 Evelien G. G. Sprenkeler
Evelien G. G. Sprenkeler Carla Guenther
Carla Guenther Imrul Faisal
Imrul Faisal Taco W. Kuijpers
Taco W. Kuijpers Susanna C. Fagerholm
Susanna C. Fagerholm- 1Department of Blood Cell Research, Sanquin Research, Amsterdam University Medical Center (AUMC), University of Amsterdam, Amsterdam, Netherlands
- 2Department of Pediatric Immunology, Rheumatology, and Infectious Diseases, Emma Children’s Hospital, Amsterdam University Medical Center (AUMC), University of Amsterdam, Amsterdam, Netherlands
- 3Molecular and Integrative Biosciences Research Programme, Faculty of Biological and Environmental Sciences, University of Helsinki, Helsinki, Finland
Megakaryoblastic leukemia 1 (MKL1) deficiency is one of the most recently discovered primary immunodeficiencies (PIDs) caused by cytoskeletal abnormalities. These immunological “actinopathies” primarily affect hematopoietic cells, resulting in defects in both the innate immune system (phagocyte defects) and adaptive immune system (T-cell and B-cell defects). MKL1 is a transcriptional coactivator that operates together with serum response factor (SRF) to regulate gene transcription. The MKL/SRF pathway has been originally described to have important functions in actin regulation in cells. Recent results indicate that MKL1 also has very important roles in immune cells, and that MKL1 deficiency results in an immunodeficiency affecting the migration and function of primarily myeloid cells such as neutrophils. Interestingly, several actinopathies are caused by mutations in genes which are recognized MKL(1/2)-dependent SRF-target genes, namely ACTB, WIPF1, WDR1, and MSN. Here we summarize these and related (ARPC1B) actinopathies and their effects on immune cell function, especially focusing on their effects on leukocyte adhesion and migration. Furthermore, we summarize recent therapeutic efforts targeting the MKL/SRF pathway in disease.
Introduction
Leukocytes constantly traffic between different compartments in the body, and need to be able to employ different types of adhesion and migration modes in different types of environments. Briefly, leukocytes adhere to endothelial cells under shear flow conditions using adhesion receptors such as selectins, integrins, and intercellular adhesion molecules, utilize 2D migration modes on the endothelial cell layer, and migrate in complex tissue environments utilizing 3D migration. Whereas 2D migration is strongly dependent on cell adhesion (i.e., ligand binding via integrins), the 3D amoeboid migration mode of leukocytes such as dendritic cells is associated with very low adhesion strength (i.e., integrin-independent) and relies on cytoskeletal deformation instead (1, 2).
Both during 2D and 3D migration, actin dynamics in migrating cells is complex and regulated by both positive and negative regulators. Actomyosin contraction at the cell rear end (uropod) aids in moving the cytoplasm and cell body, while the actin-related protein (ARP) 2/3 complex is an important mediator of actin polymerization at the leading edge (1). The small GTPases Rac and Cdc42 localize to the leading edge and regulate actin polymerization (2). In contrast, RhoA localizes to the uropod where it regulates actin cables (through the formin mDia) and actomyosin contractility (2). Proteins binding to G-actin, such as ADF/cofilin, and proteins severing and capping actin filaments, such as gelsolin, are also important factors regulating actin dynamics in migrating cells (3).
Primary immunodeficiencies (PIDs) are rare genetic disorders of the immune system, and lead to immune deficiencies of various severity. By studying primary immunodeficiencies, much has been learned about the molecular basis of immune system function, including leukocyte trafficking. In a classical example, Wiskott-Aldrich syndrome is caused by defects in the WAS protein (WASP), which plays essential roles in the regulation of the actin cytoskeleton upon cell activation. Consequently, WASP deficiency leads to fundamental defects of the immune system, including leukocyte migration, as well as a platelet defect with bleeding tendency because of reduced platelet counts (4).
Leukocyte adhesion deficiency type I-II and -III in turn are caused by defects in beta2-integrins, selectins, and kindlin-3, respectively, and result in severe defects in leukocyte (especially neutrophil) trafficking into sites of inflammation, as well as specific defects in adaptive immunity (5). Notably, all abovementioned defects (apart from LAD-II, which is a metabolic disorder which also affects leukocytes and neutrophil extravasation) represent typical hematopoietic disorders. This is explained by the lack of protein expression outside of the hematopoietic system and/or lack of redundancy in activity by homologous proteins that may substitute for the protein that is lacking or dysfunctional in activity. The selective expression of these proteins within the hematopoietic system also explains that curative treatment currently (still) consists of bone marrow transplantation.
Megakaryoblastic leukemia 1 (MKL1) deficiency is one of the most recently identified primary immunodeficiencies that causes a rare defect in actin-dependent processes, including leukocyte adhesion and migration. Here, we review what is known about MKL1 deficiency and other MKL/SRF (serum response factor)-related actin-based primary immunodeficiencies. In this review, we will focus on the proteins involved in this major pathway, their roles in immune cell migration and effector functions, and discuss potential lessons to be learned from these diseases and new opportunities with regards to therapeutic targeting of this pathway.
The MKL1/SRF Pathway
MKL1, also called MRTF-A (myocardin-related transcription factor-A) or MAL (megakaryocytic acute leukemia), is a transcriptional co-regulator expressed in many cell types. It has long been known to have important roles in regulating actin and other cytoskeleton genes in many types of cells, together with the transcription factor SRF. There are two isoforms of MKL, MKL1, and MKL2, which have similar roles in cells. However, they also have non-redundant roles, as MKL2 knockout mice are embryonic lethal (6), while MKL1 knockout mice have a milder phenotype. MKL1 knockout mice show partial embryonic lethality, abnormal mammary gland function and reduced platelet count (7).
MKL1 is an interesting transcriptional coactivator, which is itself regulated by actin cytoskeletal dynamics (8). MKL1 is normally found in the cytoplasm, where it is bound to G-actin, and therefore excluded from the nucleus, which means it cannot regulate gene transcription. When cells receive an activating stimulus, such as serum stimulation, chemokine stimulation or other types of stimuli, RhoA is activated, leading to actin polymerization into F-actin. As a consequence, MKL1 is released from G-actin and transported into the nucleus (Figure 1). There, it encounters the transcription factor SRF and together the complex initiates gene transcription. In fact, SRF recruits two families of coactivators, the MKLs and the TCFs (ternary complex factors), to couple gene transcription to growth factor signaling. The MKL/SRF complex is involved in regulating cytoskeletal genes, such as actin, in many types of cells, including leukocytes, thereby influencing actin-dependent processes in cells (Figure 1). In macrophages, the SRF pathway indeed regulates the expression of cytoskeletal genes (9). In B cells, however, deletion of SRF also led to decreased expression of IgM, CXCR4 and CD19 (10). The specific role of MKL1 in immune cells is less explored. In human MKL1-deficient neutrophils, many adhesion and actin regulators were downregulated, including CASS4, ALCAM, MYL9 (11); proteomic studies further identified WDR and actin itself. MKL1 also regulated expression of genes involved in interferon signaling, such as STAT1 and several IFIT genes, but MKL1 deficiency also resulted in the upregulation of several genes (11). We have recently shown that in murine dendritic cells, MKL1 deletion did not only impact expression of cytoskeletal genes and genes of cytoskeletal regulators (Fgr, Hck, Stmn1, Ckap2l, Anln, Tpm2, Tubb5), but also genes encoding for proteins involved in many other cellular pathways, such as lipid metabolism (12). Therefore, the effect of MKL1 on gene expression in immune cells appears to be cell type-specific. However, it is clear that many MKL/SRF-target genes in leukocytes, as in other cell types, are actin and actin regulators, for example ACTB, WIPF1, WDR1, and MSN (13), which regulate actin cytoskeletal dynamics, and therefore affect actin-dependent processes in cells (Figures 1 and 2).
