 Michelle L. Robinette
Michelle L. Robinette Deepak A. Rao
Deepak A. Rao Paul A. Monach1,2
Paul A. Monach1,2- 1Division of Rheumatology, Inflammation, and Immunity, Brigham and Women's Hospital and Harvard Medical School, Boston, MA, United States
- 2Rheumatology Section, VA Boston Healthcare System, Boston, MA, United States
Giant cell arteritis (GCA) is a granulomatous systemic vasculitis of large- and medium-sized arteries that affects the elderly. In recent years, advances in diagnostic imaging have revealed a greater degree of large vessel involvement than previously recognized, distinguishing classical cranial- from large vessel (LV)- GCA. GCA often co-occurs with the poorly understood inflammatory arthritis/bursitis condition polymyalgia rheumatica (PMR) and has overlapping features with other non-infectious granulomatous vasculitides that affect the aorta, namely Takayasu Arteritis (TAK) and the more recently described clinically isolated aortitis (CIA). Here, we review the literature focused on the immunopathology of GCA on the background of the three settings in which comparisons are informative: LV and cranial variants of GCA; PMR and GCA; the three granulomatous vasculitides (GCA, TAK, and CIA). We discuss overlapping and unique features between these conditions across clinical presentation, epidemiology, imaging, and conventional histology. We propose a model of GCA where abnormally activated circulating cells, especially monocytes and CD4+ T cells, enter arteries after an unknown stimulus and cooperate to destroy it and review the evidence for how this mechanistically occurs in active disease and improves with treatment.
Introduction
Giant cell arteritis (GCA) is a granulomatous systemic vasculitis of people age 50 or older that affects large- and medium-size arteries (1, 2). Vascular inflammation has two major patterns, which overlap in a clinical spectrum. The first and classic pattern, originally described by Horton in 1932, involves inflammation of the extracranial branches of the carotid artery with predilection for the temporal artery and is called cranial GCA. The second pattern involves the aorta and its proximal branches, particularly the axillary, subclavian, and proximal brachial branches, and is called large-vessel GCA (LV-GCA) (3). While autopsy studies in the 1970s demonstrated LV involvement in patients with cranial-GCA (4, 5), advances in imaging in the past decade have reemphasized the frequent co-occurrence of subclinical LV with cranial disease and have identified the less common entities of isolated cranial- and LV-GCA (6). Along with Takayasu arteritis (TAK), a systemic vasculitis that occurs mostly in women under age 50, and clinically isolated aortitis (CIA), a vasculitis restricted to the aorta, GCA is one of three non-infectious granulomatous vasculitides with prominent aortic involvement (1).
GCA is medical emergency due to its ability to cause irreversible vision loss and requires prompt diagnosis and initiation of treatment. Individual patient presentations vary depending on the complement of cranial or large vessels that are involved, yet patients often share common systemic features. These include laboratory evidence of systemic inflammation, constitutional symptoms, and polymyalgia rheumatica (PMR), a condition characterized by pain and stiffness in the neck, shoulders, and pelvic girdle that often co-occurs with GCA (7, 8). Mechanistic understanding of both GCA and PMR has been limited by the lack of consensus diagnostic criteria. However, GCA is better characterized than PMR due to the historical de facto diagnostic gold standard being temporal artery biopsy (TAB), which has created a more homogenous clinical group and also provided a vital source of tissue for research purposes. Immunosuppression with glucocorticoids (GC) is the cornerstone of treatment for both GCA and PMR. As most patients have disease flares with GC tapering and require prolonged treatment, steroid sparing agents have been sought, with methotrexate identified as providing benefit in PMR and likely some in GCA, and targeted blockade of IL-6R with tocilizumab (TCZ) providing benefit in GCA. Multiple other drugs are being studied in clinical trials in GCA (9–12).
Here, we review the current understanding of the immunopathology of GCA on the background of the three settings in which comparisons are informative: LV and cranial variants of GCA; PMR and GCA; and the three granulomatous vasculitides (GCA, TAK, and CIA). We also discuss clinical presentation and epidemiology of disease, and the growing role of advanced imaging for clinical and research use. We identify areas of uncertainty and discuss possible mechanisms of disease pathogenesis.
Clinical Presentation
Systemic inflammation is a cardinal feature of GCA, as well as PMR and TAK. Clinically, many patients experience non-specific constitutional symptoms including fatigue, anorexia, weight loss, fever, and night sweats. Laboratory evidence of inflammation includes anemia, thrombocytosis, and elevations in the inflammatory markers erythrocyte sedimentation rate (ESR) and/or C-reactive protein (CRP). Patients with CIA lack systemic features, according to the most commonly used definition of CIA (7, 8, 13, 14).
Cranial symptoms of GCA are the classic presentation of disease and account for the majority of the 1990 ACR classification criteria (7). Inflammation of medium-size arteries causes pain and tenderness in the artery wall itself and leads to vascular stenosis and ultimately occlusion, causing symptomatic ischemia. Ischemic symptoms include headache, jaw claudication, and acute onset visual disturbances (7), and are inversely correlated with the degree of systemic inflammation (15, 16). More rarely, scalp or tongue necrosis, sensorineural hearing loss, and even vertebrobasilar stroke can occur. The most commonly feared complication is irreversible vision loss, which occurred in 15–35% of patients prior to widespread recognition of GCA and emergent use of GC (2, 17, 18).
LV-GCA often presents with non-specific systemic symptoms, leading to delayed diagnosis (19, 20). Features suggestive of LV-GCA in patients with PMR include the need for unusually high doses of GC, bilateral diffuse lower extremity pain, pelvic girdle pain, and inflammatory low back pain (20). LV-GCA can also cause ischemic symptoms corresponding to supra-aortic vessel stenosis with resultant limb claudication or dizziness. Physical signs can include vascular bruits, loss of carotid or radial pulses, and/or discordant blood pressures (19, 21). These overlap with the symptoms and classification criteria for TAK (13). Rather than causing ischemia in downstream organs, inflammation of the aorta under the stress of high-pressure gradients generated by the heart leads to dilatation in 32% of patients with GCA, aneurysm formation in 2–10% patients, and ultimately may progress to dissection (22–24). Thus, LV-GCA is typically identified on imaging or in surgical specimens from repairs of aneurysms or dissections. In the case of surgical tissue, GCA must further be differentiated from CIA by evidence of systemic features or evidence of disease in arteries other than the aorta.
Epidemiology
GCA is the most common form of vasculitis in patients over age 50 with most being much older. PMR is 3–10 times more common than GCA and is the second most frequent rheumatic disease of elderly after rheumatoid arthritis (2). Forty–sixty percent of patients with GCA have symptoms of PMR while 16–21% PMR have GCA (25, 26). Age >50 is a defining feature of both GCA and PMR, and both peak around age 75, with the exception that patients with LV-GCA are typically younger between 50 and 65 (2, 3, 24, 27, 28). Other granulomatous vasculitides affecting the aorta also occur earlier; CIA has a mean diagnosis of age 65 while TAK peaks between 15 and 29 (14, 29). All conditions are more common in women, with increasing frequency from CIA and PMR (2:1), to cranial-GCA (almost 3:1), to LV-GCA (3:1), and finally to TAK (9:1) (3, 14, 26–30).
The incidence of cranial-GCA and PMR is most frequent in patients of Northern European ancestry. Overlapping incidence of GCA between Northern Europe at 14.6–43.5/ 100,000 and the ancestrally similar Olmstead County, Minnesota at 19.8/ 100,000 suggest a genetic predisposition (26, 27). In other populations, GCA occurs between 1.1 and 11.1/100,000 (26, 31–33), though there are no studies from Africa, South America, or the majority of continental Asia and the Middle East. It was previously thought that GCA was uncommon in African Americans (31). However, this has not consistently been shown in the literature, likely reflecting the ancestral heterogeneity within racial groups within the United States and perhaps under recognition of GCA in African Americans due to the misconception they are not affected (34–38). TAK is less common than GCA, with highest incidence in Asia, South America, and Turkey at 1–2/1,000,000 (39). The true incidence and demographics of LV-GCA and CIA are unknown but appear to be intermediate between cranial-GCA and TAK, at least in the United States (40).
While the increased frequency of GCA in patients of Northern European ancestry suggests a genetic predisposition, genetic studies have generated limited insight into pathophysiology of disease. An early reported and consistently reproduced finding is the association with MHC class II HLA DRB04, specifically the *0401 and *0404 alleles, with cranial- and LV-GCA as well as PMR (3, 41–44). Indeed, large immune-focused genotyping arrays performed on patients with TAB-confirmed cranial-GCA and TAK identified the HLA locus as the only locus to achieve genome-wide significance for association with GCA, and one of two loci with genome-wide significance in TAK (45, 46). In GCA, the majority of this association was due to HLA-DRB1 and HLA-DQA1, with a minor contribution from MHC class I HLA-B. The opposite pattern was found for TAK (47). Strong class II associations suggest a key role for antigen presentation by MHC class II to helper CD4+ T cells in GCA, and multiple studies have suggested changes to the MHC class II peptide-binding groove, however, the specific antigens recognized by CD4+ T cells in GCA remain unclear (41, 46). Likewise, TAK has more cytotoxic CD8+ T cell infiltration than GCA that may explain its association with class I (48). When data from GCA and TAK studies were combined in a meta-analysis, the only non-HLA SNP that reached significance was in IL12B, encoding the p40 portion of the IL-12 (p35p40)/IL-23 (p19p40) heterodimeric proteins that is shared by both cytokines (47). Yet, clinically targeting p40 with ustekinumab in two open-label trials has shown mixed results in GCA (49, 50). Collectively, epidemiologic data emphasizes the importance of old age, female sex, and genetics with GCA though how these factors contribute to disease pathogenesis remains largely unclear.
Imaging
In 2018 the European League Against Rheumatism (EULAR) issued guidelines for use of imaging in LVV for the first time, recommending early imaging as the diagnostic test of choice to replace TAB in all cases of clinically suspected GCA (51). Currently, there are four major imaging modalities used in clinical practice (Table 1): ultrasound, MRI, CT, and [18F]-fluorodeoxyglucose (FDG) positron emission tomography (PET) (6, 51). PET is combined with another technique, most often CT. All four modalities assess vascular wall thickness and a marker of inflammation that differs between techniques (Table 1). Ultrasound assessment is limited to superficial arteries and patients with GCA have non-compressible hypoechoic wall thickening called the “halo sign” (51). Compared to ultrasound, MRI angiography and CT angiography have increased vascular resolution, facilitating assessment of luminal irregularities such as vascular stenosis, aneurysm, and occlusion. Special MRI contrast sequences can also assess cranial vasculature (55, 56).
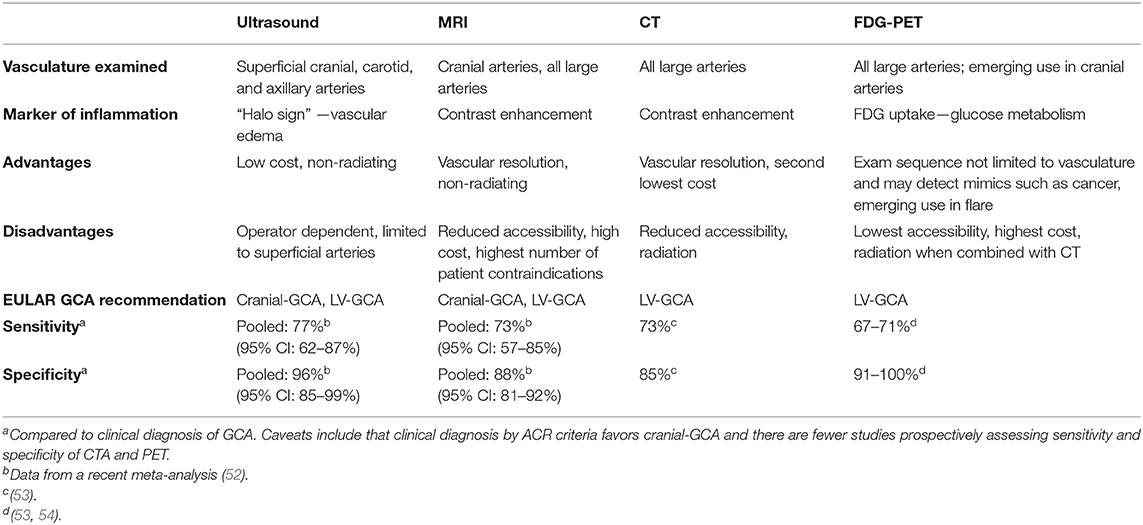
Table 1. Characteristics of imaging modalities clinically used to support a GCA diagnosis.
PET is a very sensitive technique that detects inflammation through the surrogate marker of increased glucose metabolism via FDG uptake and has been particularly important to define GCA and PMR. In 1999, a small prospective PET study first demonstrated LV enhancement in patients with GCA and, surprisingly, equally in those with PMR (57). Most subsequent studies are retrospective raising the possibility of selection bias. However, additional small prospective PET studies have demonstrated LV FDG uptake in 66.7–83% of patients with cranial GCA (54, 58) and 14–31% of patients with PMR (59, 60). Corresponding to limb girdle symptoms, patients with PMR show additional FDG uptake in periarticular regions to the hip and shoulder, ischial tuberosities, sternoclavicular joints, and trochanteric and interspinous bursa (61). Pathologic correlates to large vessel imaging studies are not intentionally obtained. Supporting the concept that imaging findings do reflect active vascular inflammation, some studies have reported that inflammatory markers correspond to the degree of LV FDG uptake (54, 62, 63), which is also reduced with treatment (58, 64, 65). However, low grade enhancement may persist with normal inflammatory markers (66). Multiple prospective serial studies have now shown this does not appear to predict clinical relapse and may rather represent vascular remodeling (58, 65, 67). Preliminary data suggests there may be an imaging cut-off that can distinguish ongoing inflammation from vascular remodeling, as well as from LVV mimics such as atherosclerosis, and is an ongoing area of research (68, 69).
