 Miklós Lipcsey1,2†
Miklós Lipcsey1,2† Barbro Persson3†
Barbro Persson3† Oskar Eriksson3
Oskar Eriksson3 Anna M. Blom4
Anna M. Blom4 Karin Fromell3
Karin Fromell3 Michael Hultström1,5
Michael Hultström1,5 Markus Huber-Lang6
Markus Huber-Lang6 Kristina N. Ekdahl3,7
Kristina N. Ekdahl3,7 Robert Frithiof1‡
Robert Frithiof1‡ Bo Nilsson3*‡
Bo Nilsson3*‡- 1Department of Surgical Sciences, Anesthesiology and Intensive Care, Uppsala University, Uppsala, Sweden
- 2Hedenstierna Laboratory, Anesthesiology and Intensive Care, Department of Surgical Sciences, Uppsala University, Uppsala, Sweden
- 3Department of Immunology Genetics and Pathology, Uppsala University, Uppsala, Sweden
- 4Department of Translational Medicine, Lund University, Malmö, Sweden
- 5Unit for Integrative Physiology, Department of Medical Cell Biology, Uppsala University, Uppsala, Sweden
- 6Institute for Clinical and Experimental Trauma-Immunology, University Hospital of Ulm, Ulm, Germany
- 7Linnaeus Centre for Biomaterials Chemistry, Linnaeus University, Kalmar, Sweden
An important manifestation of severe COVID-19 is the ARDS-like lung injury that is associated with vascular endothelialitis, thrombosis, and angiogenesis. The intravascular innate immune system (IIIS), including the complement, contact, coagulation, and fibrinolysis systems, which is crucial for recognizing and eliminating microorganisms and debris in the body, is likely to be involved in the pathogenesis of COVID-19 ARDS. Biomarkers for IIIS activation were studied in the first 66 patients with COVID-19 admitted to the ICU in Uppsala University Hospital, both cross-sectionally on day 1 and in 19 patients longitudinally for up to a month, in a prospective study. IIIS analyses were compared with biochemical parameters and clinical outcome and survival. Blood cascade systems activation leading to an overreactive conjunct thromboinflammation was demonstrated, reflected in consumption of individual cascade system components, e.g., FXII, prekallikrein, and high molecular weight kininogen and in increased levels of activation products, e.g., C4d, C3a, C3d,g, sC5b-9, TAT, and D-dimer. Strong associations were found between the blood cascade systems and organ damage, illness severity scores, and survival. We show that critically ill COVID-19 patients display a conjunct activation of the IIIS that is linked to organ damage of the lung, heart, kidneys, and death. We present evidence that the complement and in particular the kallikrein/kinin system is strongly activated and that both systems are prognostic markers of the outcome of the patients suggesting their role in driving the inflammation. Already licensed kallikrein/kinin inhibitors are potential drugs for treatment of critically ill patients with COVID-19.
Introduction
The COVID-19 pandemic caused by SARS-CoV-2, a coronavirus first reported at the end of 2019 in Wuhan, China, has had a tremendous socioeconomic impact on human society across the globe. In most cases SARS-CoV-2 causes an influenza-like disease of mild-to-medium severity, but early on it became clear that in some cases, an increased risk of deadly thromboembolism developed that required high doses of low-molecular (LMW) heparin (150-300IU/kg) to prevent thrombosis (1). In a few cases the disease worsened after 9-10 days and developed into a full-blown acute respiratory distress syndrome (ARDS)-like pulmonary inflammation with endothelialitis, thrombosis, and vascular angiogenesis that has often required intensive care with ventilator support (2) and that is characterized by a general cyto-/chemokine dysregulation (3–5), increased risk of thromboembolism, reduced oxygen saturation, and signs of severe pulmonary damage revealed by chest computed tomography, such as ground glass opacities (6, 7).
In the blood, the innate immune system consists of several cascade systems that include the complement, contact, coagulation, and fibrinolytis systems, together with cellular defense systems such as leukocytes [including polymorphonuclear cells (PMNs), monocytes and NK cells], platelets, and endothelial cells. Here we will refer to all these components together as the intravascular innate immune system (IIIS) (8, 9). The IIIS acts as a purging system that identifies and removes microorganisms and activated apoptotic and necrotic cells and debris. It also orchestrates the subsequent immune/thromboinflammatory responses that lead to the repair of injured tissue (10). In some cases the IIIS overreacts giving rise to a severe thromboinflammation that causes collateral tissue damage. Examples of such thrombotic reactions are those that occur in cardiac infarction or stroke, during hemodialysis and in the setting of transplantation during ischemia-reperfusion injury and vascular rejection (11, 12). However, the cause of the COVID-19 -linked lung injury is not yet defined, but indirect evidence speaks in favor of a thromboinflammatory reaction triggered by the virus itself and more likely by infected and damaged cells (see several recent review articles) (1, 13–21).
Negatively charged surfaces such as the biological lipid membranes of necrotic and apoptotic cells as well as viruses can activate both the complement system via the classical (C1q) and lectin [mannose binding lectin (MBL), ficolins, collectins] pathways (the CPW and LPW, respectively) as well as via the contact (FXII) system (22–25). Pre-pandemic and recent studies of CoV-1 and -2 have indicated that these viruses can trigger complement activation via their spike proteins and the virus-infected host cells (26–28). The SARS CoV-1 virus has already been reported to cause an ARDS-like condition and in animal models complement C3-knockout (KO) mice infected with SARS CoV-1 recover more easily and have a much milder disease course than do wild-type mice (29). Also, increased plasma levels of sC5b-9 complexes have been found in COVID-19 patients, deposition of C4d and C3 has been detected in the lungs of diseased patients and binding of MBL and MBL associated serine protease (MASP)-2 of the LPW to virus proteins of the SARS CoV-2 virus has been reported (26, 30, 31).
Generation of the anaphylatoxins C3a and C5a by the complement system and bradykinin (BK) production by the kallikrein/kinin system are involved in ARDS of bacterial sepsis origin and may be drivers of the COVID-19 ARDS-like lung injury (32, 33). All of these peptides have chemotactic properties, induce increased vascular permeability, and activate immune and endothelial cells leading to inflammation and cyto-/chemokine expression. In ARDS, the anaphylatoxins and BK mediate leukocyte infiltration, particularly of PMNs, and mediate the leakage of fluid into the intercellular spaces between alveolar epithelial cells and the endothelium leading to reduced gas exchange between the alveolar space and the blood. A similar mechanism may be involved in SARS-CoV-2-induced ARDS. COVID-19 patients have an increased risk of both venous and arterial thrombosis. Activation of neutrophilic granulocytes by the anaphylatoxins that cause neutrophil extracellular trap (NET) formation, exposes both tissue factor (TF) and FXII in COVID-19 and may be one mechanism by which thrombi are formed (34, 35). We also recently reported that high levels of the recognition molecule MBL is strongly associated with thrombotic disease in COVID-19 patients, perhaps by recognizing damaged cells in this milieu (36).
An important property of the IIIS is that it discriminates self from non-self and is therefore a major trigger of inflammation. The aim of our investigation was to determine to what degree the IIIS is activated in COVID-19 in a naïve population with ARDS-like disease in the beginning of the pandemic. Our goal is to pinpoint the mechanisms underlying the lung damage associated with COVID-19, identify new biomarkers and targets for therapeutic intervention, and obtain support for giving already-licensed therapeutic inhibitors of these systems (licensed for angioedema) to patients with COVID-19 ARDS. The results of our comprehensive investigation of the IIIS-associated cascade systems in COVID-19 patients support the concept that all these cascade systems (particularly the kallikrein/kinin system) and cellular responses that constitute the IIIS are engaged in a full-blown thromboinflammatory reaction that damages organ systems in COVID-19 patients, not only the lungs but also other organs such as the kidneys and heart.
Material and Methods
Patients
A prospective single-center observational study was performed in the ICU of a mixed surgical and medical unit at Uppsala University Hospital, a tertiary hospital in Uppsala, Sweden. The first 70 patients >18 years of age with confirmed or suspected COVID-19 admitted to the ICU between March 13 and April 30, 2020 were screened for inclusion. COVID-19 was diagnosed by positive reverse-transcription polymerase chain reaction (RT-PCR) on nasopharyngeal swabs. Of the screened patients, 66 were included on the basis of a positive PCR for SARS-CoV-2, informed consent, and the availability of a blood sample taken at admission to the ICU. Nineteen of the first included patients were followed for up to one month, the only exclusion criterion being extracorporeal circulation therapies since such treatments are known to induce substantial activation of the IIIS (12).
Apart from the customary ICU care and medications, all patients received thromboprophylaxis with either dalteparin sodium at 117 IU/kg [91–152 (median and interquartile range)] or apixaban at 5 mg (n = 1) to target factor Xa (target range 0.2–0.5 kIU/L) for thrombosis prophylaxis and at 200 IU/kg to target factor Xa (target range 0.5–1.0 kIU/L) for treatment after thromboembolic events. Blood was sampled in EDTA-tubes, centrifuged, and stored as plasma at −70°C until analyzed.
Clinical data were recorded prospectively, including medical history, medications, physiological data, and date of death. The Simplified Acute Physiology Score 3 (SAPS-3) (37), Sequential Organ Failure Assessment (SOFA) score (38), and renal function “Kidney disease improving global outcomes” (KDIGO) (39), circulatory support, and respiratory support data were collected as detailed in the Results section (Table 1).
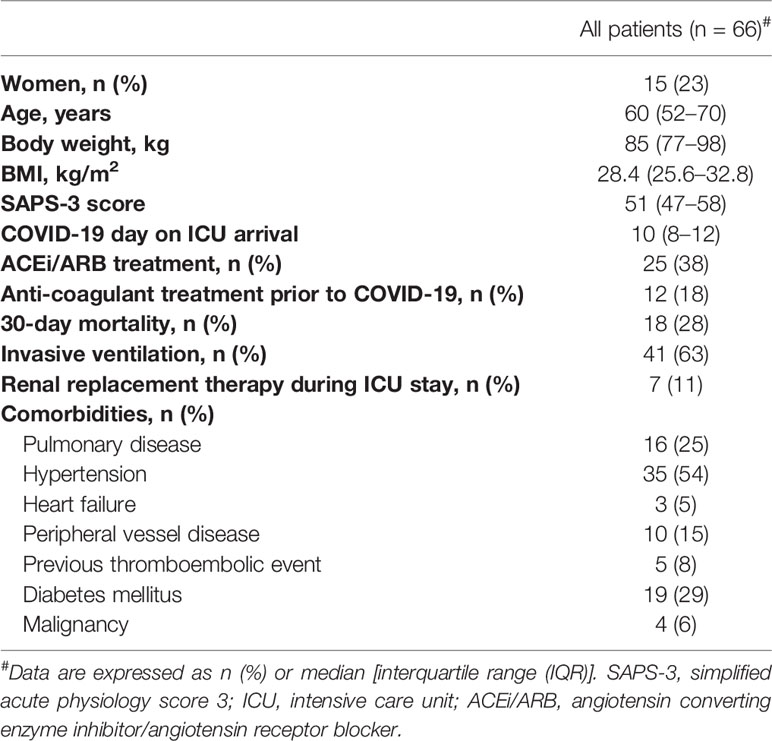
Table 1 Patient demographic characteristics and comorbidities.