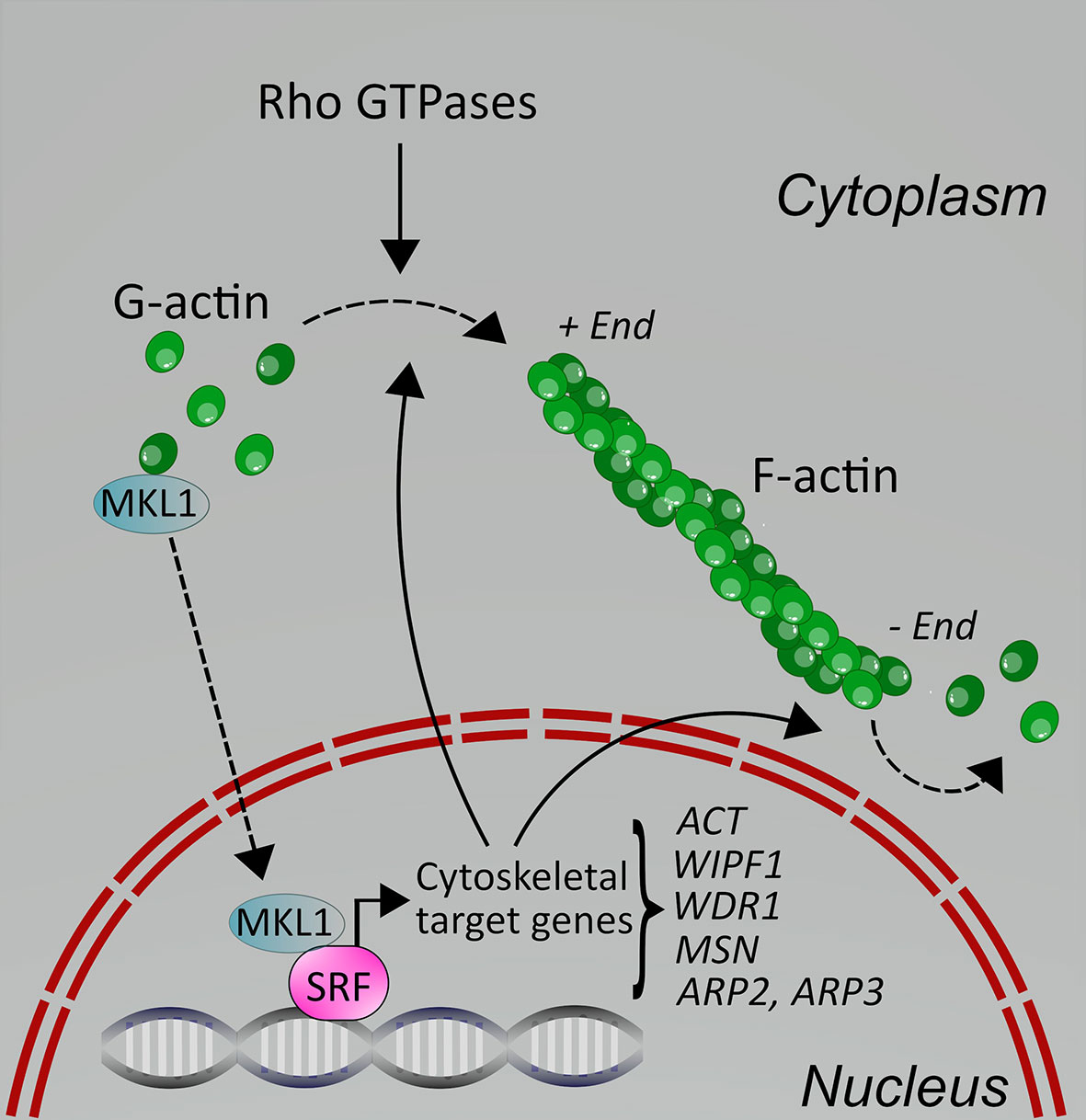
Figure 1 Schematic depicting the molecular regulation of MKL1 in cells. In resting cells, MKL1 is sequestered in the cytoplasm due to binding to G-actin through its RPEL motifs. Upon cell activation through various stimuli, including serum, chemokines and integrins, Rho GTPases are activated, leading to F-actin polymerization. This releases MKL1, allowing it to translocate into the nucleus where it can activate gene transcription together with SRF. Some of the cytoskeletal target genes are shown here; for additional details, see text. Cytoskeletal target genes impact on the actin cytoskeleton in cells, leading to changes in processes such as cell adhesion and motility.
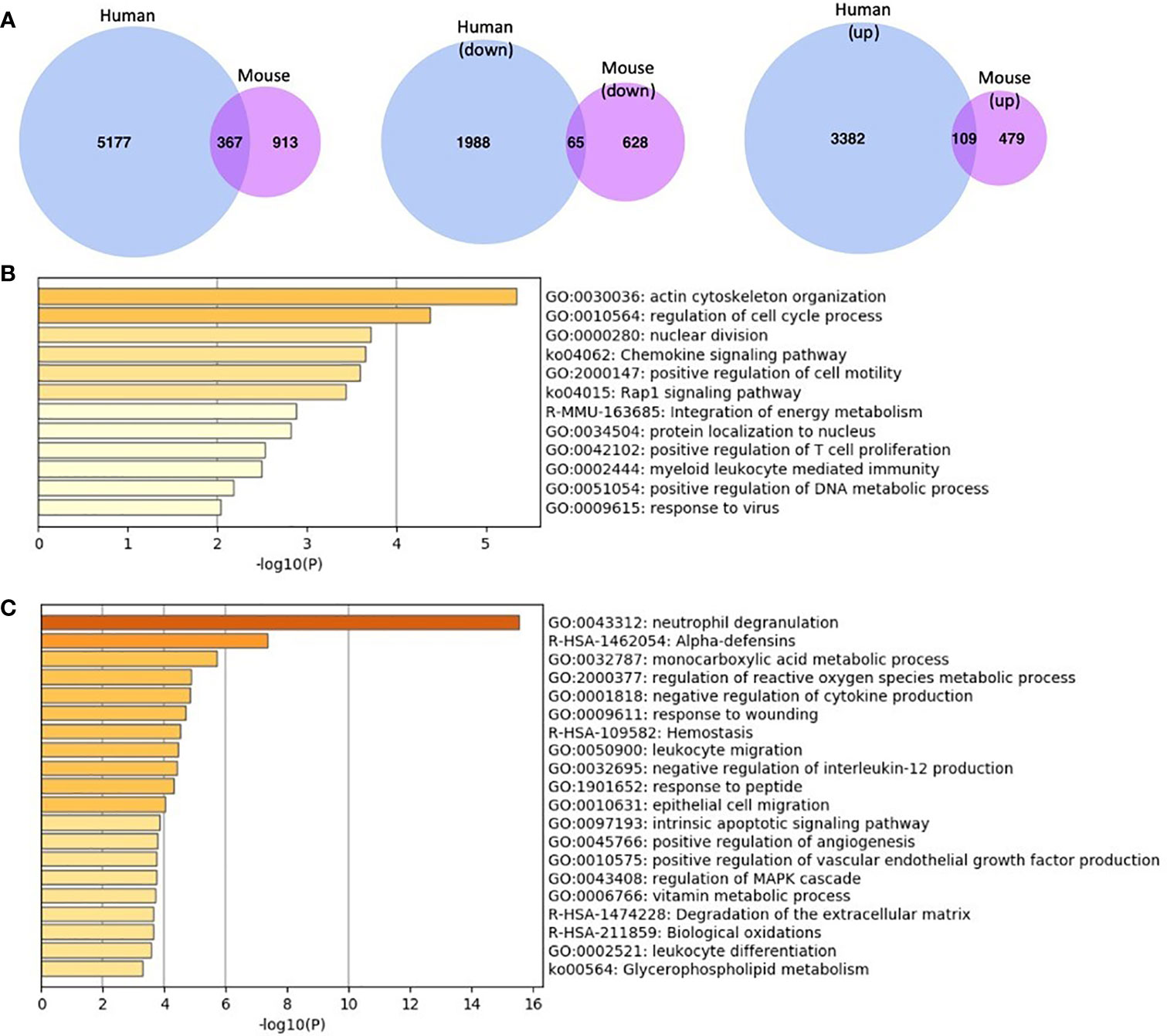
Figure 2 Comparison of gene expression profiles for MKL1-deficient leukocytes (human and murine cells) (A) Differentially expressed genes (both up- and downregulated) in MKL1-deficient cells. There were 367 genes in common between them. The second and third Venn diagrams illustrate differentially expressed genes according to expression profile, i.e., upregulated and downregulated separately. There were 65 common downregulated genes and 109 upregulated genes between human and mouse MKL1 deficient cells. The mouse genes were converted to humans by Ensembl’s BioMart package and then compared to the human genes. The Venn diagrams were created using venn.diagram package in R. (B, C) Pathway enrichment analysis using downregulated (B) and upregulated (C) common genes between human and mouse MKL1-deficient cells. This analysis was performed using Metascape.
MKL1 Deficiency
Interestingly, patients with MKL1 mutations have recently been reported. The first patient with a homozygous nonsense mutation in MKL1 was reported in 2015 (14), while two more cases (siblings) were reported in 2020 (11). The first sibling died after contracting pneumonia at an early age, the second one underwent a pre-emptive bone marrow transplant. MKL1 mutations result in primary immunodeficiency, with increased susceptibility to bacterial infections (Pseudomonas pneumonia). Lymphocyte counts and immunoglobulin levels were normal and no major defect in T- or B-cell function have been reported in these patients (14). Also no major platelet dysfunction has been reported (11, 14). Neutrophil numbers were also normal or elevated because of the concomitant infection. Phagocytosis in patient neutrophils has been reported both as normal – even of larger particles by neutrophils and macrophages (11) – and abnormal (14), while neutrophil reactive oxygen species (ROS) release and bacterial killing have been reported as being intact in MKL1 patients (11). Azurophilic granule release was found to be increased by MKL1-deficient neutrophils under suboptimal stimulation (11).
Role of MKL/SRF Signaling in Leukocyte Adhesion and Migration
MKL1 deficiency in humans results in a severe migration defect of neutrophils (reduced chemotaxis towards chemotactic agents such as formyl-methionyl-leucyl phenylalanine (fMLF) and complement component 5a (C5a) (11, 14). Also spreading of both neutrophils and dendritic cells was reduced (11, 14). Interestingly, in neutrophils, firm adhesion, and transendothelial migration under shear flow conditions were severely affected, although cell adhesion under static conditions was not reduced (11). Total actin and F-actin levels in neutrophils were reduced (11, 14). Analysis of protein expression levels by high-resolution label-free quantitative mass spectrometry showed that indeed actin and actin regulators (WDR1, ARHGAP9, PFN1) were downregulated in MKL1-deficient neutrophils (11). The dramatic effect of the MKL1 mutation on neutrophil phenotype may be explained by the fact that these cells do not express MKL2, which may compensate for the lack of MKL1 in other cell types, such as fibroblasts. Indeed, primary fibroblasts with MKL1 deficiency display normal migration (11).