Large vessel imaging patterns can also help differentiate between TAK and LV-GCA in patients who are at the border of age around 50 (Figure 1A). Indeed, a large imaging cohort study recently identified six patterns of LV involvement that were different between diseases (70). Favoring TAK were involvement of the abdominal aorta, renal, and mesenteric arteries; bilateral carotid and subclavian arteries; and isolated left subclavian artery. Favoring GCA were involvement of bilateral axillary and subclavian arteries; diffuse disease including the aorta and its proximal branches; and minimal disease without clear pattern. Additionally, vascular damage with stenosis, aneurysm, or occlusion is more common in TAK while vascular inflammation alone is more common in GCA (70). Extracranial carotid arteries involved in cranial-GCA are rarely affected by TAK. Scans obtained for other reasons may also incidentally reveal CIA in the arch and descending thoracic aorta (14).
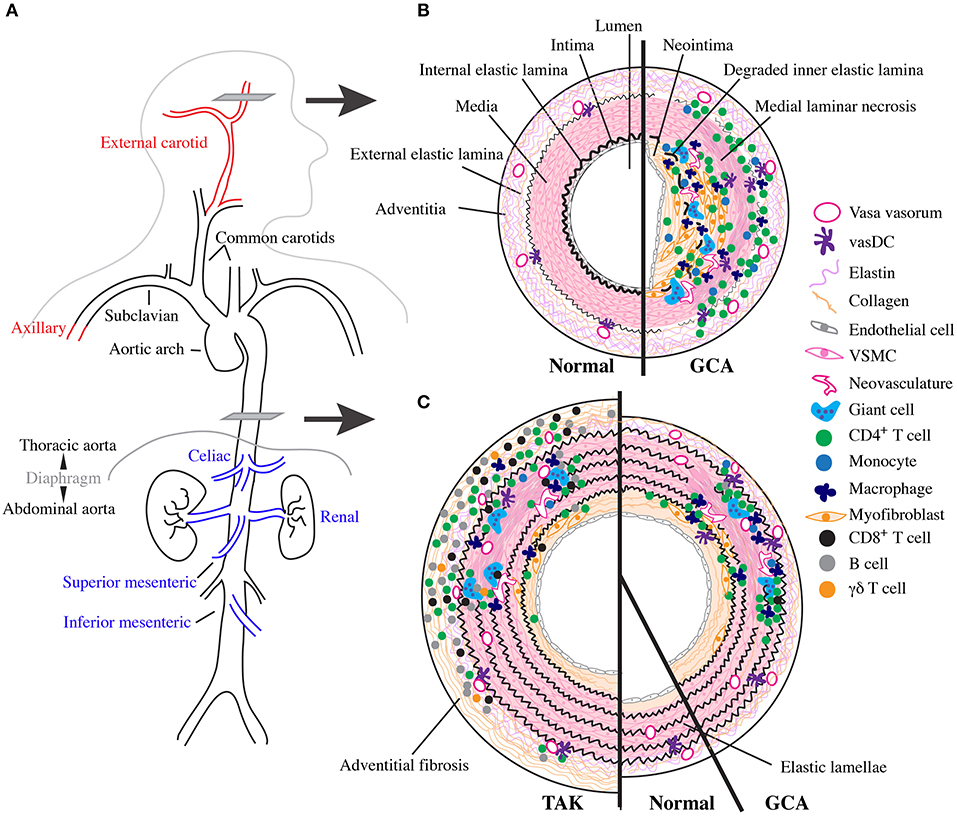
Figure 1. Vascular topology and schematic cross sections of GCA, TAK, and normal arteries. (A) Depiction of the normal human large vessel vasculature. Vascular involvement with greatest specificity for GCA shown in red and with greatest specificity for TAK in blue. (B) Cartoon representing a cross section though the temporal artery as indicated in (A) with normal vessel on the left and the inflammatory infiltrate and vascular remodeling found in cranial-GCA on the right. (C) Cartoon representing a cross section through the thoracic aorta as indicated in (A) with TAK on the left, normal in the middle, and LV-GCA on the right.
Imaging has been instrumental to define GCA but provides little insight into pathophysiology. EULAR recommends using ultrasound and MRI to diagnose cranial-GCA, with no preference in technique for LV-GCA. The optimal use and interpretation of LV imaging in clinical practice is rapidly evolving and is thus far uncertain.
Histopathology
Normal arteries have three layers separated by dense elastic fibers (Figure 1B). From the lumen outward, these include the tunica intima, internal elastic lamina (IEL), tunica media, external elastic lamina, and tunic adventitia. Intima and media are predominantly composed of endothelial cells and vascular smooth muscle cells (VSCM), respectively (71). Their thickness and complexity increases in large elastic vessels with proportional increase in stromal cells and extracellular matrix, especially within the elastic lamellae-rich media (71). The adventitia contains a dense network of elastin and collagen connective tissue produced by fibroblasts and is interdigitated with progenitor cells, adrenergic nerves, and immunosurveillant tissue resident myeloid cells called vascular dendritic cells (vasDC) (72, 73). It is also the site of the vasa vasorum, a microvascular network composed of endothelial cells and pericytes that supplies oxygen and other nutrients to the vascular wall (72). As large arteries require increased nutritional support due to their size, vasa vasorum extend further into the media in these vessels. Outside the artery proper lie more connective tissues supported by a network of small non-muscular blood vessels (74).
Temporal Arteries
GCA is a multi-focal, segmental destructive panarteritis (75–78) (Figure 1B). There is a transmural inflammatory infiltrate with greatest density between the adventitia and media that is composed predominantly of CD4+ T cells and macrophages, with few B cells and eosinophils; mature neutrophils are rare and when abundant suggest an alternative diagnosis (79, 80). Despite the name, giant cells themselves occur to a variable degree and are prominent in ~50% cases at the intima-medial junction around a deranged IEL. In the media there is laminar necrosis with loss of VSCMs and neoangiogensis; fibrinoid necrosis does not occur (79). The intima has features of vascular remodeling with hyperplasia and fibrosis, and occasionally thrombosis and recanalization, especially above sites of active inflammation. More active disease has a more diffuse and intense inflammatory infiltrate and greater number of giant cells, while quiescent disease has a scant infiltrate with fewer giant cells (81). At the end of this spectrum is “healed arteritis,” when the features of vascular damage and remodeling are seen in the absence of inflammatory cells (78).
Three other patterns of inflammation associated with GCA and PMR have also been described, together referred to as “restricted inflammation” (RI). These include small vessel vasculitis (SVV) involving the vessels in connective tissue beyond the adventitia, vasa vasorum vasculitis (VVV), and inflammation limited to adventitia (ILA) (82). Giant cells and granulomas are not present. In their limited description, SVV consisted of slightly more T than B cells and few macrophages (74), while VVV showed approximately equivalent infiltration of T cells, B cells, and macrophages (83). The extent to which these patterns are reported and their role in GCA diagnosis is controversial (82, 84). Recently, a retrospective clinicopathologic study with advanced imaging found that patients with RI had fewer cranial symptoms, less systemic inflammation, and less halo sign on ultrasound (82). However, there was no difference in visual symptoms including permanent vision loss or the degree of LV involvement between RI and classic GCA. In an accompanying systematic literature review, the positive predictive value for RI was 23%, highest for ILA with 67% for GCA and 95% for PMR (82). Notably, other forms of vasculitis, infection, and certain hematologic malignancies can present with RI and particularly with SVV (85, 86).
Large Vessels
Large vessel pathology in GCA is less well-studied. Historically, patients with LVV have often been described to have TAK and patients with CIA may be aberrantly reported as having GCA (87). However, pathologic characteristics of GCA can be disentangled by the few studies that concurrently report TAB, which confirm histopathology is largely the same across vessel sizes (4, 81, 88, 89). Compared to TAB, the aorta has reduced adventitial inflammation with the majority of inflammatory infiltrate in the media; mild adventitial fibrosis is also occasionally seen (4, 88) (Figure 1C). Interestingly, patients with aortic dissection tend to have more diffuse involvement than seen on necropsy, suggesting more robust aortic inflammation in these patients (4, 89). CIA is histopathologically identical to GCA (14, 88). Few studies have directly compared TAK and GCA histopathology, but TAK has more inflammation and fibrosis in the adventitia and media, resulting in thicker walls (40, 48, 88, 90). There is also increased invasion of CD8+ T cells, B cells, and γδT cells and more giant cells compared with GCA (48, 91) (Figure 1C).
Pathophysiology of GCA
Clinical features, epidemiology, imaging, and conventional histology give important information about GCA, PMR, TAK, and CIA, but little insight into pathophysiology. For that, we must rely on a small number of techniques, each with its own strengths and limitations, predominantly based on the characterization and manipulation of patient-derived peripheral blood mononuclear cells (PBMCs) and TAB tissue. Based on our review of data from these studies, here we envision the sequence of events that occurs in GCA, starting with initial immune activation, followed by arterial infiltration, damage, and repair response. We propose the following general model of the pathophysiology of GCA: Overlapping patterns of activation in circulating PBMCs seen between GCA and PMR suggest that immune activation precedes vascular damage. Pathologic analysis suggests vascular damage initiates in the adventitial vasa vasorum microvasculature because inflammation is never restricted to intima (85). The initial trigger for vascular injury in GCA is unknown but appears to involve interactions between pathologically activated circulating cells, especially CD4+ helper T cells and monocytes, and multiple vascular cell types. Upon breach of vascular immunoprivilege, recruitment of these abnormal monocytes and CD4+ T cells, especially IFN-γ-producing Th1 cells, cooperate to mediate vascular injury and repair. The sequence of recruitment is also unknown, but once initiated, multiple interconnected positive feedback loops sustain it in the vasculature and also likely feedback to amplify systemic immune activation.
Immune Activation and Circulating Leukocytes
Systemic inflammation is a core feature of GCA and PMR that is largely driven by IL-6, with elevated plasma levels in both conditions (92, 93). IL-6 is a pleotropic cytokine professionally produced by monocytes, macrophages, and dendritic cells within the immune system—as well as by other cells including endothelial cells, VSMCs, fibroblasts, and B cells—as an early signal of tissue damage (94, 95). Monocytes appear to be the primary source of IL-6 among PBMCs of patients with GCA and PMR (93, 96), though the contribution from other non-circulating cell types has not been assessed and is likely significant. In the immune system, IL-6 has a key role in CD4+ helper T cell differentiation, promoting the development of Th17 and T follicular helper (TFH) cells, while inhibiting that of regulatory T cells (Treg) due to opposite effects of the IL-6-induced pioneering transcription factor STAT3 in the generation of these cell types during inflammation (94, 95, 97). In the liver, IL-6 stimulates production of acute phase response proteins including CRP and fibrinogen, with resultant elevation of ESR. IL-6 levels are tied closely to clinical symptoms of GCA and are higher in patients who experience more relapses, with levels rising concurrently with symptoms during relapse (93, 98). Consistent with the negative association between systemic inflammation and cranial symptoms, patients with higher serum IL-6 have fewer ischemic complications even during relapse (98–100). Whether this reflects a biologic difference or increased clinical detection remains unclear. Beyond IL-6, other cytokines are not reproducibly systemically elevated across studies; multiple studies have shown circulating TNF and IFN-γ levels are unchanged (15, 92, 93, 101).
Patients with GCA and PMR have abnormally activated PBMCs, particularly among CD4+ T cells, which are skewed toward effector cells. Although unchanged in number, polarization of CD4+ T cells is aberrant. Both conditions share increased frequency of IFN-γ + Th1 cells and a STAT3-activation pattern with increased IL-17+ Th17 cells and reduced Treg (92, 101–103); IL-21+ TFH cells are also elevated in GCA but untested in PMR. Th17 cells are stimulated by the cytokines IL-23 and IL-1β and are pathologically associated with multiple autoimmune diseases. Th1 cells develop downstream of STAT4-activating IL-12, which also stimulates their production of the signature cytokine IFN-γ, a well-known driver of granulomatous inflammation in infections such as M. tuberculosis (104, 105). In humans, the majority of TFH cells also respond to IL-12 and can co-produce IFN-γ, like Th1 cells; these cells accumulate in a multitude of inflammatory diseases and provide B cell help. T cell production of IL-21 can also enhance cytotoxicity of CD8+ T cells and NK cells (106). In GCA, in vitro culture of patient T cells with IL-21 further engenders more Th1 and Th17 differentiation (102). Beyond polarization, GCA and PMR share an increased frequency of senescent T cells (107). Studies in PMR are more limited, but GCA patients have other evidence of increased activation. These include a shift from central memory CD4+ T cells to effector memory and terminally differentiated effector memory cells; higher expression of HLA-DR and NOTCH1, which has a pleomorphic pro-inflammatory function in mature T cells; and a gene expression signature enriched for T cell receptor signaling (102, 108–110).