The study was approved by the Swedish National Ethical Review Agency (EPM; No. 2020-01623). Informed consent was obtained from the patient or the next of kin if the patient was unable to give consent. The Declaration of Helsinki and its subsequent revisions were followed. The protocol for the study was registered (ClinicalTrials ID: NCT04316884); STROBE guidelines were followed for reporting.
General Clinical Chemistry Analyses
All routine lab tests were performed at the hospital’s clinical chemistry department. Blood samples were collected on admission to the ICU and daily during the ICU stay. Full blood counts (FBC), C-reactive protein (CRP), and kidney and liver function tests were performed in the hospital’s central laboratory. FBC was analyzed using a Sysmex XN™ instrument (Sysmex, Kobe, Japan); plasma CRP, ferritin, troponin I, procalcitonin, and kidney and liver markers were analyzed on an Architect ci16200 (Abbott Laboratories, Abbott Park, IL, USA). Acute kidney injury (AKI) was defined according to the KDIGO AKI definition. IL-6 was measured by commercial sandwich ELISA kit (D6050, R&D Systems, Minneapolis, MN, USA).
Complement Analyses
Complement function of the CPW and alternative pathway (APW) were measured by hemolytic assays (40). The concentrations of C3, C4, and factor B were quantified by nephelometry, MBL by ELISA, and C1q by a magnetic bead-based assay (41). The activation products were measured as follows: C3a by ELISA using monoclonal antibody (mAb) 4SD17.3 for capture and biotinylated polyclonal anti-C3a for detection (42), C3d,g by nephelometry after removal of high molecular weight forms of C3 by PEG precipitation (43), C4d by ELISA using an anti-human neo-C4d epitope mAb for capture (SVAR Life Science, Malmö, Sweden) (44), and sC5b-9 by an in-house ELISA using anti-human neo-C9 mAb aE11 for capture and polyclonal anti-C5 for detection (42). In order to compensate for intra-individual differences in C3 concentration due to consumption or acute phase reaction, the ratios of C3a to C3 (C3a/C3) and C3d,g to C3 (C3d,g/C3) were calculated.
Coagulation/Kallikrein/Kinin System Analyses
Thrombin-antithrombin (TAT) complexes were analyzed by ELISA using a pair of matched antibodies: anti-human thrombin for capture and anti- human antithrombin (ATIII) for detection (TAT-EIA, Enzyme Research Laboratories, South Bend, IN, USA). D-dimer was measured with STA Liatest D-Di Plus using STA-R Max2 (Diagnostica Stago, Saint-Ouen-l’Aumône, France). The kallikrein/kinin system proteins FXII, high molecular weight kininogen (HK) and prekallikein were all measured using an automated Western blot-like assay (WES). Simple Western 12- to 230-kDa assay cartridges were used under reducing conditions with a WES® analyzer (Protein Simple, Santa Clara, CA, USA) according to the manufacturer’s manual. The HK concentration represents an indirect measure of BK generation. The samples were diluted 1/250 (prekallikrein) and 1/500 (FXII, HK) in 0.1 × WES Sample Buffer, and the proteins were detected using affinity-purified, biotin-labeled goat anti-human FXII IgG (GAFXII-AP, Enzyme research Laboratories, South Bend, IN, USA) and goat anti-human kininogen IgG (BAF1396, R&D Systems, Minneapolis, MN, USA), and affinity-purified and peroxidase-conjugated anti-human prekallikrein IgG (SAPK-APHRP, Enzyme Research Laboratories). The electrophoretic protein separation and immunodetection that followed were performed using the default SimpleWestern™ settings. The quantified immune-detected signal, i.e., the area under the curve was analyzed using Compass software (version 4.0.0. ProteinSimple™), which also was used to convert the electropherograms into virtual blots. For each assay, a pool of EDTA-plasma from five healthy donors with assigned values for FXII (30 mg/L), HK (70 mg/L) and prekallikrein (50 mg/L) (45) was serially diluted and used as standard curve to calculate individual values in the patient and control samples.
Kallikrein-C1inhibitor (KK-C1INH) complexes were measured by a slightly modified sandwich ELISA as described before (46) using an affinity purified polyclonal sheep anti-human prekallikrein antibody (SAPK-AP, Enzyme research Laboratories) for capture and a biotinylated polyclonal rabbit anti-human C1INH antibody (in-house) followed by streptavidin-HRP (GE Healthcare) for detection. Standards were made from in vitro generated complexes of KK-C1INH (using a molar excess of C1INH) diluted in freshly drawn lepirudin anticoagulated plasma.
For all assays, EDTA-plasma from healthy blood donors was used as controls. The controls were handled and stored as the patients’ samples.
Statistics
GraphPad Prism 8.4 (GraphPad software, San Diego, CA, USA), Statistica (ver 13.5, TIBCO Software Inc., Palo Alto, CA, USA), or R (version 4.0.2) were used for statistical analyses and to generate graphs. Given the number of observations and their non-normal distribution, non-parametric statistical tests were used for all analyses. The Mann-Whitney U test was used to calculate differences between groups. Correlations according to Spearman were used to assess dependence between variables. Chi-square analysis was used to evaluate the risk of RAS inhibitors. Fisher´s exact test was used to evaluate patients’ results in relation to a given reference interval representing the normal range based on mean ± 2SD (as calculated in the clinic). Kaplan-Meier plots were used to estimate the probability of survival.
To investigate if and to what extent respiratory and renal failure mediate the activation of IIIS an uni- and a multivariable logistic regression was performed. Organ failure or organ support were dependent variables with IIIS activation and age as predictors to calculate odds ratios. SAPS-3 was included as a measure of illness severity. As the number of observations were limited, we used age as a surrogate for the pre COVID-19 risk of death. Moreover, if the IIIS activation variable was a predictor of outcome in the univariate model we assessed if it was still an independent predictor after adjusting for age. A p-value of <0.05 was considered significant, * <0.05, ** <0.01, *** <0.001 and **** <0.0001.
Results
COVID-19 Patients Admitted to the ICU
Blood was drawn prospectively from 66 patients when admitted to the ICU; 19 of these patients were followed longitudinally for up to 1 month. Descriptive data and the general history of the patients from the first day of admission are summarized in Table 1.
Evidence of an Established Thromboinflammation on Day 1
Table 2 is a summary of the results from the quantitation of the specific IIIS biomarkers on the day of admission to the ICU. Most of the markers showed values that were outside the reference ranges. These markers include all the cascade systems of the IIIS (the complement, coagulation, contact, and fibrinolysis systems) as well as ferritin, procalcitonin, CRP, and IL-6. In particular, the kallikrein/kinin system was strongly activated as reflected in high consumption of FXII, prekallikrein, and HK (Figure 1) (47). The consumption of prekallikrein was accompanied by generation of the corresponding complexes between the activation product KK and C1INH. Multiple correlation analyses showed that there were a number of significant correlations both within each cascade system and also between the cascade systems, immune cell counts, CRP levels, and IL-6 levels (heatmap, Figure 2). These correlations are further emphasized in Supplemental Table S1, which shows that the IL-6 and CRP levels are correlated with markers of both the complement and kallikrein/kinin systems. All these findings demonstrate that the IIIS is engaged in a response that is translated into thromboinflammation.
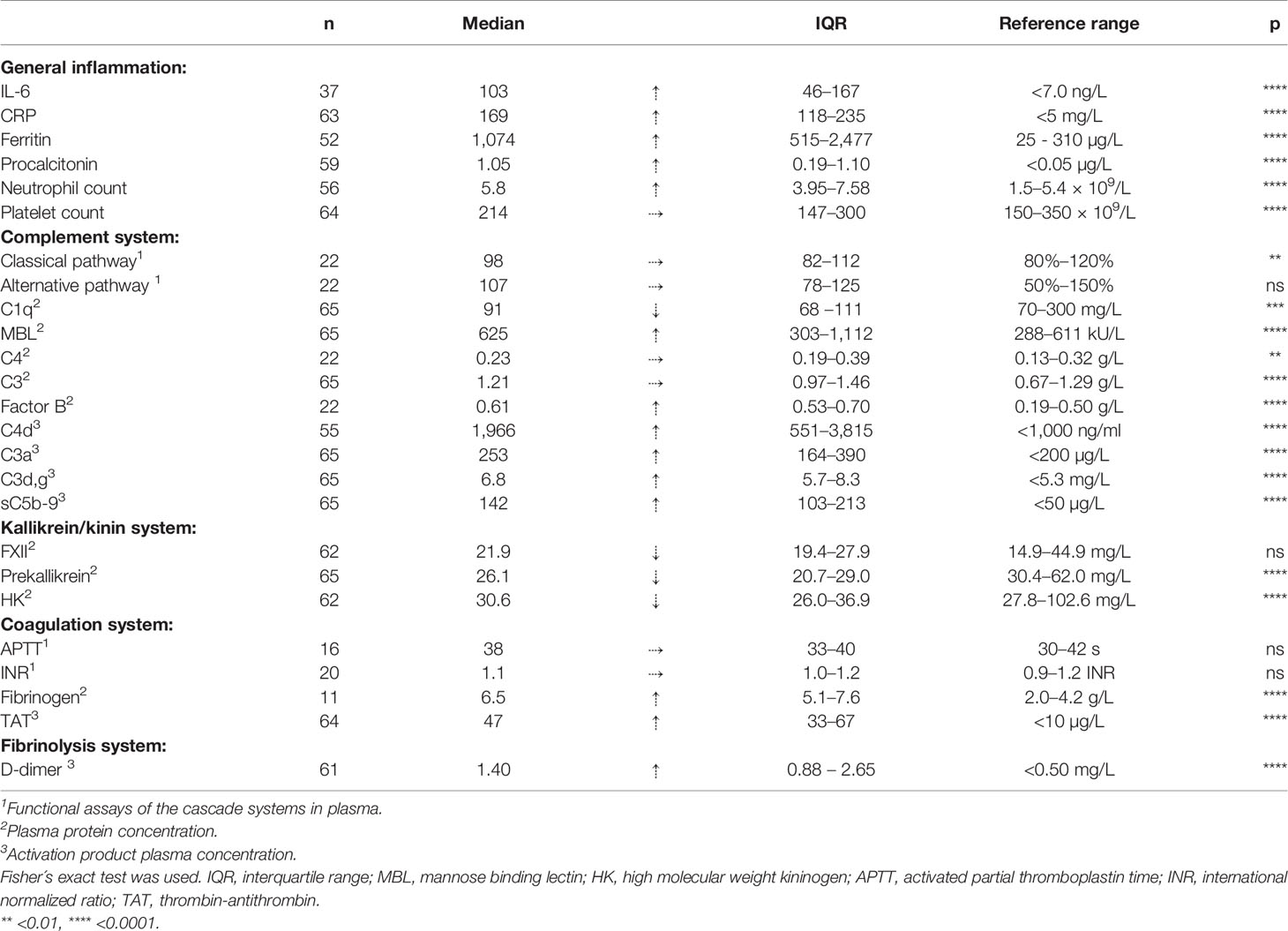
Table 2 Thromboinflammatory parameters in serum/plasma at admission to the ICU (n ≤ 65).
