 Tingjun Chen
Tingjun Chen Dale B. Bosco
Dale B. Bosco Yanlu Ying1
Yanlu Ying1 Dai-Shi Tian
Dai-Shi Tian Long-Jun Wu
Long-Jun Wu- 1Department of Neurology, Mayo Clinic, Rochester, MN, United States
- 2Department of Neurology, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Department of Neuroscience, Mayo Clinic, Jacksonville, FL, United States
- 4Department of Immunology, Mayo Clinic, Rochester, MN, United States
Neuromyelitis optica (NMO) is an autoantibody-triggered neuro-inflammatory disease which preferentially attacks the spinal cord and optic nerve. Its defining autoantibody is specific for the water channel protein, aquaporin‐4 (AQP4), which primarily is localized at the end-feet of astrocytes. Histopathology studies of early NMO lesions demonstrated prominent activation of microglia, the resident immune sentinels of the central nervous system (CNS). Significant microglial reactivity is also observed in NMO animal models induced by introducing AQP4-IgG into the CNS. Here we review the potential roles for microglial activation in human NMO patients as well as different animal models of NMO. We will focus primarily on the molecular mechanisms underlying microglial function and microglia-astrocyte interaction in NMO pathogenesis. Understanding the role of microglia in NMO pathology may yield novel therapeutic approaches for this disease.
Introduction
Neuromyelitis optica (NMO) is an autoimmune inflammatory disorder of the central nervous system (CNS) which mainly affects optic nerves and the spinal cord (1). Attacks upon optic nerves can often lead to blindness, while the extensive transverse myelitis induced within the spinal cord can cause weakness or paralysis in the legs and arms, loss of sensation, bladder and bowel function issues, and respiratory failure (2). Additionally, NMO disproportionately affects women and non-Caucasian populations and is a relapsing-remitting disease. Repeated attacks can lead to severe cumulative damage to optic nerves and the spinal cord (3). Therefore, prevention of new attacks is critical to mitigating long-term effects.
NMO was largely considered a subtype of multiple sclerosis (MS), until 2004 when Lennon and colleagues identified that autoantibodies directed towards aquaporin‐4 (AQP4-IgG, also called NMO-IgG) were a biomarker for NMO pathology (4). This key finding was used to distinguish NMO from MS and other inflammatory CNS demyelinating disorders. AQP4-IgG binds the ectodomain of AQP4 water channels that are located on the astrocytic end-feet embracing capillaries, ventricular walls, and pial-glial interfaces (4). AQP4-IgG can also sometimes be detected in a patient’s cerebrospinal fluid (CSF) (5). AQP4-IgG concentrations in the blood and CSF have been reported to correlate with clinical outcome (6–10). It should also be noted that antibodies against myelin oligodendrocyte glycoprotein (MOG-IgG) have also been suggested to induce NMO pathology (11). The idea that NMO pathology is related to pathogenic autoantibodies has lead plasma exchange and B cell depletion to be accepted as first line therapies for the acute phase of NMO (12).
The histopathology of NMO lesions also show prominent microglial reactivity in AQP4-rich CNS regions (13, 14). However, mechanisms underlying microglial activation in NMO remains largely unknown. It would be reasonable to believe that microglia are the first responders to signals emanating from astrocytes following their activation by AQP4-IgG. As such, this review focuses on the emerging evidence that microglia play an important role in AQP4-IgG related NMO pathogenesis.
Histopathology and Microglial Reactivity in NMO Patients
The key hallmark of NMO immunopathology is the striking focal loss of AQP4 protein from astrocytes. Changes to glial fibrillary acidic protein (GFAP) expression in astrocytes varies in different brain regions and disease stages (14). In early lesions, astrocytes are spared and exhibit an activated phenotype (15). In later stages, when deposits of the complement terminal membrane attack complex C5b-C9 are abundant, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining indicates astrocyte apoptosis and GFAP immunoreactivity is reduced (13–16). In some of these later lesions, microglia/macrophages contain GFAP remnants, indicating phagocytosis of astrocytes (14). Demyelination in NMO pathology, as shown by fast blue staining (14), generally accompanies astrocytic pathology (17) and local inflammation (18).
In vitro studies of NMO pathogenesis found that the binding of AQP4-IgG to its antigen induces AQP4 internalization (19, 20), reducing water permeability and spurring astrocytes to release various cytokines and chemokines (21). Coincidently, when AQP4 is internalized, excitatory amino acid transporter 2 (EAAT2) also disappears from astrocyte surface membranes (20). In the presence of requisite complement components, astrocytes undergo cytolysis during an NMO attack (13, 19, 22). Additionally, myelin loss in both grey and white matter is an immunohistochemical characteristic of fully established NMO lesions (14). However, demyelination appears to be a secondary event following the interaction of AQP4-IgG with astrocytes (17) and promotion of local inflammation (18). Neuronal loss has also been reported in the cerebral cortex of NMO patients, possibly explaining the cognitive impairment that is sometimes observed (23). As for MOG-IgG positive NMO patients, myelin loss was significant but AQP4 was preserved and dystrophic astrocytes were absent (24). Although MOG-IgG frequently co-existed with anti-N-methyl-D-aspartate (NMDA) receptor IgG, the prognosis in these patients are usually better than AQP4-IgG positive patients, indicating differences in pathogenesis (25).
In NMO patients’ CNS, perivascular regions contain accumulations of lymphocytes, neutrophils and eosinophils (14–16). The abundance of these cellular infiltrates suggests disruption of the neurovascular unit following interaction of NMO-IgG with AQP4 in the vicinity of the blood-brain-barrier (BBB) (13). This is not surprising, considering astrocyte end-feet are an integral part of the neurovasculature. Consistently, AQP4 loss in the choroid plexus coinciding with C9 neoantigen (C9neo) immunoreactivity on choroidal epithelial membranes is evidence of focal BBB pathology (13). C9neo deposition can also be detected in MOG-IgG positive patients, but on myelin sheaths (24). Although peripherally activated B cells can enter the parenchyma through intact BBB (26), loss of astrocytic end-feet may further facilitate entry of either AQP4-IgG or AQP4-IgG secreting B cells into the CNS (27).
In addition to loss of AQP4 and astrocytes, neuronal injury, demyelination, microglial activation, and macrophage infiltration are prominent in AQP4-IgG seropositive NMO pathology (14). Microglia and macrophage reactivity is indicated by both morphological criteria (i.e., amoeboid morphology) and positivity for the lysosome marker CD68 (14, 16). However, conventional histopathology cannot distinguish macrophages (infiltrating monocytes) from resident microglia in the CNS. Although there are conflicting reports as to whether the number of monocytes within circulation changes (28, 29) in response to NMO-IgG, infiltration of monocytes into the CSF is believed to happen (30). Indeed, there is an increased number of microglia-like cells within human NMO pathology. However, it is difficult to determine whether this is due to local microglial proliferation or monocyte infiltration after NMO-IgG attacks.
It should also be noted that reported CNS tissue distribution of activated microglia/macrophages is different between NMO and MS. In MS, microglial activation is wide-spread in the meninges and subpial cortex (31), while in AQP4-IgG positive NMO it is largely confined to regions that normally are AQP4-rich, like the CSF–brain interface in the corpus callosum, hypothalamic, and periventricular areas (13, 14, 32). Conversely, in MOG-IgG positive NMO, microglia/macrophages also appear in subpial, which is similar to MS; but it is associated with CD4 inflammatory infiltration and not CD8 T cells, which are abundant in MS (24). Additionally, microglia/macrophage reactivity in NMO lesions are surrounded by evidence of complement activation, which is an infrequent finding in MS lesions (14). Consistent with the role of complement in microglial function, microglia are able to secret complement components to activate astrocytes and mediate neuronal injury in a variety of neurological disorders (33–35). These complement components can also further activate microglia (36–38). Therefore, the difference in complement-related microglia activation patterns between MS and NMO indicate divergent molecular mechanisms. Nevertheless, clinical histopathology results strongly suggest the involvement of microglia in NMO pathogenesis. As such, NMO animal models are needed to investigate the precise function and mechanism of microglia in NMO.
Rodent Models of NMO
To understand the cellular and molecular mechanisms underlying the pathogenesis of NMO, several rodent models of NMO have been developed. Since AQP4-IgG was the original pathological cause of NMO, animal models that employed AQP4-IgG to develop pathology are the most common. As such, we will focus predominantly on AQP4-IgG related animal models.
