 Lilian Gomes de Oliveira1,2†
Lilian Gomes de Oliveira1,2† Yan de Souza Angelo1,2†
Yan de Souza Angelo1,2† Antonio H. Iglesias3
Antonio H. Iglesias3 Jean Pierre Schatzmann Peron1,2,3*
Jean Pierre Schatzmann Peron1,2,3*- 1Neuroimmune Interactions Laboratory, Immunology Department – Institute of Biomedical Sciences (ICB) IV, University of São Paulo (USP), São Paulo, Brazil
- 2Neuroimmunology of Arboviruses Laboratory, Scientific Platform Pasteur-USP, University of São Paulo (USP), São Paulo, Brazil
- 3Loyola University Medical Center, Stritch School of Medicine, Loyola University Chicago, Chicago, IL, United States
Neuroinflammatory and neurodegenerative diseases are a major public health problem worldwide, especially with the increase of life-expectancy observed during the last decades. For many of these diseases, we still lack a full understanding of their etiology and pathophysiology. Nonetheless their association with mitochondrial dysfunction highlights this organelle as an important player during CNS homeostasis and disease. Markers of Parkinson (PD) and Alzheimer (AD) diseases are able to induce innate immune pathways induced by alterations in mitochondrial Ca2+ homeostasis leading to neuroinflammation. Additionally, exacerbated type I IFN responses triggered by mitochondrial DNA (mtDNA), failures in mitophagy, ER-mitochondria communication and mtROS production promote neurodegeneration. On the other hand, regulation of mitochondrial dynamics is essential for CNS health maintenance and leading to the induction of IL-10 and reduction of TNF-α secretion, increased cell viability and diminished cell injury in addition to reduced oxidative stress. Thus, although previously solely seen as power suppliers to organelles and molecular processes, it is now well established that mitochondria have many other important roles, including during immune responses. Here, we discuss the importance of these mitochondrial dynamics during neuroinflammation, and how they correlate either with the amelioration or worsening of CNS disease.
Introduction
The central nervous system (CNS) depends on a complex and intricate network of molecular and cellular interactions to maintain appropriate function and homeostasis. This well-organized network when disturbed, leads to resident cells activation, inflammatory leukocyte infiltration, and further tissue damage. During recovery, counterregulatory mechanisms take place, and the activated cells return to the homeostatic state. However, in the absence of these finely tuned regulatory loops, the coordination is broken and chronic neurodegenerative and neuroinflammatory diseases may occur.
Neurodegenerative diseases represent a heterogeneous group of diseases of major public health concern. The World Health Organization (WHO) have estimated that until 2030, deaths attributed to neurological diseases will increased up to 12.22% (1). Due to their overly complex pathophysiology, interdisciplinary approaches and breakthrough science are highly needed to unravel disease mechanisms, and thus developing effective new therapies. In fact, with the extension of our life expectancy and the dramatic change in the age pyramid during the last decades, these studies are mandatory either to avoid or delay their impact on society and economy.
A common feature of neurodegenerative and neuroinflammatory diseases is the activation of CNS resident cells (2–4). Microglia and astrocytes may actively start, promote, or dampen neuroinflammation (5–8). The reason is because many immune-related receptors and molecules are extensively produced by these cells, not only during disease but also during physiological processes (3, 9). Conversely, these mechanisms demand high energy consumption, promoting important metabolic changes in the cell. In this context, the importance of mitochondria and mitochondria-related pathways is unquestionable.
More than just power houses of the cells (10), the role of mitochondria have been remarkably appreciated and revisited. Recent research has revealed important correlation of mitochondrial dynamics and the pathophysiology of brain diseases, as Alzheimer’s. Disease (AD), Parkinson’s Disease (PD) and Multiple Sclerosis (MS) (11–13). Disturbances in mitochondrial dynamics may influence many cellular and molecular pathways, as calcium-dependent immune activation, transcription factors phosphorylation, cytokine secretion, organelle transference and even cell death. Moreover, dysfunctional dynamics may also affect the release of mitochondrial damage-associated molecular patterns (mDAMPs), triggering innate immune responses in both resident and infiltrating cells (14). The release of mDAMPs leads to NOD-like receptors (NLRs), Toll-like receptors (TLRs) and cGAS-STING activation, promoting inflammatory cytokine, chemokines, and reactive oxygen species production, impacting disease outcome. However, although much has been learned regarding mitochondrial function during health and disease, mitochondrial dynamics during neuroinflammation and neurodegenerative disorders remains to be fully elucidated. Here, we aimed to summarize recent knowledge in the field, correlating dysfunctional mitochondrial dynamics with the worsening of CNS diseases.
Mitochondrial Dynamics and Neuroinflammation
Mitochondrial dynamics is a process by which this organelle changes size, location, shape, and function inside the cell (15). Mitochondrial fusion and fission greatly correlate with metabolic changes, depending on the stimuli and energy demand, as it regulates cellular functions during health as well as during disease. There is now a better understanding of the changes that occur during mitochondrial dynamics changes and its relationship with CNS resident cells.
Mitochondrial Location and Mitochondria-ER Communication
Mitochondria location within the cell is mostly regulated by the outer mitochondrial membrane (OMM), anchoring it to the cytoskeleton’s microtubules motor proteins, kinesin and dynein (16). For instance, mitochondria’s position in astrocytes influences Ca2+ levels, directly affecting astrocyte survival and communication with nearby neurons (17). Intracellular calcium level is dictated by the transferring between mitochondrial reticulum (mitRet) and endoplasmic reticulum (ER). Remarkably, several neurodegenerative diseases correlate with detrimental calcium homeostasis, evidenced by the disruption of mitRet and ER communication, as observed in Amyotrophic Lateral Sclerosis (ALS), a severe condition characterized by progressive weakness, muscle wasting and paralysis. Impaired electron transportation chain (ETC) and reduced glutamate uptake were already described in ALS. This greatly increases Ca2+ permeable activation of AMPA receptors, leading to excitotoxicity (16).
Mitochondria-ER associated membranes (MAMs) consist of around 1500 active proteins. Regardless of their fundamental importance for cellular metabolism and Ca2+ homeostasis, the molecular mechanisms that underly the recruitment and tethering of ER-mitochondria are not fully understood, and extensively debated. It has been proposed that MAMs tethering is dependent on the interaction between mitofusin 2 (MFN2) in the ER, and MFN1 and MFN2 in the OMM (16). Supporting this, ablation of MFN2 loosens ER–mitochondria interaction strongly impairing mitochondrial Ca2+ uptake (18). However, the role of MFN2 is not a consensus in the literature and some studies consider MFN2 a tethering antagonist that suppresses the excessive binding between the organelles, preventing toxic Ca2+ transfer within mit-Ret and ER (19, 20).
Tyrosine phosphatase-interacting protein 51 (PTPIP51), and the integral ER protein vesicle-associated membrane protein-associated protein B (VAPB) are also listed as tethering molecules for MAMs formation. Interestingly, during ALS, fronto-temporal dementia (FTD) and PD, disruption in the PTPIP51-VAPB interaction also induces dysregulated Ca2+ homeostasis and decreased ATP production (21, 22). Strikingly, it was orchestrated by fused in sarcoma (FUS) protein, and not by directly altering PTPIP51- VAPB expression, but by activating glycogen synthase kinase 3-beta (GSK-3b), evidencing a correlation between this molecule and ALS.
Additionally, the communication between mitochondrial voltage-dependent anion channel (VDAC) and inositol 1,4,5-trisphosphate receptor (IP3R) within the ER membrane via GRP75 was suggested as a bond of MAMs (23, 24). Curiously, the PD associated protein, DJ1, is necessary for mitochondrial Ca2+ uptake and was related with VDAC-IP3R-GRP75 complex in the maintenance of MAMS (25). Noteworthy the fact that mitochondrial Ca2+ uptake occurs in response to ER IP3R activation (19) and that fluctuations in channel activity does not affect the binding of MAM (26). Moreover, it is important to mention that IP3R activation is an important signaling pathway for immune response (27), as observed for nuclear factor of activated T cells (NFAT) (28).
NFAT activation typically leads to the transcription of inflammatory mediators that are upregulated during some neurodegenerative diseases. For instance, amyloid beta (Aβ) protein uptake by microglia induces dysregulated NFAT expression, increasing TNF-α secretion and neuronal death in vitro (29). Interestingly, Aβ and α-synuclein deposition, hallmarks of AD and PD, can both trigger inflammatory responses via TLR-2 and TLR-4, respectively (30–32). Conversely, Ma et al. (33) demonstrated that the crosstalk between TLR-4 and NFAT1 signaling into the mitochondria is a TRIF-dependent phenomenon, culminating in pro-inflammatory cytokine and ROS production, mitochondrial morphological changes and finally, prolonged microglia activation (33).
Corroborating the proinflammatory statement, it was demonstrated that cytokine activation of primary astrocytes and microglia upregulate intracellular Ca2+ mobilization and NFAT activation. NFAT upregulated genes are associated with a neurotoxic phenotype of astrocytes, known as A1 astrocytes. In A1 astrocytes (C3+GBP2+), NFAT is positively regulated by IL-1β and, in a positive feedback loop, IL-1β expression is NFAT and L-type Ca2+ channel dependent (34). Thus, bidirectional interactions between ER and mitochondrial Ca2+ levels, NFAT activation and upregulated inflammatory mediators, sustain a positive feedback loop that correlate with the chronicity of the neuroinflammatory microenvironment.
Mitochondrial Dynamics and Programmed Cell Death
Besides location and interaction with other organelles within the cell, mitochondrial fusion and fission is a crucial process for regulating cell death. Fusion is coordinated by a family of GTPase proteins with tethering activity. This family of proteins is localized on the outer mitochondria membrane (OMM), highlighting MFN1 and MFN2, and in the inner mitochondria membrane (IMM), optic atrophy 1 (OPA1) (35). The steps that orchestrate mitochondrial elongation are not fully understood, but a model suggests that the interaction of MFNs from two opposing mitochondrion is stabilized by coil-coil heptad repeat-2 (HR2) (36), increasing the surface of contact (37). Following this, at the interaction site, guanosine triphosphate (GTP) is hydrolyzed culminating in conformational changes in the MFNs and thus, OMMs fusion. Different isoforms of OPA-1 such as long membrane-bound OPA1 (L-OPA1) and short soluble OPA-1(S-OPA1), generated by proteolytic cleavage of L-OPA1, are associated with fusion and fission balance (38). Complete fusion occurs when cardiolipin (CL) interacts with L-OPA1 resulting in IMM unification following OPA1-dependent GTP hydrolysis (39). Mitochondria elongation is associated with efficient metabolism and maintenance of ATP production even during nutrient deprivation, thus increasing cellular viability (40).
