 Judith Derdelinckx
Judith Derdelinckx Patrick Cras2,3
Patrick Cras2,3 Zwi N. Berneman
Zwi N. Berneman Nathalie Cools
Nathalie Cools- 1Laboratory of Experimental Hematology, Vaccine and Infectious Disease Institute (VaxInfectio), Faculty of Medicine and Health Sciences, University of Antwerp, Antwerp, Belgium
- 2Division of Neurology, Antwerp University Hospital, Edegem, Belgium
- 3Born Bunge Institute, Translational Neurosciences, Faculty of Medicine and Health Sciences, University of Antwerp, Antwerp, Belgium
- 4Center for Cell Therapy and Regenerative Medicine, Antwerp University Hospital, Edegem, Belgium
Antigen-specific therapy for multiple sclerosis may lead to a more effective therapy by induction of tolerance to a wide range of myelin-derived antigens without hampering the normal surveillance and effector function of the immune system. Numerous attempts to restore tolerance toward myelin-derived antigens have been made over the past decades, both in animal models of multiple sclerosis and in clinical trials for multiple sclerosis patients. In this review, we will give an overview of the current approaches for antigen-specific therapy that are in clinical development for multiple sclerosis as well provide an insight into the challenges for future antigen-specific treatment strategies for multiple sclerosis.
Introduction
In autoimmune diseases, the immune system is derailed generating immunity against self. In the particular case of multiple sclerosis (MS), there are strong indications that the loss of tolerance is directed toward various myelin proteins, including myelin oligodendrocyte glycoprotein (MOG), myelin basic protein (MBP), and proteolipid protein (PLP) (1). Although the exact cause for this breach in tolerance is not yet known, it has been suggested that myelin-reactive CD4+ T lymphocytes, both of the T helper 1 (Th1) and T helper 17 (Th17) type, play a central role in the pathogenesis of MS (1–4). For instance, this is evidenced by the encephalitogenic capacity of CD4+ myelin-reactive T cells following passive transfer in experimental autoimmune encephalomyelitis (EAE) animal models (5, 6). Additionally, the fact that the strongest genetic risk factor for MS lies within the major histocompatibility complex (MHC) class II gene further underscores the importance of CD4+ T cells in MS pathogenesis (1, 7). More recently, the involvement of additional effector cells in the myelin-directed autoimmune reaction has been proposed, including myelin-reactive CD8+ T cells and B cells (4) (Box 1). Altogether, a complex autoimmune cascade, rather than a single culprit autoimmune response, appears to be driving MS pathogenesis, complicating the development of a targeted antigen-specific therapy for MS.
Box 1. The immune pathogenesis of multiple sclerosis.
MS is considered to be a predominantly T cell-mediated autoimmune disease (118), directed toward various myelin-derived antigens, including myelin basic protein (MBP), proteolipid protein (PLP), myelin oligodendrocyte glycoprotein (MOG), and αB-crystallin (1), that are expressed in the CNS. This autoimmunity is mostly mediated by CD4+ T cells, in particular T helper 1 (Th1) and Th17 cells (3), and involves further effectuation of an immune cascade involving CD8+ T cells, B cells, and NK cells. The exact mechanism by which these autoreactive T cells are initiated, has not been fully elucidated. As reviewed by Hemmer et al., two main hypotheses have been suggested for the immune-mediated development of demyelinating lesions (2). The first hypothesis—the so-called outside-in hypothesis—is based on peripheral activation of autoreactive CD4+ T cells recognizing CNS-derived antigens, e.g., due to infection-related molecular mimicry or bystander activation (119–122). Alternatively, the inside-out hypothesis states that the initial pathogenic event takes place within the CNS, namely primary oligodendrocyte damage leading to leakage of CNS antigens to the periphery and activation of autoreactive T lymphocytes in the peripheral lymph nodes (123). However, the inside-out hypothesis is controversial, with both evidence in favor (124) and against (125) primary oligodendrocyte damage as the initiating trigger for CNS auto-immunity. Hence, the origin of the autoimmune response in MS remains a matter of debate. Nonetheless, whether the initial pathogenic event takes place in the CNS or in the periphery, one of the key elements in the immune pathogenesis of MS is the escape of autoreactive T cells from tolerance control mechanisms. This allows activated encephalitogenic CD4+ T cells to migrate across the blood-brain barrier (BBB), followed by their reactivation with autoantigens in the perivascular space (126) and their release of inflammatory mediators which activate microglia (2) (Figure 1). These cells will, in turn, effectuate tissue damage and produce various chemokines leading to further recruitment of effector and antigen-presenting cells (APC).
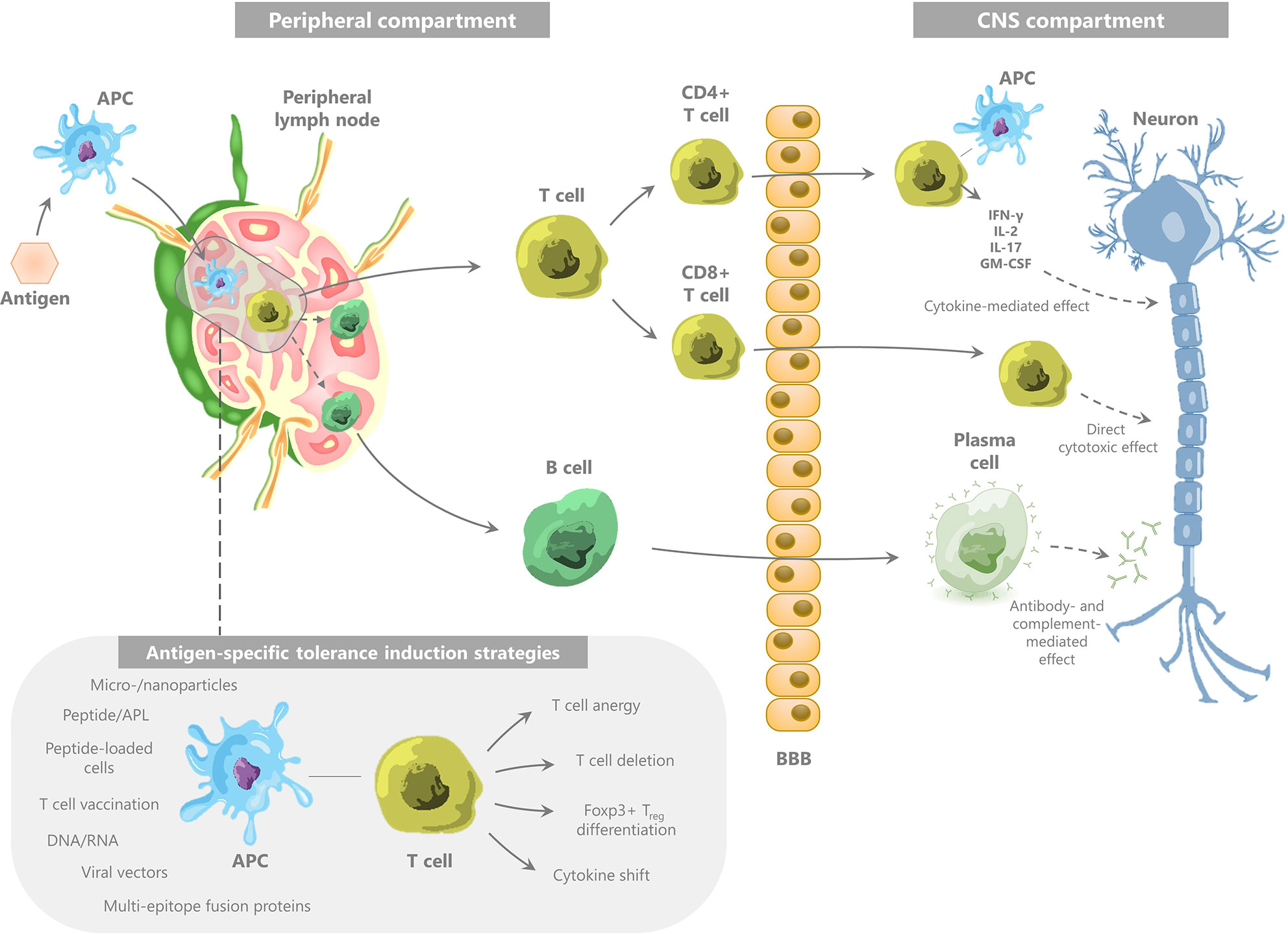
Figure 1 The immune pathogenesis of multiple sclerosis and the concept of myelin-specific tolerance induction.
Used abbreviations: APC, antigen-presenting cell; IFN, interferon; IL, interleukin; GM-CSF, granulocyte-macrophage colony-stimulating factor; APL, altered peptide ligand; Treg, regulatory T cell.
The strong increase in knowledge regarding the pathogenesis of MS has resulted in a significant expansion of the treatment armamentarium for MS over the last years. This resulted in a wide range of disease-modifying therapeutics with varying efficacy in reducing inflammation and relapse rate. However, these therapies are accompanied by various side effects, including opportunistic infections, because of the non-disease antigen-specific mode of action resulting in a more general immune modulation or immune suppression. Hence, an ideal therapy approach for MS would aim to restore the dysregulated myelin-directed immune response without hampering the normal surveillance and effector function of the immune system (Box 1).
In this review, we will first give an overview of the current approaches for antigen-specific therapy that are in clinical development for MS, summarizing the results of several phase I, II and III clinical trials. In the second part of this review, we will provide an insight into the challenges for future antigen-specific treatment strategies for MS and summarize the possible solutions for these challenges that are currently being evaluated in a preclinical setting.
Antigen-Specific Treatment in MS: Results From Clinical Trials
Peptides and Altered Peptide Ligands
Peptide-based therapy aims to restore tolerance to specific peptides or peptide mixes by repeated administration through various routes. In parallel to hyposensitization therapy for allergy, this repeated exposure to auto-antigen induces immunological alterations, including a cytokine shift away from the autoimmune Th1/Th17 profile and induction of IL-10-secreting regulatory T cells (Treg) (8–10). Disease-related peptides can be selected by different means, including i) elution from peptide-MHC complexes (representing naturally processed peptides), ii) selection of immune dominant peptide responses by use of reactivity screening assays, or iii) prediction with computer algorithms or databases (11, 12). In addition to the use of classical peptides, altered peptide ligands (APL) can be generated by subtle modification of peptide structure, mostly by amino acid substitutions at the T cell receptor (TCR) binding site. These modifications impair T cell function following TCR-ligand interaction, which can further modulate antigen-specific T cell responses. The therapeutic potential of APL has historically been underlined by the effectiveness of glatiramer acetate, which – among other working mechanisms—acts as an APL for MBP82–100, causing a shift in the MBP response from a Th1 to Th2 cytokine profile [as reviewed by Schrempf et al. (13)].