Initial animal models were derived from experimental autoimmune encephalomyelitis (EAE) where pathology was induced via adoptive transfer of AQP4-IgG into animals with preexisting myelin targeted encephalitogenic T cells (5, 39–43). However, these models did not display any of the immunology characteristics of NMO. In particular, there were no AQP4 specific immune reactions (44–47). Another early model involved transplantation of AQP4-specific T cells into naïve animals (48–53). However, this model also did not induce the characteristic AQP4 loss unless AQP4-IgG were co-injected (48, 49). More recently, new models that more closely mimic the clinical features of AQP4-IgG related NMO have been developed (Table 1).
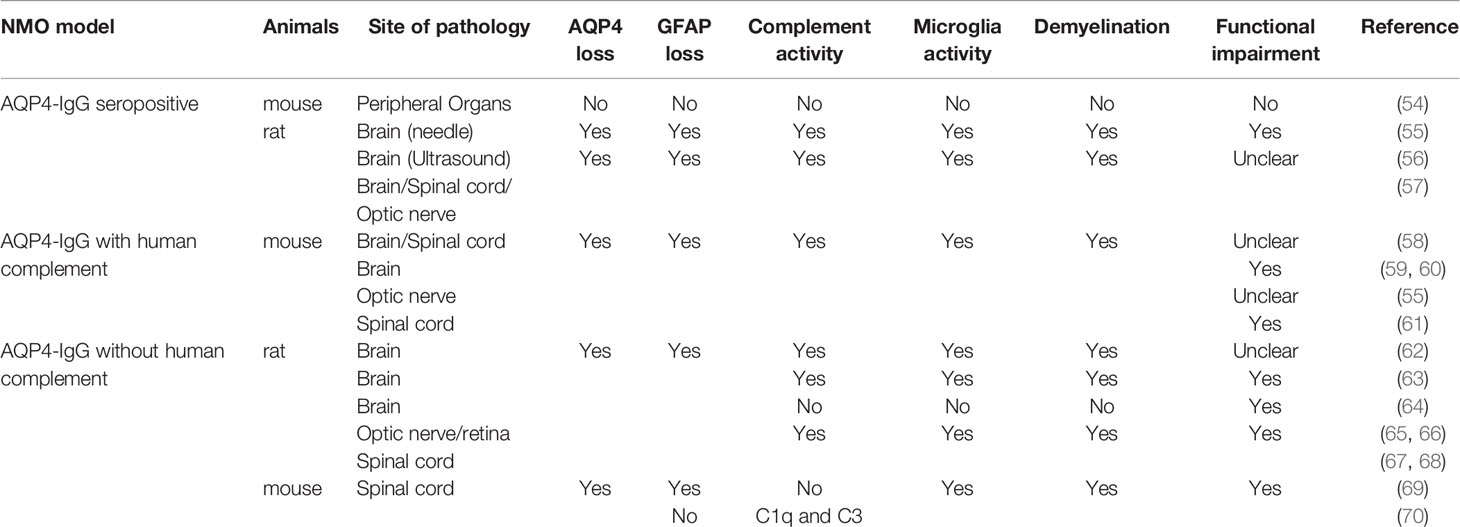
Table 1 AQP4-IgG related NMO animal models.
AQP4-IgG Seropositive Models
Some recent rodent models of NMO have utilized passive transfer of AQP4-IgG into animals to induce clinically relevant pathology and provide early evidence of microglial activation in NMO pathology (58, 61, 71). However, one complication in using passive transfer is that acute intravenous injection of AQP4-IgG has difficulty producing neurology dysfunction or NMO pathology (54, 55), as AQP4-IgGs cannot pass through an undamaged BBB. When the BBB is physically disrupted by needle puncture or ultrasound, NMO-liked pathology can be induced near leakage sites (54, 56). Repeated intraperitoneal AQP4-IgG injections has also been used to successfully induce wild-spread NMO-liked pathology in rats (57). The neurological abnormalities displayed in this model included neuropathic pain, salivation (correlates to nausea/vomiting behavior in rats/mice) (72), problems with balance, and motor impairment (57).
AQP4-IgG in seropositive models entered the CNS via three routes: (1) Circumventricular organs, which are highly vascularized structures surrounding the third and fourth ventricles and characterized by a lack of BBB (73). Using this route AQP4-IgG induces a lesion with AQP4 loss but intact GFAP staining. However, in circumventricular organs, microglia were not activated after repeated intraperitoneal AQP4-IgG injection. (2) Meningeal vessels and veins of the Virchow Robin spaces. Using this route AQP4-IgG induces meningitis 120 h after the initial intraperitoneal injection of AQP4-IgG. T cell, neutrophil and microglia activity were found in subpial lesions. Notably, microglial activity was accompanied with astrocyte damage in these areas. (3) Finally, parenchymal vessel unrelated to the meninges. Perivascular lesion deep within the parenchyma showed significant AQP4 loss and large number of activated microglia. In some lesions, strong complement deposition coincided with the loss of astrocytes and activation of microglia, which is similar with lesions of NMO medullas in patients (15).
Direct AQP4-IgG Injection Together With Human Complement
In these animal models, either AQP4-IgG derived from NMO patients or recombinant monoclonal AQP4-IgG is administered directly into the rodent CNS together with human complement (59, 60, 62). In wild type mice, when patient-derived AQP4-IgGs were intracerebrally injected acutely with human complement, matured NMO pathology including AQP4 and GFAP loss, myelin damage, complement deposition, and axon injury was observed (59). CD45+ microglia/macrophages were also widely observed in the ipsilateral brain. Moreover, animals were more likely to turn to the injection side during Y maze test, indicating neurological dysfunction in injected hemisphere of brain. In contrast, mice treated with human control IgGs and complement exhibit no behavior abnormalities, AQP4 loss, astrocyte injury, axon loss, or microglia/macrophages activation, suggesting that APQ4-IgG is the main trigger for pathology (59).
Chronic infusion of recombined AQP4-IgG with human complement was also shown to induced astrocyte damage, neutrophil infiltration, eosinophil infiltration, and microglia/macrophage activity in mice (58, 60). Additionally, deficiencies in complement regulator CD59 expression significantly increased the pathology induced by AQP4-IgG, including AQP4 loss, complement deposition, and CD45+ cell activity (55, 61, 64). These results are quite surprising, as complement proteins themselves are known to induce microglial activity (74–76) followed by neuronal damage (77) and demyelination (78). Therefore, further studies are needed to clearly delineate the respective contribution of AQP4-IgG and complement to the outcomes of NMO-like phenotypes in these models.
Direct AQP4-IgG Injection Without Human Complement
Co-injection of complement, however, may not be necessary to induce pathology. In rodents, AQP4-IgG injection without human complement was able to induce neurological dysfunction and NMO pathology within the brain, retina, and optic nerves (62, 63, 65–68, 70). Microglial reactivity was observed within areas of AQP4 loss (64, 68–70). Administration of clodronate liposomes, which efficiently depletes microglia/macrophages, significantly reduced lesion size, suggesting that microglia/macrophages contributed to NMO pathology (62).
Nevertheless, there is still debate as to whether complement has a role in these types of models. For example, in CD59 deficient mice, AQP4-IgG induced worse functional impairment, more NMO-like pathology, and stronger microglial activity (67). These results indicate that AQP4-IgG could induce endogenous complement expression, which in turn facilitates the toxicity of AQP4-IgG. However, in other reports, no complement terminal protein was detected (64, 68, 70). Additionally, pretreatment with cobra venom factor, which inhibits complement activity (79), did not rescue functional impairment and pathology in the retina (65). Therefore, it is unclear whether the pathology and neuronal dysfunction induced in these models truly involved complement related mechanisms.
Microglial Activation in Established Animal Models of NMO
As mentioned above, microglial activation has been observed in most animal models of NMO. However, it is unknown whether microglial activation is the cause or consequence of NMO pathogenesis. Moreover, it is unclear as to whether complement is involved in microglial activation, considering microglia reactivity has been observed in both complement dependent (59, 61, 67) and independent models (65, 69, 80). Serum concentrations of complement have also varied across different NMO cases, making it difficult to ascertain whether complement toxicity is initiated by AQP4-IgG (81–84). Nevertheless, the clear early events in NMO are astrocytic reactivity after AQP4-IgG binding (20) and AQP4 loss (22). AQP4-IgG activated astrocytes may then produce cytokines, which induce local inflammation and microglial activation (19–21, 70). Microglia, as the resident CNS immune cell, should typically be the first responders to reactive astrocytes following initiation of NMO pathology. However, limited direct evidence exists that illustrates microglial function and activation mechanisms in NMO. As such, a new murine model of NMO has recently been developed to determine where microglia fit into NMO pathology (70).
