . Staphylococcus aureus CcpA affects biofilm formation. Infect Immun (2008) 76(5):2044–50.
122
Seidl K Muller S Francois P Kriebitzsch C Schrenzel J Engelmann S et al . Effect of a glucose impulse on the CcpA regulon in Staphylococcus aureus. BMC Microbiol (2009) 9:95.
123
Hartmann T Zhang B Baronian G Schulthess B Homerova D Grubmuller S et al . Catabolite control protein E (CcpE) is a LysR-type transcriptional regulator of tricarboxylic acid cycle activity in Staphylococcus aureus. J Biol Chem (2013) 288(50):36116–28.
124
Ding Y Liu X Chen F Di H Xu B Zhou L et al . Metabolic sensor governing bacterial virulence in Staphylococcus aureus. Proc Natl Acad Sci USA (2014) 111(46):E4981–90.
125
Wickersham M Wachtel S Wong Fok Lung T Soong G Jacquet R Richardson A et al . Metabolic Stress Drives Keratinocyte Defenses against Staphylococcus aureus Infection. Cell Rep (2017) 18(11):2742–51.
126
Prince A Wong Fok Lung T . Consequences of Metabolic Interactions during Staphylococcus aureus Infection. Toxins (Basel) (2020) 12(9):581. doi: 10.3390/toxins12090581
127
Smit J Sogaard M Schonheyder HC Nielsen H Froslev T Thomsen RW . Diabetes and risk of community-acquired Staphylococcus aureus bacteremia: a population-based case-control study. Eur J Endocrinol (2016) 174(5):631–9.
128
Lipsky BA Tabak YP Johannes RS Vo L Hyde L Weigelt JA . Skin and soft tissue infections in hospitalised patients with diabetes: culture isolates and risk factors associated with mortality, length of stay and cost. Diabetologia (2010) 53(5):914–23.
129
Thurlow LR Stephens AC Hurley KE Richardson AR . Lack of nutritional immunity in diabetic skin infections promotes <em<Staphylococcus aureus</em< virulence. Sci Adv (2020) 6(46):eabc5569.
130
Cohen TS Takahashi V Bonnell J Tovchigrechko A Chaerkady R Yu W et al . Staphylococcus aureus drives expansion of low-density neutrophils in diabetic mice. J Clin Invest (2019) 129(5):2133–44.
131
Wong SL Demers M Martinod K Gallant M Wang Y Goldfine AB et al . Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med (2015) 21(7):815–9.
132
Sagiv JY Michaeli J Assi S Mishalian I Kisos H Levy L et al . Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep (2015) 10(4):562–73.
133
Brandau S Trellakis S Bruderek K Schmaltz D Steller G Elian M et al . Myeloid-derived suppressor cells in the peripheral blood of cancer patients contain a subset of immature neutrophils with impaired migratory properties. J Leukoc Biol (2011) 89(2):311–7.
134
Cramton SE Ulrich M Gotz F Doring G . Anaerobic conditions induce expression of polysaccharide intercellular adhesin in Staphylococcus aureus and Staphylococcus epidermidis. Infect Immun (2001) 69(6):4079–85.
135
Vuong C Kocianova S Voyich JM Yao Y Fischer ER DeLeo FR et al . A crucial role for exopolysaccharide modification in bacterial biofilm formation, immune evasion, and virulence. J Biol Chem (2004) 279(52):54881–6.
136
O’Gara JP . ica and beyond: biofilm mechanisms and regulation in Staphylococcus epidermidis and Staphylococcus aureus. FEMS Microbiol Lett (2007) 270(2):179–88.
137
Spencer JA Ferraro F Roussakis E Klein A Wu J Runnels JM et al . Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature (2014) 508(7495):269–73.
138
Mader JT Brown GL Guckian JC Wells CH Reinarz JA . A mechanism for the amelioration by hyperbaric oxygen of experimental staphylococcal osteomyelitis in rabbits. J Infect Dis (1980) 142(6):915–22.
139
Gabig TG Bearman SI Babior BM . Effects of oxygen tension and pH on the respiratory burst of human neutrophils. Blood (1979) 53(6):1133–9.
140
Colgan SP Taylor CT . Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol (2010) 7(5):281–7.
141
Wilde AD Snyder DJ Putnam NE Valentino MD Hammer ND Lonergan ZR et al . Bacterial Hypoxic Responses Revealed as Critical Determinants of the Host-Pathogen Outcome by TnSeq Analysis of Staphylococcus aureus Invasive Infection. PloS Pathog (2015) 11(12):e1005341.
142
Yarwood JM McCormick JK Schlievert PM . Identification of a novel two-component regulatory system that acts in global regulation of virulence factors of Staphylococcus aureus. J Bacteriol (2001) 183(4):1113–23.
143
Tiwari N Lopez-Redondo M Miguel-Romero L Kulhankova K Cahill MP Tran PM et al . The SrrAB two-component system regulates Staphylococcus aureus pathogenicity through redox sensitive cysteines. Proc Natl Acad Sci USA (2020) 117(20):10989–99.
144
Kinkel TL Roux CM Dunman PM Fang FC . The Staphylococcus aureus SrrAB two-component system promotes resistance to nitrosative stress and hypoxia. mBio (2013) 4(6):e00696–13.
145
Pagels M Fuchs S Pane-Farre J Kohler C Menschner L Hecker M et al . Redox sensing by a Rex-family repressor is involved in the regulation of anaerobic gene expression in Staphylococcus aureus. Mol Microbiol (2010) 76(5):1142–61.