On the opposite, mitochondrial fission is initiated following the assembly of a pre-constriction site, directing the dynamin related protein 1 (DRP1) binding site of the OMM. One of the proteins that compose the pre-constriction site is fission protein 1 (FIS1), that also inhibits fusion by preventing GTP hydrolysis of OPA1 and MFN1/2 (41). The constriction site is not randomly located, instead is pinpointed on ER-mitochondria interaction site (42). ER tubules induce actin polymerization at the narrowing site, whereas myosin mediates actin contraction and mechanical pressure to ensure pre-constriction. Then, DRP1 is recruited forming a ring-like oligomer which following the GTP hydrolysis squeezes the pre-existing constriction site. Lastly, dynamin 2 is recruited to DRP1-compression site for mitochondrial fragmentation (43). The processes that coordinate the OMM fission are better known that the ones related to IMM. So far, it was shown that prior to DRP1 recruitment, Ca2+ promotes constriction in the IMM by favoring proteolytic cleavage of OPA-1 (44). Strikingly, the pre-constriction site is also spatially linked and critical to maintain mtDNA replication in the matrix (42). Fragmented mitochondria tend to present increased stress oxidation, membrane depolarization and impaired ATP production (45).
OPA1 and MFN2 genes are essentially related to the formation of healthy mitochondrial networks. Mechanistically, total or partial loss of function of OPA1 results in fragmented mitochondria, leading to a loss of mitochondria membrane potential (ΔΨm) and thus initiating autophagic and apoptotic pathways (46, 47). Importantly, in the CNS, these alterations may lead to massive neuronal and glial cell death, as seen in optic atrophy-1 and Charcot-Marie-Tooth disease and hereditary peripheral neuropathy (46, 47). Only recently, studies have described the role of mitochondrial fusion and fission in programmed cell death due to DRP1 and MFN2 interaction with BAX and BAK, respectively. As a result of exposure to toxic levels of nitric oxide (NO), BAX interacts with DRP1 in neurons inducing mitochondria fragmentation. In this context, inhibition of DRP1 impairs BAX deposition and pore formation, improving neuronal survival (48, 49).
The degradation of damaged organelles and cytosolic components usually results in autophagy, leading to the delivery of damaged cellular components to autophagosomes for degradation (50, 51). Mitochondria specialized autophagy, named mitophagy, is triggered by OPA-1, DRP1 and MFN2. These proteins, besides their regulatory role in mitochondria dynamics, are also responsible for autophagosome formation (50, 51). Mitophagy initiation is also dependent on PTEN−induced putative kinase 1 (PINK1) and E3-ubiquitin ligase protein (Parkin). These proteins accumulate in the OMM and ubiquitinate mitochondria target proteins (52, 53). As OMM has plenty of PINK1 and MFN2, Parkin is recruited from the cytoplasm and phosphorylated, hence exerting its ubiquitin activity (54). Consequently, DRP1, NF-κB essential modulator (NEMO) and mitochondrial Rho GTPase protein 1 (MIRO1) are targeted for proteasomal degradation (55, 56). Mitophagy is consolidated when MIRO1 is degraded by the proteasome and mitochondria is detached from its anchoring microtubules (57). Lastly, mitochondria is sequestrated in a double membrane vesicle that fuses with autolysosomes that further “digests” the organelle (58). Importantly, the correlation between Parkin and PINK1 mutations to autosomal-recessive cases of PD is widely known (59). In this context, the unbalance between damaged mitochondria and its removal, importantly contributes for PD progress. This unbalanced mitochondrial dynamic correlates with impaired clearance of dysfunctional organelles through Parkin and PINK1 pathway, resulting in deleterious accumulation. The benefits of mitophagy, however, goes beyond the removal of damaged mitochondria (59). Ip et al. (60) demonstrated that PINK1 is essential for microglial secretion of IL-10 and reduction of TNF-α secretion. Remarkably, elevated IL-10 secretion correlated with mitophagy induction in macrophages via mTORC1 inhibition and consequently decreasing inflammation (60). Using a mouse microglial cell line, it was shown that mitophagy increases ΔΨm and diminishes TNF-α induced apoptosis by hampering the increase in pro-apoptotic proteins (61). Furthermore, the role of mitophagy during neurodegenerative diseases overcome the regulation of immune responses. In a mouse model of AD, microglial cells under mitophagy have elevated levels of intracellular Aβ aggregates, suggesting increased phagocytic activity, and thus clearing the harmful Aβ deposits (62).
Autophagy may be beneficial to rebuild healthy mitochondrial dynamics after pro-inflammatory responses. Following mitochondrial fragmentation, autophagy is triggered due to dysregulated respiratory chain and ROS production. Such mitochondria alterations are promoted by IFN-γ and LPS upregulation of DRP1 and LC3, an autophagy-related protein. For instance, this mechanism is essential to restore tubular mitochondrial networks after inflammatory stimulation in astrocytes, as shown in a mouse model of cortical lesion. Interestingly, astrocytes located in the core or penumbra exhibited different mitochondrial patterns, with core mitochondria prominently fragmented, as opposed to those in the penumbra (63). This evidences the importance of the neuroinflammatory microenvironment in orchestrating mitochondrial shape and size.
Astrocyte-to-Neurons Mitochondria Exchange
Many factors released from astrocytes provide neurotrophic and metabolic support for nearby neurons. These range from DNA, microRNAs, glucose-related molecules, cytokines and even organelles, such as the mitochondria (64, 65). Despite not completely understood, several reports have already demonstrated the importance of damaged and healthy mitochondria transference in between cells for neuronal metabolism and survival. During brain injury, astrocytes may release damaged mitochondria to minimize the amount of detrimental ROS and dysregulated Ca2+ balance (66). Conversely, healthy mitochondria may also be donated from astrocytes to damaged neighboring neurons, increasing its viability (67). Moreover, Davis et al. (68), firstly demonstrated that the exchange of mitochondria among neurons and astrocytes seem to work in a bidirectional way (68).
The release of mitochondria by astrocytes is a CD38/Ca2+ dependent phenomenon (69). It upregulates survival pathways in neurons after stroke, indicating a neuroprotective role for the glia-to-neuron mitochondria communication. Also, mitochondria acquired from astrocytes have a crucial role in maintaining neuronal energy production under glucose-oxygen starvation.
Joshi et al. (70) showed that previous mitochondria fragmentation is an essential step for organelle release to the extracellular space (70). They observed that inhibition of DRP1 diminishes astrocytic and microglial activation and ameliorates pro-inflammatory phenotype in mice models of AD, ALS and Huntington’s disease (HD). Interestingly, this phenotype was dependent on the release of damaged mitochondria by microglia cells, triggering neuronal death in consequence of A1-inflammatory-astrocyte activation (70).
Mitochondrial DAMPs and Neurodegeneration
Since Polly Matzinger’s “danger theory” (71), the introduction of damage-associated molecular patterns (DAMPs) has greatly broadened our understanding of how the immune system works during tissue damage and repair (71). The idea of recognizing “danger” and “alarm” signals produced by cells, as DNA, ATP and HSPs (heat-shock proteins) (72), during inflammatory events, completely changed the view of inflammatory processes. Naturally, many advances in the biology of danger signals, along with the discovery of stress-associated molecules acting as DAMPs were achieved (71, 73). Accordingly, one important source of DAMPs that has gained increased attention is the mitochondria.
Examples of mDAMPs receptors are the classical PRRs (pattern recognition receptors), such as the TLRs, NLR (NOD-like receptors), as well as STING (stimulator of interferon-genes) and RAGE (receptor for advanced glycation products) (74). It is important to note that the signaling of these receptors, ultimately lead to inflammatory responses that may promote an auspicious environment for neurodegeneration. Moreover, the activation of TLR-7/9 and STING induces a IFN-I response (75) which has been recently demonstrated by microglial single-cell analysis in mice that, during aging, three clusters of interferon-responsive microglia appear, and that they correlate with subsequent CNS disease (76). Although the role for mitochondrial dynamics in this phenomenon is still to be addressed, the existence of mDAMP-IFN pathways denotes a possible correlation, as reviewed (74).
Cardiolipin is a phospholipid located at the IMM providing the structure for the electron transportation chain (ETC), binding to Complex IV and maintaining other ETC complexes and mitochondrial content in place and sharply functional (77). Cardiolipin molecules are particularly sensitive to oxidative damage created by unbalanced mitochondrial functioning and/or ROS produced by activated microglia. Interestingly, a highly oxidative environment causes loss of Δψm and promotes the repositioning of cardiolipin molecules to the OMM (77). Then, it can be sensed by cytosolic immune receptors, as NLRP3, initiating inflammasome activation, inflammatory cytokine secretion and dysregulation of mitochondrial dynamics (78). Other effects include loss of functional ETC and release of intrinsic apoptotic molecules located between the IMM and OMM, such as cytochrome C and SMAC/Diablo (79, 80). The release of Mitochondrial Transcription Factor A (MTFA) can also initiate inflammatory processes when released extracellularly. This is because MTFA shares high homology with High Mobility Group Box (HMGB), an important DAMP, and thus proinflammatory (65). Interestingly, a subset of Gamma-delta T cells (Tγδ) increased in Multiple Sclerosis (MS) patients has been shown to be activated by cardiolipin. Although their exact role is still not clear, it suggests an important role for cardiolipin also activating adaptive immune responses during CNS disease progression (81).
Mitochondrial DNA is the most studied mDAMP, and it has a high correlation with many pathological processes. Among their distinct characteristics, mtDNA codes only 13 proteins, including mitochondrial ribosomal subunits and ETC components, essential for proper mitochondrial function (82, 83). Failures in mitochondrial dynamics often result in the accumulation of mutated mtDNA, as they lack a robust repair mechanism (83, 84). This affects the cellular capacity in producing energy and also set in motion inflammatory processes (83, 84). mtDNA is not packed and has motifs usually perceived as harmful by innate immunity receptors. During mild stressful situations when apoptotic caspases are not mobilized, loss of mitochondrial membrane action potential (Δψm) for example, facilitates both OMM and IMM permeabilization and the induction of BAX/BAK pores, enabling mtDNA release to the cytosol. Then, it can be sensed by the cyclic GMP-AMP synthase (cGAS) receptor, activating its adaptor protein STING (stimulator of interferon genes) (85). Interestingly, this mtDNA-dependent Type I interferon (IFN-I) induction is beneficial in the context of viral infection because it primes the cell into an antiviral state (86). Still, the same issue occurs when mtDNA is present extracellularly, as internalized mtDNA signals through endosomal TLR-9, resulting in NF-κB and IRFs activation, and further interferon transcription (87, 88).