Peptide- and APL-based therapy is a straightforward yet versatile approach and therefore has been the focus of many clinical trials in MS. An overview of the pivotal clinical trials focusing on peptide therapy in MS can be found in Table 1, which we will concisely discuss.
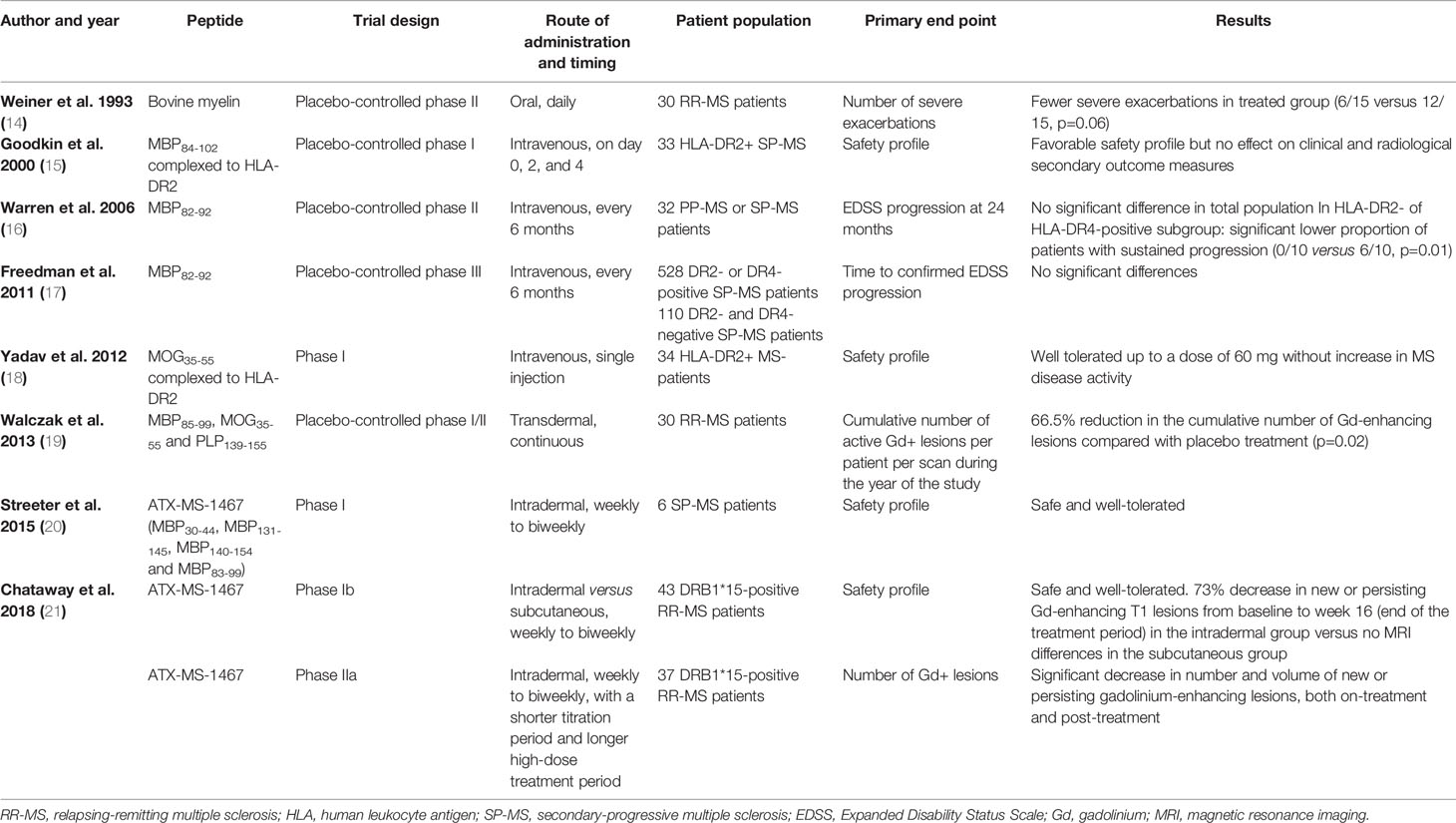
Table 1 Overview of the clinical trials using peptide therapy.
Peptide Therapy
Tolerance induction using peptide therapy was one of the first attempts for antigen-specific treatment for MS, with the first results on efficacy being available from phase II clinical trials at the end of the 90’s. In a phase II clinical trial, 30 relapsing remitting MS (RR-MS) patients were treated orally with bovine myelin or with control protein (14, 22). Although only 40% of patients in the group treated with myelin protein had at least one major exacerbation as compared to 80% of patients in the control group (p=0.06) (14), no conclusions regarding efficacy could be made based on these small numbers of patients (22).
Next, a placebo-controlled phase II clinical trial with intravenous administration of high doses of MBP82–92 was initiated by Warren et al. in 2006 (16). In this trial, 32 primary or secondary progressive MS (SP-MS) patients were treated with MBP82-92 intravenously every 6 months. No difference was found between treatment or placebo group in the primary endpoint, expanded disability status scale (EDSS) progression at 24 months. However, a subgroup analysis of the human leukocyte antigen (HLA)-DR2+ or DR4+ participants (20 subjects) revealed a significantly lower proportion of patients with sustained progression at 24 months in the treatment group (0/10) compared to the placebo group (6/10, p=0.01). Based on the finding that patients responded better depending on their HLA haplotype, a larger phase III clinical trial was initiated in DR2+ or DR4+ SP-MS patients (17). However, this phase III placebo-controlled trial in 612 study subjects failed to meet its primary outcome, i.e., time to progression by ≥1.0 EDSS point, or ≥0.5 point if baseline EDSS was 5.5 or higher (17).
Within the context of the finding of an association between HLA haplotype and clinical effect of peptide vaccination, two phase I clinical trials have been performed using fusion products with HLA molecules. First, in 2000, a phase I dose-escalating clinical trial with intravenously administered MBP84–102 complexed to HLA-DR2 (AG284) in 33 HLA-DR2+ secondary MS patients was initiated, showing a favorable safety profile but no effect on clinical and radiological secondary outcome measures (15). Secondly, in 2012, a phase I dose-escalation clinical trial in 34 HLA-DR2+ MS-patients demonstrated that a fusion product of the two outer domains of HLA-DR2 with MOG35–55 was well tolerated up to a dose of 60 mg intravenously without increase in MS disease activity (18).
In 2013, Walczak et al. reported the results of their clinical trial with transdermal myelin peptide treatment (19). In their placebo-controlled study, 30 patients with active RR-MS were treated with a skin patch, either containing a mixture of three myelin-derived peptides (MBP85–99, MOG35–55, and PLP139–155) or phosphate-buffered saline (PBS). A 66.5% reduction in the cumulative number of gadolinium (Gd)-enhancing lesions compared with placebo treatment (p=0.02) was found on 3-monthly magnetic resonance imaging (MRI) scans during the first year of treatment.
In 2015, Streeter et al. reported results from a phase I clinical trial in SP-MS patients (20), which were treated with a mix of 4 MBP-derived “apitopes” or antigen processing-independent epitopes (MBP30–44, MBP131–145, MBP140–154, and MBP83–99) called ATX-MS-1467. These apitopes mimic the naturally processed T cell epitope, binding directly onto MHC class-II on immature dendritic cells (DC). This was considered to be of importance since it was previously demonstrated that attempts to induce tolerance toward a non-naturally processed epitope, i.e., cryptic epitope, were not able to prevent EAE (23). Six SP-MS patients were treated with weekly to biweekly intradermal administrations of ATX-MS-1467, each receiving a dose escalation from 25 to 800 µg (20). Treatment was well-tolerated, with no major side effects. The phase Ib study, aiming to determine the optimal route of administration, showed a 73% decrease in new or persisting Gd-enhancing T1 lesions from baseline to week 16 (end of the treatment period) in the intradermal group, returning to baseline levels at week 48 (end of the off-treatment period), whereas no MRI differences could be detected in the subcutaneously treated group (24).
Immunomonitoring was performed in several of these clinical trials, demonstrating reduction in the frequency (14) and the proliferative capacity (25) of myelin-reactive T cells, a peripheral blood cytokine shift toward anti-inflammatory interleukin (IL)-10 secretion (25) and induction of myelin-specific transforming factor β (TGF-β)-secreting regulatory T cells (Treg) (26–28) following myelin peptide treatment.
In conclusion, clinical trials with peptide-based treatment have yielded both promising and disappointing results. Differences in administration route, patient population, and single-peptide- versus multi-peptide-based treatment may play a role in these contrasting results. At the moment, research into peptide-based therapy is continuing in MS. Currently under investigation is Neurovax®, a vaccine consisting of peptides derived from the T cell receptor (TCR) of pathogenic T cell clones of MS patients (29–31). Intramuscular administration of this vaccine aims to specifically modulate autoreactive T cells recognizing these peptides. Phase I clinical trials with this peptide product are currently ongoing in SP-MS and pediatric MS (NCT02200718, NCT02149706, and NCT02057159).