A New Murine Model of NMO That Employs Chronic AQP4-IgG Intrathecal Infusion Reveals the Important Role of Microglia
A new mouse model of NMO has been established that utilizes continuous infusing of AQP4-specific IgG into the spinal subarachnoid space, without exogenous complement (70). Recipients of monoclonal AQP4-IgG or NMO-patient-derived IgG displayed progressive motor impairment and immunohistopathology compatible to early clinical NMO pathology. The pathology showed that AQP4 levels in spinal cord tissue were decreased after AQP4-IgG infusion, but GFAP immunoreactivity was increased. Therefore, AQP4 is likely internalized but astrocytes are not undergoing apoptosis, mimicking the early stages of NMO. In AQP4 null mice, AQP4-IgG did not induce motor impairment, confirming that autoantibodies targeting AQP4 are responsible for triggering the pathology. Additionally, complement terminal proteins were not detected in this model. Instead, it was demonstrates that microglia play a central role in the observed pathology (70).
Microglia Are Required for NMO Pathogenesis
Activation of microglia/macrophages as determined by Iba1, CD11b, CD68, and C1q staining was significantly increased within the AQP4 loss lesion (70). These results are consistent with microglial/macrophages activation in human NMO patients (13, 14, 16). Interestingly, in AQP4-/- mice, AQP4-IgG did not activate microglia/macrophages, indicating that AQP4-IgG activated microglia via astrocyte signaling. Recent studies have proposed that microglia induce reactive astrocytes in many disease contexts, such as Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and MS (35, 85). The results in the mouse model of NMO demonstrated that astrocytes can induce the activation of microglial/macrophages. Astrocytic modulation of microglia is also observed in development and diseases (86, 87). For example, astrocytes release transforming growth factor (TGF)-β (88, 89) or interleukin 33 (IL-33) (86, 90) to promote microglial pruning of synapses in development. Under disease conditions, astrocyte up-regulate lipocalin-2 (LCN-2) (91) or orosomucoid (ORM) (92) to activate microglia, which leads to neuro-inflammation and degeneration.
To further investigate this proposed relationship between astrocyte-microglia communication and NMO pathology, microglia ablation approaches were employed (70). When microglia were depleted, AQP4-IgG infusion did not induce motor impairment. More strikingly, when microglial replenishment started around day 5 after ablation, motor dysfunction appeared (70), suggesting that microglia are critical mediators of the behavioral impairment. It is important to point out that microglia ablation did not prevent AQP4 internalization or astrocytic activation induced by AQP-IgG infusion. This is consistent with a previous in vitro study, which demonstrated that autoantibody-induced AQP4 internalization was microglia independent (20). Among rodent models with clinical comparable NMO pathology (40, 55, 61, 69, 70), this new study provides the direct evidence of microglia in motor dysfunction. The microgliogenic pathology separates NMO from other neuro-inflammatory diseases like MS. For example, in MOG induced experimental autoimmune encephalomyelitis (EAE) models, during disease onset, infiltrated macrophages induce inflammation (45), while microglia clear debris and limit inflammation (46). Additionally, in demyelinating diseases, microglia trigger remyelination and promote recovery (46, 93–95). Therefore, although the study suggests a damaging role for microglia in the early phases of NMO pathogenesis, microglia function could be more diverse at later phases of NMO.
Astrocyte-Microglia Interaction Is Mediated by C3-C3aR Signaling
After AQP4-IgG infusion, microglia and astrocyte cell somas were found to overlap after AQP4-IgG infusion (70). This observation suggests astrocyte-microglia physical interaction in this animal model of NMO. While microglial process convergence towards neurons has been previously reported (96, 97) this is the first time that similar events have been observed with astrocytes after AQP4-IgG infusion. Complement C3 is typically absent in astrocytes under physiological conditions, but strongly expressed under pathological conditions (35). Consistently, AQP4-IgG infusion was found to upregulate astrocytic C3. AQP4-IgG infusion also did not induce motor impairment in C3-/- animals. Similar to ATP (98, 99), the cleavage produce of C3, C3a, can act as a chemoattractant, possibly inducing the observed microglial processes convergence through C3a receptor (C3aR) expressed in microglia (100). In C3-/- and C3aR-/- mice after chronic infusion of AQP4-IgG, even though AQP4 loss still occurred, astrocyte-microglia interaction and motor deficits were largely abolished (70). Therefore, astrocyte-microglia interaction is likely mediated by C3-C3aR signaling and this interaction is a driver of this model’s NMO pathogenesis (Figure 1).
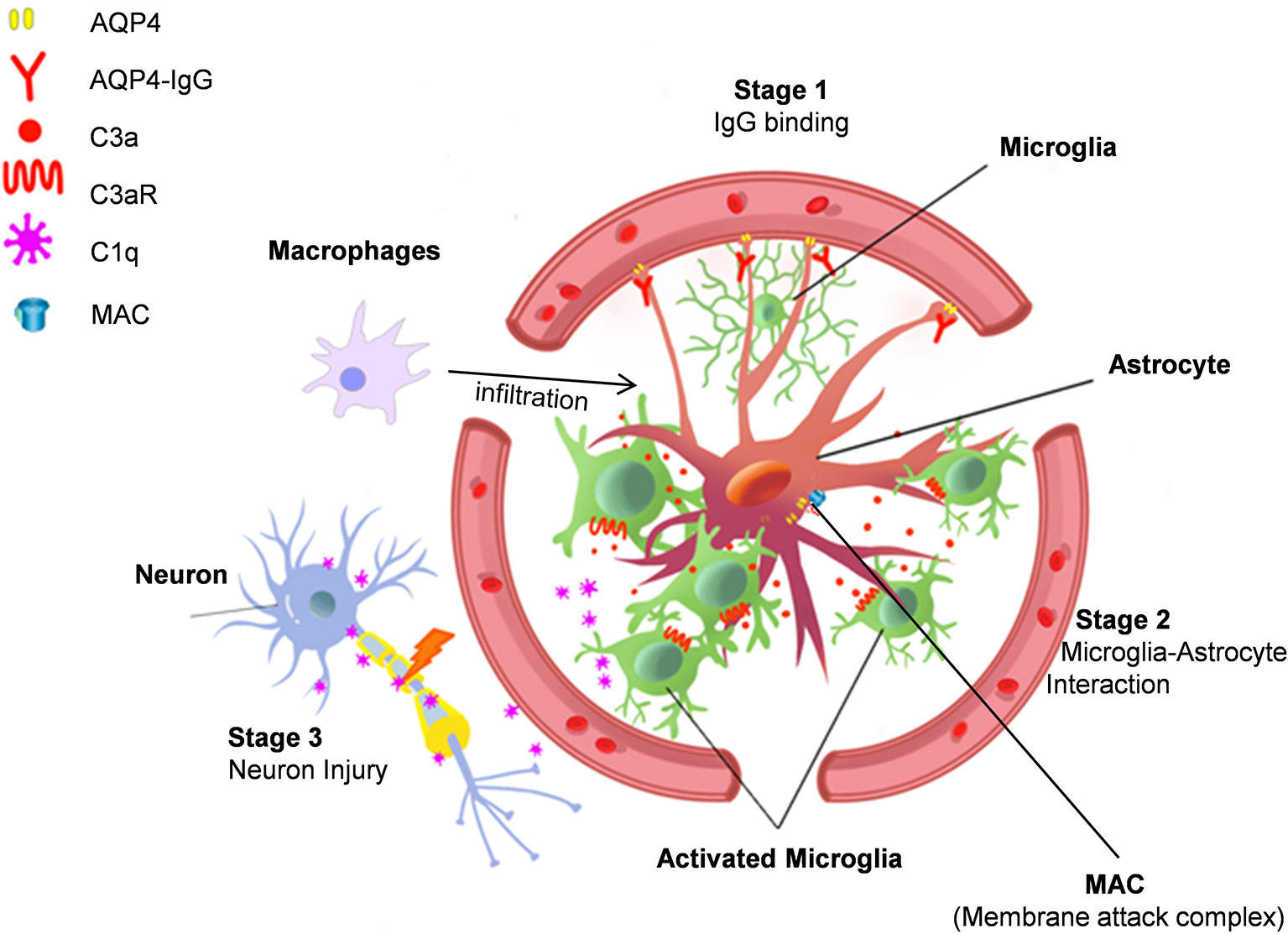
Figure 1 Astrocyte-microglia interaction drives NMO pathology. Stage 1. AQP4-IgG binds to AQP4 water channel protein in astrocyte end-feet and induces AQP4 protein internalization. Stage 2. AQP4 internalization activates astrocytes, which then increases complement C3 expression to induce microglia chemotaxis. Stage 3. Activated microglia interact with astrocytes and release C1q, which may cause neuronal injury. Additional mechanisms in NMO pathogenesis include complement dependent cytotoxicity mediated by membrane attack complexes (MAC) and focal inflammation induced by macrophage infiltration.
