 Artem Mansurkhodzhaev
Artem Mansurkhodzhaev Camila R. R. Barbosa2
Camila R. R. Barbosa2 Michele Mishto
Michele Mishto Juliane Liepe
Juliane Liepe- 1Quantitative and Systems Biology, Max-Planck-Institute for Biophysical Chemistry, Göttingen, Germany
- 2Centre for Inflammation Biology and Cancer Immunology (CIBCI) and Peter Gorer Department of Immunobiology, King's College London, London, United Kingdom
- 3Francis Crick Institute, London, United Kingdom
The human immune system relies on the capability of CD8+ T cells to patrol body cells, spot infected cells and eliminate them. This cytotoxic response is supposed to be limited to infected cells to avoid killing of healthy cells. To enable this, CD8+ T cells have T Cell Receptors (TCRs) which should discriminate between self and non-self through the recognition of antigenic peptides bound to Human Leukocyte Antigen class I (HLA-I) complexes—i.e., HLA-I immunopeptidomes—of patrolled cells. The majority of these antigenic peptides are produced by proteasomes through either peptide hydrolysis or peptide splicing. Proteasome-generated cis-spliced peptides derive from a given antigen, are immunogenic and frequently presented by HLA-I complexes. Theoretically, they also have a very large sequence variability, which might impinge upon our model of self/non-self discrimination and central and peripheral CD8+ T cell tolerance. Indeed, a large variety of cis-spliced epitopes might enlarge the pool of viral-human zwitter epitopes, i.e., peptides that may be generated with the exact same sequence from both self (human) and non-self (viral) antigens. Antigenic viral-human zwitter peptides may be recognized by CD8+ thymocytes and T cells, induce clonal deletion or other tolerance processes, thereby restraining CD8+ T cell response against viruses. To test this hypothesis, we computed in silico the theoretical frequency of zwitter non-spliced and cis-spliced epitope candidates derived from human proteome (self) and from the proteomes of a large pool of viruses (non-self). We considered their binding affinity to the representative HLA-A*02:01 complex, self-antigen expression in Medullary Thymic Epithelial cells (mTECs) and the relative frequency of non-spliced and cis-spliced peptides in HLA-I immunopeptidomes. Based on the present knowledge of proteasome-catalyzed peptide splicing and neglecting CD8+ TCR degeneracy, our study suggests that, despite their frequency, the portion of the cis-spliced peptides we investigated could only marginally impinge upon the variety of functional CD8+ cytotoxic T cells (CTLs) involved in anti-viral response.
Introduction
CD8+ T cells are the ultimate response against viral infections. Their TCRαβ selectively recognizes viral epitope-HLA-I complexes, triggering a cytotoxic attack against infected cells in order to kill the infected cells and destroy any internal viruses. To enable this crucial immunological process, CD8+ TCRαβs should ideally recognize any viral (non-self) antigen to enable a robust response against viruses, and not recognize any self-antigens to avoid an autoimmune reaction resulting from cytotoxic responses directed against non-infected parenchymal cells presenting only self-antigenic peptides at their cell surface. CD8+ T cells are able to recognize a wide variety of possible non-self-antigens due to the large variety of TCRαβ variants generated during CD8+ T Cell maturation in the thymic cortex. Here, double negative thymocytes undergo somatic rearrangement of VDJ gene segments, causing variation in the structure and thereby binding affinities of TCRαβs expressed by different thymocytes. Through subsequent sequential positive and negative selection, only thymocytes possessing TCRαβs that do not recognize self-peptide-HLA-I complexes survive, transform into naïve CD8+ T cells and migrate to periphery (1). A key step of the negative selection is the recognition, by CD8+ TCRαβ T cell clones, of self-antigenic peptide-HLA-I complexes, which are presented by professional antigen presenting cells (APCs) in the thymic medulla. These APCs, such as mTECs and thymic Dendritic cells (DCs), express transcription factors that promote the expression of a very large variety of self-antigens, thereby promoting the identification of potentially autoreactive CD8+ TCRαβ T cell clones and their elimination (2). Nonetheless, thymic deletion of self-reactive CD8+ T cells is not perfect and many potentially autoreactive CD8+ T cells are present in periphery (3–6). There, they can be controlled by peripheral tolerance mechanisms such as quiescence, ignorance, anergy, and tolerance-induced cell death (5). If some of the self-epitopes recognized by potentially autoreactive CD8+ T cells are identical to non-self-epitopes which could be generated from viral antigens, we would expect an impaired CD8+ T cell response against viruses, since these potentially autoreactive CD8+ T cell clones would have been eliminated in the thymus or pruned in periphery.
We recently named these troubling peptides, zwitter epitopes (7). Zwitter is the German word for “hybrid,” “hermaphrodites,” originating from zwi-, meaning “duplex.” For example, in chemistry, a zwitterion is an ion which possesses both positively- and negatively-charged groups.
If CD8+ T cells specific for zwitter epitopes were eliminated in the thymus, they could not recognize the virus-derived zwitter epitope during an infection, which could create “holes” in the T cell repertoire. Likewise, if the inefficient stimulation of naïve CD8+ T cells or the excessive and persistent stimulation of CD8+ effector T cells mediated by self-derived zwitter epitopes induced anergy, exhaustion or peripheral deletional tolerance, these CD8+ T cells would be eliminated and therefore unable to recognize the virus-derived zwitter epitopes and to tackle a second infection.
For example, a non-synonymous mutation in a Hepatitis C Virus (HCV), which did not affect peptide-HLA-A*02:01 binding affinity, hampered the immune response against HCV. Since this phenomenon seemed to derive from the lack of CD8+ T cells with TCRαβ recognizing the mutated peptide, Wölfl et al. (8) hypothesized that HCV exploited a “hole” in the T cell repertoire. Similarly, in mouse models of vaccinia infection, ~ one-half of the vaccinia-derived epitope candidates predicted to bind Major Histocompatibility Complexes class I (MHC-I) molecules and ~ 20% of the vaccinia-derived epitope candidates identified in MHC-I immunopeptidomes by mass spectrometry (MS) did not trigger a detectable CTL response in vaccinia-immunized mice (9, 10).
Previous studies have investigated whether zwitter epitopes could contribute to these “holes” in the T cell repertoire by computing the overlaps between self and non-self-antigens in terms of canonical non-spliced peptide sequences (11–16). Calis et al. (17) computed that just 0.15% of all theoretical 9 amino acid long (9mer) canonical peptides derived from hundreds of viral strains completely overlap with 9mer peptide sequences present in the human proteome. Likely, this ~0.15% frequency of virus-human zwitter non-spliced epitopes is not sufficient to justify the hypothesized size of “holes” in the CD8+ TCRαβ T cell repertoire. Calis et al. (17) suggested that these “holes” could arise from the degeneracy of CD8+ TCRαβ specificity, as this could lead to cross-recognition of multiple antigenic peptides, thereby increasing the immunological overlap between self and non-self-antigens. However, the immunological relevance of CD8+ TCRαβ cross-reactivity is still a matter of debate (18–20), and even largely overlapping viral epitopes can induce an independent and non-cross-reactive T cell response (21).
Alternatively, we can consider what APCs present rather than how CD8+ TCRαβs recognizes epitope-HLA-I complexes on APCs. For instance, the research in this field has so far only considered canonical “non-spliced” peptides and neglected non-canonical spliced peptides bound to HLA-I complexes. Both spliced and non-spliced peptides presented to CD8+ T cells are mainly produced by proteasomes. These proteases can cleave antigens and release non-spliced peptides as well as ligate non-contiguous peptide fragments, thereby producing spliced peptides (22). Proteasome-catalyzed peptide splicing (PCPS) can occur by combining non-contiguous peptide fragments of the same molecule—cis-PCPS—or of two distinct proteins—trans-PCPS (Figure 1A). Cis-spliced peptides are produced and presented by various cells (22). They can target CD8+ T cell responses against otherwise neglected bacterial antigens in vivo in a mouse model of Listeria monocytogenes infection (23). They can also activate CD8+ T cells specific for Listeria monocytogenes or HIV through cross-recognition in vivo (24, 25). They can be neoepitopes and present recurrent driver mutations such as KRAS G12V at the cell surface of cancer cell lines (26). While, cis-spliced epitopes derived from melanoma-associated antigens are recognized by CD8+ T cells in peripheral blood of melanoma patients (27, 28). A melanoma patient with metastasis was cured through adoptive T cell therapy using an autologous tumor-infiltrating lymphocyte clone, which was proved, in a later study, to be specific for a cis-spliced epitope rather than any non-spliced peptides derived from the melanoma-associated antigen (29, 30).

Figure 1. Proteasome-generated spliced peptides and in silico pipelines. (A) Proteasome-generated spliced peptides can be formed by: (i). cis-PCPS, when the two splice-reactants, i.e., the non-contiguous peptide fragments ligated by proteasomes, derive from the same polypeptide molecule; the ligation can occur in normal order, i.e., following the orientation from N- to C-terminus of the parental protein (normal cis-PCPS), or in the reverse order (reverse cis-PCPS); (ii). trans-PCPS, when the two splice-reactants originate from two distinct protein molecules or two distinct proteins. (B,C) In silico pipelines to estimate the frequency of zwitter epitope candidates predicted to bind HLA-A*02:01 complexes not accounting (B) or accounting (C) for non-spliced and cis-spliced peptide frequency in HLA-I immunopeptidomes.