The role of MKL1 in dendritic cell adhesion and migration has also been investigated. Interestingly, the MKL1/SRF pathway is downstream of beta2-integrins and beta2-integrin-mediated RhoA activation and F-actin polymerization in murine leukocytes (12). 3D migration of MKL1 knockout dendritic cells in response to CCL19 was normal, while (static) adhesion to integrin ligands was slightly reduced. Integrins function as mechanoreceptors in cells, transmitting mechanical force across the plasma membrane. In MKL1 knockout dendritic cells, integrin-mediated traction forces were reduced. Dendritic cells express both MKL1 and MKL2, which presumably underlies the fact that the adhesion/migration defect of MKL1 knockout dendritic cells was not as severe as in (human) neutrophils, which only express the one isoform.
MKL regulates transcription through interactions with SRF, which has been previously implicated in cell adhesion and migration in immune cells [reviewed in (15)]. SRF-deficient murine macrophages display reduced spreading, migration, and phagocytosis (9). In neutrophils, SRF plays an important role in cell trafficking in vitro and in vivo. SRF-deficient murine neutrophils failed to traffic into sites of inflammation in vivo, and displayed reduced binding to integrin ligands, and cell migration in vitro (16). Several actin regulators were downregulated in SRF knockout neutrophils, including Actb. Other SRF targets found to be downregulated also in murine MKL1 knockout dendritic cells include Lima1, Actg1, Cnn2, Tpm4, and Wdr1 (12).
MKL1 (and presumably MKL1/SRF-dependent gene transcription of genes encoding cytoskeletal elements and additional proteins involved in signaling, etc) therefore appears to be most important for integrin-mediated cell adhesion under shear flow conditions, where cells have to withstand substantial mechanical forces to resist shear forces in blood vessels. In addition, its role in integrin-mediated traction force generation further implies that MKL1-regulated gene transcription is important for integrin mechanoreceptor function. However, certain other actin-dependent functions appear to function also without MKL1-signaling (ROS release, bacterial killing) and seem intact even in the presence of a strongly reduced G-actin level. It is possible that major actin polymerization is only required for certain highly complex processes related to spreading and motility, whereas other actin-dependent activities such phagocytosis and (intraphagosomal) degranulation for the following killing process of the engulfed microbial particles may be less dependent on complex actin responses. In addition, although there are molecular similarities between the integrin/cytoskeletal processes involved in cell migration and phagocytosis, there are also differences [lack of myosin/contractility in phagocytosis, for example (17)] that may explain the differential dependence on MKL1/SRF of these processes.
Together, these human and murine data point towards an important role for MKL/SRF signaling for regulating myeloid immune cell integrin-mediated adhesion and migration in vivo, especially affecting neutrophils. Furthermore, mechanistically MKL1/SRF is probably involved in regulating neutrophil adhesion and migration through regulation of gene transcription of actin and actin-related factors/or other factors that regulate cell adhesion and migration.