Altered Peptide Ligands
Several authors demonstrated the prevention of EAE development in rodents by administration of APL for MBP (32–37) or PLP (38–40) peptides. However, clinical translation appeared to be less unequivocal. A phase II clinical trial assessing the safety and efficacy of weekly subcutaneous administration of an APL of MBP83–99 (CGP77116) was halted prematurely after treatment of 8 patients because of treatment-related occurrence of MS exacerbations in 3 patients (41). Treatment with CGP77116 carried the risk of expansion of encephalitogenic MBP83–99-reactive T cells, as demonstrated by a strong increase in frequency of MBP83–99- and CGP77116-reactive T cells in peripheral blood and cerebrospinal fluid (CSF) in two of the three patients during disease exacerbation. In the same year, a second clinical trial with a different APL of MBP83–99 (NBI5788) was suspended after hypersensitivity reactions were observed in 9.1% of treated patients (42), even though NBI5788 was shown to be safe in a phase I study (43). Hypersensitivity was Th2-driven and arose in most patients after more than 10 administrations. Nonetheless, the volume and number of Gd-enhancing lesions 4 months after the first administration was reduced in the group of patients treated with the lowest dose of 5 mg of NBI5788 (42). Hence, induction of Th2 responses toward myelin antigens appeared to be a double-edged sword, with both beneficial and adverse effects. Similar immediate hypersensitivity reactions have been reported for glatiramer acetate, making the authors conclude that APL might be a new class of therapeutics for MS, but with the need to regulate the strength of the Th2 response (42). Nevertheless, despite the success of glatiramer acetate, no clinical trials using APL have been initiated since then, even though preclinical work on APL in EAE models still continues (44–46).
Peptide-Loaded Cell Therapies
A phase I dose escalation clinical study was performed by Lutterotti et al., using autologous peripheral blood mononuclear cells (PBMC) coupled to 7 myelin peptides (MOG1–20, MOG35–55, MBP13–32, MBP83–99, MBP111–129, MBP146–170, and PLP139–154) in the presence of the chemical cross-linker 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) (47). Seven RR-MS and 2 SP-MS patients were treated with doses ranged from 1x103 to 3x109 antigen-coupled PBMC, administered in one single intravenous infusion (47). No major side effects were reported. Moreover, myelin-specific T cell responses were reduced 3 months after treatment in the four patients receiving highest doses (≥1x109 myelin-coupled PBMC). Two mechanisms appear to be driving tolerance induction through EDC-fixed peptide-loaded carrier cells, which are themselves deprived of their cellular function following fixation. Based on EAE data, a first mechanism consists of induction of apoptosis in myelin-reactive T cells upon antigen presentation without costimulation by the EDC-fixed carrier cells (48). In addition, a contribution of secondary cross-tolerance induction by presentation of peptides by host antigen-presenting cells (APC) following uptake and processing of the peptide-loaded carrier cells was demonstrated (48). Given the promising results of this phase I clinical trial, a phase I/II clinical trial focusing on peptide-loaded red blood cells, called ETIMSred, was initiated recently (49).
In addition to the use of fixed carrier cells, peptide-loaded cell therapy strategies can make use of viable APC as carrier cells to add a direct tolerogenic property to the peptide product. Recently, a phase Ib clinical trial was completed, demonstrating a favorable side effect profile of myelin antigen and aquaporine-4 antigen-loaded tolerance-inducing DC (tolDC) for the treatment of a mixed group of MS and neuromyelitis optica patients (50). Similarly, 2 phase I clinical trials using vitamin D3-treated tolerance-inducing DC (tolDC) loaded with a pool of myelin peptides are ongoing [NCT02618902 and NCT02903537 (51)]. These trials were initiated following promising results in a preclinical setting, with MOG40–55-loaded vitamin D3-treated murine tolDC showing a beneficial effect on the clinical course of EAE (52, 53).
Myelin-Specific T Cell Vaccination
Deletion of myelin-specific T cells can be aimed for by infusion of autologous anti-myelin T cells attenuated by irradiation. By exposure of the immune system to the self-antigens carried by these attenuated T cells, a T cell response leading to deletion or downregulation of autoreactive T cells is induced (54–57). This so-called myelin-specific T cell vaccination was the subject of several open-label clinical trials, followed by a first double-blind, placebo-controlled clinical trial in 2012. In this trial, 17 relapsing progressive MS patients were treated with a mixture of autologous irradiated T cells reactive to nine different myelin-derived peptides, compared to 7 placebo-treated patients (57). In the T cell-treated group, a significant reduction in Expanded Disability Status Scale (EDSS) score 1 year after treatment could be demonstrated in comparison to an increased score in the placebo-treated group, as well as a reduced relapse rate in the T cell-vaccinated group.
DNA Vaccination
Safety of and immune modulation by BHT-3009, a MBP-encoding DNA plasmid, was evaluated in a phase I/II clinical trial in 30 RR-MS and SP-MS patients and was demonstrated to be safe and well tolerated (58). Antigen-specific immune responses were evaluated in a subgroup of patients, demonstrating a significant decrease in myelin-specific proliferation of IFN-γ-producing CD4+ T cells at week 9 and 50 following BHT-3009 administration in all patients who displayed myelin-reactivity at baseline. Moreover, myelin-specific antibody titers were reduced in the CSF, pointing toward downregulation of myelin-specific immune responses both in the periphery and the central nervous system. Interestingly, tolerance induction was not only confined to MBP but spread to other myelin proteins, both in the CSF and in the peripheral blood.
In a larger phase II clinical trial, 289 RR-MS patients were randomized into three treatment groups comparing placebo, 0.5 mg BHT-3009, and 1.5 mg BHT-3009 (59). Administration was performed intramuscularly at week 0, 2, and 4, followed by 4-weekly administrations until week 44. Treatment with 0.5 mg of BHT-3009 led to a significant reduction in volume of enhancing lesions (51% reduction, p=0.02).
Overcoming Challenges of Current Antigen-Specific Treatment Approaches
Although promising results have been achieved with various of the above-mentioned approaches to induce antigen-specific tolerance in MS, several challenges remain (Table 2). It is currently generally accepted that myelin-derived proteins are the main antigens targeted by autoreactive responses in MS (1). Nonetheless, the wide variety of MS-associated myelin-derived antigens imposes difficulties for the selection of target antigens for antigen-specific therapies. Additionally, there is a high patient-to-patient variability in myelin reactivity responses (60, 61). Moreover, these responses are often dynamic in time, characterized by loss of tolerance against additional endogenous antigens released during an inflammatory or auto-immune exacerbation. This process is also known as epitope spreading. These newly released epitopes are secondary and differ from the dominant epitopes, toward which the initial autoimmune response was targeted (62). Both intramolecular spreading, i.e., development of autoreactivity against new epitopes of the initial targeted protein, and intermolecular spreading, i.e., spread of autoreactivity to other myelin-derived proteins, have been described (63). Additionally, as demonstrated in the clinical trials focusing on APL, unwanted immune responses following myelin tolerization strategies—both disease exacerbations by augmentation of the targeted Th1/Th17 immune response and hypersensitivity reactions by cytokine shift to a Th2 response—remain a matter of concern. Finally, various questions remain in the light of further clinical translation of antigen-specific therapy, including optimal antigen dose and patient stratification in order to select patients likely to benefit from a specific antigen-specific treatment approach. In the following section, we will discuss different approaches to tackle these challenges.

Table 2 Challenges for next-generation antigen-specific treatment approaches for multiple sclerosis (MS).
Lack of Target Antigen Identification, Multi-Epitope Antigen Target and Epitope Spreading
Full-Length Protein Administration by Use of Viral Vectors or Nucleic Acids
Although still requiring knowledge of the target proteins, the use of viral vectors or nucleic acids encoding full-length myelin proteins eliminates the need for prior selection of immune-dominant epitopes, which is in line with the first attempts to induce tolerance in MS using a MBP-encoding DNA vaccine (58, 59). Indeed, following translation of full-length protein encoded by viral vectors or nucleic acids, processing by APC will ensure presentation of a wide variety of naturally processed myelin peptides in a HLA-independent manner.
Viral vector transfection is a versatile method to genetically modify several cell types, including bone marrow cells or differentiated effector cells, to constitutively express myelin proteins. Historically, the use of second-generation viral vectors, such as self-inactivating lentiviral and retroviral vectors, has reduced some of the risks related to vector-based gene therapy such as insertional mutagenesis (64). This has greatly increased the translational potential of this treatment approach. In this context, several preclinical studies demonstrated successful prevention of EAE development following treatment with bone marrow, B or T cells transfected with full-length MOG-encoding retroviral (65–70) or lentiviral (71–73) vectors, as well as with vectors encoding MBP (58, 74–76) or PLP (77). However, to our knowledge, no clinical trials in MS patients using viral vectors are yet planned.
In addition to the use of nuclide acid vaccination with DNA (58, 59), the use of mRNA is gaining interest as well, given its high clinical safety profile because of the transient expression of mRNA and its inability for host genome integration (78, 79). Although direct administration of mRNA has not been investigated in the EAE model, mRNA transfection of carrier cells to induce myelin-derived antigen presentation has been attempted. Indeed, a clinical benefit of treatment with MOG mRNA-electroporated tolerogenic DC (tolDC), carrying a wide spectrum of naturally processed MOG-derived epitopes, was recently demonstrated in MOG35–55 EAE mice (80). This protective effect was accompanied by a decrease in the MOG35–55-specific pro-inflammatory response in the peripheral immune system and was likely driven by suppression of central nervous system inflammation.
Use of Multi-Epitope Fusion Proteins
Tackling of complex multi-targeted myelin reactivity which is dynamic over time—as is the case for MS—can hypothetically be achieved by broad tolerization with a mix of myelin-derived peptides, as has already been attempted in several of the clinical trials described above, however with varying success. Ideally, antigen-specific therapy should tackle all disease-related autoreactive responses concomittantly in order to downregulate pathogenic myelin reactivity. In addition to further expanding the number of peptides in the peptide mix product, the use of artificial multi-epitope fusion proteins may be a next step forward in the field of peptide-instigated tolerance induction, since they have been demonstrated to be superior to myelin peptides mixes in preventing or downregulating EAE (81). Indeed, a globular protein product of a synthetic gene encoding different MS-associated epitopes of MBP, PLP, MOG, myelin-associated oligodendrocyte basic protein and oligodendrocyte-specific protein (designated Y-MSPc), displayed stronger capacity to induce T cell anergy, a cytokine shift, and Treg induction when compared to a similar peptide mix, resulting in more effective suppression and even reversal of EAE (81). Although the mode of action behind this stronger immunomodulatory effect by the artificial protein product remains elusive, the authors suggest multiple mechanisms, including lower degradation and clearance rate, more efficient in vivo uptake of Y-MSPc, different pathways of MHC-class II presentation (81) and—more recently demonstrated—induction of a specific subset of tolerogenic myeloid CD11c+CD11b+Gr1+ DC (82).