Cis-spliced peptide identification is quite challenging. Estimation of their frequency in HLA-I immunopeptidomes varies from 1 to 34%, depending on the method used for their identification (31). While, although trans-spliced peptides have been identified in in vitro (26, 32–34), in cellulo (35) and in HLA-I immunopeptidomes (36), their immunological relevance still needs to be investigated (7).
Nevertheless, since the theoretical size of the human cis-spliced peptide database is extremely vast, they could make up a significant portion of the viral-human zwitter epitope pool and, thereby, play a role in CD8+ T cell tolerance. To test this hypothesis, we here computed the frequency of zwitter cis-spliced and non-spiced epitope candidates through comparison of human and viral proteomes. We accounted for these zwitter candidates' binding affinity to the most predominant HLA-I allele in Caucasian population, HLA-A*02:01, their estimated expression in human mTECs and their frequency in HLA-I immunopeptidomes, to accommodate these factors' potential impact on zwitter candidates' involvement in central tolerance.
Materials and Methods
Statistical Analysis
Significant difference between groups was computed by applying the Kolmogorov-Smirnov test. A p < 0.05 was considered statistically significant. The effect size of 9mer non-zwitter vs. zwitter peptides in binding HLA-A*02:01 complexes was computed via odds ratio and significance was tested using Fisher exact test, or alternatively chi square test if the sample size was too large for Fisher exact test to test significance of association. Test for association between virus length and number of zwitter peptides was based on Pearson's product moment correlation coefficient. Statistical values are reported in Supplementary Table 1.
In this study, we defined viral-human zwitter non-spiced peptides as all those non-spliced peptides from viral proteomes that completely overlapped with human non-spliced peptides. Viral-human zwitter cis-spliced peptides, on the contrary, included the following categories of peptides that completely overlapped between each other: viral cis-spliced with human non-spliced peptides, viral cis-spliced with human cis-spliced peptides, and viral non-spliced with human cis-spliced peptides.
Peptide-HLA-A*02:01 Binding Affinity Prediction
Binding of non-spliced and cis-spliced 9mers to HLA-A*02:01 molecules was predicted using Stabilized Matrix Method (SMM) (37). This predictor showed good performance in the prediction of the binding affinity to a hundred cis-spliced peptides in a previous study (38). The standalone version of prediction tool was downloaded from the IEDB Analysis Resource (39). As cut-off for peptide-HLA-A*02:01 binding affinity we set an IC50 ≤ 500 nM.
In order to assess whether zwitter 9mer peptides were more likely to be HLA-A*02:01 binders than non-zwitter 9mer peptides on a per virus basis, we separately counted the number of non-zwitter and zwitter 9mer peptides predicted to be either non-binders or binders. Based on this contingency table the odds ratios for each virus were computed.
Estimation of Viral-Human zwitter Peptides
Viral proteomes were obtained via ViralZone and the Human proteome referred to Swiss-Prot Version 2016 excluding protein isoforms (40, 41). Only viruses with human trophism were included in any downstream analysis presented here (n = 109; Supplementary Table 2). The Human proteome database contained 20,191 protein entries with a total of 11,323,862 amino acid residues.
We focused our study on 9mer peptides since they represent the majority of non-spliced and cis-spliced peptides in HLA-I immunopeptidomes (36, 38, 42). Furthermore, we focused our study on HLA-A*02:01 variant since it is likely the most studied HLA-I variant and is the predominant HLA-I allele in Caucasian population.
We defined viral-human zwitter 9mer peptides as any 9mer peptide that had a sequence that could be obtained by either peptide hydrolysis or cis-peptide splicing both from self-proteins and from viral proteins.
For viral and human proteomes, we first computed all possible 9mer sequences of non-spliced peptides by cutting proteins into fragments of length nine amino acids; normal and reverse cis-spliced peptide sequences were computed by combining splice-reactants of any length such that the resulting cis-spliced peptide sequence had a length of nine amino acids and by imposing a maximal intervening sequence length ≤25 amino acids (Figure 1A), as previously described (42). Afterwards, an alignment was performed between all resulting virus and human derived peptides. We considered two peptides as identical, i.e., as viral-human zwitter peptides, if all of their nine amino acid residues were exactly matching. The relative frequency of viral-human zwitter peptides (Fv) was calculated as:
where zv is the number of all viral-human zwitter peptides of a given virus v; and pv is the number of all possible unique 9mer peptides derived from virus v. The number of viral-human zwitter peptides, z, can be computed for the comparison of non-spliced peptides only (zv,i), of cis-spliced peptides only (zv,j), of non-spliced viral peptides compared to cis-spliced human peptides (zv,k), and of cis-spliced viral peptides compared to non-spliced human peptides (zv,l). In our analysis, we depicted either the relative frequency of viral-human non-spliced zwitter peptides (Fv,i), viral-human cis-spliced zwitter peptides (Fv,cis) or of all (non-spliced and cis-spliced) viral-human zwitter peptides (Fv,all). The latter was obtained via:
Where {} denotes the unique set of peptide sequences and pv,all are all unique non-spliced and cis-spliced peptides derived from virus v.
The above-described analysis was done based on all theoretical possible non-spliced and cis-spliced peptides. Next, we repeated the estimation of viral-human zwitter peptide frequency by restricting the analysis to human- and virus-derived non-spliced and cis-spliced peptides that efficiently bind to the HLA-A*02:01 molecule, i.e., to peptides that have a predicted IC50 ≤ 500 nM, resulting in:
where Bv is the frequency of viral-human zwitter peptide restricted to HLA-A*02:01, zv,b is the number of all viral-human zwitter peptides of a given virus v that bind HLA-A*02:01 and bv is the number of all possible unique 9mer epitope candidates derived from virus v that are predicted to bind HLA-A*02:01 with an IC50 ≤ 500 nM.
Estimation of Viral-Human zwitter Epitope Candidates Considering the Potential Antigen Repertoire of Human mTECs
To determine the potential antigen repertoire of human mTECs, we analyzed two transcriptome databases: (i) microarray gene expression values of human mTECs (43), and (ii) single-cell RNA sequencing of TECs in human embryos (44). Although mRNA expression does not perfectly mimic HLA-I immunopeptidomes (45), it was shown to be one of the strongest factors correlated with HLA-I immunopeptidomes (46).
In (43), the material was derived from patients that underwent corrective cardiac surgery. Here, we calculated average gene expression values (reported as log2 transformed fluorescence intensities) across technical replicates of each mTEC subset obtained with differing versions of microarrays provided in the dataset, and took the maximum average value.
In (44), the material was derived from healthy human fetuses as a result of medically interrupted pregnancy at weeks 8, 9, and 10. We used the subset of data that ostensibly corresponded to TECs with progenitor property of mTECs (based on the expression of the mTEC markers CLDN4 and JAG1).
We performed log-normalization of gene expression values of individual cells—reported as copy number of transcripts per individual gene—number of distinct unique molecular identifiers (UMI)—to mitigate the relationship between sequencing depth and gene expression. We then took an average normalized gene expression value between individual cells (47, 48):
where xi is the log-normalized expression of gene i, UMIij is the expression value of gene i in cell j prior to normalization expressed as UMI counts, and UMIj is the sum of UMI counts per cell j.
Afterward, we defined a crude model for antigen presentation based on the gene expression values. We assumed that the chance of an antigen being presented in mTECs' HLA-I immunopeptidomes was directly correlated with the gene expression of that antigen. The limitation of this assumption is discussed above.
We first scaled and normalized the gene expression values of the processed data obtaining weights for each antigen (wi):
where Ei is the expression value of gene i prior to normalization, and min(E) and max(E) are the minimum and maximum gene expression values in the dataset, respectively. We next sampled from the pre-computed pool of viral-human zwitter peptides a subset of peptides based on the weights (wi) of the human antigen (i), which the respective zwitter peptide was derived from. The sampling size was set at 100% of the total number of zwitter peptides to reflect the odds of presentation of each given peptide. Sampling was performed with replacement based on the calculated probabilities 60 times. Finally, the frequency of viral-human zwitter peptides considering potential antigen repertoire of mTECs compared to all viral 9mer peptides (Mv) was computed as:
where zm,v is the number of sampled viral-human zwitter peptides with weights wi and pv is the number of all possible 9mer peptides of virus v. Similarly, when we considered both predicted peptide-HLA-A*02:01 binding affinity and potential antigen repertoire of mTECs, the viral-human zwitter peptide frequency (MBv) was computed as:
where zmb,v is the number of sampled viral-human zwitter peptides restricted to HLA-A*02:01 binding with weights wi, and bv is the number of all possible 9mer peptides restricted to HLA-A*02:01 binding of virus v.
Estimation of the Frequency of Viral-Human zwitter Epitope Candidates Weighing up PCPS Frequency
Not all 9mer non-spliced and cis-spliced peptides that could derive from the human proteome are in reality produced by proteasomes and presented through HLA-I antigen processing and presentation (APP) pathway (22). Therefore, we implemented this factor in our in silico analysis of zwitter peptides. We aimed to determine the fractions of non-spliced (fnon) and cis-spliced peptides (fcis) produced and presented in HLA-I immunopeptidomes relative to all theoretically possible sequences:
where nnon and ncis is the number of presented non-spliced and cis-spliced peptides, respectively, and Nnon and Ncis is the number of all theoretically possible non-spliced and cis-spliced peptides, respectively, derived from a given antigen.