Actin-Related Primary Immunodeficiencies Involving MKL/SRF-Target Genes
Interestingly, several primary immunodeficiencies caused by cytoskeletal abnormalities [“actinopathies” (18–20)] are caused by mutations in genes which are recognized MKL(1/2)-dependent SRF-target genes, namely ACTB, WIPF1, WDR1, and MSN (13). Here, we summarize these actinopathies and their effect on immune cell function, in an attempt to dissect which MKL/SRF target genes may be important for leukocyte adhesion and trafficking.
Cytoplasmic Beta-Actin
Actin is one of the major targets of the MKL/SRF transcription axis (13) and, as observed in MKL1 deficiency (11, 14) important for homeostatic levels of actin. Actin has six isoforms, one of which is cytoplasmic beta-actin (encoded by ACTB) (21). Although loss-of-function mutations in ACTB have not been reported to cause a PID, it has been described that patients suffer from recurrent infections, which may imply an influence of these mutations on the immune system (22).
One report on immune cell function in a patient with a missense mutation in ACTB identified reduced chemotaxis and decreased ROS production of neutrophils (23). This mutation led to the expression of both normal and mutated cytoplasmic beta-actin in patient cells, including fibroblasts, leukocytes, Epstein–Barr virus-transformed B-cells, and platelets (23). Further investigation revealed that this mutated beta-actin had a slower polymerization rate than normal actin (24). More recently, a lack of beta-actin in mouse embryonic fibroblasts was shown to downregulate interferon-stimulated genes due to instability of the transcription factor interferon regulatory factor 3, thereby impairing the antiviral response (25). Likewise, SRF has been previously shown to be important for type I interferon signaling by induction of several interferon-stimulated genes (26).
Wiskott-Aldrich Syndrome Protein-Interacting Protein
Actin polymerization, i.e., the assembly of G-actin into F-actin, is mediated by the Wiskott-Aldrich syndrome protein (WASP), as it activates the actin-related proteins-2/3 (ARP2/3) complex. This complex, consisting of seven subunits, is an actin nucleator which initiates the branching of actin filaments (27). The Wiskott-Aldrich syndrome protein-interacting protein (WIP) interacts with WASP, thereby stabilizing its inactive state. When WASP gets activated, WIP is phosphorylated and dissociates from WASP (28, 29). The human gene encoding for WIP, WIPF1, has been identified to be a MKL-dependent SRF-target gene (13).
Only a handful of patients with WIP deficiency, caused by autosomal recessive mutations in WIPF1, have been identified thus far (30–32). As WIP regulates the stability and localization of WASP (33), loss of WIP will also lead to absence of WASP (30, 34). Therefore, patients with WIP deficiency have a similar clinical phenotype as WAS patients, i.e., recurrent infections, thrombocytopenia, and eczema (31, 35).
The adaptive immune cells, i.e., T-cells and B-cells, are most affected in WIP deficiency. Patients suffer from T-cell lymphopenia, where CD8+ T-cells are more affected (35). Functionally, T-cells showed impaired proliferation, defective chemotaxis, defective exocytosis, and reduced target killing, most likely because there is a failure to assemble the immunological synapse (30, 32). Also, T-cells had an abnormal morphology, and failed to elongate and assemble a leading- and trailing edge upon stimulation (32). Natural killer (NK) cell–mediated cytotoxicity was reported to be reduced (30), and B-cells showed a chemotaxis defect, which could be rescued when WIP expression was restored in these cells (32). No defects in neutrophil function were reported in WIP patients, and monocyte-derived dendritic cells showed a normal phenotype (32). As the phenotype is quite different from that seen in MKL1 deficiency, WIPF1 may not be the most important gene regulating neutrophil trafficking downstream of MKL/SRF.
WD Repeat-Containing Protein 1
Actin-interacting protein 1 (Aip1), encoded WDR1, is an actin-binding protein which enhances the disassembly and severing of actin filaments by cofilin (36, 37). WDR1 is identified as an MKL/SRF target gene (13), and expression of WDR1 is strongly reduced in murine cells lacking Srf (38), in murine dendritic cells lacking Mkl1 (12), and on the protein level in neutrophils of MKL1-deficient patients (11). Mutations in WDR1 have been reported to cause immunodeficiency (39–41), and WDR1 is considered to be the main candidate gene to cause the “Lazy Leukocyte Syndrome”, first described in 1971 (42). Patients with loss-of-function mutations in WDR1 are reported to suffer from recurrent infections, mild neutropenia and impaired wound healing (39), but also a separate syndrome of auto-inflammation, periodic fever, and thrombocytopenia has been reported (40, 41).
WDR1-deficient patient cells, including neutrophils, monocytes, dendritic cells, and lymphocytes, are defective in actin depolymerization, resulting in increased F-actin levels (39–41). About 50% of the neutrophils had an altered herniation of their nuclear lobes, which co-localized with increased F-actin staining (39, 40). Neutrophils had a severe chemotaxis defect and showed abnormal spreading on glass, while Staphylococcal aureus killing and phagocytosis of opsonized Escherichia coli were reported to be normal. ROS production was found both normal and enhanced in WDR1-deficient neutrophils (39–41). Monocytes showed increased spreading over fibronectin, and CD14+ peripheral blood mononuclear cells showed an increased caspase-1 activation, which corresponds with auto-inflammation (40, 41).
Furthermore, defects in the adaptive immune system were observed. T-cells had an increased spreading capacity and mildly impaired proliferation, but normal T-cell receptor internalization, normal migration, and normal killing of target cells by CD8+ T-cells (40). B-cells showed increased apoptosis on B-cell receptor/Toll-like receptor stimulation and abnormal spreading, but normal migration. Also, patients suffered from peripheral B-cell lymphopenia and there was a lack of switched memory B-cells, reduced clonal diversity and paucity of B-cell progenitors in the bone marrow (40).
Moesin
Moesin (encoded by MSN) is a member of the ezrin, radixin, moesin (ERM) protein family, which link cortical actin filaments to the plasma membrane and membrane receptors. ERM proteins are important for structural stability and the integrity of the cell cortex (43). MSN is recognized to be a MKL-dependent SRF-target gene (13). As MSN is located on the X chromosome, the immunodeficiency was termed X-linked moesin associated immunodeficiency (44). Patients suffer from recurrent bacterial- and viral infections, persistent eczema and lymphopenia, as well as fluctuating neutropenia (44–46).
MSN-deficient T-cells showed impaired proliferation, impaired chemotaxis, and increased adhesion to vascular cell adhesion molecule 1 (44). While no functional analysis was done on neutrophils or monocytes from these patients, neutrophils from male moesin-deficient mice (Msn -/Y) did display elevated rolling velocity in inflamed blood vessels, indicating that moesin has an important role in slow leukocyte rolling and subsequent trans-endothelial migration (47). Also, moesin-deficient mice showed a reduced neutrophil microbial killing ability towards Pseudomonas aeruginosa and reduced neutrophil-mediated vascular inflammation, while neutrophil adhesion was not affected (48).