Another important mDAMP are mtROS produced at high levels by the mitochondria (89). Mitochondrial ROS (mtROS) are mainly byproducts of the mitochondrial ETC. During respiration, O2 that does not get reduced into H2O forms the radical specially by the activity of ETC complexes I and III, which can be later converted in H2O2 mainly by enzymes that are present in the organelle (90, 91). Other mitochondrial and cellular events can also enhance the production of mtROS, such as a decreased ΔΨm, inhibition of the ETC, mitochondrial Ca2+ influx, oxygen concentration and even mitochondrial morphology (90, 92). These molecules can act both as signaling (93) or as damaging molecules, causing mtDNA mutations, oxidation in fatty acids and amino acid residues, leading to deleterious disruption of the cellular redox signaling (94–96). Specially in the brain, the damage caused by ROS are linked to protein aggregation (93, 97).
In summary, although the mitochondria represent a vital organelle, providing energy and regulating metabolic processes of the cell, it also represents a “time-bomb” capable of inflicting and propagating devastating damage to the organism. This characteristic is especially significant on the CNS, where most resident cells are extremely susceptible to mitochondrial dysfunctions, as evidenced by the metabolic linked pathogeny of the neurodegenerative diseases, that will be further described in this review.
UnDAMPening Mitochondria in Neurodegenerative Diseases
Mitochondria and Type I Interferon Responses
As mentioned, mDAMPs may signal through PRRs resulting in type I interferon responses (75, 98). Although their importance is mostly known during viral infections (99, 100), IFN-I responses in the CNS have a dual effect. In fact, there are evidences showing that IFN-I responses linked to mDAMPs in neurodegenerative diseases may also have a neuroprotective role (101, 102). Type IFN-I responses in the CNS have already been greatly reviewed by Deczkowska and colleagues (103), in which they discuss this complex signaling network duality. As an example, studies demonstrating that T cells derived IFN-I and IFN-γ are crucial to the blood brain barrier (BBB) permeability, as well as to the production of neurotrophic factors that aid the maintenance of cognitive functions. Corroborating this, the lack of IFN-β in the brain of knockout mice promotes progressive cognitive loss and impaired motor function (104).
Recognized mainly by TLR-9 and STING (75, 105), mtDNA has already been shown to be elevated in the serum of patients with ischemic brain injury (106, 107). Type I IFNs signaling rely on the interaction with Type I Interferon Receptors (IFNAR) and subsequent intracellular cascades that culminates in the phosphorylation of Jak-STATS and IRFs, and the transcription of Interferon Stimulated Genes (ISGs) (100). IFNAR are present in many cell types and, recently, transcriptome analysis in the brain showed the expression of IFNAR1 and IFNAR2 in almost every brain cell subset, including glial cells and neurons (108). In the CNS, the major IFN-αβ secreting sources are astrocytes and microglia. However, during neuroinflammation, disruption of BBB integrity facilitates the infiltration of IFN-I secreting cells, as monocytes and neutrophils (103, 108–111). Moreover, it is important to mention that the concept of CNS immune privilege is being extensively revisited, as many cell types are being described in the brain-circulation interfaces, as the choroid plexus and the meninges (112, 113), as well as within a complex network of lymphatic vessels (114).
It is also known that autocrine action of IFN-I induces significant shifts of the cellular metabolism, as augmented fatty acid oxidation and OXPHOS (65, 115, 116). These shifts are particularly important to astrocytes, as they rely on a tight controlled metabolite production to energetically supply nearby neurons, mainly by the lactate shuttle. For example, unbalanced glycolysis lowers the expression of glutathione, facilitating oxidative damage (65). Zhang et. al. 2019 recently reported that lactate, a key metabolite of the glycolytic pathway, interacts with the Mitochondrial Antiviral-Signaling protein (MAVS), preventing its oligomerization and maintaining Hexokinase 2 activity (HK2) (117). Although this was described using a model of viral infection, MAVS is increasingly showing to interact with many important enzymes beyond its role as an adaptor for cytosolic PRRs RIG-I and MDA-5. Importantly, the signaling trough MDA-5/MAVS has shown to be one of the main recognition pathways of cytosolic mtDNA, as evidenced during BAK/BAX mitochondrial disruption and failures of mtDNA turnover, being responsible for the major mtDNA induced IFN-I response (118, 119). Also, recent reports evidenced that phospholipase A2 binds to MAVS causing a disruption in the HK2 activity and increasing NF-κB phosphorylation, a novel pathway described in the experimental autoimmune encephalomyelitis (EAE) model (120). This not only give us important cues on how mtDAMPs and dysregulated type I IFN responses could accentuate neurodegenerative diseases, but also brings us new therapeutic avenues.
Mitochondria as Borrowers: Sphingolipid Metabolism and Demyelination Processes
As discussed earlier, the metabolism of sphingolipids is an important player in neuroinflammation, as they critically participate in myelin maintenance (121, 122). For example, the compromised action potential of neurons, that is caused by intracellular changes or by the progressive loss of myelin due to metabolic failure of oligodendrocytes (123), clearly evidences the importance of a proper mitochondrial functioning (124).
The myelination process is tightly regulated both during neurodevelopment and tissue repair, when oligodendrocytes keep contributing for myelin remodeling (125–127) and remyelination (128). It requires great amount of energy, leading to high oxygen and ATP consumption, evidenced by high mitochondrial content within oligodendrocyte’s interface with myelin sheets (129). Thus, oligodendrocytes support the long-term myelination by maintaining high glycolytic rates. Conversely, mtDNA mutations in mitochondrial complex IV (mCOX-IV) subunits correlate with more extensive demyelination (129, 130). This phenomenon, along with increased iron deposits in oligodendrocytes, lowers the expression of anti-oxidative enzymes, rendering this cell population exceptionally susceptible to oxidative damage (125, 131), facilitating disease progression.
These characteristics are most evident in diseases as MS and AD, where the inflammatory milieu may drastically affect oligodendrocytes. Cytokines as IL-1, TNF-α and IFN-γ, may cause important mitochondrial distress. In fact, IFN-I impair glycolysis in oligodendrocytes, which is crucial for maintaining axonal integrity through myelin remodeling (132–134).
Furthermore, myelin production itself can be targeted during CNS pathologies. The sphingomyelinase/ceramide pathways play important roles in oligodendrocyte death by promoting the release of ceramide. Ceramide is the precursor of sphingomyelin lipids, the main component of myelin (135). This molecule, like other sphingolipids, has important bioactive functions, as promoting apoptosis and cell cycle arrest (11, 135). Increasing evidences has shown that ceramide can act directly on mitochondria (122) and also activate the NLRP3 inflammasome (136). In rat liver, it has been demonstrated that ceramide can be found in intimate contact with the IMM and OMM, thus leading to loss of Δψm and activating intrinsic apoptotic pathways and mitochondrial dynamics disbalance (137). This has also been discussed during CNS inflammation, as inflammasome activation by ceramide leads to hyperphosphorylation of leptin receptor (Obr) and thus abrogating signaling pathway, as observed during obesity and metabolic syndrome (135, 138).
A STING in the Brain
Although the loss of Δψm is mostly studied during mitochondrial distress, novel data evidence that this event is also crucial for activating cytokine signaling cascades. For instance, the consequent Δψm mediated release of mtDNA can trigger cGAS activation (102, 139–141). Recent structural analysis revealed insights on how cGAS senses different dsDNA residues and, interestingly, it has a higher preference for mtDNA (142).
Discovered in the last decade (143), the cGAS-STING pathway has an important role during intracellular infections, being only recently valued under different contexts. CGAS catalyzes the production of cGAMP (cyclic GTP-AMP) in the presence of cytosolic dsDNA, serving as a second messenger for the activation of the STING adaptor protein. This promotes the phosphorylation of the Tank Binding Kinase 1 (TBK1) protein and further IRF3 nuclear translocation and IFN I transcription (144). Of note, the STING induction of type I IFN responses is a process that occurs only when the cell are not mobilizing apoptotic caspases (85). Thus, it evidences the importance of the cGAS-STING-IRF3 axis during neuropathology, as traumatic brain injury and hypoxia (145–147).
cGAS/STING also initiates NLRP3 responses by the elevation of K+ influx post lysosomal rupture (148). The NLRP3 induced IL-1β is an important acute phase cytokine that is critical to the pathophysiology of CNS diseases, as AD, PD, MS and even seizure disorders (72, 149–151). Recent research have shown that altered Δψm is dependent on IL-1R activation for further NF-κB, IRF3 and IFN-I expression (150). This novel pathway induces the release of mtDNA and further cytosolic detection by cGAS, but it is important to note that this discovery was made in monocytes and transformed lung cells (150). However, evidences indicate the presence of this axis in the CNS, as STING also modulates microglial reactivity during EAE (98, 152, 153). Moreover, the antiviral drug Ganciclovir promotes beneficial STING dependent type I IFN response in EAE model, dampening the harmful activity of activated microglia (154). Interestingly, mice knocked-out for mitophagy genes, as Parkin and PINK, that leads to inflammation and neuronal death in PD, had more prominent loss of dopaminergic neurons, which was reverted in the absence of STING. This provides an important link between STING and PD pathogenesis, evidencing the need for more studies on the biology cGAS-STING during neurodegenerative and neuroinflammatory diseases (155).
Greasing Brain Engines: Cardiolipin
Damage to the mitochondrial membrane accounts for the release of mDAMPs. Accordingly, cardiolipin holds great responsibility for the structural stability of the ETC and the functional mitochondrial shape (156, 157). Cardiolipin is mostly associated with heart diseases, mainly due to its high content and impact in this organ (130). On the other hand, its impact over the CNS is an issue that has recently gained attention. Mutations in genes involved in cardiolipin biogenesis, e.g. the trans-acylase tafazzin (TAZ), have shown to affect cognitive functions in TAZ knock-out mice, expanding their classical role in X-linked myopathies (12, 158). This lipid is found in the body in different isoforms and, despite the dominance of the tetra linoleoyl cardiolipin isoform in the periphery, studies showed that in the CNS, there is a huge number of cardiolipin isoforms, distributed among different brain regions and cellular subtypes (159). Differences in cardiolipin composition and isoforms correlate to mitochondrial position inside the cell. For instance, as total cardiolipin increases, the closer they are to the synapses (160), which seem to correlate with the capacity of cardiolipin to influence ATP production (161).