An estimate of fnon can be directly obtained from in vitro digestions of synthetic polypeptides with purified proteasomes. For this dataset, we used the peptide product database derived from 4 h digestions of 47 synthetic polypeptides with purified 20S standard proteasomes (34). This large database contains 2,429 unique non-spliced and 2,379 unique cis-spliced peptide products, which passed several quality control steps (34). We calculated the fraction of all produced 9mer non-spliced peptides (included in Specht's database) relative to all theoretically possible 9mer non-spliced peptides for each synthetic polypeptide substrate in the database. Then, we took the median value between all polypeptides as estimation of the fraction of non-spliced 9mer peptides generated by proteasomes. These calculations resulted in fnon ~ 0.27, i.e., ~27% of all possible non-spliced 9mer peptides are generated in vitro by proteasomes and detected through MS. Therefore, in the following analysis, we randomly sampled 27% of all theoretical 9mer non-spliced peptides to recompute the number of viral-human zwitter peptides in absence of reliable proteasome peptide hydrolysis and peptide cis-splicing predictors.
We could have used the same strategy to compute the fraction of cis-spliced peptides produced by proteasomes compared to all theoretical cis-spliced peptide products. However, cis-spliced peptides have been proved to be produced in significantly lower amount than non-spliced peptides (26, 33, 34). Bearing this in mind, we speculated that a large number of cis-spliced peptides produced by proteasomes in vitro could not pass all APP steps and become antigenic as compared to non-spliced peptides.
On the contrary, HLA-I immunopeptidomes should be more informative in such a matter, since the APP pathway should already have filtered out many cis-spliced peptides generated in low amount. Therefore, we used the information available about cis-spliced peptide frequency in HLA-I immunopeptidomes measured through MS and combined with the information of non-spliced peptide frequency in in vitro digestions (fnon). Indeed, the estimation of fcis based on cis-spliced peptide product frequency in vitro digestions as measured through MS could have resulted in an overestimation of fcis. Therefore, we defined the relative frequencies of cis-spliced peptides in HLA-I immunopeptidomes (f) as measured by MS as:
where ncis is the number of cis-spliced peptides detected in HLA-I immunopeptidomes and nnon is the number of non-spliced peptides detected in HLA-I immunopeptidomes. Since f was estimated to be in the range of 1–34% (31). For a given estimate of f we could then compute the number of cis-spliced peptides presented in HLA-I immunopeptidomes (ncis) as:
Furthermore, we could compute the total number of all theoretical cis-spliced peptides (Ncis) as:
where γ was estimate to have a value of 398 for proteins of length 500 amino acids or longer (42). This resulted in:
We used a range of potential frequencies of observed cis spliced peptides relative to the whole HLA-I immunopeptidome f (1–35%) to determine a range of fcis. Based on fcis and fnon, we randomly sampled non-spliced and cis spliced peptides 600 times from all viral and human proteomes without replacement. For each of the 600 samples for each fcis, we counted the number of all sampled HLA-A*02:01-restricted zwitter peptides.
HIV-Derived HLA-A*02:01-Restricted Non-immunogenic 9mer Peptides
As proof of principle, we selected a pool of HIV-derived HLA-A*02:01-restricted 9mer peptides, which were previously suggested to be non-immunogenic. This pool included non-spliced epitope candidates derived from HIV, which:
(i) were investigated by Perez et al. (49) through IFN-γ ELIspot assay in HIV- infected donor peripheral blood mononuclear cells (PBMCs) pulsed/non-pulsed with synthetic epitope candidates. We considered as non-immunogenic those peptides that did not induce immune response after peptide stimulation.
(ii) were included in a database by Ogishi and Yotsuyanagi (50). This database collected outcomes of various T cell activation assays on HLA-I-restricted non-spliced peptide sequences (8–11 mer peptides). In this database, we selected HIV-derived HLA-A*02:01-restricted 9mer peptides, which were confirmed as non-immunogenic among all studies considered in the database.
(iii) were included in the EPIMHC database (51), which collected datasets of T cell response against epitope candidates. In this database, non-immunogenic peptides were selected by applying the following parameters: Allele, HLA A0201; Length, 9mer; MHC source, Human; Peptide source organism, HIV1; Peptide Binding Level, all; T-cell activity, all; Immunogenicity level, all; Processing, all.
The pool of peptide candidates derived from these three databases were then analyzed for peptide-HLA-I bind affinity prediction—as described above—and only peptides with predicted peptide-HLA-A*02:01 IC50 ≤ 500 nM were selected (Table 1).
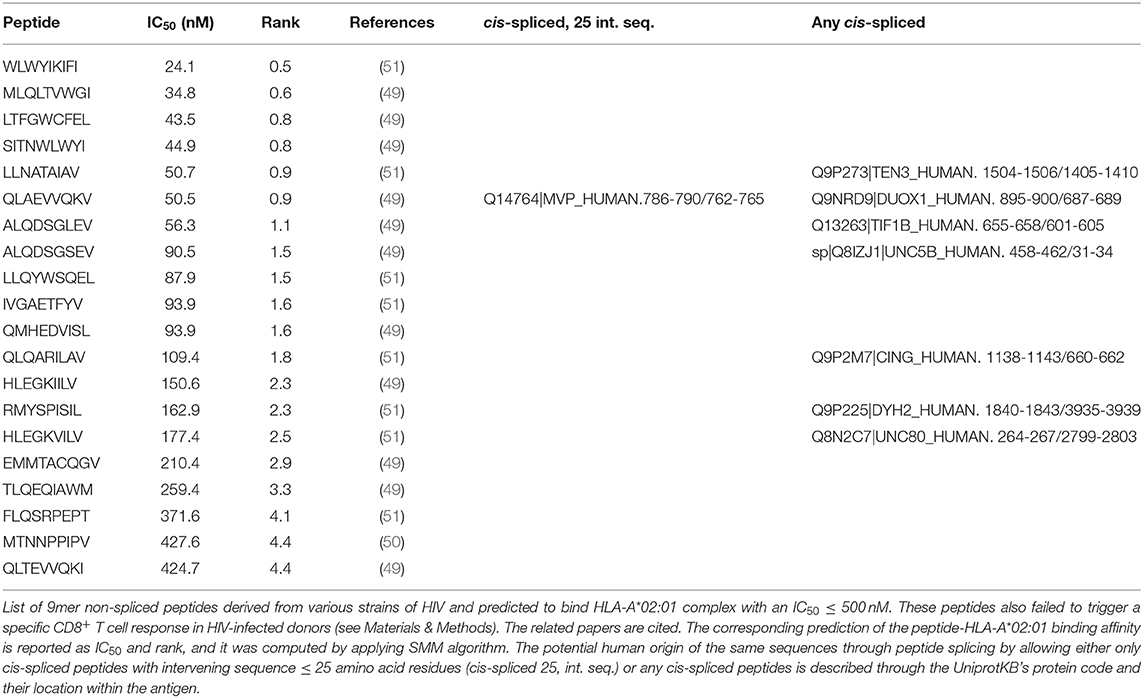
Table 1. List of HIV-derived HLA-A*02:01-restricted 9mer peptides not immunogenic and their zwitter peptide pair.
Modeling of Protein 3D Structures
For visualization purpose, the structures of Gag-Pol polyprotein of the HIV strain MVP5180 and of the human Major Vault protein (MVP) were predicted and visualized through the fully automated protein structure homology-modeling server, accessible via Expasy web server (52).
Data Availability
A summary of the files accessible via repository is reported in the following Mendeley dataset: http://dx.doi.org/10.17632/hw686hytfs.1.
The mTEC's RNA sequencing data published by Pinto et al. (43) are available at Gene Expression Omnibus (GEO) under identifier GSE49625.
The single-cell RNA sequencing of TECs in human embryos published by Zeng et al. (44) are available at Gene Expression Omnibus (GEO) under identifier GSE133341.
Results
Estimation of the Upper Bond Frequency of Viral-Human zwitter Epitope Candidates
By applying the in silico pipeline described in Figure 1B and focusing on 9mer peptides, which represent the majority of non-spliced and cis-spliced peptides in HLA-I immunopeptidomes (28, 36, 38, 42), we identified 2,340 and 9,350,135 theoretical viral-human zwitter non-spliced and cis-spliced 9mer peptides, respectively (Supplementary Table 3). On average per virus, these represent 0.06 and 2.93% of the pool of virus non-spliced and cis spliced 9mer peptides, respectively (Figure 2A). We then predicted their binding affinity to the most predominant HLA-I allele in Caucasian population, i.e., HLA-A*02:01, and filtered out all peptides with predicted IC50 > 500 nM. This step removed ~96% of the peptides (on average, only ~5% of peptides per virus are left; see Supplementary Figure 1A). This left 87 and 504,209 viral-human zwitter non-spliced and cis-spliced 9mer epitope candidates in total, which correspond, on average per virus, to 0.05 and 3.84% of the pool of HLA-A*02:01-restricted viral non-spliced and cis-spliced 9mer peptides, respectively (Figure 2B). This frequency did not account for antigen processing via the APP pathway and assumed that each and every non-spliced and cis-spliced peptide that could be produced by proteasomes was indeed produced. Therefore, it represents the upper bond of viral-human zwitter 9mer epitope candidates. Interestingly, viral-human zwitter peptides were more often predicted to bind HLA-A*02:01 with an IC50 ≤ 500 nM than non-zwitter peptides (Supplementary Figure 1B).