In comparison, other circulating lymphocytes appear to be less impacted, though comprehensive assessment using high-dimensional analyses is lacking. CD8+ T cells from GCA and PMR patients have increased oligoclonality, and many but not all studies report lower numbers; at least in GCA, they also express higher HLA-DR (102, 111–115). In some studies, there is a global reduction in the number of B cells, while NK cell numbers are unchanged (102, 116).
In the myeloid compartment, prominent changes include increased numbers of circulating monocytes and immature neutrophils. Monocytosis of classical CD14bright CD16lo cells is present in both GCA and PMR (93, 117). In GCA, these cells are phenotypically identical to healthy controls (118, 119). However, they are transcriptionally primed in circulation, expressing higher levels of pro-inflammatory cytokines IL6, IL1B, and IL-12/23 components IL23A (p19), IL12A (p35), and IL12B (p40) as well as extracellular matrix-degrading (ECM) gelatinases MMP2 and MMP9 (96, 101, 118). Surprisingly, left shift with increased circulating immature neutrophils was recently shown to be the major cellular difference by mass cytometry (CyToF) between untreated GCA patient and healthy control PBMCs (80). Collectively, these data suggest there is increased bone marrow myelopoiesis and/or recruitment in active disease. Whether PMR patients have the same transcriptional changes to monocytes or cellular distribution remains to be seen.
Initiating Arterial Inflammation
Two major challenges to understanding GCA pathogenesis are the absence of a commonly used mouse model and the lack of availability of sequential patient samples. Given these challenges, mechanistic insights rely on three human systems based on temporal artery: (1) observations from TAB; (2) manipulation of TAB or normal arteries in Matrigel (120); or (3) manipulation of TAB or normal arteries in chimeric mouse systems (121). The chimeric systems are the most complex and have evolved over time. One currently used system involves three sequential steps to produce inflammation, which may rely in part on alloreactivity (“subcutaneous-chimera”): (1) implantation of a human artery segment as a subcutaneous graft on the lower midback of a highly immunocompromised NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mouse that lacks all lymphocytes and has severely defective myeloid cells; (2) stimulation with lipopolysaccharide (LPS), activating xenograft vasDC to adopt a pro-inflammatory CD83+ CD86+ phenotype and produce T-cell recruiting chemokines; and (3) adoptive transfer of PBMCs from treatment-naïve allogeneic GCA patients, generating immune infiltration that histologically and transcriptionally resembles GCA (103, 121). Additional mechanistic insights can also be learned from another chimeric vascular allograft rejection model, where human coronary artery xenograft surgically replaces the mouse infrarenal aorta (“interposition-chimera”) (122). Similar to subcutaneous-chimeras, this model uses highly immunocompromised CB17.Cg-PrkdcscidLystbg−J/Crl mice that lack T and B cells through the same Prkdc mutation but differ in the mechanism of impaired NK and granulocyte function. After adoptive transfer of PBMCs from allogeneic blood donors, these mice develop xenograft vascular inflammation even in the absence of LPS over a similar time course. However, inflammation is more histopathologically similar to TAK, with prominent adventitial and intimal CD4+ and CD8+ T cell invasion and hyperplasia; unlike TAK and GCA, very few leukocytes invade the media, myeloid infiltration is rare, IEL are preserved, and neovascularization does not occur (123).
Arterial invasion requires activation of both circulating and vascular cells. Emphasizing the importance of pathogenic circulating cells, in subcutaneous-chimeras, normal human PBMCs cannot typically invade artery grafts even in the presence of LPS (109). Meanwhile, GCA-derived alloreactive T cells cannot invade artery grafts in the absence of LPS (121). Some of these effects may be caused by recruitment and duration of contact with the subcutaneous graft because normal human T cells—but not myeloid cells—can invade the interposition-chimera allograft without further stimulus downstream of endothelial antigen presentation to CD4+ and CD8+ T cells (122, 124). Interestingly, upon co-implantation of TAB fragments from GCA, PMR, or control patients into NSG mice, T cells recirculate from GCA arteries and invade PMR but not normal arteries in the absence of LPS, suggesting PMR vessels have lost immunoprivilege (121). PMR vasDC have a partially activated CD83+CD86− phenotype on TAB. However, whether vasDC cause the arterial leakiness in PMR or GCA vessels is unclear because adoptive transfer of GCA T cell alloreactive clones in subcutaneous-chimeras in the absence of LPS also induces a CD83+ vasDC phenotype, but T cells do not invade (121). Thus, while LPS stimulation can breach immunoprivilege in transplanted arterial sections via vasDC activation and perhaps prolonged contact with allogeneic T cells, this may not be the initiating event of vascular damage in GCA. These data also suggest—despite some overlapping phenotypes in PBMCs—that pathogenic differences occur between GCA and PMR cells that can facilitate entry into PMR primed vessels.
IFN-γ can independently break vascular immunoprivilege. Incubation of normal artery in Matrigel with IFN-γ induces VSMC expression of several chemokines including monocyte-recruiting CCL2 as well as Th1- and CD8+ T cell-recruiting CXCL9, CXCL10, and CXCL11 (125). It also induces VSMC ICAM-1 expression, an adhesion molecule that binds leukocyte integrins and, when expressed by endothelial cells, facilitates vascular transmigration. Remarkably, addition of healthy control PBMCs results in invasion of macrophages—but not healthy T cells—that subsequently become giant cells (125). Endothelial expression of HLA-DR is also induced by IFN-γ (122, 124), but whether pathogenic GCA PBMCs facilitate T cell entry has not been tested. This axis may also be important in early vascular injury in GCA, as TAB shows increased ICAM-1 expression by VSMCs in regions of structurally normal skip lesions and as well as by endothelial cells in the vasa vasorum (126, 127). Notably, other cytokines such as macrophage-derived TNF and IL-1β can also induce ICAM-1 expression on endothelial cells in vitro as well as enhance its upregulation by IFN-γ (128). Interestingly, IL1B is increased in PMR TAB compared to controls (129). Thus, local induction of cytokines from activated circulating cells may similarly prime segments of vasculature for inflammatory cell entry, though the mechanism of tissue tropism to the LV vasculature with this lens remains to be explored.
The Feed-Forward Inflammatory Infiltrate
On TAB, activated memory CD4+ T cells massively invade GCA arteries, where they polarize even further into effector cells compared to PBMC, homing mostly to the adventitial-medial border but present in all three layers. These express a broad repertoire of T cell receptors with a minimal degree of clonal expansion (130, 131). While a comprehensive assessment of infiltrating T cells is lacking, there are varying degrees of IFN-γ, IL-17, IL-21, and IL-9 produced. The balance of polarization differs between patients and is functionally relevant because cranial ischemic symptoms correspond to increased Th1 function on TAB (99, 132, 133). Indeed, ischemia positively correlations with: (1) the Th1 signature cytokine IFN-γ, (2) its activator IL-12p35, and (3) the downstream number of giant cells (99, 132, 133). In contrast, patients with higher expression of the Th17 signature cytokine IL-17A have fewer relapses and more systemic symptoms (134, 135). Consistent with this, in interposition-chimeras, IL-17 blockade does not impact intimal hyperplasia but does reduce IL6 (136). Though minimally described, TAB with RI also reflect different T cell composition. Compared to transmural inflammation, SVV has low levels of IL-17 and intermediate levels of IL-9 while VVV/ILA has the opposite pattern (135); the distribution of IFN-γ has not been described. VVV further lacks NOTCH1+ infiltrating T cells (109). Thus, T cell polarization differs between clinical and pathologic phenotypes, but how different signature cytokines affect pathogenesis largely remains to be explored.
Myeloid cells also diffusely infiltrate all three layers of the artery on TAB and densely populate granulomas around the IEL. These include three populations: a smaller CD16−CCR2+ CX3CR1− cells that produce IL-6 and IL-1ß and phenotypically resemble circulating monocytes; CD16+CCR2−CX3CR1+ macrophages that produce MMP9, MMP2, VEGF, and the potent mesenchymal mitogen PDGF; and giant cells that functionally overlap with CX3CR1+ macrophages but express the above effectors to an even greater degree by immunohistochemistry (96, 117, 137, 138). Other myeloid generated cytokines elevated in TAB that contribute to the pro-inflammatory environment include TNF, IL-12, and IL-23 (101, 139, 140). Co-culture of human peripheral blood monocytes with aortic adventitial fibroblasts induces their differentiation into macrophages that produce MMP9 (141). In other biologic conditions, various cytokines can stimulate macrophage fusion into giant cells—including IFNγ, IL-1β, and IL-6—but correlation between IFN-γ levels and number of giant cells on TAB suggest this is the primary mechanism in GCA (99, 142). Collectively, these data suggest monocytes that are transcriptionally primed to produce pro-inflammatory cytokines and gelatinases in circulation are recruited from the peripheral blood, differentiate in the inflamed vessel into macrophages, and further combine to form giant cells in response to IFN-γ. However, it is also possible that monocytes and macrophages are independently recruited to inflamed vessels from the circulation.
Multiple cell types generate positive feedback chemokine loops that enhance T cell and myeloid recruitment. In subcutaneous-chimeras, vasDC produce CCL19 and CCL21 as well as its receptor CCR7, trapping them in the artery upon activation (73). They also produce CCL20 and attract cells expressing CCR6, a phenotype shared by many infiltrating T cells on TAB as well as by Th17 and Th1/Th17 precursors in GCA patient peripheral blood (92, 143, 144). Notably, a variety of cells including DC, macrophages, Th17 cells, and VSMC can produce CCL20 and while it is overexpressed on TAB, the cellular source has not been shown (134, 136, 145). VSMC are a nexus for accentuating inflammatory signals: macrophage-generated TNF stimulates macrophage-attracting CX3CL1 in vitro; macrophage-expressed PDGF induces monocyte-recruiting CCL2 in Matrigel; Th17-produced IL-17 causes Th1/Th17-recruiting CCL20 in vitro and in interposition-chimeras; and Th1-derived IFN-γ provokes CX3CL1 plus Th1-, CD8+ T cell- and monocyte-recruiting chemokines, as previously described (125, 136, 146, 147). Finally, when co-cultured, fibroblasts induce monocyte expression of monocyte-recruiting CCL2 (141). Thus, upon entry of T cells and monocytes in the blood vessel, interactions with resident vascular cells perpetuate inflammation.
T cell interactions with other vascular cells also enhance inflammation. Endothelial cells in the vasa vasorum pathologically express Jagged1 on TAB. This can be experimentally reproduced in vitro by GCA plasma and mitigated by anti-VEGF, consistent with the increased systemic levels of VEGF in patients with GCA. In vitro and in subcutaneous-chimeras, Jagged1 ligates NOTCH1 expressed by circulating T cells and enhances their polarization to Th1 cells and, to a lesser extent, Th17 cells (103, 109). Upon entry into the vessel, T cells interact with vasDC. In normal artery specimens, these constitutively express PD-L1, a molecule that restrains PD-1+ T cells generated during chronic immune stimulation (148). In GCA TAB, vasDC upregulate antigen presentation machinery of HLA-DR, CD83, and CD86 but downregulate PD-L1; meanwhile, vascular invasive but not circulating T cells highly express PD-1 (73, 121, 148). Blocking PD-L1 in subcutaneous-chimeras results in exuberant inflammation, suggesting physiologic PD-1L+ vasDC restraint is lost in GCA immunopathology (148). Thus, inflammatory changes to other cell types augment the pathogenicity of pre-activated T cells.
Vascular Injury and Repair
Macrophages drive vascular injury largely via gelatinases. In normal vessels, VSMC constitutively produce pro-MMP2 and its inhibitor TIMP2 resulting in a quiescent vessel without proteolysis (149). With inflammation, macrophage- and giant cell-derived MMP2 and MMP9 outpace inhibitors, resulting in progressive degradation of ECM that is consequently more easily infiltrated by T cells (118, 149). Destruction occurs locally around macrophages as demonstrated by the restriction of MMP9 and proteolysis to the adventitia in ILA on TAB (118). Giant cells are gelatinase factories and, taking residence along the IEL, cleave and destroy it. The mechanisms that drive VSMC laminar necrosis are poorly described, but likely also involve myeloid mediators because, like IEL degradation, it does not occur in interposition-chimeras that lack myeloid recruitment (122, 123).
Macrophages and Th1 inflammation launch vascular remodeling, resulting in intimal hypertrophy and neovascularization. In response to a variety of mitogenic signals in Matrigel but most robustly to PDGF, healthy contractile VSMC become proliferative, invasive myointimal cells (146). These leave the media and invade the intima where they produce the vascular ECM proteins collagen I and III, generating the hypertrophic neointima (138, 146). TAB levels of PDGF and IFN-γ correspond to the degree of intimal hyperplasia, which in turn correlates with ischemic symptoms as the macrolumen becomes progressively stenotic (138). While macrophages and giant cells at the media/intimal border both produce PDGF, recombinant IFN-γ can also directly stimulate VSMC to produce PDGF as well as upregulate its receptor in interposition-chimeras that lack PBMC adoptive transfer, resulting in neointimal hyperplasia even in the absence of cellular infiltration (150). Both macrophage and T cell pathways are likely active in GCA. Patients with ischemic symptoms also have higher plasma levels of endothelin 1 (ET-1), a potent vasoconstrictor physiologically generated by endothelial cells. Interestingly, ET-1 is expressed by infiltrating immune cells on TAB and can redundantly generate intimal-invasive myointimal cells from VSMC in Matrigel (151). The degree of intimal hyperplasia further correlates with the degree of neovascularization in the intima and media, and in turn, to levels of VEGF on TAB, suggesting this process is driven by hypoxia (152). However, neovascularization co-localizes with macrophage- and giant cell-rich areas on TAB (152). Thus, the extent of neovascularization likely reflects the degree of macrophage and giant cell activation through multiple mechanisms including their production of VEGF. Collectively, vascular remodeling results in thickened blood vessels that cause symptomatic ischemia and generates a conduit for further inflammatory cell entry through leaky neovasculature (127).