Unsaturated lipids as cardiolipin are affected by mtROS and have their function compromised during oxidative stress (162). Proportional to the extension of cardiolipin peroxidation, there is a massive reduction of mitochondrial production of ATP due to Δψm loss and impairment of ETC complexes I, III, IV and V. Conversely, impaired ATP production by CNS cells is a common factor in aging and degenerative diseases (84). Studies in PD evidenced that cardiolipin interacts with α-synuclein by modifying its structure and exerting a protective role, by preventing its aggregation. Regarding this issue, divergent results demonstrated that this interaction can also affect cardiolipin functioning, resulting in increased pathology (163, 164). Moreover, α-synuclein binding to cardiolipin impairs the detection of cytosolic cytochrome c and thus inhibiting apoptotic cell death by dampening cellular oxidative stress (165).
The presence of cardiolipin in the OMM induces the recognition and further engulfment of dysfunctional mitochondria by LC3 mediated autophagy. Under conditions when the number of dysfunctional mitochondria exceeds the autophagy capacity, cardiolipin recruits BAX to form pores that release cytochrome c to the cytoplasm and triggering apoptosis. Thus, apart from the ETC complexes, cardiolipin also anchors important kinases that participate in the translocation of lipidic content through the mitochondrial membrane. When this mechanisms are impaired, it leads to a deleterious accumulation of dysfunctional mitochondria (156, 166).
Give Me Fuel, Give Me Fire: Inflammasome Activation and Neuroinflammation
In addition to the induction of type I IFN response, mDAMPs also modulate the activation of inflammasomes. Canonical inflammasome activation leads to the proteolytic cleavage of pro-caspase-1 to caspase-1 and the subsequent pro-IL-1β/IL-18 and gasdermin 1 for further extracellular release (167). Both cytokines strongly activate pro-inflammatory responses, and they have an unquestionable importance during neuroinflammation, for instance, promoting the disruption of the blood-brain-barrier (BBB) and ROS production.
NLRP3 inflammasome is activated by a wide range of molecules, including mtROS, cardiolipin and mtDNA (148, 167). Importantly, for NLRP3 complete activation, an initial priming step is required to increase the expression of inflammasome effector molecules, as the NLRP3 itself, caspase-1 and pro-IL-1β. This is mediated by the activation of TLR-4, NOD receptor 2 (NOD2) and cytokines as TNF-α and IL-1β itself. This leads to NF-κB phosphorylation and nuclear translocation to promote NLRP3-related gene transcription (167–169). However, how inflammasome senses and interacts with stressors and the details of its activation remains not fully understood.
Interestingly, inflammasome activation is closely related to ER and mitochondria communication in many ways, and mitochondrial Ca2+ imbalance may also result in inflammasome activation (170). This may occur either by directly promoting NLRP3 complex formation or by mitochondrial Ca2+ overload and further mitochondrial dysfunction. Corroborating this, blocking ER IP3R, a major regulator of ER-to-cytoplasm Ca2+exchange, effectively attenuates NLRP3 activation (171, 172). Accordingly, mitochondria calcium homeostasis is closely related to ER Ca2+ metabolism since the MAMs plays a key role in material transfer and signaling between both organelles (16).
Inactivated forms of NLRP3 are localized in the ER membrane, although under certain stimuli, NLRP3 is redistributed across the MAMs (173). Under stress conditions, cardiolipin is exposed on the OMM and serves as a bridge between NLRP3 and MAMs (78). The localization of NLRP3 over the MAMs induces clusters of mitochondria around the Golgi apparatus and the release of NLRP3 to the cytosol for inflammasome mature form assembling (173). The mitochondrial location of NLRP3 is also affected by the interaction between MFN2 and MAVS during viral infections, which recruits the inflammasome to the MAMs. However, MAVS are not essential for NLRP3 activation under other stimuli (16). Mitophagy is also an important player during inflammasome activation since the removal of impaired mitochondria reduces ROS production. The continuous production of ROS occurs during oxidative phosphorylation and several studies have demonstrated that inhibitors of complex I, II and III develop an important role in mtROS production reflecting in decreased inflammasome activation (174, 175).
Interestingly, mtROS and Ca2+ have a synergic role for pore formation within mitochondria membranes. Mitochondrial permeability transition (MPT) pores allow the release of mtDNA. Interestingly, oxidized mtDNA in the cytoplasm triggers IL-1β secretion by preferentially activating NLRP3 but not AIM-2 (176). Additionally, IL-1β production was significantly enhanced with oxidized versus normal DNA (176). Importantly, the induction of NLRP3 also induces mtDNA release in the cytosol, thus creating a positive loop in the induction of inflammasome pathway (14, 176).
CNS disorders may occur due to NLRP3 dysfunctions and its close link with mitochondrial health (177). In EAE for example, increased levels of IL-1β and NLRP3 were related to disease progression. Additionally, microglia deletion of A20, an immune suppressive protein correlated with increased NLRP3 activation and IL-1β/IL18 secretion (178). Conversely, the role of IFN-β, a well-established treatment for MS, was demonstrated to be dependent on NLRP3 activation during EAE (179).
The correlation of inflammasomes and AD pathophysiology is also debated, since increased levels of IL-1β were reported in the Aβ neighboring microglia cells. Halle et al. (180) observed that Aβ phagocytosed by microglia triggers caspase-1 and subsequent release of IL-1β, in vitro. Corroborating this, in vivo studies shown that loss of NLRP3 is associated with reduced Aβ deposition, cytokine production and lead to ameliorated spatial memory deficits in AD mouse model. As Aβ, α-synuclein also induces NLRP3 activation in mouse microglial cell line. In vivo, the administration of caspase-1 inhibitor decreases the activation of NLRP3 and induces an increase in the number of dopaminergic neurons, consequently relating a better PD prognosis (177). Interestingly, caspase-1 is also able to cleave α-synuclein, and NLRP3 inhibition abrogates synuclein aggregation, ameliorating cell damage in murine PD model (181).
Penghu and collaborators showed that mtROS induced NLRP3 activation in hippocampal microglia (182). Sarkar et al. (182) evidenced that the inhibition of mitochondrial complex I by rotenone increased ROS production, leading to augmented cleavage of caspase-1 and NLRP3 expression in microglia cells. Conversely, they also evidenced that this pathway culminates in a more prominent dopaminergic neuronal loss (183).
Concluding Remarks
Although first believed to be solely the power houses of the cell, it is now accepted that mitochondria have an active role in many cellular processes, especially during inflammation. In Figure 1 we summarize some of these relevant aspects concerning the correlation between mitochondria and brain disease. Although many of these diseases have their pathology linked to either mild or robust inflammatory responses, recent findings have unraveled that many of these mechanisms correlate with mitochondrial unbalanced function. Either because neuroinflammation can drastically impact cellular metabolism and further mitochondrial biology, for instance promoting mROS secretion or impairing ETC function, or because mitochondrial disfunction leads to the release of pro-inflammatory factors, as mtDNA. In this sense, innate immunity receptors as NLRP3 and MAVS greatly evidences the role of mitochondria as both effectors and sensors of neuroinflammation, respectively. Interestingly, structural changes in mitochondrial shape, size and turnover inside the cell, has also shown great relevance. For instance, OPA and MFN proteins defects, responsible for mitochondrial fusion are observed in Charcot-Marie-Tooth disease and optic atrophy. Mitochondria may also correlate with homeostasis and resolution of neuroinflammation. During mitophagy, for instance, there is the induction of IL-10 secretion and inflammation control, as observed in PD models. More interesting, inflammation activated astrocytes may actively transfer mitochondria to nearby neurons as an effort to avoid or reduce tissue damage, whereas damaged mitochondria are also released in order to avoid excessive ROS production, as described during stroke. In summary, the role of mitochondria during neuroinflammation and neurodegeneration has started to be better understood, not only unraveling important biological processes but also indicating that mitochondria-related immunometabolic pathways may serve as promising therapeutic targets for CNS diseases. This is corroborated by the fact that are currently 160 studies registered in www.clinicaltrials.gov found for the terms mitochondria and brain.