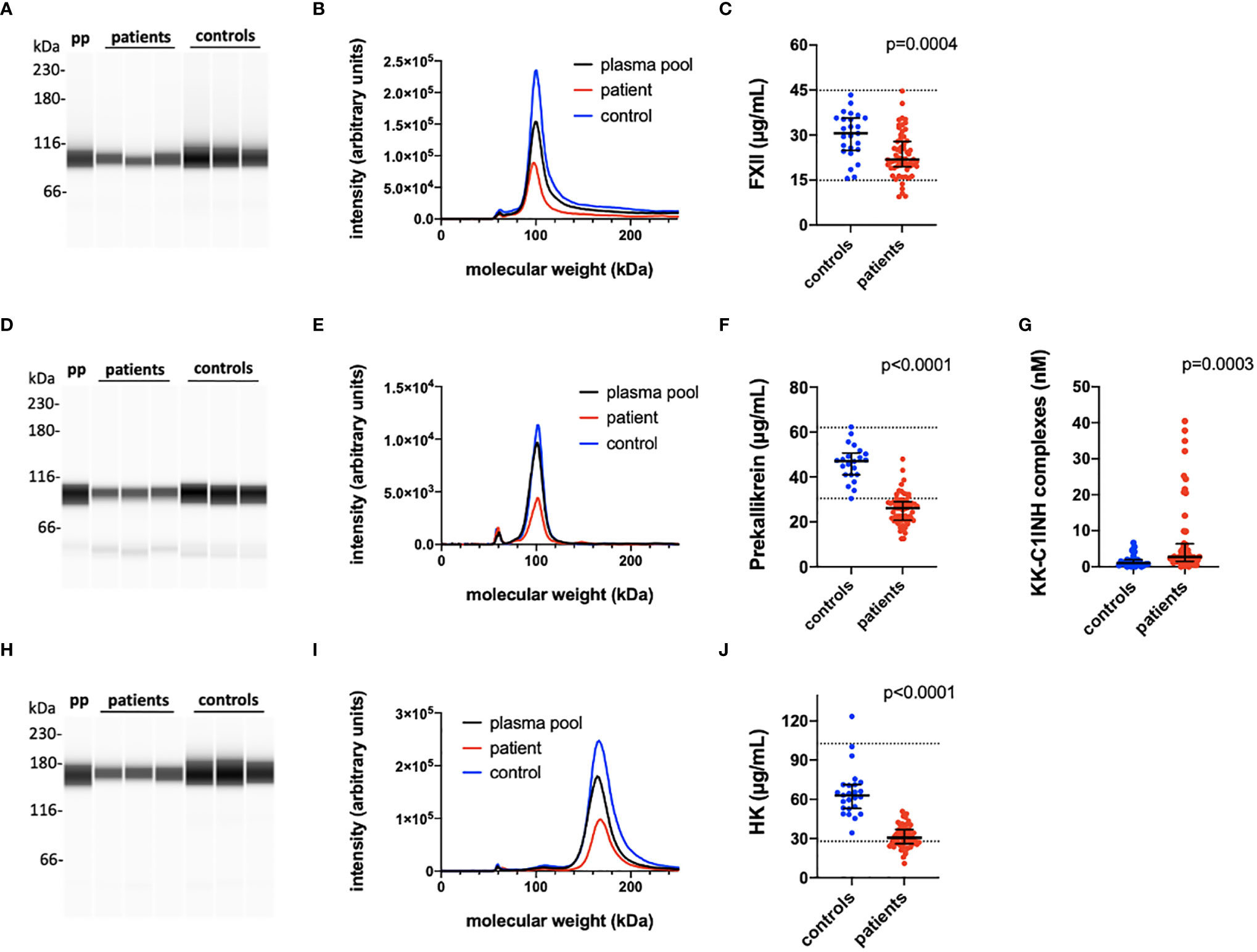
Figure 1 Quantification of the kallikrein/kinin proteins FXII, prekallikrein and high molecular weight kininogen (HK). The kallikrein/kinin system was activated as reflected in high consumption of FXII, prekallikrein, and HK. EDTA plasma from COVID-19 patients and controls were analyzed by capillary electrophoresis under reduced SDS-PAGE-like conditions and the specific kallikrein/kinin proteins detected by specific antibodies against FXII, prekallikrein, and HK, respectively (A, D, H). The area under the curves of the scan of the chemiluminescence intensities plotted in the panels (B, E, I) were interpreted to their specific concentration by comparing the value for each peak with a standard curve specific for each protein (C, F, J). The concentration of KK-C1INH complexes were analyzed by sandwich ELISA (panel G). Representative patients and controls are shown in panels (A, B, D, E, H, I). The Mann-Whitney U test was used to calculate differences between the groups. For more details see Materials and Methods. pp, plasma pool; KK, kallikrein; C1INH, C1inhibitor; HK, high molecular weight kininogen.
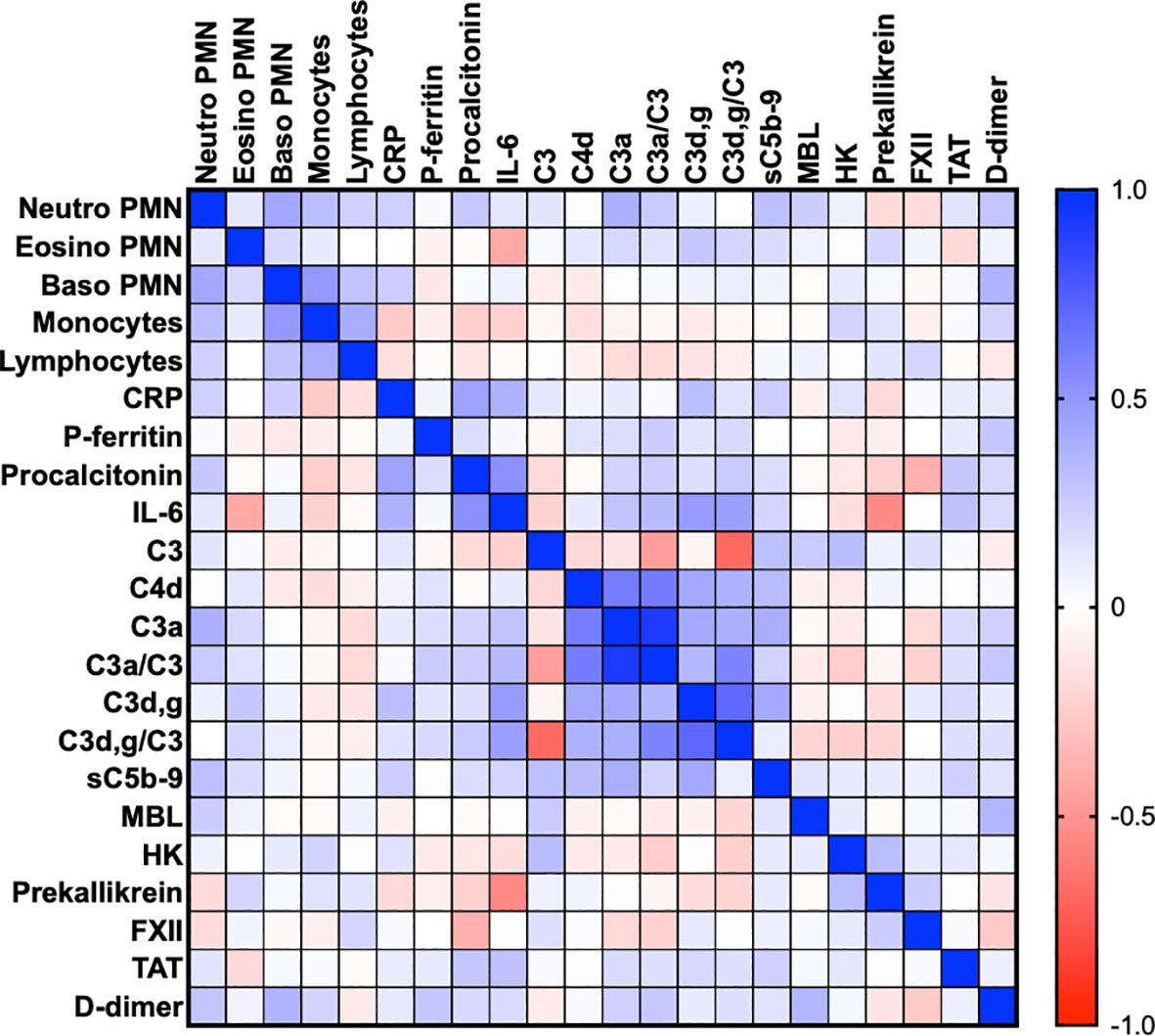
Figure 2 Thromboinflammation activation in critically ill COVID-19 patients. Thromboinflammatory parameters from 66 consecutive patients with COVID-19 were analyzed by the Spearman correlation test and presented in a heat map. Multiple significant correlations within each cascade system and also between the cascade systems, blood cell counts, CRP, and IL-6 were found. A summary of the most important correlations is presented in Table 3. TAT, thrombin-antithrombin.
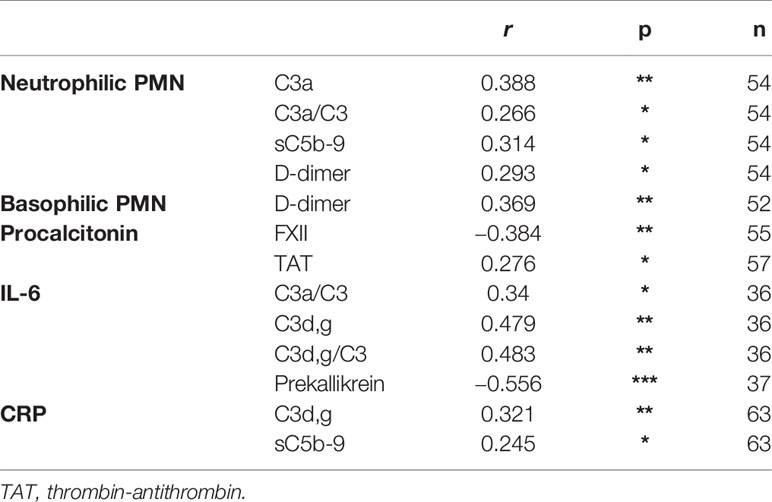
Table 3 Correlations between thromboinflammatory parameters within the IIIS (n ≤ 66).