Treatment Effects
Glucocorticoids are the standard therapy for GCA and PMR. Consistent with the need for higher doses in GCA than PMR, systemic changes occur first while local changes seen in TAB generally take much longer. Plasma IL-6 is strongly inhibited after a single dose of GC, but the median time to normalization is 4 weeks (93). Though systematic sequential immunophenotyping of PBMCs during treatment has not been reported, B cells appear to be the first to respond and normalize after 2 weeks, a time course consistent with changes in mobilization (116). After 3 months of treatment, Th17 cell frequency, and CD4+ but not CD8+ T cell HLA-DR expression return to normal (92, 101, 102). Monocyte numbers are also reduced at this time but remain higher than healthy controls (117). Furthermore, after 3–9 months of treatment, monocyte expression of IL6 and Th17-activating IL1B and IL23A normalize while expression of Th1-inducing and activating IL12A and IL12B remain elevated (101). Consistent with this, among CD4+ T cells, Th1 take longer to respond, normalizing with full disease remission (101, 102). Finally, CD8+ T cell numbers take up to 2 years to return to baseline numbers (111). Indeed, in patients diagnosed with GCA for at least 2 years, increased circulating CD4+ T cells, reduced CD8+ T cells, and the corresponding increased CD4+/CD8+ ratio but not inflammatory markers or monocytes numbers have recently been shown to be associated with thoracic aortic dilatation compared to controls (22).
Reports of TAB re-biopsy after GC treatment reveal similar results to PBMCs with initial control of Th17 pathways, and later reduction in Th1 pathways, as well as a prolonged timecourse of vascular healing. Compared to TAB with active GCA, re-biopsies at 3-9 months phenocopy peripheral blood and show profound reduction in IL6, IL1B, IL23A, and IL17 while IL12A IL12B, and INFG are unchanged (101). In another study, patients with paired re-biopsy at 1 year demonstrated a global reduction in all tested cytokines including IL1B, IL6, IL23A, IL12A, IL12B, and IFNG as well as MMP9, though patients with more relapses showed higher levels of IL12B and IFNG (139). Consistent with this prolonged time course, a prospective study of 40 patients re-biopsied at 3, 6, 9, and 12 months found active arteritis in 7/10, 9/12, 4/9, and 4/9 samples, respectively, despite normalization of inflammatory markers and clinical symptoms. There was also a time-dependent increase in vascular remodeling (153). Thus, GC quickly control Th17 signatures in circulation and TAB, likely reflecting loss of STAT3 activation from monocyte-derived IL-6. Meanwhile, Th1 pathway takes longer to respond, consistent with prolonged monocyte production of STAT4-activating IL-12, which may drive relapse and ongoing vasculitis in some individuals. Finally, vascular remodeling continues after active inflammation resolves, like the prolonged FDG signal on PET imaging.
Multiple other treatment modalities have been tested for GCA in randomized clinical trials. In the GiACTA trial, targeted blockade of IL-6R with TCZ demonstrated superiority to a course of GC alone in achieving steroid-free remission at 1 year, as defined by lack of clinical flare and normal level of IL-6-induced CRP, becoming the first non-steroid FDA-approved treatment for GCA (11). Interestingly, fewer patients with relapsing disease responded to TCZ than patients with untreated disease, raising the possibility that these patients may have more Th1 driven disease. Furthermore, one patient in the TCZ every-other-week arm developed the ischemic complication of anterior ischemic optic neuropathy. In another study, a patient with highly active disease that normalized on TCZ—but who died unrelated to GCA after 6 months of therapy—had widely active vasculitis on autopsy (154). Though GCA-related adverse events were not statistically different between groups in GiACTA, these raise the question if TCZ controls vascular-level inflammation or if it blocks systemic manifestations of flare, which may further differ between newly diagnosed and relapsed patients.
Further insights to this question are suggested by the differing results of the two open-label studies of anti-p40 ustekinumab. In the initial promising Irish trial, all patients recruited had relapsing disease and ustekinumab was successful in achieving GC-reduction without flare, albeit with persistent low GC dose in the majority of patients. Notably, of 10 patients with LV disease in this study, eight underwent reimaging by CT angiography, which demonstrated not only a halt to further vascular damage but improvement of wall thickening in all patients and complete resolution in four (49). In the American trial, both newly diagnosed and relapsing patients were recruited and all patients were required to end GC at 6 months, resulting in clinical flare across the majority of patients with elevated inflammatory markers and PMR symptoms; though data was not shown for relapse between newly diagnosed vs/ relapsing patients they were stated not to be different (50). Vascular imaging follow up was not reported (50). Consistent with molecular studies, these differing trial results suggest a degree of independence between systemic symptoms of flare downstream of IL-6 and vascular damage in relapsing patients downstream of IL-12. While GC controls both endotypes, targeted therapies directed at either can fail; however, only low dose GC may be required to control the IL-6 axis, as in PMR, at least in patients with relapsing disease. These data also emphasize that long-term follow up of TCZ-treated patients and further clinicopathologic correlation will be important and the utility of a randomized trial for ustekinumab in GCA patients with relapsing disease. Similar to GC, blocking STAT activation directly with JAK inhibitors would allow combinatorial blockade of IL-6 and IL-12/23 without GC side effects and is theoretically compelling. Indeed, multiple JAK inhibitors are currently in clinical trials (12).
Beyond IL-6, another potential emerging treatment is to target T cell overactivation directly. Indeed, abatacept (CTLA-4:Fc) was superior in achieving relapse-free survival at 1 year in a phase 2 trial (155). Other targeted therapies using TNF blockade with infliximab, adalimumab, or etanercept have been ineffective (156–158).
Comparison to TAK and CIA
Due to relative lack of tissue compared to GCA, less is known about the immunopathology of TAK, and that of the more recently-described entity CIA remains virtually unexplored. Consistent with overlapping but distinct pathology, TAK shares several features in common with GCA but differs in cytotoxic mediators (Figure 2). Like GCA, changes to circulating inflammatory cells also reflect those in the tissue. Systemically, patients with TAK share elevated systemic levels of IL-6 and increased circulating classical monocytes, Th17 cells, and Th1 cells with fewer Treg (159–161). In tissue, memory CD4+ T cells—including Th1 and Th17 subsets—are likewise the most prevalent invasive cell type, with equal macrophage infiltration between conditions (48, 162). Interestingly, in the opposite pattern of GCA, peripheral Th1 cells respond better to steroids than Th17 cells, which remain elevated despite clinical remission (162). Unlike GCA, patients with TAK also have elevated systemic TNF and consistent with this, TNF inhibitors are at least modestly clinically effective (161, 163). The major difference between TAK and GCA is among non-CD4+ lymphocytes, as B cells and CD8+ T cells are elevated in peripheral blood and tissue. As suggested by genetic HLA class I associations, CD8+ T cells seem particularly relevant, rising in circulation during flares and found actively killing vascular cells on electron microscopy (91, 159). Interestingly, GCA patients with relatively higher levels of CD8+ T cell invasion also have more severe disease, though in this condition it may also reflect the degree of Th1 inflammation given mutual dependence of CD8+ T cells on the positive-feedback IFN-γ-CXCR3 recruitment loop (114).
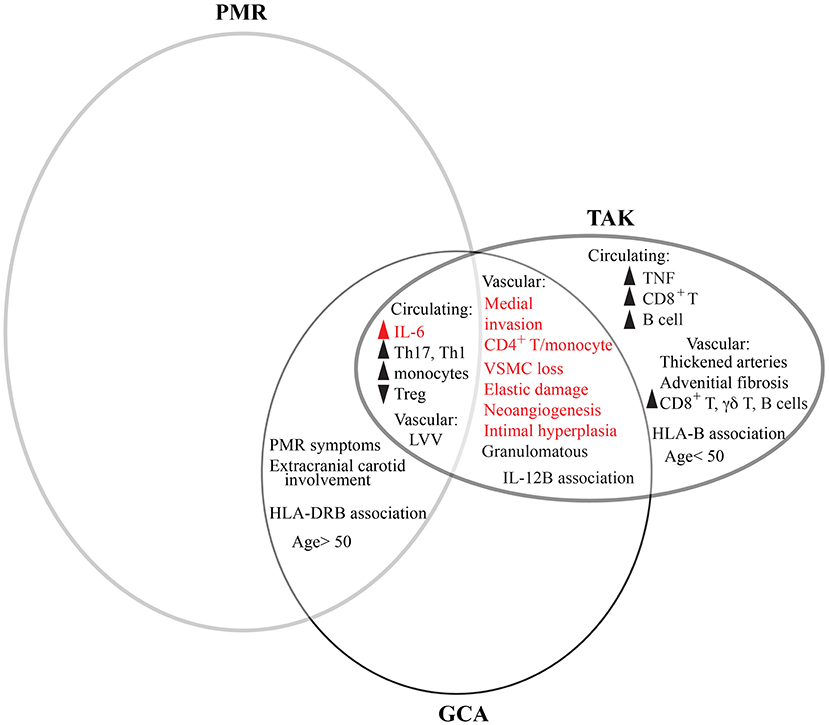
Figure 2. Comparative features between GCA, PMR, and TAK. PMR and GCA are overlapping clinical entities as patients can concurrently have both conditions. However, more patients with GCA have PMR than do patients with PMR have GCA. Meanwhile, GCA and TAK share features of vascular inflammation in large vessels. PMR, GCA, and TAK all share elevated levels of IL-6 and changes to circulating immune cells. However, distinguishing TAK from GCA are some circulating lymphocytes and cytokines, vascular pathology, and patient age. Many features are common to other types of vascular disease, such as AAA (red). There is not currently enough information to determine the extent to which CIA overlaps with other conditions.
Perspectives and Future Directions
GCA is a complex disease because it lies at the interface of two clinical spectra—the pathologically similar granulomatous vasculitides and the clinically overlapping GCA and PMR—each of which have historically been imprecisely defined based on clinical phenotypes and therefore often overlap in the literature (Figure 2). Additionally, emerging results from advanced imaging and pathologic analysis show two additional spectra—LV- and cranial-GCA and histologic RI—that demonstrate even greater overlap with PMR than previously recognized. Compounding this complexity is the clinical need to treat GCA emergently and the recent transition from pathology to imaging for diagnosis, which respectively limit the availability of untreated patient PBMCs and tissue specimens for research.
Despite phenotypic similarities between TAK and GCA, the multiple differences between affected patients—in age, demographics, vascular distribution, genetics, histopathology, and immunophenotype—suggest that these are distinct disease entities with some degree of convergence (Figure 2). Furthermore, most shared features between TAK and GCA are not unique to vasculitis. In fact, the common vascular condition of abdominal aortic aneurysm (AAA), a permanent dilatation to the aorta that affects 1–2% of men age 65 and 0.5% of women age 70, shares most features: elevated systemic levels of IL-6; monocytosis; medial invasion of memory CD4+ T cell and macrophages; and vascular remodeling with dissolution of the elastic lamellae, loss of VSCM, and neoangiogenesis (164–169) (Figure 2). This suggests despite different triggers of vascular injury, many pathways of arterial damage converge, though some differences persist and may inform our understanding of disease mechanisms. For example, granulomatous inflammation likely reflects the higher vascular IFN-γ and macrophage invasion in GCA, CIA, and TAK compared to AAA, while changes to circulating T cells reflect higher systemic levels of IL-6 (164, 167, 169, 170). Likewise, the prominent fibrosis in TAK appears to be an important distinction and may represent a novel disease target. Interestingly, the comparison of AAA is particularly relevant for GCA as they share several other epidemiologic features, including old age with rare incidence below age 50, increased prevalence in Northern Europe, and smoking as a core risk factor for aneurysm development (24, 166). Thus, these unexplained risk factors in GCA may represent common mechanisms of vascular risk.