Other examples of tolerance induction in EAE using multi-epitope fusion proteins are readily available. For instance, Elliot et al. generated a fusion protein (MP4), containing full-length MBP and the three hydrophilic domains of PLP (83). Treatment of SJL/J mice with MP4 after EAE induction completely suppressed EAE development, even when EAE induction was performed using adoptive transfer of both MBP- and PLP-reactive T cells (83). Similarly, Zhong et al. demonstrated a strong preventive and therapeutic effect on EAE of a fusion protein containing encephalitogenic epitopes of MBP, MOG, and PLP (84). Interestingly, not only PLP139–151-induced EAE was suppressed following intraperitoneal or intravenous administration of the fusion peptide, but also EAE passively induced by T cells reactive against different myelin peptides, demonstrating the ability of the fusion protein to tackle multi-targeted myelin reactivity.
Prevention of Unwanted Immune Responses
Modification of Antigen-Specific T Cell Responses
Direct influence on the T cell response following antigen recognition can be achieved by interference with the T cell-APC interaction or by creation of a tolerogenic environment for antigen presentation, either by fusion of the antigen to tolerizing factors or by antigen presentation using micro- or nanoparticles.
T cells require three signals for full antigen-specific stimulation, i.e., i) interaction of the TCR with MHC-bound antigen on the APC surface, ii) triggering of T-cell bound CD28 by costimulatory molecules CD80 and CD86, and iii) the presence of polarizing cytokines (85). Fusion of disease-specific antigens to molecules involved in this T cell-APC interaction could hypothetically result in tolerance induction by means of antigen presentation while blocking costimulatory signals. In this context, Northrup et al. generated fusion products of PLP139–151 with B7 pathway-targeting peptides mimicking CD28 and CTLA-4. This fusion protein interferes with the interaction with costimulatory molecules CD80 and CD86 (86). Subcutaneous administration of the fusion proteins at day 4, 7, and 10 post-EAE induction reduced EAE severity and suppressed weight loss. A cytokine shift was observed, with reduced splenocyte expression of pro-inflammatory IL-2 and GM-CSF, albeit dependent on the particular peptide that was used (86). To the same extent, bifunctional peptide inhibitors (BPI) have been developed to modify T cell responses. BPI consist of antigenic peptides conjugated to adhesion peptides, binding respectively to MHC and costimulatory or adhesion molecules on APC. Binding of a BPI hampers translocation and segregation of the MHC/TCR and costimulatory molecule complexes, preventing the formation of immunological synapse and subsequent T cell activation (87, 88). For instance, Kobayashi et al. demonstrated that linking of PLP139–151 to CD11a237–246, an intercellular adhesion molecule (ICAM)-1-binding peptide, suppresses PLP-induced EAE severity and incidence. The linked peptide was more effective when compared to a mixture of PLP139–151 and CD11a237–246 peptides (87). To broaden the antigen-specific immune modulation, thereby tackling epitope spreading, Badawi et al. generated a bivalent BPI consisting of both MOG38–50 and PLP139–151 bound to an adhesion molecule. In doing so, both MOG38–50- and PLP139–151-induced EAE was suppressed (88). In the same animal models, the bivalent BPI was superior to MOG38–50-BPI and PLP139–151-BPI alone for the induction of tolerance (88).
Fusion of myelin epitopes to cytokines or other active compounds by covalent binding can be used to skew the antigen-specific response toward a more tolerogenic profile. Binding of the fused cytokine to receptors on APC leads to specific targeting of the neuroantigen to these APC and enhanced antigen presentation (89). Neuroantigen-fusion proteins with granulocyte macrophage-colony stimulating factor (GM-CSF), being a major cytokine involved in development and differentiation of myeloid APC (90), displayed a more than 1000-fold increase in antigen targeting to APC compared to neuroantigen alone (91). Accordingly, subcutaneous administration of GM-CSF-neuroantigen fusion proteins has shown to be effective in the prevention and treatment of MOG35–55- (92, 93), PLP139–151- (92, 93) and MBP69–87 (89, 91)-induced EAE. Similarly, fusion proteins of myelin proteins with IFN-β (89, 94), IL-16 (89, 95), IL-13 (89, 95), IL-10 (95), IL-2 (89, 95, 96), IL-4 (89), and IL-1RA (89, 95) have been tested in Lewis rat or SJL mice EAE models. Of these, IFN-β and IL-16 gave the highest tolerogenic capacity, however still less effective than GM-CSF (89). In all settings, cytokine-neuroantigen fusion proteins were superior in terms of inhibitory capacity over neuroantigen alone (89, 95), which underlines the benefit of antigen targeting to APC.
Targeting of Antigen Expression to Specific Cell Populations
Targeting of myelin expression to specific cells can enhance tolerance induction and reduce off-target effects by specifically guiding the myelin presentation to possibly tolerogenic environments. For instance, following viral transfection, ubiquitous myelin expression can be prevented by targeting specific cell lineages by using vectors in which expression is under the transcriptional control of specific cell-type promotors. Cell lineages of interest include DC (70–72), as major APC controlling the balance between tolerance and immunity, and hepatocytes (74, 97), being part of the tolerogenic environment of the liver. Several viral vector-based cell-targeting treatment approaches have been attempted in the EAE model, which are described in Table 3.
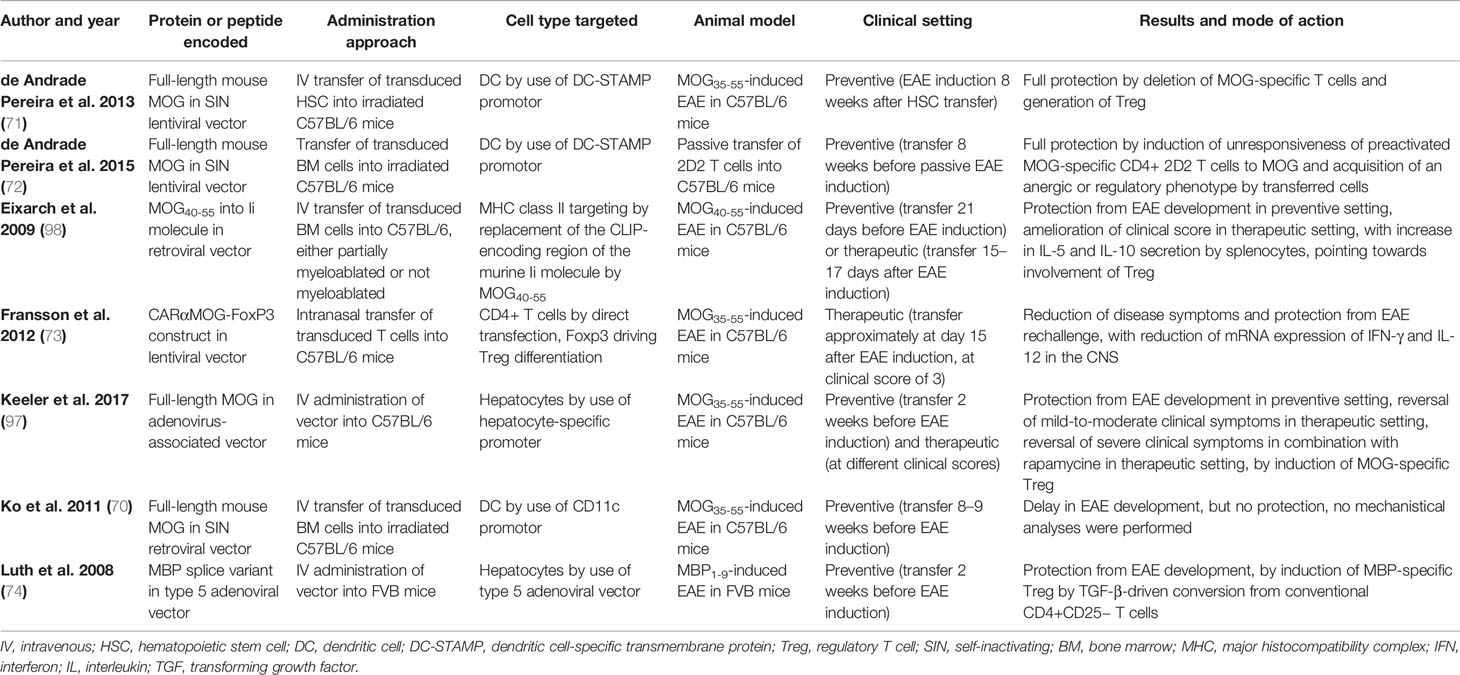
Table 3 Preclinical evaluation of viral vector transfection, targeting specific cell types, for tolerance Figinduction in EAE.
Additionally, fusion proteins can be used for direct targeting. Ring et al. generated a fusion protein of MOG35–55 and single-chain fragment variables (scFv) specific for DEC205, which is a receptor almost exclusively expressed by DC (99). Injection of this fusion protein was shown to be beneficial for both EAE development and progression when mice were treated before (preventively) or after (therapeutically) disease induction, respectively (99). MOG35–55 expression was targeted to DC, which led to significantly reduced levels of TGF-β secretion by DC and increased numbers of IL-10-producing Treg in the spleen (99). Similarly, a fusion product of MOG35–55 and anti-Siglec-H antibodies targeted MOG expression to plasmacytoid DC (pDC) and delayed or decreased clinical signs of EAE when administered in a preventive setting or therapeutic setting, respectively (100).
Micro- and Nanoparticle-Based Systems
Following the success of antigen-coupled cell therapy, micro- and nanoparticles were developed as a delivery vehicle for autoantigens, circumventing the need for autologous blood cells, thereby enhancing clinical translation (101). Micro- and nanoparticles can be used as antigen-delivering vehicles that prevent unwanted immune responses using the strategies mentioned above. Indeed, as reviewed by Kishimoto et al., three strategies can be used for tolerance induction using nanoparticles (102). First of all, nanoparticles can make use of natural tolerance processes, such as antigen presentation without costimulation, oral tolerance, or delivery to the tolerogenic liver environment. For instance, Carambia et al. demonstrated a clinical improvement in EAE mice following a single dose of autoantigen-loaded nanoparticles, specifically targeting to liver sinusoidal endothelial cells, associated with a significant higher frequency of Treg in the spleen of nanoparticle-treated mice compared to vehicle-treated mice (103). Secondly, nanoparticles can be used to specifically target tolerogenic receptors. As an example, a nanoparticle containing MOG35–55 and a plasmid containing the murine B and T lymphocyte attenuator (BTLA) was created by Yuan et al. (104). Following transfection of DC with this plasmid and subsequent administration of these transfected DC prior to induction of MOG35–55 EAE, EAE development could be prevented and was accompanied by an increased frequency of Treg (104). A final approach is to use nanoparticles to co-administer autoantigens together with tolerogenic pharmacological agents, which has been used in the context of EAE in combination with rapamycine (105, 106) and dexamethasone (107). In conclusion, micro- and nanoparticles have been shown to be a versatile treatment modality in preclinical setting, yet no clinical trials are ongoing currently.