Thromboinflammatory Reactions in Serial Samples
Several of the biomarkers were followed over time in 19 patients (Figure 3). At admission, some of their plasma protein levels and the functional tests showed signs of complement and kallikrein system consumption (e.g., CPW, C1q, and prekallikrein). Concomitantly, several of the activation markers were elevated including C4d, C3a, C3d,g, sC5b-9, D-dimer, and TAT; all these values tended to normalize over time in the ICU.
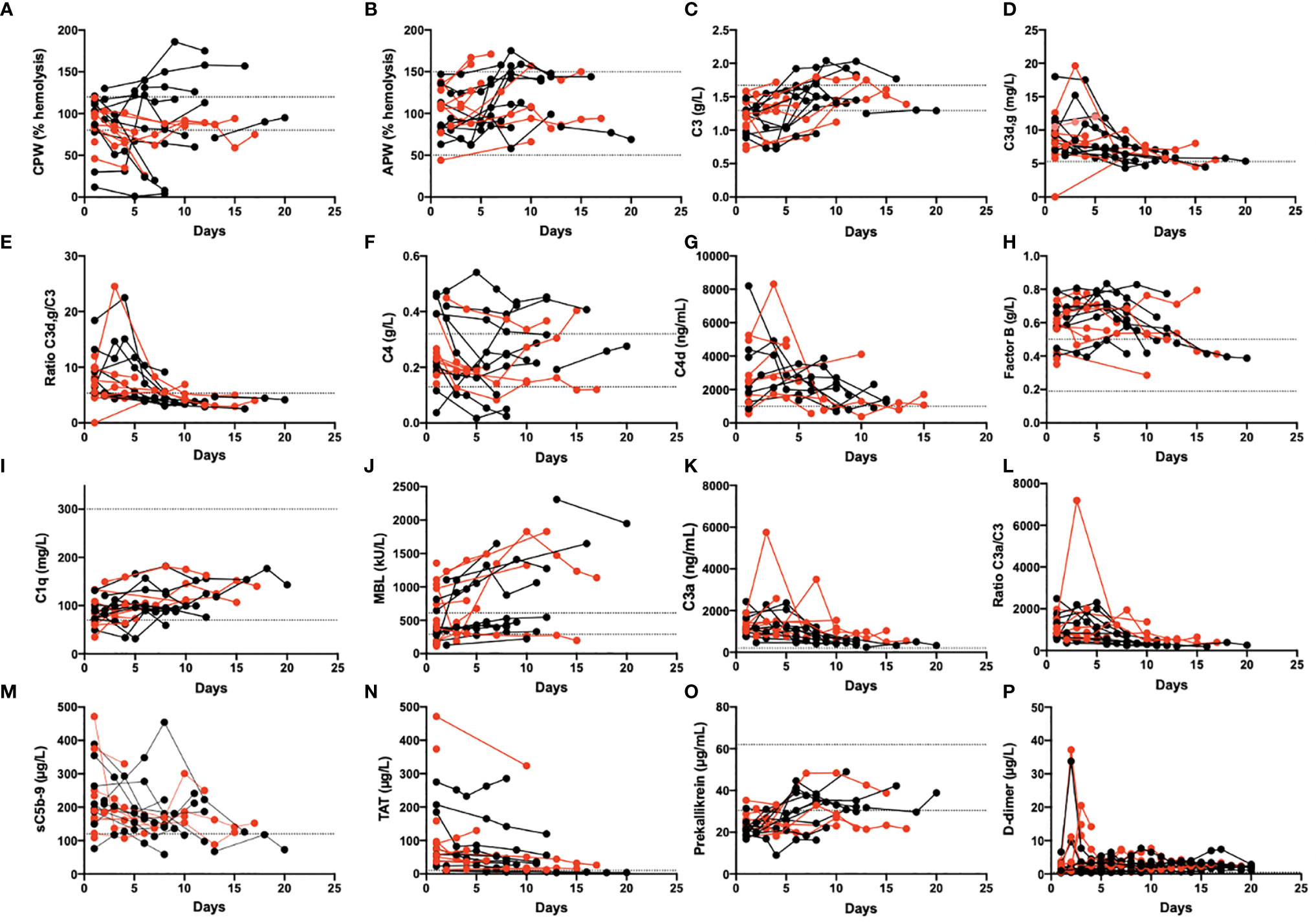
Figure 3 Longitudinal monitoring of thromboinflammation activation in critically ill COVID-19 patients. Analyzed complement related parameters included activation by the CPW and APW (A, B). levels of C3, C3d,g and the C3d,g/C3 ratio (C, D, E), C4 and C4d (F, G), Factor B, C1q, and MBL (H, I, J), the activation markers C3a, the C3a/C3 ratio, and the sC5b-9 complex (K, L, M). Additional parameters included the levels of TAT (N), prekallikrein (O), and D-dimer (P). Red dots: patients who died; black dots: patients who survived. The dotted lines mark the reference range. The presented data are from 19 of the first included patients that were followed for up to 1 month. CPW, classical pathway of complement; APW, alternative pathway of complement; MBL, mannose binding lectin; TAT, thrombin-antithrombin.
The parameters that precede C3 activation (C3d,g/C3 ratio) were investigated over time for possible correlations, and the results are presented in Table 4 and Supplemental Figure S1. Even though a varying number of observations are included from each patient, this table indicates how the CPW function, the recognition molecules C1q for the CPW and MBL for the LPW, were correlated with the first common activation step (C4) in the cascade. This supports the conclusion that activation of both the LPW and the CPW had occurred. Interestingly, levels of prekallikrein were also found to be correlated with C3d,g/C3 and factor B levels (r = −0.27; p < 0.05; Supplemental Figure S1), suggesting an association between the kallikrein/kinin system and the APW. C1q, prekallikrein, and C3d,g/C3 levels also showed an association with TAT levels (r = −0.39; p < 0.001; Supplemental Figure S1, I), suggesting a link between complement and coagulation.
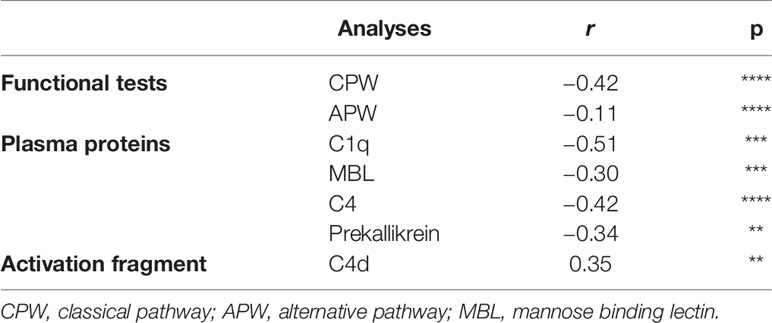
Table 4 Correlations between C3 activation (C3d,g/C3) and parameters of the various complement pathways in COVID- 19 patients that provided serial samples (n = 19).
Association of Complement and Kallikrein/Kinin Activation With Survival in the ICU
IIIS parameters at admittance were analyzed with regard to survival. The complement activation marker C3a (Supplemental Figure S2A) and the ratio C3a/C3 (not shown) were significantly higher in patients who died during their stay in the ICU (p = 0.03). Also, HK (Supplemental Figure S2B) and prekallikrein (not shown) levels were significantly or tended to be lower (p = 0.01 and p = 0.06, respectively) due to consumption (activation) in patients who died compared to those that survived during their stay in the ICU indicating that the kallikrein/kinin system was more activated in the patients who died. Thus, both the complement and the kallikrein/kinin system results predicted a poor outcome in the ICU. We then further analyzed the ratio C3a/C3, prekallikrein, and HK over time with regard to survival by using Kaplan-Meier plots (Figures 4A–C). Using the optimal cut-off values of 232 (ratio), 20 mg/L and 32mg/L for C3a, prekallikrein, and HK, respectively, we demonstrated that C3a/C3 levels tended to predict 30-day mortality, and prekallikrein and HK consumptions were strongly predictive of death in the studied cohort.
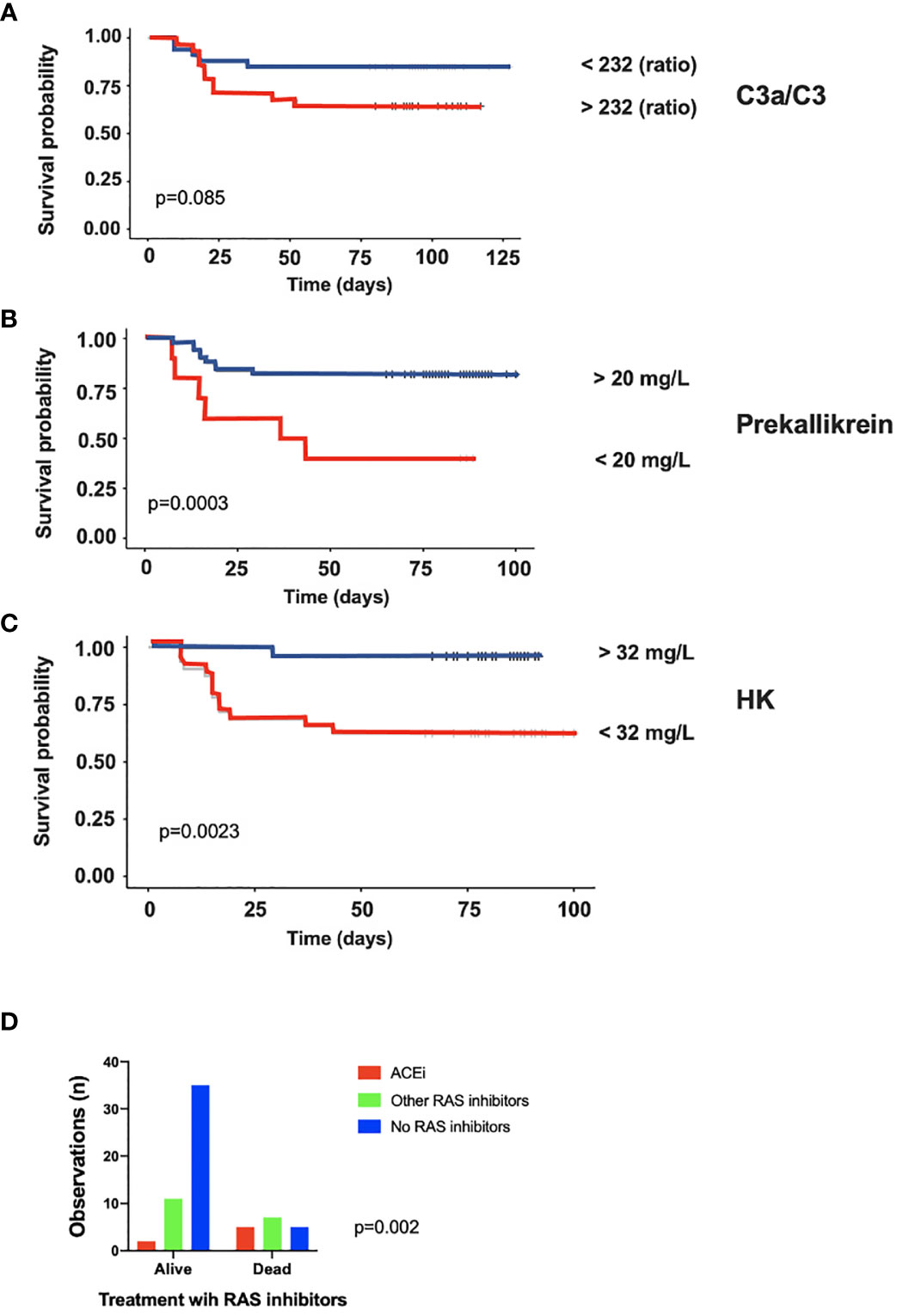
Figure 4 Association of intravascular innate immune system activation in COVID-19 ARDS with death. Kaplan-Meier plots of C3a/C3 (A), Prekallikrein (B) and HK (C) further stressed, that both the complement and the kallikrein/kinin systems are more activated in the patients who later died during their stay in the ICU. In panel (D) it is shown that the proportion of patients who died during the ICU stay was much higher in the group treated with ACE-1 inhibitors than among the patients on other types of renin-angiotensin system (RAS) inhibitors and those that did not take any RAS inhibitors at all. The presented data are from 66 consecutive patients with COVID-19 and for statistical evaluation non-parametric Mann Whitney U, Chi2 and Kaplan-Meier tests were used. HK, high molecular weight kininogen; ACEi, ACE-1 inhibitors; RAS, renin-angiotensin system.