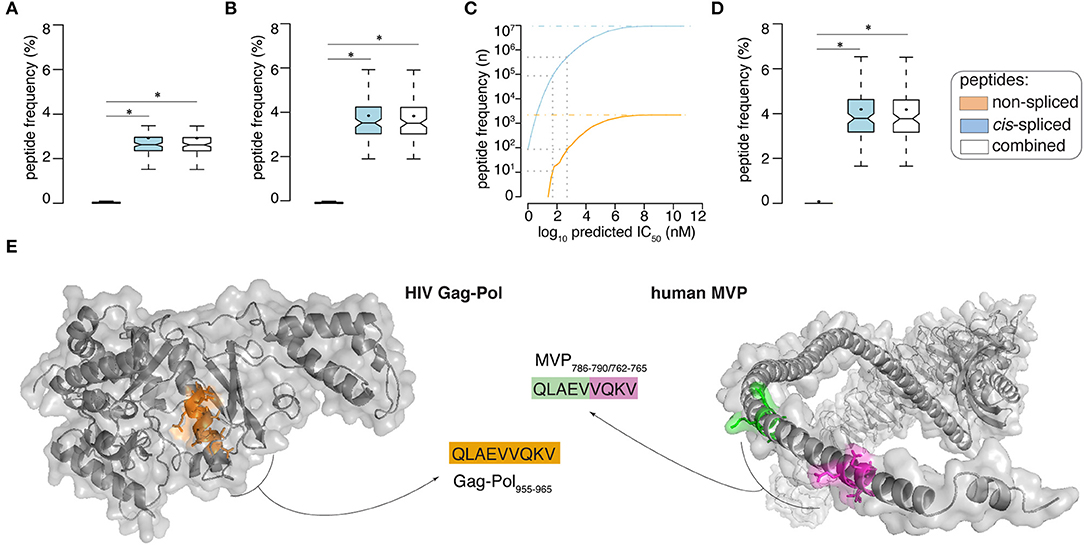
Figure 2. Viral-human zwitter epitope candidate frequency and examples. (A,B) Frequency of viral-human 9-mer (A) zwitter peptides and (B) HLA-A02:01-restricted (predicted IC50 ≤ 500 nM) zwitter epitope candidates, compared to their cognate viral peptide database and considering the whole human proteome database. (C) Number of viral-human zwitter non-spliced and cis-spliced epitope candidates depending on the peptide-HLA-A02:01predicted IC50. Gray dot lines mark the predicted IC50 of 500 nM and 50 nM. The blue and orange dot lines depict the number of viral-human zwitter cis-spliced and non-spliced peptide without peptide-HLA-A02:01predicted IC50 cut-off. (D) Frequency of HLA-A02:01-restricted (predicted IC50 ≤ 50 nM) viral-human zwitter 9mer epitope candidates, compared to their cognate viral peptide database and considering the whole human proteome database. (E) Example of HIV-human zwitter epitope candidate QLAEVVQKV, which may be derived from HIV Gag-Pol as non-spliced peptide (Gag-Pol955−963), and from the human MVP as cis-spliced peptide (MVP786−790/762−765). Both peptides are depicted in the cognate antigens. Color code corresponds to Figure 1A. In (A,B,D) Box plots depict the median and 25–75 percentiles of peptides per virus. Bars represent 5–95 percentiles. Dots represent the mean. Significant difference between groups is labeled with * (see Supplementary Table 1).
When we loosen up the IC50 cut-off, the number of viral-human zwitter non-spliced and cis-spliced 9mer epitope candidates would increase (Figure 2C). To further investigate the theoretical frequency of viral-human zwitter non-spliced and cis-spliced 9mer epitope candidates among the potentially immunodominant epitopes, we focused on a more stringent IC50 cut-off of 50 nM. For instance, Platteel et al. (23) reported a correlation between the immunogenicity of cis-spliced epitope candidates, their predicted binding affinity to H2-Kb (IC50 ≤ 2 nM) and the measured cis-spliced peptide-H2-Kb complex stability in a mouse model of Listeria monocytogenes infection. While, Assarsson et al. (9) showed that all vaccinia immunodominant HLA-A*02:01-restricted non-spliced epitopes analyzed in their study on a transgenic mouse model had a measured peptide-HLA-A*02:01 IC50 ≤ 50 nM. With this latter IC50 cut-off, 11 non-spliced and 87,154 cis-spliced peptides were left among the viral-human zwitter epitope candidates, which correspond, on average per virus, to 0.06 and 4.19% of the pool of HLA-A*02:01-restricted (predicted IC50 ≤ 50 nM) viral non-spliced and cis-spliced 9mer peptides, respectively (Figure 2D).
Example of T Cell Tolerance Against Viral-Human zwitter Epitope Candidate
As proof of principle, we selected a pool of HIV-derived HLA-A*02:01-restricted 9mer peptides, which were demonstrated to be non-immunogenic in previous studies (see Materials and Methods). Among them, we selected non-spliced peptides that were predicted to bind HLA-A*02:01 complex with IC50 ≤ 500 nM and, upon testing for CD8+ T cell response in HIV patients, were non-immunogenic (Table 1). We investigated whether any of them may also have been a viral-human zwitter 9mer epitope candidate. We considered both cis-spliced peptides with intervening sequence shorter than 26 amino acid residues, as in the rest of the study, as well as any theoretical cis-spliced peptide computed from the human proteasome. Out of twenty peptides with these characteristics, we identified the peptide QLAEVVQKV, which may derive from the Gag-Pol polyprotein of the HIV strain MVP5180 (Gag-Pol955−963). This epitope candidate has a predicted IC50 = 50 nM for HLA-A*02:01 (Table 1). Despite the good binding affinity, this epitope candidate did not trigger a PBMC response in HIV patients, according to Perez et al. (49). In their cohort of 31 HIV patients, 10 were HLA-A*02:01+ and none of them recognized the epitope candidate upon peptide stimulation. No other studies showed a recognition of this epitope candidate by CD8+ T cells, to our knowledge. According to our computation, the same peptide sequence may also derive from the Major Vault protein as a cis-spliced peptide—i.e., MVP786−790/762−765 [QLAE][VVQKV]—with intervening sequence smaller than 26 amino acid residues (Figure 2E). MVP's gene mRNA was identified in mTECs by both Pinto et al. (43) and Zeng et al. (44), thereby suggesting its expression in mTECs and, in theory, the potential presentation of the MVP786−790/762−765 cis-spliced epitope candidate to thymocytes. That might lead to negative selection of CD8+ T cell clones recognizing the peptide QLAEVVQKV, which might explain the absence of immunogenicity of the Gag-Pol955−963 [QLAEVVQKV] in HLA-A*02:01+ HIV patients.
If we expanded our research to any cis-spliced epitope candidate, regardless of the intervening sequence length, we identified six other cis-spliced epitope candidates with a sequence present in Table 1. Therefore, we should bear in mind that the pool of viral-human zwitter 9mer cis-spliced epitope candidates, which had an intervening sequence length smaller than 26 amino acid residues, represented only part of the whole theoretical cis-spliced peptides.
Estimation of Viral-Human zwitter Epitope Candidate Frequency Weighing Up mTEC Transcriptome
Viral-human zwitter non-spliced and cis-spliced 9mer epitopes may impinge upon the functional CD8+ T cell repertoire through both central and peripheral tolerance. Herein, we focused solely on the negative selection step of the central tolerance. We hypothesized that TCRαβ T cell clones that recognize self-derived zwitter epitopes bound to HLA-I complexes of mTECs and other professional APCs with high avidity are tolerized.
In tolerance, the amount of antigen presented at the cell surface is relevant to the fate of T cell clones (5). Although gene expression does not mirror the HLA-I immunopeptidomes, it appears, to some extent, to be a predictor of antigen presentation (46). Bearing this in mind, we repeated our analysis by weighing up the probability of an antigen to be represented in mTEC's HLA-I immunopeptidome, based on transcriptome data from either microarray analysis of human mTECs (43) or single-cell RNA sequencing of TECs in human embryos (44).
To this end, we transformed gene expression values of mTECs into probabilities of antigens being represented in HLA-I immunopeptidomes through a crude model for antigen presentation based on the gene expression values (see Material and Methods). Furthermore, in our analysis, the probability of an antigen to be represented in mTEC's HLA-I immunopeptidomes was weighted by the number of zwitter non-spliced and cis spliced 9mer peptides predicted to bind HLA-A*02:01 (with IC50 ≤ 500 nM) and theoretically derived from that antigen (see Material and Methods). Indeed, the chance of an antigen being presented in HLA-I immunopeptidomes also depends on the number of HLA-I-binding peptides that could be derived from that given antigen. Since we introduced a probability score in our analysis, we had to sample the viral-human zwitter non-spliced and cis-spliced 9mer epitope candidate pool, thereby estimating the average frequency rather than the absolute frequency of these peptides, which has been shown so far.