Determination of Optimal Antigen Dose
Auto-antigen dose is often extrapolated from dosing from animal models or determined by safety studies, in which the maximal tolerable dose is considered to be the dose of choice. However, the concept of low-zone tolerance, in which low antigen doses are superior in inducing tolerance compared to high doses, has already been known for several decades (108–110). Indeed, also in the context of MS, Garren et al. demonstrated in their phase II clinical trial with the DNA vaccine BHT-3009 that the 0.5 mg group was significantly superior in inducing tolerance compared to the 1.5 mg group, as demonstrated by MRI measures and in vitro PLP reactivity (59). Similarly, as demonstrated by Kappos et al. in their phase II clinical trial using an APL derived from MBP83–99, a significant decrease in the volume and number of Gd-enhancing lesions could only be detected in the patient group treated with the lowest dose (42). On the other hand, high-zone tolerance has been demonstrated for tolerance induction in other autoimmune diseases, including hemophilia (111), leaving the efficacy of low-zone versus high-zone tolerance to be determined for every tolerance-inducing strategy on an individual base. In conclusion, determination of optimal dosing should be based on both tolerability and efficacy.
In addition, optimization of the antigen product formulation to ensure sufficient antigen delivery is warranted for each particular route of administration, since delivery of an appropriate dose of the auto-antigen to the site of interest is of crucial importance for the effective induction of tolerance. For instance, upon oral administration of peptides, passage of low-dose antigen through the gut-associated lymphoid tissue (GALT) induces antigen-specific regulatory T cells (Treg) in the Peyer’s patches (22). However, suppression of ongoing autoimmune reactions, as needed in a therapeutic setting, requires large amounts of oral antigen intake, limiting the clinical applicability of this technique (112, 113). Therefore, generation of fusion proteins with higher efficacy should be aimed for, in which the antigen is either directly targeted to the GALT, e.g., by fusion to cholera toxin subunits (114, 115), or in which higher presentation efficacy can be achieved by fusion to cell membrane-associated proteins (24). Similarly, repeated nasal administration of a fusion protein consisting of cholera toxin subunit B and PLP139–151 hampered full EAE development (114). Hence, also for the nasal route of administration, formulation of the auto-antigen should be optimized.
Patient Stratification
Selection of patients likely to benefit from a particular antigen-specific therapy would aid in the development of patient-tailored therapies. Based on subgroup analyses, the HLA-DR haplotype has been demonstrated to be a parameter of importance in the immunological and clinical response to the induction of myelin-specific tolerance. This is not surprising, giving the role of APC-bound HLA-DR in the antigen presentation to CD4+ T cells. The importance of the HLA-DR haploptype is especially the case for antigen-specific tolerance induction strategies using peptides, given that some myelin peptides are HLA-restricted (116, 117), meaning that they are preferentially presented by specific HLA-molecules. However, clinical trials using HLA-DR haplotype as an inclusion parameter have yielded conflicting results. This is most likely due to confounding by other parameters, which should be taken into account for patient selection as well. This includes among others the presence of pre-treatment reactivity toward the epitopes contained in the antigen-specific therapy. Although of major importance in order to be able to assess antigen-specific immune modulation following treatment, pre-treatment myelin-specific reactivity has not been consistently determined in previously conducted clinical trials, limiting the comparative evaluation of the treatment effect on an immunological level.
Conclusion
Numerous attempts to restore tolerance toward myelin-derived antigens have been made over the past decades, both in animal models of MS and in clinical trials for MS patients. Many of these treatment approaches have shown to be safe and well-tolerated in phase I/II clinical trials, although results regarding efficacy have appeared to be less unequivocal. Given the complexity of the myelin response to be down-regulated, patient selection in terms of HLA haplotype, myelin reactivity, and previous treatment profile is warranted. This would allow efficacy analysis in a more homogeneous patient population and may guide us in the selection of patients who may potentially benefit from a particular treatment. Indeed, a one-treatment-fits-all approach is unlikely to be successful in the field of antigen-specific therapy for MS, underlying the need for more insight into parameters for patient stratification.
Additionally, current preclinical research is providing new approaches to tackle some of the challenges faced by the currently used approaches, including epitope spreading and unwanted immune responses following myelin tolerization attempts. These new findings should altogether allow to modify currently used antigen-specific approaches with the aim to enhance their clinical efficacy.
In conclusion, several decades of research into antigen-specific therapy for MS has yielded promising results and findings from currently ongoing preclinical work may add to the efficacy of this type of treatment. Ultimately, antigen-specific therapy for MS may lead to a more effective therapy for MS by induction of tolerance to a wide range of myelin-derived antigens without hampering the normal surveillance and effector function of the immune system.
Author Contributions
Conceptualization, JD and NC. Writing—original draft, JD. Writing—review and editing, JD, PC, ZB, and NC. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens of multiple sclerosis, part 1: autoreactive CD4+ T lymphocytes as pathogenic effectors and therapeutic targets. Lancet Neurol (2015) 15:198–209. doi: 10.1016/S1474-4422(15)00334-8
2. Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol (2015) 14(4):406–19. doi: 10.1016/S1474-4422(14)70305-9
3. Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol (2010) 162(1):1–11. doi: 10.1111/j.1365-2249.2010.04143.x
4. Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens of multiple sclerosis, part 2: CD8+ T cells, B cells, and antibodies in the focus of reverse-translational research. Lancet Neurol (2016) 15(3):317–31. doi: 10.1016/S1474-4422(15)00313-0
5. Ben-Nun A, Kaushansky N, Kawakami N, Krishnamoorthy G, Berer K, Liblau R, et al. From classic to spontaneous and humanized models of multiple sclerosis: impact on understanding pathogenesis and drug development. J Autoimmun (2014) 54:33–50. doi: 10.1016/j.jaut.2014.06.004
6. Robinson AP, Harp CT, Noronha A, Miller SD. The experimental autoimmune encephalomyelitis (EAE) model of MS: utility for understanding disease pathophysiology and treatment. Handb Clin Neurol (2014) 122:173–89. doi: 10.1016/B978-0-444-52001-2.00008-X
7. Hollenbach JA, Oksenberg JR. The immunogenetics of multiple sclerosis: A comprehensive review. J Autoimmun (2015) 64:13–25. doi: 10.1016/j.jaut.2015.06.010
8. Hirsch DL, Ponda P. Antigen-based immunotherapy for autoimmune disease: current status. Immunotargets Ther (2015) 4:1–11. doi: 10.2147/ITT.S49656
9. Sabatos-Peyton CA, Verhagen J, Wraith DC. Antigen-specific immunotherapy of autoimmune and allergic diseases. Curr Opin Immunol (2010) 22(5):609–15. doi: 10.1016/j.coi.2010.08.006
10. Sundstedt A, O’Neill EJ, Nicolson KS, Wraith DC. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J Immunol (2003) 170(3):1240–8. doi: 10.4049/jimmunol.170.3.1240
11. Dersh D, Yewdell JW. I’ve got algorithm: predicting tumor and autoimmune peptide targets for CD8+ T cells. J Clin Invest (2016) 126(12):4399–401. doi: 10.1172/JCI91302
12. Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res (2015) 43(Database issue):D405–12. doi: 10.1093/nar/gku938
13. Schrempf W, Ziemssen T. Glatiramer acetate: mechanisms of action in multiple sclerosis. Autoimmun Rev (2007) 6(7):469–75. doi: 10.1016/j.autrev.2007.02.003
14. Weiner HL, Mackin GA, Matsui M, Orav EJ, Khoury SJ, Dawson DM, et al. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science (1993) 259(5099):1321–4. doi: 10.1126/science.7680493
15. Goodkin DE, Shulman M, Winkelhake J, Waubant E, Andersson P, Stewart T, et al. A phase I trial of solubilized DR2:MBP84-102 (AG284) in multiple sclerosis. Neurology (2000) 54(7):1414–20. doi: 10.1212/WNL.54.7.1414