The heterogeneous and overlapping patterns of pathologic RI, LV-involvement, and PMR with GCA remain a mystery. One possibility is that RI and PMR represent more subtle degrees of vascular injury that jointly affect the microvasculature, including that of large arteries. In some patients who experience an unknown stimulus, this may progress to more fulminant disease. Supporting this, a recent report demonstrated a key role for NOTCH3 signaling between the arterial microvasculature and synovial sublining fibroblasts to generate synovitis in rheumatoid arthritis (171). Indeed, microvascular endothelial cells upregulate the NOTCH3 ligand Jagged1 in GCA, though whether this also occurs in PMR synovitis has not been tested (103). Furthermore, subtle microvascular changes may explain the ability of GCA T cells to recirculate into PMR arteries in early experiments (121). Alternatively, the regulatory logic of CD4+ T cells may differ in LV-GCA and/or RI. For example, several lines of evidence suggest that a Th1 signature favors vascular damage and ischemia. Mechanistically, this is an especially feed-forward module in the vasculature through cyclical recruitment of Th1, myeloid cells, and CD8+ T cells that ultimately propagates stenotic tissue remodeling through macrophage activation and giant cell formation. However, the role of other helper T cell modules such as Th17 in GCA is less clear—despite the evidence that they are also present systemically and in vascular tissue. Though it is possible circulating Th17 and TFH cells may simply represent off-target STAT3 activation of IL-6, another possibility is that this module corresponds more to LV inflammation. Supporting this, patients with LV disease typically have more systemic symptoms, as do patients with increased IL-17 on TAB. Furthermore, mice lacking the Rac activator Def6, a negative regulator of IRF4, spontaneously develop granulomatous aortitis due to aberrant T cell production of IL-21 and IL-17 (172). Given the more recent emphasis on LV-GCA, radio-pathologic correlation has not yet been performed but would be interesting. Comprehensive assessment of the immune infiltrate in RI is more easily achieved.
Since the discovery of GCA, the trigger for vascular inflammation has been questioned. Here, we propose a model where systemic changes in the circulation precede vascular injury and are required for disease initiation. In this model, systemic activation likely initiates in myeloid cells—perhaps monocytes—leads to circulating CD4+ T cell polarization downstream of the pioneering transcription factors STAT3 and STAT4. However, recently published data suggests myeloid activation may be even further upstream, as early as the bone marrow, given the prominent left shift seen by CyToF in patients with untreated GCA (80). Supporting this, GCA can occur as a paraneoplastic phenomenon to myelodysplastic/ myeloproliferative neoplasms as well as in the recently described somatic, myeloid-activating autoinflammatory condition VEXAS (173, 174). Upon breach of vascular immunoprivilege, pre-activated monocytes and CD4+ T cells mutually enter the vessel and cooperate to destroy it. In chimeric systems, T cell invasion likely relies on allorecognition, but in GCA, HLA associations suggest an antigenic driver that is thus far elusive and may describe tissue tropism to the microvasculature of large vessels. With GC treatment, the IL-6-STAT3 axis regulating systemic symptoms is more quickly controlled while IL-12-STAT4 axis mediating vascular damage, at least in TAB, requires prolonged treatment, consistent with the different time course needed to control these cytokines in circulating monocytes. In the absence of IL-6, Th1 cells may paradoxically initially increase as the cytokine microenvironment favors their generation and persistence (105), consistent with the weaker performance of TCZ in relapsed patients and potentially better performance by ustekinumab (11, 49). Whether flares represent reemergence of abnormally activated circulating myeloid cells, lack of control in the vessel, or some other unexpected mechanism is unclear, but comprehensive longitudinal phenotyping is likely to be informative to this end, as recently described in rheumatoid arthritis (175). In the future, integration of such longitudinal data with imaging will be particularly useful to define clinically relevant entities such as persistent subacute inflammation, flare, and remission. Ultimately, clinical trials of various immune modulators in patients with GCA will provide further insights into proposed disease mechanisms and should include dual assessment of clinical flare as well as vascular damage.
Author Contributions
MR, DR, and PM wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by NIAMS/NIH K08 AR072791 and Burroughs Wellcome Fund Career Award in Medical Sciences (to DR), and NIAMS/NIH U54-AR057319 (PM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum. (2013) 65:1–11. doi: 10.1002/art.37715
2. Dejaco C, Duftner C, Buttgereit F, Matteson EL, Dasgupta B. The spectrum of giant cell arteritis and polymyalgia rheumatica: revisiting the concept of the disease. Rheumatology. (2017) 56:506–15. doi: 10.1093/rheumatology/kew273
3. Brack A, Martinez-Taboada V, Stanson A, Goronzy JJ, Weyand CM. Disease pattern in cranial and large-vessel giant cell arteritis. Arthritis Rheum. (1999) 42:311–7. doi: 10.1002/1529-0131(199902)42:2<311::AID-ANR14>3.0.CO;2-F
4. Ostberg G. Morphological changes in the large arteries in polymyalgia arteritica. Acta Med Scand Suppl. (1972) 533:135–59. doi: 10.1111/j.0954-6820.1972.tb15615.x
5. Klein RG, Hunder GG, Stanson AW, Sheps SG. Large artery involvement in giant cell (temporal) arteritis. Ann Intern Med. (1975) 83:806–12. doi: 10.7326/0003-4819-83-6-806
6. Koster MJ, Matteson EL, Warrington KJ. Large-vessel giant cell arteritis: diagnosis, monitoring and management. Rheumatology. (2018) 57:ii32–42. doi: 10.1093/rheumatology/kex424
7. Hunder GG, Bloch DA, Michel BA, Stevens MB, Arend WP, Calabrese LH, et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. (1990) 33:1122–8. doi: 10.1002/art.1780330810
8. Dasgupta B, Cimmino MA, Kremers HM, Schmidt WA, Schirmer M, Salvarani C, et al. 2012 Provisional classification criteria for polymyalgia rheumatica: a European League Against Rheumatism/American College of Rheumatology collaborative initiative. Arthritis Rheum. (2012) 64:943–54. doi: 10.1002/art.34356
9. Mahr AD, Jover JA, Spiera RF, Hernandez-Garcia C, Fernandez-Gutierrez B, Lavalley MP, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum. (2007) 56:2789–97. doi: 10.1002/art.22754
10. Villiger PM, Adler S, Kuchen S, Wermelinger F, Dan D, Fiege V, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. (2016) 387:1921–7. doi: 10.1016/S0140-6736(16)00560-2
11. Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med. (2017) 377:317–28. doi: 10.1056/NEJMoa1613849
12. Cid MC, Rios-Garces R, Terrades-Garcia N, Espigol-Frigole G. Treatment of giant-cell arteritis: from broad spectrum immunosuppressive agents to targeted therapies. Rheumatology. (2020) 59:iii17–27. doi: 10.1093/rheumatology/kez645
13. Arend WP, Michel BA, Bloch DA, Hunder GG, Calabrese LH, Edworthy SM, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum (1990) 33:1129–34. doi: 10.1002/art.1780330811
14. Cinar I, Wang H, Stone JR. Clinically isolated aortitis: pitfalls, progress, and possibilities. Cardiovasc Pathol. (2017) 29:23–32. doi: 10.1016/j.carpath.2017.04.003
15. Hernandez-Rodriguez J, Segarra M, Vilardell C, Sanchez M, Garcia-Martinez A, Esteban MJ, et al. Elevated production of interleukin-6 is associated with a lower incidence of disease-related ischemic events in patients with giant-cell arteritis: angiogenic activity of interleukin-6 as a potential protective mechanism. Circulation. (2003) 107:2428–34. doi: 10.1161/01.CIR.0000066907.83923.32
16. O'Neill L, McCormick J, Gao W, Veale DJ, McCarthy GM, Murphy CC, et al. Interleukin-6 does not upregulate pro-inflammatory cytokine expression in an ex vivo model of giant cell arteritis. Rheumatol Adv Pract. (2019) 3:rkz011. doi: 10.1093/rap/rkz011
17. Birkhead NC, Wagener HP, Shick RM. Treatment of temporal arteritis with adrenal corticosteroids; results in fifty-five cases in which lesion was proved at biopsy. J Am Med Assoc. (1957) 163:821–7. doi: 10.1001/jama.1957.02970450023007
18. Wagener HP, Hollenhorst RW. The ocular lesions of temporal arteritis. Am J Ophthalmol. (1958) 45:617–30. doi: 10.1016/0002-9394(58)92165-2
19. Lensen KD, Voskuyl AE, Comans EF, van der Laken CJ, Smulders YM. Extracranial giant cell arteritis: a narrative review. Neth J Med. (2016) 74:182–92.
20. Prieto-Pena D, Martinez-Rodriguez I, Loricera J, Banzo I, Calderon-Goercke M, Calvo-Rio V, et al. Predictors of positive (18)F-FDG PET/CT-scan for large vessel vasculitis in patients with persistent polymyalgia rheumatica. Semin Arthritis Rheum. (2019) 48:720–7. doi: 10.1016/j.semarthrit.2018.05.007
21. Walz-Leblanc BA, Ameli FM, Keystone EC. Giant cell arteritis presenting as limb claudication. Report and review of the literature. J Rheumatol. (1991) 18:470–2.
22. Jud P, Verheyen N, Dejaco C, Haas E, Szolar D, Meinitzer A, et al. Prevalence and prognostic factors for aortic dilatation in giant cell arteritis - a longitudinal study. Semin Arthritis Rheum. (2020). doi: 10.1016/j.semarthrit.2020.11.003. [Epub ahead of print].
23. Mackie SL, Hensor EM, Morgan AW, Pease CT. Should I send my patient with previous giant cell arteritis for imaging of the thoracic aorta? A systematic literature review and meta-analysis. Ann Rheum Dis. (2014) 73:143–8. doi: 10.1136/annrheumdis-2012-202145
24. Koster MJ, Crowson CS, Labarca C, Warrington KJ. Incidence and predictors of thoracic aortic damage in biopsy-proven giant cell arteritis. Scand J Rheumatol. (2020). doi: 10.1080/03009742.2020.1786855. [Epub ahead of print].
25. Camellino D, Giusti A, Girasole G, Bianchi G, Dejaco C. Pathogenesis, diagnosis and management of polymyalgia rheumatica. Drugs Aging. (2019) 36:1015–26. doi: 10.1007/s40266-019-00705-5
26. Hysa E, Sobrero A, Camellino D, Rumi F, Carrara G, Cutolo M, et al. A seasonal pattern in the onset of polymyalgia rheumatica and giant cell arteritis? A systematic review and meta-analysis. Semin Arthritis Rheum. (2020) 50:1131–9. doi: 10.1016/j.semarthrit.2020.05.023
27. Salvarani C, Crowson CS, O'Fallon WM, Hunder GG, Gabriel SE. Reappraisal of the epidemiology of giant cell arteritis in Olmsted County, Minnesota, over a fifty-year period. Arthritis Rheum. (2004) 51:264–8. doi: 10.1002/art.20227
28. Svensson LG, Arafat A, Roselli EE, Idrees J, Clifford A, Tan C, et al. Inflammatory disease of the aorta: patterns and classification of giant cell aortitis, Takayasu arteritis, and nonsyndromic aortitis. J Thorac Cardiovasc Surg. (2015) 149:S170–5. doi: 10.1016/j.jtcvs.2014.08.003
29. Kermani TA. Takayasu arteritis and giant cell arteritis: are they a spectrum of the same disease? Int J Rheum Dis. (2019) 1(22 Suppl.):41–8. doi: 10.1111/1756-185X.13288
30. de Boysson H, Daumas A, Vautier M, Parienti JJ, Liozon E, Lambert M, et al. Large-vessel involvement and aortic dilation in giant-cell arteritis. A multicenter study of 549 patients. Autoimmun Rev. (2018) 17:391–8. doi: 10.1016/j.autrev.2017.11.029
31. Smith CA, Fidler WJ, Pinals RS. The epidemiology of giant cell arteritis. Report of a ten-year study in Shelby County, Tennessee. Arthritis Rheum. (1983) 26:1214–9. doi: 10.1002/art.1780261007
32. Kobayashi S, Yano T, Matsumoto Y, Numano F, Nakajima N, Yasuda K, et al. Clinical and epidemiologic analysis of giant cell (temporal) arteritis from a nationwide survey in 1998 in Japan: the first government-supported nationwide survey. Arthritis Rheum. (2003) 49:594–8. doi: 10.1002/art.11195
33. Mader TH, Werner RP, Chamberlain DG, Doornbos D. Giant cell arteritis in Alaska Natives. Can J Ophthalmol. (2009) 44:53–6. doi: 10.3129/i08-164
34. Gonzalez EB, Varner WT, Lisse JR, Daniels JC, Hokanson JA. Giant-cell arteritis in the southern United States. An 11-year retrospective study from the Texas Gulf Coast. Arch Intern Med. (1989) 149:1561–5. doi: 10.1001/archinte.149.7.1561
35. Garrity ST, Pistilli M, Vaphiades MS, Richards NQ, Subramanian PS, Rosa PR, et al. Ophthalmic presentation of giant cell arteritis in African-Americans. Eye. (2017) 31:113–8. doi: 10.1038/eye.2016.199
36. Gruener AM, Poostchi A, Carey AR, Eberhart CG, Henderson AD, Chang JR, et al. Association of giant cell arteritis with race. JAMA Ophthalmol. (2019) 137:1175–9. doi: 10.1001/jamaophthalmol.2019.2919
37. Chung SH, Morcos MB, Ng B. Determinants of positive temporal artery biopsies in the veterans health administration national database cohort. Arthritis Care Res. (2020) 72:699–704. doi: 10.1002/acr.23897
38. Williams JN, Ford CL, Morse M, Feldman CH. Racial disparities in rheumatology through the lens of critical race theory. Rheum Dis Clin North Am. (2020) 46:605–12. doi: 10.1016/j.rdc.2020.07.001
39. Zaldivar Villon MLF, de la Rocha JAL, Espinoza LR. Takayasu arteritis: recent developments. Curr Rheumatol Rep. (2019) 21:45. doi: 10.1007/s11926-019-0848-3
40. Pacini D, Leone O, Turci S, Camurri N, Giunchi F, Martinelli GN, et al. Incidence, etiology, histologic findings, and course of thoracic inflammatory aortopathies. Ann Thorac Surg. (2008) 86:1518–23. doi: 10.1016/j.athoracsur.2008.07.039
41. Weyand CM, Hicok KC, Hunder GG, Goronzy JJ. The HLA-DRB1 locus as a genetic component in giant cell arteritis. Mapping of a disease-linked sequence motif to the antigen binding site of the HLA-DR molecule. J Clin Invest. (1992) 90:2355–61. doi: 10.1172/JCI116125
42. Martinez-Taboda VM, Bartolome MJ, Lopez-Hoyos M, Blanco R, Mata C, Calvo J, et al. HLA-DRB1 allele distribution in polymyalgia rheumatica and giant cell arteritis: influence on clinical subgroups and prognosis. Semin Arthritis Rheum. (2004) 34:454–64. doi: 10.1016/j.semarthrit.2003.12.001
43. Weyand CM, Hunder NN, Hicok KC, Hunder GG, Goronzy JJ. HLA-DRB1 alleles in polymyalgia rheumatica, giant cell arteritis, and rheumatoid arthritis. Arthritis Rheum. (1994) 37:514–20. doi: 10.1002/art.1780370411
44. Haworth S, Ridgeway J, Stewart I, Dyer PA, Pepper L, Ollier W. Polymyalgia rheumatica is associated with both HLA-DRB1*0401 and DRB1*0404. Br J Rheumatol. (1996) 35:632–5. doi: 10.1093/rheumatology/35.7.632
45. Saruhan-Direskeneli G, Hughes T, Aksu K, Keser G, Coit P, Aydin SZ, et al. Identification of multiple genetic susceptibility loci in Takayasu arteritis. Am J Hum Genet. (2013) 93:298–305. doi: 10.1016/j.ajhg.2013.05.026
46. Carmona FD, Mackie SL, Martin JE, Taylor JC, Vaglio A, Eyre S, et al. A large-scale genetic analysis reveals a strong contribution of the HLA class II region to giant cell arteritis susceptibility. Am J Hum Genet. (2015) 96:565–80. doi: 10.1016/j.ajhg.2015.02.009
47. Carmona FD, Coit P, Saruhan-Direskeneli G, Hernandez-Rodriguez J, Cid MC, Solans R, et al. Analysis of the common genetic component of large-vessel vasculitides through a meta-Immunochip strategy. Sci Rep. (2017) 7:43953. doi: 10.1038/srep46012
48. Kurata A, Saito A, Hashimoto H, Fujita K, Ohno SI, Kamma H, et al. Difference in immunohistochemical characteristics between Takayasu arteritis and giant cell arteritis: it may be better to distinguish them in the same age. Mod Rheumatol. (2019) 29:992–1001. doi: 10.1080/14397595.2019.1570999
49. Conway R, O'Neill L, Gallagher P, McCarthy GM, Murphy CC, Veale DJ, et al. Ustekinumab for refractory giant cell arteritis: a prospective 52-week trial. Semin Arthritis Rheum. (2018) 48:523–8. doi: 10.1016/j.semarthrit.2018.04.004
50. Matza MA, Fernandes AD, Stone JH, Unizony SH. Ustekinumab for the treatment of giant cell arteritis. Arthritis Care Res. (2020). doi: 10.1002/acr.24378. [Epub ahead of print].