Compared to the whole human proteome, incorporation of potential antigen repertoire based on mTEC transcriptome resulted in a decreased average number of both zwitter non-spliced and cis-spliced epitope candidates. On average per virus, 0.04% and 2.53% of the pool of HLA-A*02:01-restricted virus non-spliced and cis-spliced 9mer epitope candidates, respectively, were zwitter peptides using Pinto's RNA sequencing database (Figure 3A). Similar results were obtained using Zeng's RNA sequencing database (Figure 3B).
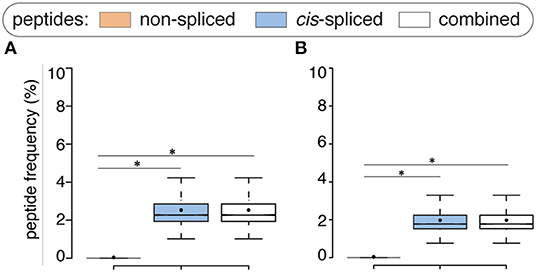
Figure 3. Viral-human zwitter epitope candidates considering mTEC's transcriptome. (A) Frequency of HLA-A02:01-restricted viral-human zwitter 9mer epitope candidates compared to their cognate viral peptide databases considering the human mTEC transcriptome computed either (A) from (43) or (B) from (44). Box plots depict the median and 25–75 percentiles. Bars represent 5–95 percentiles. Dots represent the mean. Significant difference between groups is labeled with * (see Supplementary Table 1).
Estimation of Viral-Human zwitter Epitope Candidate Frequency Weighing Up cis-PCPS Frequency
The computation done so far did not take into account the frequency of peptides produced by proteasomes through peptide hydrolysis and peptide cis-splicing and presented at the cell surface. Despite not being physiological, one of the most detailed approaches to determine what proteasomes can produce via peptide hydrolysis and peptide splicing is, in our experience, the measurement through MS of non-spliced and cis-spliced peptides produced in vitro by purified 20S proteasomes during the degradation of synthetic polypeptides recapitulating antigenic sequence. Correspondence between in vitro experiments carried out with purified 20S proteasomes and in cellulo and in vivo experiments has been demonstrated in various studies investigating both viral and tumor epitopes (23, 24, 26, 27, 30, 53–62). The analysis of in vitro digestions of synthetic polypeptides by 20S proteasomes showed that, although these proteases can cleave—and likely ligate—any amino acid, they have substrate sequence preferences (34). It also showed that cis-spliced peptides are produced, on average, in significantly smaller amount than non-spliced peptides by proteasomes (26, 32, 33). Therefore, not all non-spliced peptides, and even less cis-spliced peptides, are likely generated by proteasomes in sufficient amount to be detected in vitro by MS as well as to survive all steps of HLA-I APP pathway.
We weighed up the impact of this phenomenon in our computational analysis by gathering information from two experimental dataset sources measured by MS: a large database of non-spliced and spliced peptides produced in vitro by purified proteasomes (34) and HLA-I immunopeptidome elutions.
Through the analysis of in vitro digestion database (34), we estimated that ~27% of all theoretical non-spliced 9mer peptides that could be produced by proteasomes are in fact generated in a detectable amount. This figure is much smaller for cis-spliced peptides.
The frequency of cis-spliced peptides in HLA-I immunopeptidomes is still a controversial topic, with their frequency in HLA-I immunopeptidomes being estimated in a range from 1 to 34%, depending on the method used for their identification (31).
Using these two sets of information, we determined the relative frequency of non-spliced and cis spliced peptides generated by proteasomes and presented in HLA-I immunopeptidomes compared to all theoretical non-spliced and cis-spliced peptide products; we then implemented it into our model to better estimate viral-human zwitter peptide frequency. Based on this new analysis, we randomly selected non-spliced and cis-spliced peptides from our viral and human proteome databases, repeated sampling 600 times to reach statistical power and then repeated our entire analysis for each sample (Figure 1C).
If we assumed a ~15% cis-spliced peptide frequency in HLA-I immunopeptidomes, over all randomly sampled peptide pools, we identified, on average, a total of 7 HLA-A*02:01-restricted viral-human zwitter non-spliced 9mer epitope candidates. They correspond to 0.079% of the pool of HLA-A*02:01-restricted virus non-spliced 9mer peptides. This figure strongly varied from virus to virus. On average of sampling, 6 viruses had at least one HLA-A*02:01-restricted viral-human zwitter non-spliced 9mer peptide. No more than 5 epitope candidates per virus were estimated in this analysis (Figure 4A). In the same analysis, we identified, on average, a total of 0.3 HLA-A*02:01-restricted viral-human zwitter cis-spliced 9mer epitope candidates. They correspond to 0.0008% of the pool of HLA-A*02:01-restricted virus cis-spliced 9mer peptides, which is a frequency dramatically smaller than the 3.84% computed without accounting for cis-spliced peptide frequency in HLA-I immunopeptidomes (see Figure 2B). On average of sampling, only 1 virus had an HLA-A*02:01-restricted viral-human zwitter cis-spliced 9mer epitope candidate and no more than 2 epitope candidates per virus were estimated (Figure 4A).
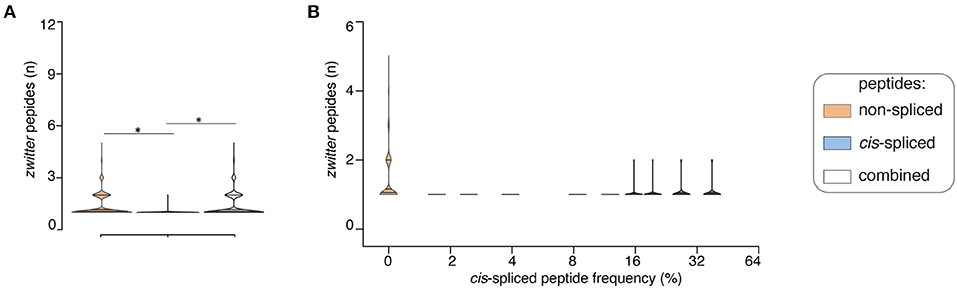
Figure 4. Viral-human zwitter epitope candidates considering cis-spliced peptide frequency in HLA-I immunopeptidomes. (A) Distribution of the number of viral-human 9mer HLA-A02:01-restricted (non-spliced, cis-spliced and combined) zwitter epitope candidates per virus across all 600 random samples presented as violin plots (rotated densities). Significant difference between groups is labeled with * (see Supplementary Table 1). This analysis was carried out by hypothesizing that cis-spliced peptides represent ~15% of peptides in HLA-I immunopeptidomes and by using the whole human proteome as database. The distribution of the number of viral-human zwitter cis-spliced epitope candidates has been displayed among viruses that had at least one zwitter peptide. (B) Number of HLA-A*02:01-restricted viral-human zwitter non-spliced and cis-spliced 9mer epitope candidates per virus per sampling iteration, depending on a broad range of theoretical cis-spliced peptide frequencies in HLA-I immunopeptidomes. Here, viral proteomes are compared to the whole human proteome database. The number of viral-human zwitter non-spliced and cis-spliced 9mer epitope candidates per virus per iteration has been computed among viruses that had at least one zwitter peptide.
Since cis-spliced peptide frequencies in HLA-I immunopeptidomes is so controversial, we repeated the non-spliced and cis-spliced peptides' sampling and downstream analysis considering a broad range of frequencies of cis-spliced peptides in HLA-I immunopeptidomes. As shown in Figure 4B, the overall picture did not change much. The average number of HLA-A*02:01-restricted viral-human zwitter non-spliced epitope candidates was estimated to be always largely higher than cis-spliced epitope candidates. Only few outliers of cis-spliced epitope candidates were identified when we assumed very large frequencies of cis-spliced peptide in HLA-I immunopeptidomes.
This phenomenon was reflected also in terms of number of viruses that, on average of sampling, had one or more HLA-A*02:01-restricted viral-human zwitter epitope candidates. The average number of viruses with one or more HLA-A*02:01-restricted viral-human zwitter epitope candidates was only increased by including cis-spliced epitope candidates if we assumed a frequency of cis-spliced peptides in HLA-I immunopeptidomes larger than 30% (Figure 5).
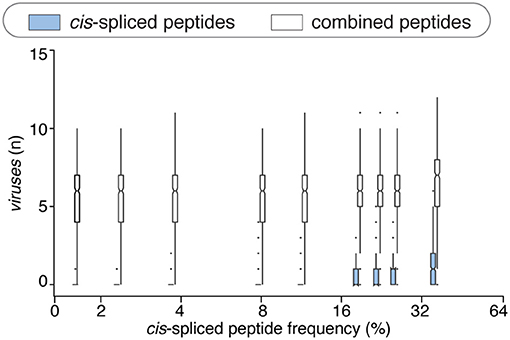
Figure 5. Viruses that have zwittter epitope candidates depending on cis-spliced peptide frequency in HLA-I immunopeptidomes. Average number of viruses that contain at least one HLA-A02:01-restricted viral-human zwitter 9mer peptide per iteration, depending on a broad range of theoretical cis-spliced peptide frequencies in HLA-I immunopeptidomes. Viral proteomes are compared to the whole human proteome database. The boxplots of the combined peptides have been slightly shifted on the x axis for representation purpose.