Complement C3 shapes brain function in healthy and diseased conditions (34, 101–103). Recent experimental evidence indicates that microglia are critical for C3-C3aR signaling in neurodegenerative diseases (104, 105). In addition, C3 signaling can also participate in neurodegeneration by activating microglial complement receptor 3 (CR3) (34, 103, 106, 107). Indeed, exogenous human complement C3 can activate murine microglia (105). Administration of AQP4-IgG with complement induces astrocytes depletion in hours (108), which skipped the initial stage of astrocytic reactions in NMO pathology (15, 19, 20). Astrocyte acute ablation likely results in neuronal damage (109), but the question is whether such models really reflect the unique pathology of NMO. In animal models with AQP4-IgG chronic infusion without complement, activation of astrocytes, microglia, and neuronal damage happen in a temporally and spatially dependent manner (68, 70). The downstream cascade might also involve microglial phagocytosis of C3 labeled neurons/synapse, which is observed during neuronal development, neuro-inflammation, and neurodegeneration diseases (35, 103, 106).
Neuronal Damage Is Possibly Mediated by Microglia-Derived C1q
After AQP4-IgG chronic infusion, microglial C3aR induces the expression of complement C1q in microglia (70), the complement cascade initiator (110). However, functional impairment does not seem related to complement induced toxicity for two reasons. First, there was no evidence of terminal complement complexes in this model. Second, astrocytes were activated but not injured. Therefore, it is possible that microglial C1q induces cell stress via different mechanisms other than classical complement cascades. Notably, C1q is able to inhibit mitochondrial function (111), which can lead to myelin and neuronal injury (107, 111, 112). Additionally, C1q can directly bind synapses and induce neuronal damage in a microglial phagocytosis dependent manner (106, 113). However, further study is needed to test whether there is complement-dependent microglial “phagoptosis” of neurons in NMO. Regardless, this model of NMO demonstrates a clear role for microglia in which microglial C1q may induce neuronal dysfunction in a complement cascade independent manner (Figure 1).
Targeting Microglia for NMO Treatment
Based on our recent understanding of critical microglial function in NMO, we propose that microglia may represent a novel therapeutic target for NMO treatment. Typically, acute NMO attacks are first treated with intravenous methylprednisolone for 3~5 days (11), combined with plasma exchange (PLEX) or immunoabsorption (114) to help reduce AQP4-IgG concentrations (115). Subsequently, low doses of prednisone/prednisolone are used for preventative immunosuppressive therapy, however this can induce serious side effects (116). Additional therapeutic treatments for NMO include Azathioprine and Rituximab, which inhibit lymphocyte and CD19+ B cell activity (117). Mycophenolate mofetil has also been useful to inhibit B cells and T cells, but patients will have elevated risk of leucopenia when combined with other immunosuppress therapies (118). It remains to be determined whether these immunosuppressive therapies reduce microglial activation when they exert their therapeutic effects.
The most exciting breakthrough in NMO therapy is eculizumab, which greatly reduces the risk of relapse in AQP4-IgG seropositive patients (3). Eculizumab neutralizes complement C5, which is mainly expressed by microglia in the CNS (74). Interestingly, C5a receptors are highly and exclusively expressed by microglia (119). Clinical evidence indicates NMO pathology is mediated by immunoglobulin and terminal complement complex (C5b-9; C9neo) (16), highlighted the importance of complement dependent cytotoxicity (CDC) in NMO. Besides immunoglobulin mediated astrocyte CDC, the demyelination in NMO lesions can be induced by production of anaphylatoxins (C3a, C4a and C5a) and opsonins (C3b and C1q) that recruit inflammatory cells (120), enhance antibody-dependent cell-mediated cytotoxicity (ADCC) (121), and facilitate phagocytosis (120). Although microglia reactivity has not been examined in eculizumab treated patients yet, early studies already implied how complement C5 inhibition modulate microglia function in CNS disease. First, inhibition of C5a receptors reduced microglia activity and provided strong benefit in mouse models of Alzheimer’s disease (112). Moreover, microglial C5a is a strong chemoattractant for polymorph nuclear cells (122), mediates eosinophil and neutrophil degranulation (123, 124). Therefore, future studies are needed to investigate whether eculizumab can reduce the relapsing risk of NMO via inhibition of microglial C5 receptor signaling.
Lastly, other microglia-targeting therapeutic approaches could be considered for NMO treatment. First, microglial inhibitor minocycline has been shown to reduce microglial activation in many CNS disorders (125–127). Clinical trials of minocycline are currently in progress for schizophrenia, autism, anxiety, and bipolar disorders (128, 129). Second, colony stimulating factor 1 (CSF1) receptor antagonists, such as PLX3397 or PLX5562, can deplete microglia via oral administration (130). PLX3397 (pexidartinib) was approved by FDA for treatment of giant-cell tumor of the tendon sheath in 2019 (131). Whether PLX compounds can be used for NMO therapy still need further investigation. However, microglia depletion/inhibition via CSF1 receptor antagonists may also result in side effects including affecting hematopoiesis and the function of macrophages in mice (132). Last but not least, C3 signaling such as C3 and C3a receptor, is critical for microglial activation and motor impairment in NMO (70). Inhibition of C3aR or neutralization of C3a may provide therapeutic effects. The C3a receptor is an especially attractive target as it is highly expressed in activated microglia (78, 104, 105).
Future Directions and Conclusion
Recent studies have demonstrated the emerging role of microglia in NMO (70). Targeting microglia for NMO treatment is promising but still challenging, considering that microglia share many common molecules with peripheral immune systems (133, 134). To develop microglia specific treatments, future studies are needed to further our understanding of the molecular mechanisms underlying the role of microglia in NMO. Additional considerations include accounting sex differences in NMO, where the female to male ratio in NMO patients is above 8:2 (135). Coincidently, microglial function show clear sex differences evidenced by studies in chronic pain (136, 137), depression (138), stroke (139), and aging (140). It would be interesting to test whether female microglia may have stronger complement or proinflammatory activation in response to AQP4-IgG. In addition, the regional heterogeneity of microglia function is emerging. Whether microglial heterogeneity contributes to preferential development of lesions in optic nerves and the spinal cord warrants further investigations. Finally, it is unknown what the role of microglia is during the remission phase of NMO. Current animal models demonstrate that motor dysfunction can partially recover after AQP4-IgG infusion stopped (68, 70). Thus, the question remains as to whether microglia initiates NMO remission due to microglial reparative function or releasing trophic factors.
In a variety of neurological disorders (e.g., stroke, chronic pain, epilepsy, and neurodegenerative diseases), microglia play beneficial or detrimental function depending on the context and timing of their activation (141–145). The novel mouse model of NMO presented in this review demonstrates a critical role for microglia in the evolution of motor impairment and the neuropathology progression initiated by AQP4-IgG activated astrocytes (70). With the emergence of new genetic tools for microglial research (146, 147), the function and mechanisms of microglia in NMO will be revealed. These will corroborate the idea that microglia may represent a novel therapeutic target in NMO treatment.
Author Contributions
TC and L-JW conceived the study and wrote the manuscript. DB, YY, D-ST, and L-JW edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The work is supported by the National Institutes of Health (R01NS110949, R01NS088627, R01NS112144, R01NS110825, R21AG064159) to L-JW.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of Wu lab at Mayo for the discussion, and Dr. Vanda Lennon, Dr. Linda Wu, Ms. Jiaying Zheng, and Mr. Lingxiao Wang, for editing the manuscript.