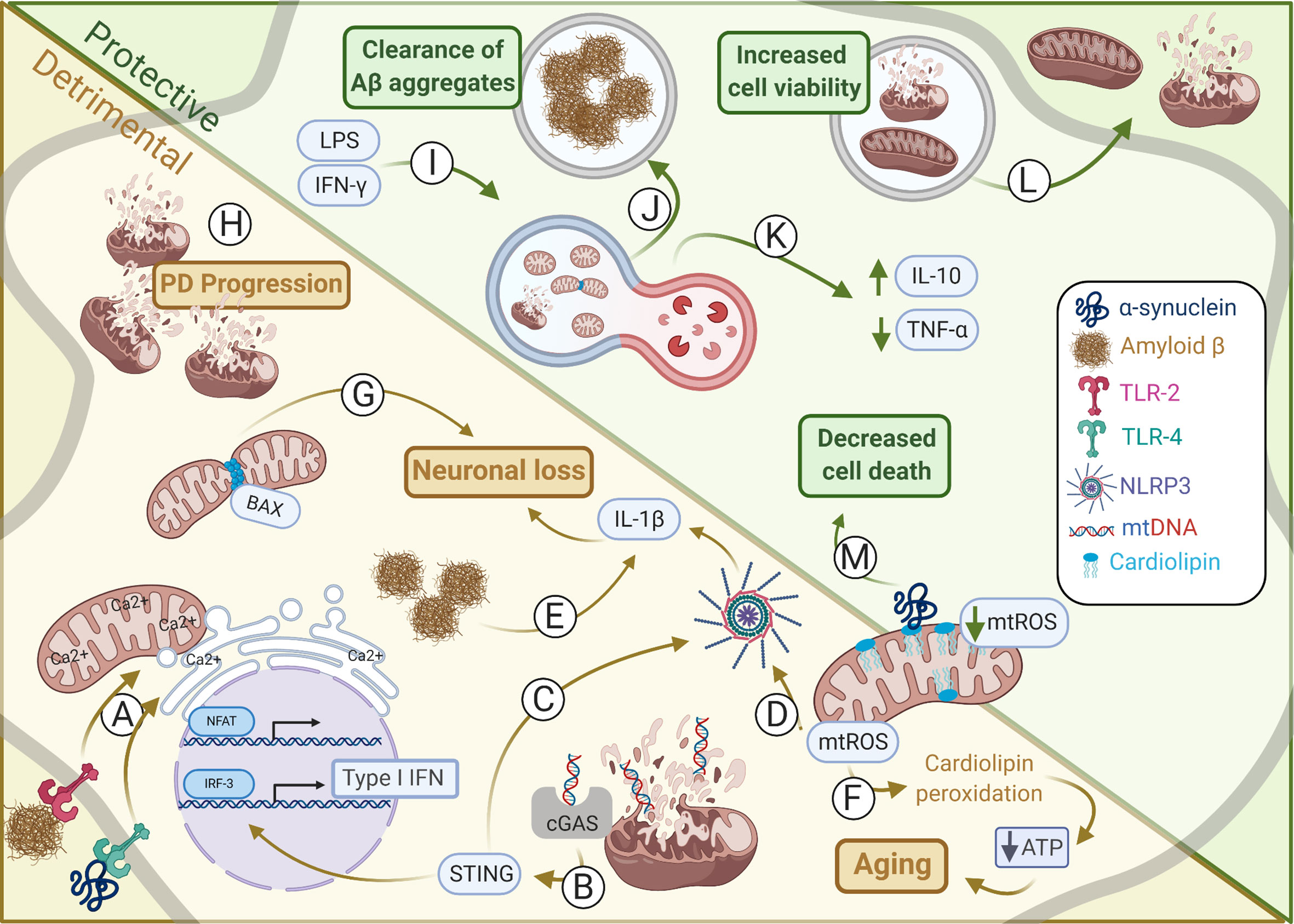
Figure 1 Mitochondrial alterations in protective and detrimental processes within CNS. Alterations in mitochondrial dynamics may induce either harmful or helpful immune responses affecting in CNS homeostasis. (A) Amyloid-β and α-synuclein induces TLR-2 and TLR-4 activation, respectively, promoting the interaction between outer mitochondria membrane (OMM) and endoplasmic reticulum (ER) membrane to synergically increase Ca2+ uptake and NFAT activation. (B) Type I IFN production is induced by mtDNA activation of cGAS-STING during sustained mitochondrial damage promoting neurodegeneration. (C) STING may also induce activation of NLRP3. (D) The NLRP3 may also be induced by mtROS thus coordinating IL-1β secretion by microglia and astrocytes promoting neuronal loss. (E) Amyloid-β aggregates induce NLRP3 inflammasome activation and IL-1β secretion by microglia. (F) mtROS induces cardiolipin peroxidation that deregulates ATP production, as observed during aging. (G) In neurons, BAX interacts with DRP1 inducing mitochondrial fragmentation. This is critical for BAX-dependent pore formation and neuronal survival. (H) Failure in mitophagy culminates in damaged mitochondria accumulation which contributes for Parkinson’s disease (PD) progress. (I) Mitophagy may be induced by IFN-γ and LPS upregulation of DRP1 and LC3, an autophagy-related protein. This is essential to restore tubular mitochondrial networks after inflammatory stimulation in astrocytes. (J) Microglial cells under mitophagy have elevated levels of intracellular Aβ aggregates, suggesting increased phagocytic activity, and thus clearing the harmful Aβ deposits. (K) PINK1 regulation of mitophagy is essential for CNS homeostasis establishment and induction of IL-10 and reduction of TNF-α secretion. (L) Instead of mitophagy the release of damage mitochondria may also minimize overall cell injury. Conversely, healthy mitochondria may also be donated from astrocytes to damaged neighboring neurons increasing its viability and maintaining its metabolism. (M) Cardiolipin can interact with α-synuclein preventing its aggregation by modifying its structure and impairing the release of cytosolic cytochrome c and thus inhibiting apoptosis and dampening cellular oxidative stress. Illustration prepared by the authors using www.biorender.com.
Author Contributions
LGO and YSA wrote the manuscript and designed the figure; AHI wrote and edited the manuscript; JPSP wrote, elaborated the topics, edited and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Neuroimmune interactions laboratory is supported by FAPESP (#2017/26170; #2017/22504-1), CAPES and CNPq.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. World Health Organization. Neurological Disorders: Public health challenges (2021). Available at: https://www.who.int/mental_health/neurology/neurodiso/en/ (Accessed February 12, 2021).
2. Mayo L, Trauger SA, Blain M, Nadeau M, Patel B, Alvarez JI, et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat Med (2014) 20:1147–56. doi: 10.1038/nm.3681
3. Peron JPS, Oliveira D, Brandão WN, Fickinger A, De Oliveira APL, Rizzo LV, et al. Central Nervous System Resident Cells in Neuroinflammation: A Brave New World. Chan James, ed. London, UK: IntechOpen (2012). 1536–77 p.
4. Chakraborty S, Kaushik DK, Gupta M, Basu A. Inflammasome signaling at the heart of central nervous system pathology. J Neurosci Res (2010) 88:1615–31. doi: 10.1002/jnr.22343
5. Shao W, Zhang S, Tang M, Zhang X, Zhou Z, Yin Y, et al. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via αB-crystallin. Nature (2013) 494:90–4. doi: 10.1038/nature11748
6. Barbierato M, Facci L, Argentini C, Marinelli C, Skaper SD, Giusti P. Astrocyte-microglia cooperation in the expression of a pro-inflammatory phenotype. CNS Neurol Disord Drug Targets (2013) 12:608–18.
7. Luheshi NM, Kovács KJ, Lopez-Castejon G, Brough D, Denes A. Interleukin-1α expression precedes IL-1β after ischemic brain injury and is localised to areas of focal neuronal loss and penumbral tissues. J Neuroinflamm (2011) 8:186. doi: 10.1186/1742-2094-8-186
8. Farez MF, Quintana FJ, Gandhi R, Izquierdo G, Lucas M, Weiner HL. Toll-like receptor 2 and poly(ADP-ribose) polymerase 1 promote central nervous system neuroinflammation in progressive EAE. Nat Immunol (2009) 10:958–64. doi: 10.1038/ni.1775
9. Hasegawa-Ishii S, Takei S, Inaba M, Umegaki H, Chiba Y, Furukawa A, et al. Defects in cytokine-mediated neuroprotective glial responses to excitotoxic hippocampal injury in senescence-accelerated mouse. Brain Behav Immun (2011) 25:83–100. doi: 10.1016/j.bbi.2010.08.006
10. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol (2017) 18:488–98. doi: 10.1038/ni.3704
11. Wentling M, Lopez-Gomez C, Park HJ, Amatruda M, Ntranos A, Aramini J, et al. A metabolic perspective on CSF-mediated neurodegeneration in multiple sclerosis. Brain (2019) 142:2756–74. doi: 10.1093/brain/awz201
12. Cole LK, Kim JH, Amoscato AA, Tyurina YY, Bayır H, Karimi B, et al. Aberrant cardiolipin metabolism is associated with cognitive deficiency and hippocampal alteration in tafazzin knockdown mice. Biochim Biophys Acta - Mol Basis Dis (2018) 1864:3353–67. doi: 10.1016/j.bbadis.2018.07.022
13. Morató L, Bertini E, Verrigni D, Ardissone A, Ruiz M, Ferrer I, et al. Mitochondrial dysfunction in central nervous system white matter disorders. Glia (2014) 1–17. doi: 10.1002/glia.22670
14. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin Xj, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature (2018) 560:198–203. doi: 10.1038/s41586-018-0372-z
15. Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem (2018) 62:341–60. doi: 10.1042/EBC20170104
16. Missiroli S, Patergnani S, Caroccia N, Pedriali G, Perrone M, Previati M, et al. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis (2018) 9. doi: 10.1038/s41419-017-0027-2
17. Jackson JG, Robinson MB. Reciprocal regulation of mitochondrial dynamics and calcium signaling in astrocyte processes. J Neurosci (2015) 35:15199–213. doi: 10.1523/JNEUROSCI.2049-15.2015
18. De Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature (2008) 456:605–10. doi: 10.1038/nature07534
19. Boitier E, Rea R, Duchen MR. Mitochondria exert a negative feedback on the propagation of intracellular Ca2+ waves in rat cortical astrocytes. J Cell Biol (1999) 145:795–808. doi: 10.1083/jcb.145.4.795
20. Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci U S A (2015) 112:E2174–81. doi: 10.1073/pnas.1504880112
21. Stoica R, Paillusson S, Gomez-Suaga P, Mitchell JC, Lau DH, Gray EH, et al. ALS/FTD-associated FUS activates GSK-3β to disrupt the VAPB–PTPIP51 interaction and ER–mitochondria associations. EMBO Rep (2016) 17:1326–42. doi: 10.15252/embr.201541726
22. Paillusson S, Gomez-Suaga P, Stoica R, Little D, Gissen P, Devine MJ, et al. α-Synuclein binds to the ER–mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol (2017) 134:129–49. doi: 10.1007/s00401-017-1704-z
23. Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca 2 microdomains to mitochondria. J Cell Biol (2002) 159:613–24. doi: 10.1083/jcb.200205091
24. Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol (2006) 175:901–11. doi: 10.1083/jcb.200608073
25. Ottolini D, Calì T, Negro A, Brini M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum Mol Genet (2013) 22:2152–68. doi: 10.1093/hmg/ddt068
26. Bartok A, Weaver D, Golenár T, Nichtova Z, Katona M, Bánsághi S, et al. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat Commun (2019) 10:1–14. doi: 10.1038/s41467-019-11646-3
27. Akimzhanov AM, Boehning D. IP3R function in cells of the immune system. Wiley Interdiscip Rev Membr Transp Signal (2012) 1:329–39. doi: 10.1002/wmts.27
28. Kim MS, Usachev YM. Mitochondrial Ca2+ cycling facilitates activation of the transcription factor NFAT in sensory neurons. J Neurosci (2009) 29:12101–14. doi: 10.1523/JNEUROSCI.3384-09.2009
29. Agostinho P, Lopes JP, Velez Z, Oliveira CR. Overactivation of calcineurin induced by amyloid-beta and prion proteins. Neurochem Int (2008) 52:1226–33. doi: 10.1016/j.neuint.2008.01.005
30. Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, et al. TLR2 Is a Primary Receptor for Alzheimer’s Amyloid β Peptide To Trigger Neuroinflammatory Activation. J Immunol (2012) 188:1098–107. doi: 10.4049/jimmunol.1101121
31. Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T. Inflammation in Alzheimer’s disease: Amyloid-β oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol (2009) 87:181–94. doi: 10.1016/j.pneurobio.2009.01.001
32. Rannikko EH, Weber SS, Kahle PJ. Exogenous α-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci (2015) 16:57. doi: 10.1186/s12868-015-0192-0
33. Ma B, Yu J, Xie C, Sun L, Lin S, Ding J, et al. Toll-like receptors promote mitochondrial translocation of nuclear transcription factor nuclear factor of activated T-cells in prolonged microglial activation. J Neurosci (2015) 35:10799–814. doi: 10.1523/JNEUROSCI.2455-14.2015