51. Dejaco C, Ramiro S, Duftner C, Besson FL, Bley TA, Blockmans D, et al. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice. Ann Rheum Dis. (2018) 77:636–43. doi: 10.1136/annrheumdis-2017-212649
52. Duftner C, Dejaco C, Sepriano A, Falzon L, Schmidt WA, Ramiro S. Imaging in diagnosis, outcome prediction and monitoring of large vessel vasculitis: a systematic literature review and meta-analysis informing the EULAR recommendations. RMD Open. (2018) 4:e000612. doi: 10.1136/rmdopen-2017-000612
53. Sammel AM, Hsiao E, Schembri G, Nguyen K, Brewer J, Schrieber L, et al. Diagnostic accuracy of positron emission tomography/computed tomography of the head, neck, and chest for giant cell arteritis: a prospective, double-blind, cross-sectional study. Arthritis Rheumatol. (2019) 71:1319–28. doi: 10.1002/art.40864
54. Lariviere D, Benali K, Coustet B, Pasi N, Hyafil F, Klein I, et al. Positron emission tomography and computed tomography angiography for the diagnosis of giant cell arteritis: a real-life prospective study. Medicine. (2016) 95:e4146. doi: 10.1097/MD.0000000000004146
55. Klink T, Geiger J, Both M, Ness T, Heinzelmann S, Reinhard M, et al. Giant cell arteritis: diagnostic accuracy of MR imaging of superficial cranial arteries in initial diagnosis-results from a multicenter trial. Radiology. (2014) 273:844–52. doi: 10.1148/radiol.14140056
56. Rheaume M, Rebello R, Pagnoux C, Carette S, Clements-Baker M, Cohen-Hallaleh V, et al. High-resolution magnetic resonance imaging of scalp arteries for the diagnosis of giant cell arteritis: results of a prospective cohort study. Arthritis Rheumatol. (2017) 69:161–8. doi: 10.1002/art.39824
57. Blockmans D, Maes A, Stroobants S, Nuyts J, Bormans G, Knockaert D, et al. New arguments for a vasculitic nature of polymyalgia rheumatica using positron emission tomography. Rheumatology. (1999) 38:444–7. doi: 10.1093/rheumatology/38.5.444
58. Blockmans D, de Ceuninck L, Vanderschueren S, Knockaert D, Mortelmans L, Bobbaers H. Repetitive 18F-fluorodeoxyglucose positron emission tomography in giant cell arteritis: a prospective study of 35 patients. Arthritis Rheum. (2006) 55:131–7. doi: 10.1002/art.21699
59. Blockmans D, De Ceuninck L, Vanderschueren S, Knockaert D, Mortelmans L, Bobbaers H. Repetitive 18-fluorodeoxyglucose positron emission tomography in isolated polymyalgia rheumatica: a prospective study in 35 patients. Rheumatology. (2007) 46:672–7. doi: 10.1093/rheumatology/kel376
60. Yamashita H, Kubota K, Takahashi Y, Minaminoto R, Morooka M, Ito K, et al. Whole-body fluorodeoxyglucose positron emission tomography/computed tomography in patients with active polymyalgia rheumatica: evidence for distinctive bursitis and large-vessel vasculitis. Mod Rheumatol. (2012) 22:705–11. doi: 10.3109/s10165-011-0581-x
61. Rehak Z, Sprlakova-Pukova A, Kazda T, Fojtik Z, Vargova L, Nemec P. (18)F-FDG PET/CT in polymyalgia rheumatica-a pictorial review. Br J Radiol. (2017) 90:20170198. doi: 10.1259/bjr.20170198
62. Lavado-Perez C, Martinez-Rodriguez I, Martinez-Amador N, Banzo I, Quirce R, Jimenez-Bonilla J, et al. (18)F-FDG PET/CT for the detection of large vessel vasculitis in patients with polymyalgia rheumatica. Rev Esp Med Nucl Imagen Mol. (2015) 34:275–81. doi: 10.1016/j.remn.2015.05.011
63. Prieto-Gonzalez S, Depetris M, Garcia-Martinez A, Espigol-Frigole G, Tavera-Bahillo I, Corbera-Bellata M, et al. Positron emission tomography assessment of large vessel inflammation in patients with newly diagnosed, biopsy-proven giant cell arteritis: a prospective, case-control study. Ann Rheum Dis. (2014) 73:1388–92. doi: 10.1136/annrheumdis-2013-204572
64. Nielsen BD, Gormsen LC, Hansen IT, Keller KK, Therkildsen P, Hauge EM. Three days of high-dose glucocorticoid treatment attenuates large-vessel 18F-FDG uptake in large-vessel giant cell arteritis but with a limited impact on diagnostic accuracy. Eur J Nucl Med Mol Imaging. (2018) 45:1119–28. doi: 10.1007/s00259-018-4021-4
65. Banerjee S, Quinn KA, Gribbons KB, Rosenblum JS, Civelek AC, Novakovich E, et al. Effect of treatment on imaging, clinical, and serologic assessments of disease activity in large-vessel vasculitis. J Rheumatol. (2020) 47:99–107. doi: 10.3899/jrheum.181222
66. Henes JC, Muller M, Krieger J, Balletshofer B, Pfannenberg AC, Kanz L, et al. [18F] FDG-PET/CT as a new and sensitive imaging method for the diagnosis of large vessel vasculitis. Clin Exp Rheumatol. (2008) 26:S47–52.
67. Sammel AM, Hsiao E, Schembri G, Bailey E, Nguyen K, Brewer J, et al. Cranial and large vessel activity on positron emission tomography scan at diagnosis and 6 months in giant cell arteritis. Int J Rheum Dis. (2020) 23:582–8. doi: 10.1111/1756-185X.13805
68. Grayson PC, Alehashemi S, Bagheri AA, Civelek AC, Cupps TR, Kaplan MJ, et al. (18) F-fluorodeoxyglucose-positron emission tomography as an imaging biomarker in a prospective, longitudinal cohort of patients with large vessel vasculitis. Arthritis Rheumatol. (2018) 70:439–49. doi: 10.1002/art.40379
69. Quinn KA, Ahlman MA, Malayeri AA, Marko J, Civelek AC, Rosenblum JS, et al. Comparison of magnetic resonance angiography and (18)F-fluorodeoxyglucose positron emission tomography in large-vessel vasculitis. Ann Rheum Dis. (2018) 77:1165–71. doi: 10.1136/annrheumdis-2018-213102
70. Gribbons KB, Ponte C, Carette S, Craven A, Cuthbertson D, Hoffman GS, et al. Patterns of arterial disease in Takayasu's arteritis and giant cell arteritis. Arthritis Care Res. (2019) 72:1615–24. doi: 10.1093/rheumatology/kez058.023
71. Milutinovic A, Suput D, Zorc-Pleskovic R. Pathogenesis of atherosclerosis in the tunica intima, media, and adventitia of coronary arteries: an updated review. Bosn J Basic Med Sci. (2020) 20:21–30. doi: 10.17305/bjbms.2019.4320
72. Stenmark KR, Yeager ME, El Kasmi KC, Nozik-Grayck E, Gerasimovskaya EV, Li M, et al. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol. (2013) 75:23–47. doi: 10.1146/annurev-physiol-030212-183802
73. Krupa WM, Dewan M, Jeon MS, Kurtin PJ, Younge BR, Goronzy JJ, et al. Trapping of misdirected dendritic cells in the granulomatous lesions of giant cell arteritis. Am J Pathol. (2002) 161:1815–23. doi: 10.1016/S0002-9440(10)64458-6
74. Belilos E, Maddox J, Kowalewski RM, Kowalewska J, Turi GK, Nochomovitz LE, et al. Temporal small-vessel inflammation in patients with giant cell arteritis: clinical course and preliminary immunohistopathologic characterization. J Rheumatol. (2011) 38:331–8. doi: 10.3899/jrheum.100455
75. Hamilton CR Jr, Shelley WM, Tumulty PA. Giant cell arteritis: including temporal arteritis and polymyalgia rheumatica. Medicine. (1971) 50:1–27. doi: 10.1097/00005792-197101000-00001
76. Chemnitz J, Christensen BC, Christoffersen P, Garbarsch C, Hansen TM, Lorenzen I. Giant-cell arteritis. Histological, immunohistochemical and electronmicroscopic studies. Acta Pathol Microbiol Immunol Scand A. (1987) 95:251–62. doi: 10.1111/j.1699-0463.1987.tb00039_95A.x
77. Lie JT. Illustrated histopathologic classification criteria for selected vasculitis syndromes. American College of Rheumatology Subcommittee on Classification of Vasculitis. Arthritis Rheum. (1990) 33:1074–87. doi: 10.1002/art.1780330804
78. McDonnell PJ, Moore GW, Miller NR, Hutchins GM, Green WR. Temporal arteritis. A clinicopathologic study. Ophthalmology. (1986) 93:518–30. doi: 10.1016/S0161-6420(86)33706-0
79. Genereau T, Lortholary O, Pottier MA, Michon-Pasturel U, Ponge T, de Wazieres B, et al. Temporal artery biopsy: a diagnostic tool for systemic necrotizing vasculitis. French Vasculitis Study Group. Arthritis Rheum. (1999) 42:2674–81. doi: 10.1002/1529-0131(199912)42:12<2674::AID-ANR25>3.0.CO;2-A
80. Wang L, Ai Z, Khoyratty T, Zec K, Eames HL, van Grinsven E, et al. ROS-producing immature neutrophils in giant cell arteritis are linked to vascular pathologies. JCI Insight. (2020) 5:139163. doi: 10.1172/jci.insight.139163
81. Lie JT. Aortic and extracranial large vessel giant cell arteritis: a review of 72 cases with histopathologic documentation. Semin Arthritis Rheum. (1995) 24:422–31. doi: 10.1016/S0049-0172(95)80010-7
82. Galli E, Muratore F, Boiardi L, Restuccia G, Cavazza A, Catanoso M, et al. Significance of inflammation restricted to adventitial/periadventitial tissue on temporal artery biopsy. Semin Arthritis Rheum. (2020) 50:1064–72. doi: 10.1016/j.semarthrit.2020.05.021
83. Disdier P, Pellissier JF, Harle JR, Figarella-Branger D, Bolla G, Weiller PJ. Significance of isolated vasculitis of the vasa vasorum on temporal artery biopsy. J Rheumatol. (1994) 21:258–60.