References
1. Jarius S, Wildemann B. AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol (2010) 6:383–92. doi: 10.1038/nrneurol.2010.72
2. Zekeridou A, Lennon VA. Aquaporin-4 autoimmunity. Neurol Neuroimmunol Neuroinflamm (2015) 2:e110. doi: 10.1212/NXI.0000000000000110
3. Pittock SJ, Berthele A, Fujihara K, Kim HJ, Levy M, Palace J, et al. Eculizumab in Aquaporin-4-Positive Neuromyelitis Optica Spectrum Disorder. N Engl J Med (2019) 381:614–25. doi: 10.1056/NEJMoa1900866
4. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet (2004) 364:2106–12. doi: 10.1016/S0140-6736(04)17551-X
5. Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol (2009) 66:617–29. doi: 10.1002/ana.21802
6. Bergamaschi R, Pichiecchio A, Persico A, Bastianello S. Relevance of clinical findings for diagnosis of neuromyelitis optica in MRI and NMO-IgG era. Eur J Neurol (2009) 16:e76–7. doi: 10.1111/j.1468-1331.2009.02561.x
7. Jarius S, Franciotta D, Bergamaschi R, Wright H, Littleton E, Palace J, et al. NMO-IgG in the diagnosis of neuromyelitis optica. Neurology (2007) 68:1076–7. doi: 10.1212/01.wnl.0000256822.01222.bd
8. Matiello M, Lennon VA, Jacob A, Pittock SJ, Lucchinetti CF, Wingerchuk DM, et al. NMO-IgG predicts the outcome of recurrent optic neuritis. Neurology (2008) 70:2197–200. doi: 10.1212/01.wnl.0000303817.82134.da
9. Klawiter EC, Alvarez E 3rd, Xu J, Paciorkowski AR, Zhu L, Parks BJ, et al. NMO-IgG detected in CSF in seronegative neuromyelitis optica. Neurology (2009) 72:1101–3. doi: 10.1212/01.wnl.0000345066.57745.50
10. Weinshenker BG, Wingerchuk DM, Vukusic S, Linbo L, Pittock SJ, Lucchinetti CF, et al. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol (2006) 59:566–9. doi: 10.1002/ana.20770
11. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflamm (2016) 13:280. doi: 10.1186/s12974-016-0718-0
12. Kim SH, Jeong IH, Hyun JW, Joung A, Jo HJ, Hwang SH, et al. Treatment Outcomes With Rituximab in 100 Patients With Neuromyelitis Optica: Influence of FCGR3A Polymorphisms on the Therapeutic Response to Rituximab. JAMA Neurol (2015) 72:989–95. doi: 10.1001/jamaneurol.2015.1276
13. Guo Y, Weigand SD, Popescu BF, Lennon VA, Parisi JE, Pittock SJ, et al. Pathogenic implications of cerebrospinal fluid barrier pathology in neuromyelitis optica. Acta Neuropathol (2017) 133:597–612. doi: 10.1007/s00401-017-1682-1
14. Lucchinetti CF, Guo Y, Popescu BF, Fujihara K, Itoyama Y, Misu T. The pathology of an autoimmune astrocytopathy: lessons learned from neuromyelitis optica. Brain Pathol (2014) 24:83–97. doi: 10.1111/bpa.12099
15. Roemer SF, Parisi JE, Lennon VA, Benarroch EE, Lassmann H, Bruck W, et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain (2007) 130:1194–205. doi: 10.1093/brain/awl371
16. Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain (2002) 125:1450–61. doi: 10.1093/brain/awf151
17. Bukhari W, Barnett MH, Prain K, Broadley SA. Molecular pathogenesis of neuromyelitis optica. Int J Mol Sci (2012) 13:12970–93. doi: 10.3390/ijms131012970
18. Hoftberger R, Lassmann H. Inflammatory demyelinating diseases of the central nervous system. Handb Clin Neurol (2017) 145:263–83. doi: 10.1016/B978-0-12-802395-2.00019-5
19. Hinson SR, Romero MF, Popescu BF, Lucchinetti CF, Fryer JP, Wolburg H, et al. Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc Natl Acad Sci USA (2012) 109:1245–50. doi: 10.1073/pnas.1109980108
20. Hinson SR, Clift IC, Luo N, Kryzer TJ, Lennon VA. Autoantibody-induced internalization of CNS AQP4 water channel and EAAT2 glutamate transporter requires astrocytic Fc receptor. Proc Natl Acad Sci USA (2017) 114:5491–6. doi: 10.1073/pnas.1701960114
21. Howe CL, Kaptzan T, Magana SM, Ayers-Ringler JR, LaFrance-Corey RG, Lucchinetti CF. Neuromyelitis optica IgG stimulates an immunological response in rat astrocyte cultures. Glia (2014) 62:692–708. doi: 10.1002/glia.22635
22. Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology (2007) 69:2221–31. doi: 10.1212/01.WNL.0000289761.64862.ce
23. Saji E, Arakawa M, Yanagawa K, Toyoshima Y, Yokoseki A, Okamoto K, et al. Cognitive impairment and cortical degeneration in neuromyelitis optica. Ann Neurol (2013) 73:65–76. doi: 10.1002/ana.23721
24. Hoftberger R, Guo Y, Flanagan EP, Lopez-Chiriboga AS, Endmayr V, Hochmeister S, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol (2020) 139:875–92. doi: 10.1007/s00401-020-02132-y
25. Fan S, Xu Y, Ren H, Guan H, Feng F, Gao X, et al. Comparison of myelin oligodendrocyte glycoprotein (MOG)-antibody disease and AQP4-IgG-positive neuromyelitis optica spectrum disorder (NMOSD) when they co-exist with anti-NMDA (N-methyl-D-aspartate) receptor encephalitis. Mult Scler Relat Disord (2018) 20:144–52. doi: 10.1016/j.msard.2018.01.007
26. Alter A, Duddy M, Hebert S, Biernacki K, Prat A, Antel JP, et al. Determinants of human B cell migration across brain endothelial cells. J Immunol (2003) 170:4497–505. doi: 10.4049/jimmunol.170.9.4497
27. Ikeshima-Kataoka H. Neuroimmunological Implications of AQP4 in Astrocytes. Int J Mol Sci (2016) 17:1306–23. doi: 10.3390/ijms17081306
28. Zeng Q, Dong X, Ruan C, Hu B, Luo Y, Luo Z, et al. CD14(+)CD16(++) monocytes are increased in patients with NMO and are selectively suppressed by glucocorticoids therapy. J Neuroimmunol (2016) 300:1–8. doi: 10.1016/j.jneuroim.2016.09.011
29. Yandamuri SS, Jiang R, Sharma A, Cotzomi E, Zografou C, Ma AK, et al. Charitable Foundation, High-throughput investigation of molecular and cellular biomarkers in NMOSD. Neurol Neuroimmunol Neuroinflamm (2020) 7(5):e852. doi: 10.1212/NXI.0000000000000852
30. Shimizu M, Okuno T, Kinoshita M, Sumi H, Fujimura H, Yamashita K, et al. Mitochondrial DNA enhance innate immune responses in neuromyelitis optica by monocyte recruitment and activation. Sci Rep (2020) 10:13274. doi: 10.1038/s41598-020-70203-x
31. Moll NM, Rietsch AM, Ransohoff AJ, Cossoy MB, Huang D, Eichler FS, et al. Cortical demyelination in PML and MS: Similarities and differences. Neurology (2008) 70:336–43. doi: 10.1212/01.wnl.0000284601.54436.e4
32. Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol (2006) 63:964–8. doi: 10.1001/archneur.63.7.964
33. Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, et al. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflamm (2017) 14:48. doi: 10.1186/s12974-017-0814-9
34. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science (2016) 352:712–6. doi: 10.1126/science.aad8373
35. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature (2017) 541:481–7. doi: 10.1038/nature21029
36. Moller T, Nolte C, Burger R, Verkhratsky A, Kettenmann H. Mechanisms of C5a and C3a complement fragment-induced [Ca2+]i signaling in mouse microglia. J Neurosci (1997) 17:615–24. doi: 10.1523/JNEUROSCI.17-02-00615.1997
37. Song D, Sulewski ME Jr., Wang C, Song J, Bhuyan R, Sterling J, et al. Complement C5a receptor knockout has diminished light-induced microglia/macrophage retinal migration. Mol Vis (2017) 23:210–8.