16. Warren KG, Catz I, Ferenczi LZ, Krantz MJ. Intravenous synthetic peptide MBP8298 delayed disease progression in an HLA Class II-defined cohort of patients with progressive multiple sclerosis: results of a 24-month double-blind placebo-controlled clinical trial and 5 years of follow-up treatment. Eur J Neurol (2006) 13(8):887–95. doi: 10.1111/j.1468-1331.2006.01533.x
17. Freedman MS, Bar-Or A, Oger J, Traboulsee A, Patry D, Young C, et al. A phase III study evaluating the efficacy and safety of MBP8298 in secondary progressive MS. Neurology (2011) 77(16):1551–60. doi: 10.1212/WNL.0b013e318233b240
18. Yadav V, Bourdette DN, Bowen JD, Lynch SG, Mattson D, Preiningerova J, et al. Recombinant T-cell receptor ligand (RTL) for treatment of multiple sclerosis: a double-blind, placebo-controlled, phase 1, dose-escalation study. Autoimmune Dis (2012) 2012:954739. doi: 10.1155/2012/954739
19. Walczak A, Siger M, Ciach A, Szczepanik M, Selmaj K. Transdermal application of myelin peptides in multiple sclerosis treatment. JAMA Neurol (2013) 70(9):1105–9. doi: 10.1001/jamaneurol.2013.3022
20. Streeter HB, Rigden R, Martin KF, Scolding NJ, Wraith DC. Preclinical development and first-in-human study of ATX-MS-1467 for immunotherapy of MS. Neurol Neuroimmunol Neuroinflamm (2015) 2(3):e93. doi: 10.1212/NXI.0000000000000093
21. Chataway J, Martin K, Barrell K, Sharrack B, Stolt P, Wraith DC. Effects of ATX-MS-1467 immunotherapy over 16 weeks in relapsing multiple sclerosis. Neurology (2018) 2:e93. doi: 10.1212/WNL.0000000000005118
22. Hohol MJ, Khoury SJ, Cook SL, Orav EJ, Hafler DA, Weiner HL. Three-year open protocol continuation study of oral tolerization with myelin antigens in multiple sclerosis and design of a phase III pivotal trial. Ann N Y Acad Sci (1996) 778:243–50. doi: 10.1111/j.1749-6632.1996.tb21132.x
23. Anderton SM, Viner NJ, Matharu P, Lowrey PA, Wraith DC. Influence of a dominant cryptic epitope on autoimmune T cell tolerance. Nat Immunol (2002) 3(2):175–81. doi: 10.1038/ni756
24. Buerth C, Mausberg AK, Heininger MK, Hartung HP, Kieseier BC, Ernst JF. Oral tolerance induction in experimental autoimmune encephalomyelitis with Candida utilis expressing the immunogenic MOG35-55 peptide. PloS One (2016) 11(5):e0155082. doi: 10.1371/journal.pone.0155082
25. Jurynczyk M, Walczak A, Jurewicz A, Jesionek-Kupnicka D, Szczepanik M, Selmaj K. Immune regulation of multiple sclerosis by transdermally applied myelin peptides. Ann Neurol (2010) 68(5):593–601. doi: 10.1002/ana.22219
26. Fukaura H, Kent SC, Pietrusewicz MJ, Khoury SJ, Weiner HL, Hafler DA. Antigen-specific TGF-beta1 secretion with bovine myelin oral tolerization in multiple sclerosis. Ann N Y Acad Sci (1996) 778:251–7. doi: 10.1111/j.1749-6632.1996.tb21133.x
27. Fukaura H, Kent SC, Pietrusewicz MJ, Khoury SJ, Weiner HL, Hafler DA. Induction of circulating myelin basic protein and proteolipid protein-specific transforming growth factor-beta1-secreting Th3 T cells by oral administration of myelin in multiple sclerosis patients. J Clin Invest (1996) 98(1):70–7. doi: 10.1172/JCI118779
28. Hafler DA, Kent SC, Pietrusewicz MJ, Khoury SJ, Weiner HL, Fukaura H. Oral administration of myelin induces antigen-specific TGF-beta 1 secreting T cells in patients with multiple sclerosis. Ann N Y Acad Sci (1997) 835:120–31. doi: 10.1111/j.1749-6632.1997.tb48623.x
29. Bourdette DN, Edmonds E, Smith C, Bowen JD, Guttmann CR, Nagy ZP, et al. A highly immunogenic trivalent T cell receptor peptide vaccine for multiple sclerosis. Mult Scler (2005) 11(5):552–61. doi: 10.1191/1352458505ms1225oa
30. Vandenbark AA, Culbertson NE, Bartholomew RM, Huan J, Agotsch M, LaTocha D, et al. Therapeutic vaccination with a trivalent T-cell receptor (TCR) peptide vaccine restores deficient FoxP3 expression and TCR recognition in subjects with multiple sclerosis. Immunology (2008) 123(1):66–78. doi: 10.1111/j.1365-2567.2007.02703.x
31. Vandenbark AA. TCR peptide vaccination in multiple sclerosis: boosting a deficient natural regulatory network that may involve TCR-specific CD4+CD25+ Treg cells. Curr Drug Targets Inflammation Allergy (2005) 4(2):217–29. doi: 10.2174/1568010053586327
32. Gaur A, Boehme SA, Chalmers D, Crowe PD, Pahuja A, Ling N, et al. Amelioration of relapsing experimental autoimmune encephalomyelitis with altered myelin basic protein peptides involves different cellular mechanisms. J Neuroimmunol (1997) 74(1-2):149–58. doi: 10.1016/S0165-5728(96)00220-2
33. Karin N, Mitchell DJ, Brocke S, Ling N, Steinman L. Reversal of experimental autoimmune encephalomyelitis by a soluble peptide variant of a myelin basic protein epitope: T cell receptor antagonism and reduction of interferon gamma and tumor necrosis factor alpha production. J Exp Med (1994) 180(6):2227–37. doi: 10.1084/jem.180.6.2227
34. Sakai K, Zamvil SS, Mitchell DJ, Hodgkinson S, Rothbard JB, Steinman L. Prevention of experimental encephalomyelitis with peptides that block interaction of T cells with major histocompatibility complex proteins. Proc Natl Acad Sci USA (1989) 86(23):9470–4. doi: 10.1073/pnas.86.23.9470
35. Smilek DE, Wraith DC, Hodgkinson S, Dwivedy S, Steinman L, McDevitt HO. A single amino acid change in a myelin basic protein peptide confers the capacity to prevent rather than induce experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA (1991) 88(21):9633–7. doi: 10.1073/pnas.88.21.9633
36. Wraith DC, Smilek DE, Mitchell DJ, Steinman L, McDevitt HO. Antigen recognition in autoimmune encephalomyelitis and the potential for peptide-mediated immunotherapy. Cell (1989) 59(2):247–55. doi: 10.1016/0092-8674(89)90287-0
37. Wauben MH, Joosten I, Schlief A, van der Zee R, Boog CJ, van Eden W. Inhibition of experimental autoimmune encephalomyelitis by MHC class II binding competitor peptides depends on the relative MHC binding affinity of the disease-inducing peptide. J Immunol (1994) 152(8):4211–20.
38. Franco A, Southwood S, Arrhenius T, Kuchroo VK, Grey HM, Sette A, et al. T cell receptor antagonist peptides are highly effective inhibitors of experimental allergic encephalomyelitis. Eur J Immunol (1994) 24(4):940–6. doi: 10.1002/eji.1830240424
39. Santambrogio L, Lees MB, Sobel RA. Altered peptide ligand modulation of experimental allergic encephalomyelitis: immune responses within the CNS. J Neuroimmunol (1998) 81(1-2):1–13. doi: 10.1016/S0165-5728(97)00138-0
40. Nicholson LB, Greer JM, Sobel RA, Lees MB, Kuchroo VK. An altered peptide ligand mediates immune deviation and prevents autoimmune encephalomyelitis. Immunity (1995) 3(4):397–405. doi: 10.1016/1074-7613(95)90169-8
41. Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med (2000) 6(10):1167–75. doi: 10.1038/80516
42. Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. Altered Pept Ligand Relapsing MS Study Group Nat Med (2000) 6(10):1176–82. doi: 10.1038/80525
43. Crowe PD, Qin Y, Conlon PJ, Antel JP. NBI-5788, an altered MBP83-99 peptide, induces a T-helper 2-like immune response in multiple sclerosis patients. Ann Neurol (2000) 48(5):758–65. doi: 10.1002/1531-8249(200011)48:5<758::AID-ANA9>3.0.CO;2-2
44. Deraos G, Rodi M, Kalbacher H, Chatzantoni K, Karagiannis F, Synodinos L, et al. Properties of myelin altered peptide ligand cyclo(87-99)(Ala91,Ala96)MBP87-99 render it a promising drug lead for immunotherapy of multiple sclerosis. Eur J Med Chem (2015) 101:13–23. doi: 10.1016/j.ejmech.2015.06.015
45. Leech MD, Chung CY, Culshaw A, Anderton SM. Peptide-based immunotherapy of experimental autoimmune encephalomyelitis without anaphylaxis. Eur J Immunol (2007) 37(12):3576–81. doi: 10.1002/eji.200737148
46. Tselios T, Apostolopoulos V, Daliani I, Deraos S, Grdadolnik S, Mavromoustakos T, et al. Antagonistic effects of human cyclic MBP(87-99) altered peptide ligands in experimental allergic encephalomyelitis and human T-cell proliferation. J Med Chem (2002) 45(2):275–83. doi: 10.1021/jm0102147
47. Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, et al. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med (2013) 5(188):188ra75. doi: 10.1126/scitranslmed.3006168
48. Turley DM, Miller SD. Peripheral tolerance induction using ethylenecarbodiimide-fixed APCs uses both direct and indirect mechanisms of antigen presentation for prevention of experimental autoimmune encephalomyelitis. J Immunol (2007) 178(4):2212–20. doi: 10.4049/jimmunol.178.4.2212
49. Zurich W. Establishing immune tolerance in multiple sclerosis with peptide-coupled red blood cells. (2018). Available at: https://www.wysszurich.uzh.ch/projects/wyss-zurich-projects/etimsred.