There are various factors that can impinge upon the number of viral-human zwitter epitope candidates that could be derived from a given virus. One of them is the number of amino acid residues present in viral proteomes. The direct correlation between viral-human zwitter epitope candidates and the size of virus proteome databases was however stronger if we did not consider the frequencies of cis-spliced peptide in HLA-I immunopeptidomes (Figure 5, Supplementary Table 1). Another factor can be the sequence motifs of viral proteome, which may not favor the presentation of viral-human zwitter epitope candidates through a specific HLA-I allele. For example, this is the case of the Hepatitis delta virus I, which has an underrepresentation of viral-human zwitter epitope candidates among those that are predicted to bind HLA-A*02:01 molecules as compared to the total number of its theoretical viral-human zwitter peptides (Figure 6).
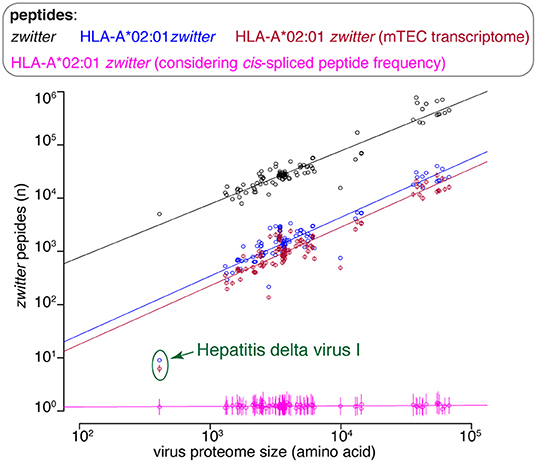
Figure 6. Viral-human zwitter epitope candidate frequency depends on virus length and sequence motifs. Number of viral-human zwitter combined (i.e., non-spliced + cis spliced peptides) 9mer peptides per virus, depending on the number of amino acid residues in its proteome. For the groups labeled in pink, we considered a cis-spliced peptide frequency of ~15%, as in Figure 3A. Viral-human zwitter 9mer peptides and HLA-A*02:01-restricted viral-human zwitter 9mer epitope candidates are represented with a dot each virus. HLA-A*02:01-restricted viral-human zwitter 9mer epitope candidates either using mTEC's RNA-based proteome database (43) or considering the theoretical cis-spliced peptide frequency in HLA-I immunopeptidomes are represented with a dot (mean) and bars (SD) of sampling iterations. Regression lines are shown. The Hepatitis delta virus I has an underrepresented number of HLA-A*02:01-restricted viral-human zwitter 9mer epitope candidates, which are here labeled.
Discussion
Despite proteasome-generated spliced epitopes being known about for more than a decade (61, 63), the potential implications of their presentation by HLA-I complexes only started to concern the scientific community in recent years when we and others showed that spliced peptides represented a sizeable portion of HLA-I immunopeptidomes (28, 36, 38, 42). One of these concerns was the hypothetical impact of spliced peptides on central and peripheral tolerance and on the repertoire of CD8+ T cells recognizing viruses. Indeed, the theoretical substantial sequence variability of cis-spliced peptides may strongly increase the number of viral-human zwitter epitope candidates, thereby reducing the ability of the CD8+ T cell repertoire able to recognize viruses (7, 64). Here we showed in silico evidence that cis-spliced peptides might not play such an unsettling role in the central and peripheral tolerance of the CD8+ T cell repertoire. The main reason is that cis-spliced peptides produced and presented through APP pathway represent just a tiny fraction of all theoretical cis-spliced peptide sequences, as suggested by biochemical and immunopeptidomics studies. According to our preliminary estimations, zwitter cis-spliced epitopes would only significantly impinge upon the virus-specific repertoire of CD8+ T cells if we assumed a very large frequency of these unconventional peptides in HLA-I immunopeptidomes. Although, our analysis was restricted to cis-spliced epitope candidates with intervening sequence shorter than 26 amino acid residues, which may represent only part of HLA-I spliced immunopeptidomes (36).
Additionally, we should bear in mind that our analysis did not consider two potentially important factors: CD8+ TCR specificity degeneracy and driving forces that can restrict the variety of non-spliced and cis-spliced peptides produced by proteasomes.
The former has already been investigated in a seminal work of Calis et al. (17), who focused on non-spliced epitope candidates. Some examples of TCR cross-recognition of pathogen-derived cis-spliced and non-spliced epitopes have been already reported (24, 25). However, we think that we would need data on a larger pool of TCRs before accounting for this factor in our model. To note, this aspect would be even more relevant if we wanted to extend this investigation to CD4+ T cell repertoire, bearing in mind that CD4+ TCR degeneracy is more pronounced than in CD8+ T cells, and trans-spliced peptides are under the spotlight in type 1 Diabetes (65–68).
The latter factor is the impact that substrate sequences have on both peptide hydrolysis and splicing. Proteasomes can cleave and likely splice after any amino acid, as confirmed by a large database of non-spliced and spliced peptides produced in vitro by these enzymes (34). However, peptide sequence motifs seem to impinge upon proteasome dynamics (69) as well as the variety and quantity of non-spliced and cis-spliced peptides that they generate (26, 33, 34, 70–72). This factor may reduce the variety of non-spliced and cis-spliced peptides that are finally presented through HLA-I complexes to CD8+ T cells, and thus alter the frequency of viral-human zwitter epitope candidates.
Finally, in future studies we might also consider the impact that proteasome isoforms might have on the frequency of zwitter epitope candidates. Indeed, standard proteasomes, immunoproteasomes and thymoproteasomes seem to have, at least from a quantitative perspective, different dynamics and substrate sequence preferences for both peptide hydrolysis and splicing (27, 33, 55, 56, 59, 69, 70, 73–75). This can impinge upon the proteome and antigenic landscape of both professional APCs and infected cells (28, 76), and ultimately upon central and peripheral tolerance of CD8+ T cells potentially specific for viral-human zwitter epitopes.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: A summary of the files accessible via repository is reported in the following Mendeley dataset: http://dx.doi.org/10.17632/hw686hytfs.1. The mTEC's RNA sequencing data published by Pinto et al. (43) are available at Gene Expression Omnibus (GEO) under identifier GSE49625. The single-cell RNA sequencing of TECs in human embryos published by Zeng et al. (44) are available at Gene Expression Omnibus (GEO) under identifier GSE133341.
Author Contributions
JL conceived the project. AM implemented the in silico pipelines and carried out the data analysis and figure preparation, which were supervised by JL and MM. CRRB identified the epitope candidates reported in Table 1. MM critically revised the immunological implication of the study and the project development. MM, JL, AM, and CRRB wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was in part supported by: (i) MPI-BPC collaboration agreement 2018, Cancer Research UK [C67500; A29686] and National Institute for Health Research (NIHR) Biomedical Research Center based at Guy's and St Thomas' NHS Foundation Trust and King's College London and/or the NIHR Clinical Research Facility to MM. AM was supported by the International Max-Planck Research School (IMPRS) for Genome Sciences.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank A.C. Graham (KCL) for proofreading the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.614276/full#supplementary-material
References
1. Huseby E, White J, Crawford F, Vass T, Becker D, Pinilla C, et al. How the T cell repertoire becomes peptide and MHC specific. Cell. (2005) 122:247–69. doi: 10.1016/j.cell.2005.05.013
2. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see). Nat Rev Immunol. (2014) 14:377–91. doi: 10.1038/nri3667
3. Bouneaud C, Kourilsky P, Bousso P. Impact of negative selection on the T cell repertoire reactive to a self-peptide: a large fraction of T cell clones escapes clonal deletion. Immunity. (2000) 13:829–40. doi: 10.1016/S1074-7613(00)00080-7
4. Culina S, Lalanne AI, Afonso G, Cerosaletti K, Pinto S, Sebastiani G, et al. Islet-reactive CD8(+) T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci Immunol. (2018) 3:eaao4013. doi: 10.1126/sciimmunol.aao4013
5. ElTanbouly MA, Noelle RJ. Rethinking peripheral T cell tolerance: checkpoints across a T cell's journey. Nat Rev Immunol. (2020). doi: 10.1038/s41577-020-00454-2. [Epub ahead of print].