Actin-Related Proteins 2/3 Complex Subunit 1B
The ARP2/3 complex, consisting of seven subunits, plays an essential role in the formation of branched actin networks by nucleating a daughter filament to the side of a pre-existing actin filament (27). These branched actin networks are especially of importance for generating a protrusive force that aids in cellular adhesion and motility (49). Almost all subunits of the ARP2/3 complex have been recognized to be MKL/SRF-target genes [ARP2, ARP3, ARP2/3 complex subunit 2 (ARPC2), ARPC4, and ARPC5] (13), suggesting that MKL1/SRF is involved in actin branching by regulating transcription of ARP2/3 complex family members. ARPC1 is present in two isoforms in humans, ARPC1A and ARPC1B, the latter being the dominant form in hematopoietic cells (50, 51). Although it is not (yet) identified as a MKL/SRF-target gene, patients with ARPC1B deficiency have a very similar neutrophil phenotype compared to MKL1-deficient patients.
Patients with ARPC1B deficiency suffer from a combined immunodeficiency including a neutrophil defect, resulting clinically in bacterial- and viral infections, bleeding tendency, eczema, allergy, and vasculitis (50–55). Similar to MKL1 deficiency, neutrophils have a severe migration defect due to an actin polymerization defect. Also, ROS production and the phagocytosis and killing of bacteria were found to be intact, and azurophilic granule release was increased under suboptimal stimulation (50).
Although no major defects in T- or B-cell function have been reported in MKL1 deficiency, ARPC1B deficiency does result in lymphocytes defects, including defective migration and proliferation, defective immunological synapse assembly by T-cells (53, 55), impaired regulatory T-cell suppressor activity and impaired NK-cell degranulation (52). Primary fibroblasts showed normal migration, most likely due to expression of ARPC1A in these cells (similar to possible MKL2 compensation in MKL1 deficiency) (50).
Thus, MKL1-deficient neutrophils have an almost identical phenotype as ARPC1B-deficient neutrophils, which could be explained by the involvement of MKL/SRF in transcription of ARP2/3 complex family members. However, ARPC1B deficiency also affects lymphocytes, which has not been observed in MKL1 deficiency. A possible explanation might be redundancy of MKL2 in lymphocytes, which express low levels of this isoform.
In conclusion, analysis of primary immunodeficiencies involving MKL/SRF targets and putative targets (Table 1) implicate ARP-subunits as possible targets downstream of MKL/SRF that could be involved in regulating leukocyte adhesion and migration in vivo.

Table 1 List of MKL/SRF-related actinopathies with corresponding protein function, clinical symptoms, and reported functionally affected hematopoietic cells in these patients.
Targeting of MKL/SRF in Immune Cell-Mediated Inflammation—New Possibilities?
MKL/SRF signaling and MKL/SRF regulated gene expression of actin-related factors are important in leukocyte adhesion and migration, as shown by the actin-related primary immunodeficiencies described above. Interestingly, genetic syndromes also exist where actin dynamics is upregulated, eg X-linked neutropenia. In this disease, neutrophil levels in blood are low, but there is increased migration of leukocytes into tissues. X-linked neutropenia is caused by gain-of-function mutations in WASP, leading to increased actin dynamics, and, as a consequence, to upregulation of actin-dependent neutrophil adhesion, migration, and recruitment into tissues (57).
As MKL/SRF signaling and downstream targets of this pathway are clearly associated with leukocyte migration, targeting this pathway in inflammatory diseases where leukocyte migration into tissues is upregulated may offer a new therapeutic strategy. MKL/SRF signaling is important in many cell types and tissues, and is also implicated in disease in different settings. Indeed, the MKL/SRF pathway has already been targeted in a number of different diseases, including several with an immune/inflammatory cell component, such as fibrosis and corneal wound healing. Interestingly, MKL/SRF inhibitors have been used to treat fibrosis in lung, skin, colon, liver, joints, and the eye (both the external cornea and the internal subretina) (Table 2). The target for these therapeutic approaches are myofibroblasts and the excess extracellular matrix (ECM) production by these cells, as MKL1 has been found to be important for myofibroblast differentiation (74). However, in scleroderma/systemic sclerosis patient skin samples, a disease associated with internal organ stiffening due to fibrosis, nuclear MKL1 has also been found in infiltrating macrophages (65), where it has been linked to proinflammatory gene expression (75, 76), indicating that also macrophages could be a target cell type for SRF inhibition in fibrosis.

Table 2 MKL/SRF targeting in disease.
Targeting the MKL/SRF pathway to improve corneal wound healing has emerged as an attractive strategy. The cornea is a transparent endothelial cell layer in the eye and its thinning as well as replacement with connective and corneal stroma is associated with blindness. In the cornea, stromal cells produce cytokines like interleukin-1 (IL-1) and transforming growth factor-β (TGF-β) to induce inflammatory and degradative processes or to induce ECM deposition (77). TGF-β1 activates the MKL/SRF pathway (67, 78), indicating MKL/SRF could play a role in corneal wound healing (79). Indeed, eye drops supplemented with the inhibitor Y-27632 to inhibit Rho Associated Kinase (ROCK) upstream of MKL/SRF, have been used to improve corneal endothelial wound healing in vivo (80).
Summary and Future Prospects
MKL1 deficiency is one of the most recently identified primary immunodeficiencies and is associated with globally impaired actin regulation, defective cell adhesion and abnormal trafficking of myeloid leukocytes. Studies in both mouse and man have provided novel insights into its complex role in immune cell migration and function in the context of infections. The main cell types affected by MKL1 deficiency and MKL/SRF signaling are neutrophils and macrophages. Neutrophils are involved in a wide variety of inflammatory processes including acute organ injury, cystic fibrosis, ischemia reperfusion injury, atherosclerosis, and autoimmunity (such as rheumatoid arthritis). It is tempting to speculate that MKL/SRF, which is already being targeted in several diseases (Table 2) could also be used to reduce neutrophil and macrophage trafficking in these and other inflammatory disorders.
Author Contributions
ES, CG, TK, and SF wrote the manuscript on behalf of the LADOMICS consortium. IF analyzed bioinformatics data and made Figure 2. All authors contributed to the article and approved the submitted version.
Funding
The work in the authors’ laboratories is supported by the Academy of Finland, Svenska Kulturfonden, Liv och Hälsa, HiLIFE, and E-Rare/Academy of Finland (LADOMICS) (to SF), Magnus Ehrnrooth foundation (to CG). TK and ES were partially funded by the European Union’s Horizon 2020 research and innovation programme under Grant Agreement No.668303, Program on Prevention Outcomes Practices Grant PPOP-12-001, the Center of Immunodeficiencies Amsterdam Grant CIDA-2015, and the E-Rare ZonMW grant #90030376506.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Professor Ronen Alon (Weizmann Institute) for helpful comments on the manuscript.