50. Zubizarreta I, Florez-Grau G, Vila G, Cabezon R, Espana C, Andorra M, et al. Immune tolerance in multiple sclerosis and neuromyelitis optica with peptide-loaded tolerogenic dendritic cells in a phase 1b trial. Proc Natl Acad Sci USA (2019) 116(17):8463–70. doi: 10.1073/pnas.1820039116
51. Willekens B, Presas-Rodriguez S, Mansilla MJ, Derdelinckx J, Lee WP, Nijs G, et al. Tolerogenic dendritic cell-based treatment for multiple sclerosis (MS): a harmonised study protocol for two phase I clinical trials comparing intradermal and intranodal cell administration. BMJ Open (2019) 9(9):e030309. doi: 10.1136/bmjopen-2019-030309
52. Mansilla MJ, Contreras-Cardone R, Navarro-Barriuso J, Cools N, Berneman Z, Ramo-Tello C, et al. Cryopreserved vitamin D3-tolerogenic dendritic cells pulsed with autoantigens as a potential therapy for multiple sclerosis patients. J Neuroinflamm (2016) 13(1):113. doi: 10.1186/s12974-016-0584-9
53. Mansilla MJ, Selles-Moreno C, Fabregas-Puig S, Amoedo J, Navarro-Barriuso J, Teniente-Serra A, et al. Beneficial effect of tolerogenic dendritic cells pulsed with MOG autoantigen in experimental autoimmune encephalomyelitis. CNS Neurosci Ther (2015) 21(3):222–30. doi: 10.1111/cns.12342
54. Vandenbark AA, Abulafia-Lapid R. Autologous T-cell vaccination for multiple sclerosis: a perspective on progress. BioDrugs (2008) 22(4):265–73. doi: 10.2165/00063030-200822040-00006
55. Hellings N, Raus J, Stinissen P. T-cell-based immunotherapy in multiple sclerosis: induction of regulatory immune networks by T-cell vaccination. Expert Rev Clin Immunol (2006) 2(5):705–16. doi: 10.1586/1744666X.2.5.705
56. Achiron A, Lavie G, Kishner I, Stern Y, Sarova-Pinhas I, Ben-Aharon T, et al. T cell vaccination in multiple sclerosis relapsing-remitting nonresponders patients. Clin Immunol (2004) 113(2):155–60. doi: 10.1016/j.clim.2004.06.004
57. Karussis D, Shor H, Yachnin J, Lanxner N, Amiel M, Baruch K, et al. T cell vaccination benefits relapsing progressive multiple sclerosis patients: a randomized, double-blind clinical trial. PloS One (2012) 7(12):e50478. doi: 10.1371/journal.pone.0050478
58. Bar-Or A, Vollmer T, Antel J, Arnold DL, Bodner CA, Campagnolo D, et al. Induction of antigen-specific tolerance in multiple sclerosis after immunization with DNA encoding myelin basic protein in a randomized, placebo-controlled phase 1/2 trial. Arch Neurol (2007) 64(10):1407–15. doi: 10.1001/archneur.64.10.nct70002
59. Garren H, Robinson WH, Krasulova E, Havrdova E, Nadj C, Selmaj K, et al. Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann Neurol (2008) 63(5):611–20. doi: 10.1002/ana.21370
60. Saez-Torres I, Brieva L, Espejo C, Barrau MA, Montalban X, Martinez-Caceres EM. Specific proliferation towards myelin antigens in patients with multiple sclerosis during a relapse. Autoimmunity (2002) 35(1):45–50. doi: 10.1080/08916930290005927
61. Greer JM. Autoimmune T-cell reactivity to myelin proteolipids and glycolipids in multiple sclerosis. Mult Scler Int (2013) 2013:151427. doi: 10.1155/2013/151427
62. Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol (2002) 2(2):85–95. doi: 10.1038/nri724
63. Tuohy VK, Yu M, Yin L, Kawczak JA, Johnson JM, Mathisen PM, et al. The epitope spreading cascade during progression of experimental autoimmune encephalomyelitis and multiple sclerosis. Immunol Rev (1998) 164:93–100. doi: 10.1111/j.1600-065X.1998.tb01211.x
64. David RM, Doherty AT. Viral vectors: the road to reducing genotoxicity. Toxicol Sci (2017) 155(2):315–25. doi: 10.1093/toxsci/kfw220
65. Chan J, Ban EJ, Chun KH, Wang S, Backstrom BT, Bernard CC, et al. Transplantation of bone marrow transduced to express self-antigen establishes deletional tolerance and permanently remits autoimmune disease. J Immunol (2008) 181(11):7571–80. doi: 10.4049/jimmunol.181.11.7571
66. Chen XT, Chan ST, Hosseini H, Layton D, Boyd R, Alderuccio F, et al. Transplantation of retrovirally transduced bone marrow prevents autoimmune disease in aged mice by peripheral tolerance mechanisms. Autoimmunity (2011) 44(5):384–93. doi: 10.3109/08916934.2010.541173
67. Chung JY, Figgett W, Fairfax K, Bernard C, Chan J, Toh BH, et al. Gene therapy delivery of myelin oligodendrocyte glycoprotein (MOG) via hematopoietic stem cell transfer induces MOG-specific B cell deletion. J Immunol (2014) 192(6):2593–601. doi: 10.4049/jimmunol.1203563
68. Hosseini H, Oh DY, Chan ST, Chen XT, Nasa Z, Yagita H, et al. Non-myeloablative transplantation of bone marrow expressing self-antigen establishes peripheral tolerance and completely prevents autoimmunity in mice. Gene Ther (2012) 19(11):1075–84. doi: 10.1038/gt.2011.179
69. Nasa Z, Chung JY, Chan J, Toh BH, Alderuccio F, et al. Nonmyeloablative conditioning generates autoantigen-encoding bone marrow that prevents and cures an experimental autoimmune disease. Am J Transplant (2012) 12(8):2062–71. doi: 10.1111/j.1600-6143.2012.04068.x
70. Ko HJ, Chung JY, Nasa Z, Chan J, Siatskas C, Toh BH, et al. Targeting MOG expression to dendritic cells delays onset of experimental autoimmune disease. Autoimmunity (2011) 44(3):177–87. doi: 10.3109/08916934.2010.515274
71. de Andrade Pereira B, Fraefel C, Hilbe M, Ackermann M, Dresch C, et al. Transcriptional targeting of DCs with lentiviral vectors induces antigen-specific tolerance in a mouse model of multiple sclerosis. Gene Ther (2013) 20(5):556–66. doi: 10.1038/gt.2012.73
72. de Andrade Pereira B, Ackermann M, Chaudhary S, Vogel R, Vogt B, Dresch C, et al. Tolerance of activated pathogenic CD4+ T cells by transcriptional targeting of dendritic cells. Gene Ther (2015) 22(5):382–90. doi: 10.1038/gt.2015.6
73. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflamm (2012) 9:112. doi: 10.1186/1742-2094-9-112
74. Luth S, Huber S, Schramm C, Buch T, Zander S, Stadelmann C, et al. Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen-specific Tregs. J Clin Invest (2008) 118(10):3403–10. doi: 10.1172/JCI32132
75. Melo ME, Qian J, El-Amine M, Agarwal RK, Soukhareva N, Kang Y, et al. Gene transfer of Ig-fusion proteins into B cells prevents and treats autoimmune diseases. J Immunol (2002) 168(9):4788–95. doi: 10.4049/jimmunol.168.9.4788
76. Xu B, Scott DW. A novel retroviral gene therapy approach to inhibit specific antibody production and suppress experimental autoimmune encephalomyelitis induced by MOG and MBP. Clin Immunol (2004) 111(1):47–52. doi: 10.1016/j.clim.2003.12.013
77. Xu B, Haviernik P, Wolfraim LA, Bunting KD, Scott DW. Bone marrow transplantation combined with gene therapy to induce antigen-specific tolerance and ameliorate EAE. Mol Ther (2006) 13(1):42–8. doi: 10.1016/j.ymthe.2005.09.002
78. Van Tendeloo VF, Ponsaerts P, Lardon F, Nijs G, Lenjou M, Van Broeckhoven C, et al. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood (2001) 98(1):49–56. doi: 10.1182/blood.V98.1.49
79. Van Tendeloo VF, Ponsaerts P, Berneman ZN. mRNA-based gene transfer as a tool for gene and cell therapy. Curr Opin Mol Ther (2007) 9(5):423–31.
80. Derdelinckx J, Mansilla MJ, De Laere M, Lee WP, Navarro-Barriuso J, Wens I, et al. Clinical and immunological control of experimental autoimmune encephalomyelitis by tolerogenic dendritic cells loaded with MOG-encoding mRNA. J Neuroinflamm (2019) 16(1):167. doi: 10.1186/s12974-019-1541-1
81. Kaushansky N, Kerlero de Rosbo N, Zilkha-Falb R, Yosef-Hemo R, Cohen L, Ben-Nun A. ‘Multi-epitope-targeted’ immune-specific therapy for a multiple sclerosis-like disease via engineered multi-epitope protein is superior to peptides. PloS One (2011) 6(11):e27860. doi: 10.1371/journal.pone.0027860
82. Kaushansky N, Kaminitz A, Allouche-Arnon H, Ben-Nun A. Modulation of MS-like disease by a multi epitope protein is mediated by induction of CD11c(+)CD11b(+)Gr1(+) myeloid-derived dendritic cells. J Neuroimmunol (2019) 333:476953. doi: 10.1016/j.jneuroim.2019.04.013
83. Elliott EA, McFarland HI, Nye SH, Cofiell R, Wilson TM, Wilkinset al. JA. Treatment of experimental encephalomyelitis with a novel chimeric fusion protein of myelin basic protein and proteolipid protein. J Clin Invest (1996) 98(7):1602–12. doi: 10.1172/JCI118954
84. Zhong MC, Kerlero de Rosbo N, Ben-Nun A. Multiantigen/multiepitope-directed immune-specific suppression of “complex autoimmune encephalomyelitis” by a novel protein product of a synthetic gene. J Clin Invest (2002) 110(1):81–90. doi: 10.1172/JCI0215692
85. Corthay A. A three-cell model for activation of naive T helper cells. Scand J Immunol (2006) 64(2):93–6. doi: 10.1111/j.1365-3083.2006.01782.x
86. Northrup L, Sestak JO, Sullivan BP, Thati S, Hartwell BL, Siahaan TJ, et al. Co-delivery of autoantigen and b7 pathway modulators suppresses experimental autoimmune encephalomyelitis. AAPS J (2014) 16(6):1204–13. doi: 10.1208/s12248-014-9671-y
87. Kobayashi N, Kobayashi H, Gu L, Malefyt T, Siahaan TJ. Antigen-specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. J Pharmacol Exp Ther (2007) 322(2):879–86. doi: 10.1124/jpet.107.123257
88. Badawi AH, Siahaan TJ. Suppression of MOG- and PLP-induced experimental autoimmune encephalomyelitis using a novel multivalent bifunctional peptide inhibitor. J Neuroimmunol (2013) 263(1-2):20–7. doi: 10.1016/j.jneuroim.2013.07.009