34. Sama MA, Mathis DM, Furman JL, Abdul HM, Artiushin IA, Kraner SD, et al. Interleukin-1β-dependent signaling between astrocytes and neurons depends critically on astrocytic calcineurin/NFAT activity. J Biol Chem (2008) 283:21953–64. doi: 10.1074/jbc.M800148200
35. Brandt T, Cavellini L, Kühlbrandt W, Cohen MM. A mitofusin-dependent docking ring complex triggers mitochondrial fusion in vitro. Elife (2016) 5:1–23. doi: 10.7554/eLife.14618
36. Huang X, Zhou X, Hu X, Joshi AS, Guo X, Zhu Y, et al. Sequences flanking the transmembrane segments facilitate mitochondrial localization and membrane fusion by mitofusin. Proc Natl Acad Sci U S A (2017) 114:E9863–72. doi: 10.1073/pnas.1708782114
37. Eura Y, Ishihara N, Yokota S, Mihara K. Two Mitofusin Proteins, Mammalian Homologues of FZO, with Distinct Functions Are Both Required for Mitochondrial Fusion. J Biochem (2003) 134:333–44. doi: 10.1093/jb/mvg150
38. Liu YJ, McIntyre RL, Janssens GE, Houtkooper RH. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech Ageing Dev (2020) 186:111212. doi: 10.1016/j.mad.2020.111212
39. DeVay RM, Dominguez-Ramirez L, Lackner LL, Hoppins S, Stahlberg H, Nunnari J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J Cell Biol (2009) 186:793–803. doi: 10.1083/jcb.200906098
40. Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A (2011) 108:10190–5. doi: 10.1073/pnas.1107402108
41. Liesa M, Van Der Bliek A, Shirihai OS. To Fis or not to Fuse? This is the question!. EMBO J (2019) 38(8):1–3. doi: 10.15252/embj.2019101839
42. Lewis SC, Uchiyama LF, Nunnari J. ER-mitochondria contacts couple mtDNA synthesis with Mitochondrial division in human cells. Sci (80- ) (2016) 353(6296):aaf5549. doi: 10.1126/science.aaf5549
43. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Sci (80- ) (2011) 334:358–62. doi: 10.1126/science.1207385
44. Cho B, Cho HM, Jo Y, Kim HD, Song M, Moon C, et al. Constriction of the mitochondrial inner compartment is a priming event for mitochondrial division. Nat Commun (2017) 8:1–17. doi: 10.1038/ncomms15754
45. Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab (2013) 17:491–506. doi: 10.1016/j.cmet.2013.03.002
46. Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet (2000) 26:207–10. doi: 10.1038/79936
47. Züchner S, Noureddine M, Kennerson M, Verhoeven K, Claeys K, De Jonghe P, et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet (2005) 37:289–94. doi: 10.1038/ng1514
48. Maes ME, Grosser JA, Fehrman RL, Schlamp CL, Nickells RW. Completion of BAX recruitment correlates with mitochondrial fission during apoptosis. Sci Rep (2019) 9:16565. doi: 10.1038/s41598-019-53049-w
49. Duan C, Kuang L, Xiang X, Zhang J, Zhu Y, Wu Y, et al. Drp1 regulates mitochondrial dysfunction and dysregulated metabolism in ischemic injury via Clec16a-, BAX-, and GSH- pathways. Cell Death Dis (2020) 11:251. doi: 10.1038/s41419-020-2461-9
50. Kanki T, Wang K, Klionsky DJ. A genomic screen for yeast mutants defective in mitophagy. Autophagy (2010) 6:278–80. doi: 10.4161/auto.6.2.10901
51. Yamashita SI, Jin X, Furukawa K, Hamasaki M, Nezu A, Otera H, et al. Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J Cell Biol (2016) 215:649–65. doi: 10.1083/jcb.201605093
52. Wang L, Chen R-F, Liu J-W, Lee I-K, Lee C-P, Kuo H-C, et al. DC-SIGN (CD209) Promoter –336 A/G Polymorphism Is Associated with Dengue Hemorrhagic Fever and Correlated to DC-SIGN Expression and Immune Augmentation. PloS Negl Trop Dis (2011) 5:e934. doi: 10.1371/journal.pntd.0000934
53. Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ. PINK1 drives parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol (2013) 200:163–72. doi: 10.1083/jcb.201210111
54. Tang MY, Vranas M, Krahn AI, Pundlik S, Trempe JF, Fon EA. Structure-guided mutagenesis reveals a hierarchical mechanism of Parkin activation. Nat Commun (2017) 8:1–14. doi: 10.1038/ncomms14697
55. Wang H, Song P, Du L, Tian W, Yue W, Liu M, et al. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: Implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem (2011) 286:11649–58. doi: 10.1074/jbc.M110.144238
56. Henn IH, Bouman L, Schlehe JS, Schlierf A, Schramm JE, Wegener E, et al. Parkin mediates neuroprotection through activation of IκB kinase/nuclear factor-κB signaling. J Neurosci (2007) 27:1868–78. doi: 10.1523/JNEUROSCI.5537-06.2007
57. Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, et al. PINK1 and Parkin target miro for phosphorylation and degradation to arrest mitochondrial motility. Cell (2011) 147:893–906. doi: 10.1016/j.cell.2011.10.018
58. Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Sci (80- ) (2011) 334:1141–4. doi: 10.1126/science.1210333
60. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Sci (80- ) (2017) 356:513–9. doi: 10.1126/science.aal3535
61. Lei Q, Tan J, Yi S, Wu N, Wang Y, Wu H. Mitochonic acid 5 activates the MAPK–ERK–yap signaling pathways to protect mouse microglial BV-2 cells against TNFα-induced apoptosis via increased Bnip3-related mitophagy. Cell Mol Biol Lett (2018) 23:14. doi: 10.1186/s11658-018-0081-5
62. Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci (2019) 22:401–12. doi: 10.1038/s41593-018-0332-9
63. Motori E, Puyal J, Toni N, Ghanem A, Angeloni C, Malaguti M, et al. Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab (2013) 18:844–59. doi: 10.1016/j.cmet.2013.11.005
64. Sriram S. Role of glial cells in innate immunity and their role in CNS demyelination. J Neuroimmunol (2011) 239:13–20. doi: 10.1016/j.jneuroim.2011.08.012
65. Fiebig C, Keiner S, Ebert B, Schäffner I, Jagasia R, Lie DC, et al. Mitochondrial dysfunction in astrocytes impairs the generation of reactive astrocytes and enhances neuronal cell death in the cortex upon photothrombotic lesion. Front Mol Neurosci (2019) 12:40. doi: 10.3389/fnmol.2019.00040
66. Verkhratsky A, Matteoli M, Parpura V, Mothet J, Zorec R. Astrocytes as secretory cells of the central nervous system: idiosyncrasies of vesicular secretion. EMBO J (2016) 35:239–57. doi: 10.15252/embj.201592705
67. Quintana DD, Garcia JA, Sarkar SN, Jun S, Engler-Chiurazzi EB, Russell AE, et al. Hypoxia-reoxygenation of primary astrocytes results in a redistribution of mitochondrial size and mitophagy. Mitochondrion (2019) 47:244–55. doi: 10.1016/j.mito.2018.12.004
68. Davis CHO, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, et al. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A (2014) 111:9633–8. doi: 10.1073/pnas.1404651111
69. Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature (2016) 535:551–5. doi: 10.1038/nature18928
70. Joshi AU, Minhas PS, Liddelow SA, Haileselassie B, Andreasson KI, Dorn GW, et al. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci (2019) 22:1635–48. doi: 10.1038/s41593-019-0486-0
71. Pradeu T, Cooper EL. The danger theory: 20 years later. Front Immunol (2012) 3:287. doi: 10.3389/fimmu.2012.00287
72. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw (2018) 18:1635–48. doi: 10.4110/in.2018.18.e27
73. Harris HE, Raucci A. Alarmin(g) news about danger: Workshop on Innate Danger Signals and HMGB1. EMBO Rep (European Mol Biol Organization) (2006) 774–8. doi: 10.1038/sj.embor.7400759
74. Grazioli S, Pugin J. Mitochondrial damage-associated molecular patterns: From inflammatory signaling to human diseases. Front Immunol (2018) 9:832. doi: 10.3389/fimmu.2018.00832
75. Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H, et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep (2019) 29:1261–73.e6. doi: 10.1016/j.celrep.2019.09.050
76. Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity (2019) 50:253–71.e6. doi: 10.1016/j.immuni.2018.11.004
77. Nguyen HM, Mejia EM, Chang W, Wang Y, Watson E, On N, et al. Reduction in cardiolipin decreases mitochondrial spare respiratory capacity and increases glucose transport into and across human brain cerebral microvascular endothelial cells. J Neurochem (2016) 139:68–80. doi: 10.1111/jnc.13753
78. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity (2013) 39:311–23. doi: 10.1016/j.immuni.2013.08.001
79. Wang C, Youle RJ. The Role of Mitochondria. Annu Rev Genet (2009) 43:95–118. doi: 10.1146/annurev-genet-102108-134850
80. Elmore S. Apoptosis: A Review of Programmed Cell Death. Toxicol Pathol (2007) 35:495–516. doi: 10.1080/01926230701320337
81. Migalovich Sheikhet H, Villacorta Hidalgo J, Fisch P, Balbir-Gurman A, Braun-Moscovici Y, Bank I. Dysregulated CD25 and Cytokine Expression by γδ T Cells of Systemic Sclerosis Patients Stimulated With Cardiolipin and Zoledronate. Front Immunol (2018) 9:753. doi: 10.3389/fimmu.2018.00753
82. Madsen PM, Pinto M, Patel S, Mccarthy S, Gao H, Taherian M, et al. Neurobiology of Disease Mitochondrial DNA Double-Strand Breaks in Oligodendrocytes Cause Demyelination, Axonal Injury, and CNS Inflammation. J Neurosci (2017) 10185–99. doi: 10.1523/JNEUROSCI.1378-17.2017
83. Chinnery PF, Hudson G. Mitochondrial genetics. Br Med Bull (2013) 106:135–59. doi: 10.1093/bmb/ldt017
84. Cabral-Costa JV, Kowaltowski AJ. Neurological disorders and mitochondria. Mol Aspects Med (2020) 71:100826. doi: 10.1016/j.mam.2019.10.003