84. Le Pendu C, Meignin V, Gonzalez-Chiappe S, Hij A, Galateau-Salle F, Mahr A. Poor predictive value of isolated adventitial and periadventitial infiltrates in temporal artery biopsies for diagnosis of giant cell arteritis. J Rheumatol. (2017) 44:1039–43. doi: 10.3899/jrheum.170061
85. Cavazza A, Muratore F, Boiardi L, Restuccia G, Pipitone N, Pazzola G, et al. Inflamed temporal artery: histologic findings in 354 biopsies, with clinical correlations. Am J Surg Pathol. (2014) 38:1360–70. doi: 10.1097/PAS.0000000000000244
86. Delaval L, Samson M, Schein F, Agard C, Trefond L, Deroux A, et al. Temporal arteritis revealing antineutrophil cytoplasmic antibody-associated vasculitides: case-control study of 50 cases. Arthritis Rheumatol. (2020). doi: 10.1002/art.41527. [Epub ahead of print].
87. Burke AP, Tavora F, Narula N, Tomaszewski JE, Virmani R. Aortitis and ascending aortic aneurysm: description of 52 cases and proposal of a histologic classification. Hum Pathol. (2008) 39:514–26. doi: 10.1016/j.humpath.2007.08.018
88. Miller DV, Isotalo PA, Weyand CM, Edwards WD, Aubry MC, Tazelaar HD. Surgical pathology of noninfectious ascending aortitis: a study of 45 cases with emphasis on an isolated variant. Am J Surg Pathol. (2006) 30:1150–8. doi: 10.1097/01.pas.0000213293.04026.ec
89. Liu G, Shupak R, Chiu BK. Aortic dissection in giant-cell arteritis. Semin Arthritis Rheum. (1995) 25:160–71. doi: 10.1016/S0049-0172(95)80028-X
90. Watanabe R, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Pathogenesis of giant cell arteritis and takayasu arteritis-similarities and differences. Curr Rheumatol Rep. (2020) 22:68. doi: 10.1007/s11926-020-00948-x
91. Seko Y, Minota S, Kawasaki A, Shinkai Y, Maeda K, Yagita H, et al. Perforin-secreting killer cell infiltration and expression of a 65-kD heat-shock protein in aortic tissue of patients with Takayasu's arteritis. J Clin Invest. (1994) 93:750–8. doi: 10.1172/JCI117029
92. Samson M, Audia S, Fraszczak J, Trad M, Ornetti P, Lakomy D, et al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum. (2012) 64:3788–98. doi: 10.1002/art.34647
93. Roche NE, Fulbright JW, Wagner AD, Hunder GG, Goronzy JJ, Weyand CM. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis Rheum. (1993) 36:1286–94. doi: 10.1002/art.1780360913
94. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
95. Murakami M, Kamimura D, Hirano T. Pleiotropy and specificity: insights from the interleukin 6 family of cytokines. Immunity. (2019) 50:812–31. doi: 10.1016/j.immuni.2019.03.027
96. Wagner AD, Goronzy JJ, Weyand CM. Functional profile of tissue-infiltrating and circulating CD68+ cells in giant cell arteritis. Evidence for two components of the disease. J Clin Invest. (1994) 94:1134–40. doi: 10.1172/JCI117428
97. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. (2010) 32:605–15. doi: 10.1016/j.immuni.2010.05.003
98. Garcia-Martinez A, Hernandez-Rodriguez J, Espigol-Frigole G, Prieto-Gonzalez S, Butjosa M, Segarra M, et al. Clinical relevance of persistently elevated circulating cytokines (tumor necrosis factor alpha and interleukin-6) in the long-term followup of patients with giant cell arteritis. Arthritis Care Res. (2010) 62:835–41. doi: 10.1002/acr.20043
99. Weyand CM, Tetzlaff N, Bjornsson J, Brack A, Younge B, Goronzy JJ. Disease patterns and tissue cytokine profiles in giant cell arteritis. Arthritis Rheum. (1997) 40:19–26. doi: 10.1002/art.1780400105
100. Cid MC, Font C, Oristrell J, de la Sierra A, Coll-Vinent B, Lopez-Soto A, et al. Association between strong inflammatory response and low risk of developing visual loss and other cranial ischemic complications in giant cell (temporal) arteritis. Arthritis Rheum. (1998) 41:26–32. doi: 10.1002/1529-0131(199801)41:1<26::AID-ART4>3.0.CO;2-0
101. Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation. (2010) 121:906–15. doi: 10.1161/CIRCULATIONAHA.109.872903
102. Terrier B, Geri G, Chaara W, Allenbach Y, Rosenzwajg M, Costedoat-Chalumeau N, et al. Interleukin-21 modulates Th1 and Th17 responses in giant cell arteritis. Arthritis Rheum. (2012) 64:2001–11. doi: 10.1002/art.34327
103. Wen Z, Shen Y, Berry G, Shahram F, Li Y, Watanabe R, et al. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci Transl Med. (2017) 9:aal3322. doi: 10.1126/scitranslmed.aal3322
104. Patel DD, Kuchroo VK. Th17 cell pathway in human immunity: lessons from genetics and therapeutic interventions. Immunity. (2015) 43:1040–51. doi: 10.1016/j.immuni.2015.12.003
105. Sungnak W, Wang C, Kuchroo VK. Multilayer regulation of CD4 T cell subset differentiation in the era of single cell genomics. Adv Immunol. (2019) 141:1–31. doi: 10.1016/bs.ai.2018.12.001
106. Rao DA. T cells that help B cells in chronically inflamed tissues. Front Immunol. (2018) 9:1924. doi: 10.3389/fimmu.2018.01924
107. Dejaco C, Duftner C, Al-Massad J, Wagner AD, Park JK, Fessler J, et al. NKG2D stimulated T-cell autoreactivity in giant cell arteritis and polymyalgia rheumatica. Ann Rheum Dis. (2013) 72:1852–9. doi: 10.1136/annrheumdis-2012-201660
108. Brandstadter JD, Maillard I. Notch signalling in T cell homeostasis and differentiation. Open Biol. (2019) 9:190187. doi: 10.1098/rsob.190187
109. Piggott K, Deng J, Warrington K, Younge B, Kubo JT, Desai M, et al. Blocking the NOTCH pathway inhibits vascular inflammation in large-vessel vasculitis. Circulation. (2011) 123:309–18. doi: 10.1161/CIRCULATIONAHA.110.936203
110. De Smit E, Lukowski SW, Anderson L, Senabouth A, Dauyey K, Song S, et al. Longitudinal expression profiling of CD4+ and CD8+ cells in patients with active to quiescent giant cell arteritis. BMC Med Genom. (2018) 11:61. doi: 10.1186/s12920-018-0376-4
111. Dasgupta B, Duke O, Timms AM, Pitzalis C, Panayi GS. Selective depletion and activation of CD8+ lymphocytes from peripheral blood of patients with polymyalgia rheumatica and giant cell arteritis. Ann Rheum Dis. (1989) 48:307–11. doi: 10.1136/ard.48.4.307
112. Elling H, Elling P. Decreased level of suppressor/cytotoxic T cells (OKT8+) in polymyalgia rheumatica and temporal arteritis: relation to disease activity. J Rheumatol. (1985) 12:306–9.
113. Martinez-Taboada VM, Goronzy JJ, Weyand CM. Clonally expanded CD8 T cells in patients with polymyalgia rheumatica and giant cell arteritis. Clin Immunol Immunopathol. (1996) 79:263–70. doi: 10.1006/clin.1996.0078
114. Samson M, Ly KH, Tournier B, Janikashvili N, Trad M, Ciudad M, et al. Involvement and prognosis value of CD8(+) T cells in giant cell arteritis. J Autoimmun. (2016) 72:73–83. doi: 10.1016/j.jaut.2016.05.008
115. Martinez-Taboada VM, Blanco R, Fito C, Pacheco MJ, Delgado-Rodriguez M, Rodriguez-Valverde V. Circulating CD8+ T cells in polymyalgia rheumatica and giant cell arteritis: a review. Semin Arthritis Rheum. (2001) 30:257–71. doi: 10.1053/sarh.2001.9734
116. van der Geest KS, Abdulahad WH, Chalan P, Rutgers A, Horst G, Huitema MG, et al. Disturbed B cell homeostasis in newly diagnosed giant cell arteritis and polymyalgia rheumatica. Arthritis Rheumatol. (2014) 66:1927–38. doi: 10.1002/art.38625
117. van Sleen Y, Wang Q, van der Geest KSM, Westra J, Abdulahad WH, Heeringa P, et al. Involvement of monocyte subsets in the immunopathology of giant cell arteritis. Sci Rep. (2017) 7:6553. doi: 10.1038/s41598-017-06826-4
118. Watanabe R, Maeda T, Zhang H, Berry GJ, Zeisbrich M, Brockett R, et al. MMP (Matrix Metalloprotease)-9-producing monocytes enable T cells to invade the vessel wall and cause vasculitis. Circ Res. (2018) 123:700–15. doi: 10.1161/CIRCRESAHA.118.313206
119. Riley JL PD-1 signaling in primary T cells. Immunol Rev. (2009) 229:114–25. doi: 10.1111/j.1600-065X.2009.00767.x
120. Corbera-Bellalta M, Garcia-Martinez A, Lozano E, Planas-Rigol E, Tavera-Bahillo I, Alba MA, et al. Changes in biomarkers after therapeutic intervention in temporal arteries cultured in Matrigel: a new model for preclinical studies in giant-cell arteritis. Ann Rheum Dis. (2014) 73:616–23. doi: 10.1136/annrheumdis-2012-202883
121. Ma-Krupa W, Jeon MS, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J Exp Med. (2004) 199:173–83. doi: 10.1084/jem.20030850
122. Lorber MI, Wilson JH, Robert ME, Schechner JS, Kirkiles N, Qian HY, et al. Human allogeneic vascular rejection after arterial transplantation and peripheral lymphoid reconstitution in severe combined immunodeficient mice. Transplantation. (1999) 67:897–903. doi: 10.1097/00007890-199903270-00018
123. Wang Y, Burns WR, Tang PC, Yi T, Schechner JS, Zerwes HG, et al. Interferon-gamma plays a nonredundant role in mediating T cell-dependent outward vascular remodeling of allogeneic human coronary arteries. FASEB J. (2004) 18:606–8. doi: 10.1096/fj.03-0840fje
124. Koh KP, Wang Y, Yi T, Shiao SL, Lorber MI, Sessa WC, et al. T cell-mediated vascular dysfunction of human allografts results from IFN-gamma dysregulation of NO synthase. J Clin Invest. (2004) 114:846–56. doi: 10.1172/JCI21767
125. Corbera-Bellalta M, Planas-Rigol E, Lozano E, Terrades-Garcia N, Alba MA, Prieto-Gonzalez S, et al. Blocking interferon gamma reduces expression of chemokines CXCL9, CXCL10 and CXCL11 and decreases macrophage infiltration in ex vivo cultured arteries from patients with giant cell arteritis. Ann Rheum Dis. (2016) 75:1177–86. doi: 10.1136/annrheumdis-2015-208371
126. Wawryk SO, Ayberk H, Boyd AW, Rode J. Analysis of adhesion molecules in the immunopathogenesis of giant cell arteritis. J Clin Pathol. (1991) 44:497–501. doi: 10.1136/jcp.44.6.497
127. Cid MC, Cebrian M, Font C, Coll-Vinent B, Hernandez-Rodriguez J, Esparza J, et al. Cell adhesion molecules in the development of inflammatory infiltrates in giant cell arteritis: inflammation-induced angiogenesis as the preferential site of leukocyte-endothelial cell interactions. Arthritis Rheum. (2000) 43:184–94. doi: 10.1002/1529-0131(200001)43:1<184::AID-ANR23>3.0.CO
128. Melrose J, Tsurushita N, Liu G, Berg EL. IFN-gamma inhibits activation-induced expression of E- and P-selectin on endothelial cells. J Immunol. (1998) 161:2457–64.