In order to evaluate the possible effect of ACE-1 inhibitors on survival in the cohort, patients on ACE-1 inhibitors (n = 7) were compared to those receiving other renin-angiotensin system (RAS) inhibitors (n = 19) or other hypertensive drugs and those not receiving RAS inhibitors (n = 40; Figure 4D). The risk of death was highest for those on ACE inhibitors, whereas those on other RAS inhibitors or non-RAS drugs all had a substantially lower risk (OR 8.8 (range 1.5–46); p < 0.01).
Association of Complement and Kallikrein/Kinin System Activation With Organ Function
Increased C3a/C3 ratio was associated with impaired renal function as assessed by estimated glomerular filtration rate [eGFR (creatinine)] (Figure 5A), but this effect was not seen after adjustment for age. Decreased prekallikrein level was a predictor of severe ARDS and of mechanical ventilation, and this was consistent also after adjusting for age (Figure 5B). These findings could suggest that the association between the C3a/C3 ratio and death is mediated through renal failure (36), and strongly suggest that the association between prekallikrein and death is mediated through respiratory failure.
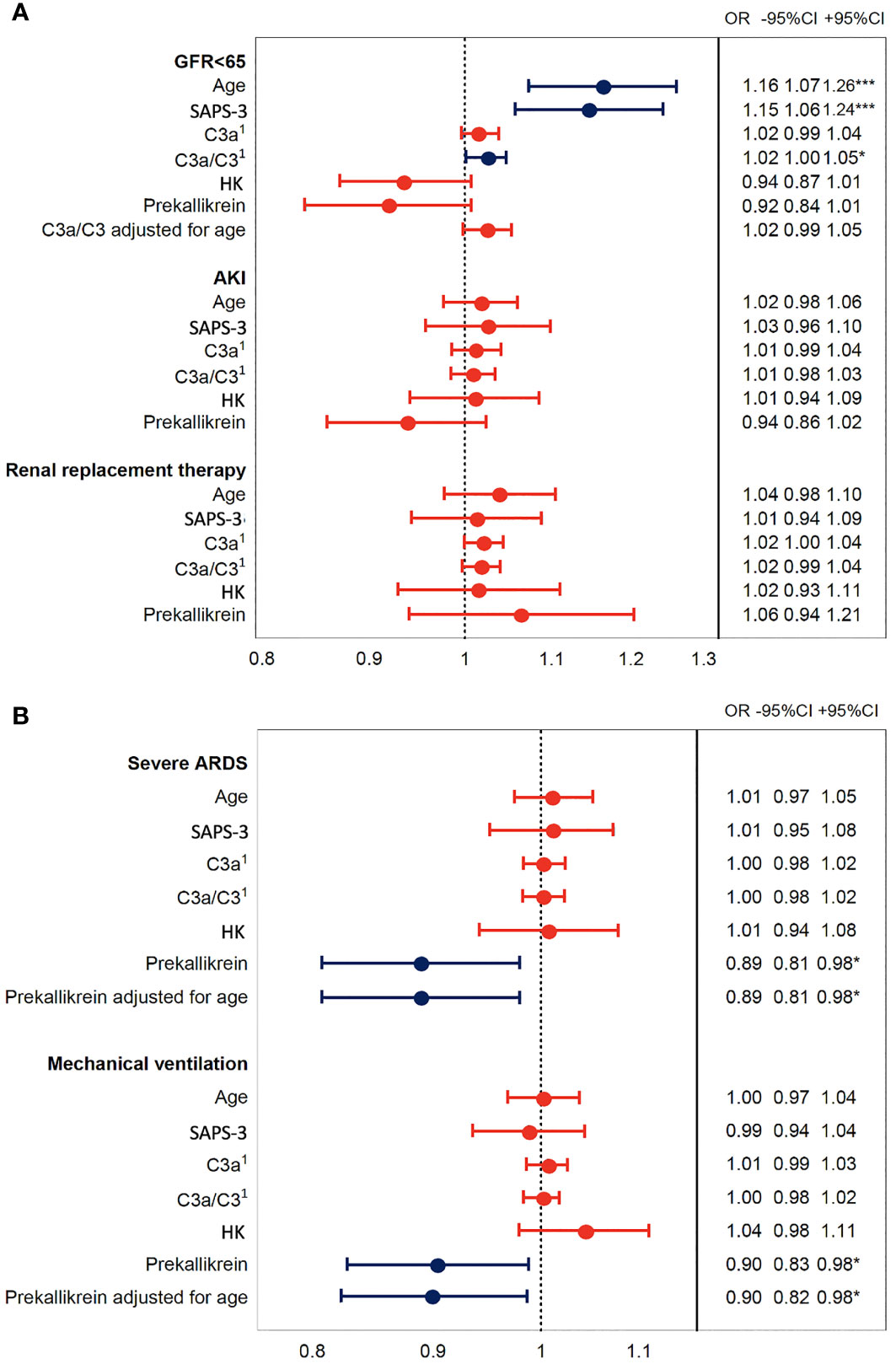
Figure 5 Association of IIIS activation in COVID-19 ARDS with organ damage. The plot shows odds ratios for IIIS activation variables C3a, C3a/C3, HK, and prekallikrein levels that were associated with death in Figure 4. Age is included as a surrogate for the pre COVID-19 risk of death and SAPS-3 as a marker of illness severity. Panel (A) depicts the kidney failure related outcomes, low glomerular filtration rate (GFR < 65 ml/min), acute kidney injury (AKI, vs. no AKI), and renal replacement therapy, i.e., dialysis. Panel (B) depicts the respiration failure related outcomes severe acute respiratory distress syndrome (ARDS, vs. mild or moderate ARDS) and mechanical ventilation. Blue intervals indicate statistical significance (*, *** right hand table), while red intervals do not. Increasing C3a/C3 ratios were predictors of low GFR but not after adjustment for age, while decreased prekallikrein levels were associated with respiratory failure also after adjustment for age. 1C3a has the unit of 10 ng/ml and C3a/C3 of ×10 in this figure. IIIS, intravascular innate immune system; HK, high molecular weight kininogen; SAPS-3, Simplified Acute Physiology Score 3.
Association of Complement and Kallikrein/Kinin System Activation With ICU Disease Indices
In Supplemental Table S2 it is shown that markers of complement and kallikrein/kinin activation correlate significantly to indices of kidney injury (AKI) and eGFR[creatinine], eGFR[cystatin C], cardiovascular disease (P-N-termpBNP, troponin I, heart rate), pulmonary function (pO2/FiO2), and with the two ICU indices SAPS-3 (probability of death) and SOFA (organ failure). D-dimer was also linked to heart rate and troponin I levels.
Discussion
The ARDS-like COVID-19-mediated lung injury is a severe and life-threatening condition that develops in a subset of SARS-CoV-2-infected patients. The pathophysiological mechanism involved in the development of this condition is not fully elucidated, but several reports have suggested the involvement of disturbances in the cascade systems of the blood (7, 27, 30, 48–50). In the present investigation we report the results of biomarker measurements reflecting IIIS activation and thromboinflammation in a single-center study of the first eligible 66 patients admitted to our ICU at the beginning of the pandemic. The investigational patients represent essentially naïve COVID-19 patients who, in addition to receiving established ICU treatment, had only been treated with LMW heparin, minimal extracorporeal treatment (hemodialysis), and no steroids. The patients’ clinical parameters were analyzed prospectively both cross-sectionally at admission (Table 2) and longitudinally (Figure 3). The most prominent aberration in the IIIS analyses was a strong kallikrein/kinin systems activation where individual components of the kallikrein/kinin system (FXII, prekallikrein and HK) were found to be very low in patients on day 1 reflecting a very strong activation (Figure 1). Over time, the prekallikrein concentrations tended to normalize. The complement parameters showed mixed results, with complement function (CPW and APW hemolytic assays) and individual components ranging from high to low values, reflecting both acute-phase reactions and complement consumption (activation). Complement activation products (C3a, C3d,g, and sC5b-9) tended to be high at the beginning of the ICU treatment but normalized over time; the coagulation system activation product TAT and the fibrinolysis marker D-dimer behaved in the same way. Thus, we found a general activation of the blood cascade systems (contact, complement, coagulation, and fibrinolysis systems), indicating a complete thromboinflammatory reaction mediated by the IIIS. General cross-cascade system correlations showed multiple associations between the cascade systems. Correlations between several markers of the complement and kallikrein/kinin systems, platelets, PMNs, ferritin, IL-6, and CRP indicate that the activation of the cascade systems had been translated into inflammation. Consistent with such an important role for inflammation in COVID-19, is the finding that both IL-6 and CRP are strong markers of COVID-19 disease activity (51). Correlation of the longitudinal parameters implies that the complement activation (C3d,g/C3) was triggered by both the classical (C1q) and the lectin (MBL) pathways. Interestingly, prekallikrein consumption also correlated with APW activation (which is reflected in C3d,g/C3 generation and the factor B concentration), consistent with previous in vitro findings in which kallikrein was found to cleave factor B to Ba and Bb in a manner similar that of factor D (52, 53).
We found that changes in the levels of individual components and activation products of the complement and kallikrein/kinin systems were associated with survival/death, organ function, and scores of illness severity. Within the complement system, there are several strong inflammatory products, such as C3d,g and the anaphylatoxins C3a and C5a. We measured C3a and C3d,g as well as sC5b-9 as an indirect measure of C5a generation, since C5a tends to bind to its highly expressed receptors and compromise the interpretation of its measurement (54). In particular, C3a and C3a/C3 at the time of admission showed a statistically significant and strong link to death and respiratory failure, assessed by the pO2/FiO2 ratio; to eGFR(creatinine); and to heart rate (Supplemental Figure S2 and Table S2). In addition, C3d,g and sC5b-9 were associated with the SAPS-3 score, with more favorable SAPS-3 levels being seen at low levels of complement activation. This finding suggests that complement activation reflects the outcome and damage to organs such as the lungs, the kidneys and the heart. A Kaplan-Meier analysis of C3a/C3 also tended to predict survival in the present cohort and logistic regression showed that C3a/C3 was associated with eGFR(creatinine) albeit not after adjustment for age.