85. Rongvaux A, Jackson R, Harman CCD, Li T, West AP, De Zoete MR, et al. Apoptotic caspases prevent the induction of type i interferons by mitochondrial DNA. Cell (2014) 159:1563–77. doi: 10.1016/j.cell.2014.11.037
86. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature (2015) 520:553–7. doi: 10.1038/nature14156
87. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol (2004) 5:987–95. doi: 10.1038/ni1112
88. Schumacker PT, Gillespie MN, Nakahira K, Choi AMK, Crouser ED, Piantadosi CA, et al. Mitochondria in lung biology and pathology: More than just a powerhouse. Am J Physiol - Lung Cell Mol Physiol (2014) 306:L962. doi: 10.1152/ajplung.00073.2014
89. Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochem (2005) 70:200–14. doi: 10.1007/s10541-005-0102-7
90. Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic Biol Med (2009) 47:333–43. doi: 10.1016/j.freeradbiomed.2009.05.004
91. Fridavich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem (1995) 64:97–112. doi: 10.1146/annurev.bi.64.070195.000525
92. Angelova PR, Abramov AY. Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett (2018) 592:692–702. doi: 10.1002/1873-3468.12964
93. Lee YM, He W, Liou Y-C. The redox language in neurodegenerative diseases: oxidative post-translational modifications by hydrogen peroxide. Cell Death Dis (2021) 12:58. doi: 10.1038/s41419-020-03355-3
94. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev (2014) 94:909–50. doi: 10.1152/physrev.00026.2013
95. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J (2009) 417:1–13. doi: 10.1042/BJ20081386
96. Nissanka N, Moraes CT. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett (2018) 592:728–42. doi: 10.1002/1873-3468.12956
97. Lévy E, El Banna N, Baïlle D, Heneman-Masurel A, Truchet S, Rezaei H, et al. Causative links between protein aggregation and oxidative stress: A review. Int J Mol Sci (2019) 20. doi: 10.3390/ijms20163896
98. Yang K, Huang R, Fujihira H, Suzuki T, Yan N. N-glycanase NGLY1 regulates mitochondrial homeostasis and inflammation through NRF1. J Exp Med (2018) 215:2600–16. doi: 10.1084/jem.20180783
99. Giovannoni F, Bosch I, Polonio CM, Torti MF, Wheeler MA, Li Z, et al. AHR is a Zika virus host factor and a candidate target for antiviral therapy. Nat Neurosci (2020) 23:939–51. doi: 10.1038/s41593-020-0664-0
100. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15:87–103. doi: 10.1038/nri3787
101. Main BS, Zhang M, Brody KM, Kirby FJ, Crack PJ, Taylor JM. Type-I interferons mediate the neuroinflammatory response and neurotoxicity induced by rotenone. J Neurochem (2017) 141:75–85. doi: 10.1111/jnc.13940
102. Nazmi A, Field RH, Griffin EW, Haugh O, Hennessy E, Cox D, et al. Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia (2019) 67:1254–76. doi: 10.1002/glia.23592
103. Deczkowska A, Baruch K, Schwartz M. Type I/II Interferon Balance in the Regulation of Brain Physiology and Pathology. Trends Immunol (2016) 37:181–92. doi: 10.1016/j.it.2016.01.006
104. Hosseini S, Michaelsen-Preusse K, Grigoryan G, Chhatbar C, Kalinke U, Korte M. Type I Interferon Receptor Signaling in Astrocytes Regulates Hippocampal Synaptic Plasticity and Cognitive Function of the Healthy CNS. Cell Rep (2020) 31:107666. doi: 10.1016/j.celrep.2020.107666
105. Ward GA, McGraw K, McLemore AF, Lam NB, Hou H-A, Meyer BS, et al. Oxidized Mitochondrial DNA Engages TLR9 to Activate the NLRP3 Inflammasome in Myelodysplastic Syndromes. Blood (2019) 134:774–4. doi: 10.1182/blood-2019-122358
106. Bruno DCF, Donatti A, Martin M, Almeida VS, Geraldis JC, Oliveira FS, et al. Circulating nucleic acids in the plasma and serum as potential biomarkers in neurological disorders. Braz J Med Biol Res = Rev Bras Pesqui medicas e Biol (2020) 53:e9881. doi: 10.1590/1414-431x20209881
107. Otandault A, Abraham JD, Al Amir Dache Z, Khalyfa A, Jariel-Encontre I, Forné T, et al. Hypoxia differently modulates the release of mitochondrial and nuclear DNA. Br J Cancer (2020) 122:715–25. doi: 10.1038/s41416-019-0716-y
108. Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, et al. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Sci (80- ) (2014) 346:89–93. doi: 10.1126/science.1252945
109. Klein RS, Garber C, Funk KE, Salimi H, Soung A, Kanmogne M, et al. Neuroinflammation During RNA Viral Infections. Annu Rev Immunol (2019) 37:73–95. doi: 10.1146/annurev-immunol-042718-041417
110. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev (2004) 202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x
111. Vainchtein ID, Molofsky AV. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci (2020) 43:144–54. doi: 10.1016/j.tins.2020.01.003
112. Niederkorn JY. See no evil, hear no evil, do no evil: The lessons of immune privilege. Nat Immunol (2006) 7:354–9. doi: 10.1038/ni1328
113. Forrester JV, McMenamin PG, Dando SJ. CNS infection and immune privilege. Nat Rev Neurosci (2018) 19:655–71. doi: 10.1038/s41583-018-0070-8
114. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523:337–41. doi: 10.1038/nature14432
115. Wu D, Sanin DE, Everts B, Chen Q, Qiu J, Buck MD, et al. Type 1 Interferons Induce Changes in Core Metabolism that Are Critical for Immune Function. Immunity (2016) 44:1325–36. doi: 10.1016/j.immuni.2016.06.006
116. Buck MDD, O’Sullivan D, Klein Geltink RII, Curtis JDD, Chang CH, Sanin DEE, et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell (2016) 166:63–76. doi: 10.1016/j.cell.2016.05.035
117. Zhang W, Wang G, Xu Z-G, Zhang X, Li H, Lin H-K. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell (2019) 178:176–89. doi: 10.1016/j.cell.2019.05.003
118. Reikine S, Nguyen JB, Modis Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front Immunol (2014) 5:342. doi: 10.3389/fimmu.2014.00342
119. Ren Z, Ding T, Zuo Z, Xu Z, Deng J, Wei Z. Regulation of MAVS Expression and Signaling Function in the Antiviral Innate Immune Response. Front Immunol (2020) 11:1030. doi: 10.3389/fimmu.2020.01030
120. Chao CC, Gutiérrez-Vázquez C, Rothhammer V, Mayo L, Wheeler MA, Tjon EC, et al. Metabolic Control of Astrocyte Pathogenic Activity via cPLA2-MAVS. Cell (2019) 179:1483–98.e22. doi: 10.1016/j.cell.2019.11.016
121. Fecher C, Trovò L, Müller SA, Snaidero N, Wettmarshausen J, Heink S, et al. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat Neurosci (2019) 22:1731–42. doi: 10.1038/s41593-019-0479-z
122. Hernández-Corbacho MJ, Salama MF, Canals D, Senkal CE, Obeid LM. Sphingolipids in mitochondria. Biochim Biophys Acta - Mol Cell Biol Lipids (2017) 1862:56–68. doi: 10.1016/j.bbalip.2016.09.019
123. Bradl M, Lassmann H. Oligodendrocytes: Biology and pathology. Acta Neuropathol (2010) 119:37–53. doi: 10.1007/s00401-009-0601-5
124. Peterson LK, Fujinami RS. Inflammation, demyelination, neurodegeneration and neuroprotection in the pathogenesis of multiple sclerosis. J Neuroimmunol (2007) 184:37–44. doi: 10.1016/j.jneuroim.2006.11.015
125. Rose J, Brian C, Woods J, Pappa A, Panayiotidis MI, Powers R, et al. Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology (2017) 391:109–15. doi: 10.1016/j.tox.2017.06.011
126. Pelvig DP, Pakkenberg H, Stark AK, Pakkenberg B. Neocortical glial cell numbers in human brains. Neurobiol Aging (2008) 29:1754–62. doi: 10.1016/j.neurobiolaging.2007.04.013
127. Jellinger KA. Basic mechanisms of neurodegeneration: A critical update. J Cell Mol Med (2010) 14:457–87. doi: 10.1111/j.1582-4934.2010.01010.x
128. Jarjour A a, Zhang H, Bauer N, Ffrench-Constant C, Williams A. In vitro modeling of central nervous system myelination and remyelination. Glia (2012) 60:1–12. doi: 10.1002/glia.21231
129. Rinholm JE, Vervaeke K, Tadross MR, Tkachuk AN, Kopek BG, Brown TA, et al. Movement and structure of mitochondria in oligodendrocytes and their myelin sheaths. Glia (2016) 64:810–25. doi: 10.1002/glia.22965
130. Pope S, Land JM, Heales SJR. Oxidative stress and mitochondrial dysfunction in neurodegeneration; cardiolipin a critical target? Biochim Biophys Acta - Bioenerg (2008) 1777:794–9. doi: 10.1016/j.bbabio.2008.03.011
131. Philips T, Rothstein JD. Oligodendroglia: Metabolic supporters of neurons. J Clin Invest (2017) 127:3271–80. doi: 10.1172/JCI90610
132. Minchenberg SB, Massa PT. The control of oligodendrocyte bioenergetics by interferon-gamma (IFN-γ) and Src homology region 2 domain-containing phosphatase-1 (SHP-1). J Neuroimmunol (2019) 331:46–57. doi: 10.1016/j.jneuroim.2017.10.015
133. Luo F, Herrup K, Qi X, Yang Y, Yang Y. Inhibition of Drp1 hyper-activation is protective in animal models of experimental multiple sclerosis Correspondence to HHS Public Access. Exp Neurol (2017) 292:21–34. doi: 10.1016/j.expneurol.2017.02.015
134. Balabanov R, Strand K, Goswami R, McMahon E, Begolka W, Miller SD, et al. Interferon-γ-oligodendrocyte interactions in the regulation of experimental autoimmune encephalomyelitis. J Neurosci (2007) 27:2013–24. doi: 10.1523/JNEUROSCI.4689-06.2007
135. Jana A, Hogan EL, Pahan K. Ceramide and neurodegeneration: Susceptibility of neurons and oligodendrocytes to cell damage and death. J Neurol Sci (2009) 278:5–15. doi: 10.1016/j.jns.2008.12.010
136. Vandanmagsar B, Youm Y-H, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med (2011) 17:179–88. doi: 10.1038/nm.2279