38. Wing MG, Seilly DJ, Nicholas RS, Rahman S, Zajicek J, Lachmann PJ, et al. Comparison of C1q-receptors on rat microglia and peritoneal macrophages. J Neuroimmunol (1999) 94:74–81. doi: 10.1016/S0165-5728(98)00227-6
39. Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T, et al. Neuromyelitis optica: Passive transfer to rats by human immunoglobulin. Biochem Biophys Res Commun (2009) 386:623–7. doi: 10.1016/j.bbrc.2009.06.085
40. Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol (2009) 66:630–43. doi: 10.1002/ana.21837
41. Saini H, Rifkin R, Gorelik M, Huang H, Ferguson Z, Jones MV, et al. Passively transferred human NMO-IgG exacerbates demyelination in mouse experimental autoimmune encephalomyelitis. BMC Neurol (2013) 13:104. doi: 10.1186/1471-2377-13-104
42. Lee CL, Wang KC, Chen SJ, Chen CM, Tsai CP, Chen SY. Repetitive intrathecal injection of human NMO-IgG with complement exacerbates disease severity with NMO pathology in experimental allergic encephalomyelitis mice. Mult Scler Relat Disord (2019) 30:225–30. doi: 10.1016/j.msard.2019.02.025
43. Kurosawa K, Misu T, Takai Y, Sato DK, Takahashi T, Abe Y, et al. Severely exacerbated neuromyelitis optica rat model with extensive astrocytopathy by high affinity anti-aquaporin-4 monoclonal antibody. Acta Neuropathol Commun (2015) 3:82. doi: 10.1186/s40478-015-0259-2
44. Saligrama N, Zhao F, Sikora MJ, Serratelli WS, Fernandes RA, Louis DM, et al. Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature (2019) 572:481–7. doi: 10.1038/s41586-019-1467-x
45. Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci (2011) 14:1142–9. doi: 10.1038/nn.2887
46. Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med (2014) 211:1533–49. doi: 10.1084/jem.20132477
47. Monaghan KL, Zheng W, Hu G, Wan ECK. Monocytes and Monocyte-Derived Antigen-Presenting Cells Have Distinct Gene Signatures in Experimental Model of Multiple Sclerosis. Front Immunol (2019) 10:2779. doi: 10.3389/fimmu.2019.02779
48. Zeka B, Hastermann M, Hochmeister S, Kogl N, Kaufmann N, Schanda K, et al. Highly encephalitogenic aquaporin 4-specific T cells and NMO-IgG jointly orchestrate lesion location and tissue damage in the CNS. Acta Neuropathol (2015) 130:783–98. doi: 10.1007/s00401-015-1501-5
49. Zeka B, Hastermann M, Kaufmann N, Schanda K, Pende M, Misu T, et al. Aquaporin 4-specific T cells and NMO-IgG cause primary retinal damage in experimental NMO/SD. Acta Neuropathol Commun (2016) 4:82. doi: 10.1186/s40478-016-0355-y
50. Sagan SA, Winger RC, Cruz-Herranz A, Nelson PA, Hagberg S, Miller CN, et al. Tolerance checkpoint bypass permits emergence of pathogenic T cells to neuromyelitis optica autoantigen aquaporin-4. Proc Natl Acad Sci USA (2016) 113:14781–6. doi: 10.1073/pnas.1617859114
51. Jones MV, Huang H, Calabresi PA, Levy M. Pathogenic aquaporin-4 reactive T cells are sufficient to induce mouse model of neuromyelitis optica. Acta Neuropathol Commun (2015) 3:28. doi: 10.1186/s40478-015-0207-1
52. Pohl M, Kawakami N, Kitic M, Bauer J, Martins R, Fischer MT, et al. T cell-activation in neuromyelitis optica lesions plays a role in their formation. Acta Neuropathol Commun (2013) 1:85. doi: 10.1186/2051-5960-1-85
53. Cruz-Herranz A, Sagan SA, Sobel RA, Green AJ, Zamvil SS. T cells targeting neuromyelitis optica autoantigen aquaporin-4 cause paralysis and visual system injury. J Nat Sci (2017) 3(5):e358.
54. Ratelade J, Bennett JL, Verkman AS. Intravenous neuromyelitis optica autoantibody in mice targets aquaporin-4 in peripheral organs and area postrema. PLoS One (2011) 6:e27412. doi: 10.1371/journal.pone.0027412
55. Asavapanumas N, Verkman AS. Neuromyelitis optica pathology in rats following intraperitoneal injection of NMO-IgG and intracerebral needle injury. Acta Neuropathol Commun (2014) 2:48. doi: 10.1186/2051-5960-2-48
56. Yao X, Adams MS, Jones PD, Diederich CJ, Verkman AS. Noninvasive, Targeted Creation of Neuromyelitis Optica Pathology in AQP4-IgG Seropositive Rats by Pulsed Focused Ultrasound. J Neuropathol Exp Neurol (2019) 78:47–56. doi: 10.1093/jnen/nly107
57. Hillebrand S, Schanda K, Nigritinou M, Tsymala I, Bohm D, Peschl P, et al. Circulating AQP4-specific auto-antibodies alone can induce neuromyelitis optica spectrum disorder in the rat. Acta Neuropathol (2019) 137:467–85. doi: 10.1007/s00401-018-1950-8
58. Zhang H, Verkman AS. Eosinophil pathogenicity mechanisms and therapeutics in neuromyelitis optica. J Clin Invest (2013) 123:2306–16. doi: 10.1172/JCI67554
59. Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain (2010) 133:349–61. doi: 10.1093/brain/awp309
60. Saadoun S, Waters P, Owens GP, Bennett JL, Vincent A, Papadopoulos MC. Neuromyelitis optica MOG-IgG causes reversible lesions in mouse brain. Acta Neuropathol Commun (2014) 2:35. doi: 10.1186/2051-5960-2-35
61. Zhang H, Verkman AS. Longitudinally extensive NMO spinal cord pathology produced by passive transfer of NMO-IgG in mice lacking complement inhibitor CD59. J Autoimmun (2014) 53:67–77. doi: 10.1016/j.jaut.2014.02.011
62. Asavapanumas N, Ratelade J, Verkman AS. Unique neuromyelitis optica pathology produced in naive rats by intracerebral administration of NMO-IgG. Acta Neuropathol (2014) 127:539–51. doi: 10.1007/s00401-013-1204-8
63. Matsumoto Y, Kanamori A, Nakamura M, Takahashi T, Nakashima I, Negi A. Sera from patients with seropositive neuromyelitis optica spectral disorders caused the degeneration of rodent optic nerve. Exp Eye Res (2014) 119:61–9. doi: 10.1016/j.exer.2013.12.010
64. Marignier R, Ruiz A, Cavagna S, Nicole A, Watrin C, Touret M, et al. Neuromyelitis optica study model based on chronic infusion of autoantibodies in rat cerebrospinal fluid. J Neuroinflamm (2016) 13:111. doi: 10.1186/s12974-016-0577-8
65. Felix CM, Levin MH, Verkman AS. Complement-independent retinal pathology produced by intravitreal injection of neuromyelitis optica immunoglobulin G. J Neuroinflamm (2016) 13:275. doi: 10.1186/s12974-016-0746-9
66. Zhang Y, Bao Y, Qiu W, Peng L, Fang L, Xu Y, et al. Structural and visual functional deficits in a rat model of neuromyelitis optica spectrum disorders related optic neuritis. Exp Eye Res (2018) 175:124–32. doi: 10.1016/j.exer.2018.06.011
67. Yao X, Verkman AS. Marked central nervous system pathology in CD59 knockout rats following passive transfer of Neuromyelitis optica immunoglobulin G. Acta Neuropathol Commun (2017) 5:15. doi: 10.1186/s40478-017-0417-9
68. Geis C, Ritter C, Ruschil C, Weishaupt A, Grunewald B, Stoll G, et al. The intrinsic pathogenic role of autoantibodies to aquaporin 4 mediating spinal cord disease in a rat passive-transfer model. Exp Neurol (2015) 265:8–21. doi: 10.1016/j.expneurol.2014.12.015
69. Yick LW, Ma OK, Ng RC, Kwan JS, Chan KH. Aquaporin-4 Autoantibodies From Neuromyelitis Optica Spectrum Disorder Patients Induce Complement-Independent Immunopathologies in Mice. Front Immunol (2018) 9:1438. doi: 10.3389/fimmu.2018.01438
70. Chen T, Lennon VA, Liu YU, Bosco DB, Li Y, Yi MH, et al. Astrocyte-microglia interaction drives evolving neuromyelitis optica lesion. J Clin Invest (2020) 130:4025–38. doi: 10.1172/JCI134816
71. Ratelade J, Zhang H, Saadoun S, Bennett JL, Papadopoulos MC, Verkman AS. Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathol (2012) 123:861–72. doi: 10.1007/s00401-012-0986-4
72. Piddlesden SJ, Lassmann H, Zimprich F, Morgan BP, Linington C. The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am J Pathol (1993) 143:555–64.