129. Weyand CM, Hicok KC, Hunder GG, Goronzy JJ. Tissue cytokine patterns in patients with polymyalgia rheumatica and giant cell arteritis. Ann Intern Med. (1994) 121:484–91. doi: 10.7326/0003-4819-121-7-199410010-00003
130. Martinez-Taboada V, Hunder NN, Hunder GG, Weyand CM, Goronzy JJ. Recognition of tissue residing antigen by T cells in vasculitic lesions of giant cell arteritis. J Mol Med. (1996) 74:695–703. doi: 10.1007/s001090050074
131. Weyand CM, Schonberger J, Oppitz U, Hunder NN, Hicok KC, Goronzy JJ. Distinct vascular lesions in giant cell arteritis share identical T cell clonotypes. J Exp Med. (1994) 179:951–60. doi: 10.1084/jem.179.3.951
132. Armstrong AT, Tyler WB, Wood GC, Harrington TM. Clinical importance of the presence of giant cells in temporal arteritis. J Clin Pathol. (2008) 61:669–71. doi: 10.1136/jcp.2007.049049
133. Conway R, O'Neill L, McCarthy GM, Murphy CC, Fabre A, Kennedy S, et al. Interleukin 12 and interleukin 23 play key pathogenic roles in inflammatory and proliferative pathways in giant cell arteritis. Ann Rheum Dis. (2018) 77:1815–24. doi: 10.1136/annrheumdis-2018-213488
134. Espigol-Frigole G, Corbera-Bellalta M, Planas-Rigol E, Lozano E, Segarra M, Garcia-Martinez A, et al. Increased IL-17A expression in temporal artery lesions is a predictor of sustained response to glucocorticoid treatment in patients with giant-cell arteritis. Ann Rheum Dis. (2013) 72:1481–7. doi: 10.1136/annrheumdis-2012-201836
135. Ciccia F, Rizzo A, Guggino G, Cavazza A, Alessandro R, Maugeri R, et al. Difference in the expression of IL-9 and IL-17 correlates with different histological pattern of vascular wall injury in giant cell arteritis. Rheumatology. (2015) 54:1596–604. doi: 10.1093/rheumatology/kev102
136. Rao DA, Eid RE, Qin L, Yi T, Kirkiles-Smith NC, Tellides G, et al. Interleukin (IL)-1 promotes allogeneic T cell intimal infiltration and IL-17 production in a model of human artery rejection. J Exp Med. (2008) 205:3145–58. doi: 10.1084/jem.20081661
137. Rittner HL, Kaiser M, Brack A, Szweda LI, Goronzy JJ, Weyand CM. Tissue-destructive macrophages in giant cell arteritis. Circ Res. (1999) 84:1050–8. doi: 10.1161/01.RES.84.9.1050
138. Kaiser M, Weyand CM, Bjornsson J, Goronzy JJ. Platelet-derived growth factor, intimal hyperplasia, and ischemic complications in giant cell arteritis. Arthritis Rheum. (1998) 41:623–33. doi: 10.1002/1529-0131(199804)41:4<623::AID-ART9>3.0.CO;2-6
139. Visvanathan S, Rahman MU, Hoffman GS, Xu S, Garcia-Martinez A, Segarra M, et al. Tissue and serum markers of inflammation during the follow-up of patients with giant-cell arteritis–a prospective longitudinal study. Rheumatology. (2011) 50:2061–70. doi: 10.1093/rheumatology/ker163
140. Espigol-Frigole G, Planas-Rigol E, Lozano E, Corbera-Bellalta M, Terrades-Garcia N, Prieto-Gonzalez S, et al. Expression and function of IL12/23 related cytokine subunits (p35, p40, and p19) in giant-cell arteritis lesions: contribution of p40 to Th1- and Th17-mediated inflammatory pathways. Front Immunol. (2018) 9:809. doi: 10.3389/fimmu.2018.00809
141. Tieu BC, Lee C, Sun H, Lejeune W, Recinos A 3rd, Ju X, et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. (2009) 119:3637–51. doi: 10.1172/JCI38308
142. Brooks PJ, Glogauer M, McCulloch CA. An overview of the derivation and function of multinucleated giant cells and their role in pathologic processes. Am J Pathol. (2019) 189:1145–58. doi: 10.1016/j.ajpath.2019.02.006
143. Deng J, Ma-Krupa W, Gewirtz AT, Younge BR, Goronzy JJ, Weyand CM. Toll-like receptors 4 and 5 induce distinct types of vasculitis. Circ Res. (2009) 104:488–95. doi: 10.1161/CIRCRESAHA.108.185777
144. Wagner AD, Bjornsson J, Bartley GB, Goronzy JJ, Weyand CM. Interferon-gamma-producing T cells in giant cell vasculitis represent a minority of tissue-infiltrating cells and are located distant from the site of pathology. Am J Pathol. (1996) 148:1925–33.
145. Lee AY, Eri R, Lyons AB, Grimm MC, Korner H. CC chemokine ligand 20 and its cognate receptor CCR6 in mucosal T cell immunology and inflammatory bowel disease: odd couple or axis of evil? Front Immunol. (2013) 4:194. doi: 10.3389/fimmu.2013.00194
146. Lozano E, Segarra M, Garcia-Martinez A, Hernandez-Rodriguez J, Cid MC. Imatinib mesylate inhibits in vitro and ex vivo biological responses related to vascular occlusion in giant cell arteritis. Ann Rheum Dis. (2008) 67:1581–8. doi: 10.1136/ard.2007.070805
147. Ludwig A, Berkhout T, Moores K, Groot P, Chapman G. Fractalkine is expressed by smooth muscle cells in response to IFN-gamma and TNF-alpha and is modulated by metalloproteinase activity. J Immunol. (2002) 168:604–12. doi: 10.4049/jimmunol.168.2.604
148. Zhang H, Watanabe R, Berry GJ, Vaglio A, Liao YJ, Warrington KJ, et al. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc Natl Acad Sci USA. (2017) 114:E970–9. doi: 10.1073/pnas.1616848114
149. Segarra M, Garcia-Martinez A, Sanchez M, Hernandez-Rodriguez J, Lozano E, Grau JM, et al. Gelatinase expression and proteolytic activity in giant-cell arteritis. Ann Rheum Dis. (2007) 66:1429–35. doi: 10.1136/ard.2006.068148
150. Tellides G, Tereb DA, Kirkiles-Smith NC, Kim RW, Wilson JH, Schechner JS, et al. Interferon-gamma elicits arteriosclerosis in the absence of leukocytes. Nature. (2000) 403:207–11. doi: 10.1038/35003221
151. Lozano E, Segarra M, Corbera-Bellalta M, Garcia-Martinez A, Espigol-Frigole G, Pla-Campo A, et al. Increased expression of the endothelin system in arterial lesions from patients with giant-cell arteritis: association between elevated plasma endothelin levels and the development of ischaemic events. Ann Rheum Dis. (2010) 69:434–42. doi: 10.1136/ard.2008.105692
152. Kaiser M, Younge B, Bjornsson J, Goronzy JJ, Weyand CM. Formation of new vasa vasorum in vasculitis. Production of angiogenic cytokines by multinucleated giant cells. Am J Pathol. (1999) 155:765–74. doi: 10.1016/S0002-9440(10)65175-9
153. Maleszewski JJ, Younge BR, Fritzlen JT, Hunder GG, Goronzy JJ, Warrington KJ, et al. Clinical and pathological evolution of giant cell arteritis: a prospective study of follow-up temporal artery biopsies in 40 treated patients. Mod Pathol. (2017) 30:788–96. doi: 10.1038/modpathol.2017.10
154. Unizony S, Arias-Urdaneta L, Miloslavsky E, Arvikar S, Khosroshahi A, Keroack B, et al. Tocilizumab for the treatment of large-vessel vasculitis (giant cell arteritis, Takayasu arteritis) and polymyalgia rheumatica. Arthritis Care Res. (2012) 64:1720–9. doi: 10.1002/acr.21750
155. Langford CA, Cuthbertson D, Ytterberg SR, Khalidi N, Monach PA, Carette S, et al. A randomized, double-blind trial of abatacept (CTLA-4Ig) for the treatment of giant cell arteritis. Arthritis Rheumatol. (2017) 69:837–45. doi: 10.1002/art.40044
156. Hoffman GS, Cid MC, Rendt-Zagar KE, Merkel PA, Weyand CM, Stone JH, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. (2007) 146:621–30. doi: 10.7326/0003-4819-146-9-200705010-00004
157. Martinez-Taboada VM, Rodriguez-Valverde V, Carreno L, Lopez-Longo J, Figueroa M, Belzunegui J, et al. A double-blind placebo controlled trial of etanercept in patients with giant cell arteritis and corticosteroid side effects. Ann Rheum Dis. (2008) 67:625–30. doi: 10.1136/ard.2007.082115
158. Seror R, Baron G, Hachulla E, Debandt M, Larroche C, Puechal X, et al. Adalimumab for steroid sparing in patients with giant-cell arteritis: results of a multicentre randomised controlled trial. Ann Rheum Dis. (2014) 73:2074–81. doi: 10.1136/annrheumdis-2013-203586
159. Matsumoto K, Suzuki K, Yoshimoto K, Seki N, Tsujimoto H, Chiba K, et al. Significant association between clinical characteristics and changes in peripheral immuno-phenotype in large vessel vasculitis. Arthritis Res Ther. (2019) 21:304. doi: 10.1186/s13075-019-2068-7
160. Kong X, Sun Y, Ma L, Chen H, Wei L, Wu W, et al. The critical role of IL-6 in the pathogenesis of Takayasu arteritis. Clin Exp Rheumatol. (2016) 34:S21–7.
161. Tamura N, Maejima Y, Tezuka D, Takamura C, Yoshikawa S, Ashikaga T, et al. Profiles of serum cytokine levels in Takayasu arteritis patients: potential utility as biomarkers for monitoring disease activity. J Cardiol. (2017) 70:278–85. doi: 10.1016/j.jjcc.2016.10.016
162. Saadoun D, Garrido M, Comarmond C, Desbois AC, Domont F, Savey L, et al. Th1 and Th17 cytokines drive inflammation in Takayasu arteritis. Arthritis Rheumatol. (2015) 67:1353–60. doi: 10.1002/art.39037
163. Barra L, Yang G, Pagnoux C, Canadian Vasculitis N. Non-glucocorticoid drugs for the treatment of Takayasu's arteritis: a systematic review and meta-analysis. Autoimmun Rev. (2018) 17:683–93. doi: 10.1016/j.autrev.2018.01.019
164. Koch AE, Haines GK, Rizzo RJ, Radosevich JA, Pope RM, Robinson PG, et al. Human abdominal aortic aneurysms. Immunophenotypic analysis suggesting an immune-mediated response. Am J Pathol. (1990) 137:1199–213.
165. Juvonen J, Surcel HM, Satta J, Teppo AM, Bloigu A, Syrjala H, et al. Elevated circulating levels of inflammatory cytokines in patients with abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. (1997) 17:2843–7. doi: 10.1161/01.ATV.17.11.2843
166. Sakalihasan N, Michel JB, Katsargyris A, Kuivaniemi H, Defraigne JO, Nchimi A, et al. Abdominal aortic aneurysms. Nat Rev Dis Primers. (2018) 4:34. doi: 10.1038/s41572-018-0030-7
167. Ocana E, Bohorquez JC, Perez-Requena J, Brieva JA, Rodriguez C. Characterisation of T and B lymphocytes infiltrating abdominal aortic aneurysms. Atherosclerosis. (2003) 170:39–48. doi: 10.1016/S0021-9150(03)00282-X
168. Raffort J, Lareyre F, Clement M, Hassen-Khodja R, Chinetti G, Mallat Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. (2017) 14:457–71. doi: 10.1038/nrcardio.2017.52
169. Satta J, Laurila A, Paakko P, Haukipuro K, Sormunen R, Parkkila S, et al. Chronic inflammation and elastin degradation in abdominal aortic aneurysm disease: an immunohistochemical and electron microscopic study. Eur J Vasc Endovasc Surg. (1998) 15:313–9. doi: 10.1016/S1078-5884(98)80034-8
170. Schonbeck U, Sukhova GK, Gerdes N, Libby P. T(H)2 predominant immune responses prevail in human abdominal aortic aneurysm. Am J Pathol. (2002) 161:499–506. doi: 10.1016/S0002-9440(10)64206-X
171. Wei K, Korsunsky I, Marshall JL, Gao A, Watts GFM, Major T, et al. A. Accelerating Medicines Partnership Rheumatoid, C. Systemic Lupus Erythematosus, Siebel CW, Buckley CD, Raychaudhuri S, and Brenner MB, Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature. (2020) 582:259–64. doi: 10.1038/s41586-020-2222-z
172. Chen Q, Yang W, Gupta S, Biswas P, Smith P, Bhagat G, et al. IRF-4-binding protein inhibits interleukin-17 and interleukin-21 production by controlling the activity of IRF-4 transcription factor. Immunity. (2008) 29:899–911. doi: 10.1016/j.immuni.2008.10.011
173. Roupie AL, de Boysson H, Thietart S, Carrat F, Seguier J, Terriou L, et al. Giant-cell arteritis associated with myelodysplastic syndrome: French multicenter case control study and literature review. Autoimmun Rev. (2020) 19:102446. doi: 10.1016/j.autrev.2019.102446
174. Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. (2020) 383:2628–38. doi: 10.1056/NEJMoa2026834
Keywords: vasculitis, CIA, LVV, Takayasu, PMR, temporal arteritis, GCA, giant cell arteritis
Citation: Robinette ML, Rao DA and Monach PA (2021) The Immunopathology of Giant Cell Arteritis Across Disease Spectra. Front. Immunol. 12:623716. doi: 10.3389/fimmu.2021.623716
Received: 30 October 2020; Accepted: 04 February 2021;
Published: 25 February 2021.
Edited by:
Joshua Daniel Ooi, Monash University, AustraliaReviewed by:
Carlo Chizzolini, Université de Genève, SwitzerlandDelamo Bekele, Mayo Clinic, United States
Philipp Jud, Medical University of Graz, Austria
Copyright © 2021 Robinette, Rao and Monach. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michelle L. Robinette, bXJvYmluZXR0ZUBwYXJ0bmVycy5vcmc=