137. Novgorodov SA, Wu BX, Gudz TI, Bielawski J, Ovchinnikova TV, Hannun YA, et al. Novel pathway of ceramide production in mitochondria: Thioesterase and neutral ceramidase produce ceramide from sphingosine and acyl-CoA. J Biol Chem (2011) 286:25352–62. doi: 10.1074/jbc.M110.214866
138. Singh I, Pahan K, Khan M, Singh AK. Cytokine-mediated induction of ceramide production is redox-sensitive: Implications to proinflammatory cytokine-mediated apoptosis in demyelinating diseases. J Biol Chem (1998) 273:20354–62. doi: 10.1074/jbc.273.32.20354
139. Pal S, Rao GN, Pal A. High glucose-induced ROS accumulation is a critical regulator of ERK1/2-Akt-tuberin-mTOR signalling in RGC-5 cells. Life Sci (2020) 256:117914. doi: 10.1016/j.lfs.2020.117914
140. Harris J, Deen N, Zamani S, Hasnat MA. Mitophagy and the release of inflammatory cytokines. Mitochondrion (2018) 41:2–8. doi: 10.1016/j.mito.2017.10.009
141. Bader V, Winklhofer KF. Mitochondria at the interface between neurodegeneration and neuroinflammation. Semin Cell Dev Biol (2019) 99:163–71. doi: 10.1016/j.semcdb.2019.05.028
142. Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, et al. Structure-function analysis of STING activation by c[G(2′,5′) pA(3′,5′)p] and targeting by antiviral DMXAA. Cell (2013) 154:748–62. doi: 10.1016/j.cell.2013.07.023
143. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455:674–8. doi: 10.1038/nature07317
144. Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS–STING pathway in health and disease. Nat Rev Genet (2019) 20:657–74. doi: 10.1038/s41576-019-0151-1
145. Reshi L, Wang H-V, Hong J-R. Modulation of Mitochondria During Viral Infections. In: Mitochondrial Diseases. London, UK: InTech. doi: 10.5772/intechopen.73036
146. Chin AC. PERK-STING signaling drives neuroinflammation in traumatic brain injury. J Neurosci (2020) 40:2384–6. doi: 10.1523/JNEUROSCI.2881-19.2020
147. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol (2016) 17:1142–9. doi: 10.1038/ni.3558
148. Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell (2017) 171:1110–24.e18. doi: 10.1016/j.cell.2017.09.039
149. Banerjee I, Behl B, Mendonca M, Sarkar SN, Fitzgerald KA, Rathinam Correspondence VAK. Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity (2018) 49:413–26.e5. doi: 10.1016/j.immuni.2018.07.006
150. Aarreberg LD, Esser-Nobis K, Driscoll C, Shuvarikov A, Roby JA, Gale M. Interleukin-1β Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol Cell (2019) 74:801–15.e6. doi: 10.1016/j.molcel.2019.02.038
151. Hewett SJ, Jackman NA, Claycomb RJ. Interleukin-1β in Central Nervous System Injury and Repair. Eur J Neurodegener Dis (2012) 1:195–211.
152. Xu Q, Xu W, Cheng H, Yuan H, Tan X. Efficacy and mechanism of cGAMP to suppress Alzheimer’s disease by elevating TREM2. Brain Behav Immun (2019) 81:495–508. doi: 10.1016/j.bbi.2019.07.004
153. Wuertz KMG, Treuting PM, Hemann EA, Esser-Nobis K, Snyder AG, Graham JB, et al. STING is required for host defense against neuropathological West Nile virus infection. PloS Pathog (2019) 15. doi: 10.1371/journal.ppat.1007899
154. Mathur V, Burai R, Vest RT, Bonanno LN, Lehallier B, Zardeneta ME, et al. Activation of the STING-Dependent Type I Interferon Response Reduces Microglial Reactivity and Neuroinflammation. Neuron (2017) 96:1290–302.e6. doi: 10.1016/j.neuron.2017.11.032
155. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation HHS Public Access. Nature (2018) 561:258–62. doi: 10.1038/s41586-018-0448-9
156. Li XX, Tsoi B, Li YF, Kurihara H, He RR. Cardiolipin and Its Different Properties in Mitophagy and Apoptosis. J Histochem Cytochem (2015) 63:301–11. doi: 10.1369/0022155415574818
157. Nagashima S, Takeda K, Ohno N, Ishido S, Aoki M, Saitoh Y, et al. MITOL deletion in the brain impairs mitochondrial structure and ER tethering leading to oxidative stress. Life Sci Alliance (2019) 2. doi: 10.26508/lsa.201900308
158. Chao H, Anthonymuthu TS, Kenny EM, Amoscato AA, Cole LK, Hatch GM, et al. Disentangling oxidation/hydrolysis reactions of brain mitochondrial cardiolipins in pathogenesis of traumatic injury. JCI Insight (2018) 3. doi: 10.1172/jci.insight.97677
159. Pointer CB, Wenzel TJ, Klegeris A. Extracellular cardiolipin regulates select immune functions of microglia and microglia-like cells. Brain Res Bull (2019) 146:153–63. doi: 10.1016/j.brainresbull.2019.01.002
160. Pointer CB, Klegeris A. Cardiolipin in Central Nervous System Physiology and Pathology. Cell Mol Neurobiol (2017) 37:1161–72. doi: 10.1007/s10571-016-0458-9
161. Paradies G, Paradies V, De Benedictis V, Ruggiero FM, Petrosillo G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim Biophys Acta - Bioenerg (2014) 1837:408–17. doi: 10.1016/j.bbabio.2013.10.006
162. Aitken RJ, Wingate JK, De Iuliis GN, Koppers AJ, McLaughlin EA. Cis-unsaturated fatty acids stimulate reactive oxygen species generation and lipid peroxidation in human spermatozoa. J Clin Endocrinol Metab (2006) 91:4154–63. doi: 10.1210/jc.2006-1309
163. Ghio S, Camilleri A, Caruana M, Ruf VC, Schmidt F, Leonov A, et al. Cardiolipin Promotes Pore-Forming Activity of Alpha-Synuclein Oligomers in Mitochondrial Membranes. ACS Chem Neurosci (2019) 10:3815–29. doi: 10.1021/acschemneuro.9b00320
164. Ryan T, Bamm VV, Stykel MG, Coackley CL, Humphries KM, Jamieson-Williams R, et al. Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat Commun (2018) 9:1–17. doi: 10.1038/s41467-018-03241-9
165. Díaz-Quintana A, Pérez-Mejías G, Guerra-Castellano A, De La Rosa MA, Díaz-Moreno I. Wheel and Deal in the Mitochondrial Inner Membranes: The Tale of Cytochrome c and Cardiolipin. Oxid Med Cell Longev (2020) 2020:1–20. doi: 10.1155/2020/6813405
166. Chu CT, Bayir H, Kagan VE. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: Implications for Parkinson disease. Autophagy (2014) 10:376–8. doi: 10.4161/auto.27191
167. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
168. He Y, Hara H, Núñez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
169. Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
170. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A (2012) 109:11282–7. doi: 10.1073/pnas.1117765109
171. Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat Commun (2012) 3:1–9. doi: 10.1038/ncomms2339
172. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature (2012) 492:123–7. doi: 10.1038/nature11588
173. Zhang Z, Meszaros G, He WT, Xu Y, Magliarelli H de F, Mailly L, et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J Exp Med (2017) 214:2671–93. doi: 10.1084/jem.20162040
174. Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial Reactive Oxygen Species Induces NLRP3-Dependent Lysosomal Damage and Inflammasome Activation. J Immunol (2013) 191:5230–8. doi: 10.4049/jimmunol.1301490
175. Won JH, Park S, Hong S, Son S, Yu JW. Rotenone-induced Impairment of Mitochondrial Electron Transport Chain Confers a Selective Priming Signal for NLRP3 Inflammasome Activation. J Biol Chem (2015) 290:27425–37. doi: 10.1074/jbc.M115.667063
176. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
177. Voet S, Srinivasan S, Lamkanfi M, Van Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med (2019) 11:10248. doi: 10.15252/emmm.201810248
178. Voet S, Mc Guire C, Hagemeyer N, Martens A, Schroeder A, Wieghofer P, et al. A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat Commun (2018) 9:1–15. doi: 10.1038/s41467-018-04376-5
179. Inoue M, Shinohara ML. The role of interferon-β in the treatment of multiple sclerosis and experimental autoimmune encephalomyelitis - in the perspective of inflammasomes. Immunology (2013) 139:11–8. doi: 10.1111/imm.12081
180. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol (2008) 9(8):857–65. doi: 10.1038/ni.1636
181. Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, et al. Inflammasome inhibition prevents -synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med (2018) 10. doi: 10.1126/scitranslmed.aah4066
182. Wei P, Yang F, Zheng Q, Tang W, Li J. The potential role of the NLRP3 inflammasome activation as a link between mitochondria ROS generation and neuroinflammation in postoperative cognitive dysfunction. Front Cell Neurosci (2019) 13:73. doi: 10.3389/fncel.2019.00073
Keywords: mitochondria, neuroinflammation, neurodegenerative diseases, Alzheimer disease, Parkinson disease, multiple sclerosis
Citation: de Oliveira LG, Angelo YdS, Iglesias AH and Peron JPS (2021) Unraveling the Link Between Mitochondrial Dynamics and Neuroinflammation. Front. Immunol. 12:624919. doi: 10.3389/fimmu.2021.624919
Received: 01 November 2020; Accepted: 25 February 2021;
Published: 16 March 2021.
Edited by:
Christophe Chevillard, INSERM U1090 Technologies Avancées pour le Génome et la Clinique, FranceReviewed by:
Paola Italiani, National Research Council (CNR), ItalyEva Bartok, University Hospital Bonn, Germany
Copyright © 2021 de Oliveira, Angelo, Iglesias and Peron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean Pierre Schatzmann Peron, amVhbnBpZXJyZUB1c3AuYnI=
†These authors have contributed equally to this work