73. Czosnyka M, Czosnyka Z, Momjian S, Pickard JD. Cerebrospinal fluid dynamics. Physiol Meas (2004) 25:R51–76. doi: 10.1088/0967-3334/25/5/R01
74. Griffin RS, Costigan M, Brenner GJ, Ma CH, Scholz J, Moss A, et al. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J Neurosci (2007) 27:8699–708. doi: 10.1523/JNEUROSCI.2018-07.2007
75. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
76. Holers VM, Kinoshita T, Molina H. The evolution of mouse and human complement C3-binding proteins: divergence of form but conservation of function. Immunol Today (1992) 13:231–6. doi: 10.1016/0167-5699(92)90160-9
77. Simonetti M, Hagenston AM, Vardeh D, Freitag HE, Mauceri D, Lu J, et al. Nuclear calcium signaling in spinal neurons drives a genomic program required for persistent inflammatory pain. Neuron (2013) 77:43–57. doi: 10.1016/j.neuron.2012.10.037
78. Zhang LY, Pan J, Mamtilahun M, Zhu Y, Wang L, Venkatesh A, et al. Microglia exacerbate white matter injury via complement C3/C3aR pathway after hypoperfusion. Theranostics (2020) 10:74–90. doi: 10.7150/thno.35841
79. Kock MA, Hew BE, Bammert H, Fritzinger DC, Vogel CW. Structure and function of recombinant cobra venom factor. J Biol Chem (2004) 279:30836–43. doi: 10.1074/jbc.M403196200
80. Duan T, Smith AJ, Verkman AS. Complement-independent bystander injury in AQP4-IgG seropositive neuromyelitis optica produced by antibody-dependent cellular cytotoxicity. Acta Neuropathol Commun (2019) 7:112. doi: 10.1186/s40478-019-0766-7
81. Nytrova P, Potlukova E, Kemlink D, Woodhall M, Horakova D, Waters P, et al. Complement activation in patients with neuromyelitis optica. J Neuroimmunol (2014) 274:185–91. doi: 10.1016/j.jneuroim.2014.07.001
82. Veszeli N, Fust G, Csuka D, Trauninger A, Bors L, Rozsa C, et al. A systematic analysis of the complement pathways in patients with neuromyelitis optica indicates alteration but no activation during remission. Mol Immunol (2014) 57:200–9. doi: 10.1016/j.molimm.2013.09.010
83. Chen Y, Li R, Wu AM, Shu YQ, Lu ZQ, Hu XQ. The complement and immunoglobulin levels in NMO patients. Neurol Sci (2014) 35:215–20. doi: 10.1007/s10072-013-1481-y
84. Jitprapaikulsan J, Fryer JP, Majed M, Smith CY, Jenkins SM, Cabre P, et al. Clinical utility of AQP4-IgG titers and measures of complement-mediated cell killing in NMOSD. Neurol - Neuroimmunol Neuroinflamm (2020) 7:e727. doi: 10.1212/NXI.0000000000000727
85. Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med (2018) 24:931–8. doi: 10.1038/s41591-018-0051-5
86. Vainchtein ID, Molofsky AV. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci (2020) 43:144–54. doi: 10.1016/j.tins.2020.01.003
87. Jha MK, Jo M, Kim JH, Suk K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist (2019) 25:227–40. doi: 10.1177/1073858418783959
88. Bialas AR, Stevens B. TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci (2013) 16:1773–82. doi: 10.1038/nn.3560
89. Norden DM, Fenn AM, Dugan A, Godbout JP. TGFbeta produced by IL-10 redirected astrocytes attenuates microglial activation. Glia (2014) 62:881–95. doi: 10.1002/glia.22647
90. Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science (2018) 359:1269–73. doi: 10.1126/science.aal3589
91. Kim JH, Ko PW, Lee HW, Jeong JY, Lee MG, Kim JH, et al. Astrocyte-derived lipocalin-2 mediates hippocampal damage and cognitive deficits in experimental models of vascular dementia. Glia (2017) 65:1471–90. doi: 10.1002/glia.23174
92. Jo M, Kim JH, Song GJ, Seo M, Hwang EM, Suk K. Astrocytic Orosomucoid-2 Modulates Microglial Activation and Neuroinflammation. J Neurosci (2017) 37:2878–94. doi: 10.1523/JNEUROSCI.2534-16.2017
93. Lampron A, Larochelle A, Laflamme N, Prefontaine P, Plante MM, Sanchez MG, et al. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med (2015) 212:481–95. doi: 10.1084/jem.20141656
94. Sariol A, Mackin S, Allred MG, Ma C, Zhou Y, Zhang Q, et al. Microglia depletion exacerbates demyelination and impairs remyelination in a neurotropic coronavirus infection. Proc Natl Acad Sci USA (2020) 117:24464–74. doi: 10.1073/pnas.2007814117
95. Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci (2013) 16:1211–8. doi: 10.1038/nn.3469
96. Eyo UB, Gu N, De S, Dong H, Richardson JR, Wu LJ. Modulation of microglial process convergence toward neuronal dendrites by extracellular calcium. J Neurosci (2015) 35:2417–22. doi: 10.1523/JNEUROSCI.3279-14.2015
97. Eyo UB, Peng J, Murugan M, Mo M, Lalani A, Xie P, et al. Regulation of Physical Microglia-Neuron Interactions by Fractalkine Signaling after Status Epilepticus. eNeuro (2016) 3:0209-16. doi: 10.1523/ENEURO.0209-16.2016
98. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci (2005) 8:752–8. doi: 10.1038/nn1472
99. Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu LJ. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J Neurosci (2014) 34:10528–40. doi: 10.1523/JNEUROSCI.0416-14.2014
100. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci (2014) 34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014
101. Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature (2016) 534:538–43. doi: 10.1038/nature18283
102. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
103. Werneburg S, Jung J, Kunjamma RB, Ha SK, Luciano NJ, Willis CM, et al. Targeted Complement Inhibition at Synapses Prevents Microglial Synaptic Engulfment and Synapse Loss in Demyelinating Disease. Immunity (2020) 52:167–182 e7. doi: 10.1016/j.immuni.2019.12.004
104. Lian H, Litvinchuk A, Chiang AC, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J Neurosci (2016) 36:577–89. doi: 10.1523/JNEUROSCI.2117-15.2016
105. Litvinchuk A, Wan YW, Swartzlander DB, Chen F, Cole A, Propson NE, et al. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron (2018) 100:1337–53.e5. doi: 10.1016/j.neuron.2018.10.031
106. Anderson SR, Zhang J, Steele MR, Romero CO, Kautzman AG, Schafer DP, et al. Complement Targets Newborn Retinal Ganglion Cells for Phagocytic Elimination by Microglia. J Neurosci (2019) 39:2025–40. doi: 10.1523/JNEUROSCI.1854-18.2018
107. Dejanovic B, Huntley MA, De Maziere A, Meilandt WJ, Wu T, Srinivasan K, et al. Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron (2018) 100:1322–1336 e7. doi: 10.1016/j.neuron.2018.10.014
108. Herwerth M, Kalluri SR, Srivastava R, Kleele T, Kenet S, Illes Z, et al. In vivo imaging reveals rapid astrocyte depletion and axon damage in a model of neuromyelitis optica-related pathology. Ann Neurol (2016) 79:794–805. doi: 10.1002/ana.24630
109. Schreiner B, Romanelli E, Liberski P, Ingold-Heppner B, Sobottka-Brillout B, Hartwig T, et al. Astrocyte Depletion Impairs Redox Homeostasis and Triggers Neuronal Loss in the Adult CNS. Cell Rep (2015) 12:1377–84. doi: 10.1016/j.celrep.2015.07.051
110. Nesargikar PN, Spiller B, Chavez R. The complement system: history, pathways, cascade and inhibitors. Eur J Microbiol Immunol (Bp) (2012) 2:103–11. doi: 10.1556/EuJMI.2.2012.2.2
111. Ten VS, Yao J, Ratner V, Sosunov S, Fraser DA, Botto M, et al. Complement component c1q mediates mitochondria-driven oxidative stress in neonatal hypoxic-ischemic brain injury. J Neurosci (2010) 30:2077–87. doi: 10.1523/JNEUROSCI.5249-09.2010
112. Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM, et al. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J Immunol (2009) 183:1375–83. doi: 10.4049/jimmunol.0901005
113. Bie B, Wu J, Foss JF, Naguib M. Activation of mGluR1 Mediates C1q-Dependent Microglial Phagocytosis of Glutamatergic Synapses in Alzheimer’s Rodent Models. Mol Neurobiol (2019) 56:5568–85. doi: 10.1007/s12035-019-1467-8
114. Faissner S, Nikolayczik J, Chan A, Gold R, Yoon MS, Haghikia A. Immunoadsorption in patients with neuromyelitis optica spectrum disorder. Ther Adv Neurol Disord (2016) 9:281–6. doi: 10.1177/1756285616646332
115. Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke KD, Wegner B, et al. Neuromyelitis optica: Evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol (2016) 79:206–16. doi: 10.1002/ana.24554
116. Patt H, Bandgar T, Lila A, Shah N. Management issues with exogenous steroid therapy. Indian J Endocrinol Metab (2013) 17:S612–7. doi: 10.4103/2230-8210.123548
117. Nikoo Z, Badihian S, Shaygannejad V, Asgari N, Ashtari F. Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: a randomized clinical trial. J Neurol (2017) 264:2003–9. doi: 10.1007/s00415-017-8590-0
118. Jacob A, Matiello M, Weinshenker BG, Wingerchuk DM, Lucchinetti C, Shuster E, et al. Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Arch Neurol (2009) 66:1128–33. doi: 10.1001/archneurol.2009.175
119. Martin CB, Ingersoll SA, Martin BK. Regulation of the C5a receptor promoter in glial cells: Minimal dependence upon the CCAAT element in astrocytes. Mol Immunol (2007) 44:713–21. doi: 10.1016/j.molimm.2006.04.016
120. Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci (2012) 35:369–89. doi: 10.1146/annurev-neuro-061010-113810
121. Derer S, Cossham M, Rosner T, Kellner C, Beurskens FJ, Schwanbeck R, et al. A Complement-Optimized EGFR Antibody Improves Cytotoxic Functions of Polymorphonuclear Cells against Tumor Cells. J Immunol (2015) 195:5077–87. doi: 10.4049/jimmunol.1501458
122. Sewell DL, Nacewicz B, Liu F, Macvilay S, Erdei A, Lambris JD, et al. Complement C3 and C5 play critical roles in traumatic brain cryoinjury: blocking effects on neutrophil extravasation by C5a receptor antagonist. J Neuroimmunol (2004) 155:55–63. doi: 10.1016/j.jneuroim.2004.06.003
124. Saadoun S, Waters P, MacDonald C, Bell BA, Vincent A, Verkman AS, et al. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain. Ann Neurol (2012) 71:323–33. doi: 10.1002/ana.22686
125. Chini M, Popplau JA, Lindemann C, Carol-Perdiguer L, Hnida M, Oberlander V, et al. Resolving and Rescuing Developmental Miswiring in a Mouse Model of Cognitive Impairment. Neuron (2020) 105:60–74 e7. doi: 10.1016/j.neuron.2019.09.042
126. Manso Y, Holland PR, Kitamura A, Szymkowiak S, Duncombe J, Hennessy E, et al. Minocycline reduces microgliosis and improves subcortical white matter function in a model of cerebral vascular disease. Glia (2018) 66:34–46. doi: 10.1002/glia.23190
127. Park D, Kim S, Kim H, Shin J, Jung H, Um JW. Seizure progression triggered by IQSEC3 loss is mitigated by reducing activated microglia in mice. Glia (2020) 68:2661–73. doi: 10.1002/glia.23876
128. Delorme R, Ey E, Toro R, Leboyer M, Gillberg C, Bourgeron T. Progress toward treatments for synaptic defects in autism. Nat Med (2013) 19:685–94. doi: 10.1038/nm.3193
129. Chaudhry IB, Hallak J, Husain N, Minhas F, Stirling J, Richardson P, et al. Minocycline benefits negative symptoms in early schizophrenia: a randomised double-blind placebo-controlled clinical trial in patients on standard treatment. J Psychopharmacol (2012) 26:1185–93. doi: 10.1177/0269881112444941
130. Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron (2014) 82:380–97. doi: 10.1016/j.neuron.2014.02.040
131. Denny WA, Flanagan JU. Small-molecule CSF1R kinase inhibitors; review of patents 2015-present. Expert Opin Ther Pat (2020) 30:3510–27. doi: 10.1080/13543776.2021.1839414
132. Lei F, Cui N, Zhou C, Chodosh J, Vavvas DG, Paschalis EI. CSF1R inhibition by a small-molecule inhibitor is not microglia specific; affecting hematopoiesis and the function of macrophages. Proc Natl Acad Sci USA (2020) 117:23336–8. doi: 10.1073/pnas.1922788117
133. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci (2014) 17:131–43. doi: 10.1038/nn.3599
134. Ajami B, Samusik N, Wieghofer P, Ho PP, Crotti A, Bjornson Z, et al. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat Neurosci (2018) 21:541–51. doi: 10.1038/s41593-018-0100-x
135. Borisow N, Kleiter I, Gahlen A, Fischer K, Wernecke KD, Pache F, et al. Influence of female sex and fertile age on neuromyelitis optica spectrum disorders. Mult Scler (2017) 23:1092–103. doi: 10.1177/1352458516671203
136. Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci (2015) 18:1081–3. doi: 10.1038/nn.4053
137. Chen G, Luo X, Qadri MY, Berta T, Ji RR. Sex-Dependent Glial Signaling in Pathological Pain: Distinct Roles of Spinal Microglia and Astrocytes. Neurosci Bull (2018) 34:98–108. doi: 10.1007/s12264-017-0145-y
138. Liu LL, Li JM, Su WJ, Wang B, Jiang CL. Sex differences in depressive-like behaviour may relate to imbalance of microglia activation in the hippocampus. Brain Behav Immun (2019) 81:188–97. doi: 10.1016/j.bbi.2019.06.012
139. Villa A, Gelosa P, Castiglioni L, Cimino M, Rizzi N, Pepe G, et al. Sex-Specific Features of Microglia from Adult Mice. Cell Rep (2018) 23:3501–11. doi: 10.1016/j.celrep.2018.05.048
140. Olmedillas Del Moral M, Frohlich N, Figarella K, Mojtahedi N, Garaschuk O. Effect of Caloric Restriction on the in vivo Functional Properties of Aging Microglia. Front Immunol (2020) 11:750. doi: 10.3389/fimmu.2020.00750
141. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med (2017) 23:1018–27. doi: 10.1038/nm.4397
142. Qin C, Zhou LQ, Ma XT, Hu ZW, Yang S, Chen M, et al. Dual Functions of Microglia in Ischemic Stroke. Neurosci Bull (2019) 35:921–33. doi: 10.1007/s12264-019-00388-3
143. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest (2012) 122:1164–71. doi: 10.1172/JCI58644
144. Eyo UB, Wu LJ. Microglia: Lifelong patrolling immune cells of the brain. Prog Neurobiol (2019) 179:101614. doi: 10.1016/j.pneurobio.2019.04.003
145. Peng J, Gu N, Zhou L, U BE, Murugan M, Gan WB, et al. Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat Commun (2016) 7:12029. doi: 10.1038/ncomms12029
146. Umpierre AD, Wu LJ. Microglia Research in the 100th Year Since Its Discovery. Neurosci Bull (2020) 36:303–6. doi: 10.1007/s12264-020-00477-8
Keywords: microglia, neuromyelitis optica, complement C3, aquaporin-4, astrocyte-microglia communication, autoimmune, C3a receptor
Citation: Chen T, Bosco DB, Ying Y, Tian D-S and Wu L-J (2021) The Emerging Role of Microglia in Neuromyelitis Optica. Front. Immunol. 12:616301. doi: 10.3389/fimmu.2021.616301
Received: 11 October 2020; Accepted: 04 January 2021;
Published: 19 February 2021.
Edited by:
Amanda Sierra, Achucarro Basque Center for Neuroscience, SpainReviewed by:
José Luis Marín-Teva, University of Granada, SpainFrederike Cosima Oertel, University of California, San Francisco, United States
Copyright © 2021 Chen, Bosco, Ying, Tian and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Long-Jun Wu, d3UubG9uZ2p1bkBtYXlvLmVkdQ==