Even more pronounced correlations and links were found for the kallikrein/kinin system. Both prekallikrein and HK were consumed (i.e., activated) to a significantly greater extent in the patients who later died during their stay in the ICU. Further supporting the predictive value of kallikrein/kinin system parameters, a Kaplan-Meier analysis demonstrated that prekallikrein and HK consumption was strongly predictive of death in the cohort. The kallikrein/kinin system was correlated with organ failure, reflected by eGFR (cystatin C), and injury reflected by troponin I levels and the propensity to receive mechanical ventilation, indicating that the lungs, kidneys and heart were affected. Logistic regression analysis of prekallikrein strongly suggested that the association with death was due to is respiratory failure. The strong correlation between FXII, prekallikrein, and HK indicated that the HK activation product BK is involved in the organ damage (34, 36, 50).
C3a, C5a and, in particular, BK formation could explain many of the symptoms and signs elicited by cell destruction, leukocyte infiltration, increased vascular permeability, thrombotic reactions and angiogenesis, which together cause poor gas exchange. This would explain the endothelialitis, thrombi, and increased vascular angiogenesis observed in pulmonary biopsies in COVID-19 patients (2). A recent in vitro mechanistic study of the RAS in cells obtained from bronchial alveolar lavage of COVID-19 patients strongly corroborates this concept. Due to low expression of ACE-1 and increased levels of ACE-2, the BK activity is predicted to be highly elevated (55). In turn, it is also tempting to speculate, that enhanced levels of BK and its interaction with both the BK-receptor 1 and 2 may result in disassembly and loss of intercellular adherence and tight junction molecules and consequently in an air-blood-barrier or remote organ-blood-barrier dysfunction (56, 57). Also, in this context hypertension has been suspected to be associated with an increased risk for severe COVID-19 infection, and particular drugs that act within the RAS have been suspected to increase the risk for severe disease. However, when the renin, ACE-1, and angiotensin receptor inhibitors have been grouped together in studies, this link to COVID-19 severity has not been confirmed. Given the disparate mechanisms of action of these drugs, it is necessary to compare the effects of these drugs separately. ACE-1 inhibitors lower the blood pressure by blocking ACE-1 from metabolizing angiotensin I to angiotensin II. ACE-1 also acts as an inactivator of BK, which makes it feasible that an inhibitor of ACE-1 might increase the levels of active BK and thereby aggravate the ARDS condition in COVID-19 patients. In our small cohort, we have separated our patients into those treated with ACE-1 inhibitors alone and those who are on other RAS inhibitors or on other hypertensive drugs. When we do so, we find that the risk for death substantially increases in patients on ACE-1 inhibitors, as compared to the other group without ACE-1 inhibitors. However, a larger population is required to confirm this preliminary finding.
Although the activation of IIIS in COVID-19 induced ARDS has many common features with ARDS of other ethiologies, such as sepsis, there are several differences both in terms of activation pathways and the focus of inflammation. We report that the IIIS is primarily activated through the kallikrein/kinin system and the CPW and LPW of complement (Figure 6). Others have reported the APW and later the CPW to be the key steps in complement activation in septic shock (58). These differences may be at least partly explained by the specific pathophysiologic process in SARS-CoV-2 infection, compared to the heterogeneous microbiology in different forms of sepsis. Another important dissimilarity between COVID-19 and sepsis induced ARDS is the focus of inflammation. ARDS in COVID-19 originates from the lung, as opposed to sepsis induced ARDS that is, unless the infection focus is in the lung, caused by a systemic inflammatory response in a distant organ (59). In the former, usually termed pulmonary ARDS, the lung is the motor of IIIS activation with highest IIIS activation locally, while in the latter, usually termed extrapulmonary ARDS, the lung is only secondarily affected and IIIS activation measured in plasma is less dependent on the severity of ARDS.
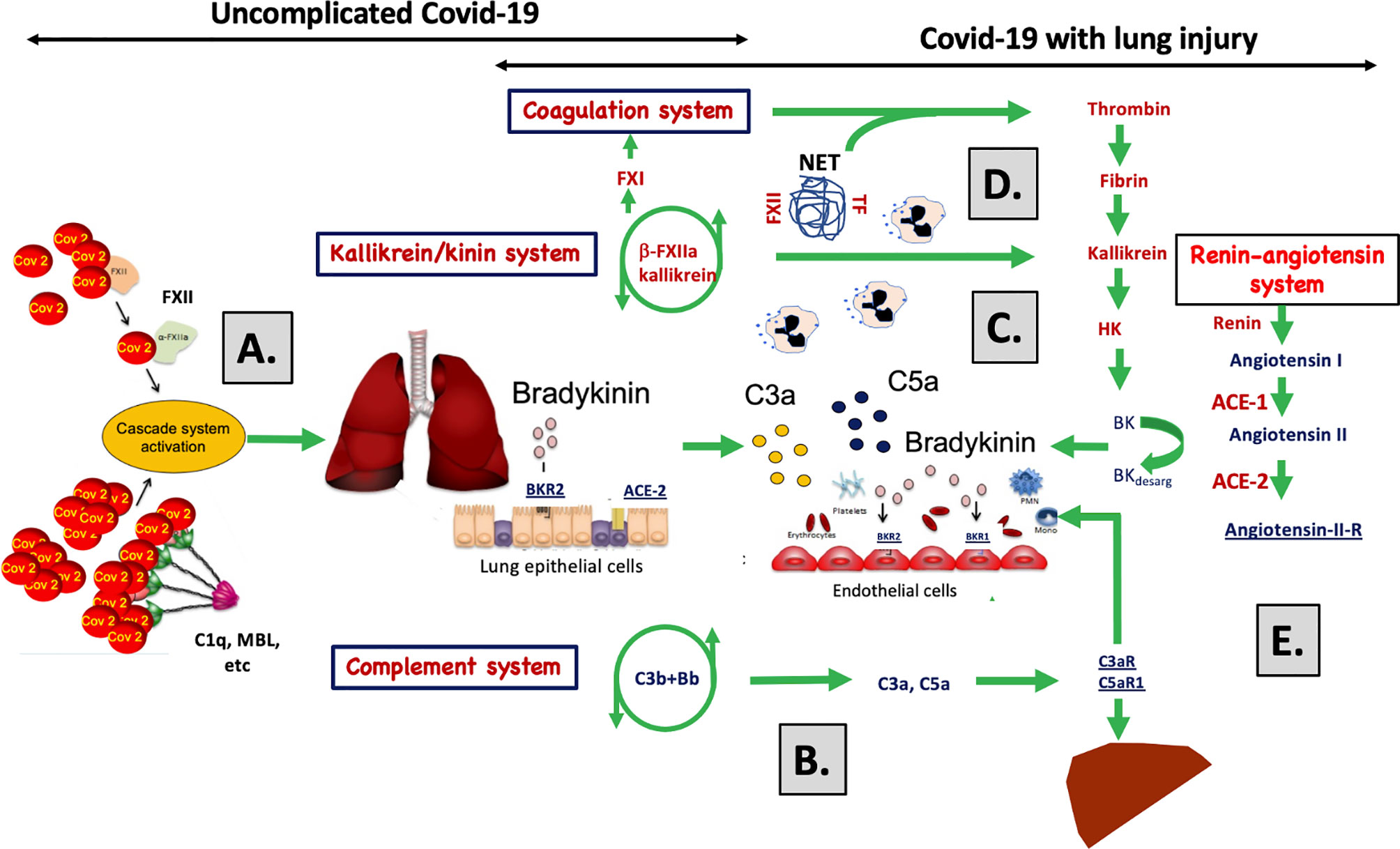
Figure 6 A graphical abstract summarizing the discussion of the data obtained in the study. (A) SARS-CoV-2 infects cells via ACE-2 and C1q, MBL and FXII recognize virus particles and their products, and recognize apoptotic and necrotic cells. These reactions are tentative triggers of the classical and lectin pathways of complement and of the kallikrein/kinin system described in the present study and by others (1, 13–21). The elicited C3a, C5a, and bradykinin (BK) generation induce, increased vascular permeability, endothelial activation, and nerve end stimulation via bradykinin receptor (BKR) 2 (15). (B) In response to C3a and C5a, proinflammatory cyto-/chemokines (e.g., IL-1β, IL-6) are produced that induce an acute inflammation and an acute phase reaction that increases the plasma protein (e.g., fibrinogen, MBL, C4, C3, Factor B) concentrations multi-fold and aggravate cascade system activation. (C) C5a triggers the up-regulation of BKR1, which is not constitutively expressed. BK can now act via BKR1 and BKR2 on both the pulmonary epithelium and endothelium. Together C3a, C5a, and BK have the potential to cause vascular leakage, edema, leukocyte chemotaxis, and activation that disturb oxygenation of the blood. (D) Local thrombi and pulmonary emboli further aggravate the oxygenation and cause collateral damage in other organs. Activated endothelial cells can induce thrombosis by binding MBL, FXII and by exposing tissue factor (TF) (34, 36, 50). Activation of neutrophilic granulocytes cause formation of neutrophil extracellular traps (NETs) that bind TF and triggers FXII activation (34, 35) and thereby initiating both the extrinsic and intrinsic pathways of the coagulation system. (E) The docking protein for SARS-CoV-2 on human cells is ACE-2 of the renin-angiotensin system (RAS) may cause dysregulation of the kallikrein/kinin system since BK and its metabolites are regulated directly and indirectly by the enzymes (ACE-1 and -2) of the RAS. In support of this, two of the signs and symptoms of COVID-19 can be induced as side effects during treatment of hypertension with ACE-1 inhibitors: these side effects include dry cough and increased vascular permeability (angioedema). Both of these symptoms can be alleviated by giving the patient icatibant (Firazyr®), an inhibitor of BKR2, suggesting that these symptoms may be caused by the kallikrein/kinin system.
The concept that activation of the IIIS is either caused by the virus directly or, more likely, caused by NET formation (34, 50) in combination with the large amounts of activated and damaged (apoptotic and necrotic) cells produced, may imply that individual components of the IIIS can be targeted in order to treat COVID-19 patients. There are several licensed drugs that affect components of the IIIS. The complement system has already been targeted (31). Initial trials with a licensed anti-C5 antibody (eculizumab) have been performed, with potentially promising results (16, 60–63). Also, several anti-complement drugs are under development for use in COVID-19, such as C3 inhibitors of the compstatin family, which recently was shown to induce recovery and a drop in several major inflammatory parameters (63, 64). From the results of the present study, inhibition of the kallikrein/kinin system seems to be an important step in the search for therapeutic alternatives for treatment of COVID-19. Drugs for the treatment of angioedema target the kallikrein/kinin system: the effect of BK can be inhibited by the bradykinin receptor (BKR)-2 inhibitor icatibant (65), and the kallikrein inhibitory antibody lanadelumab (13). The kallikrein inhibitor ecallantide, which is licensed in the US, and purified and recombinant C1INH are other examples of drugs that could potentially be used in COVID-19 patients (65). The successful treatment of ARDS with icatibant in a hantavirus infection has already shown that the kallikrein/kinin system played a major role in this patient’s infection and that a BKR2 inhibitor alleviated the virus-induced ARDS (66).