References
1. Lammermann T, Germain RN. The multiple faces of leukocyte interstitial migration. Semin Immunopathol (2014) 36(2):227–51. doi: 10.1007/s00281-014-0418-8
2. Moreau HD, Piel M, Voituriez R, Lennon-Dumenil AM. Integrating Physical and Molecular Insights on Immune Cell Migration. Trends Immunol (2018) 39(8):632–43. doi: 10.1016/j.it.2018.04.007
3. Lee SH, Dominguez R. Regulation of actin cytoskeleton dynamics in cells. Mol Cells (2010) 29(4):311–25. doi: 10.1007/s10059-010-0053-8
4. Bouma G, Burns SO, Thrasher AJ. Wiskott-Aldrich Syndrome: Immunodeficiency resulting from defective cell migration and impaired immunostimulatory activation. Immunobiology (2009) 214(9-10):778–90. doi: 10.1016/j.imbio.2009.06.009
5. Fagerholm SC, Guenther C, Llort Asens M, Savinko T, Uotila LM. Beta2-Integrins and Interacting Proteins in Leukocyte Trafficking, Immune Suppression, and Immunodeficiency Disease. Front Immunol (2019) 10:254. doi: 10.3389/fimmu.2019.00254
6. Oh J, Richardson JA, Olson EN. Requirement of myocardin-related transcription factor-B for remodeling of branchial arch arteries and smooth muscle differentiation. Proc Natl Acad Sci U S A (2005) 102(42):15122–7. doi: 10.1073/pnas.0507346102
7. Sun Y, Boyd K, Xu W, Ma J, Jackson CW, Fu A, et al. Acute myeloid leukemia-associated Mkl1 (Mrtf-a) is a key regulator of mammary gland function. Mol Cell Biol (2006) 26(15):5809–26. doi: 10.1128/MCB.00024-06
8. Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol (2010) 11:353–65. doi: 10.1038/nrm2890
9. Sullivan AL, Benner C, Heinz S, Huang W, Xie L, Miano JM, et al. Serum Response Factor Utilizes Distinct Promoter- and Enhancer-Based Mechanisms to Regulate Cytoskeletal Gene Expression in Macrophages. Mol Cell Biol (2011) 31(4):861–75. doi: 10.1128/MCB.00836-10
10. Fleige A, Alberti S, Gröbe L, Frischmann U, Geffers R, ller WM, et al. Serum Response Factor Contributes Selectively to Lymphocyte Development. J Biol Chem (2007) 282(33):24320–8. doi: 10.1074/jbc.M703119200
11. Sprenkeler EGG, Henriet SSV, Tool ATJ, Kreft IC, van der Bijl I, Aarts CEM, et al. MKL1 deficiency results in a severe neutrophil motility defect due to impaired actin polymerization. Blood (2020) 135:2171–81. doi: 10.1182/blood.2019002633
12. Guenther C, Faisal I, Uotila LM, Asens ML, Harjunpaa H, Savinko T, et al. A beta2-Integrin/MRTF-A/SRF Pathway Regulates Dendritic Cell Gene Expression, Adhesion, and Traction Force Generation. Front Immunol (2019) 10:1138. doi: 10.3389/fimmu.2019.01138
13. Esnault C, et al. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev (2014) 28(9):943–58. doi: 10.1101/gad.239327.114
14. Record J, Malinova D, Zenner HL, Plagnol V, Nowak K, Syed F, et al. Immunodeficiency and severe susceptibility to bacterial infection associated with a loss-of-function homozygous mutation of MKL1. Blood (2015) 126(13):1527–35. doi: 10.1182/blood-2014-12-611012
15. Taylor A, Halene S. The regulatory role of serum response factor pathway in neutrophil inflammatory response. Curr Opin Hematol (2015) 22(1):67–73. doi: 10.1097/MOH.0000000000000099
16. Taylor A, Tang W, Bruscia EM, Zhang PX, Lin A, Gaines P, et al. SRF is required for neutrophil migration in response to inflammation. Blood (2014) 123(19):3027–36. doi: 10.1182/blood-2013-06-507582
17. Jaumouille V, Cartagena-Rivera AX, Waterman CM. Coupling of beta2 integrins to actin by a mechanosensitive molecular clutch drives complement receptor-mediated phagocytosis. Nat Cell Biol (2019) 21(11):1357–69. doi: 10.1038/s41556-019-0414-2
18. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
19. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
20. Papa R, Picco P, Gattorno M. The expanding pathways of autoinflammation: a lesson from the first 100 genes related to autoinflammatory manifestations. Adv Protein Chem Struct Biol (2020) 120:1–44. doi: 10.1016/bs.apcsb.2019.11.001
21. Vedula P, Kashina A. The makings of the ‘actin code’: regulation of actin’s biological function at the amino acid and nucleotide level. J Cell Sci (2018) 131(9):jcs215509. doi: 10.1242/jcs.215509
22. Cuvertino S, Stuart HM, Chandler KE, Roberts NA, Armstrong R, Bernardini L, et al. ACTB Loss-of-Function Mutations Result in a Pleiotropic Developmental Disorder. Am J Hum Genet (2017) 101(6):1021–33. doi: 10.1016/j.ajhg.2017.11.006
23. Nunoi H, Yamazaki T, Tsuchiya H, Kato S, Malech HL, Matsuda I, et al. A heterozygous mutation of beta-actin associated with neutrophil dysfunction and recurrent infection. Proc Natl Acad Sci U S A (1999) 96(15):8693–8. doi: 10.1073/pnas.96.15.8693
24. Hundt N, Preller M, Swolski O, Ang AM, Mannherz HG, Manstein DJ, et al. Molecular mechanisms of disease-related human beta-actin mutations p.R183W and p.E364K. FEBS J (2014) 281(23):5279–91. doi: 10.1111/febs.13068
25. Xie X, Endara-Coll M, Mahmood R, Jankauskas R, Gjorgjieva T, Percipalle P. Mitochondria-localized beta-actin is essential for priming innate antiviral immune signaling by regulating IRF3 protein stability. Cell Mol Immunol (2019) 16(10):837–40. doi: 10.1038/s41423-019-0269-2
26. Xie L, Sullivan AL, Collier JG, Glass CK. Serum response factor indirectly regulates type I interferon-signaling in macrophages. J Interferon Cytokine Res (2013) 33(10):588–96. doi: 10.1089/jir.2012.0065
27. Mullins RD, Heuser JA, Pollard TD. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci U S A (1998) 95(11):6181–6. doi: 10.1073/pnas.95.11.6181
28. Vijayakumar V, Monypenny J, Chen XJ, Machesky LM, Lilla S, Thrasher AJ, et al. Tyrosine phosphorylation of WIP releases bound WASP and impairs podosome assembly in macrophages. J Cell Sci (2015) 128(2):251–65. doi: 10.1242/jcs.154880
29. Kim AS, Kakalis LT, Abdul-Manan N, Liu GA, Rosen MK. Autoinhibition and activation mechanisms of the Wiskott-Aldrich syndrome protein. Nature (2000) 404(6774):151–8. doi: 10.1038/35004513
30. Lanzi G, Moratto D, Vairo D, Masneri S, Delmonte O, Paganini T, et al. A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. J Exp Med (2012) 209(1):29–34. doi: 10.1084/jem.20110896