6. Yu W, Jiang N, Ebert PJ, Kidd BA, Muller S, Lund PJ, et al. Clonal deletion prunes but does not eliminate self-specific alphabeta CD8(+) T Lymphocytes. Immunity. (2015) 42:929–41. doi: 10.1016/j.immuni.2015.05.001
7. Liepe J, Ovaa H, Mishto M. Why do proteases mess up with antigen presentation by re-shuffling antigen sequences? Curr Opin Immunol. (2018) 52:81–6. doi: 10.1016/j.coi.2018.04.016
8. Wolfl M, Rutebemberwa A, Mosbruger T, Mao Q, Li HM, Netski D, et al. Hepatitis C virus immune escape via exploitation of a hole in the T cell repertoire. J Immunol. (2008) 181:6435–46. doi: 10.4049/jimmunol.181.9.6435
9. Assarsson E, Sidney J, Oseroff C, Pasquetto V, Bui HH, Frahm N, et al. A quantitative analysis of the variables affecting the repertoire of T cell specificities recognized after vaccinia virus infection. J Immunol. (2007) 178:7890–901. doi: 10.4049/jimmunol.178.12.7890
10. Croft NP, Smith SA, Pickering J, Sidney J, Peters B, Faridi P, et al. Most viral peptides displayed by class I MHC on infected cells are immunogenic. Proc Natl Acad Sci USA. (2019) 116:3112–7. doi: 10.1073/pnas.1815239116
11. Kanduc D, Stufano A, Lucchese G, Kusalik A. Massive peptide sharing between viral and human proteomes. Peptides. (2008) 29:1755–66. doi: 10.1016/j.peptides.2008.05.022
12. Kusalik A, Bickis M, Lewis C, Li Y, Lucchese G, Marincola FM, et al. Widespread and ample peptide overlapping between HCV and Homo sapiens proteomes, Peptides, United States. Peptides. (2007) 28:1260–7. doi: 10.1016/j.peptides.2007.04.001
13. Ricco R, Kanduc D. Hepatitis B virus and Homo sapiens proteome-wide analysis: a profusion of viral peptide overlaps in neuron-specific human proteins. Biologics. (2010) 4:75–81. doi: 10.2147/BTT.S8890
14. Trost B, Kusalik A, Lucchese G, Kanduc D. Bacterial peptides are intensively present throughout the human proteome. Self Nonself. (2010) 1:71–4. doi: 10.4161/self.1.1.9588
15. Trost B, Lucchese G, Stufano A, Bickis M, Kusalik A, Kanduc D. No human protein is exempt from bacterial motifs, not even one. Self Nonself. (2010) 1:328–34. doi: 10.4161/self.1.4.13315
16. Frankild S., de Boer RJ, Lund O, Nielsen M, Kesmir C. Amino acid similarity accounts for T cell cross-reactivity and for “holes” in the T cell repertoire. PLoS ONE. (2008) 3:e1831. doi: 10.1371/journal.pone.0001831
17. Calis JJ, de Boer RJ, Kesmir C. Degenerate T-cell recognition of peptides on MHC molecules creates large holes in the T-cell repertoire. PLoS Comput Biol. (2012) 8:e1002412. doi: 10.1371/journal.pcbi.1002412
18. Ishizuka J, Grebe K, Shenderov E, Peters B, Chen Q, Peng Y, et al. Quantitating T cell cross-reactivity for unrelated peptide antigens. J Immunol. (2009) 183:4337–45. doi: 10.4049/jimmunol.0901607
19. Rossjohn J, Gras S, Miles JJ, Turner SJ, Godfrey DI, McCluskey J. T cell antigen receptor recognition of antigen-presenting molecules. Annu Rev Immunol. (2015) 33:169–200. doi: 10.1146/annurev-immunol-032414-112334
20. Whalley T, Dolton G, Brown PE, Wall A, Wooldridge L, van den Berg H, et al. GPU-accelerated discovery of pathogen-derived molecular mimics of a T-cell insulin epitope. Front Immunol. (2020) 11:296. doi: 10.3389/fimmu.2020.00296
21. Assmus LM, Guan J, Wu T, Farenc C, Sng XYX, Zareie P, et al. Overlapping peptides elicit distinct CD8(+) T cell responses following influenza a virus infection. J Immunol. (2020) 205:1731–42. doi: 10.4049/jimmunol.2000689
22. Mishto M, Liepe J. Post-translational peptide splicing and T cell responses. Trends Immunol. (2017) 38:904–15. doi: 10.1016/j.it.2017.07.011
23. Platteel ACM, Liepe J, Textoris-Taube K, Keller C, Henklein P, Schalkwijk HH, et al. Multi-level strategy for identifying proteasome-catalyzed spliced epitopes targeted by CD8+ T cells during bacterial infection. Cell Rep. (2017) 20:1242–53. doi: 10.1016/j.celrep.2017.07.026
24. Platteel AC, Mishto M, Textoris-Taube K, Keller C, Liepe J, Busch DH, et al. CD8(+) T cells of Listeria monocytogenes-infected mice recognize both linear and spliced proteasome products. Eur J Immunol. (2016) 46:1109–18. doi: 10.1002/eji.201545989
25. Paes W, Leonov G, Partridge T, Chikata T, Murakoshi H, Frangou A, et al. Contribution of proteasome-catalyzed peptide cis-splicing to viral targeting by CD8(+) T cells in HIV-1 infection. Proc Natl Acad Sci USA. (2019) 116:24748–59. doi: 10.1073/pnas.1911622116
26. Mishto M, Mansurkhodzhaev A, Ying G, Bitra A, Cordfunke RA, Henze S, et al. An in silico-in vitro pipeline identifying an HLA-A(*)02:01(+) KRAS G12V(+) spliced epitope candidate for a broad tumor-immune response in cancer patients. Front Immunol. (2019) 10:2572. doi: 10.3389/fimmu.2019.02572
27. Ebstein F, Textoris-Taube K, Keller C, Golnik R, Vigneron N, Van den Eynde BJ, et al. Proteasomes generate spliced epitopes by two different mechanisms and as efficiently as non-spliced epitopes. Sci Rep. (2016) 6:24032. doi: 10.1038/srep24032
28. Faridi P, Woods K, Ostrouska S, Deceneux C, Aranha R, Duscharla D, et al. Spliced peptides and cytokine-driven changes in the immunopeptidome of melanoma. Cancer Immunol Res. (2020) 8:1322–34. doi: 10.1158/2326-6066.CIR-19-0894
29. Robbins PF, el-Gamil M, Kawakami Y, Stevens E, Yannelli JR, Rosenberg SA. Recognition of tyrosinase by tumor-infiltrating lymphocytes from a patient responding to immunotherapy. Cancer Res. (1994)54:3124–6.
30. Dalet A, Robbins PF, Stroobant V, Vigneron N, Li YF, El-Gamil M, et al. An antigenic peptide produced by reverse splicing and double asparagine deamidation. Proc Natl Acad Sci USA. (2011) 108:E323–31. doi: 10.1073/pnas.1101892108
31. Mishto M. What we see, what we do not see and what we do not want to see in HLA class I Immunopeptidomes. Proteomics. (2020). doi: 10.1002/pmic.202000112. [Epub ahead of print].
32. Berkers CR, de Jong A, Schuurman KG, Linnemann C, Meiring HD, Janssen L, et al. Definition of proteasomal peptide splicing rules for high-efficiency spliced peptide presentation by MHC class i molecules. J Immunol. (2015) 195:4085–95. doi: 10.4049/jimmunol.1402455
33. Mishto M, Goede A, Taube KT, Keller C, Janek K, Henklein P, et al. Driving forces of proteasome-catalyzed peptide splicing in yeast and humans. Mol Cell Proteomics. (2012) 11:1008–23. doi: 10.1074/mcp.M112.020164
34. Specht G, Roetschke HP, Mansurkhodzhaev A, Henklein P, Textoris-Taube K, Urlaub H, et al. Large database for the analysis and prediction of spliced and non-spliced peptide generation by proteasomes. Sci Data. (2020) 7:146. doi: 10.1038/s41597-020-0487-6
35. Dalet A, Vigneron N, Stroobant V, Hanada K, Van den Eynde BJ. Splicing of distant Peptide fragments occurs in the proteasome by transpeptidation and produces the spliced antigenic peptide derived from fibroblast growth factor-5. J Immunol. (2010) 184:3016–24. doi: 10.4049/jimmunol.0901277
36. Faridi P, Li C, Ramarathinam SH, Vivian JP, Illing PT, Mifsud NA, et al. A subset of HLA-I peptides are not genomically templated: evidence for cis- and trans-spliced peptide ligands. Sci Immunol. (2018) 3:eaar3947. doi: 10.1126/sciimmunol.aar3947
37. Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics. (2005) 6:132. doi: 10.1186/1471-2105-6-132
38. Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, Sette A, et al. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science. (2016) 354:354–358. doi: 10.1126/science.aaf4384
39. Peters B, Tong W, Sidney J, Sette A, Weng Z. Examining the independent binding assumption for binding of peptide epitopes to MHC-I molecules. Bioinformatics. (2003) 2003:1765–72. doi: 10.1093/bioinformatics/btg247
40. Hulo C, Castro E, Masson P, Bougueleret L., Bairoch A, Xenarios I, et al. ViralZone: a knowledge resource to understand virus diversity. Nucleic Acids Res. (2011) 39:D576–82. doi: 10.1093/nar/gkq901
41. Bairoch A, Apweiler R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Res. (1999) 27:49–54. doi: 10.1093/nar/27.1.49
42. Liepe J, Sidney J, Lorenz FKM, Sette A, Mishto M. Mapping the MHC class I-spliced immunopeptidome of cancer cells. Cancer Immunol Res. (2019) 7:62–76. doi: 10.1158/2326-6066.CIR-18-0424
43. Pinto S, Michel C, Schmidt-Glenewinkel H, Harder N, Rohr K, Wild S, et al. Overlapping gene coexpression patterns in human medullary thymic epithelial cells generate self-antigen diversity. Proc Natl Acad Sci USA. (2013) 110:E3497–505. doi: 10.1073/pnas.1308311110
44. Zeng Y, Liu C, Gong Y, Bai Z, Hou S, He J, et al. Single-cell RNA sequencing resolves spatiotemporal development of pre-thymic lymphoid progenitors and thymus organogenesis in human embryos. Immunity. (2019) 51:930–48 e6. doi: 10.1016/j.immuni.2019.09.008
45. Weinzierl AO, Lemmel C, Schoor O, Muller M, Kruger T, Wernet D, et al. Distorted relation between mRNA copy number and corresponding major histocompatibility complex ligand density on the cell surface. Mol Cell Proteomics. (2007) 6:102–13. doi: 10.1074/mcp.M600310-MCP200
46. Pearson H, Daouda T, Granados DP, Durette C, Bonneil E, Courcelles M, et al. MHC class I-associated peptides derive from selective regions of the human genome. J Clin Invest. (2016) 126:4690–701. doi: 10.1172/JCI88590
47. Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. (2015) 33:495–502. doi: 10.1038/nbt.3192
48. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck W. Comprehensive integration of single-cell data. Cell. (2019) 177:1888–902. doi: 10.1016/j.cell.2019.05.031
49. Perez CL, Larsen MV, Gustafsson R, Norstrom MM, Atlas A, Nixon DF, et al. Broadly immunogenic HLA class I supertype-restricted elite CTL epitopes recognized in a diverse population infected with different HIV-1 subtypes. J Immunol. (2008) 180:5092–100. doi: 10.4049/jimmunol.180.7.5092
50. Ogishi M, Yotsuyanagi H. Quantitative prediction of the landscape of T cell epitope immunogenicity in sequence space. Front Immunol. (2019) 10:827. doi: 10.3389/fimmu.2019.00827
51. Molero-Abraham M, Lafuente EM, Reche P. Customized predictions of peptide-MHC binding and T-cell epitopes using EPIMHC. Methods Mol Biol. (2014) 1184:319–32. doi: 10.1007/978-1-4939-1115-8_18
52. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. (2018) 46:W296–303. doi: 10.1093/nar/gky427
53. Chapiro J, Claverol S, Piette F, Ma W, Stroobant V, Guillaume B, et al. Destructive cleavage of antigenic peptides either by the immunoproteasome or by the standard proteasome results in differential antigen presentation. J Immunol. (2006) 176:1053–61. doi: 10.4049/jimmunol.176.2.1053
54. Deol P, Zaiss DM, Monaco JJ, Sijts AJ. Rates of processing determine the immunogenicity of immunoproteasome-generated epitopes. J Immunol. (2007) 178:7557–62. doi: 10.4049/jimmunol.178.12.7557
55. Guillaume B, Chapiro J, Stroobant V, Colau D, Van Holle B, Parvizi G, et al. Two abundant proteasome subtypes that uniquely process some antigens presented by HLA class I molecules. Proc Natl Acad Sci USA. (2010) 107:18599–604. doi: 10.1073/pnas.1009778107
56. Guillaume B, Stroobant V, Bousquet-Dubouch MP, Colau D, Chapiro J, Parmentier N, et al. Dalet, and B.J. Van den Eynde, Analysis of the processing of seven human tumor antigens by intermediate proteasomes. J Immunol. (2012) 189:3538–47. doi: 10.4049/jimmunol.1103213
57. Tenzer S, Wee E, Burgevin A, Stewart-Jones G, Friis L, Lamberth K, et al. Antigen processing influences HIV-specific cytotoxic T lymphocyte immunodominance. Nat Immunol. (2009) 10:636–46. doi: 10.1038/ni.1728
58. Zanker D, Waithman J, Yewdell JW, Chen W. Mixed proteasomes function to increase viral peptide diversity and broaden antiviral CD8+ T cell responses. J Immunol. (2013) 191:52–9. doi: 10.4049/jimmunol.1300802
59. Dalet A, Stroobant V, Vigneron N, Van den Eynde BJ. Differences in the production of spliced antigenic peptides by the standard proteasome and the immunoproteasome. Eur J Immunol. (2011) 41:39–46. doi: 10.1002/eji.201040750
60. Michaux A, Larrieu P, Stroobant V, Fonteneau JF, Jotereau F, Van den Eynde BJ, et al. A spliced antigenic peptide comprising a single spliced amino acid is produced in the proteasome by reverse splicing of a longer peptide fragment followed by trimming. J Immunol. (2014) 192:1962–71. doi: 10.4049/jimmunol.1302032
61. Vigneron N, Stroobant V, Chapiro J, Ooms A, Degiovanni G, Morel S, et al. An antigenic peptide produced by peptide splicing in the proteasome. Science. (2004) 304:587–90. doi: 10.1126/science.1095522
62. Warren EH, Vigneron NJ, Gavin MA, Coulie PG, Stroobant V, Dalet A, et al. An antigen produced by splicing of noncontiguous peptides in the reverse order. Science. (2006) 313:1444–7. doi: 10.1126/science.1130660
63. Hanada K, Yewdell JW, Yang JC. Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature. (2004) 427:252–6. doi: 10.1038/nature02240
64. Grignolio A, Mishto M, Faria AM, Garagnani P, Franceschi C, Tieri P. Towards a liquid self: how time, geography, and life experiences reshape the biological identity. Front Immunol. (2014) 5:153. doi: 10.3389/fimmu.2014.00153
65. Arribas-Layton D, Guyer P, Delong T, Dang M, Chow IT, Speake C. Hybrid insulin peptides are recognized by human T cells in the context of DRB1*04:01. Diabetes. (2020) 69:1492–502. doi: 10.2337/db19-0620
66. Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med. (2016) 22:1482–7. doi: 10.1038/nm.4203
67. Wang Y, Sosinowski T, Novikov A, Crawford F, White J, Jin N, et al. How C-terminal additions to insulin B-chain fragments create superagonists for T cells in mouse and human type 1 diabetes. Sci Immunol. (2019) 4:eaav7517. doi: 10.1126/sciimmunol.aav7517
68. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science. (2016) 351:711–4. doi: 10.1126/science.aad2791
69. Liepe J, Holzhutter HG, Bellavista E, Kloetzel PM, Stumpf MP, Mishto M. Quantitative time-resolved analysis reveals intricate, differential regulation of standard- and immuno-proteasomes. Elife. (2015) 4:e07545. doi: 10.7554/eLife.07545
70. Mishto M, Liepe J, Textoris-Taube K, Keller C, Henklein P, Weberruss M, et al. Proteasome isoforms exhibit only quantitative differences in cleavage and epitope generation. Eur J Immunol. (2014) 44:3508–21. doi: 10.1002/eji.201444902
71. Textoris-Taube K, Keller C, Liepe J, Henklein P, Sidney J, Sette A, et al. The T210M substitution in the HLA-a*02:01 gp100 epitope strongly affects overall proteasomal cleavage site usage and antigen processing. J Biol Chem. (2015) 290:30417–28. doi: 10.1074/jbc.M115.695189
72. Toes RE, Nussbaum AK, Degermann S, Schirle M, Emmerich NP, Kraft M, et al. Discrete cleavage motifs of constitutive and immunoproteasomes revealed by quantitative analysis of cleavage products. J Exp Med. (2001) 194:1–12. doi: 10.1084/jem.194.1.1
73. Dianzani C, Vecchio D, Clemente N, Chiocchetti A, Martinelli Boneschi F, Galimberti D, et al. Untangling extracellular proteasome-osteopontin circuit dynamics in multiple sclerosis. Cells. (2019) 8:262. doi: 10.3390/cells8030262
74. Kuckelkorn U, Stubler S, Textoris-Taube K, Kilian C, Niewienda A, Henklein P, et al. Proteolytic dynamics of human 20S thymoproteasome. J Biol Chem. (2019) 294:7740–54. doi: 10.1074/jbc.RA118.007347
75. Fabre B, Lambour T, Garrigues L, Amalric F, Vigneron N, Menneteau T, et al. Deciphering preferential interactions within supramolecular protein complexes: the proteasome case. Mol Syst Biol. (2015) 11:771. doi: 10.15252/msb.20145497
Keywords: bioinformatics, antigen presentation, MHC-I, peptide splicing, negative selection, T-cell repertoire, T-cell tolerance
Citation: Mansurkhodzhaev A, Barbosa CRR, Mishto M and Liepe J (2021) Proteasome-Generated cis-Spliced Peptides and Their Potential Role in CD8+ T Cell Tolerance. Front. Immunol. 12:614276. doi: 10.3389/fimmu.2021.614276
Received: 05 October 2020; Accepted: 28 January 2021;
Published: 24 February 2021.
Edited by:
Eddie A. James, Benaroya Research Institute, United StatesReviewed by:
Anthony Wayne Purcell, Monash University, AustraliaDietmar M. W. Zaiss, University of Edinburgh, United Kingdom
Copyright © 2021 Mansurkhodzhaev, Barbosa, Mishto and Liepe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michele Mishto, bWljaGVsZS5taXNodG9Aa2NsLmFjLnVr; Juliane Liepe, amxpZXBlQG1waWJwYy5tcGcuZGU=
†These authors have contributed equally to this work