89. Mannie MD, Blanchfield JL, Islam SM, Abbott DJ. Cytokine-neuroantigen fusion proteins as a new class of tolerogenic, therapeutic vaccines for treatment of inflammatory demyelinating disease in rodent models of multiple sclerosis. Front Immunol (2012) 3:255. doi: 10.3389/fimmu.2012.00255
90. Conti L, Gessani S. GM-CSF in the generation of dendritic cells from human blood monocyte precursors: recent advances. Immunobiology (2008) 213(9-10):859–70. doi: 10.1016/j.imbio.2008.07.017
91. Blanchfield JL, Mannie MD. A GMCSF-neuroantigen fusion protein is a potent tolerogen in experimental autoimmune encephalomyelitis (EAE) that is associated with efficient targeting of neuroantigen to APC. J Leukoc Biol (2010) 87(3):509–21. doi: 10.1189/jlb.0709520
92. Abbott DJ, Blanchfield JL, Martinson DA, Russell SC, Taslim N, Curtis AD, et al. Neuroantigen-specific, tolerogenic vaccines: GM-CSF is a fusion partner that facilitates tolerance rather than immunity to dominant self-epitopes of myelin in murine models of experimental autoimmune encephalomyelitis (EAE). BMC Immunol (2011) 12:72. doi: 10.1186/1471-2172-12-72
93. Islam SM, Curtis AD2, Taslim N, Wilkinson DS, Mannie MD. GM-CSF-neuroantigen fusion proteins reverse experimental autoimmune encephalomyelitis and mediate tolerogenic activity in adjuvant-primed environments: association with inflammation-dependent, inhibitory antigen presentation. J Immunol (2014) 193(5):2317–29. doi: 10.4049/jimmunol.1303223
94. Mannie MD, Abbott DJ, Blanchfield JL. Experimental autoimmune encephalomyelitis in Lewis rats: IFN-beta acts as a tolerogenic adjuvant for induction of neuroantigen-dependent tolerance. J Immunol (2009) 182(9):5331–41. doi: 10.4049/jimmunol.0803756
95. Mannie MD, Abbott DJ. A fusion protein consisting of IL-16 and the encephalitogenic peptide of myelin basic protein constitutes an antigen-specific tolerogenic vaccine that inhibits experimental autoimmune encephalomyelitis. J Immunol (2007) 179(3):1458–65. doi: 10.4049/jimmunol.179.3.1458
96. Mannie MD, Clayson BA, Buskirk EJ, DeVine JL, Hernandez JJ, Abbott DJ. IL-2/neuroantigen fusion proteins as antigen-specific tolerogens in experimental autoimmune encephalomyelitis (EAE): correlation of T cell-mediated antigen presentation and tolerance induction. J Immunol (2007) 178(5):2835–43. doi: 10.4049/jimmunol.178.5.2835
97. Keeler GD, Kumar S, Palaschak B, Silverberg EL, Markusic DM, Jones NT, et al. Gene therapy-induced antigen-specific Tregs inhibit neuro-inflammation and reverse disease in a mouse model of multiple sclerosis. Mol Ther (2017) 187:6346–56. doi: 10.1016/j.ymthe.2017.09.001
98. Eixarch H, Espejo C, Gomez A, Mansilla MJ, Castillo M, Mildner A, et al. Tolerance induction in experimental autoimmune encephalomyelitis using non-.myeloablative hematopoietic gene therapy with autoantigen. Mol Ther (2009) 17(5):897–905. doi: 10.1038/mt.2009.42
99. Ring S, Maas M, Nettelbeck DM, Enk AH, Mahnke K. Targeting of autoantigens to DEC205(+) dendritic cells in vivo suppresses experimental allergic encephalomyelitis in mice. J Immunol (2013) 191(6):2938–47. doi: 10.4049/jimmunol.1202592
100. Loschko J, Heink S, Hackl D, Dudziak D, Reindl W, Korn T, et al. Antigen targeting to plasmacytoid dendritic cells via Siglec-H inhibits Th cell-dependent autoimmunity. J Immunol (2011) 187(12):6346–56. doi: 10.4049/jimmunol.1102307
101. Quintana FJ. Nanoparticles for the induction of antigen-specific Tregs. Immunotherapy (2013) 5(5):437–40. doi: 10.2217/imt.13.25
102. Kishimoto TK, Maldonado RA. Nanoparticles for the induction of antigen-specific immunological tolerance. Front Immunol (2018) 9:230. doi: 10.3389/fimmu.2018.00230
103. Carambia A, Freund B, Schwinge D, Bruns OT, Salmen SC, Ittrich H, et al. Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice. J Hepatol (2015) 62(6):1349–56. doi: 10.1016/j.jhep.2015.01.006
104. Yuan B, Zhao L, Fu F, Liu Y, Lin C, Wu X, et al. A novel nanoparticle containing MOG peptide with BTLA induces T cell tolerance and prevents multiple sclerosis. Mol Immunol (2014) 57(2):93–9. doi: 10.1016/j.molimm.2013.08.006
105. Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci USA (2015) 112(2):E156–65. doi: 10.1073/pnas.1408686111
106. Tostanoski LH, Chiu YC, Gammon JM, Simon T, Andorko JI, Bromberg JS, et al. Reprogramming the local lymph node microenvironment promotes tolerance that is systemic and antigen specific. Cell Rep (2016) 16(11):2940–52. doi: 10.1016/j.celrep.2016.08.033
107. Peine KJ, Guerau-de-Arellano M, Lee P, Kanthamneni N, Severin M, Probst GD, et al. Treatment of experimental autoimmune encephalomyelitis by codelivery of disease associated Peptide and dexamethasone in acetalated dextran microparticles. Mol Pharm (2014) 11(3):828–35. doi: 10.1021/mp4005172
108. Seidel-Guyenot W, Perschon S, Dechant N, Alt R, Knop J, Steinbrink K. Low zone tolerance induced by systemic application of allergens inhibits Tc1-mediated skin inflammation. J Allergy Clin Immunol (2006) 117(5):1170–7. doi: 10.1016/j.jaci.2006.01.014
109. Schmidt T, Lorenz N, Raker V, Reissig S, Waisman A, Weigmann B, et al. Epicutaneous and oral low-zone tolerance protects from colitis in mice. J Invest Dermatol (2016) 136(9):1831–9. doi: 10.1016/j.jid.2016.04.037
110. Kolsch E, Stumpf R, Weber G. Low zone tolerance and suppressor T cells. Transplant Rev (1975) 26:56–86. doi: 10.1111/j.1600-065X.1975.tb00175.x
111. Steinman L, Ho PP, Robinson WH, Utz PJ, Villoslada P. Antigen-specific tolerance to self-antigens in protein replacement therapy, gene therapy and autoimmunity. Curr Opin Immunol (2019) 61:46–53. doi: 10.1016/j.coi.2019.07.011
112. Thompson HS, Staines NA. Could specific oral tolerance be a therapy for autoimmune disease? Immunol Today (1990) 11(11):396–9. doi: 10.1016/0167-5699(90)90158-6
113. Sun JB, Rask C, Olsson T, Holmgren J, Czerkinsky C. Treatment of experimental autoimmune encephalomyelitis by feeding myelin basic protein conjugated to cholera toxin B subunit. Proc Natl Acad Sci USA (1996) 93(14):7196–201. doi: 10.1073/pnas.93.14.7196
114. Yuki Y, Byun Y, Fujita M, Izutani W, Suzuki T, Udaka S, et al. Production of a recombinant hybrid molecule of cholera toxin-B-subunit and proteolipid-protein-peptide for the treatment of experimental encephalomyelitis. Biotechnol Bioeng (2001) 74(1):62–9. doi: 10.1002/bit.1095
115. Kap YS, van Driel N, Arends R, Rouwendal G, Verolin M, Blezer E, et al. Immune modulation by a tolerogenic myelin oligodendrocyte glycoprotein (MOG)10-60 containing fusion protein in the marmoset experimental autoimmune encephalomyelitis model. Clin Exp Immunol (2015) 180(1):28–39. doi: 10.1111/cei.12487
116. Berthelot L, Laplaud DA, Pettre S, Ballet C, Michel L, Hillion S, et al. Blood CD8+ T cell responses against myelin determinants in multiple sclerosis and healthy individuals. Eur J Immunol (2008) 38(7):1889–99. doi: 10.1002/eji.200838023
117. Martin R, Jaraquemada D, Flerlage M, Richert J, Whitaker J, Long EO, et al. Fine specificity and HLA restriction of myelin basic protein-specific cytotoxic T cell lines from multiple sclerosis patients and healthy individuals. J Immunol (1990) 145(2):540–8.
118. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med (2004) 199(7):971–9. doi: 10.1084/jem.20031579
119. Libbey JE, McCoy LL, Fujinami RS. Molecular mimicry in multiple sclerosis. Int Rev Neurobiol (2007) 79:127–47. doi: 10.1016/S0074-7742(07)79006-2
120. Mallucci G, Peruzzotti-Jametti L, Bernstock JD, Pluchino S. The role of immune cells, glia and neurons in white and gray matter pathology in multiple sclerosis. Prog Neurobiol (2015) 127-128:1–22. doi: 10.1016/j.pneurobio.2015.02.003
121. Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol (2005) 23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707
122. Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell (1995) 80(5):695–705. doi: 10.1016/0092-8674(95)90348-8
123. Henderson AP, Barnett MH, Parratt JD, Prineas JW. Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol (2009) 66(6):739–53. doi: 10.1002/ana.21800
124. Traka M, Podojil JR, McCarthy DP, Miller SD, Popko B. Oligodendrocyte death results in immune-mediated CNS demyelination. Nat Neurosci (2016) 19(1):65–74. doi: 10.1038/nn.4193
125. Locatelli G, Wortge S, Buch T, Ingold B, Frommer F, Sobottka B, et al. Primary oligodendrocyte death does not elicit anti-CNS immunity. Nat Neurosci (2012) 15(4):543–50. doi: 10.1038/nn.3062
Keywords: multiple sclerosis, antigen-specific therapy, tolerance induction, myelin, experimental autoimmune encephalomyelitis
Citation: Derdelinckx J, Cras P, Berneman ZN and Cools N (2021) Antigen-Specific Treatment Modalities in MS: The Past, the Present, and the Future. Front. Immunol. 12:624685. doi: 10.3389/fimmu.2021.624685
Received: 31 October 2020; Accepted: 04 January 2021;
Published: 19 February 2021.
Edited by:
Fang Zhou, CAS Lamvac Biotech Co., Ltd., ChinaReviewed by:
Daniel Benitez-Ribas, Hospital Clínic de Barcelona, SpainJun Peng, Shandong University, China
Pablo Villoslada, Institut de Recerca Biomèdica August Pi i Sunyer (IDIBAPS), Spain
Copyright © 2021 Derdelinckx, Cras, Berneman and Cools. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Judith Derdelinckx, SnVkaXRoLmRlcmRlbGluY2t4QHVhbnR3ZXJwZW4uYmU=