146
Heim CE Bosch ME Yamada KJ Aldrich AL Chaudhari SS Klinkebiel D et al . Lactate production by Staphylococcus aureus biofilm inhibits HDAC11 to reprogramme the host immune response during persistent infection. Nat Microbiol (2020) 5(10):1271–84.
147
Zhang D Tang Z Huang H Zhou G Cui C Weng Y et al . Metabolic regulation of gene expression by histone lactylation. Nature (2019) 574(7779):575–80.
148
Liberti MV Locasale JW . Histone Lactylation: A New Role for Glucose Metabolism. Trends Biochem Sci (2020) 45(3):179–82.
149
Williams ER . Lactylation drives resolution. Sci Signaling (2019) 12(606):eaba0502.
150
Netea MG Joosten LA Latz E Mills KH Natoli G Stunnenberg HG et al . Trained immunity: A program of innate immune memory in health and disease. Science (2016) 352(6284):aaf1098.
151
Arts RJ Joosten LA Netea MG . Immunometabolic circuits in trained immunity. Semin Immunol (2016) 28(5):425–30.
152
van der Heijden C Noz MP Joosten LAB Netea MG Riksen NP Keating ST . Epigenetics and Trained Immunity. Antioxid Redox Signal (2018) 29(11):1023–40.
153
Peignier A Parker D . Trained immunity and host-pathogen interactions. Cell Microbiol (2020) 22:e13261.
154
Saeed S Quintin J Kerstens HH Rao NA Aghajanirefah A Matarese F et al . Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science (2014) 345(6204):1251086.
155
Feuerstein R Forde AJ Lohrmann F Kolter J Ramirez NJ Zimmermann J et al . Resident macrophages acquire innate immune memory in staphylococcal skin infection. Elife (2020) 9:e55602. doi: 10.7554/eLife.55602
156
Chan LC Rossetti M Miller LS Filler SG Johnson CW Lee HK et al . Protective immunity in recurrent Staphylococcus aureus infection reflects localized immune signatures and macrophage-conferred memory. Proc Natl Acad Sci USA (2018) 115(47):E11111–9.
157
Chan LC Chaili S Filler SG Miller LS Solis NV Wang H et al . Innate Immune Memory Contributes to Host Defense against Recurrent Skin and Skin Structure Infections Caused by Methicillin-Resistant Staphylococcus aureus. Infect Immun (2017) 85(2):e00876–16.
158
Lu C Ward PS Kapoor GS Rohle D Turcan S Abdel-Wahab O et al . IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature (2012) 483(7390):474–8.
159
Xiao M Yang H Xu W Ma S Lin H Zhu H et al . Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev (2012) 26(12):1326–38.
160
Kleinnijenhuis J Quintin J Preijers F Joosten LA Ifrim DC Saeed S et al . Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA (2012) 109(43):17537–42.
161
Arts RJ Blok BA Aaby P Joosten LA de Jong D van der Meer JW et al . Long-term in vitro and in vivo effects of gamma-irradiated BCG on innate and adaptive immunity. J Leukoc Biol (2015) 98(6):995–1001.
162
Arts RJ Novakovic B Ter Horst R Carvalho A Bekkering S Lachmandas E et al . Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab (2016) 24(6):807–19.
163
Wong Fok Lung T Monk IR Acker KP Mu A Wang N Riquelme SA et al . Staphylococcus aureus small colony variants impair host immunity by activating host cell glycolysis and inducing necroptosis. Nat Microbiol (2020) 5(1):141–53.
164
Kriegeskorte A Grubmuller S Huber C Kahl BC von Eiff C Proctor RA et al . Staphylococcus aureus small colony variants show common metabolic features in central metabolism irrespective of the underlying auxotrophism. Front Cell Infect Microbiol (2014) 4:141.
165
Acker KP Wong Fok Lung T West E Craft J Narechania A Smith H et al . Strains of Staphylococcus aureus that Colonize and Infect Skin Harbor Mutations in Metabolic Genes. iScience (2019) 19:281–90.
166
Kitur K Wachtel S Brown A Wickersham M Paulino F Penaloza HF et al . Necroptosis Promotes Staphylococcus aureus Clearance by Inhibiting Excessive Inflammatory Signaling. Cell Rep (2016) 16(8):2219–30.
167
Lagziel S Gottlieb E Shlomi T . Mind your media. Nat Metab (2020) 2:1369–72.
168
Artyomov MN Van den Bossche J . Immunometabolism in the Single-Cell Era. Cell Metab (2020) 32:710–25.
169
Arguello RJ Combes AJ Char R Gigan JP Baaziz AI Bousiquot E et al . SCENITH: A Flow Cytometry-Based Method to Functionally Profile Energy Metabolism with Single-Cell Resolution. Cell Metab (2020) 32(6):1063–75.e7.
Summary
Keywords
Staphylococcus aureus , biofilm, immunometabolism, macrophage, myeloid-derived suppressor cell, lactate
Citation
Horn CM and Kielian T (2021) Crosstalk Between Staphylococcus aureus and Innate Immunity: Focus on Immunometabolism. Front. Immunol. 11:621750. doi: 10.3389/fimmu.2020.621750
Received
26 October 2020
Accepted
21 December 2020
Published
05 February 2021
Volume
11 - 2020
Edited by
Rachel McLoughlin, Trinity College Dublin, Ireland
Reviewed by
Frederick J. Sheedy, Trinity College Dublin, Ireland; Alice Prince, Columbia University, United States
Updates
Copyright
© 2021 Horn and Kielian.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tammy Kielian, tkielian@unmc.edu
This article was submitted to Vaccines and Molecular Therapeutics, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.