A limitation in this hypothesis-generating study is the relatively low number of patients. Likewise, as our study is exploratory, we did not correct for multiple testing in the statistical analysis, which could lead to spurious associations. Importantly, the main findings of our study, e.g., kallikrein/kinin system activation and its relation to survival, were confirmed by multiple individual biomarkers, corroborating these results.
Furthermore, a clear strength is the extensive work-up leading to high resolution investigation of the IIIS. We therefore suggest that the newly described strong activation of the kallikrein/kinin system is a main driver of the ARDS-like condition in COVID-19, which is part of a conjunct activation of the IIIS. The strong link to respiratory failure and death implicates activation of the IIIS and the kallikrein/kinin system as potential mechanism that mediates the tissue damage in COVID-19 patients (Discussion summarized in Figure 6). The biomarkers of the IIIS, particularly within the kallikrein/kinin system, are likely to have a predictive value and pinpoint potential new targets for the treatment of COVID-19 disease with already licensed drugs.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by the Swedish National Ethical Review Agency (EPM; No. 2020-01623). Informed consent was obtained from the patient or the next of kin if the patient was unable give consent. The Declaration of Helsinki and its subsequent revisions were followed. The protocol for the study was registered (ClinicalTrials ID: NCT04316884); STROBE guidelines were followed for reporting. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
ML, BP, OE, MH, MHL, KE, RF, and BN were responsible for the design of the study. ML, MH, and RF were responsible for the patients and for the collection of the patient samples. BP, OE, KE, and BN were responsible for laboratory data. BP performed the WES assays and KF performed the assessment of KK-C1INH complexes. AB provided the C4d analysis. BN wrote the first original draft of the manuscript. All authors participated in the data collection and in revision of the manuscript. ML and BP share the first authorship and RF and BN share the last authorship. ML appears first and BN appears last in the author list due to applying a strict alphabetical order for these positions in the author list. All authors contributed to the article and approved the submitted version.
Funding
The study was funded by grants from SciLifeLab/The Knut and Alice Wallenberg Foundation (KAW2020.0182), the Swedish Research Council (2014-02569, 2014-07606, 2015-06429, 2016-01060, 2016-04519, 2020-05762), the Swedish Heart-Lung Foundation (HLF 2020-0398), and by faculty grants form Linnaeus University as well as in part by the DFG-grant CRC1149 A01 (INST 40/479-2).
Conflict of Interest
AB is named as inventor in a patent application including claims to use of C4d as biomarker.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
Acknowledgments
The technical expertise of Sliva Abdalla, Amanda Åman, and David Eikrem is greatly appreciated. The authors thank the study nurses Elin Söderberg and Joanna Wessbergh, and the biobank research assistants Philip Karlsson and Erik Danielsson.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.627579/full#supplementary-material
Abbreviations
ACE, Angiotensin converting enzyme; AKI, Acute kidney injury; APTT, Activated partial thromboplastin time; APW, Alternative pathway; ARB, Angiotensin receptor blocker; ARDS, Acute respiratory distress syndrome; BK, Bradykinin; BKR, Bradykinin receptor; C1INH, C1 inhibitor; CPW, Classical pathway; eGFR, Estimated glomerular filtration rate; FBC, Full blood count; HK, High molecular weight kininogen; ICU, Intensive care unit; IIIS, Intravascular innate immune system; INR, International normalized ratio; IQR, Interquartile range; KDIGO, Kidney Disease: Improving Global Outcomes; KK, Kallikrein; KO, knock-out; LMW, Low-molecular weight; LPW, Lectin pathway; mAb, Monoclonal antibody; MASP, Mannose binding lectin Associated Serine Protease; MBL, Mannose binding lectin; NET, Neutrophil extracellular trap; PMN, Polymorphonuclear cells; RAS, Renin-angiotensin system; RT-PCR, Reverse transcription polymerase chain reaction; SAPS-3, Simplified Acute Physiology Score 3; SOFA, Sequential Organ Failure Assessment; TAT, Thrombin–antithrombin; TF, Tissue factor; WES, Western blot-like assay.
References
1. Marietta M, Coluccio V, Luppi M. COVID-19, coagulopathy and venous thromboembolism: more questions than answers. Intern Emerg Med (2020) 8:1375–87. doi: 10.1007/s11739-020-02432-x
2. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med (2020) 383:120–8. doi: 10.1056/nejmoa2015432
3. Skevaki C, Fragkou PC, Cheng C, Xie M, Renz H. Laboratory characteristics of patients infected with the novel SARS-CoV-2 virus. J Infect (2020) 81:205–12. doi: 10.1016/j.jinf.2020.06.039
4. Remy KE, Mazer M, Striker DA, Ellebedy AH, Walton AH, Unsinger J, et al. Severe immunosuppression and not a cytokine storm characterize COVID-19 infections. JCI Insight (2020) 5(17):e140329. doi: 10.1172/jci.insight.140329
5. Valle DMD, Kim-Schulze S, Huang H-H, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med (2020) 26:1636–43. doi: 10.1038/s41591-020-1051-9
6. Cavagna E, Muratore F, Ferrari F. Pulmonary Thromboembolism in COVID-19: Venous Thromboembolism or Arterial Thrombosis? Radiol Cardiothorac Imaging (2020) 2:e200289. doi: 10.1148/ryct.2020200289
7. Mackman N, Antoniak S, Wolberg AS, Kasthuri R, Key NS. Coagulation Abnormalities and Thrombosis in Patients Infected With SARS-CoV-2 and Other Pandemic Viruses. Arterioscler Thromb Vasc Biol (2020) 40(9):2033–44. doi: 10.1161/atvbaha.120.314514
8. Geng Y-J, Wei Z-Y, Qian H-Y, Huang J, Lodato R, Castriotta RJ. Pathophysiological Characteristics and Therapeutic Approaches for Pulmonary Injury and Cardiovascular Complications of Coronavirus Disease 2019. Cardiovasc Pathol (2020) 47:107228. doi: 10.1016/j.carpath.2020.107228
9. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol (2012) 13:34–45. doi: 10.1038/nri3345
10. Ekdahl KN, Teramura Y, Hamad OA, Asif S, Duehrkop C, Fromell K, et al. Dangerous liaisons: complement, coagulation, and kallikrein/kinin cross-talk act as a linchpin in the events leading to thromboinflammation. Immunol Rev (2016) 274:245–69. doi: 10.1111/imr.12471
11. Biglarnia A-R, Huber-Lang M, Mohlin C, Ekdahl KN, Nilsson B. The multifaceted role of complement in kidney transplantation. Nat Rev Nephrol (2018) 14:767–81. doi: 10.1038/s41581-018-0071-x
12. Ekdahl KN, Soveri I, Hilborn J, Fellstrom B, Nilsson B. Cardiovascular disease in haemodialysis: Role of the intravascular innate immune system. Nat Rev Nephrol (2017) 13:285–96. doi: 10.1038/nrneph.2017.17
13. Colarusso C, Terlizzi M, Pinto A, Sorrentino R. A lesson from a saboteur: high molecular weight kininogen (HMWK) impact in COVID-19. Brit J Pharmacol (2020) 177(21):4866–72. doi: 10.1111/bph.15154
14. Maglakelidze N, Manto KM, Craig TJ. A Review: Does Complement or the Contact System Have a Role in Protection or Pathogenesis of COVID-19? Pulm Ther (2020) 2:169–276. doi: 10.1007/s41030-020-00118-5
15. Thomson TM, Toscano E, Casis E, Paciucci R. C1-INH and the contact system in COVID-19. Brit J Haematol (2020) 190(4):520–4. doi: 10.1111/bjh.16938
16. Risitano AM, Mastellos DC, Huber-Lang M, Yancopoulou D, Garlanda C, Ciceri F, et al. Complement as a target in COVID-19? Nat Rev Immunol (2020) 20:343–4. doi: 10.1038/s41577-020-0320-7
17. Lo MW, Kemper C, Woodruff TM. COVID-19: Complement, Coagulation, and Collateral Damage. J Immunol Baltim Md 1950 (2020) 205(6):1488–95. doi: 10.4049/jimmunol.2000644
18. Henry BM, Vikse J, Benoit S, Favaloro EJ, Lippi G. Hyperinflammation and Derangement of Renin-Angiotensin-Aldosterone System in COVID-19: a novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin Chim Acta (2020) 507:167–73. doi: 10.1016/j.cca.2020.04.027
19. Shatzel JJ, DeLoughery EP, Lorentz CU, Tucker EI, Aslan JE, Hinds MT, et al. The contact activation system as a potential therapeutic target in patients with COVID-19. Res Pract Thromb Haemostasis (2020) 4:500–5. doi: 10.1002/rth2.12349
20. Campbell CM, Kahwash R. Will Complement Inhibition Be the New Target in Treating COVID-19-Related Systemic Thrombosis? Circulation (2020) 141:1739–41. doi: 10.1161/circulationaha.120.047419
21. Java A, Apicelli AJ, Liszewski MK, Coler-Reilly A, Atkinson JP, Kim AHJ, et al. The complement system in COVID-19: friend and foe? JCI Insight (2020) 5(15):e140711. doi: 10.1172/jci.insight.140711
22. Lu J, Kishore U. C1 Complex: An Adaptable Proteolytic Module for Complement and Non-Complement Functions. Front Immunol (2017) 8:592. doi: 10.3389/fimmu.2017.00592
23. Kaplan AP, Joseph K, Silverberg M. Pathways for bradykinin formation and inflammatory disease. J Allergy Clin Immun (2002) 109:195–209. doi: 10.1067/mai.2002.121316
24. Joseph K, Shibayama Y, Ghebrehiwet B, Kaplan AP. Factor XII-dependent contact activation on endothelial cells and binding proteins gC1qR and cytokeratin 1. Thromb Haemostasis (2001) 85:119 124.