31. Al-Mousa H, Hawwari A, Al-Ghonaium A, Al-Saud B, Al-Dhekri H, Al-Muhsen S, et al. Hematopoietic stem cell transplantation corrects WIP deficiency. J Allergy Clin Immunol (2017) 139(3):1039–40.e4. doi: 10.1016/j.jaci.2016.08.036
32. Pfajfer L, Seidel MG, Houmadi R, Rey-Barroso J, Hirschmugl T, Salzer E, et al. WIP deficiency severely affects human lymphocyte architecture during migration and synapse assembly. Blood (2017) 130(17):1949–53. doi: 10.1182/blood-2017-04-777383
33. Chou HC, Anton IM, Holt MR, Curcio C, Lanzardo S, Worth A, et al. WIP regulates the stability and localization of WASP to podosomes in migrating dendritic cells. Curr Biol (2006) 16(23):2337–44. doi: 10.1016/j.cub.2006.10.037
34. Konno A, Kirby M, Anderson SA, Schwartzberg PL, Candotti F. The expression of Wiskott-Aldrich syndrome protein (WASP) is dependent on WASP-interacting protein (WIP). Int Immunol (2007) 19(2):185–92. doi: 10.1093/intimm/dxl135
35. Schwinger W, Urban C, Ulreich R, Sperl D, Karastaneva A, Strenger V, et al. The Phenotype and Treatment of WIP Deficiency: Literature Synopsis and Review of a Patient With Pre-transplant Serial Donor Lymphocyte Infusions to Eliminate CMV. Front Immunol (2018) 9:2554. doi: 10.3389/fimmu.2018.02554
36. Chen Q, Courtemanche N, Pollard TD. Aip1 promotes actin filament severing by cofilin and regulates constriction of the cytokinetic contractile ring. J Biol Chem (2015) 290(4):2289–300. doi: 10.1074/jbc.M114.612978
37. Ono S. Functions of actin-interacting protein 1 (AIP1)/WD repeat protein 1 (WDR1) in actin filament dynamics and cytoskeletal regulation. Biochem Biophys Res Commun (2018) 506(2):315–22. doi: 10.1016/j.bbrc.2017.10.096
38. Randrianarison-Huetz V, Papaefthymiou A, Herledan G, Noviello C, Faradova U, Collard L, et al. Srf controls satellite cell fusion through the maintenance of actin architecture. J Cell Biol (2018) 217(2):685–700. doi: 10.1083/jcb.201705130
39. Kuhns DB, Fink DL, Choi U, Sweeney C, Lau K, Priel DL, et al. Cytoskeletal abnormalities and neutrophil dysfunction in WDR1 deficiency. Blood (2016) 128(17):2135–43. doi: 10.1182/blood-2016-03-706028
40. Pfajfer L, Mair NK, Jimenez-Heredia R, Genel F, Gulez N, Ardeniz O, et al. Mutations affecting the actin regulator WD repeat-containing protein 1 lead to aberrant lymphoid immunity. J Allergy Clin Immunol (2018) 142(5):1589–604.e11. doi: 10.1016/j.jaci.2018.04.023
41. Standing AS, Malinova D, Hong Y, Record Y, Moulding D, Blundell MP, et al. Autoinflammatory periodic fever, immunodeficiency, and thrombocytopenia (PFIT) caused by mutation in actin-regulatory gene WDR1. J Exp Med (2017) 214(1):59–71. doi: 10.1084/jem.20161228
42. Miller ME, Oski FA, Harris MB. Lazy-leucocyte syndrome. A new disorder of neutrophil function. Lancet (1971) 1(7701):665–9. doi: 10.1016/S0140-6736(71)92679-1
43. Ponuwei GA. A glimpse of the ERM proteins. J BioMed Sci (2016) 23:35. doi: 10.1186/s12929-016-0246-3
44. Lagresle-Peyrou C, Luce S, Ouchani F, Soheili TS, Sadek H, Chouteau M, et al. X-linked primary immunodeficiency associated with hemizygous mutations in the moesin (MSN) gene. J Allergy Clin Immunol (2016) 138(6):1681–9.e8. doi: 10.1016/j.jaci.2016.04.032
45. Bradshaw G, et al. Exome Sequencing Diagnoses X-Linked Moesin-Associated Immunodeficiency in a Primary Immunodeficiency Case. Front Immunol (2018) 9:420. doi: 10.3389/fimmu.2018.00420
46. Delmonte OM, Biggs CM, Hayward A, Comeau AM, Kuehn HS, Rosenzweig SD, et al. First Case of X-Linked Moesin Deficiency Identified After Newborn Screening for SCID. J Clin Immunol (2017) 37(4):336–8. doi: 10.1007/s10875-017-0391-9
47. Matsumoto M, Hirata T. Moesin regulates neutrophil rolling velocity in vivo. Cell Immunol (2016) 304-305:59–62. doi: 10.1016/j.cellimm.2016.04.007
48. Liu X, Yang T, Suzuki K, Tsukita S, Ishii M, Zhou S, et al. Moesin and myosin phosphatase confine neutrophil orientation in a chemotactic gradient. J Exp Med (2015) 212(2):267–80. doi: 10.1084/jem.20140508
49. Machesky LM, Reeves E, Wientjes F, Mattheyse FJ, Grogan A, Totty NF, et al. Mammalian actin-related protein 2/3 complex localizes to regions of lamellipodial protrusion and is composed of evolutionarily conserved proteins. Biochem J (1997) 328( Pt 1):105–12. doi: 10.1042/bj3280105
50. Kuijpers TW, Tool ATJ, van der Bijl I, de Boer M, van Houdt M, de Cuyper IM, et al. Combined immunodeficiency with severe inflammation and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol (2017) 140(1):273–277 e10. doi: 10.1016/j.jaci.2016.09.061
51. Kahr WH, Pluthero FG, Elkadri A, Warner N, Drobac M, Chen CH, et al. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat Commun (2017) 8:14816. doi: 10.1038/ncomms14816
52. Volpi S, Cicalese MP, Tuijnenburg P, Tool ATJ, Cuadrado E, Abu-Halaweh M, et al. A combined immunodeficiency with severe infections, inflammation, and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol (2019) 143(6):2296–9. doi: 10.1016/j.jaci.2019.02.003
53. Randzavola LO, Strege K, Juzans M, Asano Y, Stinchcombe JC, Gawden-Bone CM, et al. Loss of ARPC1B impairs cytotoxic T lymphocyte maintenance and cytolytic activity. J Clin Invest (2019) 129(12):5600–14. doi: 10.1172/JCI129388
54. Somech R, Lev A, Lee YN, Simon AJ, Barel O, Schiby G, et al. Disruption of Thrombocyte and T Lymphocyte Development by a Mutation in ARPC1B. J Immunol (2017) 199(12):4036–45. doi: 10.4049/jimmunol.1700460
55. Brigida I, Zoccolillo M, Cicalese MP, Pfajfer L, Barzaghi F, Scala S, et al. T-cell defects in patients with ARPC1B germline mutations account for combined immunodeficiency. Blood (2018) 132(22):2362–74. doi: 10.1182/blood-2018-07-863431
56. Sprenkeler EGG, Webbers SDS, Kuijpers TW. When Actin is Not Actin’ Like It Should: A New Category of Distinct Primary Immunodeficiency Disorders. J Innate Immun (2020) 13:3–25. doi: 10.1159/000509717
57. Keszei M, Record J, Kritikou JS, Wurzer H, Geyer C, Thiemann M, et al. Constitutive activation of WASp in X-linked neutropenia renders neutrophils hyperactive. J Clin Invest (2018) 128(9):4115–31. doi: 10.1172/JCI64772
58. Yu-Wai-Man C, Spencer-Dene B, Lee RMH, Hutchings K, Lisabeth EM, Treisman R, et al. Local delivery of novel MRTF/SRF inhibitors prevents scar tissue formation in a preclinical model of fibrosis. Sci Rep (2017) 7:518. doi: 10.1038/s41598-017-00212-w
59. Kobayashi M, Tokuda K, Kobayashi Y, Yamashiro C, Uchi S-H, Hatano M, et al. Suppression of Epithelial-Mesenchymal Transition in Retinal Pigment Epithelial Cells by an MRTF-A Inhibitor. Retina (2019) 60:528–37. doi: 10.1167/iovs.18-25678
60. Haak AJ, et al. Targeting the Myofibroblast Genetic Switch: Inhibitors of Myocardin-Related Transcription Factor/Serum Response Factor–Regulated Gene Transcription Prevent Fibrosis in a Murine Model of Skin Injury. J Pharmacol Exp Ther (2014) 349(3):480–6. doi: 10.1124/jpet.114.213520
61. Bond JE, et al. Wound Contraction is Attenuated by Fasudil Inhibition of Rho-Kinase. Plast Reconstr Surg (2011) 128(5):438e–50e. doi: 10.1097/PRS.0b013e31822b7352
62. Johnson LA, Rodansky ES, Haak AJ, Larsen SD, Neubig RR, Higgins PDR, et al. Novel Rho/MRTF/SRF Inhibitors Block Matrix-stiffness and TGF-β–Induced Fibrogenesis in Human Colonic Myofibroblasts. Inflamm Bowel Dis (2016) 20(1):154–65. doi: 10.1097/01.MIB.0000437615.98881.31
63. Lu Y, Lv F, Kong M, Chen X, Duan Y, Chen X, et al. A cAbl-MRTF-A Feedback Loop Contributes to Hepatic Stellate Cell Activation. Front Cell Dev Biol (2019) 7:243. doi: 10.3389/fcell.2019.00243
64. Yokota S, Chosa N, Kyakumoto S, Kimura H, Ibi M, Kamo M, et al. ROCK/actin/MRTF signaling promotes the fibrogenic phenotype of fibroblast-like synoviocytes derived from the temporomandibular joint. Int J Mol Med (2017) 39(4):799–808. doi: 10.3892/ijmm.2017.2896
65. Shiwen X, Stratton R, Nikitorowicz-Buniak J, Ahmed-Abdi B, Ponticos M, Denton C, et al. A Role of Myocardin Related Transcription Factor-A (MRTF-A) in Scleroderma Related Fibrosis. PloS One (2015) 10:e0126015. doi: 10.1371/journal.pone.0126015
66. Sisson TH, Ajayi IO, Subbotina N, Dodi AE, Rodansky ES, Chibucos LN, et al. Inhibition of myocardin-related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am J Pathol (2015) 185(4):969–86. doi: 10.1016/j.ajpath.2014.12.005
67. Sandbo N, Kregel S, Taurin S, Bhorade S, Dulin NO. Critical Role of Serum Response Factor in Pulmonary Myofibroblast Differentiation Induced by TGF-beta. Am J Respir Cell Mol Biol (2009) 41:32–338. doi: 10.1165/rcmb.2008-0288OC
68. Xiong Y, Bedi K, Berritt S, Attipoe BK, Brooks TG, Wang K, et al. Targeting MRTF/SRF in CAP2-dependent dilated cardiomyopathy delays disease onset. JCI Insight (2019) 4:6. doi: 10.1172/jci.insight.124629
69. Minami T, Kuwahara K, Nakagawa Y, Takaoka M, Kinoshita H, Nakao K, et al. Reciprocal expression of MRTF-A and myocardin is crucial for pathological vascular remodelling in mice. EMBO J (2012) 31(23):4428–40. doi: 10.1038/emboj.2012.296
70. Hays TT, Ma B, Zhou N, Stoll S, Pearce WJ, Qiu H. Vascular smooth muscle cells direct extracellular dysregulation in aortic stiffening of hypertensive rats. Aging Cell (2018) 17(e12748.):1–13. doi: 10.1111/acel.12748
71. Zhou N, Lee J-J, Stoll S, Ma B, Wiener R, Wang C, et al. Inhibition of SRF/myocardin reduces aortic stiffness by targeting vascular smooth muscle cell stiffening in hypertension. Cardiovasc Res (2017) 113(2):171–82. doi: 10.1093/cvr/cvw222
72. Zhou N, Lee J-J, Stoll S, Ma B, Costa KD, Qiu H. Rho Kinase Regulates Aortic Vascular Smooth Muscle Cell Stiffness Via Actin/SRF/Myocardin in Hypertension. Cell Physiol Biochem (2018) 44(2):701–15. doi: 10.1159/000485284
73. Zabini D, Granton E, Hu Y, Miranda MZ, Weichelt U, Bonnet SB, et al. Loss of SMAD3 Promotes Vascular Remodeling in Pulmonary Arterial Hypertension via MRTF Disinhibition. Am J Respir Crit Care Med (2017) 197(2). doi: 10.1164/rccm.201702-0386OC
74. Velasquez LS, Sutherland LB, Liu Z, Grinnell F, Kamm KE, Schneider JW, et al. Activation of MRTF-A–dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. PNAS (2013) 110(42):16850–5. doi: 10.1073/pnas.1316764110
75. Yu L, Weng X, Liang P, Dai X, Wu X, Xu H, et al. MRTF-A mediates LPS-induced pro-inflammatory transcription by interacting with the COMPASS complex. J Cell Sci (2014) 127:4645–57. doi: 10.1242/jcs.152314
76. Xie L. MKL1/2 and ELK4 co-regulate distinct serum response factor (SRF) transcription programs in macrophages. BMC Genomics (2014) 15(301):1–15. doi: 10.1186/1471-2164-15-301
77. Fini ME. Keratocyte and fibroblast phenotypes in the repairing cornea. Prog Retin Eye Res (1999) 18(4):529–51. doi: 10.1016/S1350-9462(98)00033-0
78. Miranda MZ, Bialik JF, Speight P, Dan Q, Yeung T, Szászi K, et al. TGF-β1 regulates the expression and transcriptional activity of TAZ protein via a Smad3-independent, myocardin-related transcription factor-mediated mechanism. J Biol Chem (2017) 292:14902–20. doi: 10.1074/jbc.M117.780502
79. Weinl C, Riehle H, Park D, Stritt C, Beck S, Huber G, et al. Endothelial SRF/MRTF ablation causes vascular disease phenotypes in murine retinae. J Clin Invest (2013) 123(5):2193–206. doi: 10.1172/JCI64201
Keywords: MKL1, SRF, neutrophil, migration, immunodeficiency—primary
Citation: Sprenkeler EGG, Guenther C, Faisal I, Kuijpers TW and Fagerholm SC (2021) Molecular Mechanisms of Leukocyte Migration and Its Potential Targeting—Lessons Learned From MKL1/SRF-Related Primary Immunodeficiency Diseases. Front. Immunol. 12:615477. doi: 10.3389/fimmu.2021.615477
Received: 09 October 2020; Accepted: 04 January 2021;
Published: 22 February 2021.
Edited by:
Helen Michelle McGettrick, University of Birmingham, United KingdomReviewed by:
Giorgia Santilli, University College London, United KingdomMarie-Dominique Filippi, Cincinnati Children’s Research Foundation, United States
Copyright © 2021 Sprenkeler, Guenther, Faisal, Kuijpers and Fagerholm. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susanna C. Fagerholm, c3VzYW5uYS5mYWdlcmhvbG1AaGVsc2lua2kuZmk=