25. Trouw LA, Blom AM, Gasque P. Role of complement and complement regulators in the removal of apoptotic cells. Mol Immunol (2008) 45:1199 1207. doi: 10.1016/j.molimm.2007.09.008
26. Ip WKE, Chan KH, Law HKW, Tso GHW, Kong EKP, Wong WHS, et al. Mannose-Binding Lectin in Severe Acute Respiratory Syndrome Coronavirus Infection. J Infect Dis (2005) 191:1697–704. doi: 10.1086/429631
27. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl Res J Lab Clin Med (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
28. Holter JC, Pischke SE, de Boer E, Lind A, Jenum S, Holten AR, et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc Natl Acad Sci (2020) 117(40):25018–25. doi: 10.1073/pnas.2010540117
29. Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, et al. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. Mbio (2018) 9:e01753–18. doi: 10.1128/mbio.01753-18
30. Cugno M, Meroni PL, Gualtierotti R, Griffini S, Grovetti E, Torri A, et al. Complement activation in patients with Covid-19: a novel therapeutic target. J Allergy Clin Immunol (2020) 146:215–7. doi: 10.1016/j.jaci.2020.05.006
31. Polycarpou A, Howard M, Farrar CA, Greenlaw R, Fanelli G, Wallis R, et al. Rationale for targeting complement in COVID-19. EMBO Mol Med (2020) 12:e12642. doi: 10.15252/emmm.202012642
32. Schapira M, Gardaz JP, Py P, Lew PD, Perrin LH, Suter PM. Prekallikrein activation in the adult respiratory distress syndrome. Bull Européen Physiopathologie Respir (1985) 21:237–41.
33. Hammerschmidt D, Hudson L, Weaver LJ, Craddock P, Jacob H. ASSOCIATION OF COMPLEMENT ACTIVATION AND ELEVATED PLASMA-C5a WITH ADULT RESPIRATORY DISTRESS SYNDROME Pathophysiological Relevance and Possible Prognostic Value. Lancet (1980) 315:947–9. doi: 10.1016/s0140-6736(80)91403-8
34. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest (2020) 130(11):6151–7. doi: 10.1172/jci141374
35. Renné T, Stavrou EX. Roles of Factor XII in Innate Immunity. Front Immunol (2019) 10:2011. doi: 10.3389/fimmu.2019.02011
36. Eriksson O, Persson B, Lipcsey M, Ekdahl K, Nilsson B. Mannose-Binding Lectin is Associated with Thrombosis and Coagulopathy in Critically Ill COVID-19 Patients. Thromb Haemostasis (2020) 120(12):1720–4. doi: 10.1055/s-0040-1715835
37. Moreno RP, Metnitz PGH, Almeida E, Jordan B, Bauer P, Campos RA, et al. SAPS 3—From evaluation of the patient to evaluation of the intensive care unit. Part 2: Development of a prognostic model for hospital mortality at ICU admission. Intens Care Med (2005) 31:1345–55. doi: 10.1007/s00134-005-2763-5
38. Vincent J-L, Moreno R, Takala J, Willatts S, Mendonça AD, Bruining H, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure: On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine (see contributors to the project in the appendix). Intens Care Med (1996) 22:707–10. doi: 10.1007/bf01709751
39. Khwaja A. KDIGO Clinical Practice Guidelines for Acute Kidney Injury. Nephron (2012) 120:c179–84. doi: 10.1159/000339789
40. Nilsson UR, Nilsson B. Simplified assays of hemolytic activity of the classical and alternative complement pathways. J Immunol Methods (2002) 72:49–59. doi: 10.1016/0022-1759(84)90432-0
41. Sandholm K, Persson B, Skattum L, Eggertsen G, Nyman D, Gunnarsson I, et al. Evaluation of a Novel Immunoassay for Quantification of C1q for Clinical Diagnostic Use. Front Immunol (2019) 10:7. doi: 10.3389/fimmu.2019.00007
42. Ekdahl KN, Nilsson B, Pekna M, Nilsson UR. Generation of iC3 at the interface between blood and gas. Scand J Immunol (1992) 35:85–91. doi: 10.1111/j.1365-3083-1992.tb02837.x
43. Ekdahl KN, Norberg D, Bengtsson AA, Sturfelt G, Nilsson UR, Nilsson B. Use of serum or buffer-changed EDTA-Plasma in a rapid, inexpensive, and easy-to-perform hemolytic complement assay for differential diagnosis of systemic lupus erythematosus and monitoring of patients with the disease. Clin Vaccine Immunol (2007) 14:549–55. doi: 10.1128/CVI.00486-06
44. Martin M, Trattner R, Nilsson SC, Björk A, Zickert A, Blom AM, et al. Plasma C4d Correlates With C4d Deposition in Kidneys and With Treatment Response in Lupus Nephritis Patients. Front Immunol (2020) 11:582737. doi: 10.3389/fimmu.2020.582737
45. Kaplan AP, Ghebrehiwet B. The plasma bradykinin-forming pathways and its interrelationships with complement. Mol Immunol (2010) 47:2161–9. doi: 10.1016/j.molimm.2010.05.010
46. Bäck J, Lang MH, Elgue G, Kalbitz M, Sanchez J, Ekdahl KN, et al. Distinctive regulation of contact activation by antithrombin and C1-inhibitor on activated platelets and material surfaces. Biomaterials (2009) 30:6573–80. doi: 10.1016/j.biomaterials.2009.07.052
47. Etscheid M, Kreß J, Seitz R, Dodt J. The hyaluronic acid-binding protease: A novel vascular and inflammatory mediator? Int Immunopharmacol (2008) 8:166–70. doi: 10.1016/j.intimp.2007.10.012
48. van Dam LF, Kroft LJM, van der Wal LI, Cannegieter SC, Eikenboom J, de Jonge E, et al. Clinical and computed tomography characteristics of COVID-19 associated acute pulmonary embolism: A different phenotype of thrombotic disease? Thromb Res (2020) 193:86–9. doi: 10.1016/j.thromres.2020.06.010
49. Gibson PG, Qin L, Puah SH. COVID-19 acute respiratory distress syndrome (ARDS): clinical features and differences from typical pre-COVID-19 ARDS. Med J Aust (2020) 213:54. doi: 10.5694/mja2.50674
50. Busch MH, Timmermans SAMEG, Nagy M, Visser M, Huckriede J, Aendekerk JP, et al. Neutrophils and Contact Activation of Coagulation as Potential Drivers of COVID-19. Circulation (2020) 142:1787–90. doi: 10.1161/circulationaha.120.050656
51. Liu F, Li L, Xu M, Wu J, Luo D, Zhu Y, et al. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J Clin Virol (2020) 127:104370. doi: 10.1016/j.jcv.2020.104370
52. DiScipio RG. The activation of the alternative pathway C3 convertase by human plasma kallikrein. Immunology (1982) 45:587–95.
53. Irmscher S, Döring N, Halder LD, Jo EAH, Kopka I, Dunker C, et al. Kallikrein Cleaves C3 and Activates Complement. J Innate Immun (2018) 10:94–105. doi: 10.1159/000484257
54. Hetland G, Moen O, Bergh K, Högåsen K, Hack CE, Mollnes TE, et al. Both Plasma- and Leukocyte-Associated C5a Are Essential for Assessment of C5a Generation In Vivo. Ann Thorac Surg (1997) 63:1076–80. doi: 10.1016/s0003-4975(96)01255-6
55. Garvin MR, Alvarez C, Miller JI, Prates ET, Walker AM, Amos BK, et al. A mechanistic model and therapeutic interventions for COVID-19 involving a RAS-mediated bradykinin storm. Elife (2020) 9:e59177. doi: 10.7554/elife.59177
56. Mugisho OO, Robilliard LD, Nicholson LFB, Graham ES, O’Carroll SJ. Bradykinin receptor-1 activation induces inflammation and increases the permeability of human brain microvascular endothelial cells. Cell Biol Int (2020) 44:343–51. doi: 10.1002/cbin.11232
57. Terzuoli E, Corti F, Nannelli G, Giachetti A, Donnini S, Ziche M. Bradykinin B2 Receptor Contributes to Inflammatory Responses in Human Endothelial Cells by the Transactivation of the Fibroblast Growth Factor Receptor FGFR-1. Int J Mol Sci (2018) 19:2638. doi: 10.3390/ijms19092638
58. Charchaflieh J, Wei J, Labaze G, Hou YJ, Babarsh B, Stutz H, et al. The Role of Complement System in Septic Shock. Clin Dev Immunol (2012) 2012:407324. doi: 10.1155/2012/407324
59. Pelosi P, D’Onofrio D, Chiumello D, Paolo S, Chiara G, Capelozzi VL, et al. Pulmonary and extrapulmonary acute respiratory distress syndrome are different. Eur Respir J (2003) 22:48s–56s. doi: 10.1183/09031936.03.00420803
60. Laurence J, Mulvey JJ, Seshadri M, Racanelli A, Harp J, Schenck EJ, et al. Anti-complement C5 therapy with eculizumab in three cases of critical COVID-19. Clin Immunol (2020) 219:108555. doi: 10.1016/j.clim.2020.108555
61. Carvelli J, Demaria O, Vély F, Batista L, Benmansour NC, Fares J, et al. Association of COVID-19 inflammation with activation of the C5a-C5aR1 axis. Nature (2020) 588(7836):146–50. doi: 10.1038/s41586-020-2600-6
62. de Latour RP, Bergeron A, Lengline E, Dupont T, Marchal A, Galicier L, et al. Complement C5 inhibition in patients with COVID-19 - a promising target? Haematologica (2020) 105(12):2847–50. doi: 10.3324/haematol.2020.260117
63. Mastellos DC, da Silva BGPP, Fonseca BAL, Fonseca NP, Martins MA, Mastaglio S, et al. Complement C3 vs C5 inhibition in severe COVID-19: Early clinical findings reveal differential biological efficacy. Clin Immunol (2020) 220:108598. doi: 10.1016/j.clim.2020.108598
64. Mastaglio S, Ruggeri A, Risitano AM, Angelillo P, Yancopoulou D, Mastellos DC, et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol (2020) 215:108450. doi: 10.1016/j.clim.2020.108450
65. LoVerde D, Files DC, Krishnaswamy G. Angioedema. Crit Care Med (2017) 45:725–35. doi: 10.1097/ccm.0000000000002281
Keywords: thromboinflammation, kallikrein/kinin system, complement system, coagulation system, fibrinolysis system, COVID-19, prognosis
Citation: Lipcsey M, Persson B, Eriksson O, Blom AM, Fromell K, Hultström M, Huber-Lang M, Ekdahl KN, Frithiof R and Nilsson B (2021) The Outcome of Critically Ill COVID-19 Patients Is Linked to Thromboinflammation Dominated by the Kallikrein/Kinin System. Front. Immunol. 12:627579. doi: 10.3389/fimmu.2021.627579
Received: 09 November 2020; Accepted: 18 January 2021;
Published: 22 February 2021.
Edited by:
Hans A. R. Bluyssen, Adam Mickiewicz University, PolandReviewed by:
Malgorzata Wygrecka, University of Giessen, GermanyCecilia Garlanda, Humanitas University, Italy
Ronald Paul Taylor, University of Virginia, United States
Peter Garred, University of Copenhagen, Denmark
Copyright © 2021 Lipcsey, Persson, Eriksson, Blom, Fromell, Hultström, Huber-Lang, Ekdahl, Frithiof and Nilsson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Nilsson, Ym8ubmlsc3NvbkBpZ3AudXUuc2U=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship